 Rajani M. George
Rajani M. George Anthony B. Firulli
Anthony B. Firulli- Herman B Wells Center for Pediatric Research Department of Pediatrics, Anatomy, Biochemistry, and Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN, United States
Epigenetic control of gene expression during cardiac development and disease has been a topic of intense research in recent years. Advances in experimental methods to study DNA accessibility, transcription factor occupancy, and chromatin conformation capture technologies have helped identify regions of chromatin structure that play a role in regulating access of transcription factors to the promoter elements of genes, thereby modulating expression. These chromatin structures facilitate enhancer contacts across large genomic distances and function to insulate genes from cis-regulatory elements that lie outside the boundaries for the gene of interest. Changes in transcription factor occupancy due to changes in chromatin accessibility have been implicated in congenital heart disease. However, the factors controlling this process and their role in changing gene expression during development or disease remain unclear. In this review, we focus on recent advances in the understanding of epigenetic factors controlling cardiac morphogenesis and their role in diseases.
Introduction
The heart is the first functional organ to develop during embryogenesis (Christoffels and Jensen, 2020). The earliest cardiac progenitors are specified from the emerging mesoderm during gastrulation and form the cardiac crescent. Cardiac progenitor cells (CPCs) within the cardiac crescent come together and are distinguished into two populations of progenitor cells: the primary and secondary heart fields (Kelly et al., 2014). CPCs expressing transcription factors (TFs) Nkx2-5, Gata4, and Tbx5 coalesce at midline and form a linear tube that circulates blood in the developing embryo (Bruneau, 2013). As cells within the primary heart field proliferate to form the left ventricle, progenitor cells from the second heart field, expressing the markers Isl1 and Tbx1, move into the developing heart at the arterial and venous poles and eventually give rise to the right ventricle, outflow tract, and the majority of the interventricular septum (Black, 2007). Differential addition of CPCs from the two poles causes the heart to undergo rightward looping, which juxtaposes the atria and ventricles in their final orientation, and through septation, the heart forms its final four-chambered structure (Sylva et al., 2014). The adult mammalian heart is composed of a multitude of cell types: atrial and ventricular cardiomyocytes (CMs) make up most of the heart volume; endocardial cells line the chambers, the lumen of arteries, and specialized valve cells; smooth muscle cells form the aorta and coronary arteries; the epicardium sheaths the heart; and specialized cells form the cardiac conduction system that drives chamber contraction (Pinto et al., 2016).
Cardiac development requires finely tuned gene expression within these various cell types (Olson, 2006). Gene expression is modulated by TFs that bind to DNA regulatory elements to activate or repress transcription. TFs need to have access to consensus DNA binding sequences to recruit the transcriptional machinery, which then initiates transcription. DNA within the nucleus is highly organized into chromatin, a complex of DNA and proteins. Chromatin organization is dynamic and undergoes changes in its structure from loose and open to tightly condensed and closed. Conformational changes to chromatin modulate gene expression by controlling the TFs’ accessibility to the DNA binding sites (Voss and Hager, 2014). Epigenetic modifications do not alter the DNA sequence itself but can change the chromatin structure to modulate its accessibility (Allfrey et al., 1964). The need to expand our understanding of the role that epigenetic regulation plays in gene expression has led scientists to study the structure of the three-dimensional (3D) genome and the hierarchy of chromatin organization (Yu and Ren, 2017). The study of the epigenome has led to the characterization of DNA base methylation, posttranslational histone modifications, the interactions of long non-coding RNA molecules, and changes in chromatin folding. These epigenetic events allow (or block) trans-acting factors to interact with specific cis-elements located within transcriptional enhancers, thus fine-tuning gene expression during development. Studies examining epigenetic changes have given new insight into gene expression changes that take place during disease conditions such as dilated cardiomyopathy. Recent work has also shed more light on diseases referred to as cohesinopathies, which are caused by disruptions to cohesin, a protein complex that helps form the 3D chromatin structure. In this review, we give an overview of the various mechanisms of epigenetic regulation and recent developments examining regulatory mechanisms in the context of cardiac development and disease.
Mechanisms of Epigenetic Regulation of Gene Expression
DNA Methylation
DNA methylation is an early epigenomic change, occurring during DNA replication to mark the daughter strand (Riggs, 1975; Bird, 1978). Methylation of DNA, specifically at the fifth carbon of the cytosine base, occurs on CpG dinucleotides and, in the context of epigenetic regulation, most commonly leads to transcriptional repression (Greenberg and Bourc’his, 2019). Analysis of DNA methylation status during mouse cardiac development (comparing E11.5 to E14.5) shows >50% change in methylation status within a subset of genes involved in heart development and cardiac tissue growth (Chamberlain et al., 2014). When Dnmt3b (DNA methyltransferase 3b), an enzyme that catalyzes transfer of methyl groups to CpG motifs, is deleted within the endocardium using Nfatc1Cre (Wu et al., 2011), qRT-PCR analysis at E11.5 and E14.5 reveals significantly increased levels of Has3 (hyaluronan synthase 3). Has3 is an extracellular matrix remodeling enzyme and, in the context of heart development, is required for endothelial-to-mesenchymal transition and valve formation, therefore suggesting a link between the HAS3 methylation status and regulation during cardiac development (Chamberlain et al., 2014). Knockdown of another member of the same family of DNA methyltransferases, Dnmt3a, using siRNA in mouse embryonic CMs resulted in an observed decrease in beating frequency, defective contractile movement, and disrupted sarcomere assembly (Fang et al., 2016). RNA-seq and methylome analyses identified increased expression and associated decreased CpG methylation at promoters of the following CM structural genes: Myh7, Myh7b, Tnni3, and Tnnt2 (Fang et al., 2016). The importance of CpG methylation in regulating the switch between fetal and adult CM gene expression program is further illustrated by the conditional deletion of Dnmt3a/b in CMs using the Mlc2a promoter to drive Cre recombinase expression (Gilsbach et al., 2014). MethylC-seq analysis in these mutant CMs reveal reduced postnatal de novo methylation of fetal Troponin1 (Tnni1), which partially relieves repression of Tnni1 (Gilsbach et al., 2014). CpG methylation and gene expression analysis of purified embryonic human CMs from fetal (16–24 weeks of pregnancy), infant (1–12 months), and adult (46–60 years) patient samples show dynamic changes in CpG profile and genomic regions that exhibit low levels of CpG-marked enhancers or silencers that lie in cis with genes involved in CM maturation, reflecting the change in their mitotic ability through development (Gilsbach et al., 2018). Work by Li et al. (2016) examining another family of CpG modifiers, Tet1/2/3, which facilitates demethylation by oxidizing CpG residues, in mouse embryonic stem cells (mESCs) shows that loss of Tet3 alone or Tet1, Tet2, and Tet3 triple knockout leads to improper adoption of cardiac mesodermal fate at the expense of neural cell fate as determined by qRT-PCR analysis of neuronal markers Sox1 and Foxg1 and CM markers Nkx2-5, Myh6, Myh7, and Tnnt2.
In human disease conditions, variability in methylome status is observed (Movassagh et al., 2011; Haas et al., 2013). Using the Illumina 450K methylation assay on tissues from dilated cardiomyopathy patients reveals significant changes in the methylation status of cardiac disease-associated genes such as NPPA and NPPB, with methylation status validation by MassARRAY (Meder et al., 2017). Analysis of non-failing donor human hearts and cardiac patients for DNA methylation signature using array-based Illumina Infinium HumanMethylation450 BeadChips reveals 168 differentially methylated CpG loci in atrial and ventricular heart tissues, with 24 of these loci in predicted human heart-specific enhancers (Hoff et al., 2019). In human patients with ischemic cardiomyopathy due to coronary heart disease, methylome analysis using Illumina Infinium HumanMethylation450 BeadChips shows significant increase in CpG methylation and transcriptional repression of the citric acid (TCA) cycle and respiratory electron transport gene network (Pepin et al., 2019). Authors identify KLF15, a Krüppel-like factor, as a target for EZH2 that facilitates a metabolic reprogramming as shown by increased CpG methylation, EZH2 binding at the KLF15 promoter, and KLF15-mediated suppression of key oxidative metabolic genes (Pepin et al., 2019). Thus, CpG methylation is established as a key epigenetic signature in CMs that distinguishes their gene expression and function including an important role for CpG methylation in the metabolic switch that occurs in human heart CMs during failure.
Chromatin Remodelers
The 2 m of naked DNA found inside a 5-μm mammalian nucleus would be highly suspectable to breaks and damage if it was not packaged into an organized structure that allows for reliable replication, transcription, and repair (Felsenfeld and Groudine, 2003). Chromatin is a complex of negatively charged DNA, which weakly interacts with positively charged residues found in histone proteins that allow for the tight and safe packaging of genetic information within the nucleus (McGhee and Felsenfeld, 1980). The functional unit of chromatin is the nucleosome, which is formed by a core of histone proteins H2A, H2B, H3, and H4, around which DNAs (147 base pairs) are wrapped (Kornberg, 1974). These core histone proteins are subject to various posttranslational modifications, including acetylation, methylation, phosphorylation, and ubiquitination at specific residues within the amino-terminal histone tails or within the globular/core domains of histones (Clapier and Cairns, 2009). Various combinations of these posttranslational modifications make up the epigenetic code or histone code that marks regions of open or closed chromatin (Karlic et al., 2010). Transcriptionally active (open) regions of chromatin contain high levels of histone 3 (H3) monoacetylated (ac) at Lys-9 and Lys-14 (H3K9ac and H3K14ac). Trimethylated (me3) Lys-4 (H3K4me3) is present within promoter regions. Other modifications, such as dimethylated (me2) Lys-79 (H3K79me2) and trimethylated Lys-36 (H3K36me3), mark transcriptionally active coding regions. Repressive epigenetic signals are regions of deacetylation and histone H3 trimethylation of Lys-9 (H3K9me3) and Lys-27 (H3K27me3) (Barski et al., 2007; Ernst et al., 2011). Enhancer sequences (evolutionarily conserved non-coding regions of DNA that TFs bind to) are required to drive gene expression. These enhancer regions exhibit unique epigenetic signals as well as increased H3K27me3 and lack of H3K27ac, which poises them for gene activation. The H3K27ac modification is indicative of active enhancers (Rada-Iglesias et al., 2011). These epigenetic marks have been employed to help identify novel enhancers involved in cardiac development (Nord et al., 2013).
Histone marks alone are not always a predictor for enhancer activity; a comparison of chromatin immunoprecipitation using next-generation sequencing (ChIP-seq) profiles of cardiac-specific TFs (GATA4, MEF2A, MEF2C, NKX2-5, SRF, TBX5, and TEAD1) showed that only 16% of regions with TF occupancy overlapped with H3K27ac chromatin marks within fetal cardiac tissues (Akerberg et al., 2019). Analysis of chromatin accessibility using ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) revealed that multiple TF binding regions strongly corelated with the ATAC-seq signal; 100% of regions binding all seven TFs have a strong ATAC-seq signal (Akerberg et al., 2019). These genome-wide screening experiments examining chromatin accessibility and the associated histone code in CMs show the importance of epigenetic remodeling, driving TF access to reprogram the CM transcriptome during the transition from fetal to adult stages. Experiments seeking to correlate the differences in the distribution histone modifications and gene expression were performed by first subjecting adult mice to transverse aortic constriction inducing cardiac hypertrophy and then subsequently heart failure, followed by screening of the diseased hearts for changes in seven different histone modifications and gene expression (Papait et al., 2013). Of 1,109 differentially regulated genes, 596 exhibit at least one altered histone modification at their promoter and 325 genes observed an upregulation or downregulation of gene expression coincident with the histone change (Papait et al., 2013).
A well-studied epigenetic modifier during heart development, EP300, is a histone acetyltransferase (HAT) that acetylates H3 on lysine 27 (H3K27ac) and binds within the promoter regions of a number of critical heart genes such as Gata4, Nkx2-5, and Mef2c to activate the transcription of these cardiac TFs (Takaya et al., 2008; Sun et al., 2010; Trivedi et al., 2010). Indeed, a point mutation in EP300 leads to atrial septal defects and ventricular septal defects (Shikama et al., 2003). Conditional inactivation of a subunit (Ezh2) of PRC2, which establishes the chromatin mark H3K27me3, using Nkx2-5Cre (Ezh2fl/fl Nkx2-5Cre) causes perinatal lethality with only 2% of pups surviving to postnatal day 20 (He et al., 2012). In this study, no significant apoptosis is detected via terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL); however, CM proliferation as measured by phosphorylated histone H3 immunohistochemistry is twofold reduced in mutant CMs at E16.5. Transcriptomic analysis via qRT-PCR and RNA-seq indicates that Pax6, Isl1, and Six1, genes that are expressed in early cardiac progenitors and downregulated in differentiated CMs, are significantly overexpressed in E12.5 mutant ventricles, suggesting that effective cardiac progenitor differentiation to CMs requires repressive H3K27me3 activity (He et al., 2012).
Another class of proteins regulate chromatin by non-covalent enzymatic activity. The SWI/SNF complexes consist of a core ATPase that utilizes energy from ATP hydrolysis to modify chromatin by changing nucleosome DNA contacts, moving nucleosomes along the DNA, and removing or exchanging the DNA from nucleosomes (Ho and Crabtree, 2010; Hargreaves and Crabtree, 2011). Early cardiac development requires finely tuned epigenetic regulation by these complexes as demonstrated by ectopic cardiogenesis when BAF60C (SMARCD3), a cardiac-specific ATP-dependent chromatin remodeling protein, and GATA4 and TBX5 are expressed in the non-cardiogenic mouse embryo mesoderm (Lickert et al., 2004; Takeuchi and Bruneau, 2009). RNA-seq analysis of hearts of mice, where Baf60c is conditionally knocked out from CMs using Myh6-Cre, showed mis-regulation of structural CM proteins, not TFs involved in cardiac development, possibly mediated by the direct interaction between myocardin (MYOCD) and BAF60C (Sun et al., 2018). Expression of the core ATPase subunit of the SWI/SNF complex, Brg1, is required in CMs for their maturation, as determined by isoform switching of αMHC and βMHC in the mouse myocardium (Hang et al., 2010), as well as in the endocardium to drive trabeculation (Stankunas et al., 2008). Knockout of Arid2/BAF200 leads to embryonic lethality by E12.5–E14.5 due to improper myocardial development leading to thinning of ventricular walls and improper coronary formation (He et al., 2014). This family of chromatin remodelers is also implicated in disease, covered in further detail in section “Cardiac Diseases and the Epigenome”.
Topologically Associating Domain (TAD)
Work published concurrently in 2012 used chromatin conformation capture in mammalian cells to identify large loops at the megabase scale: TADs (Dixon et al., 2012; Nora et al., 2012; Figure 1). TADs are not defined by a specific chromatin state alone but by the increased frequency of DNA interactions within a genomic region. Chromatin conformation capture experiments indirectly measure these contact frequencies at the whole-genome level using next-generation sequencing (Kempfer and Pombo, 2020). Live and single-molecule imaging at TADs shows that these structures are dynamic (Hansen et al., 2017). The generally accepted model for TAD formation is loop extrusion, where cohesin protein complexes are loaded and extrude chromatin into progressively larger loops until they either dissociate from chromatin, bump into each other, or run into insulator molecules such as the zinc finger CCCTC binding factor (CTCF) (Alipour and Marko, 2012; Sanborn et al., 2015; Fudenberg et al., 2016). These loops allow genomic sites that lie far apart in the linear genome to come into close spatial proximity to each other (Figure 1). Transcriptionally active and inactive TADs are organized in compartments A and B, respectively, which have different folding and compartmentalization configurations (Lieberman-Aiden et al., 2009; Wang et al., 2016). TAD compartment organization overlaps with histone modification: compartment A is enriched in markers for active chromatin (H3K27ac, H3K4me1/me3, H3K9me1, and the repressive H3K27me3 mark), while compartment B is enriched in the heterochromatin mark H3K9me3 (Rao et al., 2014). The mechanisms of TAD looping, their role in regulating gene expression, and their biological relevance remain unclear.
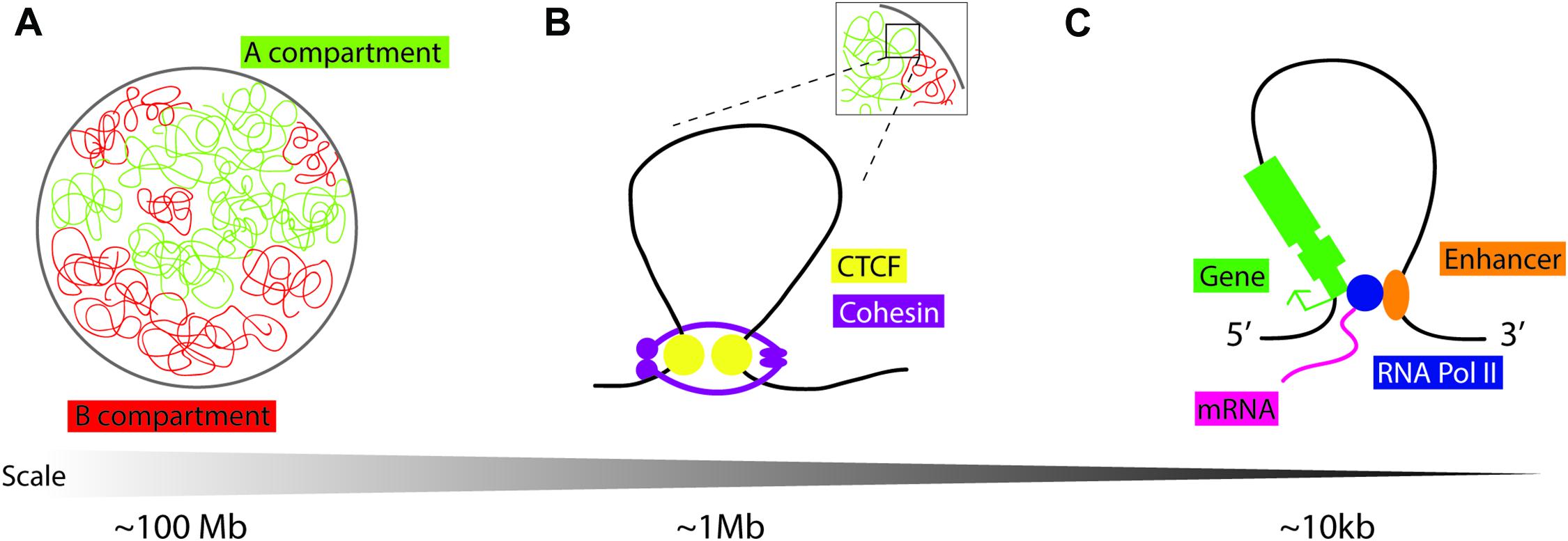
Figure 1. Schematic representation of chromatin loops. (A) Chromatin in the nucleus is distinguished into two compartments on the basis of histone modifications and transcriptional activity: compartment A is “open/active,” compartment B is “closed/repressed.” (B) Topologically associating domains (TADs) are dynamic chromatin structures that are extruded by cohesin, a protein complex consisting of SMC1, SMC3, RAD21, and STAG1/2. Loop extrusion is stabilized by the boundary molecule CTCF. (C) “Open” chromatin is permissive to loop formation to enable cis-enhancer binding to RNA Pol II and interaction with the gene promoter to facilitate tissue and temporally specific transcription.
Disruption of TADs during development is implicated in ectopic gene expression and disease (Ibn-Salem et al., 2014; Lupianez et al., 2015, 2016; Muro et al., 2019). Comparison of genome architecture in primate, mouse, cattle, opossum, chicken, clawed frog, and zebrafish showed conservation of non-coding regions that coincide with TAD boundaries (Harmston et al., 2017; Krefting et al., 2018). However, the extent of TAD conservation across species and its functional significance are still unclear (Eres and Gilad, 2020). Although there is some variation in reports, mainly due to differences in resolution arising from experimental limitations and the types of computing tools used to generate models, the average size of a TAD is approximately 1 Mb (Dali and Blanchette, 2017; Zufferey et al., 2018).
CTCF protein is enriched at the TAD boundaries with pairs of CTCF DNA binding sites preferentially found in a convergent orientation, allowing the CTCF protein to act as a domain boundary molecule (Rao et al., 2014; Grubert et al., 2020; Figure 1). To study the function of CTCF in mediating TAD formation, researchers may employ an in vitro inducible knockout system, using the auxin-inducible degron (AID) tag with an eGFP cassette at the 3′ end of the CTCF coding sequence (Morawska and Ulrich, 2013). Cells are also transfected with Tir1 F-box from Oryza sativa, which can bind to the AID tag in the presence of auxin, triggering targeted proteasome-dependent degradation of CTCF. Thus, adding auxin to cell culture media depletes CTCF protein to levels undetectable by western blot, and removal of auxin from growth media results in recovery of CTCF protein. The Bruneau lab targeted CTCF in mESCs by using this system, reporting that higher-order chromosome folding, i.e., compartments A and B, remain intact, as determined by Hi-C contact frequency mapping, with a limited effect on transcriptional state across the genome (Nora et al., 2017). Genes most affected by CTCF depletion tend to have enhancers/promoter regions in close proximity to CTCF sites. Reversal of CTCF depletion, by removal of auxin from growth media, leads to the re-formation of TADs (Nora et al., 2017), although the caveat to this result is that low levels of background CTCF protein can also facilitate TAD formation. More recently, the Bruneau lab has also uncovered the molecular basis for convergent CTCF motif orientation at TAD boundaries; the N-terminal portion of CTCF is responsible for its stabilization at one side of non-palindromic CTCF DNA binding sites during Cohesin-driven loop extrusion (Nora et al., 2020). In vivo knockout experiments have provided further information on the role of CTCF in TAD formation and maintenance.
In the mouse embryonic heart, conditional deletion of CTCF using Nkx2-5Cre leads to embryonic lethality by embryonic day (E)12.5 and myocardial thinning (Gomez-Velazquez et al., 2017). Conditional knockout embryos are phenotypically normal until E9.5, but by E10.5, the interventricular septum appears disorganized and progressively worsens in E11.5 embryos. The four chambers and atrioventricular canal form normally. No difference in apoptosis (TUNEL) or proliferation (phosphorylated histone H3) is observed. Transcriptomic analysis using RNA-seq of Ctcffl/fl;Nkx2-5Cre embryos at E10.5 showed limited change in gene expression with genes being involved with mitochondrial function as the largest functional group showing differences (Gomez-Velazquez et al., 2017). Authors then go on to show changes in gene expression (in situ hybridization) and chromatin structure (4C-seq) at the Iroquois (Irx) gene cluster and adjoining genes, including a mitochondrial subunit gene Ndufs6. Mitochondria in Ctcffl/fl;Nkx2-5Cre E11.5 CMs appear swollen and disorganized (Gomez-Velazquez et al., 2017). Work from the Vondriska lab employs a α-Myosin Heavy chain tamoxifen-inducible Cre recombinase (MerCreMer) mouse to generate CTCF conditional knockout (CTCF-CKO) in adult CMs (Rosa-Garrido et al., 2017). Depletion of CTCF levels after tamoxifen treatment led to a decrease in survivability, with impaired ejection fraction, left ventricular chamber dilation, and muscle hypertrophy at the organ and cell levels, with 100% mortality observed in conditional knockout mice 7 weeks after tamoxifen administration. Surprisingly, Hi-C analysis of hearts from these CTCF-CKO mice shows little change (<2%) in TAD boundaries and A/B compartmentalization as compared to controls. However, Fit Hi-C, a method to analyze genome-wide chromosomal contacts that are statistically significant, revealed that interactions and accessibility at a large number of enhancer regions are changed in CTCF-CKO mice: 4,037 increase/decrease in contact compared to 1,013 unchanged. The genes in the surrounding chromosomal regions are enriched for cardiac pathology pathways (Rosa-Garrido et al., 2017).
Taken together, these experiments suggest that the preexisting chromatin landscape, which has existed before the loss of TADs, is sufficient to retain the DNA-TF accessibility even when the TADs are lost. TADs and their associated CTCF sites alone might not be essential for correct developmental gene expression; however, they can cause misexpression when redirected with different loci being more or less sensitive to these changes, leading to embryonic lethality in the mouse models tested.
An important facilitator of TAD formation is the four-subunit protein complex, cohesin, well studied for its role in chromatid formation and chromosome segregation during mitosis (Lica et al., 1986; Cooke et al., 1987). The cohesin complex forms a ring-like structure and consists of core proteins: structural maintenance of chromosomes (SMC)1, SMC3, RAD21, and stromal antigen (STAG)1/2. Auxin-induced degradation of the cohesin subunit RAD21 in human cancer cell lines leads to a loss of all TADs, but transcriptional activity remains largely unchanged (Rao et al., 2017). Another subunit of the cohesin core complex, STAG2, is required for heart morphogenesis; Stag2-null embryos die by E10.5 and exhibit observable heart abnormalities by E9.5 (De Koninck et al., 2020). Histological analysis of Stag2 knockout embryos at E9.5 shows significantly smaller right ventricles and shorter outflow tracts, with decreased anti-phosphohistone H3 staining associated with reduced CM proliferation when compared to controls (De Koninck et al., 2020). Based on observed right ventricle and outflow tract defects, authors examined SHF progenitor populations in Stag2-knockout embryos, and RNA-seq analysis indicated downregulation of important SHF regulators, Fgf8, Hand2, and Wnt5a (De Koninck et al., 2020). Further work is required to understand the molecular function of the cohesin complex of proteins, their contribution to TAD formation, and their role in cardiac disease.
Non-coding RNAs
Long non-coding RNAs (lncRNAs) are greater than 200 nucleotides (nt) in length and function by binding DNA, other RNAs, or RNA binding proteins or can contain their own transcriptional start site to make micropeptides (<100 amino acids) (Ulitsky and Bartel, 2013). There are also reports of lncRNAs interacting with established chromatin modifiers (Mishra and Kanduri, 2019). The first instance of this is HOTAIR (HOX transcript antisense intergenic RNA), which is transcribed from the human HOXC locus to trans regulate transcriptional silencing of the HOXD locus in a tissue-dependent manner by direct interaction with the histone methyltransferase PRC2 (Rinn et al., 2007). Further experiments reveal that 20% of human lncRNAs are associated with PRC2 (Khalil et al., 2009), including the well-characterized lncRNAs Xist, RepA, Kcnq1ot1, Braveheart, and Malat-1, suggesting that lncRNAs serve to modulate a scanning of the genome for genes that require silencing (Davidovich and Cech, 2015), which could be facilitated by PRC2 binding nascent RNA (Beltran et al., 2016). The biological relevance of lncRNA-mediated chromatin modulation has also been demonstrated by mis-expression of lncRNAs in human cancer (Begolli et al., 2019).
Perhaps more interesting are a number of studies that indicate that lncRNAs directly interact with the zinc finger TF CTCF and that these RNA–CTCF interactions are required for some TADs to form (Saldana-Meyer et al., 2014; Kung et al., 2015; Hansen et al., 2019). Work from the Reinberg lab generated a CTCF mutant protein that retains the DNA binding capability but has decreased RNA binding by deleting 14 amino acids from zinc finger 1 and nine amino acids from zinc finger 10 (Saldana-Meyer et al., 2019). Using mESCs with an AID system (Nora et al., 2017) to knock out CTCF and express the RNA binding-deficient CTCF mutant protein, single-cell RNA-seq experiments along with Hi-C analysis identified only modest changes in both gene expression and 3D chromatin structure within CTCF RNA binding-deficient cell lines, with 60% of TF sites exhibiting decreased CTCF binding within gene promoters and no changes in A/B compartmentalization (Saldana-Meyer et al., 2019). The Tjian lab addressed the same question by deleting the internal RNA binding region (RBRi) of CTCF protein in endogenous mESC lines (Hansen et al., 2019). RNA-seq analysis indicates that RBRi deleted cell lines show modest gene expression changes (∼500 genes mis-regulated with an average fold change of 2.7) and no change in A/B compartmentalization similar to what was seen by Saldana-Meyer et al. Chromatin conformation capture experiments find that almost half of all CTCF loops are lost in RBRi deletion mutants, suggesting that there are two kinds of CTCF-mediated loops: RBRi dependent and RBRi independent. Even with modest gene expression changes in RBRi deletion mutants, some TAD formation requires RNA binding with CTCF (Hansen et al., 2019). Although these direct RNA–CTCF interactions are still controversial in the field, it is possible that these interactions might explain how lncRNAs help modulate gene expression.
In the context of cardiac development, a specific role of lncRNAs has been uncovered (Martens et al., 2017; Hobuss et al., 2019). The Myh-associated RNA transcripts, or Myheart (Mhrt), are alternatively spliced lncRNA transcripts that lie within the myosin gene locus (Han et al., 2014). This well-characterized lncRNA is downregulated in hearts pressure-overloaded by transaortic constriction (TAC). Overexpression of Mhrt led to cardioprotective effects with minimal/absent fibrosis, improvement in fractional shortening, normalized left ventricle size, and reduced change in Nppa expression. Luciferase assays and ChIP experiments show that the Mhrt promoter is directly regulated by the chromatin remodeling factor Brg1, the ATPase subunit of SWI2/SNF2-like chromatin-remodeling complexes. RNA immunoprecipitation experiments demonstrate that Mhrt directly binds to BRG1 to reduce its occupancy at target genes (Han et al., 2014).
Recent work from the Bruneau lab employed RNA-seq reads of mESCs differentiated into CMs to screen for lncRNAs on the basis of their epigenetic regulation, clear splice structure, homology to human and/or mammalian genomes, cardiac progenitor specificity, and expression in the developing embryo (George et al., 2019). Six novel lncRNAs were identified: Rubie, Handlr, Atcayos, HrtLincR4, HrtLincR5, and HrtLincRX. Knockout mouse lines were generated using CRISPR/Cas9 genome editing and assayed for loss of expression or cardiac phenotypes. Authors report that none of the tested lncRNAs are required for viable mouse development, suggesting a lack of function or that, within the context of cardiac development, further understanding of lncRNA biology would require manipulation of additional molecular compensatory mechanisms (George et al., 2019).
Short (<22-nt) single-stranded non-coding RNA molecules known as microRNAs (miRNAs) have also been implicated in epigenomic regulation (Yao et al., 2019). The role that miRNAs play in cardiovascular diseases has been extensively covered by recent reviews (Colpaert and Calore, 2021).
Cardiac Diseases and the Epigenome
Epigenetic changes have been identified as a causative agent for disease in multiple organ systems and cell types (Zoghbi and Beaudet, 2016). Germline mutations in genes encoding part of the cohesin complex and its regulatory factors are collectively referred to as cohesinopathies (Piche et al., 2019). The most common of these is the Cornelia de Lange syndrome (CdLS, OMIM 122470) where patients present with growth retardation, intellectual disability, and facial dysmorphism (Sarogni et al., 2020). Of the CdLS patients, 14–70% also present with congenital heart defects (Chatfield et al., 2012). Sixty percent of patients carry heterozygous mutations in NIPBL, a protein that loads cohesin onto chromatin (Liu et al., 2009). Inducible pluripotent stem cells (iPSCs) derived from CdLS patients were differentiated to CMs, and RNA-seq analysis identified altered gene expression in several critical cardiac development genes: GATA4/6, MYH6/7, MYH7, ACTN2, HAND2, TBX1/5, and TDGF1, within the NIPBL haploinsufficient samples as compared to control patient samples (Mills et al., 2018).
The DiGeorge syndrome, a 1.5- to 3.0-Mb heterozygous deletion of chromosome 22q11 (OMIM 188400), causes congenital heart defects and is linked to TBX1 haploinsufficiency (Lindsay et al., 1999). Tbx1 is expressed in the SHF, and patients with this disease show outflow tract defects and persistent truncus arteriosus (lack of septation between the aorta and pulmonary trunk) (Du et al., 2019). TBX1 regulates chromatin by interacting with various epigenetic modifiers: the BAF60A/SMARCD1 subunits, the Setd7 histone H3K4 monomethyltransferase, and the histone demethylase LSD1 (Chen et al., 2012; Fulcoli et al., 2016). The importance of epigenetic regulation in this disease is demonstrated by treating pregnant mice with a histone demethylase inhibitor, thus increasing levels of methylated H3K4, which partially rescues the cardiovascular anomalies in Tbx1+/KO embryos (Fulcoli et al., 2016).
Children with Down syndrome (trisomy 21; OMIM 190685) present with a higher-than-normal incidence rate (>50%) of ventricular septal and atrial septal defects (Antonarakis, 2017). Transcriptomic analysis of 45 trisomy 21 patient samples identified significant misexpression of 247 genes not located on chromosome 21, compared to only 77 genes dysregulated that are located on chromosome 21 (Vilardell et al., 2011). Using patient samples from a pair of monozygotic twins, one of which had trisomy 21, Letourneau et al. (2014) performed transcriptomic analysis and derived iPSC cell lines. Domains of dysregulated genes are identified throughout the genome that overlaps with nuclear lamina associating regions of low gene expression, suggesting that chromatin modulation might be responsible for gene mis-regulation in this syndrome (Letourneau et al., 2014). However, it is still unclear how cardiac defects arise from these global chromatin landscape changes.
CHD7, a chromatin remodeling factor that is a member of the chromodomain helicase DNA-binding family of ATP-dependent chromatin remodeling enzymes, is mutated in the CHARGE syndrome (coloboma of the eye, heart defects, atresia of the choanae, severe retardation of growth/development, genital abnormalities, and ear abnormalities; OMIM 214800) (Vissers et al., 2004). Nonsense and frameshift indel mutations in CHD7 occur de novo, resulting in the generation of a loss-of-function protein (Basson and van Ravenswaaij-Arts, 2015). Seventy-five percent of patients present with a congenital heart defect (Lalani et al., 2006). ChIP-qPCR analysis on NkL-Tag, a mouse cardiac cell line, indicates that CHD7 binds directly to Nkx2-5 enhancers in vitro (Liu et al., 2014). Further ChIP analysis demonstrates that recruitment to Nkx2-5 enhancers is mediated via the CHD7 interaction with SMAD1 downstream of BMP2 signaling (Liu et al., 2014).
Other than congenital cardiac disease, postnatal epigenetic changes are also correlated with gene expression changes leading to cardiovascular aging, a complex process characterized by decreased heart function and ventricular and atrial remodeling (Zhang et al., 2018). These epigenetic changes are thought to be induced by changes in reactive oxygen species (ROS) or metabolite levels (Etchegaray and Mostoslavsky, 2016). Myocardial infarction or pressure overload conditions in the heart result in fibroblast activation and inflammation, leading to cardiac fibrosis, which causes changes in epigenetic regulation (Felisbino and McKinsey, 2018). We have reviewed these changes in earlier relevant sections, but the etiology of how these epigenetic changes lead to cardiac dysfunction requires further study.
Conclusion
Extensive work in recent years has uncovered some of the molecular mechanisms which control chromatin regulation and disease conditions arising from epigenetic mis-regulation. The conserved nature of chromatin domains and the regulatory mechanisms controlling their establishment and maintenance would suggest that changes to chromatin landscape would lead to dramatic changes in gene expression. However, for the most part, this has not been the case, suggesting that in an epigenetic context, overlapping layers of modifications regulate transcription. Although our understanding of chromatin biology has vastly advanced in recent years with chromatin conformation capture technologies, much remains unclear about what the functional role is of these domains in regulating enhancer accessibility or enabling/repressing transcription. In adult disease conditions, epigenetic changes play a more complex role with a large variability of phenotypes between patients, further confounding the analysis of causative factors. Alterations to epigenetic factors that correlate with disease suggest that these aberrant proteins undergo changes in subunit function and lack biological redundancies. Interestingly, cardiomyopathy phenotypes in human patients appear to be sensitive to these epigenetic aberrations, suggesting specificity in both tissue and type of epigenetic factor expressed during heart development. Refining our understanding of the epigenetic mechanisms at play in cardiac development is required to parse out these phenotypes.
Author Contributions
Both authors conceptualized, wrote, and edited the manuscript.
Funding
Infrastructural support to the Herman B Wells Center for Pediatric Research is in part supported by grants from the National Institutes of Health (P01 HL134599-04 and R01 HL145060-02 to AF), the American Heart Association (19POST34381038 to RG), and the Riley Children’s Foundation, Division of Pediatric Cardiology and the Carleton Buehl McCulloch Chair of Pediatrics.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all the scientists whose work has contributed to the understanding of chromatin biology. We apologize to researchers whose work we could not discuss here due to space limitations. We also thank Beth Firulli for their critical reading of the manuscript.
References
Akerberg, B. N., Gu, F., VanDusen, N. J., Zhang, X., Dong, R., Li, K., et al. (2019). A reference map of murine cardiac transcription factor chromatin occupancy identifies dynamic and conserved enhancers. Nat. Commun. 10:4907. doi: 10.1038/s41467-019-12812-3
Alipour, E., and Marko, J. F. (2012). Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Res. 40, 11202–11212. doi: 10.1093/nar/gks925
Allfrey, V. G., Faulkner, R., and Mirsky, A. E. (1964). Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc. Natl. Acad. Sci. U S A. 51, 786–794. doi: 10.1073/pnas.51.5.786
Antonarakis, S. E. (2017). Down syndrome and the complexity of genome dosage imbalance. Nat. Rev. Genet. 18, 147–163. doi: 10.1038/nrg.2016.154
Barski, A., Cuddapah, S., Cui, K., Roh, T. Y., Schones, D. E., Wang, Z., et al. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. doi: 10.1016/j.cell.2007.05.009
Basson, M. A., and van Ravenswaaij-Arts, C. (2015). Functional Insights into Chromatin Remodelling from Studies on CHARGE Syndrome. Trends Genet. 31, 600–611. doi: 10.1016/j.tig.2015.05.009
Begolli, R., Sideris, N., and Giakountis, A. (2019). LncRNAs as Chromatin Regulators in Cancer: From Molecular Function to Clinical Potential. Cancers 11:11101524. doi: 10.3390/cancers11101524
Beltran, M., Yates, C. M., Skalska, L., Dawson, M., Reis, F. P., Viiri, K., et al. (2016). The interaction of PRC2 with RNA or chromatin is mutually antagonistic. Genome Res. 26, 896–907. doi: 10.1101/gr.197632.115
Bird, A. P. (1978). Use of restriction enzymes to study eukaryotic DNA methylation: II. The symmetry of methylated sites supports semi-conservative copying of the methylation pattern. J. Mol. Biol. 118, 49–60. doi: 10.1016/0022-2836(78)90243-7
Black, B. L. (2007). Transcriptional pathways in second heart field development. Semin. Cell Dev. Biol. 18, 67–76. doi: 10.1016/j.semcdb.2007.01.001
Bruneau, B. G. (2013). Signaling and transcriptional networks in heart development and regeneration. Cold Spring Harb. Perspect. Biol. 5:a008292. doi: 10.1101/cshperspect.a008292
Chamberlain, A. A., Lin, M., Lister, R. L., Maslov, A. A., Wang, Y., Suzuki, M., et al. (2014). DNA methylation is developmentally regulated for genes essential for cardiogenesis. J. Am. Heart. Assoc. 3:e000976. doi: 10.1161/JAHA.114.000976
Chatfield, K. C., Schrier, S. A., Li, J., Clark, D., Kaur, M., Kline, A. D., et al. (2012). Congenital heart disease in Cornelia de Lange syndrome: phenotype and genotype analysis. Am. J. Med. Genet. A 158A, 2499–2505. doi: 10.1002/ajmg.a.35582
Chen, L., Fulcoli, F. G., Ferrentino, R., Martucciello, S., Illingworth, E. A., and Baldini, A. (2012). Transcriptional control in cardiac progenitors: Tbx1 interacts with the BAF chromatin remodeling complex and regulates Wnt5a. PLoS Genet. 8:e1002571. doi: 10.1371/journal.pgen.1002571
Christoffels, V., and Jensen, B. (2020). Cardiac Morphogenesis: Specification of the Four-Chambered Heart. Cold Spring Harb. Perspect. Biol. 12:37143. doi: 10.1101/cshperspect.a037143
Clapier, C. R., and Cairns, B. R. (2009). The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 78, 273–304. doi: 10.1146/annurev.biochem.77.062706.153223
Colpaert, R. M. W., and Calore, M. (2021). Epigenetics and microRNAs in cardiovascular diseases. Genomics 113, 540–551. doi: 10.1016/j.ygeno.2020.12.042
Cooke, C. A., Heck, M. M., and Earnshaw, W. C. (1987). The inner centromere protein (INCENP) antigens: movement from inner centromere to midbody during mitosis. J. Cell Biol. 105, 2053–2067. doi: 10.1083/jcb.105.5.2053
Dali, R., and Blanchette, M. (2017). A critical assessment of topologically associating domain prediction tools. Nucleic Acids Res. 45, 2994–3005. doi: 10.1093/nar/gkx145
Davidovich, C., and Cech, T. R. (2015). The recruitment of chromatin modifiers by long noncoding RNAs: lessons from PRC2. RNA 21, 2007–2022. doi: 10.1261/rna.053918.115
De Koninck, M., Lapi, E., Badia-Careaga, C., Cossio, I., Gimenez-Llorente, D., Rodriguez-Corsino, M., et al. (2020). Essential Roles of Cohesin STAG2 in Mouse Embryonic Development and Adult Tissue Homeostasis. Cell Rep. 32:108014. doi: 10.1016/j.celrep.2020.108014
Dixon, J. R., Selvaraj, S., Yue, F., Kim, A., Li, Y., Shen, Y., et al. (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. doi: 10.1038/nature11082
Du, Q., de la Morena, M. T., and van Oers, N. S. C. (2019). The Genetics and Epigenetics of 22q11.2 Deletion Syndrome. Front. Genet. 10:1365. doi: 10.3389/fgene.2019.01365
Eres, I. E., and Gilad, Y. (2020). A TAD Skeptic: Is 3D Genome Topology Conserved? Trends Genet. 37, 216–223. doi: 10.1016/j.tig.2020.10.009
Ernst, J., Kheradpour, P., Mikkelsen, T. S., Shoresh, N., Ward, L. D., Epstein, C. B., et al. (2011). Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49. doi: 10.1038/nature09906
Etchegaray, J. P., and Mostoslavsky, R. (2016). Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 62, 695–711. doi: 10.1016/j.molcel.2016.05.029
Fang, X., Poulsen, R. R., Wang-Hu, J., Shi, O., Calvo, N. S., Simmons, C. S., et al. (2016). Knockdown of DNA methyltransferase 3a alters gene expression and inhibits function of embryonic cardiomyocytes. FASEB J. 30, 3238–3255. doi: 10.1096/fj.201600346R
Felisbino, M. B., and McKinsey, T. A. (2018). Epigenetics in Cardiac Fibrosis: Emphasis on Inflammation and Fibroblast Activation. JACC Basic Transl. Sci. 3, 704–715. doi: 10.1016/j.jacbts.2018.05.003
Felsenfeld, G., and Groudine, M. (2003). Controlling the double helix. Nature 421, 448–453. doi: 10.1038/nature01411
Fudenberg, G., Imakaev, M., Lu, C., Goloborodko, A., Abdennur, N., and Mirny, L. A. (2016). Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 15, 2038–2049. doi: 10.1016/j.celrep.2016.04.085
Fulcoli, F. G., Franzese, M., Liu, X., Zhang, Z., Angelini, C., and Baldini, A. (2016). Rebalancing gene haploinsufficiency in vivo by targeting chromatin. Nat. Commun. 7:11688. doi: 10.1038/ncomms11688
George, M. R., Duan, Q., Nagle, A., Kathiriya, I. S., Huang, Y., Rao, K., et al. (2019). Minimal in vivo requirements for developmentally regulated cardiac long intergenic non-coding RNAs. Development 146, 185314. doi: 10.1242/dev.185314
Gilsbach, R., Preissl, S., Gruning, B. A., Schnick, T., Burger, L., Benes, V., et al. (2014). Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat. Commun. 5:5288. doi: 10.1038/ncomms6288
Gilsbach, R., Schwaderer, M., Preissl, S., Gruning, B. A., Kranzhofer, D., Schneider, P., et al. (2018). Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun. 9:391. doi: 10.1038/s41467-017-02762-z
Gomez-Velazquez, M., Badia-Careaga, C., Lechuga-Vieco, A. V., Nieto-Arellano, R., Tena, J. J., Rollan, I., et al. (2017). CTCF counter-regulates cardiomyocyte development and maturation programs in the embryonic heart. PLoS Genet. 13:e1006985. doi: 10.1371/journal.pgen.1006985
Greenberg, M. V. C., and Bourc’his, D. (2019). The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 20, 590–607. doi: 10.1038/s41580-019-0159-6
Grubert, F., Srivas, R., Spacek, D. V., Kasowski, M., Ruiz-Velasco, M., Sinnott-Armstrong, N., et al. (2020). Landscape of cohesin-mediated chromatin loops in the human genome. Nature 583, 737–743. doi: 10.1038/s41586-020-2151-x
Haas, J., Frese, K. S., Park, Y. J., Keller, A., Vogel, B., Lindroth, A. M., et al. (2013). Alterations in cardiac DNA methylation in human dilated cardiomyopathy. EMBO Mol. Med. 5, 413–429. doi: 10.1002/emmm.201201553
Han, P., Li, W., Lin, C. H., Yang, J., Shang, C., Nuernberg, S. T., et al. (2014). A long noncoding RNA protects the heart from pathological hypertrophy. Nature 514, 102–106. doi: 10.1038/nature13596
Hang, C. T., Yang, J., Han, P., Cheng, H. L., Shang, C., Ashley, E., et al. (2010). Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature 466, 62–67. doi: 10.1038/nature09130
Hansen, A. S., Hsieh, T. S., Cattoglio, C., Pustova, I., Saldana-Meyer, R., Reinberg, D., et al. (2019). Distinct Classes of Chromatin Loops Revealed by Deletion of an RNA-Binding Region in CTCF. Mol. Cell 76:39. e313. doi: 10.1016/j.molcel.2019.07.039
Hansen, A. S., Pustova, I., Cattoglio, C., Tjian, R., and Darzacq, X. (2017). CTCF and cohesin regulate chromatin loop stability with distinct dynamics. Elife 6:25776. doi: 10.7554/eLife.25776
Hargreaves, D. C., and Crabtree, G. R. (2011). ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 21, 396–420. doi: 10.1038/cr.2011.32
Harmston, N., Ing-Simmons, E., Tan, G., Perry, M., Merkenschlager, M., and Lenhard, B. (2017). Topologically associating domains are ancient features that coincide with Metazoan clusters of extreme noncoding conservation. Nat. Commun. 8:441. doi: 10.1038/s41467-017-00524-5
He, A., Ma, Q., Cao, J., von Gise, A., Zhou, P., Xie, H., et al. (2012). Polycomb repressive complex 2 regulates normal development of the mouse heart. Circ. Res. 110, 406–415. doi: 10.1161/CIRCRESAHA.111.252205
He, L., Tian, X., Zhang, H., Hu, T., Huang, X., Zhang, L., et al. (2014). BAF200 is required for heart morphogenesis and coronary artery development. PLoS One 9:e109493. doi: 10.1371/journal.pone.0109493
Ho, L., and Crabtree, G. R. (2010). Chromatin remodelling during development. Nature 463, 474–484. doi: 10.1038/nature08911
Hobuss, L., Bar, C., and Thum, T. (2019). Long Non-coding RNAs: At the Heart of Cardiac Dysfunction? Front. Physiol. 10:30. doi: 10.3389/fphys.2019.00030
Hoff, K., Lemme, M., Kahlert, A. K., Runde, K., Audain, E., Schuster, D., et al. (2019). DNA methylation profiling allows for characterization of atrial and ventricular cardiac tissues and hiPSC-CMs. Clin. Epigenet. 11:89. doi: 10.1186/s13148-019-0679-0
Ibn-Salem, J., Kohler, S., Love, M. I., Chung, H. R., Huang, N., Hurles, M. E., et al. (2014). Deletions of chromosomal regulatory boundaries are associated with congenital disease. Genome Biol. 15:423. doi: 10.1186/s13059-014-0423-1
Karlic, R., Chung, H. R., Lasserre, J., Vlahovicek, K., and Vingron, M. (2010). Histone modification levels are predictive for gene expression. Proc. Natl. Acad. Sci. U S A. 107, 2926–2931. doi: 10.1073/pnas.0909344107
Kelly, R. G., Buckingham, M. E., and Moorman, A. F. (2014). Heart fields and cardiac morphogenesis. Cold Spring Harb. Perspect. Med. 4:a015750. doi: 10.1101/cshperspect.a015750
Kempfer, R., and Pombo, A. (2020). Methods for mapping 3D chromosome architecture. Nat. Rev. Genet. 21, 207–226. doi: 10.1038/s41576-019-0195-2
Khalil, A. M., Guttman, M., Huarte, M., Garber, M., Raj, A., Rivea Morales, D., et al. (2009). Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. U S A. 106, 11667–11672. doi: 10.1073/pnas.0904715106
Kornberg, R. D. (1974). Chromatin structure: a repeating unit of histones and DNA. Science 184, 868–871. doi: 10.1126/science.184.4139.868
Krefting, J., Andrade-Navarro, M. A., and Ibn-Salem, J. (2018). Evolutionary stability of topologically associating domains is associated with conserved gene regulation. BMC Biol. 16:87. doi: 10.1186/s12915-018-0556-x
Kung, J. T., Kesner, B., An, J. Y., Ahn, J. Y., Cifuentes-Rojas, C., Colognori, D., et al. (2015). Locus-specific targeting to the X chromosome revealed by the RNA interactome of CTCF. Mol. Cell 57, 361–375. doi: 10.1016/j.molcel.2014.12.006
Lalani, S. R., Safiullah, A. M., Fernbach, S. D., Harutyunyan, K. G., Thaller, C., Peterson, L. E., et al. (2006). Spectrum of CHD7 mutations in 110 individuals with CHARGE syndrome and genotype-phenotype correlation. Am. J. Hum. Genet. 78, 303–314. doi: 10.1086/500273
Letourneau, A., Santoni, F. A., Bonilla, X., Sailani, M. R., Gonzalez, D., Kind, J., et al. (2014). Domains of genome-wide gene expression dysregulation in Down’s syndrome. Nature 508, 345–350. doi: 10.1038/nature13200
Li, X., Yue, X., Pastor, W. A., Lin, L., Georges, R., Chavez, L., et al. (2016). Tet proteins influence the balance between neuroectodermal and mesodermal fate choice by inhibiting Wnt signaling. Proc. Natl. Acad. Sci. U S A. 113, E8267–E8276. doi: 10.1073/pnas.1617802113
Lica, L. M., Narayanswami, S., and Hamkalo, B. A. (1986). Mouse satellite DNA, centromere structure, and sister chromatid pairing. J. Cell Biol. 103, 1145–1151. doi: 10.1083/jcb.103.4.1145
Lickert, H., Takeuchi, J. K., Von Both, I., Walls, J. R., McAuliffe, F., Adamson, S. L., et al. (2004). Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature 432, 107–112. doi: 10.1038/nature03071
Lieberman-Aiden, E., van Berkum, N. L., Williams, L., Imakaev, M., Ragoczy, T., Telling, A., et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. doi: 10.1126/science.1181369
Lindsay, E. A., Botta, A., Jurecic, V., Carattini-Rivera, S., Cheah, Y. C., Rosenblatt, H. M., et al. (1999). Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature 401, 379–383. doi: 10.1038/43900
Liu, J., Zhang, Z., Bando, M., Itoh, T., Deardorff, M. A., Clark, D., et al. (2009). Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol. 7:e1000119. doi: 10.1371/journal.pbio.1000119
Liu, Y., Harmelink, C., Peng, Y., Chen, Y., Wang, Q., and Jiao, K. (2014). CHD7 interacts with BMP R-SMADs to epigenetically regulate cardiogenesis in mice. Hum. Mol. Genet. 23, 2145–2156. doi: 10.1093/hmg/ddt610
Lupianez, D. G., Kraft, K., Heinrich, V., Krawitz, P., Brancati, F., Klopocki, E., et al. (2015). Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025. doi: 10.1016/j.cell.2015.04.004
Lupianez, D. G., Spielmann, M., and Mundlos, S. (2016). Breaking TADs: How Alterations of Chromatin Domains Result in Disease. Trends Genet. 32, 225–237. doi: 10.1016/j.tig.2016.01.003
Martens, L., Ruhle, F., and Stoll, M. (2017). LncRNA secondary structure in the cardiovascular system. Noncoding RNA Res. 2, 137–142. doi: 10.1016/j.ncrna.2017.12.001
McGhee, J. D., and Felsenfeld, G. (1980). Nucleosome structure. Annu. Rev. Biochem. 49, 1115–1156. doi: 10.1146/annurev.bi.49.070180.005343
Meder, B., Haas, J., Sedaghat-Hamedani, F., Kayvanpour, E., Frese, K., Lai, A., et al. (2017). Epigenome-Wide Association Study Identifies Cardiac Gene Patterning and a Novel Class of Biomarkers for Heart Failure. Circulation 136, 1528–1544. doi: 10.1161/CIRCULATIONAHA.117.027355
Mills, J. A., Herrera, P. S., Kaur, M., Leo, L., McEldrew, D., Tintos-Hernandez, J. A., et al. (2018). NIPBL(+/-) haploinsufficiency reveals a constellation of transcriptome disruptions in the pluripotent and cardiac states. Sci. Rep. 8:1056. doi: 10.1038/s41598-018-19173-9
Mishra, K., and Kanduri, C. (2019). Understanding Long Noncoding RNA and Chromatin Interactions: What We Know So Far. Noncoding RNA 5:5040054. doi: 10.3390/ncrna5040054
Morawska, M., and Ulrich, H. D. (2013). An expanded tool kit for the auxin-inducible degron system in budding yeast. Yeast 30, 341–351. doi: 10.1002/yea.2967
Movassagh, M., Vujic, A., and Foo, R. (2011). Genome-wide DNA methylation in human heart failure. Epigenomics 3, 103–109. doi: 10.2217/epi.10.70
Muro, E. M., Ibn-Salem, J., and Andrade-Navarro, M. A. (2019). The distributions of protein coding genes within chromatin domains in relation to human disease. Epigenet. Chromatin 12:72. doi: 10.1186/s13072-019-0317-2
Nora, E. P., Caccianini, L., Fudenberg, G., So, K., Kameswaran, V., Nagle, A., et al. (2020). Molecular basis of CTCF binding polarity in genome folding. Nat. Commun. 11:5612. doi: 10.1038/s41467-020-19283-x
Nora, E. P., Goloborodko, A., Valton, A. L., Gibcus, J. H., Uebersohn, A., Abdennur, N., et al. (2017). Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–944e922. doi: 10.1016/j.cell.2017.05.004
Nora, E. P., Lajoie, B. R., Schulz, E. G., Giorgetti, L., Okamoto, I., Servant, N., et al. (2012). Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385. doi: 10.1038/nature11049
Nord, A. S., Blow, M. J., Attanasio, C., Akiyama, J. A., Holt, A., Hosseini, R., et al. (2013). Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell 155, 1521–1531. doi: 10.1016/j.cell.2013.11.033
Olson, E. N. (2006). Gene regulatory networks in the evolution and development of the heart. Science 313, 1922–1927. doi: 10.1126/science.1132292
Papait, R., Cattaneo, P., Kunderfranco, P., Greco, C., Carullo, P., Guffanti, A., et al. (2013). Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc. Natl. Acad. Sci. U S A. 110, 20164–20169. doi: 10.1073/pnas.1315155110
Pepin, M. E., Ha, C. M., Crossman, D. K., Litovsky, S. H., Varambally, S., Barchue, J. P., et al. (2019). Genome-wide DNA methylation encodes cardiac transcriptional reprogramming in human ischemic heart failure. Lab. Invest. 99, 371–386. doi: 10.1038/s41374-018-0104-x
Piche, J., Van Vliet, P. P., Puceat, M., and Andelfinger, G. (2019). The expanding phenotypes of cohesinopathies: one ring to rule them all! Cell Cycle 18, 2828–2848. doi: 10.1080/15384101.2019.1658476
Pinto, A. R., Ilinykh, A., Ivey, M. J., Kuwabara, J. T., D’Antoni, M. L., Debuque, R., et al. (2016). Revisiting Cardiac Cellular Composition. Circ. Res. 118, 400–409. doi: 10.1161/CIRCRESAHA.115.307778
Rada-Iglesias, A., Bajpai, R., Swigut, T., Brugmann, S. A., Flynn, R. A., and Wysocka, J. (2011). A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283. doi: 10.1038/nature09692
Rao, S. S. P., Huang, S. C., Glenn, St Hilaire, B., Engreitz, J. M., Perez, E. M., et al. (2017). Cohesin Loss Eliminates All Loop Domains. Cell 171, 305–320e324. doi: 10.1016/j.cell.2017.09.026
Rao, S. S., Huntley, M. H., Durand, N. C., Stamenova, E. K., Bochkov, I. D., Robinson, J. T., et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. doi: 10.1016/j.cell.2014.11.021
Riggs, A. D. (1975). X inactivation, differentiation, and DNA methylation. Cytogenet. Cell Genet. 14, 9–25. doi: 10.1159/000130315
Rinn, J. L., Kertesz, M., Wang, J. K., Squazzo, S. L., Xu, X., Brugmann, S. A., et al. (2007). Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129, 1311–1323. doi: 10.1016/j.cell.2007.05.022
Rosa-Garrido, M., Chapski, D. J., Schmitt, A. D., Kimball, T. H., Karbassi, E., Monte, E., et al. (2017). High-Resolution Mapping of Chromatin Conformation in Cardiac Myocytes Reveals Structural Remodeling of the Epigenome in Heart Failure. Circulation 136, 1613–1625. doi: 10.1161/CIRCULATIONAHA.117.029430
Saldana-Meyer, R., Gonzalez-Buendia, E., Guerrero, G., Narendra, V., Bonasio, R., Recillas-Targa, F., et al. (2014). CTCF regulates the human p53 gene through direct interaction with its natural antisense transcript, Wrap53. Genes Dev. 28, 723–734. doi: 10.1101/gad.236869.113
Saldana-Meyer, R., Rodriguez-Hernaez, J., Escobar, T., Nishana, M., Jacome-Lopez, K., Nora, E. P., et al. (2019). RNA Interactions Are Essential for CTCF-Mediated Genome Organization. Mol. Cell 76, 412–422e415. doi: 10.1016/j.molcel.2019.08.015
Sanborn, A. L., Rao, S. S., Huang, S. C., Durand, N. C., Huntley, M. H., Jewett, A. I., et al. (2015). Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. U S A. 112, E6456–E6465. doi: 10.1073/pnas.1518552112
Sarogni, P., Pallotta, M. M., and Musio, A. (2020). Cornelia de Lange syndrome: from molecular diagnosis to therapeutic approach. J. Med. Genet. 57, 289–295. doi: 10.1136/jmedgenet-2019-106277
Shikama, N., Lutz, W., Kretzschmar, R., Sauter, N., Roth, J. F., Marino, S., et al. (2003). Essential function of p300 acetyltransferase activity in heart, lung and small intestine formation. EMBO J. 22, 5175–5185. doi: 10.1093/emboj/cdg502
Stankunas, K., Hang, C. T., Tsun, Z. Y., Chen, H., Lee, N. V., Wu, J. I., et al. (2008). Endocardial Brg1 represses ADAMTS1 to maintain the microenvironment for myocardial morphogenesis. Dev. Cell 14, 298–311. doi: 10.1016/j.devcel.2007.11.018
Sun, H., Yang, X., Zhu, J., Lv, T., Chen, Y., Chen, G., et al. (2010). Inhibition of p300-HAT results in a reduced histone acetylation and down-regulation of gene expression in cardiac myocytes. Life Sci. 87, 707–714. doi: 10.1016/j.lfs.2010.10.009
Sun, X., Hota, S. K., Zhou, Y. Q., Novak, S., Miguel-Perez, D., Christodoulou, D., et al. (2018). Cardiac-enriched BAF chromatin-remodeling complex subunit Baf60c regulates gene expression programs essential for heart development and function. Biol. Open 7:29512. doi: 10.1242/bio.029512
Sylva, M., van den Hoff, M. J., and Moorman, A. F. (2014). Development of the human heart. Am. J. Med. Genet. A 164A, 1347–1371. doi: 10.1002/ajmg.a.35896
Takaya, T., Kawamura, T., Morimoto, T., Ono, K., Kita, T., Shimatsu, A., et al. (2008). Identification of p300-targeted acetylated residues in GATA4 during hypertrophic responses in cardiac myocytes. J. Biol. Chem. 283, 9828–9835. doi: 10.1074/jbc.M707391200
Takeuchi, J. K., and Bruneau, B. G. (2009). Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature 459, 708–711. doi: 10.1038/nature08039
Trivedi, C. M., Zhu, W., Wang, Q., Jia, C., Kee, H. J., Li, L., et al. (2010). Hopx and Hdac2 interact to modulate Gata4 acetylation and embryonic cardiac myocyte proliferation. Dev. Cell 19, 450–459. doi: 10.1016/j.devcel.2010.08.012
Ulitsky, I., and Bartel, D. P. (2013). lincRNAs: genomics, evolution, and mechanisms. Cell 154, 26–46. doi: 10.1016/j.cell.2013.06.020
Vilardell, M., Rasche, A., Thormann, A., Maschke-Dutz, E., Perez-Jurado, L. A., Lehrach, H., et al. (2011). Meta-analysis of heterogeneous Down Syndrome data reveals consistent genome-wide dosage effects related to neurological processes. BMC Genomics 12:229. doi: 10.1186/1471-2164-12-229
Vissers, L. E., van Ravenswaaij, C. M., Admiraal, R., Hurst, J. A., de Vries, B. B., Janssen, I. M., et al. (2004). Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 36, 955–957. doi: 10.1038/ng1407
Voss, T. C., and Hager, G. L. (2014). Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat. Rev. Genet. 15, 69–81. doi: 10.1038/nrg3623
Wang, S., Su, J. H., Beliveau, B. J., Bintu, B., Moffitt, J. R., Wu, C. T., et al. (2016). Spatial organization of chromatin domains and compartments in single chromosomes. Science 353, 598–602. doi: 10.1126/science.aaf8084
Wu, B., Wang, Y., Lui, W., Langworthy, M., Tompkins, K. L., Hatzopoulos, A. K., et al. (2011). Nfatc1 coordinates valve endocardial cell lineage development required for heart valve formation. Circ. Res. 109, 183–192. doi: 10.1161/CIRCRESAHA.111.245035
Yao, Q., Chen, Y., and Zhou, X. (2019). The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 51, 11–17. doi: 10.1016/j.cbpa.2019.01.024
Yu, M., and Ren, B. (2017). The Three-Dimensional Organization of Mammalian Genomes. Annu. Rev. Cell Dev. Biol. 33, 265–289. doi: 10.1146/annurev-cellbio-100616-060531
Zhang, W., Song, M., Qu, J., and Liu, G. H. (2018). Epigenetic Modifications in Cardiovascular Aging and Diseases. Circ. Res. 123, 773–786. doi: 10.1161/CIRCRESAHA.118.312497
Zoghbi, H. Y., and Beaudet, A. L. (2016). Epigenetics and Human Disease. Cold Spring Harb. Perspect. Biol. 8:a019497. doi: 10.1101/cshperspect.a019497
Keywords: epigenetics, cardiac development, CTCF, TADs, heart
Citation: George RM and Firulli AB (2021) Epigenetics and Heart Development. Front. Cell Dev. Biol. 9:637996. doi: 10.3389/fcell.2021.637996
Received: 04 December 2020; Accepted: 26 March 2021;
Published: 06 May 2021.
Edited by:
Duan Ma, Fudan University, ChinaReviewed by:
Hamish W. King, Queen Mary University of London, United KingdomDeqiang Li, University of Maryland, Baltimore, United States
Copyright © 2021 George and Firulli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthony B. Firulli, dGZpcnVsbGlAaXUuZWR1