Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurological disorder characterized by progressive degeneration of motor neurons in the brain and spinal cord. Spinal motor neurons align along the spinal cord length within the vertebral column, and extend long axons to connect with skeletal muscles covering the body surface. Due to this anatomy, spinal motor neurons are among the most difficult cells to observe in vivo. Larval zebrafish have transparent bodies that allow non-invasive visualization of whole cells of single spinal motor neurons, from somas to the neuromuscular synapses. This unique feature, combined with its amenability to genome editing, pharmacology, and optogenetics, enables functional analyses of ALS-associated proteins in the spinal motor neurons in vivo with subcellular resolution. Here, we review the zebrafish skeletal neuromuscular system and the optical methods used to study it. We then introduce a recently developed optogenetic zebrafish ALS model that uses light illumination to control oligomerization, phase transition and aggregation of the ALS-associated DNA/RNA-binding protein called TDP-43. Finally, we will discuss how this disease-in-a-fish ALS model can help solve key questions about ALS pathogenesis and lead to new ALS therapeutics.
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurological disorder in which motor neurons in the brain and spinal cord are selectively degenerated, leading to progressive muscle weakness. Approximately 1–2 individuals per 100,000 are diagnosed with ALS each year, with motor symptoms typically appearing in mid-adulthood (average age 55) (Taylor et al., 2016). About 10% of ALS cases are heritable (familial ALS) and linked to single-gene pathogenic mutations. The remaining 90% of ALS cases occur without family history (sporadic ALS) and are thought to involve multiple genetic mutations and/or environmental factors. While the root cause of sporadic ALS is unknown, its common pathological hallmark is deposition of ubiquitin-positive cytoplasmic inclusions containing aggregated forms of DNA/RNA-binding protein TDP-43, encoded by the TARDBP gene, in the degenerating motor neurons (Arai et al., 2006; Neumann et al., 2006; Mackenzie et al., 2007). Better understanding of the causes and consequences of TDP-43 aggregation will increase our understanding of ALS pathogenesis and aid in the development of therapeutics.
There is emerging evidence that an intricate intracellular network of biomolecular condensates or membraneless organelles underlies the physiological functions of cells, including neurons (Shin and Brangwynne, 2017). Many membraneless organelles are enriched in proteins containing regions of low sequence complexity, called low-complexity domains (LCDs) or intrinsically disordered regions (IDRs), which drive liquid–liquid phase separation through homotypic and heterotypic protein–protein or protein–RNA interactions (Mathieu et al., 2020). Aberrant phase separation of IDR proteins can generate solid-like fibers, which are candidate origins for irreversible aggregates that accumulate in neurodegenerative diseases (Lin et al., 2015; Molliex et al., 2015; Murakami et al., 2015; Patel et al., 2015; Zhang et al., 2015). TDP-43 is equipped with an IDR at its C-terminus, and has been identified as a component of membraneless organelles in the nucleus (Dammer et al., 2012; Gasset-Rosa et al., 2019) and cytoplasm (Dewey et al., 2011; Alami et al., 2014; Gopal et al., 2017; McGurk et al., 2018; Wang et al., 2020) under both normal and stressed conditions. The fact that ALS-associated mutations of TARDBP mostly occur in the IDR has led to the idea that pathological phase transitions of TDP-43 mediated by altered homotypic and heterotypic interactions through IDR contribute to ALS pathogenesis. Cytoplasmic aggregation of TDP-43 has been observed by overexpressing wild-type and mutant TDP-43 in cellular and animal models of ALS (Barmada et al., 2010; Li et al., 2010; Walker et al., 2015; De Giorgio et al., 2019; Watanabe et al., 2020). Since exogenously expressed TDP-43 can be toxic in the absence of TDP-43 aggregates, it is difficult to determine whether the TDP-43 toxicity stems from dosage increase, aggregation, or both (Barmada et al., 2010; Arnold et al., 2013; Asakawa et al., 2020). Therefore, dissecting oligomerization- or phase transition-dependent toxicity from overexpression-dependent toxicity has remained a challenging but important task in understanding the mechanism of TDP-43 neurotoxicity.
The zebrafish is a unique vertebrate model that offers access to spatiotemporal dynamics of disease-related proteins in in vivo motor neurons. With the high translucency of larval zebrafish body tissue, fluorescently tagged probes are visible in a single spinal motor neuron, allowing for direct visualization of specific proteins, organelles, and cytoskeletons in real time. In addition, genetic amenability and high genetic homology to humans have allowed for rapid establishment of stable zebrafish lines relevant for the functional exploration of ALS-related genes (Ramesh et al., 2010; Sakowski et al., 2012; Hewamadduma et al., 2013; Schmid et al., 2013; Da Costa et al., 2014; Armstrong et al., 2016; Lebedeva et al., 2017; Ohki et al., 2017; Lissouba et al., 2018; Shaw et al., 2018; Bercier et al., 2019; Bose et al., 2019; Kim et al., 2019; Asakawa et al., 2020; Bourefis et al., 2020). Recent studies have achieved optogenetic induction of ALS pathology by controlling the biophysics of disease-associated proteins with an unprecedented spatiotemporal precision by light illumination (Shin et al., 2017; Mann et al., 2019; Zhang et al., 2019; Asakawa et al., 2020). In this mini-review, we describe the skeletal neuromuscular system of larval zebrafish and optogenetic approaches for controlling in vivo phase transition of TDP-43 in motor neurons pertinent to the study of ALS pathogenesis.
The Crystal-Clear Neuromuscular System of Zebrafish Larvae
Zebrafish grow to about 4 mm in length in the first 5 days of life, developing over 30 axial muscle segments in a fusiform, laterally compressed body (Schroter et al., 2008). In parallel, the spinal motor neurons are generated in the segmentally iterated spinal cord and innervate corresponding skeletal muscle segments (Figure 1A). A 5 day-old larva performs free swimming and is already an efficient visually guided predator (Budick and O’Malley, 2000), indicating a growing, but mature multimodal sensory-guided motor circuit. Generation of the spinal motor neurons largely ceases in the second day of development (Reimer et al., 2013), with 63–71 spinal motor neurons per spinal hemisegment present in the larval stage and remaining largely constant until the adult stage (van Raamsdonk et al., 1983; Asakawa et al., 2013; Svara et al., 2018). These motor neurons innervate peripheral muscles consisting of fast-twitch fibers that occupy majority of the muscle mass and slow-twitch fibers that cover the superficial muscle layer (Figures 1B,C). Spinal motor neurons are categorized into primary and secondary motor neurons, both of which start to differentiate at 1 day post-fertilization (Myers et al., 1986). There are five primary motor neurons in each spinal hemisegment (MiP, dRoP, vRoP, CaP, and VaP). These are the earlier differentiating population, possess relatively large-sized somas that align rostro-caudally, and innervate distinct areas of fast-twitch muscle fibers along the dorsoventral myotomal axis (Asakawa et al., 2013; Bello-Rojas et al., 2019). The remaining ∼60 neurons in a hemisegment are the secondary motor neurons that develop next: these possess relatively small-sized somas and innervate the deeper fast twitch muscle or superficial slow muscle fibers (Menelaou and McLean, 2012; Asakawa et al., 2013; Bello-Rojas et al., 2019). Overlapping patterns of innervation observed within and between these motor neuron types indicate polyneuronal innervation of muscle fibers (Lefebvre et al., 2007; Bello-Rojas et al., 2019). Zebrafish motor neurons, which vary in cell-size and physiology (McLean et al., 2007; Menelaou and McLean, 2012), might model selective vulnerability of motor neurons in ALS; large-sized, fast fatigable motor neurons are the most vulnerable to degeneration (Roselli and Caroni, 2014). Remarkably, unlike mammals, both larval (Ohnmacht et al., 2016) and adult (Reimer et al., 2008) zebrafish retain the ability to regenerate spinal motor neurons from the pMN progenitor domain in the ventral spinal cord after spinal cord injury or genetic ablation. This ability is worth further study for its potential contribution to regenerative therapy of motor neurons in humans.
FIGURE 1
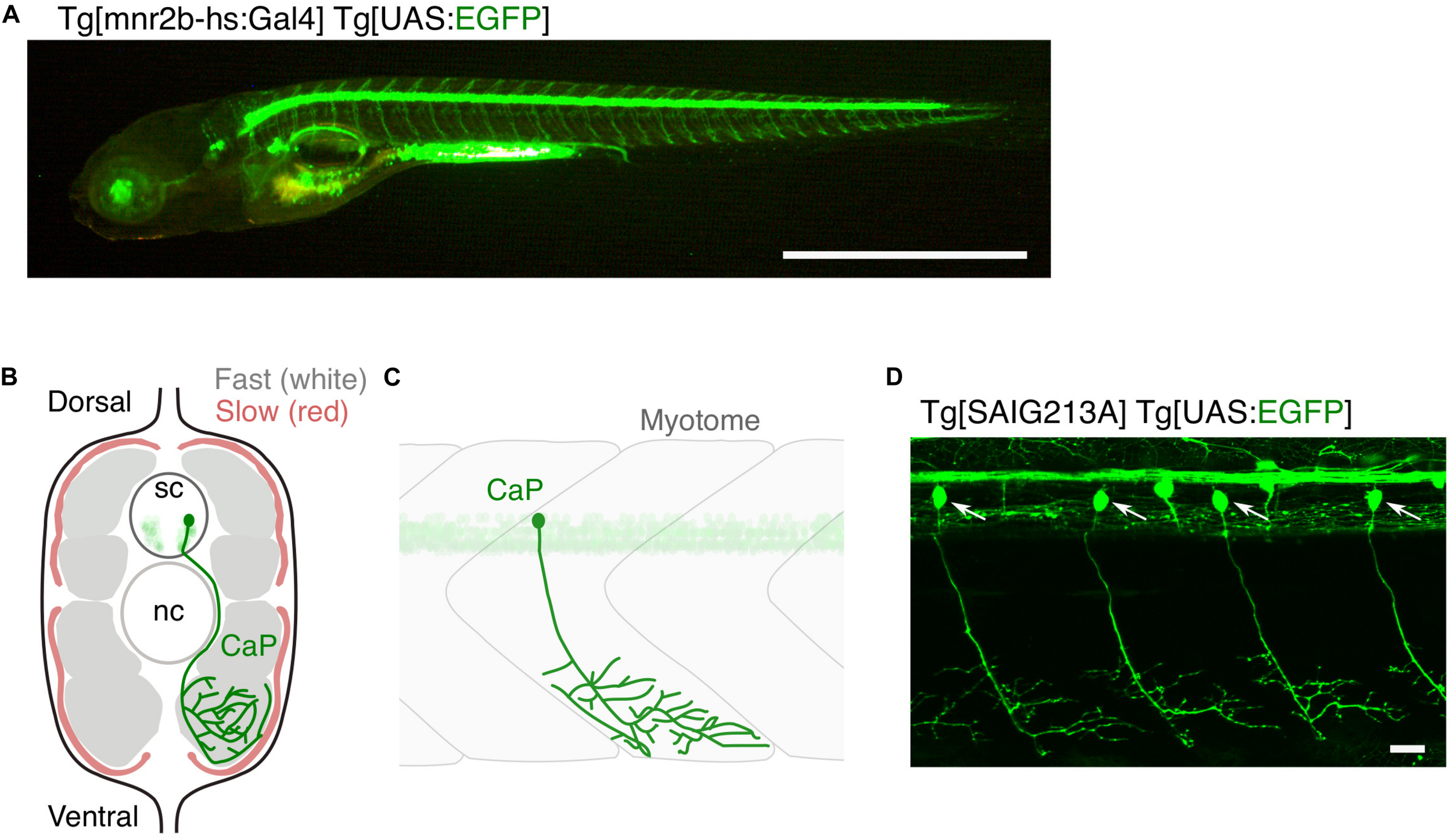
Large and small populations of spinal motor neurons can be manipulated with the Gal4/UAS system in zebrafish. (A) Tg[mnr2b-hs:Gal4]; Tg[UAS:EGFP] larva at 5 day post-fertilization. In the Gal4 driver Tg[mnr2b-hs:Gal4], the Gal4FF transcription factor (Asakawa et al., 2008) is expressed from the bacterial artificial chromosome (BAC) transgene carrying the mnr2b locus (encoding the Mnx-type homeobox protein, which promotes motor neuron differentiation) and drives expression of a gene downstream of upstream activation sequence (UAS) in the most of the spinal motor neurons. (B) Schematic illustration of a transverse section of the middle trunk of a 5 day-old wild-type zebrafish larva, with the dorsal side up. Fast-twitch muscle and slow muscle are shown in gray and red, respectively. A CaP innervating the ventral myotome is shown in green. sc, spinal cord. nc, notochord. Illustration modified from Bello-Rojas et al. (2019). (C) Schematic illustration of a wild-type CaP innervating the ventral myotome, from lateral view. (D) Among the spinal motor neurons, CaPs (arrows) are selectively labeled in Tg[SAIG213A] Tg[UAS:EGFP] fish. Bars are 1 mm (A) and 20 μm (D).
A single motor neuron or a population of motor neurons can be non-invasively manipulated in zebrafish larvae with targeted expression of genetically encoded fluorophore and photo-responsive probes using the cis-elements of motor neuron-specific genes, such as Mnx-family homeobox genes (Wendik et al., 2004; Asakawa et al., 2012), and/or Gal4/UAS gene expression systems (Figures 1A,D; Zelenchuk and Bruses, 2011; Asakawa et al., 2013). Laser-assisted transection of a fluorescently labeled motor neuron (Rosenberg et al., 2012) was used to directly monitor, in real time, the sequence of events involving motor nerve degeneration following injury (Wallerian degeneration) (Waller, 1850), as well as the resultant dynamic interactions between the injured nerves and macrophages. Engulfment of the TDP-43-expressing motor neurons by microglia after UV-induced damage was directly observed (Svahn et al., 2018). Motor neurons can be more precisely manipulated by a method called optogenetics, in which genetically-encoded proteins change conformation in the presence of light (Deisseroth et al., 2006). Targeted expression and photostimulation of KillerRed (KR) generates reactive oxygen species (ROS) and induce motor neuron death, followed by microglial activation (Bulina et al., 2006; Formella et al., 2018). Motor neuron physiology can be manipulated by controlling membrane potential with photostimulation of the light-gated ion channels (Volgraf et al., 2006; Gorostiza et al., 2007; Wyart et al., 2009; Bernal Sierra et al., 2018; Antinucci et al., 2020). Further, development of motor neurons, such as axon guidance, can be optically controlled by light-activatable cytoskeletal regulator Rac1, promoting axon guidance of the caudal primary motor neuron (CaP) in the wild-type fish, as well as of plod3-/- mutant fish defective in axon guidance (Harris et al., 2020). In addition to these optogenetic methods, expression of appropriate fluorescent marker proteins enables the visualization of key subcellular structures of motor neurons, including pre- and post-synapses (Flanagan-Steet et al., 2005; Asakawa and Kawakami, 2018; Bello-Rojas et al., 2019), cytoskeletal components (Asakawa and Kawakami, 2010; Bercier et al., 2019), and mitochondria (Bergamin et al., 2016), in live animals.
Optogenetic Induction of TDP-43 Aggregation in in vivo Motor Neurons
The optoDroplet approach, recently introduced by Shin et al. (2017), adopts the photolyase homology region (PHR) of Arabidopsis cryptochrome 2/CRY2 (CRY2PHR) that self-associates upon blue light exposure (Bugaj et al., 2013) to induce oligomerization and phase transition of client proteins tagged with CRY2PHR (Shin et al., 2017). This technique has been successfully applied to TDP-43 in cultured cells (Mann et al., 2019; Zhang et al., 2019), and has been used to discriminate cellular events that are triggered by TDP-43 oligomerization from those triggered by dosage increase of TDP-43. In these studies, CRY2PHR or a point mutant version of Cry2 (Cry2olig) exhibiting enhanced clustering (Taslimi et al., 2014) was fused to the N-terminus (Mann et al., 2019; Zhang et al., 2019; Otte et al., 2020; Figure 2A). These optogenetic TDP-43 effectively displayed clustering upon blue light illumination, leading to cytoplasmic deposition of TDP-43 aggregates with the pathological signature of S409/S410 phosphorylation typically detected in degenerating motor neurons in ALS (Mann et al., 2019; Zhang et al., 2019), suggesting that the optogenetically induced TDP-43 aggregation mimics at least some of the TDP-43 pathology occurring ALS. Photostimulation of optoTDP-43 increases cell death rate in the cultured human neurons, with cytoplasmic shift and aggregation of optoTDP-43, demonstrating that optoTDP-43 phase transition is neurotoxic (Mann et al., 2019). The light-driven phase transition of optoTDP-43 does not recruit major stress granule (SG) markers, suggesting that TDP-43 is not a core component of SGs (Zhang et al., 2019). This result is consistent with the recent observations that TDP-43 that failed to be recruited to RNA-rich granules, such as SGs, is prone to aberrant phase transition (McGurk et al., 2018; Gasset-Rosa et al., 2019; Mann et al., 2019; Zhang et al., 2019).
FIGURE 2
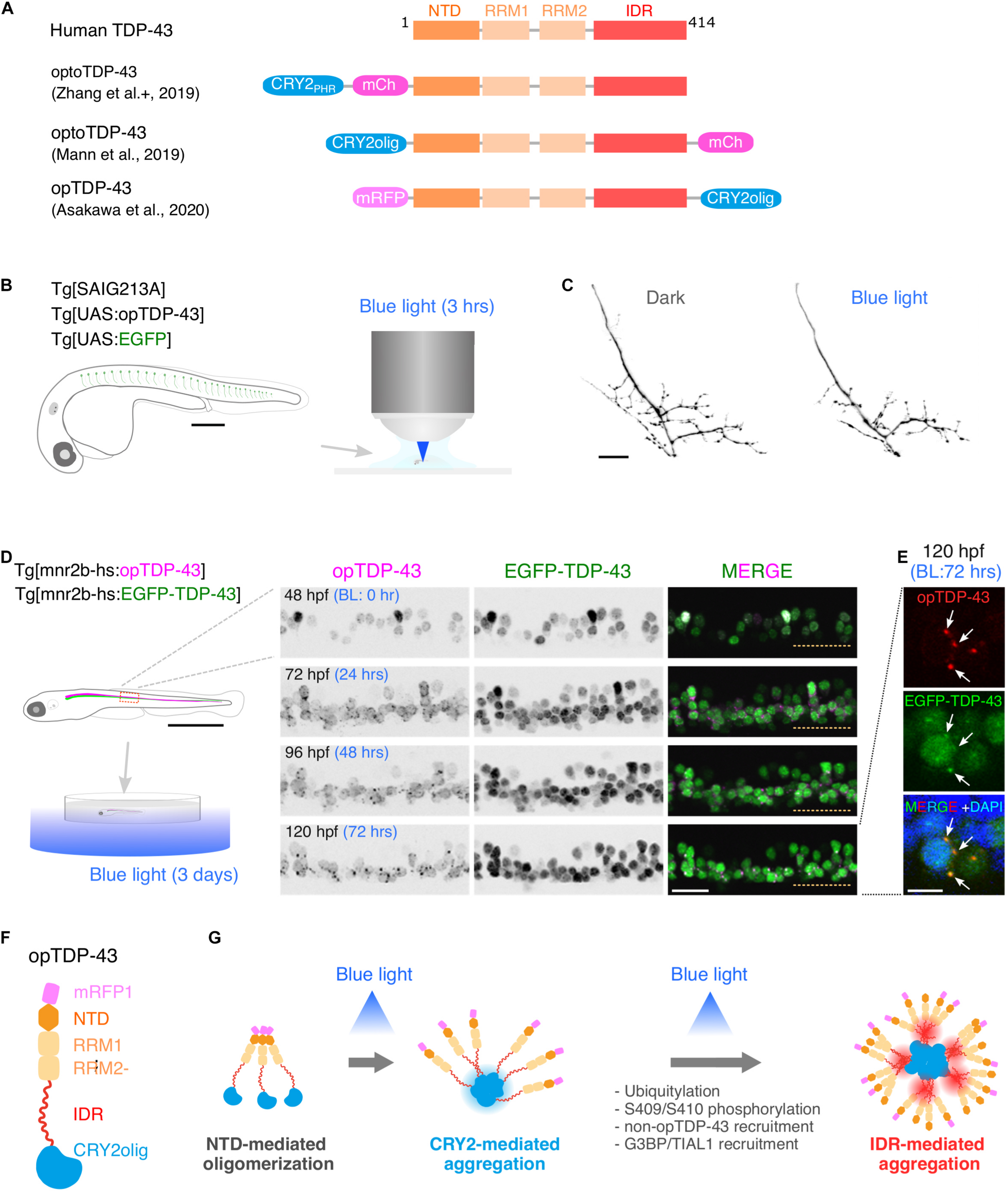
Optogenetic induction of TDP-43 aggregates in in vivo spinal motor neurons. (A) Structures of optogenetic TDP-43. The human TDP-43 (top) consists of 414 amino acid residues subdivided into the N-terminal domain (NTD), 2 RNA recognition motifs (RRM1 and RRM2), and a C-terminus intrinsically disordered region (IDR). The optoTDP-43 (Mann et al., 2019; Zhang et al., 2019) and opTDP-43 (Asakawa et al., 2020) constructs carry the CRY2-modules at their N- and C-terminus, respectively. mCherry (237 aa), mRFP1 (225 aa), CRY2PHR (498 aa), and CRY2olig (498 aa) not to scale with TDP-43. (B) An agarose-embedded zebrafish embryo expressing both EGFP and opTDP-43 in CaPs (Tg[SAIG213A] Tg[UAS:opTDP-43] Tg[UAS:EGFP] triple transgenic) is illuminated with a confocal blue laser light for 3 h, 28–31 h post-fertilization (hpf). (C) The total axonal length at 48 hpf was reduced in the CaP irradiated with the blue light (Blue light) compared to the CaP grown in the dark (Dark). The figure panels are adapted from Asakawa et al. (2020). (D) An unrestrained zebrafish larva expressing both opTDP-43 and non-optogenetic EGFP-TDP-43 was irradiated with blue LED light. The spinal motor column was scanned every 24 h for 3 days (from 48 to 120 hpf). The duration of the blue light illumination is indicated in blue letters. Horizontal dashed line demarcates the ventral limit of the spinal cord. The figure panels are adapted from the study by Asakawa et al. (2020). (E) Cytoplasmic opTDP-43 foci colocalize with non-optogenetic EGFP-TDP-43 (arrows) in the spinal motor neurons at 120 hpf in (D). BL, Blue light. (F) Schematic drawing of opTDP-43 protein. (G) Blue light illumination drives CRY2olig-dependent opTDP-43 oligomerization and aggregation. A short-term illumination induces the oligomerization of opTDP-43, whereas a long-term illumination causes cytoplasmic aggregation of opTDP-43. Non-optogenetic TDP-43 is incorporated into the opTDP-43 aggregates [e.g., EGFP-TDP-43 in (E)] possibly through IDR-mediated intermolecular interactions. Cytoplasmic opTDP-43 aggregates are partially positive for immunoreactivities against ubiquitin, phosphorylation at S409/S410, and classical stress granule components (G3BP and TIAL1) (Asakawa et al., 2020). The bars indicate 250 μm (B), 20 μm (C), 1 mm (D, left), 20 μm (D, panels), and 5 μm (E).
A key step toward understanding of motor neuron degeneration in ALS is to induce TDP-43 oligomerization and phase transition in motor neurons and evaluate their cellular outcome in in vivo contexts (Asakawa et al., 2020; Otte et al., 2020). In zebrafish, expression of an optogenetic TDP-43 (opTDP-43), with which CRY2olig is harnessed at the C-terminus of TDP-43 (Figure 2A; Asakawa et al., 2020), can be targeted to CaPs among other motor neurons by using the prdm14-Gal4 driver (Figure 1C), enabling detailed analyses of opTDP-43 dynamics, as well as its cellular consequences, at subcellular resolution (Asakawa et al., 2020). opTDP-43 is primarily nuclear under normal conditions, as is the wild-type TDP-43, but dispersed throughout the CaP in response to blue light illumination for 3 h. Crucially, CaP axon outgrowth was halted even after the illumination ceased prior to cytoplasmic accumulation of opTDP-43 inclusion and the nuclear opTDP-43 localization was restored, showing that opTDP-43 toxicity precedes deposition of its cytoplasmic aggregates (Asakawa et al., 2020; Figures 2B,C). Live imaging analyses of the illuminated CaPs expressing opTDP-43 revealed an enhanced myofiber denervation frequency compared to the wild-type CaPs, underscoring the importance of normal TDP-43 phase behavior in formation and maintenance of neuromuscular synapses (Asakawa et al., 2020). The precise molecular mechanism of this oligomerization-triggered but aggregation-independent opTDP-43 toxicity is currently unclear. Another intriguing observation from the zebrafish model is that the light-dependent cytoplasmic opTDP-43 relocation occurred in the spinal motor neurons and the tactile sensing Rohon-Beard sensory cells in the spinal cord, but was almost absent in the embryonic epithelial cells and differentiated myofibers in a time frame examined (Asakawa et al., 2020). This observation suggests that the oligomerization-triggered cytoplasmic opTDP-43 relocation is a cell-type-dependent phenomenon (Vogler et al., 2018). Knowledge of the mechanisms underlying this neuron-specific cytoplasmic opTDP-43 relocation could aid in our understanding of cytoplasmic relocation of TDP-43 in the context of ALS motor neurons, whose mechanism is almost entirely unknown at present.
A major advantage of optogenetics is that it allows precise temporal control of a photo-responsive probe. In zebrafish larvae expressing opTDP-43 in most of the spinal motor neurons, the extension of the photostimulation duration from 3 h to 3 days resulted in the accumulation of cytoplasmic opTDP-43 aggregates in the motor neurons (Figures 2D,E; Asakawa et al., 2020). The cytoplasmic opTDP-43 aggregates contained non-optogenetic TDP-43 and were recognized to varying degrees by the antibodies against ubiquitin, phospho-S409/410, G3BP, and TIAL1 (Figures 2F,G; Asakawa et al., 2020), indicating that the opTDP-43 aggregates are heterogenous in protein composition and some species of the opTDP-43 aggregates recapitulate ALS pathology. While the pan-motor neuronal cytoplasmic opTDP-43 aggregation induced by the 3 day light illumination did not lead to overt motor deficit, the same set of conditions with opTDP-43 carrying ALS-associated IDR mutation (A315T) led to the failure to inflate the swim bladder and an impaired motor activity in a small population (13%) of larvae (Asakawa et al., 2020). Consistent with this observation, in fly, photostimulation of optoTDP-43 eventually leads to the formation of detergent-insoluble aggregates in the motor neurons that persist with age and cause larval and adult motor deficits (Otte et al., 2020). Overall, the temporally regulated induction of TDP-43 oligomerization and phase transition have demonstrated that TDP-43 exerts its toxicity through multiple mechanisms depending on its multimerization and phase status. Although a fraction of non-optogenetic TDP-43 was recruited to cytoplasmic opTDP-43 aggregates in the zebrafish optogenetic ALS model, most of it remained in the nucleus after 3 days of illumination during the larval stage (Asakawa et al., 2020). To determine if the cytoplasmic opTDP-43 aggregates eventually deplete the nuclear TDP-43 pool as observed in ALS cases, it is imperative to establish an illumination condition where the physiology of juvenile and adult fish is minimally affected by the blue light while the light-dependent opTDP-43 phase transition is fully controllable in time and space. In this regard, it is worth exploring other optogenetic probes for regulation of protein interactions that can be activated by different light wave lengths, such as a bacterial phytochrome-based probe sensitive to near-infrared light (Redchuk et al., 2020).
Perspectives
The optoDroplet approach has explicitly demonstrated the causal relationship between TDP-43 phase transition and neurotoxicity in vivo in motor neurons. The accumulation of cytoplasmic TDP-43 aggregates is a hallmark of degenerating neurons in ALS. However, how TDP-43 is localized in the nucleus under normal conditions and the mechanisms by which it gradually loses its normal state and forms cytoplasmic aggregates are not clear. Thus, it is tempting to speculate that light-dependent opTDP-43 phase transition is a fast-forward replay of the changes occurring in TDP-43 in ALS, especially as TDP-43 dynamics is currently anatomically inaccessible in human motor neurons. Uncovering intermediate stages of the opTDP-43 phase transition would provide mechanistic insights into TDP-43 cytotoxicity and potential therapeutic targets. The illumination time-dependent formation of cytoplasmic opTDP-43 aggregates suggests that oligomerization and phase separation of opTDP-43 is tunable and can be used to reveal the temporal sequence of downstream events driven by pathological TDP-43 phase transition in the spinal motor neurons. This opTDP-43 phase transition can be targeted with subcellular resolution to explore spatial sources of TDP-43 toxicity, such as the nucleus, axon, and pre-/post-synaptic terminals. The ability to observe diseased motor neurons in a transparent animal also allows us to directly investigate systemic responses of neighboring cell types, including interneurons, myofibers, glial cells, and immune cells, to aberrant TDP-43 transitions occurring in the motor neurons. Moreover, since the IDR of TDP-43 is classified as a prion-like domain and TDP-43 has a prion-like aggregation-seeding ability (Nonaka et al., 2013), potential intercellular propagation of opTDP-43-driven TDP-43 aggregation could be directly tested within an intact CNS. Finally, we suggest that future studies take full advantage of zebrafish in a whole organism compound screening, advancing the opTDP-43-based zebrafish ALS model into a system for screening small molecules that mitigate toxic TDP-43 phase transition for developing effective ALS therapeutics.
Statements
Author contributions
KA wrote the manuscript and generated the figures, with inputs from HH and KK. All authors approved the submitted version.
Funding
This work was supported by the SERIKA FUND (KA) and KAKENHI (JP19K06933 and JP20H05345) (KA).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AlamiN. H.SmithR. B.CarrascoM. A.WilliamsL. A.WinbornC. S.HanS. S. W.et al (2014). Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations.Neuron.81536–543. 10.1016/j.neuron.2013.12.018
2
AntinucciP.DumitrescuA.DeleuzeC.MorleyH. J.LeungK.HagleyT.et al (2020). A calibrated optogenetic toolbox of stable zebrafish opsin lines.Elife9:54937. 10.7554/eLife.54937
3
AraiT.HasegawaM.AkiyamaH.IkedaK.NonakaT.MoriH.et al (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis.Biochem. Biophys. Res. Commun.351602–611. 10.1016/j.bbrc.2006.10.093
4
ArmstrongG. A.LiaoM.YouZ.LissoubaA.ChenB. E.DrapeauP. (2016). Homology Directed Knockin of Point Mutations in the Zebrafish tardbp and fus Genes in ALS Using the CRISPR/Cas9 System.PLoS One11:e0150188. 10.1371/journal.pone.0150188
5
ArnoldE. S.LingS. C.HuelgaS. C.Lagier-TourenneC.PolymenidouM.DitsworthD.et al (2013). ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43.Proc. Natl. Acad. Sci. U S A.110E736–E745. 10.1073/pnas.1222809110
6
AsakawaK.AbeG.KawakamiK. (2013). Cellular dissection of the spinal cord motor column by BAC transgenesis and gene trapping in zebrafish.Front. Neural. Circuits.7:100. 10.3389/fncir.2013.00100
7
AsakawaK.HandaH.KawakamiK. (2020). Optogenetic modulation of TDP-43 oligomerization accelerates ALS-related pathologies in the spinal motor neurons.Nat. Commun.11:1004. 10.1038/s41467-020-14815-x
8
AsakawaK.HigashijimaS.KawakamiK. (2012). An mnr2b/hlxb9lb enhancer trap line that labels spinal and abducens motor neurons in zebrafish.Dev. Dyn.241327–332. 10.1002/dvdy.22781
9
AsakawaK.KawakamiK. (2010). A transgenic zebrafish for monitoring in vivo microtubule structures.Dev. Dyn.2392695–2699. 10.1002/dvdy.22400
10
AsakawaK.KawakamiK. (2018). Protocadherin-Mediated Cell Repulsion Controls the Central Topography and Efferent Projections of the Abducens Nucleus.Cell Rep.241562–1572. 10.1016/j.celrep.2018.07.024
11
AsakawaK.SusterM. L.MizusawaK.NagayoshiS.KotaniT.UrasakiA.et al (2008). Genetic dissection of neural circuits by Tol2 transposon-mediated Gal4 gene and enhancer trapping in zebrafish.Proc. Natl. Acad. Sci. U S A.1051255–1260. 10.1073/pnas.0704963105
12
BarmadaS. J.SkibinskiG.KorbE.RaoE. J.WuJ. Y.FinkbeinerS. (2010). Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis.J. Neurosci.30639–649. 10.1523/JNEUROSCI.4988-09.2010
13
Bello-RojasS.IstrateA. E.KishoreS.McLeanD. L. (2019). Central and peripheral innervation patterns of defined axial motor units in larval zebrafish.J. Comp. Neurol.5272557–2572. 10.1002/cne.24689
14
BercierV.HubbardJ. M.FidelinK.DuroureK.AuerT. O.RevenuC.et al (2019). Dynactin1 depletion leads to neuromuscular synapse instability and functional abnormalities.Mol. Neurodegener.14:27. 10.1186/s13024-019-0327-3
15
BergaminG.CieriD.VazzaG.ArgentonF.MostacciuoloM. L. (2016). Zebrafish Tg(hb9:MTS-Kaede): a new in vivo tool for studying the axonal movement of mitochondria.Biochim. Biophys. Acta.18601247–1255. 10.1016/j.bbagen.2016.03.007
16
Bernal SierraY. A.RostB. R.PofahlM.FernandesA. M.KoptonR. A.MoserS.et al (2018). Potassium channel-based optogenetic silencing.Nat. Commun.9:4611. 10.1038/s41467-018-07038-8
17
BoseP.ArmstrongG. A. B.DrapeauP. (2019). Neuromuscular junction abnormalities in a zebrafish loss-of-function model of TDP-43.J. Neurophysiol.121285–297. 10.1152/jn.00265.2018
18
BourefisA. R.CampanariM. L.Buee-ScherrerV.KabashiE. (2020). Functional characterization of a FUS mutant zebrafish line as a novel genetic model for ALS.Neurobiol. Dis.142:104935. 10.1016/j.nbd.2020.104935
19
BudickS. A.O’MalleyD. M. (2000). Locomotor repertoire of the larval zebrafish: swimming, turning and prey capture.J. Exp. Biol.2032565–2579.
20
BugajL. J.ChoksiA. T.MesudaC. K.KaneR. S.SchafferD. V. (2013). Optogenetic protein clustering and signaling activation in mammalian cells.Nat. Methods.10249–252. 10.1038/nmeth.2360
21
BulinaM. E.ChudakovD. M.BritanovaO. V.YanushevichY. G.StaroverovD. B.ChepurnykhT. V.et al (2006). A genetically encoded photosensitizer.Nat. Biotechnol.2495–99. 10.1038/nbt1175
22
Da CostaM. M.AllenC. E.HigginbottomA.RameshT.ShawP. J.McDermottC. J. (2014). A new zebrafish model produced by TILLING of SOD1-related amyotrophic lateral sclerosis replicates key features of the disease and represents a tool for in vivo therapeutic screening.Dis. Model Mech.773–81. 10.1242/dmm.012013
23
DammerE. B.FalliniC.GozalY. M.DuongD. M.RossollW.XuP.et al (2012). Coaggregation of RNA-binding proteins in a model of TDP-43 proteinopathy with selective RGG motif methylation and a role for RRM1 ubiquitination.PLoS One7:e38658. 10.1371/journal.pone.0038658
24
De GiorgioF.MaduroC.FisherE. M. C.Acevedo-ArozenaA. (2019). Transgenic and physiological mouse models give insights into different aspects of amyotrophic lateral sclerosis.Dis. Model Mech.12:037424. 10.1242/dmm.037424
25
DeisserothK.FengG.MajewskaA. K.MiesenbockG.TingA.SchnitzerM. J. (2006). Next-generation optical technologies for illuminating genetically targeted brain circuits.J. Neurosci.2610380–10386. 10.1523/JNEUROSCI.3863-06.2006
26
DeweyC. M.CenikB.SephtonC. F.DriesD. R.MayerP.IIIGoodS. K.et al (2011). TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor.Mol. Cell Biol.311098–1108. 10.1128/MCB.01279-10
27
Flanagan-SteetH.FoxM. A.MeyerD.SanesJ. R. (2005). Neuromuscular synapses can form in vivo by incorporation of initially aneural postsynaptic specializations.Development.1324471–4481. 10.1242/dev.02044
28
FormellaI.SvahnA. J.RadfordR. A. W.DonE. K.ColeN. J.HoganA.et al (2018). Real-time visualization of oxidative stress-mediated neurodegeneration of individual spinal motor neurons in vivo.Redox Biol.19226–234. 10.1016/j.redox.2018.08.011
29
Gasset-RosaF.LuS.YuH.ChenC.MelamedZ.GuoL.et al (2019). Cytoplasmic TDP-43 De-mixing Independent of Stress Granules Drives Inhibition of Nuclear Import. Loss of Nuclear TDP-43, and Cell Death.Neuron.102:e337. 10.1016/j.neuron.2019.02.038
30
GopalP. P.NirschlJ. J.KlinmanE.HolzbaurE. L. (2017). Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons.Proc. Natl. Acad. Sci. U S A.114E2466–E2475. 10.1073/pnas.1614462114
31
GorostizaP.VolgrafM.NumanoR.SzobotaS.TraunerD.IsacoffE. Y. (2007). Mechanisms of photoswitch conjugation and light activation of an ionotropic glutamate receptor.Proc. Natl. Acad. Sci. U S A.10410865–10870. 10.1073/pnas.0701274104
32
HarrisJ. M.WangA. Y.Boulanger-WeillJ.SantorielloC.FoianiniS.LichtmanJ. W.et al (2020). Long-Range Optogenetic Control of Axon Guidance Overcomes Developmental Boundaries and Defects.Dev. Cell.53:e577. 10.1016/j.devcel.2020.05.009
33
HewamaddumaC. A.GriersonA. J.MaT. P.PanL.MoensC. B.InghamP. W.et al (2013). Tardbpl splicing rescues motor neuron and axonal development in a mutant tardbp zebrafish.Hum. Mol. Genet.222376–2386. 10.1093/hmg/ddt082
34
KimS.ChungA. Y.NaJ. E.LeeS. J.JeongS. H.KimE.et al (2019). Myelin degeneration induced by mutant superoxide dismutase 1 accumulation promotes amyotrophic lateral sclerosis.Glia671910–1921. 10.1002/glia.23669
35
LebedevaS.de Jesus DominguesA. M.ButterF.KettingR. F. (2017). Characterization of genetic loss-of-function of Fus in zebrafish.RNA Biol.1429–35. 10.1080/15476286.2016.1256532
36
LefebvreJ. L.JingL.BecaficcoS.Franzini-ArmstrongC.GranatoM. (2007). Differential requirement for MuSK and dystroglycan in generating patterns of neuromuscular innervation.Proc. Natl. Acad. Sci. U S A.1042483–2488. 10.1073/pnas.0610822104
37
LiY.RayP.RaoE. J.ShiC.GuoW.ChenX.et al (2010). A Drosophila model for TDP-43 proteinopathy.Proc. Natl. Acad. Sci. U S A.1073169–3174. 10.1073/pnas.0913602107
38
LinY.ProtterD. S.RosenM. K.ParkerR. (2015). Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins.Mol. Cell.60208–219. 10.1016/j.molcel.2015.08.018
39
LissoubaA.LiaoM.KabashiE.DrapeauP. (2018). Transcriptomic Analysis of Zebrafish TDP-43 Transgenic Lines.Front. Mol. Neurosci.11:463. 10.3389/fnmol.2018.00463
40
MackenzieI. R.BigioE. H.InceP. G.GeserF.NeumannM.CairnsN. J.et al (2007). Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations.Ann. Neurol.61427–434. 10.1002/ana.21147
41
MannJ. R.GleixnerA. M.MaunaJ. C.GomesE.DeChellis-MarksM. R.NeedhamP. G.et al (2019). RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43.Neuron.102:e328. 10.1016/j.neuron.2019.01.048
42
MathieuC.PappuR. V.TaylorJ. P. (2020). Beyond aggregation: Pathological phase transitions in neurodegenerative disease.Science37056–60. 10.1126/science.abb8032
43
McGurkL.GomesE.GuoL.Mojsilovic-PetrovicJ.TranV.KalbR. G.et al (2018). Poly(ADP-Ribose) Prevents Pathological Phase Separation of TDP-43 by Promoting Liquid Demixing and Stress Granule Localization.Mol. Cell.71e709. 10.1016/j.molcel.2018.07.002
44
McLeanD. L.FanJ.HigashijimaS.HaleM. E.FetchoJ. R. (2007). A topographic map of recruitment in spinal cord.Nature44671–75. 10.1038/nature05588
45
MenelaouE.McLeanD. L. (2012). A gradient in endogenous rhythmicity and oscillatory drive matches recruitment order in an axial motor pool.J. Neurosci.3210925–10939. 10.1523/JNEUROSCI.1809-12.2012
46
MolliexA.TemirovJ.LeeJ.CoughlinM.KanagarajA. P.KimH. J.et al (2015). Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization.Cell163123–133. 10.1016/j.cell.2015.09.015
47
MurakamiT.QamarS.LinJ. Q.SchierleG. S.ReesE.MiyashitaA.et al (2015). ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function.Neuron88678–690. 10.1016/j.neuron.2015.10.030
48
MyersP. Z.EisenJ. S.WesterfieldM. (1986). Development and axonal outgrowth of identified motoneurons in the zebrafish.J. Neurosci.62278–2289.
49
NeumannM.SampathuD. M.KwongL. K.TruaxA. C.MicsenyiM. C.ChouT. T.et al (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis.Science314130–133. 10.1126/science.1134108
50
NonakaT.Masuda-SuzukakeM.AraiT.HasegawaY.AkatsuH.ObiT.et al (2013). Prion-like properties of pathological TDP-43 aggregates from diseased brains.Cell Rep.4124–134. 10.1016/j.celrep.2013.06.007
51
OhkiY.Wenninger-WeinzierlA.HruschaA.AsakawaK.KawakamiK.HaassC.et al (2017). Glycine-alanine dipeptide repeat protein contributes to toxicity in a zebrafish model of C9orf72 associated neurodegeneration.Mol. Neurodegener.12:6. 10.1186/s13024-016-0146-8
52
OhnmachtJ.YangY.MaurerG. W.Barreiro-IglesiasA.TsarouchasT. M.WehnerD.et al (2016). Spinal motor neurons are regenerated after mechanical lesion and genetic ablation in larval zebrafish.Development1431464–1474. 10.1242/dev.129155
53
OtteC. G.FortunaT. R.MannJ. R.GleixnerA. M.RameshN.PylesN. J.et al (2020). Optogenetic TDP-43 nucleation induces persistent insoluble species and progressive motor dysfunction in vivo.Neurobiol. Dis.146:105078. 10.1016/j.nbd.2020.105078
54
PatelA.LeeH. O.JawerthL.MaharanaS.JahnelM.HeinM. Y.et al (2015). A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation.Cell1621066–1077. 10.1016/j.cell.2015.07.047
55
RameshT.LyonA. N.PinedaR. H.WangC.JanssenP. M.CananB. D.et al (2010). A genetic model of amyotrophic lateral sclerosis in zebrafish displays phenotypic hallmarks of motoneuron disease.Dis. Model Mech.3652–662. 10.1242/dmm.005538
56
RedchukT. A.KarasevM. M.VerkhushaP. V.DonnellyS. K.HulsemannM.VirtanenJ.et al (2020). Optogenetic regulation of endogenous proteins.Nat. Commun.11:605. 10.1038/s41467-020-14460-4
57
ReimerM. M.NorrisA.OhnmachtJ.PataniR.ZhongZ.DiasT. B.et al (2013). Dopamine from the brain promotes spinal motor neuron generation during development and adult regeneration.Dev. Cell.25478–491. 10.1016/j.devcel.2013.04.012
58
ReimerM. M.SorensenI.KuschaV.FrankR. E.LiuC.BeckerC. G.et al (2008). Motor neuron regeneration in adult zebrafish.J. Neurosci.288510–8516. 10.1523/JNEUROSCI.1189-08.2008
59
RoselliF.CaroniP. (2014). Modeling neuronal vulnerability in ALS.Neuron.83758–760. 10.1016/j.neuron.2014.08.010
60
RosenbergA. F.WolmanM. A.Franzini-ArmstrongC.GranatoM. (2012). In vivo nerve-macrophage interactions following peripheral nerve injury.J. Neurosci.323898–3909. 10.1523/JNEUROSCI.5225-11.2012
61
SakowskiS. A.LunnJ. S.BustaA. S.OhS. S.Zamora-BerridiG.PalmerM.et al (2012). Neuromuscular effects of G93A-SOD1 expression in zebrafish.Mol. Neurodegener.744. 10.1186/1750-1326-7-44
62
SchmidB.HruschaA.HoglS.Banzhaf-StrathmannJ.StreckerK.van der ZeeJ.et al (2013). Loss of ALS-associated TDP-43 in zebrafish causes muscle degeneration, vascular dysfunction, and reduced motor neuron axon outgrowth.Proc. Natl. Acad. Sci. U S A.1104986–4991. 10.1073/pnas.1218311110
63
SchroterC.HerrgenL.CardonaA.BrouhardG. J.FeldmanB.OatesA. C. (2008). Dynamics of zebrafish somitogenesis.Dev. Dyn.237545–553. 10.1002/dvdy.21458
64
ShawM. P.HigginbottomA.McGownA.CastelliL. M.JamesE.HautbergueG. M.et al (2018). Stable transgenic C9orf72 zebrafish model key aspects of the ALS/FTD phenotype and reveal novel pathological features.Acta Neuropathol. Commun.6:125. 10.1186/s40478-018-0629-7
65
ShinY.BerryJ.PannucciN.HaatajaM. P.ToettcherJ. E.BrangwynneC. P. (2017). Spatiotemporal Control of Intracellular Phase Transitions Using Light-Activated optoDroplets.Cell168159–171e114. 10.1016/j.cell.2016.11.054
66
ShinY.BrangwynneC. P. (2017). Liquid phase condensation in cell physiology and disease.Science357:aaf4382. 10.1126/science.aaf4382
67
SvahnA. J.DonE. K.BadrockA. P.ColeN. J.GraeberM. B.YerburyJ. J.et al (2018). Nucleo-cytoplasmic transport of TDP-43 studied in real time: impaired microglia function leads to axonal spreading of TDP-43 in degenerating motor neurons.Acta Neuropathol.136445–459. 10.1007/s00401-018-1875-2
68
SvaraF. N.KornfeldJ.DenkW.BollmannJ. H. (2018). Volume EM Reconstruction of Spinal Cord Reveals Wiring Specificity in Speed-Related Motor Circuits.Cell Rep.232942–2954. 10.1016/j.celrep.2018.05.023
69
TaslimiA.VranaJ. D.ChenD.BorinskayaS.MayerB. J.KennedyM. J.et al (2014). An optimized optogenetic clustering tool for probing protein interaction and function.Nat. Commun.5:4925. 10.1038/ncomms5925
70
TaylorJ. P.BrownR. H.Jr.ClevelandD. W. (2016). Decoding ALS: from genes to mechanism.Nature539197–206. 10.1038/nature20413
71
van RaamsdonkW.MosW.Smit-OnelM. J.van der LaarseW. J.FehresR. (1983). The development of the spinal motor column in relation to the myotomal muscle fibers in the zebrafish (Brachydanio rerio). I. Posthatching development.Anat Embryol.167125–139. 10.1007/BF00304606
72
VoglerT. O.WheelerJ. R.NguyenE. D.HughesM. P.BritsonK. A.LesterE.et al (2018). TDP-43 and RNA form amyloid-like myo-granules in regenerating muscle.Nature563508–513. 10.1038/s41586-018-0665-2
73
VolgrafM.GorostizaP.NumanoR.KramerR. H.IsacoffE. Y.TraunerD. (2006). Allosteric control of an ionotropic glutamate receptor with an optical switch.Nat. Chem. Biol.247–52. 10.1038/nchembio756
74
WalkerA. K.SpillerK. J.GeG.ZhengA.XuY.ZhouM.et al (2015). Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43.Acta Neuropathol.130643–660. 10.1007/s00401-015-1460-x
75
WallerA. (1850). Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog and observations of the alteration produced thereby in the structure of their primitive fibers.Philos. Trans. R. Soc. Lond.140423–429.
76
WangC.DuanY.DuanG.WangQ.ZhangK.DengX.et al (2020). Stress Induces Dynamic. Cytotoxicity-Antagonizing TDP-43 Nuclear Bodies via Paraspeckle LncRNA NEAT1-Mediated Liquid-Liquid Phase Separation.Mol Cell.79:e447. 10.1016/j.molcel.2020.06.019
77
WatanabeS.InamiH.OiwaK.MurataY.SakaiS.KomineO.et al (2020). Aggresome formation and liquid-liquid phase separation independently induce cytoplasmic aggregation of TAR DNA-binding protein 43.Cell Death Dis.11:909. 10.1038/s41419-020-03116-2
78
WendikB.MaierE.MeyerD. (2004). Zebrafish mnx genes in endocrine and exocrine pancreas formation.Dev. Biol.268372–383. 10.1016/j.ydbio.2003.12.026
79
WyartC.Del BeneF.WarpE.ScottE. K.TraunerD.BaierH.et al (2009). Optogenetic dissection of a behavioural module in the vertebrate spinal cord.Nature461407–410. 10.1038/nature08323
80
ZelenchukT. A.BrusesJ. L. (2011). In vivo labeling of zebrafish motor neurons using an mnx1 enhancer and Gal4/UAS.Genesis49546–554. 10.1002/dvg.20766
81
ZhangH.Elbaum-GarfinkleS.LangdonE. M.TaylorN.OcchipintiP.BridgesA. A.et al (2015). RNA Controls PolyQ Protein Phase Transitions.Mol. Cell.60220–230. 10.1016/j.molcel.2015.09.017
82
ZhangP.FanB.YangP.TemirovJ.MessingJ.KimH. J.et al (2019). Chronic optogenetic induction of stress granules is cytotoxic and reveals the evolution of ALS-FTD pathology.Elife8:39578. 10.7554/eLife.39578
Summary
Keywords
RNA metabolism, phase transition, neurodegenarative disease, optogenetics, protein aggregation
Citation
Asakawa K, Handa H and Kawakami K (2021) Illuminating ALS Motor Neurons With Optogenetics in Zebrafish. Front. Cell Dev. Biol. 9:640414. doi: 10.3389/fcell.2021.640414
Received
11 December 2020
Accepted
22 February 2021
Published
18 March 2021
Volume
9 - 2021
Edited by
Yasuhito Shimada, Mie University, Japan
Reviewed by
Angela S. Laird, Macquarie University, Australia; Alison L. Reynolds, University College Dublin, Ireland
Updates
Copyright
© 2021 Asakawa, Handa and Kawakami.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazuhide Asakawa, kasakawa@nig.ac.jp
This article was submitted to Molecular Medicine, a section of the journal Frontiers in Cell and Developmental Biology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.