 Qi Lou1,2,3†
Qi Lou1,2,3† Minyi Zhao1†
Minyi Zhao1† Quanhui Xu4,5†
Quanhui Xu4,5† Siyu Xie4Yingying Liang2,3Jian Chen4Lisha Yuan5Lingling Wang6Linjia Jiang4
Siyu Xie4Yingying Liang2,3Jian Chen4Lisha Yuan5Lingling Wang6Linjia Jiang4 Lisha Mou3*
Lisha Mou3* Dongjun Lin1*
Dongjun Lin1* Meng Zhao1,2,4,5*
Meng Zhao1,2,4,5*- 1Department of Hematology, The Seventh Affiliated Hospital, Sun Yat-sen University, Shenzhen, China
- 2Shenzhen Lansi Institute of Artificial Intelligence in Medicine, Shenzhen, China
- 3Health Science Center, The First Affiliated Hospital of Shenzhen University, Shenzhen Second People’s Hospital, Shenzhen, China
- 4Sun Yat-sen Memorial Hospital, RNA Biomedical Institute, Sun Yat-sen University, Guangzhou, China
- 5Key Laboratory of Stem Cells and Tissue Engineering, Zhongshan School of Medicine, Ministry of Education, Sun Yat-sen University, Guangzhou, China
- 6The Fifth Affiliated Hospital of Sun Yat-sen University, Zhuhai, China
Bone marrow mesenchymal stem/stromal cells (BMSCs) can be transformed into tumor-associated MSCs (TA-MSCs) within the tumor microenvironment to facilitate tumor progression. However, the underline mechanism and potential therapeutic strategy remain unclear. Here, we explored that interleukin 17 (IL-17) cooperating with IFNγ transforms BMSCs into TA-MSCs, which promotes tumor progression by recruiting macrophages/monocytes and myeloid-derived suppressor cells (MDSCs) in murine melanoma. IL-17 and IFNγ transformed TA-MSCs have high expression levels of myelocyte-recruiting chemokines (CCL2, CCL5, CCL7, and CCL20) mediated by activated NF-κB signaling pathway. Furthermore, retinoic acid inhibits NF-κB signaling, decreases chemokine expression, and suppresses the tumor-promoting function of transformed TA-MSCs by prohibiting the recruitment of macrophages/monocytes and MDSCs in the tumor microenvironment. Overall, our findings demonstrate that IL-17 collaborating with IFNγ to induce TA-MSC transformation, which can be targeted by RA for melanoma treatment.
Introduction
Mesenchymal stem/stromal cells (MSCs) self-renew and differentiate into adipocytes, osteoblasts, and chondroblasts in bone marrow (BM) (Friedenstein et al., 1966, 1976, 1987), where BM MSCs (BMSCs) produce multiple growth factors, including SCF, CXCL12, Ang, and Wnt ligands, to support hematopoiesis (Kfoury and Scadden, 2015). Furthermore, MSCs reside in various tissues, such as liver, heart, adipose tissue, and lymph node to support their tissue homeostasis and regeneration (Uccelli et al., 2008). Additionally, tissue-resident MSCs can regulate immune response by producing various immunoregulatory molecules, such as TGF-β, NOS2, PEG2, and PD-L1 (Jiang and Xu, 2020).
MSCs are also involved in tumor progression (Pietras and Ostman, 2010). Tumor-associated MSCs (TA-MSCs) support tumor cell growth and angiogenesis by secreting multiple growth factors, such as BMP and VEGF (Beckermann et al., 2008; McLean et al., 2011). TA-MSCs also suppress immunosurveillance in the tumor microenvironment by inhibiting adaptive and innate immune cells. TA-MSCs suppress T cells by producing immune suppressive factors, such as NOS2, IDO, and PD-L1 (Ren et al., 2008). More importantly, TA-MSCs can recruit macrophages and myeloid-derived suppressor cells (MDSCs) into the tumor microenvironment through CC-chemokine receptor 2 (CCR2) ligands, including CC-chemokine ligand 2 (CCL2), CCL7, and CCL12 (Ren et al., 2012). The recruited macrophages and MDSCs further suppress immune surveillance and promote tumor growth within the tumor microenvironment to promote tumor growth (Qian and Pollard, 2010; Kumar et al., 2016).
TNFα, a proinflammatory cytokine highly expressed in tumor inflammatory environment, can transform BMSCs to TA-MSCs, which produce high-level CCR2 ligands to promote tumor growth by recruiting monocytes/macrophages (Ren et al., 2012). Interleukin 17 (IL-17) is an important pro-inflammatory cytokine secreted by CD4+ Th17 and CD8+ Tc17 cells and highly expressed in tumor microenvironment (Miossec et al., 2009). Deletion of IL-17 reduces MDSCs in tumor microenvironment and inhibits tumor growth (He et al., 2010; Wu et al., 2014). However, whether IL-17 participates in TA-MSC transformation to support tumor growth within tumor microenvironment remains unknown. Retinoic acid (RA), a metabolite of vitamin A (Cunningham and Duester, 2015), can induce differentiation of acute promyelocytic leukemia cells (de Thé, 2018). Studies suggest that RA could inhibit solid tumor growth and regulate the tumor microenvironment (Abu et al., 2005; Bolis et al., 2020; Sun et al., 2020). Here, we found that IL-17 incorporating with IFNγ transforms BMSCs into TA-MSCs to promote tumor growth, which is inhibited by RA treatment in melanoma.
Results
IL-17 and IFNγ Transform BMSCs Into TA-MSCs to Facilitate Melanoma Progress in vivo
To explore the role of IL-17 in transforming BMSCs to TA-MSCs, we investigated the tumor growth co-engrafted with BMSCs and IL-17 transformed MSCs. We subcutaneously inoculated B16F0 melanoma cells with normal BMSCs or BMSCs pretreated with IL-17 and IFNγ, respectively, or jointly into C57BL/6 mice (Figure 1A). The B16F0 melanoma cells with BMSCs, which were pretreated with IL-17 and IFNγ jointly, gave more aggressive tumor growth compared to control B16F0 melanoma cells (5.2-fold increase in tumor weight, and 4-fold increase in tumor size). However, B16F0 melanoma cells with normal BMSCs or BMSCs treated with IL-17 and IFNγ respectively, did not show a significant difference in tumor weight or volume compared to control B16F0 melanoma cells (Figures 1B,C). This indicated that IL-17 incorporated with IFNγ to stimulate the BMSC to TA- MSC transformation, which promoted tumor growth in melanoma. Furthermore, we investigated that whether IL-17 and IFNγ transformed TA-MSCs can recruit monocytes/macrophages and MDSCs. Our FACS assay showed that the myelocytes, including macrophages, monocytes, and neutrophils, were dramatically increased in peripheral blood when melanoma mice were co-inoculated with IL-17 and IFNγ transformed TA-MSCs (1. 5-, 2. 5-, and 1.6-fold increase, respectively) (Figure 1D). However, no significant increase of circulating T cells was observed in mice co- engrafted with pretreated TA-MSCs compared to mice with control melanoma cells (Figure 1E). No significant increase of either myelocytes or T cells was observed in peripheral blood when melanoma mice were co-inoculated with normal BMSCs or BMSCs pretreated with IL-17 or IFNγ individually (Figure 1D). More importantly, the increased macrophages, monocytes, and MDSCs were observed in the tumor microenvironment (2. 5-, 3. 5-, and 1.6-fold increase, respectively) when melanoma mice were co-engrafted with IL-17 and IFNγ transformed TA-MSCs (Figure 1F). However, co-inoculation of normal BMSCs or BMSCs pretreated with IL-17 or IFNγ individually did not increase the numbers of macrophages and monocytes in the tumor microenvironment, and the BMSC induced slight increase of MDSCs was not statistically significant (Figure 1F). Furthermore, tumor-resident T cells were not regulated by control BMSCs or by transformed TA-MSCs (Figure 1G).
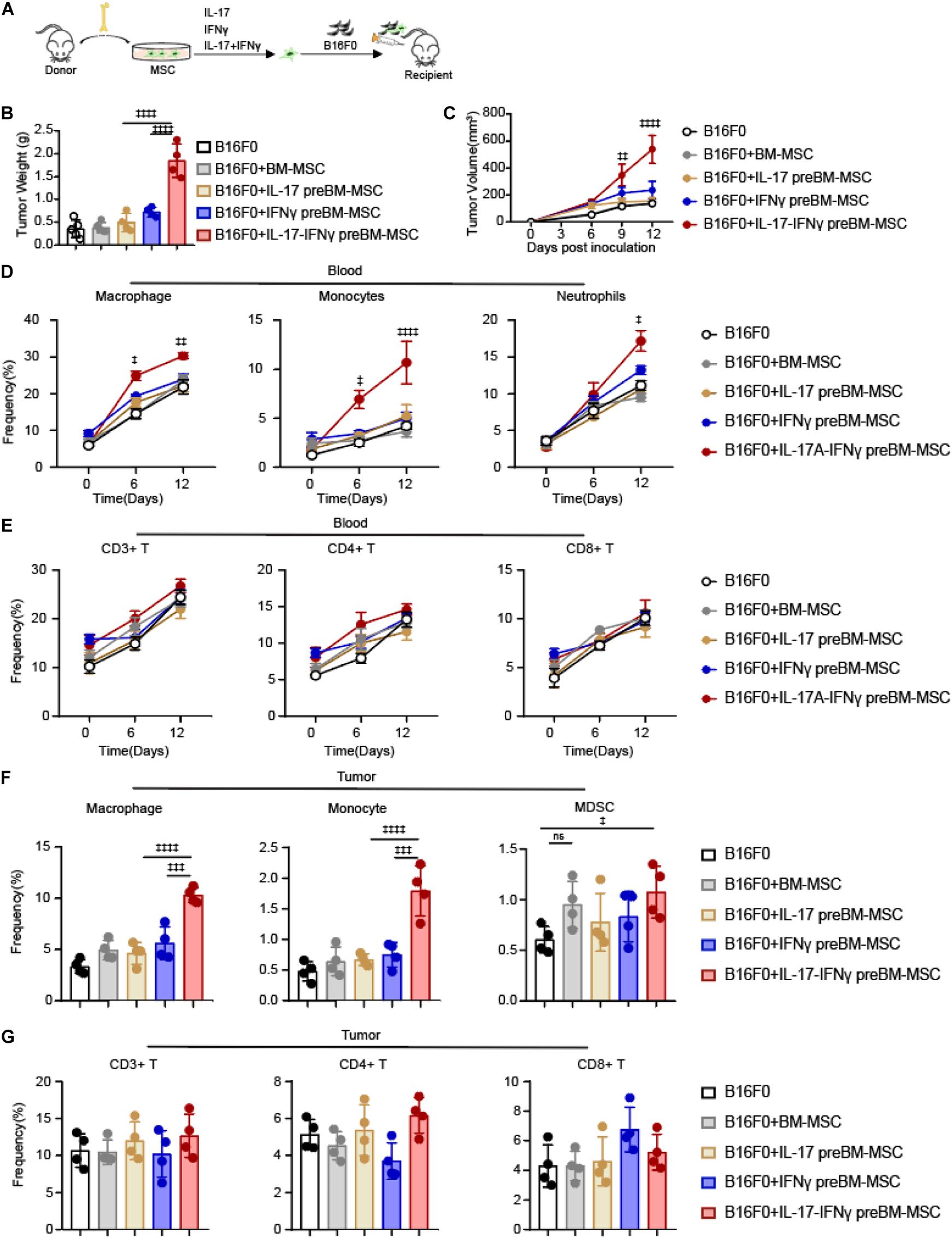
Figure 1. IL-17 and IFNγ transform BMSCs into TA-MSCs to facilitate melanoma progress in vivo. (A) Schematic depicting the strategy to investigate the tumor-promotion function of IL-17 and IFNγ transformed BMSCs in murine melanoma. (B,C) Tumor weight (B) and tumor growth curve (C) in melanoma mice inoculated with B16F0 cells alone or with IL-17 and/or IFNγ pre-treated BMSCs (n = 4–5). (D,E) The percentage of circulated macrophages, monocytes, neutrophils (D), and T cells (E) in the peripheral blood. (F,G) The frequency of resident macrophages, monocytes, MDSCs (F), and T cells (G) in the tumor tissues in melanoma mice inoculated with B16F0 cells alone or with IL-17 and/or IFNγ pre-treated BMSCs (n = 4). Data represent mean ± SD of 3 independent experiments. ‡p < 0.05, ‡‡p < 0.01, ‡‡‡p < 0.001, ‡‡‡‡p < 0.0001. ns, not significant.
Overall, our observations showed that IL-17 and IFNγ jointly but not individually transformed normal BMSCs into TA-MSCs, and the IL-17 and IFNγ transformed TA-MSCs can recruit myelocytes into the tumor microenvironment to promote tumor growth.
IL-17 and IFNγ Synergistically Increase Immunoregulatory Genes in BMSCs
To investigated the underlining mechanism that IL-17 and IFNγ transform BMSCs to TA-MSCs, we analyzed the expression of immunoregulatory molecules in BMSCs after IL-17 and/or IFNγ stimulation. Transcriptional analysis showed that IL-17 and IFNγ synergistically increased the expression level of immunosuppressors, such as NOS2, PD-L1, CXCL9, and CXCL10 (1, 711-, 280-, 1, 742-, and 2,035-fold increase, respectively) in transformed TA-MSCs compared to control BMSCs. IFNγ individual treatment also significantly increased the expression of immunosuppressors (106-, 62-, 573-, and 218-fold increase, respectively), however, IL-17 treatment did not show a significant effect on these immunosuppressors (Figure 2A). Furthermore, IL-17 and IFNγ synergistically stimulate a dramatic increase of myelocyte recruiting chemokines, including CCL2, CCL5, CCL7, and CCL20 (218-, 8-, 27-, and 13-fold increase, respectively) in transformed TA-MSCs, although a slight increase was observed in BMSCs after IL-17 treatment (6-, 2-, 4-, and 3-fold increase, respectively) (Figure 2B). No significant increase of myelocyte recruiting chemokines was observed in BMSCs after IFNγ treatment (Figure 2B).
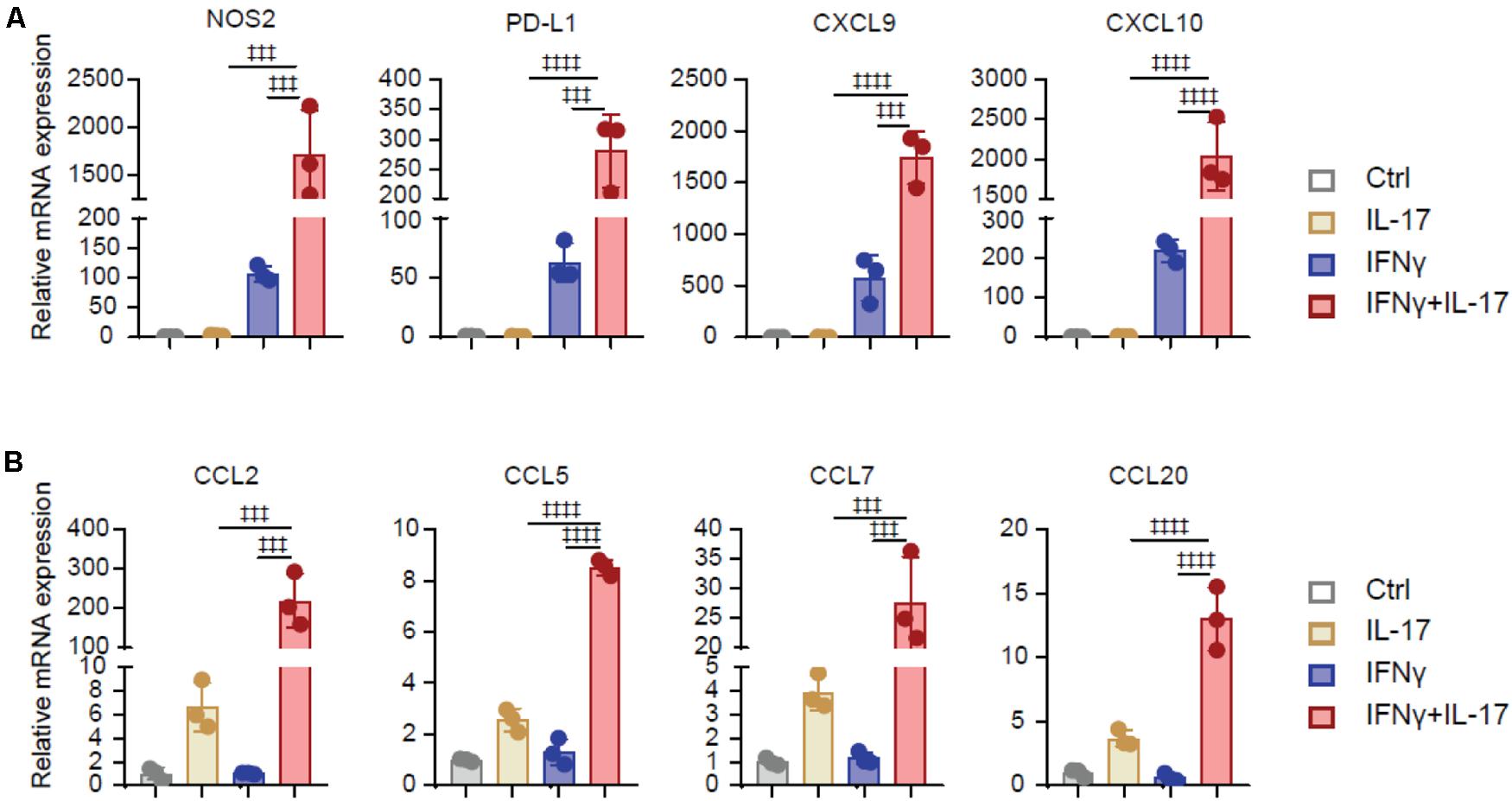
Figure 2. IL-17 and IFNγ synergistically increase immunoregulatory genes in BMSCs. (A,B) The relative mRNA expression level of immunoregulatory molecules (NOS2, PD-L1, CXCL9, and CXCL10) (A) and myelocyte-recruiting chemokines (CCL2, CCL5, CCL7, and CCL20) (B) in the BMSCs treated with IL-17 and IFNγ, respectively, or jointly. Data represent mean ± SD of 3 independent experiments. ‡‡‡p < 0.001, ‡‡‡‡p < 0.0001. ns, not significant.
Taken together, these data illustrated that IL-17 and IFNγ synergistically increased the expression of immunosuppressive factors and myelocyte recruiting factors in BMSCs, but IL17 or IFNγ individual treatment had a limited effect on TA-MSC transformation.
RA Inhibits TA-MSC Transformation and Further Suppresses Melanoma Progress in vivo
RA inhibits Th17 differentiation (Mucida et al., 2007; Elias et al., 2008), which suggested that RA might inhibit IL-17 signaling. Therefore, we investigated whether RA regulates IL-17 mediated TA-MSC transformation. We simultaneously supplied RA during IL-17 and IFNγ mediated TA-MSC transformation, and further performed co-engrafted cell-derived xenograft experiments with B16F0 melanoma cells and transformed TA-MSCs or RA treated TA-MSCs (Figure 3A). Intriguingly, RA supplement dramatically inhibited the tumor-promoting capacity of IL-17 and IFNγ transformed TA-MSCs, which had 68% reduction in tumor weight (Figure 3B) and 53% reduction in tumor volume compared to TA-MSCs without RA treatment (Figure 3C). To explore the underline mechanism, we further analyzed the myelocytes in peripheral blood and tumor microenvironment. We surprisingly found that RA remarkably inhibited the myelocyte recruiting function of TA-MSCs, with 64% decrease of macrophages, 53% decrease of monocytes, and 89% decrease of neutrophils in peripheral blood (Figure 3D). More importantly, RA- treated TA-MSCs completely failed to recruit macrophages, monocytes, and MDSCs into the tumor microenvironment (85, 83, and 108% decrease, respectively) (Figure 3F). Consistent with our previous observation, no significant change of circulating T cells (Figure 3E) or tumor-resident T cells (Figure 3G) was observed in mice co-engrafted with TA-MSCs or RA treated TA-MSCs compared to mice with control BMSCs.
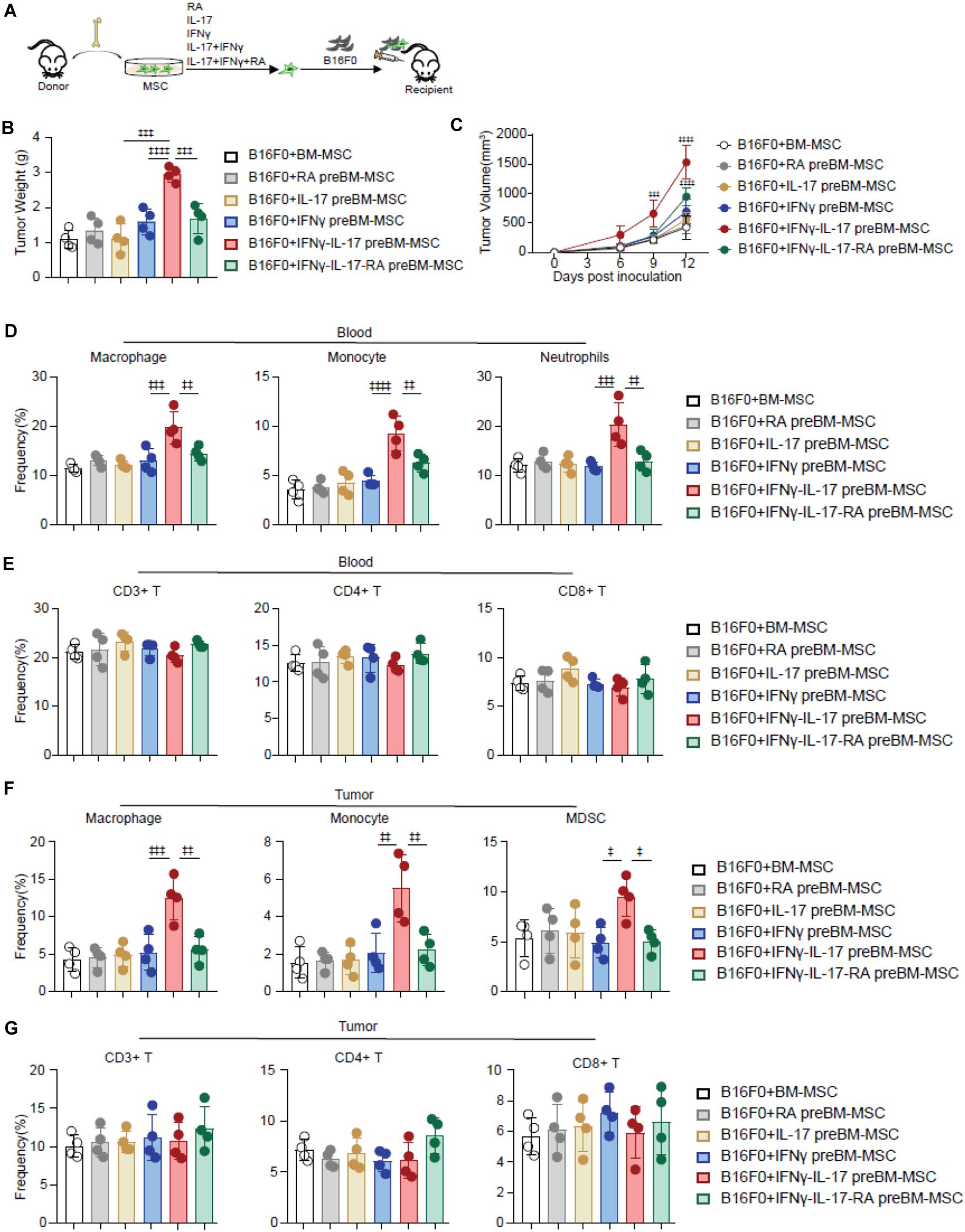
Figure 3. RA inhibits TA-MSC transformation and suppresses TA-MSC mediated melanoma progress in vivo. (A) Schematic depicting the strategy to investigate the inhibition role of RA on IL-17 and IFNγ mediated TA-MSC transformation. (B,C) Tumor weight (B) and tumor growth curve (C) of mice inoculated with B16F0 cells, and B16F0 cells with BMSCs with indicated treatment (n = 4–5). (D,E) The percentage of circulated macrophages, monocytes, neutrophils (D), and T cells (E) in the peripheral blood. (F,G) The frequency of macrophages, monocytes, MDSCs (F) and T cells (G) in the tumor microenvironment of mice inoculated with B16F0 cells, and B16F0 cells with BMSCs with indicated treatment at 12 days after tumor cell inoculation (n = 4). Data represent mean ± SD of 3 independent experiments. ‡p < 0.05, ‡‡p < 0.01, ‡‡‡p < 0.001, ‡‡‡‡p < 0.0001. ns, not significant.
Overall, our data demonstrated RA treatment significantly blocked IL-17 and IFNγ mediated TA-MSC transformation in promoting tumor growth in melanoma.
RA Inhibits IL-17-Stimulated Myelocyte-Recruiting Chemokine Expression in BMSCs Through Inhibiting NF-κB Signaling Pathway
To explore the molecular mechanism that RA suppressed IL-17 and IFNγ mediated TA-MSC transformation, we first analyzed the expression of immunoregulatory molecules in IL-17 and IFNγ transformed TA-MSCs. Intriguingly, RA completely blocked the increase of myelocyte recruiting chemokines expression, including CCL2, CCL5, CCL7, and CCL20, in IL-17 and IFNγ transformed TA-MSCs (Figure 4A). We also noticed that IL-17 treatment alone also slightly increased myelocyte recruiting chemokines expression, which was also completely blocked by RA treatment (87, 89, 90, and 83% reduction) (Figure 4B). However, RA did not inhibit the immunosuppressive molecule expression, including NOS2, PD-L1, CXCL9, CXCL10 (Figure 4C). This indicated that RA inhibited TA-MSC transformation mainly by blocking their ability to recruit myelocytes for tumor-promoting.
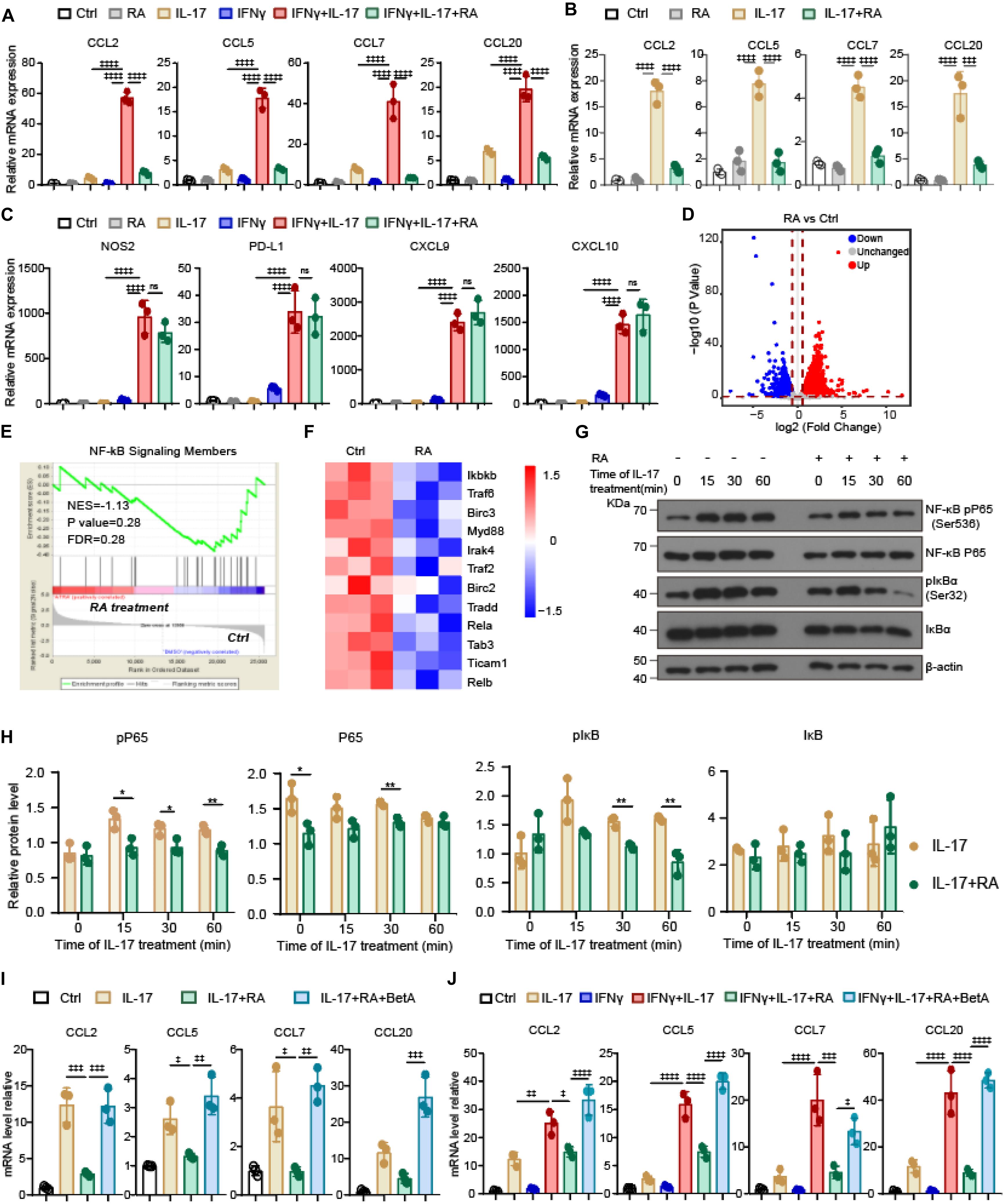
Figure 4. RA inhibits IL-17-stimulated myelocyte-recruiting chemokine expression in BMSCs through inhibiting NF-κB signaling pathway. (A) The relative mRNA expression level of myelocyte-recruiting chemokines (CCL2, CCL5, CCL7, and CCL20) in the IL-17 and IFNγ pre-treated BMSCs with or without RA treatment as indicated. (B) The relative mRNA expression level of myelocyte-recruiting chemokines in the BMSCs after IL-17 treatment with or without the RA treatment as indicated. (C) The relative mRNA expression level of immunoregulatory molecules (NOS2, PD-L1, CXCL9, and CXCL10) in the IL-17 and IFNγ pre-treated BMSCs with or without RA treatment as indicated. (D) Volcano plots illustrating the biological and statistical significance of genes between BMSCs with and without RA treatment. Upregulated genes are highlighted in red, unchanged genes are highlighted in gray, and downregulated genes are highlighted in blue. (E) Signature enrichment plots from GSEA using NF-κB pathway gene-set in BMSCs treated with and without RA. The enriched p-value is derived from Fisher’s exact test. (F) A heatmap revealed NF-κB-pathway gene expression changes after RA treatment in BMSCs. The rows show Z-scores calculated for each gene. (G,H) Immunoblotting analysis of key NF-κB pathway elements in the BMSCs exposed to IL-17 with or without the RA treatment as indicated. (I) The relative mRNA expression of myelocyte-recruiting chemokines in BMSCs treated by IL-17, RA, and BetA as indicated. (J) The relative mRNA expression of myelocyte-recruiting chemokines in BMSCs treated by IFNγ, IL-17, RA, and BetA as indicated. Data represent mean ± SD of 3 independent experiments. * or ‡p < 0.05, ** or ‡‡p < 0.01, ‡‡‡p < 0.001, ‡‡‡‡p < 0.0001. ns, not significant.
To understand the molecular mechanism that RA inhibits IL-17 signaling to suppress myelocyte recruiting chemokine expression, we performed transcription analysis for BMSCs under RA treatment. Our RNA sequencing (RNA-seq) analysis successfully detected 14,423 genes, in which 1,474 genes were upregulated and 1,393 genes were downregulated in BMSCs after RA treatment (Figure 4D). We noticed that NF-κB pathway, which stimulates CCL2 release in TA-MSCs (Katanov et al., 2015), was inhibited in BMSCs upon RA treatment (Figure 4E). Strikingly, major NF-κB pathway elements were downregulated in BMSCs after RA treatment (Figure 4F). Furthermore, RA treatment significantly inhibited IL-17 stimulated phosphorylation of RalA-p65 and IκBα, two key molecules in NF-κB pathway, in BMSCs (Figures 4G,H). This indicated that RA might inhibit NF-κB pathway to suppress the expression of myelocyte recruiting chemokines in IL-17 transformed TA-MSCs. To confirm this, we employed NF-κB specific activator, betulinic acid (BetA) (Kasperczyk et al., 2005), to rescue the suppressed NF-κB signaling in IL-17 transformed TA-MSCs upon RA treatment. Notably, BetA treatment completely rescued the expression of CCL2, CCL5, CCL7, and CCL20 in IL-17 transformed TA-MSCs under RA treatment (Figure 4I). The rescue effect was also observed in IL-17 and IFNγ transformed TA-MSCs (Figure 4J).
Taken together, our data showed that IL-17 activates NF-κB pathway to upregulate myelocyte recruiting chemokines in TA-MSCs, and the TA-MSC transformation was significantly blocked by RA treatment due to inhibition of NF-κB signaling pathway.
RA Inhibits TNFα Mediated Chemokine Expression in TA-MSCs by Blocking NF-κB Signaling Pathway
TNFα transforms TA-MSCs through upregulating myelocyte recruiting chemokines (Ren et al., 2012), therefore, we asked whether RA inhibits TNFα mediated TA-MSC transformation. Interestingly, RA treatment significantly blocked the upregulation of CCL2, CCL5, CCL7, and CCL20 in TNFα treated BMSCs (61, 85, 52, and 58% decrease, respectively) (Figure 5A). Consistently, the RA mediated myelocyte recruiting chemokine expression inhibition was also observed in TNFα and IFNγ treated BMSCs (65, 77, 79, and 76%, decrease, respectively) (Figure 5B). However, we did not observe that RA significantly inhibited immunoregulatory molecules, including NOS2, PD-L1, CXCL9, and CXCL10, which were stimulated by TNFα and IFNγ in BMSCs (Figure 5C). This is consistent with the previous report that TNFα educated TA-MSCs to recruit macrophages to promote tumor growth (Ren et al., 2012).
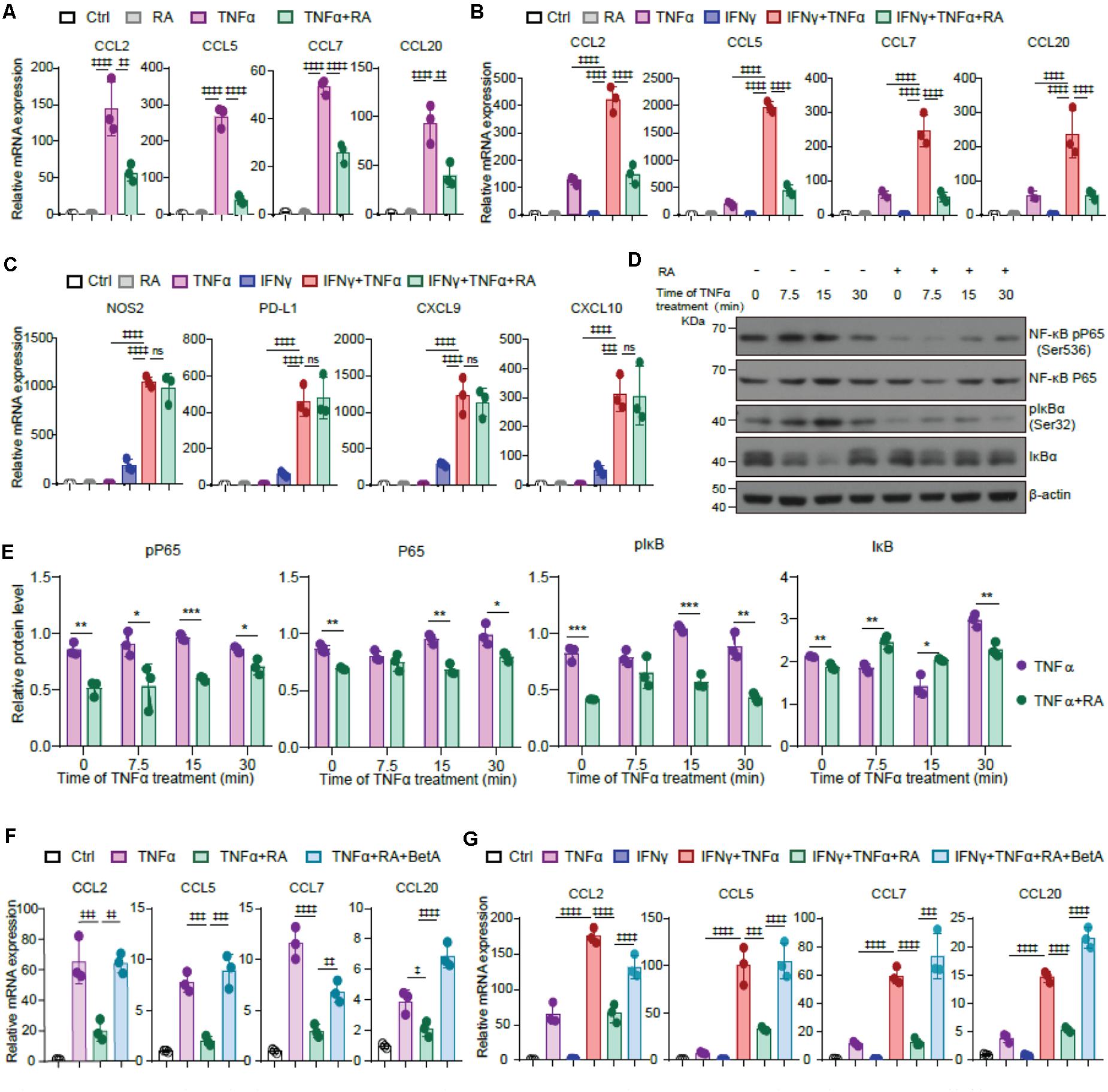
Figure 5. RA inhibits TNFα mediated chemokine expression in TA-MSCs by blocking NF-κB signaling pathway. (A) The mRNA expression of myelocyte-recruiting chemokines (CCL2, CCL5, CCL7, and CCL20) in BMSCs treated with TNFα and RA as indicated. (B,C) The relative mRNA expression of myelocyte-recruiting chemokines (B) and immunoregulatory molecules (NOS2, PD-L1, CXCL9, and CXCL10) (C) in the TNFα and IFNγ treated BMSCs with or without the RA treatment as indicated. (D,E) Immunoblotting analysis of NF-κB RelA p65, IκBa, and pIκBa (Ser32) in the BMSCs exposed to TNFα and RA as indicated. (F) The relative mRNA expression of myelocyte-recruiting chemokines in BMSCs treated by TNFα and RA with or without the BetA as indicated. (G) The relative mRNA expression of myelocyte-recruiting chemokines in BMSCs treated by IFNγ, TNFα, RA, and BetA as indicated. Data represent mean ± SD of 3 independent experiments. ∗p < 0.05, ∗∗ or ‡‡p < 0.01, ∗∗∗ or ‡‡‡p < 0.001, ‡‡‡‡p < 0.0001. ns, not significant.
As NF-κB pathway is critical to CCL family regulation in IL-17 transformed TA-MSCs, we next investigated the role of NF-κB pathway in TNFα mediated TA-MSC transformation. Consistently, we observed that TNFα treatment activated NF-κB pathway in BMSCs, which was evidenced by the activation of NK-κB pP65 and pIκBα, which was coupled with the reduction of IκBα, at 7.5–15 min after TNFα treatment. Notably, RA treatment remarkably attenuated the activation of NFκB pathway in BMSCs under TNFα treatment (Figures 5D,E). More importantly, NF-κB activator, BetA, completely rescued the decrease of CCL2, CCL5, CCL7, and CCL20 under RA treatment in TNFα educated BMSCs (Figure 5F) and BMSCs treated with TNFα and IFNγ simultaneously (Figure 5G).
Taken together, these data demonstrated that RA inhibited NF-κB pathway to suppress the expression of myelocyte chemokines in TNFα educated TA-MSCs.
Discussion
BMSCs can be transformed into TA-MSCs, which is featured by producing high-level CCR2 ligands to recruit monocytes/macrophages and MDSCs in promoting tumor growth (Ren et al., 2012; Wang Y. et al., 2014; Lin et al., 2016). Tumor proinflammatory cytokine, TNFα, efficiently transforms BMSCs into TA-MSCs and promotes tumor growth in lymphoma, melanoma, and breast carcinoma (Ren et al., 2012; Katanov et al., 2015). IL-17 is involved in inflammatory process and enhances the expression of an immunosuppressive molecule, NOS2, in murine hepatitis (Oukka, 2008; Han et al., 2014). However, unlike innate immune cell generated TNFα, IL-17 is derived from T cells (Oukka, 2008). IL-17 has both pro-tumor and anti-tumor effects. IL-17 inhibits tumor progression and metastasis in melanoma and colon cancer by promoting the function of T cells and NK cells (Kryczek et al., 2009; Martin-Orozco et al., 2009). However, growing evidences show that IL-17 promotes tumor growth in various solid tumors, including melanoma, breast cancer, colon cancer, and hepatocellular carcinoma (Wang et al., 2009; Grivennikov et al., 2012; Coffelt et al., 2015; Gomes et al., 2016). Genetic evidence shows that IL-17 can directly promote proliferation of transformed colonic epithelial cells tumor through its type A receptor (IL-17RA) (Wang K. et al., 2014). Here, we found that IL-17 cooperating with IFNγ to transform TA-MSCs in supporting tumor growth in melanoma. These suggested that blocking IL-17 signaling may inhibit melanoma cells through multiple mechanisms. Out work also suggested that adaptive immune cells can modulate the protumorigenic function of TA-MSCs, which recruits macrophages to support tumor growth (Ren et al., 2012). Intriguingly, we observed that IL-17 treatment alone cannot efficiently transform BMSCs to TA-MSCs, due to the less myelocyte recruiting chemokine expression, and limited ability to recruit macrophages and MDSCs. However, IFNγ remarkably strengthened the myelocyte recruiting chemokines upregulation ability of IL-17, therefore IFNγ and IL-17 synergistically promoted BMSC to TA-MSC transformation. Moreover, TNFα induces cell necroptosis and apoptosis (Locksley et al., 2001; Kalliolias and Ivashkiv, 2016), therefore TNFα transformed TA-MSCs may have limited ability to promote tumor growth. However, IL-17 promotes cell proliferation (Wu et al., 2014), presumably, IL-17 transformed TA-MSCs may have greater efficiency to promote tumor growth. Accordingly, high IL-17 level is observed in colon cancer, skin cancer, and lung cancer patients with poor clinical outcome (Marshall et al., 2016; Razi et al., 2019; Bellone et al., 2020). Recent work showed that IL-17 also regulates the protumorigenic function of cancer-associated fibroblasts (CAFs) (Mucciolo et al., 2021), which are also transformed from normal BMSCs (Quante et al., 2011). Therefore, further studies are warranted to determine the discrepancy between TA-MSCs and CAF in regulating melanoma.
ATRA has revolutionized the treatment of acute promyelocytic leukemia (de Thé, 2018). However, the application of ATRA in solid tumors remains to be explored. RA can inhibit tumor cell proliferation in melanoma (Edward and MacKie, 1989; Zhang and Rosdahl, 2005; Li and Han, 2020) and promote immune surveillance in breast cancer, colorectal cancer, and melanoma by influencing the metabolism of MDSCs, upregulating genes related to immune response, and supporting the survival of tumor-specific CD8+ T cells (Guo et al., 2012; Paroni et al., 2020; Sun et al., 2020). Conversely, RA treatment was also reported to benefits tumor progression in sarcoma and chronic lymphocytic leukemia (CLL) by promoting the pro-tumoral differentiation of intertumoral monocytes in sarcoma and increasing CD38 expression in CLL cells (Chen et al., 2018; Devalaraja et al., 2020). Our study showed that RA treatment almost completely inhibited the increase of myelocyte recruiting ability of IL-17 and IFNγ transformed TA-MSCs, although it barely influenced the expression of the immunosuppressive molecules induced by IFNγ. RA treatment successfully inhibited the BMSC to TA-MSC transformation and significantly inhibited tumor growth in melanoma, which opens an avenue for tumor microenvironment targeting therapy.
Both IL-17 and TNFα can activate NF-κB signaling pathway (Sugita et al., 2002; Taniguchi and Karin, 2018) and we confirmed that NF-κB signaling pathway was activated in BMSCs under IL-17 or TNFα treatment. Moreover, NF-κB pathway activation is proved to be important for the paracrine function of tumor-derived-MSCs and cancer-associated fibroblasts in lung cancer and breast cancer in secreting CCL2, IL-6, and IL-8 in the tumor microenvironment (Katanov et al., 2015; Li et al., 2016; Bai et al., 2017; Su et al., 2018). RA is shown to inhibit NF-κB pathway in LPS-stimulated monocytes and renal cells through RARα-STAT1-dependent or TLR4-dependent mechanisms (Austenaa et al., 2009; Sierra-Mondragon et al., 2018). Our finding showed that RA inhibited NF-κB pathway in IL-17- or TNFα-treated BMSCs, which indicates that RA inhibits proinflammatory-factor-mediated BMSC to TA-MSC transformation by inhibiting NF-κB pathway. Indeed, NF-κB pathway activator completely recovered the TA-MSC transformation, which was inhibited by RA treatment in TNFα or IL-17 transformed TA-MSCs. NF-κB promotes tumor growth (Barcellos-de-Souza et al., 2016; Yu et al., 2017). Consistently, our work showed that NF-κB stimulated the tumor supporting function of TA-MSCs. Although IL-17 is considered as a modest activator of NF-κB pathway (Shen and Gaffen, 2008), evidence suggest that IL-17 activates NF-κB through multiple avenues, such as mitogen-activated protein kinase (MAPK) pathway and transforming growth factor β-activated kinase (TAK)1 (Amatya et al., 2017).
Collectively, our study identified IL-17 can educate healthy BMSCs into TA-MSCs, and uncovered a new therapeutic approach to target TA-MSCs by RA. This finding may extend the mechanism and application of RA in tumor therapy.
Materials and Methods
Reagents and Mice
Murine IFNγ (315-05-100), IL-17A (210-17) were purchased from PEPROTECH. Murine TNFα (410-MT) was purchased from R&D Systems. Retinoic acid (PHR1187) was purchased from Sigma-Aldrich. Betulinic acid (BetA, HY-10529) was purchased from MedChemExpress. Monoclonal antibodies to CD11b (M1/70), F4/80 (BM8), and Ly6G (17-5931-82) were purchased from eBioscience and Gr-1 (RB6-8C5) was purchased from BioLegend. Primary antibodies for western blotting against P65 (rabbit, 1:1,000, 8,242), pP65 (Ser536) (rabbit, 1:1,000, 3,033), IκBα (rabbit, 1:1,000, 4,812), pIκBα (Ser32) (rabbit, 1:1,000, 2,859), β-actin (rabbit, 1:1,000, 4,970) were purchased from Cell Signaling Technology.
C57BL/6 mice were bred under specific pathogen-free conditions in the animal facility of Sun Yat-sen university. All animal protocols were approved by our Institutional Animal Care and Use Committee.
Cell Culture
BMSCs were isolated from the tibia and femur bone marrow of C57BL/6 mice following the protocol described in the previous reference (Ren et al., 2012). Cells were maintained in DMEM low-glucose medium (10-014-CVR, CORNING) supplemented with 20% fetal bovine serum (12483020, Gibco), 2% penicillin-streptomycin (SV30010, Invitrogen) and 10 μM ROCK inhibitor (S1049, Selleck) in the adhesive petri dishes. All non-adherent cells were removed after 24 h, and adherent cells were maintained. To obtain MSC clones, cells maintained in 10 cm dishes at 80–90% density were harvested and seeded into 6-well plates at a density of 5 × 105 cells/well. Cells were used before the 3rd passage. B16F0 cells were maintained in DMEM high-glucose medium (10-013-CVR, CORNING) supplemented with 10% FBS and 1% penicillin-streptomycin.
RNA Isolation and Gene Expression Assay
Before RNA isolation, BMSCs were incubated with or without cytokines of (50 ng ml–1 IL-17, 10 ng ml–1 IFNγ, and 10 ng ml–1 TNFα) or drugs (100 nM RA and 10 μg ml–1 BetA), respectively, or jointly for 6 h (Han et al., 2014; Song et al., 2015). Total mRNA was isolated with MagZolTM Reagent (R4801-03, Magen) according to the manufacturer’s instruction. mRNA purity and quantity were determined with NanoDrop (Thermo Scientific) before qPCR and RNA-seq analysis. For Real-Time qPCR, cDNA was synthesized from mRNA by using the TransScript All-in-One First-Strand cDNA Synthesis SuperMix for qPCR (One-Step gDNA Removal) Kit (AT341, Transgen). Quantitative Real-Time PCR was performed on Bio-Rad CFX96 TouchTM Real-Time PCR Detection system with SYBR Green I Master Mix reagent (11203ES03, YEASEN). Sequences of forward and reverse primer pairs are as follows:
| Gene | Forward primer | Reverse primer |
| sequence (5′-3′) | sequence (5′-3′) | |
| Nos2 | GTTCTCAGCCCAACAATACAAGA | GTGGACGGGTCGATGTCAC |
| PD-L1 | GCTCCAAAGGACTTGTACGTG | TGATCTGAAGGGCAGCATTTC |
| Cxcl9 | TCCTTTTGGGCATCATCTTCC | TTTGTAGTGGATCGTGCCTCG |
| Cxcl10 | CCAAGTGCTGCCGTCATTTTC | GGCTCGCAGGGATGATTTCAA |
| CCL2 | TTAAAAACCTGGATCGGAACCAA | GCATTAGCTTCAGATTTACGGGT |
| CCL5 | GCTGCTTTGCCTACCTCTCC | TCGAGTGACAAACACGACTGC |
| CCL7 | GCTGCTTTCAGCATCCAAGTG | CCAGGGACACCGACTACTG |
| CCL20 | GCCTCTCGTACATACAGACGC | CCAGTTCTGCTTTGGATCAGC |
| β-actin | GGCTGTATTCCCCTCCATCG | CCAGTTGGTAACAATGCCATGT |
Tumor Transplantation
1 × 105 BMSCs which were pre-treated with or without 50 ng ml–1 IL-17, 10 ng ml–1 IFNγ, and 100 nM RA, respectively, or jointly for 12 h before subcutaneously injection with 2.5 × 105 B16F0 into recipient C57BL/6 mice. Tumor size and weight were measured at various time points. Peripheral blood was collected on the 6th and 12th day and resultant tumors were harvested on the 12th day after tumor cell inoculation for further analysis.
Flow Cytometry
For cell population analysis, cells isolated from peripheral blood and tumors were suspended in staining buffer (PBS, 2% FBS) at a concentration of 2 × 106 cells ml–1 and 100 ml of suspension was incubated with fluorescently labeled antibodies for 1 h on ice. Macrophages were gated as CD11b+ F4/80+. Neutrophils were gated as CD11b+ Ly6G+. Monocytes were gated as CD11b+Gr-1+ Ly6G–. MDSCs were gated as CD11b+Gr-1+. Analyses were performed using a flow cytometer (Attune NxT; Thermo Fisher). The immune cell frequency was calculated as the frequency of each immune cell population in total nucleated cells in peripheral blood or total resident nucleated blood cells from tumor site.
Western Blotting
For immunoblotting analysis, BMSCs incubated with 50 ng ml–1 IL-17 for 15, 30, and 60 min or 10 ng ml–1 TNFα for 7.5, 15, 30 min were pre-challenged by 100 nM RA for 6 h. Cells were washed with ice-cold PBS, harvested and lysed for 15 min by lysis buffer containing 0.5% TritonX-100 (T9284, Sigma), 20 mM Hepes pH7.4 (H-4034, Sigma), 150 mM NaCl (A100241, Sangon Biotech), 12.5 mM β-glycerophosphate (A500486, Sangon Biotech), 1.5 mM MgCl2 (M4880, Sigma), 2 mM EGTA (A600077, Sangon Biotech), and a cocktail of protease inhibitors, Na3VO4 (A600869, Sangon Biotech), NaF (A500850, Sangon Biotech), and PMSF (A610425, Sangon Biotech). Equal amounts of protein extracts were resolved in 10% SDS-PAGE and transferred to PVDF membranes (IPVH00010, Merck Millipore). The membranes were blocked with 5% non-fat milk in Tris-buffered saline with Tween-20 (TBST, pH 7.6) for 1 h at room temperature before incubated overnight with the primary antibodies (p65 1:1,000, pp65 (Ser536) 1:1,000, IκBα 1:1,000, pIκBα (Ser32) 1:1,000, β-actin 1:1,000) at 4°C and then incubated with the secondary antibodies (rabbit, 1:10,000, W401B, Promega) for 1 h at room temperature. Finally, the blots were detected by enhanced chemiluminescent reagents (Millipore).
Gene Set Enrichment Analysis (GSEA)
RNA of control MSC and RA pretreated MSCs (100 nM RA for 24 h) were used for RNAseq analysis. Raw data.QZ files were imported into GSEA 3.0 software where background correction and normalization were performed with standard default settings. The.QZ files were combined into one.gct file in GenePattern, then imported into GSEA along with a matching phenotype label file (.cls). GSEA analysis was run with the following parameters: number of permutations = 1,000, collapse dataset to gene symbols = false, permutation type = gene_set, plot graphs for the top sets of each phenotype = 150 (default = 20), gene sets database = h.all.v6.0 symbols.gmt (all hallmarks, version 6), with a phenotype comparison of RA pre-treatment vs. control BMSCs. Leading edge analysis was completed on the Hallmark GSEA output with NF-κB signaling hallmark gene set.
Statistical Analysis
The statistical analysis was performed using GraphPad Prism 8.0 software. Two-tailed Student’s t tests were used for the comparison between two groups (*p < 0.05, **p < 0.01, and ***p < 0.001) and the one-way ANOVAs with Tukey’s multiple comparison tests were used for the comparison between more than two groups (‡p < 0.05, ‡‡p < 0.01, ‡‡‡p < 0.001, and ‡‡‡‡p < 0.0001). The two-way ANOVAs with Tukey’s multiple comparison tests were used for comparison between more than two groups at various time points (‡p < 0.05, ‡‡p < 0.01, ‡‡‡p < 0.001, and ‡‡‡‡p < 0.0001). All data are expressed as mean ± SD.
Data Availability Statement
The accession number for the RNA-seq data reported in our manuscript is GEO: GSE169145.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee, SYSU.
Author Contributions
QL, MiZ, and QX designed and performed most of the experiments and analyzed the data. SX, YL, JC, LY, and LW contributed to animal experiments and all the transcriptional assay. LM, DL, and LJ contributed to the discussion. QL, MiZ, and MeZ wrote the manuscript. MeZ supervised the project. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the National Key Research and Development Program of China (2017YFA0103403 and 2018YFA0107200), the Key Research and Development Program of Guangdong Province (2019B020234002), NSFC (81822001, 81900101, 81800164, 81870127, 82000838, and 31871467), Shenzhen Foundation of Science and Technology (JCYJ20170818103626421 and JCYJ20190806164009212), the National Science and Technology Major Project of China (2017YFA0105500), Guangdong Innovative and Entrepreneurial Research Team Program (2016ZT06S029 and 2019ZT08Y485), Sanming Project of Medicine in Shenzhen (No. SZSM201911004), and 111 Project (B13037) for their generous support.
References
Abu, J., Batuwangala, M., Herbert, K., and Symonds, P. (2005). Retinoic acid and retinoid receptors: potential chemopreventive and therapeutic role in cervical cancer. Lancet Oncol. 6, 712–720. doi: 10.1016/s1470-2045(05)70319-3
Amatya, N., Garg, A., and Gaffen, S. (2017). IL-17 signaling: the Yin and the Yang. Trends Immunol. 38, 310–322. doi: 10.1016/j.it.2017.01.006
Austenaa, L., Carlsen, H., Hollung, K., Blomhoff, H., and Blomhoff, R. (2009). Retinoic acid dampens LPS-induced NF-kappaB activity: results from human monoblasts and in vivo imaging of NF-kappaB reporter mice. J. Nutr. Biochem. 20, 726–734. doi: 10.1016/j.jnutbio.2008.07.002
Bai, X., Xi, J., Bi, Y., Zhao, X., Bing, W., Meng, X., et al. (2017). TNF-α promotes survival and migration of MSCs under oxidative stress via NF-κB pathway to attenuate intimal hyperplasia in vein grafts. J. Cell. Mol. Med. 21, 2077–2091. doi: 10.1111/jcmm.13131
Barcellos-de-Souza, P., Comito, G., Pons-Segura, C., Taddei, M., Gori, V., Becherucci, V., et al. (2016). Mesenchymal stem cells are recruited and activated into carcinoma-associated fibroblasts by prostate cancer microenvironment-derived TGF-β1. Stem Cells 34, 2536–2547. doi: 10.1002/stem.2412
Beckermann, B., Kallifatidis, G., Groth, A., Frommhold, D., Apel, A., Mattern, J., et al. (2008). VEGF expression by mesenchymal stem cells contributes to angiogenesis in pancreatic carcinoma. Br. J. Cancer 99, 622–631. doi: 10.1038/sj.bjc.6604508
Bellone, M., Brevi, A., and Huber, S. (2020). Microbiota-propelled T helper 17 cells in inflammatory diseases and cancer. Microbiol. Mol. Biol. Rev. 84:e00064-19. doi: 10.1128/MMBR.00064-19
Bolis, M., Paroni, G., Fratelli, M., Vallerga, A., Guarrera, L., Zanetti, A., et al. (2020). All-trans retinoic acid stimulates viral mimicry, interferon responses and antigen presentation in breast-cancer cells. Cancers 12:1169. doi: 10.3390/cancers12051169
Chen, L., Diao, L., Yang, Y., Yi, X., Rodriguez, B., Li, Y., et al. (2018). CD38-mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discov. 8, 1156–1175. doi: 10.1158/2159-8290.cd-17-1033
Coffelt, S., Kersten, K., Doornebal, C., Weiden, J., Vrijland, K., Hau, C., et al. (2015). IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522, 345–348. doi: 10.1038/nature14282
Cunningham, T., and Duester, G. (2015). Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 16, 110–123. doi: 10.1038/nrm3932
de Thé, H. (2018). Differentiation therapy revisited. Nat. Rev. Cancer 18, 117–127. doi: 10.1038/nrc.2017.103
Devalaraja, S., To, T., Folkert, I., Natesan, R., Alam, M., Li, M., et al. (2020). Tumor-derived retinoic acid regulates intratumoral monocyte differentiation to promote immune suppression. Cell 180, 1098–1114.e1016. doi: 10.1016/j.cell.2020.02.042
Edward, M., and MacKie, R. (1989). Retinoic acid-induced inhibition of lung colonization and changes in the synthesis and properties of glycosaminoglycans of metastatic B16 melanoma cells. J. Cell Sci. 94(Pt 3), 537–543.
Elias, K., Laurence, A., Davidson, T., Stephens, G., Kanno, Y., Shevach, E., et al. (2008). Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood 111, 1013–1020. doi: 10.1182/blood-2007-06-096438
Friedenstein, A., Chailakhyan, R., and Gerasimov, U. (1987). Bone marrow osteogenic stem cells: in vitro cultivation and transplantation in diffusion chambers. Cell Tissue Kinet. 20, 263–272. doi: 10.1111/j.1365-2184.1987.tb01309.x
Friedenstein, A., Gorskaja, J., and Kulagina, N. (1976). Fibroblast precursors in normal and irradiated mouse hematopoietic organs. Exp. Hematol. 4, 267–274.
Friedenstein, A., Piatetzky-Shapiro, I., and Petrakova, K. (1966). Osteogenesis in transplants of bone marrow cells. J. Embryol. Exp. Morphol. 16, 381–390.
Gomes, A., Teijeiro, A., Burén, S., Tummala, K., Yilmaz, M., Waisman, A., et al. (2016). Metabolic inflammation-associated IL-17A causes non-alcoholic steatohepatitis and hepatocellular carcinoma. Cancer Cell 30, 161–175. doi: 10.1016/j.ccell.2016.05.020
Grivennikov, S., Wang, K., Mucida, D., Stewart, C., Schnabl, B., Jauch, D., et al. (2012). Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491, 254–258. doi: 10.1038/nature11465
Guo, Y., Pino-Lagos, K., Ahonen, C., Bennett, K., Wang, J., Napoli, J., et al. (2012). A retinoic acid–rich tumor microenvironment provides clonal survival cues for tumor-specific CD8(+) T cells. Cancer Res. 72, 5230–5239. doi: 10.1158/0008-5472.can-12-1727
Han, X., Yang, Q., Lin, L., Xu, C., Zheng, C., Chen, X., et al. (2014). Interleukin-17 enhances immunosuppression by mesenchymal stem cells. Cell Death Differ. 21, 1758–1768. doi: 10.1038/cdd.2014.85
He, D., Li, H., Yusuf, N., Elmets, C., Li, J., Mountz, J., et al. (2010). IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid-derived suppressor cells. J. Immunol. 184, 2281–2288. doi: 10.4049/jimmunol.0902574
Jiang, W., and Xu, J. (2020). Immune modulation by mesenchymal stem cells. Cell Prolif. 53:e12712. doi: 10.1111/cpr.12712
Kalliolias, G., and Ivashkiv, L. (2016). TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 12, 49–62. doi: 10.1038/nrrheum.2015.169
Kasperczyk, H., La Ferla-Brühl, K., Westhoff, M., Behrend, L., Zwacka, R., Debatin, K., et al. (2005). Betulinic acid as new activator of NF-kappaB: molecular mechanisms and implications for cancer therapy. Oncogene 24, 6945–6956. doi: 10.1038/sj.onc.1208842
Katanov, C., Lerrer, S., Liubomirski, Y., Leider-Trejo, L., Meshel, T., Bar, J., et al. (2015). Regulation of the inflammatory profile of stromal cells in human breast cancer: prominent roles for TNF-α and the NF-κB pathway. Stem Cell Res. Ther. 6:87. doi: 10.1186/s13287-015-0080-7
Kfoury, Y., and Scadden, D. (2015). Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell 16, 239–253. doi: 10.1016/j.stem.2015.02.019
Kryczek, I., Wei, S., Szeliga, W., Vatan, L., and Zou, W. (2009). Endogenous IL-17 contributes to reduced tumor growth and metastasis. Blood 114, 357–359. doi: 10.1182/blood-2008-09-177360
Kumar, V., Patel, S., Tcyganov, E., and Gabrilovich, D. (2016). The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 37, 208–220. doi: 10.1016/j.it.2016.01.004
Li, C., and Han, X. (2020). Co-delivery of dacarbazine and all-trans retinoic acid (ATRA) using lipid nanoformulations for synergistic antitumor efficacy against malignant melanoma. Nanoscale Res. Lett. 15:113. doi: 10.1186/s11671-020-3293-3
Li, X., Wang, S., Zhu, R., Li, H., Han, Q., and Zhao, R. (2016). Lung tumor exosomes induce a pro-inflammatory phenotype in mesenchymal stem cells via NFκB-TLR signaling pathway. J. Hematol. Oncol. 9:42. doi: 10.1186/s13045-016-0269-y
Lin, L., Du, L., Cao, K., Huang, Y., Yu, P., Zhang, L., et al. (2016). Tumour cell-derived exosomes endow mesenchymal stromal cells with tumour-promotion capabilities. Oncogene 35, 6038–6042. doi: 10.1038/onc.2016.131
Locksley, R., Killeen, N., and Lenardo, M. (2001). The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104, 487–501. doi: 10.1016/s0092-8674(01)00237-9
Marshall, E. A., Ng, K. W., Kung, S. H., Conway, E. M., Martinez, V. D., Halvorsen, E. C., et al. (2016). Emerging roles of T helper 17 and regulatory T cells in lung cancer progression and metastasis. Mol. Cancer 15:27. doi: 10.1186/s12943-016-0551-1
Martin-Orozco, N., Muranski, P., Chung, Y., Yang, X., Yamazaki, T., Lu, S., et al. (2009). T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity 31, 787–798. doi: 10.1016/j.immuni.2009.09.014
McLean, K., Gong, Y., Choi, Y., Deng, N., Yang, K., Bai, S., et al. (2011). Human ovarian carcinoma–associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J. Clin. Invest. 121, 3206–3219. doi: 10.1172/jci45273
Miossec, P., Korn, T., and Kuchroo, V. (2009). Interleukin-17 and type 17 helper T cells. N. Engl. J. Med. 361, 888–898. doi: 10.1056/NEJMra0707449
Mucciolo, G., Curcio, C., Roux, C., Li, W., Capello, M., Curto, R., et al. (2021). IL17A critically shapes the transcriptional program of fibroblasts in pancreatic cancer and switches on their protumorigenic functions. Proc. Natl. Acad. Sci. U.S.A. 118:e2020395118. doi: 10.1073/pnas.2020395118
Mucida, D., Park, Y., Kim, G., Turovskaya, O., Scott, I., Kronenberg, M., et al. (2007). Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317, 256–260. doi: 10.1126/science.1145697
Oukka, M. (2008). Th17 cells in immunity and autoimmunity. Ann. Rheum. Dis. 67(Suppl. 3), iii26–iii29. doi: 10.1136/ard.2008.098004
Paroni, G., Zanetti, A., Barzago, M., Kurosaki, M., Guarrera, L., Fratelli, M., et al. (2020). Retinoic acid sensitivity of triple-negative breast cancer cells characterized by constitutive activation of the notch1 pathway: the role of Rarβ. Cancers 12:3027. doi: 10.3390/cancers12103027
Pietras, K., and Ostman, A. (2010). Hallmarks of cancer: interactions with the tumor stroma. Exp. Cell Res. 316, 1324–1331. doi: 10.1016/j.yexcr.2010.02.045
Qian, B., and Pollard, J. (2010). Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51. doi: 10.1016/j.cell.2010.03.014
Quante, M., Tu, S., Tomita, H., Gonda, T., Wang, S., Takashi, S., et al. (2011). Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 19, 257–272. doi: 10.1016/j.ccr.2011.01.020
Razi, S., Baradaran Noveiry, B., Keshavarz-Fathi, M., and Rezaei, N. (2019). IL-17 and colorectal cancer: from carcinogenesis to treatment. Cytokine 116, 7–12. doi: 10.1016/j.cyto.2018.12.021
Ren, G., Zhang, L., Zhao, X., Xu, G., Zhang, Y., Roberts, A., et al. (2008). Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell 2, 141–150. doi: 10.1016/j.stem.2007.11.014
Ren, G., Zhao, X., Wang, Y., Zhang, X., Chen, X., Xu, C., et al. (2012). CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell 11, 812–824. doi: 10.1016/j.stem.2012.08.013
Shen, F., and Gaffen, S. (2008). Structure-function relationships in the IL-17 receptor: implications for signal transduction and therapy. Cytokine 41, 92–104. doi: 10.1016/j.cyto.2007.11.013
Sierra-Mondragon, E., Molina-Jijon, E., Namorado-Tonix, C., Rodríguez-Muñoz, R., Pedraza-Chaverri, J., and Reyes, J. (2018). All-trans retinoic acid ameliorates inflammatory response mediated by TLR4/NF-κB during initiation of diabetic nephropathy. J. Nutr. Biochem. 60, 47–60. doi: 10.1016/j.jnutbio.2018.06.002
Song, X., Dai, D., He, X., Zhu, S., Yao, Y., Gao, H., et al. (2015). Growth factor FGF2 cooperates with interleukin-17 to repair intestinal epithelial damage. Immunity 43, 488–501. doi: 10.1016/j.immuni.2015.06.024
Su, S., Chen, J., Yao, H., Liu, J., Yu, S., Lao, L., et al. (2018). CD10GPR77 cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell 172, 841–856.e816. doi: 10.1016/j.cell.2018.01.009
Sugita, S., Kohno, T., Yamamoto, K., Imaizumi, Y., Nakajima, H., Ishimaru, T., et al. (2002). Induction of macrophage-inflammatory protein-3alpha gene expression by TNF-dependent NF-kappaB activation. J. Immunol. 168, 5621–5628. doi: 10.4049/jimmunol.168.11.5621
Sun, H., Chen, J., Wu, W., Yang, Y., Xu, Y., Yu, X., et al. (2020). Retinoic acid synthesis deficiency fosters the generation of polymorphonuclear myeloid-derived suppressor cells in colorectal cancer. Cancer Immunol. Res. 9, 20–33. doi: 10.1158/2326-6066.cir-20-0389
Taniguchi, K., and Karin, M. (2018). NF-κB, inflammation, immunity and cancer: coming of age. Nat. Rev. Immunol. 18, 309–324. doi: 10.1038/nri.2017.142
Uccelli, A., Moretta, L., and Pistoia, V. (2008). Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 8, 726–736. doi: 10.1038/nri2395
Wang, K., Kim, M., Di Caro, G., Wong, J., Shalapour, S., Wan, J., et al. (2014). Interleukin-17 receptor a signaling in transformed enterocytes promotes early colorectal tumorigenesis. Immunity 41, 1052–1063. doi: 10.1016/j.immuni.2014.11.009
Wang, L., Yi, T., Kortylewski, M., Pardoll, D., Zeng, D., and Yu, H. (2009). IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J. Exp. Med. 206, 1457–1464. doi: 10.1084/jem.20090207
Wang, Y., Chen, X., Cao, W., and Shi, Y. (2014). Plasticity of mesenchymal stem cells in immunomodulation: pathological and therapeutic implications. Nat. Immunol. 15, 1009–1016. doi: 10.1038/ni.3002
Wu, P., Wu, D., Ni, C., Ye, J., Chen, W., Hu, G., et al. (2014). γδT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity 40, 785–800. doi: 10.1016/j.immuni.2014.03.013
Yu, P., Huang, Y., Xu, C., Lin, L., Han, Y., Sun, W., et al. (2017). Downregulation of CXCL12 in mesenchymal stromal cells by TGFβ promotes breast cancer metastasis. Oncogene 36, 840–849. doi: 10.1038/onc.2016.252
Keywords: MSC, tumor associated MSC, retinoic acid, interleukin-17, interferon-γ, tumor microenvironment
Citation: Lou Q, Zhao M, Xu Q, Xie S, Liang Y, Chen J, Yuan L, Wang L, Jiang L, Mou L, Lin D and Zhao M (2021) Retinoic Acid Inhibits Tumor-Associated Mesenchymal Stromal Cell Transformation in Melanoma. Front. Cell Dev. Biol. 9:658757. doi: 10.3389/fcell.2021.658757
Received: 26 January 2021; Accepted: 26 February 2021;
Published: 06 April 2021.
Edited by:
Shu-Heng Jiang, Shanghai Cancer Institute, ChinaCopyright © 2021 Lou, Zhao, Xu, Xie, Liang, Chen, Yuan, Wang, Jiang, Mou, Lin and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisha Mou, bGlzaGFtb3VAZ21haWwuY29t; Dongjun Lin, bGluZGpAbWFpbC5zeXN1LmVkdS5jbg==; Meng Zhao, emhhb20zOEBtYWlsLnN5c3UuZWR1LmNu
†These authors have contributed equally to this work and share first authorship