 Rebecca J. Noort
Rebecca J. Noort Grace A. Christopher
Grace A. Christopher Jessica L. Esseltine
Jessica L. Esseltine- Division of BioMedical Sciences, Faculty of Medicine, Memorial University of Newfoundland, St. John’s, NL, Canada
Every single cell in the body communicates with nearby cells to locally organize activities with their neighbors and dysfunctional cell-cell communication can be detrimental during cell lineage commitment, tissue patterning and organ development. Pannexin channels (PANX1, PANX2, and PANX3) facilitate purinergic paracrine signaling through the passage of messenger molecules out of cells. PANX1 is widely expressed throughout the body and has recently been identified in human oocytes as well as 2 and 4-cell stage human embryos. Given its abundance across multiple adult tissues and its expression at the earliest stages of human development, we sought to understand whether PANX1 impacts human induced pluripotent stem cells (iPSCs) or plays a role in cell fate decisions. Western blot, immunofluorescence and flow cytometry reveal that PANX1 is expressed in iPSCs as well as all three germ lineages derived from these cells: ectoderm, endoderm, and mesoderm. PANX1 demonstrates differential glycosylation patterns and subcellular localization across the germ lineages. Using CRISPR-Cas9 gene ablation, we find that loss of PANX1 has no obvious impact on iPSC morphology, survival, or pluripotency gene expression. However, PANX1 gene knockout iPSCs exhibit apparent lineage specification bias under 3-dimensional spontaneous differentiation into the three germ lineages. Indeed, loss of PANX1 increases representation of endodermal and mesodermal populations in PANX1 knockout cells. Importantly, PANX1 knockout iPSCs are fully capable of differentiating toward each specific lineage when exposed to the appropriate external signaling pressures, suggesting that although PANX1 influences germ lineage specification, it is not essential to this process.
Introduction
Pluripotent stem cells (PSCs), including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) are characterized by the ability to self-renew indefinitely and the capacity to differentiate into theoretically any cell type in the embryo (Nagy et al., 1993; Itskovitz-Eldor et al., 2000). Differentiation is the process by which stem cells are assigned a cell fate. At each successive stage of lineage commitment cells become more and more specialized and lose the capacity to become other cell types. Terminal differentiation refers to the final stage of cell fate specification, when a cell is locked in form and function. One of the first cell fate decisions in development occurs during gastrulation when the embryo patterns the three embryonic germ layers: ectoderm, endoderm, mesoderm. The ectoderm eventually gives rise to tissues including skin and brain while the endoderm patterns for internal organs and the mesoderm contributes to muscle, bones, and connective tissues. Embryonic germ layer specification can be mimicked in vitro through active or passive differentiation paradigms of human PSCs.
Pluripotent stem cells (and epiblast cells in the embryo) maintain their stemness through activation of POU5F1, NANOG, and transforming growth factor beta (TGFβ) signaling pathways (reviewed in Tam and Loebel, 2007). Gastrulation and the subsequent emergence of the three germ layers from pluripotent stem cells initiates when Activin/Nodal, bone morphogenic protein (BMP), and WNT signaling pathways are activated (Camacho-Aguilar and Warmflash, 2020). WNT3 signaling enables formation of the primitive streak and specification of the bi-potent mesendoderm (Tada et al., 2005; Tam and Loebel, 2007; Tan et al., 2013). The mesoderm subsequently specifies through additional WNT3 pathway activation and BMP signaling, whereas endoderm is specified by elevated activation of Nodal/Activin A signaling pathways (Tam and Loebel, 2007; ten Berge et al., 2008). The ectoderm germ lineage is formed when remaining epiblast cells that did not ingress through the primitive streak are subjected to TGFβ/Nodal and BMP pathway inhibition (Tam and Loebel, 2007; Tchieu et al., 2017). Germ lineage specification can be mimicked in vitro through exogenous exposure to the aforementioned signaling molecules in directed differentiation of PSCs. In contrast to directed differentiation, spontaneous differentiation enables passive, cell-guided lineage commitment and generally results in cell populations from all three germ layers (Desbaillets et al., 2000; Itskovitz-Eldor et al., 2000; Takahashi et al., 2007).
Cells do not undertake differentiation in isolation; rather communication among neighboring cells, and between cells and the niche, is essential to ensure appropriate cell fate specification (Perrimon et al., 2012). In addition to secreted factors, small signaling molecules released from cellular channels also play increasingly recognized roles in these processes. For example, purinergic signaling through release of extracellular ATP participates in neural precursor cell and mesenchymal stem cell self-renewal, migration, and differentiation (Glaser et al., 2014; Cavaliere et al., 2015; Kaebisch et al., 2015; Jiang et al., 2017). Pannexin proteins (PANX1, PANX2, and PANX3) form large-pore channels, permeable to ions and metabolites less than 1 kDa in size (Penuela et al., 2013). PANX1 is widely expressed across multiple tissues in the body while PANX2 and PANX3 are more restricted in tissue expression (Penuela et al., 2013). The most well-defined role of PANX1 is ATP release (Chekeni et al., 2010). In healthy cells PANX1 channels normally remain in a closed state until induced to transiently open in response to a variety of stimuli including membrane deformation, receptor activation, and intracellular calcium release (Sandilos et al., 2012; Bao et al., 2013; Furlow et al., 2015; Sanchez-Arias et al., 2019). PANX1 signaling can also influence the self-renewal and differentiation of multiple somatic (adult) stem cell types including osteoprogenitor cells, skeletal myoblasts, and postnatal neural precursor cells (Wicki-Stordeur et al., 2012; Wicki-Stordeur and Swayne, 2013; Wicki-Stordeur et al., 2016; Ishikawa and Yamada, 2017; Pham et al., 2018). However, much less is known about the impact of PANX1 signaling in the early embryo or in pluripotent stem cells (Hainz et al., 2018).
Recent reports have revealed that PANX1 is highly expressed at the earliest stages of human development, and localizes to the plasma membrane of human oocytes as well as 2- and 4-cell stage human embryos (Sang et al., 2019). The high expression of PANX1 in human oocytes and embryos suggests a fundamental role for PANX1 in human development (Esseltine and Laird, 2016; Shao et al., 2016; Hainz et al., 2018). Indeed, several human disease-causing germline PANX1 variants have now been identified. The first human patient identified to harbor a homozygous genetic variant in PANX1 (PANX1-R217H) suffers from a staggering number of maladies in several of the organs most highly enriched in PANX1, including severe neurological deficits and primary ovarian failure (Shao et al., 2016). This mutation was shown to decrease PANX1 channel function while not affecting trafficking. Recently, four independent families were reported in which different heterozygous PANX1 variants cause female infertility due to primary oocyte death (Sang et al., 2019). These four human variants interfered with PANX1 posttranslational modification and plasma membrane trafficking, decreased PANX1 protein abundance in cells, and aberrant channel function.
Because PANX1 is expressed at the very earliest stages of human development, and because human mutations negatively impact human oocyte survival, we sought to uncover whether PANX1 also impacts human pluripotent stem cells or stem cell fate decisions. Here we find that PANX1 protein is expressed at the cell surface of human iPSCs. PANX1 knockout (PANX-/-) iPSCs generated through CRISPR-Cas9 gene ablation appear morphologically indistinguishable from control. Interestingly, we find enhanced representation of endodermal and mesodermal cells from spontaneously differentiated PANX1-/- iPSCs compared to control. Therefore, we conclude that PANX1 protein expression influences PSC commitment to the three embryonic germ layers.
Materials and Methods
Induced Pluripotent Stem Cell Lines
All studies were approved by the Human Ethics Research Board (HREB # 2018.210). Female wildtype human induced pluripotent stem cells (iPSCs) were created from dermal fibroblasts isolated from an apparently healthy 30-year-old female as described in Esseltine et al. (2017) and obtained through a material transfer agreement with The University of Western Ontario.
iPSCs were routinely cultured as colonies in feeder-free conditions in a humidified 37°C cell culture incubator buffered with 5% CO2 and atmospheric oxygen. iPSCs were grown on dishes coated with Geltrex (Cat# A141330, ThermoFisher, Waltham, MA, United States) and fed daily with Essential 8 medium (Cat# A1517001, ThermoFisher). Colonies were passaged every 4-5 days when they exhibited tight cell packing, smooth borders, and phase-bright smattering at colony centers. Individual iPSCs within the colonies exhibited prominent nucleoli and high nucleus-to-cytoplasm ratio as is characteristic for human pluripotent stem cells (Thomson et al., 1998; Takahashi et al., 2007). For passaging, iPSCs were incubated with gentle cell dissociation buffer (Cat# 13151014, ThermoFisher) at room temperature until colonies were visibly broken apart, approximately 3-5 min (Beers et al., 2012). Gentle cell dissociation buffer was then replaced with 1 mL of Essential 8 to stop the reaction. Colonies were then scraped from the dish surface and broken into small aggregates of cells (roughly 50 – 200 μm in diameter). The resultant aggregates were seeded into fresh Geltrex-coated wells containing Essential 8 at split ratios of 1:5 to 1:20. iPSCs were maintained in culture for 20 weeks after thawing at which point the culture was terminated and a fresh vial of low-passage iPSCs was thawed from the liquid nitrogen. We confirmed our iPSC cell banks have normal copy number at various mutation hotspots using the hPSC Genetic Analysis Kit (Cat # 07550, STEMCELL Technologies, Vancouver, BC, Canada).
Single cell iPSC passaging was achieved using StemPro Accutase (Cat# A1110501, ThermoFisher). iPSCs were treated with Accutase at 37°C for 8-10 min and triturated to create a single cell suspension. Single cells were plated in medium supplemented with the Rho-associated kinase inhibitor (ROCKi), Y-27632 (Cat# Y-5301, LC Laboratories, Woburn, MA, United States) to promote single cell iPSC survival (Watanabe et al., 2007).
CRISPR-Cas9 Gene Ablation
PANX1 knockout iPSCs were created as described previously (Esseltine et al., 2020). Briefly, iPSCs were transfected using the Mirus TransIT®-LT1 Transfection Reagent (Cat# MIR-2304, Mirus Bio LLC, Madison, WI, United States) with the pSpCas9(BB)-2A-GFP plasmid (Cat# 48138, Addgene, Cambridge, MA, United States) according to the manufacturer’s instructions (Ran et al., 2013). The sgRNA was designed using the Sanger Institute CRISPR finder1 and was selected based on its low exonic off-target prediction [human PANX1: Sanger sgRNA ID 1087081842 (5′-GCTGCGAAACGCCAGAACAG-3′)]. After transfection, GFP-expressing single cells were sorted using fluorescence activated cell sorting (FACS) and re-plated at low density to permit easy isolation of individual clones. The resulting individual clones were examined for ablation of the target gene at the genomic level via PCR and Sanger sequencing while ablation of the PANX1 protein was assessed via immunofluorescence, Western blotting, and flow cytometry.
Embryoid Body Generation for 3D Spontaneous Differentiation
Embryoid bodies (EBs) of 9000 cells each were created in 96-well round-bottom plates coated with 1% agarose prepared in deionized water to confer a non-adherent surface which promotes iPSC self-aggregation (Friedrich et al., 2009; Dahlmann et al., 2013). A single-cell iPSC suspension was created via Accutase dissociation as described above and re-suspended in Essential 6, which lacks the essential pluripotency factors TGFβ and FGF2 (Cat# A1516401, ThermoFisher), supplemented with 10 μM Y-27632 to promote cell survival (Lin and Chen, 2014). Essential 6 media was replenished every other day to promote spontaneous differentiation.
Monolayer Directed Differentiation to the Three Germ Layers
Directed differentiation into the three germ layers was achieved using the STEMdiffTM Trilineage Differentiation Kit (Cat# 05230, STEMCELL Technologies) according to the manufacturer’s instructions.
Quantitative Reverse Transcription Polymerase Chain Reaction
Undifferentiated iPSCs, along with differentiated cells and embryoid bodies were collected for gene expression analysis. RNA was extracted using the PureLinkTM RNA isolation kit (Cat # 12183018A, ThermoFisher) with DNase I treatment according to the manufacturers’ instructions. Purified RNA was quantified using a NanoDropTM 2000 spectrophotometer (Cat# ND-2000, ThermoFisher), and stored at −80°C until use. High quality RNA was identified by a λ260/280 of ≥ 2.0 and λ260/230 of ≥ 2.0.
RNA was converted into complementary DNA (cDNA) using the High-Capacity cDNA Reverse Transcription Kit (Cat# 4368814, ThermoFisher) according to the manufacturer’s instructions. Typically, 500 ng of RNA were used per cDNA reaction. The resulting cDNA was stored at −30°C until use.
Quantitative reverse transcription polymerase chain reaction (qPCR) was performed using intercalating dye chemistry (Zipper et al., 2004). Oligonucleotide sets listed in Table 1 were designed for specific target amplification and minimal primer dimer formation using NCBI Primer-BLAST (NIH, Bethesda, MD, United States2) and IDT’s Oligo Analyzer Tool (IDT, Newark, NJ, United States). Bio-Rad SsoAdvancedTM Universal SYBR® Green Supermix (Cat# 1725274, Bio-Rad, Hercules, CA, United States) was utilized and oligonucleotides (all from IDT) were used at 10 μM in each reaction. Standard run time cycling parameters were as follows: one cycle of 50°C for 2 min, one cycle of 95°C for 30 s, 40 cycles of 95°C for 10 s, 60°C for 1 min, followed by a melt curve from 60°C to 95°C. Data was analyzed using QuantStudioTM real-time PCR software (Version1.3, ThermoFisher). Gene expression for each sample were normalized to the reference gene (GAPDH or 18SrRNA) to generate a deltaCT (ΔCT) (Schmittgen and Livak, 2008). Stable expression of GAPDH throughout differentiation was confirmed by 1-way analysis of variance (ANOVA) across all cell lines and tissue types generated in this study according to the methodology described in Schmittgen and Livak (2008) (data not shown) (Schmittgen and Livak, 2008). Samples where CT-values were ≥ the CT-value of the no-template control were considered qPCR non-detects and were excluded from further analysis.

Table 1. Oligo sets for qPCR.
The heatmap in Figure 5C was generated from the average 2–ΔCT value (fold change from GAPDH expression) of each condition using R Studio (version 3.6.1) software with the ggplots.2 package, heatmap.2 function, and row scaling. For all other qPCR analysis, fold change expression of genes relative to a control sample (such as the control (wildtype) iPSC cell population) were evaluated using the ΔΔCT method as described (Schmittgen and Livak, 2008). Fold change represents the standard error of the mean of 2-8 technical replicates were plotted in GraphPad PRISM (Version 6.07, GraphPad, San Diego, CA, United States).
Immunofluorescence
Embryoid bodies and monolayer cultures were fixed in 10% buffered formalin (Cat# CA71007-344, VWR, Radnor, PA, United States) for 1 h at room temperature. Fourteen day old (day 14) EBs were cryogenically prepared and immunolabelled according to the methodology described in STEMCELL Technologies’ Document #27171, Version 1.0.0, Nov 2019. In summary, day 14 EBs were first dehydrated in Ca2+/Mg2+-free PBS supplemented with 30% sucrose for 1-4 days at 4°C until the EBs sank. Dehydrated EBs were then incubated for 1 hour at 37°C in gelatin embedding solution consisting of 10% sucrose and 7.5% gelatin prepared in Ca2+/Mg2+-free PBS. The EBs were then transferred to a cryopreservation mold and snap frozen in a slurry of dry ice and isopentane followed by cryosectioning at 14 μm slice thickness. For antigen retrieval, slides were placed into a plastic container with citrate buffer, pH 6.0: 0.294% tri-sodium citrate (dihydrate) (Cat# A12274, Alfa Aesar, Tewksbury, MA, United States) with 0.05% Tween®20. Samples were heated in a rice steamer for 20 minutes. Five day old (day 5) EBs were immunostained as whole-mount preparations. Immunostaining with primary antibodies, dyes, and stains indicated in Table 2 was performed as described in the figure legends. AlexaFluor-conjugated secondary antibodies were all purchased from ThermoFisher. Slides were mounted using Mowial®488 reagent with 1,4-diazabicyclo[2.2.2]octane (DABCO) antifade compound (Cold Spring Harbor Laboratory, 2007).
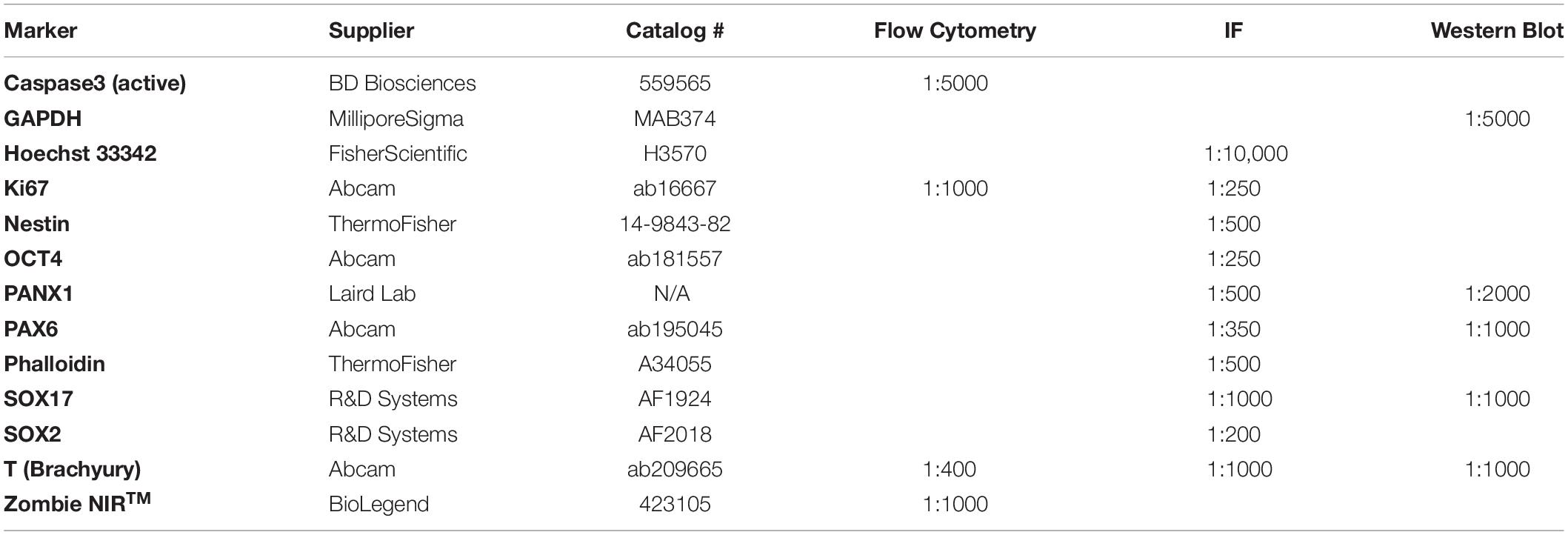
Table 2. Primary antibodies and dyes.
Phase Contrast Imaging
Phase contrast images of monolayer cells and embryoid bodies were taken on a Zeiss AxioObserver microscope using 5X/0.12 NA A-Plan and 10X/0.25 NA Ph1 objectives. Images from these microscopes were taken in 8-bit greyscale using an Axiocam MRm camera and AxioVision Version 4.8.2 software. All phase contrast imaging equipment is from Carl Zeiss Microscopy (Jena, DEU).
Confocal Microscopy
Fluorescent confocal images were acquired on an Olympus Fluoview FV10i—W3 confocal microscope (Olympus, Tokyo, JPN) fitted with a 10X/0.4, or 60X/1.2 NA lens and Fluoview version 2.1.17 software. The following lasers were used to visualize fluorophores: DAPI/Hoechst (405 nm laser); Alexa Fluor 488 (473 nm laser); Alexa Fluor 555 (559 nm laser); Alexa Fluor 647 (635 nm laser). Other images were acquired using an Olympus Fluoview FV1000 confocal microscope fitted with 10X/0.4 NA, 20X/0.75NA or 40X/0.95NA and the following lasers: 405 nm, 458 nm, 568 nm, 633 nm. Images were analyzed using FIJI open source software (Schindelin et al., 2012). To facilitate better visualization, fluorescent confocal images were occasionally subjected to uniform brightness/contrast enhancement.
Flow Cytometry
Flow cytometry was performed on a CytoFLEX (Beckman Coulter, Brea, CA, United States) flow cytometer. Antibodies for flow cytometry were titrated over a range of concentrations prior to use. The following controls were included in all flow cytometry runs: unstained control, fluorescence-minus-one (FMO) controls, secondary-only controls, and single-color compensation controls for fluorochromes. UltraComp compensation beads (Cat# 01-2222-43, ThermoFisher) were used with antibodies raised in mice.
Live single-cell suspensions were labelled with Zombie NIRTM fixable viability dye (Cat# 423105, BioLegend®, San Diego, CA, United States) to eliminate dead cells during the analysis stage. Next, the cells were fixed in 10% buffered formalin for 10 min at 4°C in the dark. After fixation, the cells were permeabilized (Ca2+Mg2+-free PBS with 0.5% BSA supplemented with 0.1% Triton X-100) for 15 min at room temperature in the dark. Primary and secondary antibodies (used at dilutions according to Table 2) were incubated for 30 min at 4°C in the dark. Flow cytometric analysis was performed using FlowJo software (version 10.7.1).
SDS–PAGE and Western Blot
Cells were lysed with a solution comprising 50 mM Tris–HCl pH 8, 150 mM NaCl, 0.02% NaN3, 1% Triton X-100, 1 mM NaVO4, 10 mM NaF, 2 μg/mL leupeptin, and 2 μg/mL aprotinin. Soluble proteins were separated using SDS-PAGE and transferred to a 0.45 μm nitrocellulose membrane (Cat# 1620115, Bio-Rad). Primary antibodies (Table 2) were prepared in tris buffered saline + Tween®20 (TBST) + 3% skim milk and incubated overnight at 4°C. Secondary antibodies conjugated to HRP were prepared in TBST + 3% skim milk and incubated for 1 h at room temperature. Proteins were visualized with Bio-Rad ClarityTM Western ECL Substrate (Cat# 1705061, Bio-Rad) using a GE ImageQuant LAS 400 (Cat# 28955810, GE Healthcare, Chicago, IL, United States).
Statistics
Statistical analysis was performed in GraphPad PRISM Version 6.07. Error bars depict ± standard error of the mean (SEM) with n ≥ 3 biological replicates (independent experiments) unless otherwise stated. Statistical significance for comparisons between 2 groups was determined by unpaired Student’s t-test. Statistical significance for comparisons between 3 or more groups was determined by Analysis of Variance (ANOVA) followed by a Tukey’s multiple comparisons test. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ****p ≤ 0.0001.
Results
Human iPSCs Express PANX1
Previous research has indicated that human pluripotent stem cells express transcripts of all three pannexin family members (Hainz et al., 2018). Our qPCR analysis suggests that human iPSCs express mRNA for PANX1 and PANX2, however we are unable to detect PANX3 transcripts in these cells (Figure 1A). qPCR also reveals that PANX1 gene expression is significantly higher in iPSCs compared to the dermal fibroblasts from which they were derived (Figure 1B). Immunofluorescence localizes PANX1 protein primarily to the cell surface of human iPSCs with lesser populations of intracellular staining (Figure 1D). Although we identified PANX2 mRNA in our iPSCs, we were unable to find a reliable antibody to detect the PANX2 protein. Therefore, we focused our subsequent studies on PANX1.
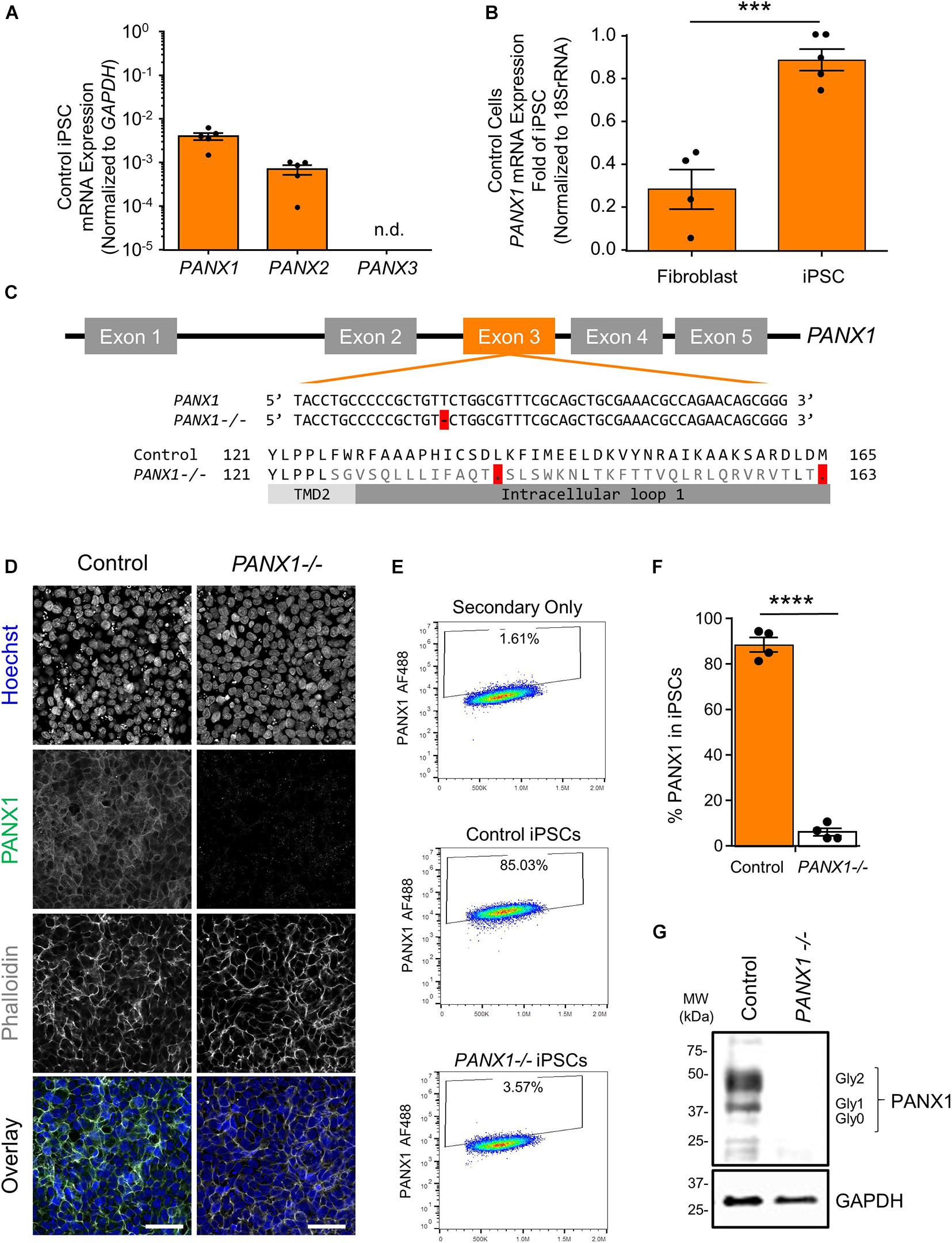
Figure 1. PANX1 expression and CRISPR-Cas9 gene ablation in human iPSCs. (A) Quantitative RT-PCR (qPCR) demonstrates the presence of PANX1 and PANX2 transcripts in human iPSCs, but not PANX3. n.d. qPCR non-detect. (B) PANX1 mRNA is significantly upregulated in human iPSCs compared to the dermal fibroblasts from which they were derived. Data represent the standard error of the mean of 5 independent experiments. ***, p < 0.001 as assessed through Student’s T-test. (C) CRISPR-Cas9 guide RNA was designed targeting the third exon of the human PANX1 gene. Resulting CRISPR-Cas9 engineering induced a single base pair deletion, thus disrupting the reading frame. After gene ablation, human iPSCs no longer express PANX1 protein as shown through immunofluorescence (D), flow cytometry (E,F), and Western blot (G). In the Western blot, control human iPSCs express PANX1 protein as several discreet band sizes corresponding with the non-glycosylated protein (Gly0), high mannose (Gly1) and complex carbohydrate addition (Gly2). Immunofluorescence images were acquired using consistent parameters and are representative of more than 5 replicate experiments. Equal adjustments for brightness or contrast in FIJI were applied to both conditions. PANX1 (green); Nuclei (Hoechst, blue); Actin (Phalloidin, gray). Data represent the standard error of the mean of 4 independent experiments. ****, p < 0.0001. Scale bar = 50 um.
PANX1-/- iPSCs Are Morphologically Comparable to WT Control
Because PANX1 mRNA is upregulated after iPSC reprogramming, and human PANX1 mutations are linked to primary human oocyte death, we sought to determine whether PANX1 protein was essential for human iPSC survival, growth, or pluripotency. CRISPR-Cas9 was used to genetically ablate PANX1 in iPSCs (Figure 1). The resulting clonal knockout iPSCs have a single base pair deletion in the third PANX1 exon resulting in a frameshift mutation and up to 15 early stop codons within the PANX1 transcript (Figure 1C). At the protein level, the mutation alters the amino acid sequence starting from the second transmembrane domain (Figure 1C). Western blot analysis shows PANX1 protein in control iPSCs expressed as three distinct bands relating to the three glycosylation states where Gly2 corresponds with complex carbohydrate modification, Gly1 is the addition of a high mannose species, and Gly0 PANX1 lacks glycosylation (Figure 1G; Penuela et al., 2007). After CRISPR-Cas9 gene ablation, PANX1-/- iPSCs no longer express PANX1 protein as shown through Western blot, flow cytometry, and immunofluorescence (Figures 1D–G).
PANX1-/- iPSCs appear morphologically indistinguishable from control cells and continue to grow as large colonies of tightly packed cells characteristic of human pluripotent stem cells (Figure 2A). PANX1-/- iPSCs continue to express similar amounts of the pluripotency markers POU5F1 (encoding for OCT4) and NANOG compared to control iPSCs (Figure 2B). Furthermore, PANX1-/- iPSCs do not upregulate PANX2, PANX3, or GJA1 (encoding for Cx43) in response to loss of the PANX1 protein (Figure 2C). Flow cytometry demonstrates equivalent expression of the proliferation marker, Ki67 as well as equally low expression of the apoptosis marker, cleaved caspase 3 (CC3) (Figure 2D). Therefore, PANX1 genetic ablation does not appear to negatively impact human iPSC survival, proliferation, morphology, or pluripotency marker expression.
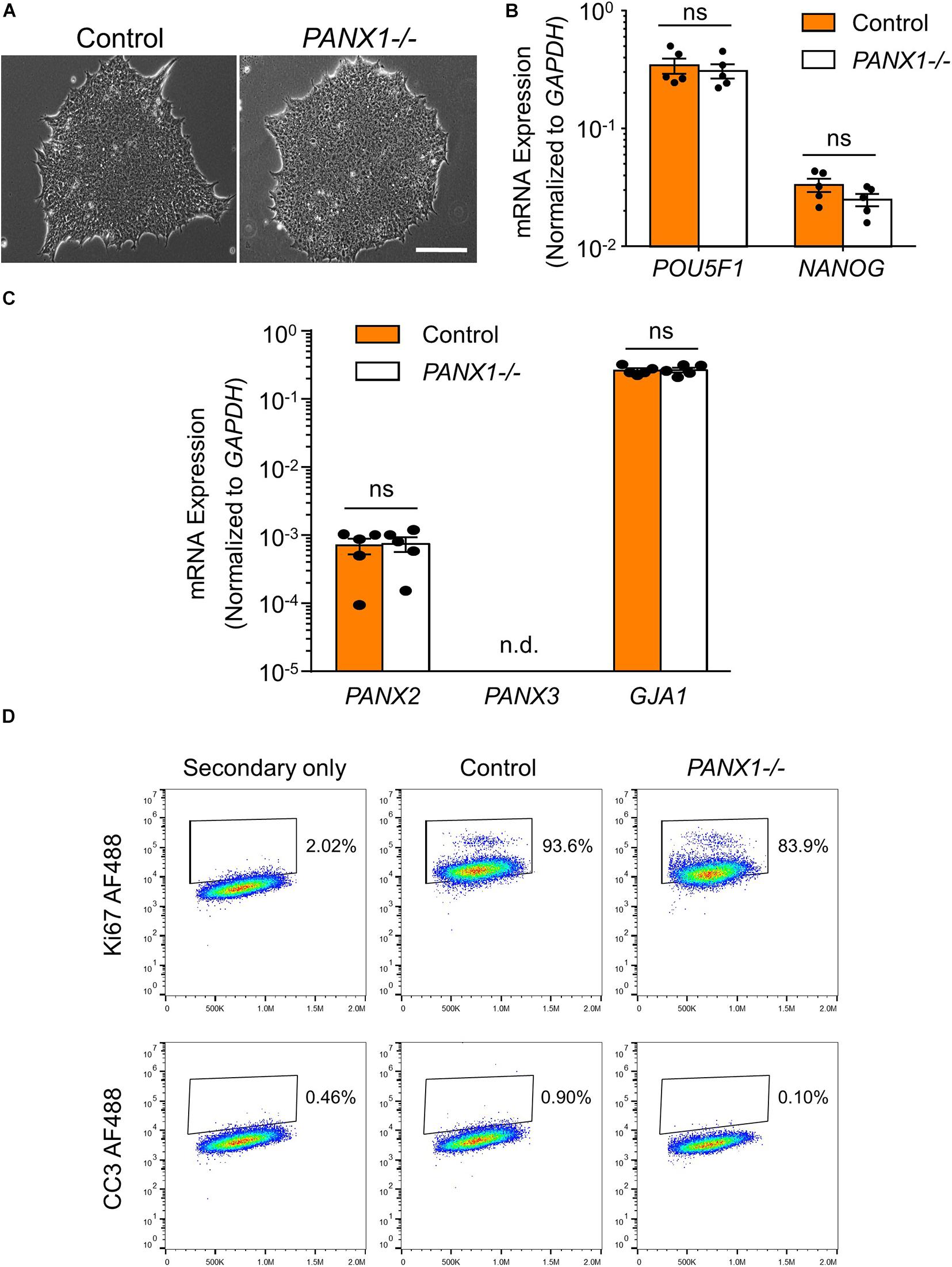
Figure 2. PANX1-/- iPSCs are morphologically comparable to control. (A) Phase contrast micrographs demonstrate that control and PANX1-/- iPSCs each grow as large colonies of tightly packed cells with refractive borders and little differentiation. Scale bar = 200 um. qPCR analysis of (B) the key pluripotency genes POU5F1 (encoding for OCT4) and NANOG as well as (C) PANX2, PANX3 (qPCR non-detect) or GJA1 (encoding for Cx43) expression in control and PANX1-/- iPSCs. Data represent the standard error of the mean of 5 independent experiments. ns, non-significant. n.d. qPCR non-detect. (D) Flow cytometry evaluation of the proliferation marker Ki67 as well as the apoptotic marker cleaved caspase 3 (CC3) in control and PANX1-/- iPSCs.
PANX1 Is Alternatively Glycosylated and Differentially Localized in Cells From the Three Embryonic Germ Layers
Although PANX1 genetic ablation was well tolerated in human iPSCs, we investigated whether this protein plays a role in cell fate specification. Given the wide expression of PANX1 across many tissues of the body, we examined PANX1 expression in each of the three germ layers: endoderm, ectoderm, and mesoderm (Figure 3). PANX1 was indeed expressed in each germ layer as shown through qPCR and Western blot (Figures 3B–D). PANX1 transcript abundance was significantly elevated in mesoderm cells compared to undifferentiated iPSCs (Figure 3B). However, this increased mRNA abundance did not translate into a similar increase in PANX1 protein expression in mesoderm cells (Figures 3C,D). Similar to what was observed in iPSCs, densitometric analysis of the three glycosylation species revealed that 37.15 ± 2.47% of ectoderm PANX1 is fully glycosylated while 25.59 ± 2.91% exists as high mannose and 37.24 ± 3.28% is unglycosylated (Figures 3D,E). Interestingly, PANX1 in mesoderm cells is significantly more glycosylated than iPSCs (61.25 ± 1.29% Gly2, 22.71 ± 7.58% Gly1 and 16.01 ± 6.43% Gly0). On the other hand, endoderm cells appeared to have a significant reduction in the Gly1 and Gly2 species. Endodermal PANX1 exists as 61.09 ± 5.84% unglycosylated species while only 22.38 ± 2.35% is fully glycosylated with complex carbohydrate species (Figure 3E).
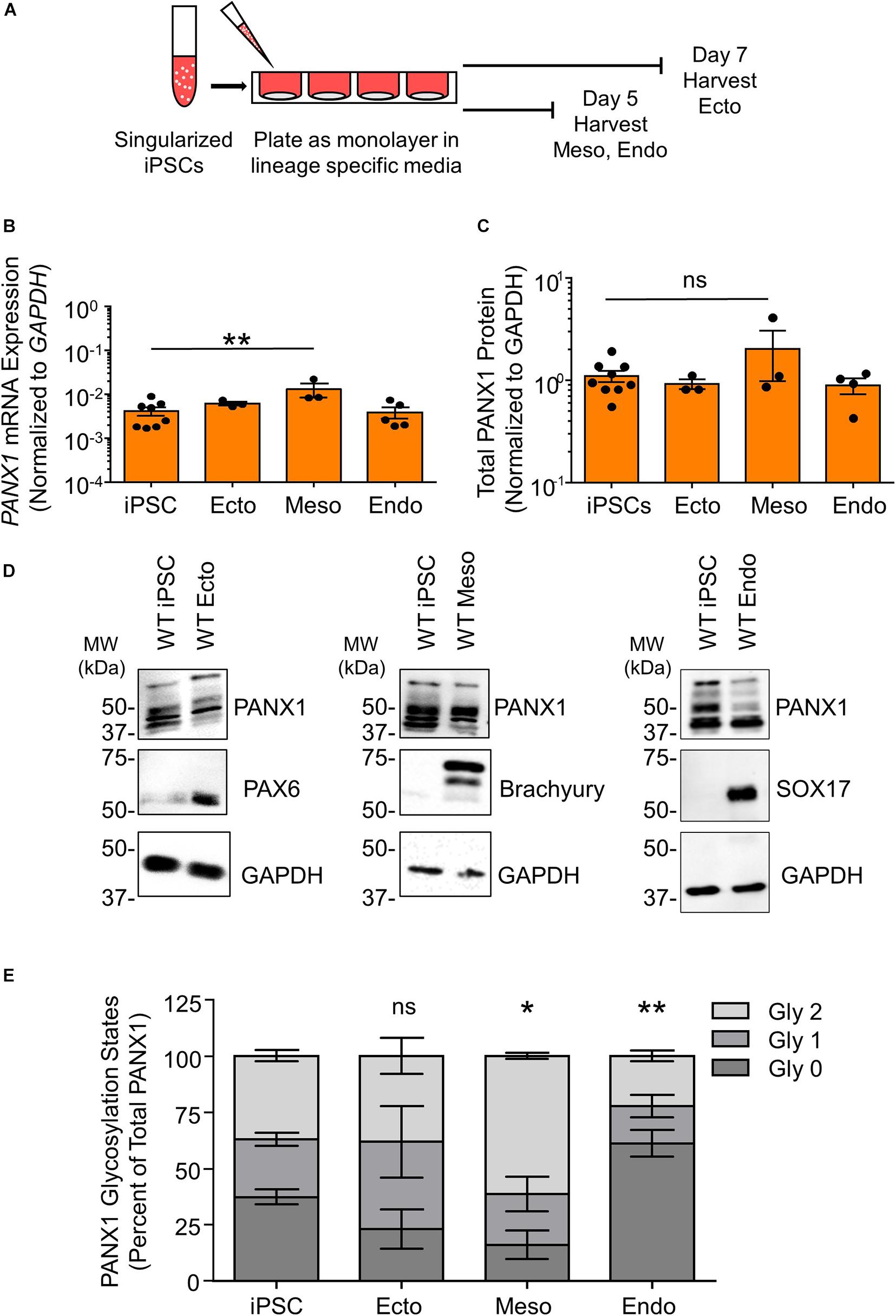
Figure 3. PANX1 is expressed in cells from all three embryonic germ layers. (A) Schematic depicting 2D directed germ lineage differentiation. (B) qPCR analysis of PANX1 mRNA expression in control human iPSCs as well as after directed differentiation into each of the three embryonic germ layers, ectoderm (ecto), mesoderm (meso), and endoderm (endo). (C) Densitometric analysis of total PANX1 protein expression in iPSCs, ectoderm, mesoderm, and endoderm cells. (D) Representative Western blots and (E) densitometric analysis of the three PANX1 glycosylation states. Gly0, non-glycosylated protein; Gly1, high mannose; Gly2, complex carbohydrate addition. Data represent the standard error of the mean of 3-10 independent experiments. ns, no significance; *, p < 0.05; **, p < 0.01 compared to iPSCs.
Because glycosylation has been reported to play a role in PANX1 trafficking to the plasma membrane, we also evaluated the subcellular distribution of PANX1 in the three germ lineages. Immunofluorescence shows PANX1 protein localization at the cell surface in Nestin positive ectoderm cells and Brachyury positive mesoderm cells (Figure 4A, inset). However, in SOX17 positive endoderm cells, PANX1 localized mainly to intracellular compartments and overlapped with EEA1, indicating PANX1 localization in early endosomes (Figure 4B inset, yellow arrowheads).
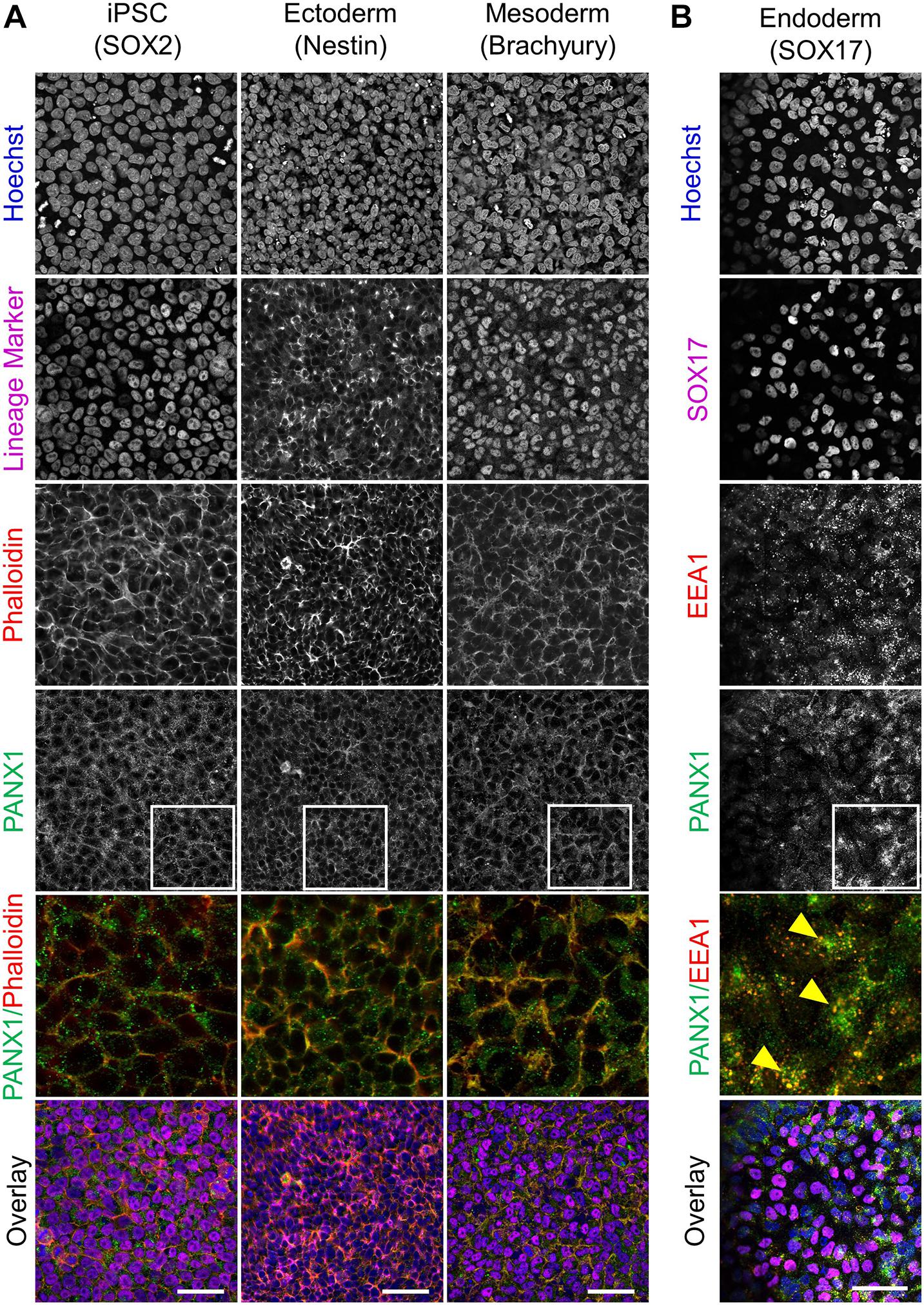
Figure 4. PANX1 is differentially localized in the three embryonic germ layers. (A) Immunofluorescent evaluation of PANX1 (green) demonstrates primarily cell surface localization in iPSCs, ectoderm and mesoderm with lesser intracellular pools. Nuclei (Hoechst, blue); lineage markers (SOX2, Nestin, Brachyury, magenta); Actin (phalloidin, red). (B) In endoderm cells PANX1 is localized intracellularly where it partially overlaps with early endosomes (EEA1, red; SOX17, magenta). Inset: regions of interest zoomed in to highlight regional PANX1 localization. Brightness and contrast were equally adjusted across conditions in FIJI. Scale bar = 50 μm.
PANX1 Knockout Embryoid Bodies Exhibit Skewed Lineage Specification
Spontaneous differentiation enables passive, cell-guided cell fate specification and generally results in cell populations from all three germ layers (ectoderm, endoderm, and mesoderm). In order to determine whether loss of PANX1 altered inherent lineage specification of human iPSCs, we investigated the spontaneous differentiation potential of control and PANX1-/- iPSCs cultured as 3-dimensional embryoid bodies (EBs) (Figure 5). Control and PANX1-/- iPSCs self-aggregated into embryoid bodies of comparable size and shape after 24 h in culture (Figure 5B). As expected, both control and PANX1-/- EBs downregulated genes associated with undifferentiated state (POU5F1 and NANOG) relative to starting iPSCs, indicating that the cells within the EBs were losing pluripotent stemness and were committing to downstream lineages (Figure 5C). PANX1 mRNA expression significantly increased in spontaneously differentiated EBs compared to undifferentiated iPSCs (Figure 5D) while PANX2 mRNA remained constant throughout differentiation in both control and PANX1-/- EBs. On the other hand, GJA1 (Cx43) mRNA significantly decreased throughout EB differentiation, although there was no difference in GJA1 expression between control and PANX1-/- EBs (Figure 5D).
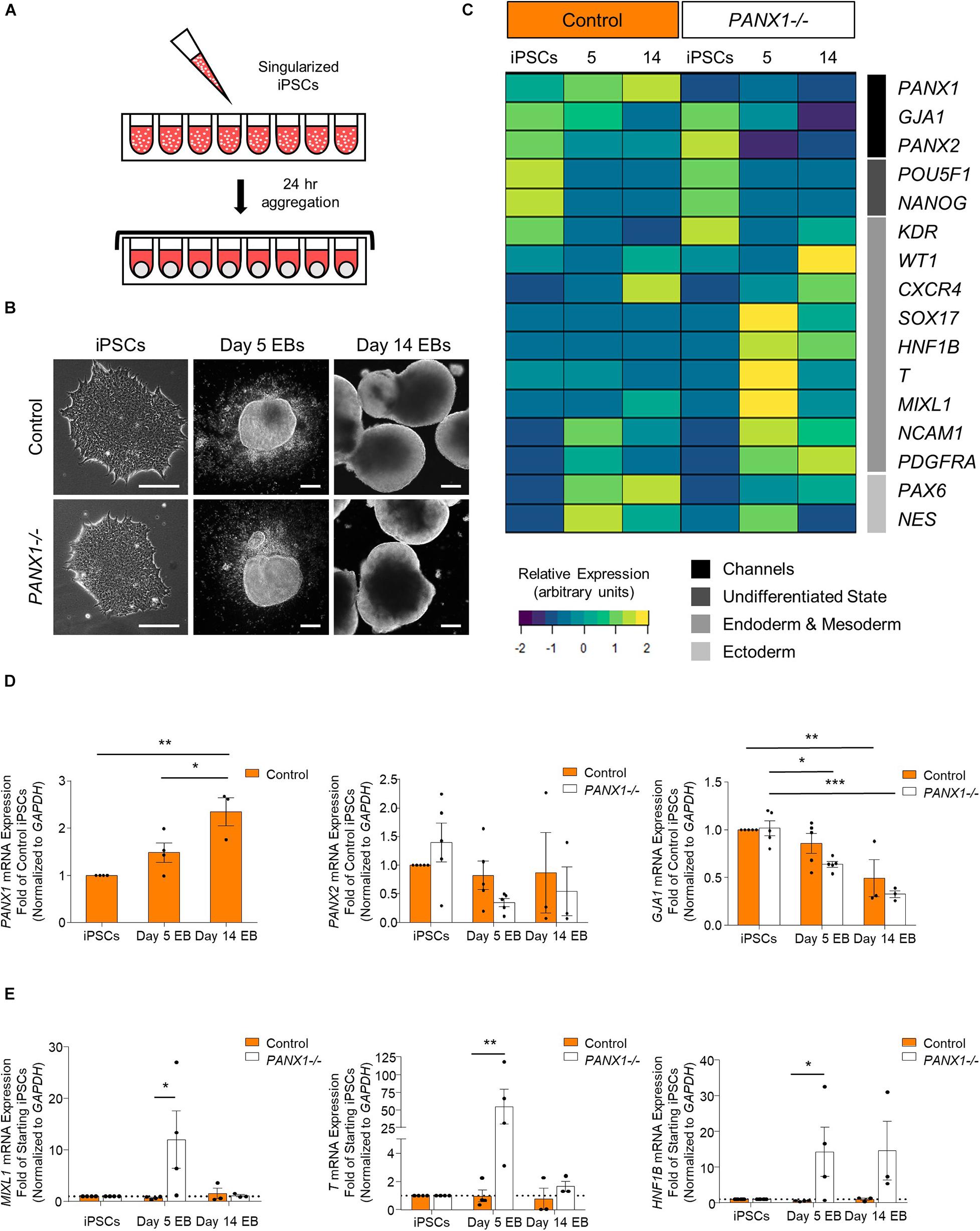
Figure 5. PANX1 knockout embryoid bodies exhibit skewed lineage specification. (A) Schematic depicting 3D embryoid body (EB) formation. (B) Control and PANX1-/- iPSCs self-aggregate and form embryoid bodies. Scale bar = 200 um. (C) Gene expression analysis of control and PANX1-/- iPSCs as well as embryoid bodies after 5 and 14 days of spontaneous differentiation. The heatmap was generated using the average 2–ΔCT data value (normalized to GAPDH) for each condition using R Studio software with the ggplots. 2 package and row clustering. Transcripts for PANX3 were not detected at any point during differentiation. (D) PANX1, PANX2 and GJA1 (Cx43) qPCR gene expression analysis of control and PANX1-/- iPSCs and embryoid bodies after 5 and 14 days of spontaneous differentiation. (E) Lineage-specific gene expression of control and PANX1-/- iPSCs and embryoid bodies after 5 and 14 days of spontaneous differentiation. Bar graph data presented as 2–ΔΔCT (fold change of iPSCs). Data represents the mean of 3-5 independent experiments. ∗, p < 0.05; ∗∗, p < 0.001; ∗∗∗, p < 0.0001 compared to starting iPSCs.
Comprehensive gene expression analysis shows altered expression of genes associated with the germ layers in PANX1-/- EBs compared to control (Figures 5C,E). Indeed, after 5 days of spontaneous differentiation, expression of mesendoderm (MIXL1), mesoderm (T, PDGFRA, NCAM1) and endoderm (SOX17, HNF1B) markers were elevated in PANX1-/- EBs compared to control (Figures 5C,E). Time course analysis suggests that at 5 days of spontaneous differentiation, expression of endoderm and mesoderm lineage genes are higher in PANX1-/- EBs compared to control, and decline by day 14 (Figure 5E) consistent with mesendoderm commitment and subsequent downstream lineage progression.
We corroborated our qPCR data using immunofluorescence imaging of control and PANX1-/- embryoid bodies at day 5 (Figure 6A) and day 14 (Figure 6B). Consistent with our gene expression analysis, we observed a greater proportion of PANX1-/- cells expressing Brachyury (mesoderm) and SOX17 (endoderm) relative to control (Figure 6). On the other hand, both control and PANX1-/- EBs expressed the ectoderm markers PAX6 and Nestin (Figure 6). Taken together, spontaneously differentiated PANX1-/- EBs appear to favor formation of mesoderm and endoderm germ layers while the capacity to form ectodermal cells is minimally impacted.
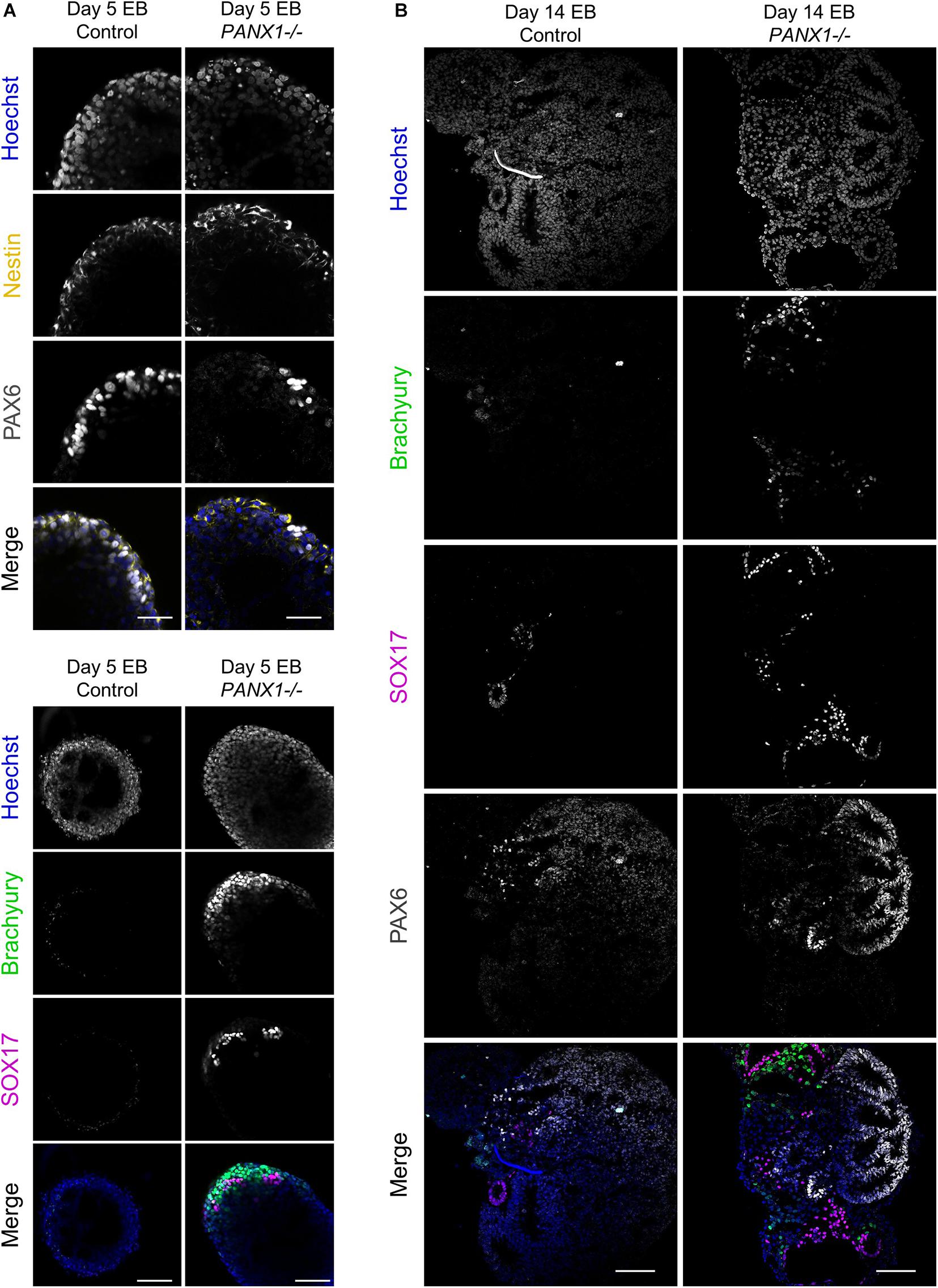
Figure 6. Immunofluorescent analysis of PANX1-/- embryoid bodies. (A) Immunofluorescence of ectoderm (Nestin, yellow; PAX6, gray), mesoderm (Brachyury, green) and endoderm (SOX17, magenta) in control and PANX1-/- day 5 whole mount EBs. (B) After 14 days of differentiation, control and PANX1-/- EBs were cryosectioned and evaluated for germ lineage expression. Equal brightness contrast enhancements were made in FIJI for picture clarity. Nuclei (Hoechst, blue). Scale bar = 100 μm.
Exogenous Pressures Override PANX1-/- Lineage Bias
We determined above that PANX1-/- iPSCs exhibit apparent lineage specification bias when permitted to spontaneously differentiate. However, we also wanted to determine whether loss of PANX1 impacted directed germ lineage differentiation promoted through the application of exogenous growth factors and small molecules. To that end, we used the STEMdiffTM Trilineage Differentiation Kit (STEMCELL Technologies) to evaluate the ability of PANX1-/- iPSCs to differentiate into cells from all three germ layers in response to external pressures. Despite the demonstrated lineage biases outlined above in the passive embryoid body cultures, both qPCR and Western blot demonstrate that PANX1-/- iPSCs effectively differentiate into cells of all three germ lineages when cultured with the Trilineage Differentiation Kit (Figure 7). Similar to what we observed in the undifferentiated iPSCs, both PANX2 and GJA1 (Cx43) transcripts are expressed in the three germ layers but are not upregulated in compensation for the loss of PANX1 during directed differentiation (Figure 8). Furthermore, PANX3 transcripts remained undetectable by qPCR in cells from all three germ layers (data not shown). Therefore, although we find that PANX1 influences germ layer specification, it is not essential to this process.
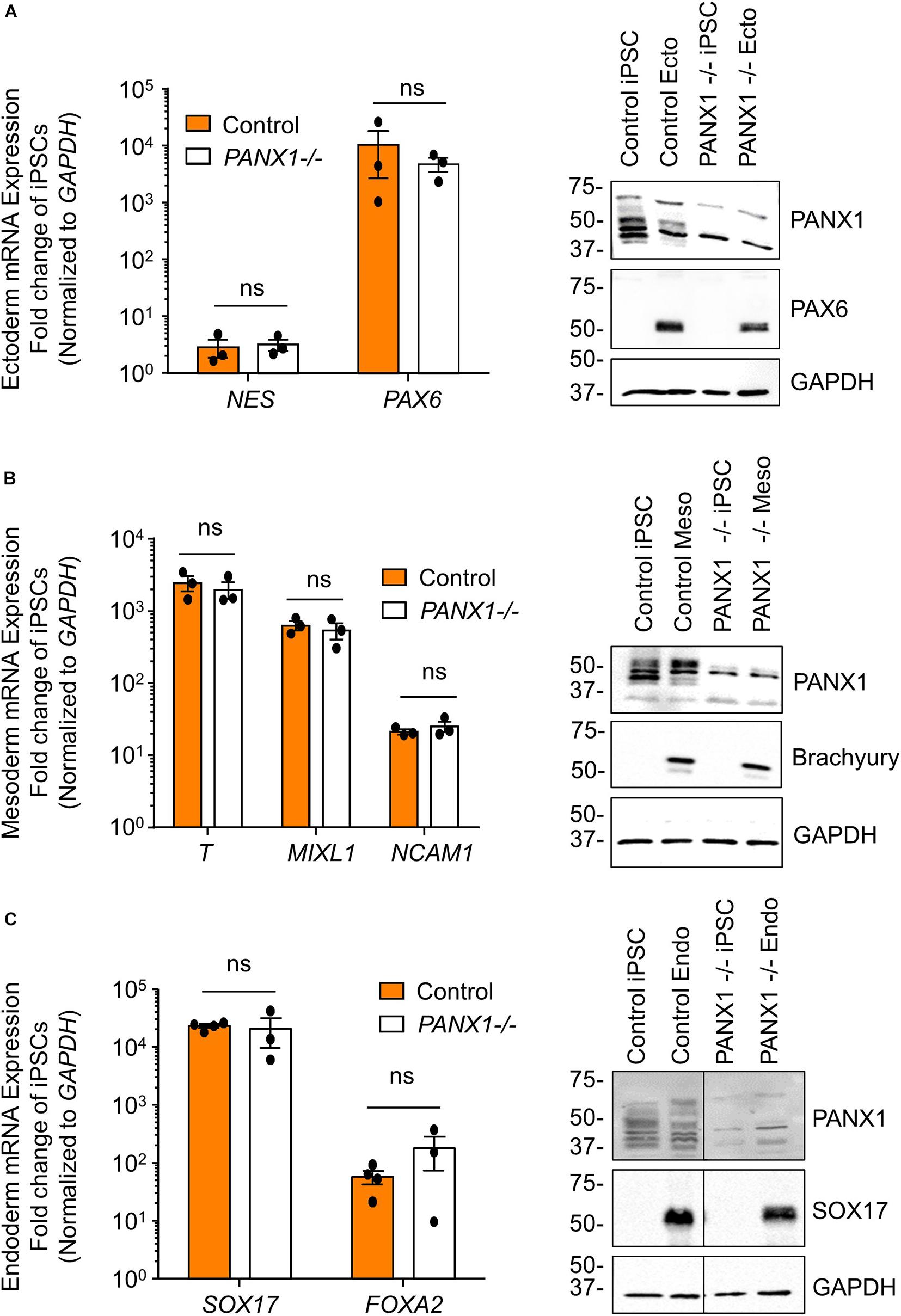
Figure 7. Exogenous pressures override PANX1-/- lineage bias. qPCR gene expression analysis and Western blot of lineage-specific markers after directed differentiation of control and PANX1-/- iPSCs into (A) ectoderm (NES, PAX6), (B) mesoderm (T (Brachyury), MIXL1, NCAM1) and (C) endoderm (SOX17, FOXA2). Data represent the standard error of the mean of 3-4 independent experiments. ns, non-significant compared to control.
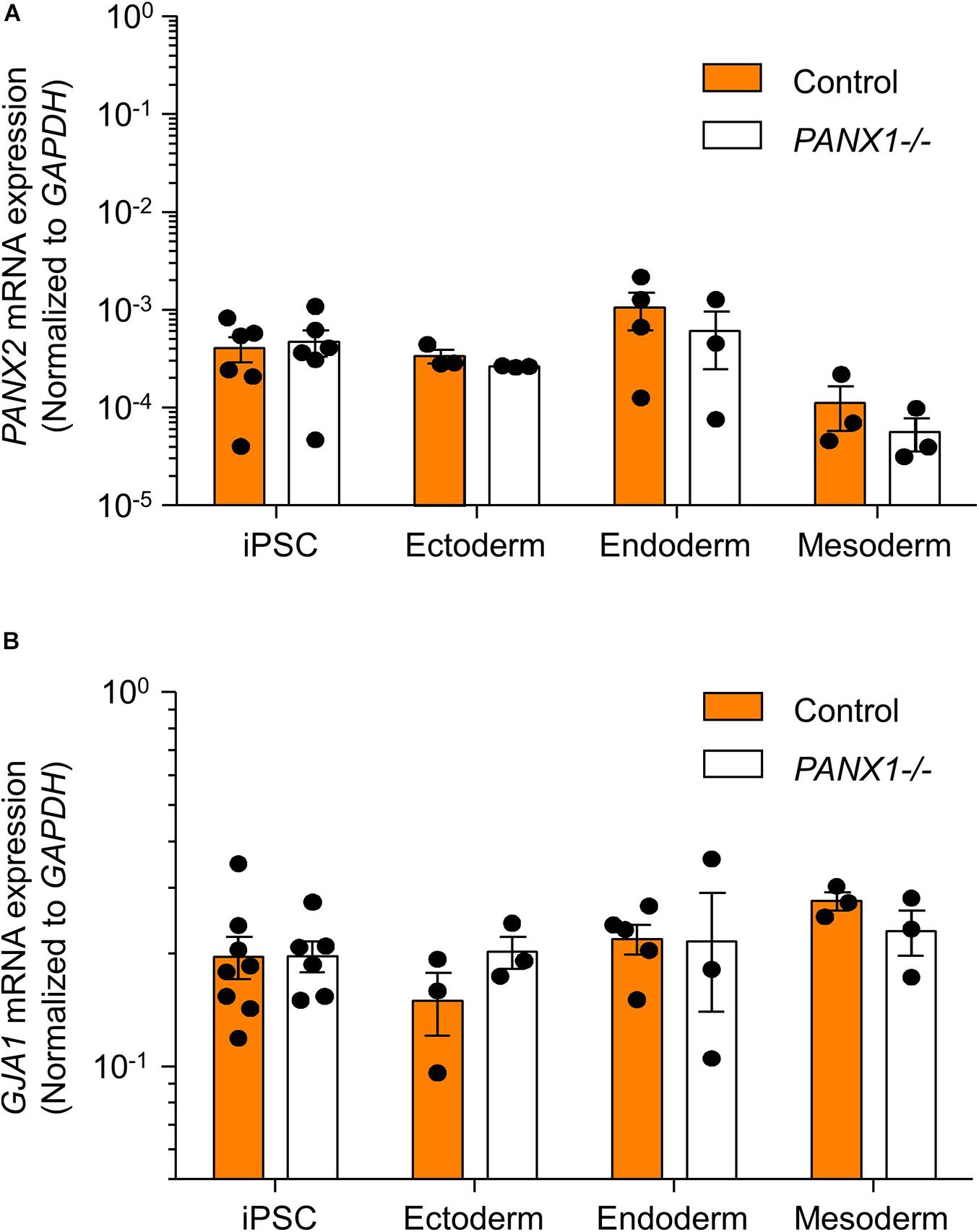
Figure 8. PANX2 and Cx43 do not compensate for PANX1 loss during germ lineage differentiation. qPCR analysis of (A) PANX2 and (B) GJA1 (Cx43) mRNA expression in directed ectoderm, mesoderm, and endoderm cultures. Data presented as 2– ΔCT (fold change from GAPDH) and represent the standard error of the mean of 3-8 independent experiments.
Discussion
Here we find that PANX1 protein is expressed in human iPSCs as well as in cells of all three embryonic germ lineages: ectoderm, endoderm, and mesoderm, further underlying the potential for this protein to participate in cell fate specification. Despite being dispensable for stem cell morphology, proliferation, and survival, PANX1 does contribute to cell fate specification with exaggerated mesendoderm cell abundance in spontaneously differentiated PANX1-/- cultures compared to control. Having confirmed PANX1 gene ablation through genotyping, immunofluorescence, Western blot, flow cytometry and qPCR, we are confident that our clonal knockout cells are indeed deficient in PANX1 protein. However, we do occasionally detect non-specific bands in our PANX1 Western blots. Depending on PANX1 expression and total protein loaded onto gels, we have found that these non-specific bands generally appear when longer exposure times were necessary to capture sufficient PANX1 signal. However, these bands were consistent across different samples and thus considered non-specific.
The human protein atlas reports that PANX1 is widely expressed in tissues arising from all three germ layers3 and the many mouse studies conducted around the world have highlighted how important murine PANX1 is in postnatal health and disease. For example, PANX1 is reported to exacerbate the spread of ischemic injury in mice following stroke via the disruption of electrochemical gradients in neurons and glial cell types (Chekeni et al., 2010; Bond and Naus, 2014). Human PANX1 also has reported involvement in pathogen-mediated activation of the caspase cascade by releasing ATP which attracts phagocytic cells, resulting in the clearance of the damaged/infected cells (Chekeni et al., 2010; Bond and Naus, 2014). On the other hand, HIV can use the PANX1 channel to enter lymphocytes (Orellana et al., 2013) and once inside the cell, the virus can elicit PANX1-mediated ATP release to destabilize the cell membrane and ultimately facilitate viral spread (Séror et al., 2011; Penuela et al., 2014). Surprisingly, few studies have focused on human pannexin proteins or the role of pannexins in early development. We are now able to uncover how PANX1 signaling influences the earliest developmental decisions through spontaneous and directed differentiation of human iPSCs, and by modeling human tissue development using PSC-derived organoids.
PANX1 is expressed in the human oocyte as well as the 2- and 4-cell stage embryo (Sang et al., 2019). Human embryonic stem cells and induced pluripotent stem cells have previously been shown to express all three PANX transcripts (Hainz et al., 2018). However, we only detect expression of PANX1 and PANX2 and were unable to detect PANX3 in any of the stem cell types that we evaluated. We also show that human iPSCs express PANX1 protein, which is concentrated at the cell surface of undifferentiated iPSCs with lesser amounts of intracellular PANX1 populations. Furthermore, PANX1 expression persists in cells of all three embryonic germ layers. Interestingly, while PANX1 was primarily at the cell surface of iPSCs, ectoderm and mesoderm cells, it was redistributed to intracellular compartments in endoderm cells where it partially overlapped with early endosomes. Prolonged increases in ATP can induce PANX1 internalization to endosomal compartments (Boyce et al., 2015). It remains unclear whether a similar mechanism triggers PANX1 internalization during endoderm differentiation. Intracellular PANX1 localization has been shown to correlate with reduced PANX1 glycosylation. We see a similar trend here, with PANX1 in endoderm cells exhibiting both decreased Gly1 and Gly2 and corresponding intracellular localization. However, in the absence of glycosylation mutant studies, we are unable to say definitively that the reduction in PANX1 glycosylation drives intracellular redistribution. It remains to be seen what role this intracellular PANX1 pool plays in endodermal tissues, but given that our PANX1-/- iPSCs exhibit enhanced endodermal differentiation, removing this protein is clearly beneficial for endodermal lineage commitment. One might suppose that because endoderm cells internalize PANX1 they do not need a functional PANX1 pool and might therefore benefit from PANX1 knockout. However, the same cannot be said for the mesoderm cells, which retain PANX1 at the cell surface, and this germ lineage is equally enhanced in PANX1-/- iPSCs.
Most of what is currently understood about pannexins in stem cell fate specification arises from somatic (adult or tissue-resident) stem cell studies. Pannexin signaling can influence the self-renewal and differentiation of multiple somatic stem cell types including osteoprogenitor cells, skeletal myoblasts, and postnatal neuronal stem cells (Swayne and Bennett, 2016; Ishikawa and Yamada, 2017; Pham et al., 2018). PANX1 is implicated in neural precursor cell maintenance and proliferation, while neuronal differentiation involves both PANX1 and PANX2 (reviewed in Swayne and Bennett, 2016). PANX1 specifically regulates neurite growth and dendritic spine density in mice whereas PANX2 influences retinoic acid-induced neurite extension (Swayne et al., 2010; Wicki-Stordeur et al., 2012, 2016; Wicki-Stordeur and Swayne, 2013; Horton et al., 2017; Xu et al., 2018). Meanwhile, PANX1 and PANX3 are both expressed in mesodermal tissues such as bone and cartilage and they have both been implicated in the regulation and commitment of resident progenitor cell populations in mice (Bond et al., 2011; Ishikawa and Yamada, 2017; Lee et al., 2018; Pham et al., 2018). PANX1 is expressed in murine bone marrow-derived stromal cells while PANX3 inhibits osteoprogenitor cell proliferation and contributes to chondrocyte differentiation (Bond et al., 2011; Ishikawa et al., 2014). In contrast to these studies on adult progenitor cell populations, very little is understood about pannexins in pluripotent stem cells, including mouse and human ESCs or iPSCs.
Human germline PANX1 mutations which cause decreased protein abundance and trafficking defects lead to human oocyte death and female infertility (Sang et al., 2019). This effect can be mimicked using isolated mouse oocytes injected with PANX1 mutant complementary RNA (cRNA). On the other hand, PANX1 knockout mice remain fertile and continue to birth average litter sizes. These observed differences in fecundity may be due to inherent species differences, or in vitro versus in vivo manipulations. We find that CRISPR-Cas9 genetic ablation of PANX1 does not negatively impact human iPSC proliferation, survival, or morphology. It is possible that PANX1 missense mutations are more impactful than complete ablation due to as-yet unknown gain of function properties or changes in protein partner interactions. It would be interesting to examine whether human iPSCs are amenable to insertion of human missense mutations via gene editing or whether PANX1-/- iPSCs can differentiate to primordial germ cells.
The most well-defined role of PANX1 is in the regulated release of ATP through several mechanisms including mechanical stress, membrane depolarization, changes in intracellular ion concentration and others (Penuela et al., 2013). Autocrine and paracrine signaling mechanisms triggered through cellular release of ATP and ADP have reported trophic, differentiating, and immunomodulatory effects and ATP signaling has been linked to proliferation of mouse embryonic stem cell and several postnatal progenitor cell populations (Heo and Han, 2006; Cavaliere et al., 2015). Activated pannexin channels appear to play a supporting role in augmenting purinergic receptor activity through the release of extracellular nucleotides and nucleosides. Additionally, PANX1 has been widely implicated in cell death signaling (Chekeni et al., 2010; Penuela et al., 2014; Imamura et al., 2020). Apoptotic induction through caspase activation leads to cleavage of the PANX1 carboxyl-terminal tail, which subsequently results in release of ATP and other small molecules into the extracellular space. Although we have found that iPSCs tolerate the loss of PANX1, follow up investigations are necessary to determine whether the lineage bias that we have presented here actually results from compromised ectodermal lineage specification or due to altered proliferation or apoptosis of certain germ layers within PANX1 null cells. The effect of PANX1 on lineage specification may in fact be combinatorial as we see increase mesendoderm gene expression while others have reported selective loss of multipotent ectodermal progenitor cells in PANX1 knockout mice (Wicki-Stordeur et al., 2016).
Human developmental disorders arise in part due to flawed cell fate specification which can contribute to organ and tissue dysfunction. Until we have a comprehensive understanding of human development and cell fate decisions our capacity to treat developmental disorders remains incomplete. Given the ubiquitous expression of PANX1 in adult tissues, we expect one or more aspects of human stem cell pluripotency or early lineage specification to be affected by the loss of PANX1. Future studies will uncover protein interacting partners and specific messenger molecules involved in this process. Furthermore, it will be interesting to determine whether PANX1 is similarly involved in downstream specification of various terminally differentiated cells or 3-dimensional organoid development.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Newfoundland and Labrador Human Research Ethics Board #2018.210. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author Contributions
JE used CRISPR/Cas9 to generate the PANX1-/- iPSCs and oversaw the project and wrote the manuscript. RN and GC performed all the experiments, analyzed the data, and assembled the figures. All authors reviewed and edited the manuscript.
Funding
This study was supported through the Natural Sciences and Engineering Research Council Discovery Grant RGPIN-2019-04345, Memorial University Faculty of Medicine startup funds and the Medical Research Endowment Fund to JE. RN is supported by a Faculty of Medicine Dean’s Fellowship, the F.A. Aldrich Graduate Fellowship and the Natural Sciences and Engineering Research Council Canadian Graduate Scholarship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Dale Laird for generously providing us with the control and PANX1-/- iPSCs as well as the human PANX1 antibody used in this study.
Footnotes
- ^ http://www.sanger.ac.uk/htgt/wge/
- ^ https://www.ncbi.nlm.nih.gov/tools/primer-blast/
- ^ https://www.proteinatlas.org/ENSG00000110218-PANX1/tissue
References
Bao, J., Wang, J., Yao, Y., Wang, Y., Fan, X., Sun, K., et al. (2013). Correlation of ventricular arrhythmias with genotype in arrhythmogenic right ventricular cardiomyopathy. Circ. Cardiovasc. Genet. 6, 552–556. doi: 10.1161/CIRCGENETICS.113.000122
Beers, J., Gulbranson, D. R., George, N., Siniscalchi, L. I., Jones, J., Thomson, J. A., et al. (2012). Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nat. Protoc. 7, 2029–2040. doi: 10.1038/nprot.2012.130
Bond, S. R., and Naus, C. C. (2014). The pannexins: past and present. Front. Physiol. 5:58. doi: 10.3389/fphys.2014.00058
Bond, S. R., Lau, A., Penuela, S., Sampaio, A. V., Underhill, T. M., Laird, D. W., et al. (2011). Pannexin 3 is a novel target for Runx2, expressed by osteoblasts and mature growth plate chondrocytes. J. Bone Miner. Res. 26, 2911–2922. doi: 10.1002/jbmr.509
Boyce, A. K., Kim, M. S., Wicki-Stordeur, L. E., and Swayne, L. A. (2015). ATP stimulates pannexin 1 internalization to endosomal compartments. Biochem. J. 470, 319–330. doi: 10.1042/BJ20141551
Camacho-Aguilar, E., and Warmflash, A. (2020). Insights into mammalian morphogen dynamics from embryonic stem cell systems. Curr. Top Dev. Biol. 137, 279–305. doi: 10.1016/bs.ctdb.2019.11.010
Cavaliere, F., Donno, C., and D’Ambrosi, N. (2015). Purinergic signaling: a common pathway for neural and mesenchymal stem cell maintenance and differentiation. Front. Cell. Neurosci. 9:211. doi: 10.3389/fncel.2015.00211
Chekeni, F. B., Elliott, M. R., Sandilos, J. K., Walk, S. F., Kinchen, J. M., and Lazarowski, E. R. (2010). Pannexin 1 channels mediate ‘find-me’ signal release and membrane permeability during apoptosis. Nature 467, 863–867. doi: 10.1038/nature09413
Cold Spring Harbor Laboratory (2007). Mowial-DABCO stock solution. Cold Spring Harb. Protoc. doi: 10.1101/pdb.rec10913
Dahlmann, J., Kensah, G., Kempf, H., Skvorc, D., Gawol, A., Elliott, D. A., et al. (2013). The use of agarose microwells for scalable embryoid body formation and cardiac differentiation of human and murine pluripotent stem cells. Biomaterials 34, 2463–2471. doi: 10.1016/j.biomaterials.2012.12.024
Desbaillets, I., Ziegler, U., Groscurth, P., and Gassmann, M. (2000). Embryoid bodies: an in vitro model of mouse embryogenesis. Exp. Physiol. 85, 645–651. doi: 10.1111/j.1469-445x.2000.02104.x
Esseltine, J. L., and Laird, D. W. (2016). Next-generation connexin and pannexin cell biology. Trends Cell Biol. 26, 944–955. doi: 10.1016/j.tcb.2016.06.003
Esseltine, J. L., Brooks, C. R., Edwards, N. A., Subasri, M., Sampson, J., Séguin, C., et al. (2020). Dynamic regulation of connexins in stem cell pluripotency. Stem Cells 38, 52–66. doi: 10.1002/stem.3092
Esseltine, J. L., Shao, Q., Brooks, C., Sampson, J., Betts, D. H., Séguin, C. A., et al. (2017). Connexin43 mutant patient-derived induced pluripotent stem cells exhibit altered differentiation potential. J. Bone Miner. Res. 32, 1368–1385. doi: 10.1002/jbmr.3098
Friedrich, J., Seidel, C., Ebner, R., and Kunz-Schughart, L. A. (2009). Spheroid-based drug screen: considerations and practical approach. Nat. Protoc. 4, 309–324. doi: 10.1038/nprot.2008.226
Furlow, P. W., Zhang, S., Soong, T. D., Halberg, N., Goodarzi, H., Mangrum, C., et al. (2015). Mechanosensitive pannexin-1 channels mediate microvascular metastatic cell survival. Nat. Cell Biol. 17, 943–952. doi: 10.1038/ncb3194
Glaser, T., de Oliveira, S. L., Cheffer, A., Beco, R., Martins, P., Fornazari, M., et al. (2014). Modulation of mouse embryonic stem cell proliferation and neural differentiation by the P2X7 receptor. PLoS One 9:e96281. doi: 10.1371/journal.pone.0096281
Hainz, N., Beckmann, A., Schubert, M., Haase, A., Martin, U., Tschernig, T., et al. (2018). Human stem cells express pannexins. BMC Res. Notes 11:54. doi: 10.1186/s13104-018-3125-z
Heo, J. S., and Han, H. J. (2006). ATP stimulates mouse embryonic stem cell proliferation via protein kinase C, phosphatidylinositol 3-kinase/Akt, and mitogen-activated protein kinase signaling pathways. Stem Cells 24, 2637–2648. doi: 10.1634/stemcells.2005-0588
Horton, S. M., Luna Lopez, C., Blevins, E., Howarth, H., Weisberg, J., Shestopalov, V. I., et al. (2017). Pannexin 1 modulates axonal growth in mouse peripheral nerves. Front. Cell. Neurosci. 11:365. doi: 10.3389/fncel.2017.00365
Imamura, H., Sakamoto, S., Yoshida, T., Matsui, Y., Penuela, S., Laird, D. W., et al. (2020). Single-cell dynamics of pannexin-1-facilitated programmed ATP loss during apoptosis. Elife 9:e61960. doi: 10.7554/eLife.61960
Ishikawa, M., and Yamada, Y. (2017). The role of pannexin 3 in bone biology. J. Dent. Res. 96, 372–379. doi: 10.1177/0022034516678203
Ishikawa, M., Iwamoto, T., Fukumoto, S., and Yamada, Y. (2014). Pannexin 3 inhibits proliferation of osteoprogenitor cells by regulating Wnt and p21 signaling. J. Biol. Chem. 289, 2839–2851. doi: 10.1074/jbc.M113.523241
Itskovitz-Eldor, J., Schuldiner, M., Karsenti, D., Eden, A., Yanuka, O., Amit, M., et al. (2000). Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol. Med. 6, 88–95. doi: 10.1007/bf03401776
Jiang, L. H., Hao, Y., Mousawi, F., Peng, H., and Yang, X. (2017). Expression of P2 purinergic receptors in mesenchymal stem cells and their roles in extracellular nucleotide regulation of cell functions. J. Cell. Physiol. 232, 287–297. doi: 10.1002/jcp.25484
Kaebisch, C., Schipper, D., Babczyk, P., and Tobiasch, E. (2015). The role of purinergic receptors in stem cell differentiation. Comput. Struct. Biotechnol. J. 13, 75–84. doi: 10.1016/j.csbj.2014.11.003
Lee, V. R., Barr, K. J., Kelly, J. J., Johnston, D., Brown, C. F. C., Robb, K. P., et al. (2018). Pannexin 1 regulates adipose stromal cell differentiation and fat accumulation. Sci. Rep. 8:16166. doi: 10.1038/s41598-018-34234-9
Lin, Y., and Chen, G. (2014). “StemBook,” in StemBook, ed. The Stem Cell Research Community (Amsterdam: IOS Press).
Nagy, A., Rossant, J., Nagy, R., Abramow-Newerly, W., and Roder, J. C. (1993). Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 90, 8424–8428. doi: 10.1073/pnas.90.18.8424
Orellana, J. A., Velasquez, S., Williams, D. W., Sáez, J. C., Berman, J. W., and Eugenin, E. A. (2013). Pannexin1 hemichannels are critical for HIV infection of human primary CD4+ T lymphocytes. J. Leukoc. Biol. 94, 399–407. doi: 10.1189/jlb.0512249
Penuela, S., Bhalla, R., Gong, X. Q., Cowan, K. N., Celetti, S. J., Cowan, B. J., et al. (2007). Pannexin 1 and pannexin 3 are glycoproteins that exhibit many distinct characteristics from the connexin family of gap junction proteins. J. Cell Sci. 120(Pt 21), 3772–3783. doi: 10.1242/jcs.009514
Penuela, S., Gehi, R., and Laird, D. W. (2013). The biochemistry and function of pannexin channels. Biochim. Biophys. Acta 1828, 15–22. doi: 10.1016/j.bbamem.2012.01.017
Penuela, S., Harland, L., Simek, J., and Laird, D. W. (2014). Pannexin channels and their links to human disease. Biochem. J. 461, 371–381. doi: 10.1042/BJ20140447
Perrimon, N., Pitsouli, C., and Shilo, B. Z. (2012). Signaling mechanisms controlling cell fate and embryonic patterning. Cold Spring Harb. Perspect. Biol. 4:a005975. doi: 10.1101/cshperspect.a005975
Pham, T. L., St-Pierre, M. E., Ravel-Chapuis, A., Parks, T. E. C., Langlois, S., Penuela, S., et al. (2018). Expression of Pannexin 1 and Pannexin 3 during skeletal muscle development, regeneration, and Duchenne muscular dystrophy. J. Cell. Physiol. 233, 7057–7070. doi: 10.1002/jcp.26629
Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., and Zhang, F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308. doi: 10.1038/nprot.2013.143
Sanchez-Arias, J. C., Liu, M., Choi, C. S. W., Ebert, S. N., Brown, C. E., and Swayne, L. A. (2019). Pannexin 1 regulates network ensembles and dendritic spine development in cortical neurons. eNeuro 6, 0503–0518. doi: 10.1523/ENEURO.0503-18.2019
Sandilos, J. K., Chiu, Y. H., Chekeni, F. B., Armstrong, A. J., Walk, S. F., Ravichandran, K. S., et al. (2012). Pannexin 1, an ATP release channel, is activated by caspase cleavage of its pore-associated C-terminal autoinhibitory region. J. Biol. Chem. 287, 11303–11311. doi: 10.1074/jbc.M111.323378
Sang, Q., Zhang, Z., Shi, J., Sun, X., Li, B., Yan, Z., et al. (2019). A pannexin 1 channelopathy causes human oocyte death. Sci. Transl. Med. 11:eaav8731. doi: 10.1126/scitranslmed.aav8731
Schindelin, J., Arganda-Carreras, I., Frise, E., Kaynig, V., Longair, M., Pietzsch, T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. doi: 10.1038/nmeth.2019
Schmittgen, T. D., and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108. doi: 10.1038/nprot.2008.73
Séror, C., Melki, M. T., Subra, F., Raza, S. Q., Bras, M., Saïdi, H., et al. (2011). Extracellular ATP acts on P2Y2 purinergic receptors to facilitate HIV-1 infection. J. Exp. Med. 208, 1823–1834. doi: 10.1084/jem.20101805
Shao, Q., Lindstrom, K., Shi, R., Kelly, J., Schroeder, A., Juusola, J., et al. (2016). A germline variant in the PANX1 gene has reduced channel function and is associated with multisystem dysfunction. J. Biol. Chem. 291, 12432–12443. doi: 10.1074/jbc.M116.717934
Swayne, L. A., and Bennett, S. A. (2016). Connexins and pannexins in neuronal development and adult neurogenesis. BMC Cell Biol. 17(Suppl. 1):10. doi: 10.1186/s12860-016-0089-5
Swayne, L. A., Sorbara, C. D., and Bennett, S. A. (2010). Pannexin 2 is expressed by postnatal hippocampal neural progenitors and modulates neuronal commitment. J. Biol. Chem. 285, 24977–24986. doi: 10.1074/jbc.M110.130054
Tada, S., Era, T., Furusawa, C., Sakurai, H., Nishikawa, S., Kinoshita, M., et al. (2005). Characterization of mesendoderm: a diverging point of the definitive endoderm and mesoderm in embryonic stem cell differentiation culture. Development 132, 4363–4374. doi: 10.1242/dev.02005
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. doi: 10.1016/j.cell.2007.11.019
Tam, P. P., and Loebel, D. A. (2007). Gene function in mouse embryogenesis: get set for gastrulation. Nat. Rev. Genet. 8, 368–381. doi: 10.1038/nrg2084
Tan, J. Y., Sriram, G., Rufaihah, A. J., Neoh, K. G., and Cao, T. (2013). Efficient derivation of lateral plate and paraxial mesoderm subtypes from human embryonic stem cells through GSKi-mediated differentiation. Stem Cells Dev. 22, 1893–1906. doi: 10.1089/scd.2012.0590
Tchieu, J., Zimmer, B., Fattahi, F., Amin, S., Zeltner, N., Chen, S., et al. (2017). A modular platform for differentiation of human PSCs into all major ectodermal lineages. Cell Stem Cell 21, 399–410.e7. doi: 10.1016/j.stem.2017.08.015
ten Berge, D., Koole, W., Fuerer, C., Fish, M., Eroglu, E., and Nusse, R. (2008). Wnt signaling mediates self-organization and axis formation in embryoid bodies. Cell Stem Cell 3, 508–518. doi: 10.1016/j.stem.2008.09.013
Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S., et al. (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147. doi: 10.1126/science.282.5391.1145
Watanabe, K., Ueno, M., Kamiya, D., Nishiyama, A., Matsumura, M., Wataya, T., et al. (2007). A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 25, 681–686. doi: 10.1038/nbt1310
Wicki-Stordeur, L. E., and Swayne, L. A. (2013). Panx1 regulates neural stem and progenitor cell behaviours associated with cytoskeletal dynamics and interacts with multiple cytoskeletal elements. Cell Commun. Signal. 11:62. doi: 10.1186/1478-811X-11-62
Wicki-Stordeur, L. E., Dzugalo, A. D., Swansburg, R. M., Suits, J. M., and Swayne, L. A. (2012). Pannexin 1 regulates postnatal neural stem and progenitor cell proliferation. Neural Dev. 7:11. doi: 10.1186/1749-8104-7-11
Wicki-Stordeur, L. E., Sanchez-Arias, J. C., Dhaliwal, J., Carmona-Wagner, E. O., Shestopalov, V. I., Lagace, D. C., et al. (2016). Pannexin 1 differentially affects neural precursor cell maintenance in the ventricular zone and peri-infarct cortex. J. Neurosci. 36, 1203–1210. doi: 10.1523/JNEUROSCI.0436-15.2016
Xu, X., Wicki-Stordeur, L. E., Sanchez-Arias, J. C., Liu, M., Weaver, M. S., Choi, C. S. W., et al. (2018). Probenecid disrupts a novel pannexin 1-collapsin response mediator protein 2 interaction and increases microtubule stability. Front. Cell. Neurosci. 12:124. doi: 10.3389/fncel.2018.00124
Keywords: human pluripotent stem cells, pannexin channels, germ lineage specification, differentiation, CRISPR-Cas9 gene ablation
Citation: Noort RJ, Christopher GA and Esseltine JL (2021) Pannexin 1 Influences Lineage Specification of Human iPSCs. Front. Cell Dev. Biol. 9:659397. doi: 10.3389/fcell.2021.659397
Received: 27 January 2021; Accepted: 22 March 2021;
Published: 16 April 2021.
Edited by:
Delilah Hendriks, Hubrecht Institute (KNAW), NetherlandsReviewed by:
Marie Billaud, Brigham and Women’s Hospital, Harvard Medical School, United StatesLeigh Anne Swayne, University of Victoria, Canada
Copyright © 2021 Noort, Christopher and Esseltine. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jessica L. Esseltine, amVzc2VsdGluZUBtdW4uY2E=
†These authors have contributed equally to this work and share first authorship