 Jingjing Fa1,2†
Jingjing Fa1,2† Xiaoqing Zhang1†
Xiaoqing Zhang1† Xiaoping Zhang1†
Xiaoping Zhang1† Ming Qi1†
Ming Qi1† Xingyu Zhang1Qihua Fu1,2,3Zhuoming Xu4*
Xingyu Zhang1Qihua Fu1,2,3Zhuoming Xu4* Yunqian Gao1,2*
Yunqian Gao1,2* Bo Wang1,3*
Bo Wang1,3*- 1Pediatric Translational Medicine Institute, Shanghai Children’s Medical Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2Department of Laboratory Medicine, Shanghai Children’s Medical Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 3Faculty of Medical Science, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 4Cardiac Intensive Care Unit, Department of Thoracic and Cardiovascular Surgery, Shanghai Children’s Medical Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China
Congenital heart defects (CHDs) are the most common birth defects worldwide. 22q11.2 deletion syndrome is the most common microdeletion disorder that has been frequently associated with conotruncal malformations. By now, the dosage-sensitive gene TBX1 has been adopted as the major pathogenic gene responsible for 22q11.2 deletion, which is regulated by canonical Wnt/β-catenin signaling pathway in heart outflow tract development. Here, we report the long noncoding RNA (lncRNA) lnc-TSSK2-8, which is encompassed in the 22q11.2 region, that can activate canonical Wnt/β-catenin signaling by protecting β-catenin from degradation, which could result from decreased ubiquitination. Such effects were mediated by two short heat shock proteins HSPA6 and α-β-crystallin (CRYAB), whose expression was regulated by lnc-TSSK2-8 through a competing endogenous RNA (ceRNA) mechanism. In clinical practice, the pathogenesis of copy number variation (CNV) was always attributed to haploinsufficiency of protein-coding genes. Here, we report that the 22q11.2 lncRNA lnc-TSSK2-8 significantly activated canonical Wnt/β-catenin signaling, which has major roles in cardiac outflow tract development and should act upstream of TBX1. Our results suggested that lncRNAs should contribute to the etiology of CNV-related CHD.
Introduction
Congenital heart defects (CHDs) are the most common birth defects worldwide, with an incidence of nearly 10∼12 per 1,000 live infants (1∼1.2%) (Hoffman and Kaplan, 2002; Van Der Linde et al., 2011). As a consequence of improvement in surgical and medical management, more and more patients with severe forms of CHDs can survive until their 30s and beyond. Genetic variation, environmental causes, and their interactions could contribute to the disruption of heart development, thus leading to formation of CHDs (Pierpont et al., 2018; Zu et al., 2020). Genetic variations associated with CHDs have been revealed extensively in recent years. Single gene disorder and gross chromosomal anomalies/aneuploidy can be identified in up to 5 and 10% CHD cases, respectively. Copy number variations (CNVs) are segmental deletions or duplications ranging widely in size of millions of base pairs; pathogenic CNVs could explain about 3∼25% syndromic CHD and 3∼10% isolated CHD cases (Cowan and Ware, 2015).
22q11.2 deletion syndrome (22q11.2DS) is the most common microdeletion disorder with an estimated prevalence ranging from 1 per 3,000 to 1 per 6,000 live births (Tezenas Du Montcel et al., 1996; Devriendt et al., 1998; Goodship et al., 1998; Botto et al., 2003; Oskarsdottir et al., 2004). It is well documented that 22q11.2DS involves 0.7∼3 million base pairs, resulting in multiorgan dysfunction including CHDs, palatal abnormalities, immunodeficiency, developmental delays, cognitive deficits, and neuropsychiatric illnesses. 22q11.2 deletion is the second most common cause of CHDs; conotruncal malformations [malformations of the outflow tract (OFT) such as the tetralogy of Fallot (TOF), truncus arteriosus, interrupted aortic arch type B, and ventricular septal defect (VSD)] account for ∼70% of the heart defects associated this CNV (Agergaard et al., 2012). On the other hand, CHD is also the main cause of mortality (∼87%) in 22q11.2DS patients (McDonald-McGinn et al., 2001; Repetto et al., 2014).
Since the function of proteins had been well annotated, most clinical pathogenic CNVs were interpreted based on their effect on gene dosage of protein-coding genes. There are 46 protein-coding genes located in the typical 3M region of 22q11.2 locus, among which most studies of interest relate to TBX1, which encodes a T-box transcription factor. TBX1 was found to be a crucial gene in the LCR22A–LCR22B region; heterozygous loss of function mutations of Tbx1 in the mouse resulted in penetrant defects that are reminiscent of CHD in 22q11.2 (Lindsay et al., 2001; Chapnik et al., 2012). Another gene of interest is DGCR8, which encodes a double-stranded RNA-binding protein that is involved in microRNA (miRNA) biogenesis. Heterozygosity of Dgcr8 results in neuronal deficits (Stark et al., 2008), whereas ablation of both alleles in neural crest cells causes heart defects (Chapnik et al., 2012). Additionally, evidence of other individual protein-coding genes in 22q11.2 region responsible for major cardiac phenotypes of 22q11.2DS has also accumulated. For example, haploinsufficiency of CRKL could account for the cardiac anomalies in individuals with nested distal deletions (Zheng et al., 2015).
Long noncoding RNAs (lncRNAs), which contain >200 nucleotides, have been revealed to participate in the regulation of cellular and tissue function and play a great part in heart development (Devaux et al., 2015). LncRNAs can repress or activate gene expression epigenetically with diverse mechanisms: e.g., most lncRNA molecules localize in the nucleus and regulate gene expression epigenetically; a proportion (∼15%) of lncRNA are located in the cytoplasm where they function posttranscriptionally (Devaux et al., 2015). Several lncRNAs have been identified as key regulatory molecules involved in cardiac development, such as Braveheart (Klattenhoff et al., 2013), Fendrr (Grote and Herrmann, 2013), Upperhand (Anderson et al., 2016), and BANCR (Wilson et al., 2020). Several examples of CHD-associated lncRNAs have also been reported. Gu et al. (2016) indicated that circulating plasma lncRNAs might serve as novel biomarkers for CHD. Jiang et al. (2018) discovered that HOTAIR is upregulated in cardiac tissues and plasma of patients with CHD; thus, it could serve as a potential diagnostic biomarker for CHD. Wang et al. (2018) suggested the relationship between HA117 and TOF, although the molecular basis of HA117 remains unclear. Jiang et al. (2019) reported upregulated SNHG6 expression in fetal heart tissues of patients with VSDs. Ma et al. (2020) reported that TBX5-AS1 should be involved in TOF by affecting cell proliferation by regulating TBX5. This evidence signifies the importance of lncRNA in CHD. Haploinsufficiency of TBX1 is regarded as the major candidate for cardiac OFT malformation. Recently, canonical Wnt/β-catenin has been revealed to play major roles in cardiac OFT development upstream of TBX1 (Racedo et al., 2017). It is unknown whether lncRNAs participate in this process. The canonical Wnt/β-catenin signaling pathway has been revealed to play developmental stage-specific roles in early heart development. The Wnt family proteins serve as secreted signaling molecules that initiate a series of intracellular pathways. In the canonical Wnt/β-catenin signaling pathway, Wnt signaling starts with the generation of Wnt ligands into the extracellular space. The Wnt molecules then bind to the Frizzled receptor (Frz) and its coreceptor LRP5/6 to form trimeric complexes. The formation of such complexes inhibits the glycogen synthase kinase 3β (GSK-3β) destruction complex, which functions in stabilizing the cytosolic β-catenin (Blankesteijn et al., 2008). Cytosolic accumulation of β-catenin leads to its translocation into the nucleus. Subsequently, the Wnt proteins bind to the T-cell factor (TCF)/lymphoid enhancer-binding factor (LEF) transcription factors to activate Wnt-responsive genes. Activation of Wnt/β-catenin signaling during embryoid body (EB) formation promotes embryonic stem cell differentiation into cardiomyocytes. Whereas the activation of Wnt/β-catenin signaling in a later stage (during gastrulation) results in inhibition of cardiomyocyte formation (Naito et al., 2006; Ueno et al., 2007).
In the present study, we discovered that the lncRNA lnc-TSSK2-8, which is located in the 22q11.2 region, significantly promotes the stabilization of β-catenin. Therefore, we propose that lnc-TSSK2-8 should contribute to heart development by regulating Wnt/β-catenin signaling pathway. In contrast to earlier studies that emphasize the crucial roles of protein-coding genes, these data suggest that the relationship between disease and noncoding transcripts should be further explored.
Materials and Methods
Rapid Amplification of Complementary DNA Ends
Here, 5′ and 3′ rapid amplification of complementary DNA (cDNA) ends (RACE) was performed on RNA isolated from HEK293 cells using a SMARTer RACE 5′/3′ Kit (TAKARA, Dalian, China), following the manufacturer’s instructions. The PCR products were separated on a 1% agarose gel and validated by Sanger sequencing. The following gene-specific primers were used for PCR:
GACTGAAGGAGTAGAAA (5′ RACE primer)
GTCCAGGTGTCCCTGCCTCCCATTG (3′ RACE primer 1)
GGGGGAAGCCCACAATGAGCAG (3′ RACE primer 2)
Isolation of Nuclear and Cytoplasmic RNA
Nuclear RNA and cytoplasmic RNA of HEK293 cells were extracted and purified using the PARISTM Kit (Invitrogen, Thermo Fisher Scientific, Inc., Waltham, MA, United States) according to the manufacturer’s instructions.
Cell Culture and Long Noncoding RNA Overexpression
The HEK293 cell line was purchased from the Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China. Cells were cultured with Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Thermo Fisher Scientific, Inc., Waltham, MA, United States) containing 10% fetal bovine serum (FBS; Gibco, Thermo Fisher Scientific, Inc., Waltham, MA, United States) and 0.1% penicillin/streptomycin (NCM Biotech, Suzhou, Jiangsu, China) at 37°C with 5% CO2.
The cDNA sequence of lnc-TSSK2-8 was inserted into pcDNA3.1 (pcDNA3.1-lncTSSK2.8) for overexpression. HEK293 cells were transfected with pcDNA3.1-lncTSSK2.8 by using Lipofectamine 3000 (Invitrogen, Thermo Fisher Scientific, Inc., Waltham, MA, United States) according to the manufacturer’s instruction.
RNA Sequencing Analysis
Cell samples were lysed in TRIzol (Ambion, Austin, TX, United States). Total RNA was extracted using the RNeasy kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
A total amount of 2 μg RNA per sample was used as input material for the RNA preparations. Firstly, ribosomal RNA was removed using Epicenter Ribo-zeroTM Kit (Illumina Inc., San Diego, CA, United States), and rRNA-free residue was cleaned up by ethanol precipitation. By using the fragmentation buffer, the mRNA and noncoding RNAs were fragmented into short fragments (about 200∼700 bp), then the first-strand cDNA was synthesized by random hexamer-primer using the fragments as templates. Buffer, dNTPs, RNase H, and DNA polymerase I were added to synthesize the second strand cDNA. Subsequently, A-Tailing Mix (QIAGEN, Hilden, Germany) and RNA Index Adapters were added to perform end-repair, and the resulting cDNA was amplified by PCR. The double-strand cDNA was purified with QiaQuick PCR extraction kit and then used for end-polishing. Sequencing adapters were ligated to the fragments, then the second strand was degraded using Uracil-N-Glyosylase (UNG) finally. The quantity and quality of cDNA libraries were evaluated using Agilent 2100 Bioanalyzer (ABI, New York, NY, United States). The double-stranded PCR products were heat denatured and circularized by the splint oligo sequence to obtain the final library. The single-stranded circular DNA was amplified using phi29 (Thermo Fisher Scientific, Waltham, MA, United States) to generate a DNA nanoball. The library preparations were sequenced on BGISEQ-500 platform, and 150-bp paired-end reads were generated.
Raw sequencing reads with low quality (e.g., proportion of bases with sQ ≤ 5 greater than 50%; proportion of N greater >10%; 5′adaptor contamination) were removed using customized scripts. The remaining clean data were aligned to the human reference genome (version GRCh38) using STAR (Dobin et al., 2013) with standard options for long RNA sequencing (RNAseq) pipeline of Encyclopedia of DNA Elements (ENCODE;1): –outFilterType BySJout, –outFilterMultimapNmax 20, –alignSJoverhangMin 8, –alignSJDBoverhangMin1, –outFilterMismatchNmax 999, –outFilterMismatchNoverReadLmax 0.04, –alignIntronMin 20, –alignIntronMax 1000000, –alignMatesGapMax 1000000. Gene level expression was estimated using the featureCounts software (Liao et al., 2014). Differential gene expression analysis was performed on the raw counts using the DESeq2 (Love et al., 2014) package. Genes with fold change greater than 2 and P value lower than 0.05 were identified as differentially expressed genes (DEGs). Functional enrichment analysis was performed with the clusterProfiler (Yu et al., 2012) package; gene set enrichment analysis was performed for Gene Ontology (GO) items [Biological Process (BP), Molecular Function (MF), Cell Component (CC)] and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway items. We used the genes with fold change greater than 1.2 for functional enrichment analysis, since the DEG number is too small for such analysis. BH adjustments were used for multiple testing of functional enrichment analysis; p < 0.05 and q < 0.05 were used as thresholds.
RT-qPCR
Total RNA was extracted with TRIzol reagent (Ambion, Austin, TX, United States) and reverse transcribed into cDNA with PrimeScript RT reagent Kit (Takara, Dalian, China). RT-qPCR was conducted with TB Green Premix Ex Taq II kit (Takara, Dalian, China) on the CFX 9600 Real-Time PCR detection system (Bio-Rad Laboratories, Inc., Hercules, CA, United States). Fold changes of the lncRNA and mRNAs were calculated with small nuclear RNA U6 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as internal controls, respectively, based on 2–Δ Δ Ct method. All the Bulge-Loop RT primers for both microRNAs and U6 were purchased from RiboBio (Guangzhou, Guangdong, China).
Luciferase Reporter Assay
Lnc-TSSK2-8 wild type/mutation (WT/MUT) and heat shock protein (HSP)A6 UTR WT/MUT were inserted into the BamH I and Hind III sites of the pMIR dual-luciferase vector (Promega, Madison, WI, United States). These plasmids as well as the pMIR vector were transfected with the miR-6721-5p mimics or NC-mimics into HEK293 cells for 48 h, respectively. Finally, luciferase activities were detected using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, United States) according to the manufacturer’s protocol.
Western Blotting
The transfected cells were disintegrated using Western blot (WB) IP lysis buffer (Beyotime Biotechnology, Shanghai, China). All protein extracts were separated with 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; Beyotime, Nantong, Jiangsu, China), transferred onto polyvinylidene fluoride (PVDF) membranes (Roche Diagnostic Corporation, Indianapolis, IN, United States) and blotted with primary antibodies (HSPA6, Abcam, Cat. No. ab69408; β-Catenin, Cell Signaling Technology, Cat. No. 8480S; p-β-Catenin, Cell Signaling Technology, Cat. No. 5651S; GAPDH, Cell Signaling Technology, Cat. No. 5174S; Tubulin, Cell Signaling Technology, Cat. No. 5335S). Sequentially, membranes were incubated with secondary antibodies (1:10,000; Cell Signaling Technology, Cat. No. 7074). At last, proteins were visualized using ECL detection kits (Millipore Corp., Millipore Billerica, MA, United States).
BrdU Cell Proliferation Assay
The cell proliferation assay was performed using TransDetect EdU Flow Cytometry Kit-647 Fluorophore (TransGen Biotech, Beijing, China). Cells were cultured with EdU solution for 2 h and fixed by 1× EdU permeabilization buffer. The assay was visualized by the FACScan system (Bio-Rad Laboratories, Inc., Hercules, CA, United States).
Statistical Analysis
The SPSS 16.0 software was used for statistical analysis. Differences between two groups were analyzed by implementing Student t-test, and P-value < 0.05 was regarded as statistically significant. All data were expressed as mean ± SD.
Results
Full-Length Characterization and Subcellular Localization of lnc-TSSK2-8
The full-length sequence of lnc-TSSK2-8 was successfully obtained from HEK293 cells by rapid amplification of the 5′ and 3′ cDNA assays. We confirmed the sequence by performing PCR and Sanger sequencing. The full-length transcript of lnc-TSSK2-8 was 1,128 in size with a polyadenylated tail (Figure 1A). To determine the subcellular localization of lnc-TSSK2-8, we separated the nuclear and cytoplasm fractions of HEK293 cells and quantified its expression with RT-qPCR. The results suggested that lnc-TSSK2-8 is located both in the nucleus and cytoplasm, although the cytoplasmic composition was much more abundant (Figure 1B).
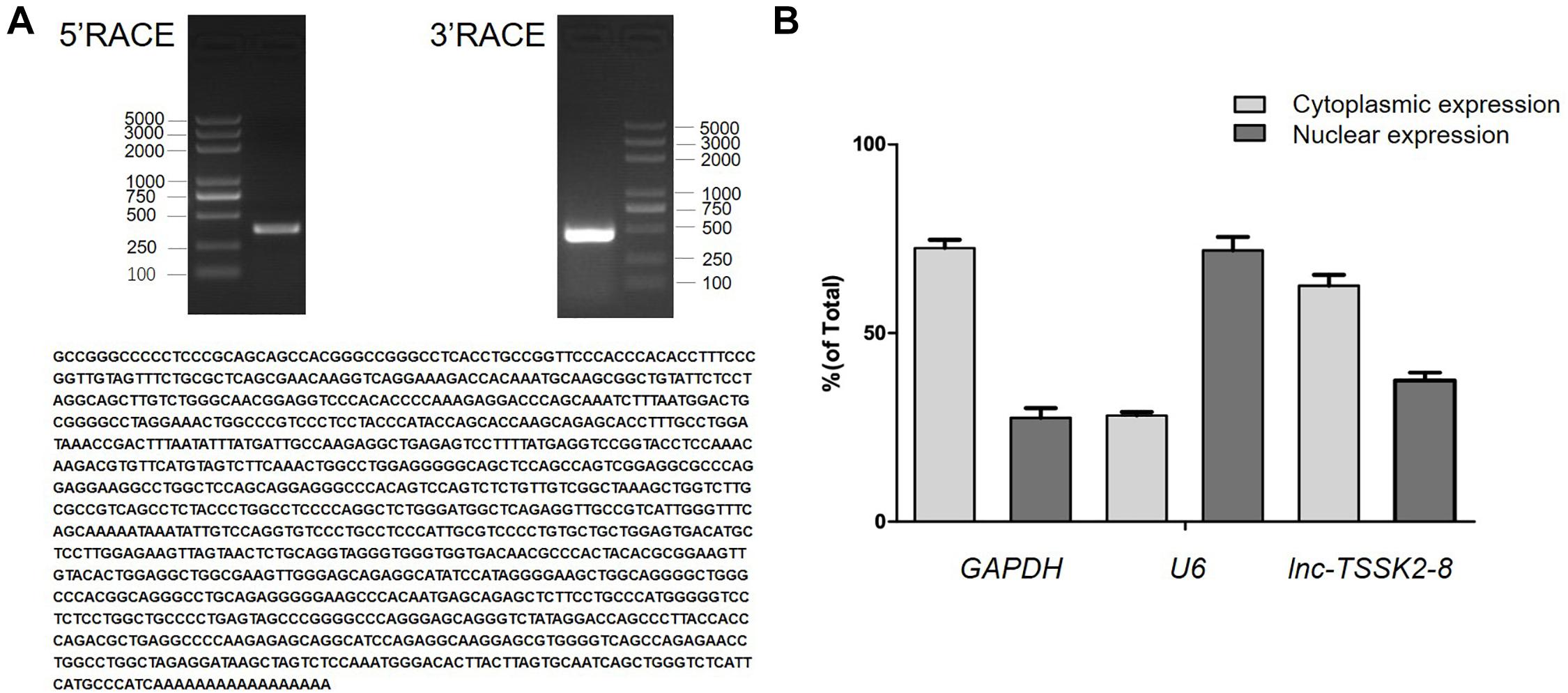
Figure 1. Full-length characterization and subcellular expression analysis of lnc-TSSK2-8. (A) Electrophoresis analysis of rapid amplification of cDNA ends (RACE) PCR and the obtained full-length cDNA sequence of lnc-TSSK2-8. (B) Subcellular distribution of lnc-TSSK2-8. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and U6 were used for references of cytoplasmic expression and nuclear expression, respectively.
lnc-TSSK2-8 Activates Canonical Wnt/β-Catenin Signaling
We overexpressed the full-length transcript of lnc-TSSK2-8 in HEK293 cells (Figure 2A) and examined the expression of the key components of canonical Wnt/β-catenin signaling pathway. Overexpression of lnc-TSSK2-8 increased cell proliferation by ∼20% (Figure 2B) and activated the transcription of the LEF (LEF1) (Figure 2C), whereas the transcriptional level of β-catenin (CTNNB1) did not change (Figure 2D). Further exploration suggested that the protein expression level of β-catenin increased significantly; such an increase should be associated with its decreased phosphorylation (Figures 2E,G). Subsequently, we revealed that the phosphorylation of GSK-3β (Figure 2F) was enhanced and established that ubiquitination of β-catenin could be inhibited by lnc-TSSK2-8.
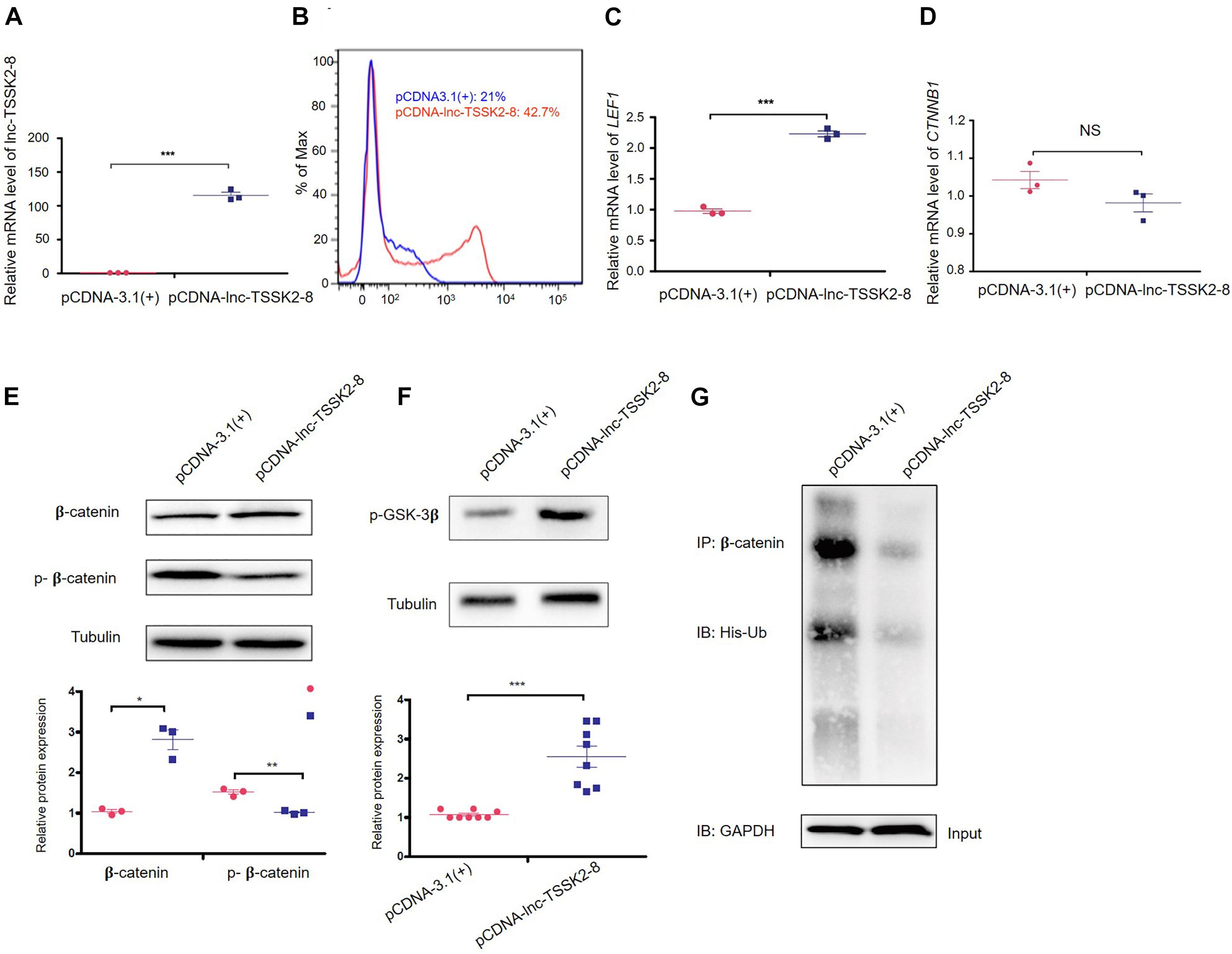
Figure 2. Overexpression of lnc-TSSK2-8 activates the Wnt/β-catenin signaling pathway. (A) RT-qPCR quantification of overexpression lnc-TSSK2-8 in HEK293 cells. (B) Flow cytometry analysis of cell proliferation. The transcriptional level of lymphoid enhancer-binding factor 1 (LEF1) (C) and β-catenin (CTNNB1) (D) in HEK293 cells with lnc-TSSK2-8 overexpression. (E) The influence of lnc-TSSK2-8 overexpression on protein expression and phosphorylation of β-catenin. (F) The influence of lnc-TSSK2-8 overexpression on the activity of glycogen synthase kinase 3β (GSK-3β). (G) The influence of lnc-TSSK2-8 overexpression on ubiquitination of β-catenin. *p < 0.05, **p < 0.001, **p < 0.0001.
Transcriptomic Analysis of lnc-TSSK2-8 Overexpression
To explore the molecular basis of Wnt/β-catenin signaling regulation, we performed transcriptomic analysis. Our results indicated that overexpression of lnc-TSSK2-8 resulted in differential expression of 62 genes/lncRNAs, among which HSPA6 was the most significantly upregulated (p = 1.27 × 10–5) (Figures 3A,B). GO gene set enrichment analysis indicated that the DEGs were enriched in MFs such as transmembrane transporter activities (Figure 3C). KEGG enrichment analysis indicated that the DEGs were enriched in beta-Alanine metabolism (p < 0.05).
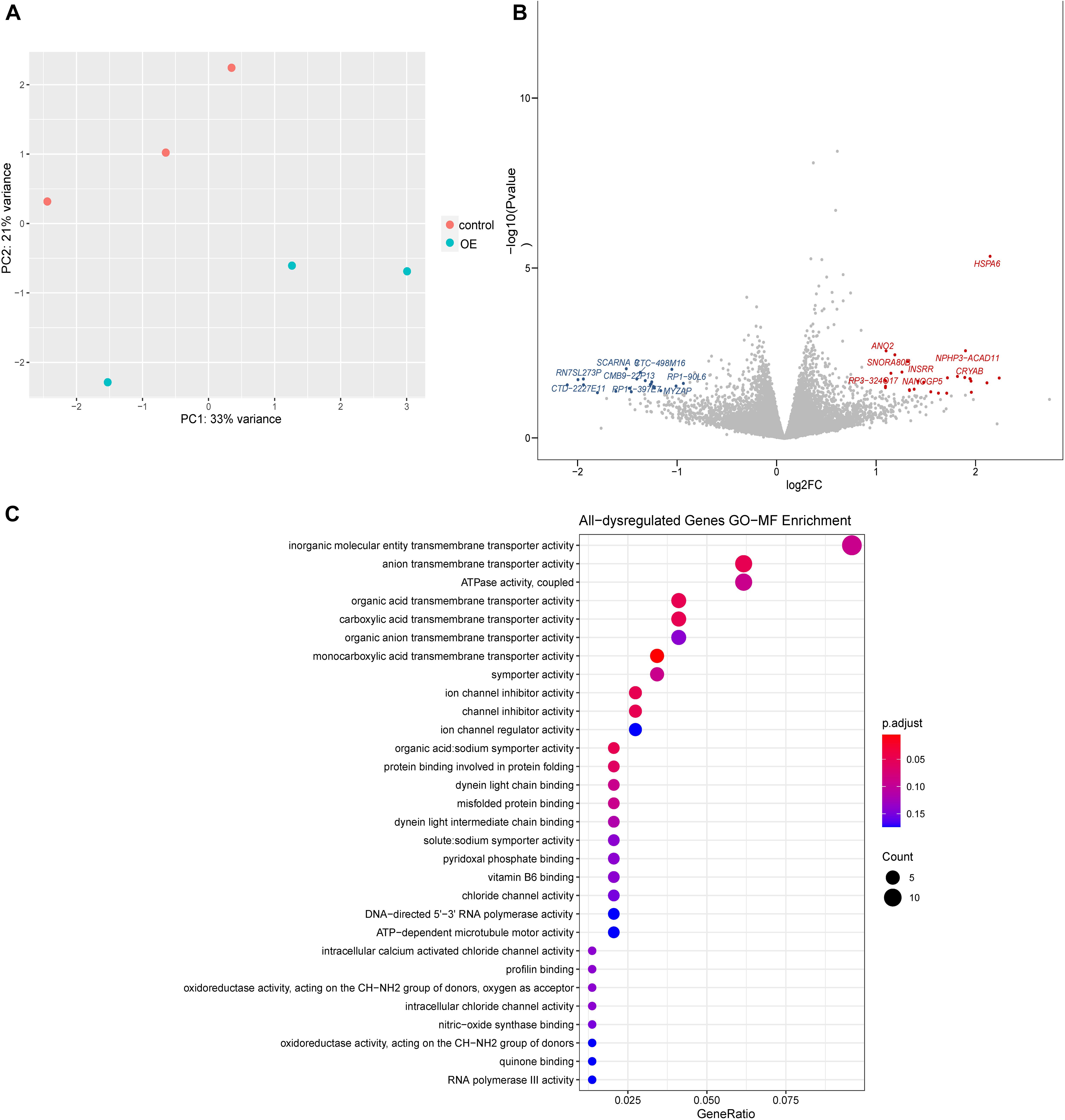
Figure 3. Differential gene expression analysis of lnc-TSSK2-8 overexpression in HEK293 cells. (A) Principal component analysis of RNAseq data derived from lnc-TSSK2-8 overexpression and control cell samples. (B) Volcano plot of differential gene expression analysis; symbols of the top 15 for each of upregulated and downregulated genes were labeled. (C) Gene Ontology (GO) gene set functional enrichment analysis of DEGs.
lnc-TSSK2-8 Activates Wnt/β-Catenin Signaling Through a Competing Endogenous RNA Mechanism
Since lnc-TSSK2-8 was mainly distributed in the cytoplasm, we explored whether it could exert its function as a competing endogenous RNA (ceRNA) on DEGs. We predicted that miR-6721-5p targets both the HSPA6 3′ UTR and the lnc-TSSK2-8 full-length sequence (Figure 4A) with the miRDB2 (Liu and Wang, 2019; Chen and Wang, 2020). We then validated the interaction between miR-6721-5p and HSPA6-3′UTR/lnc-TSSK2-8 using a dual-luciferase reporter assay. The results suggested that ectopic expression of miR-6721-5p leads to notably reduced luciferase activity of WT HSPA6 3′ UTR, while the luciferase activity could not be changed if the binding sites of HSPA6 3′UTR were mutated (Figure 4B). Similarly, the interaction of miR-6721-5p and lnc-TSSK2-8 was also validated (Figure 4C). Overexpression of miR-6721-5p decreased the transcriptional level of lnc-TSSK2-8 (Figure 4D) but did not affect the gene expression of β-catenin (Figure 4E), while the stabilization of β-catenin was impaired, since its phosphorylation and protein expression were upregulated and downregulated, respectively (Figure 4F). Furthermore, overexpression of lnc-TSSK2-8 greatly activated the transcription of HSPA6 (Figure 4G). We overexpressed HSPA6 to verify its effect on Wnt/β-catenin signaling. The result indicated that HSPA6 (Figures 4H,I) activated transcription of LEF1 (Figure 4J) and exerted an inverse effect of miR-6721-5p on β-catenin expression (Figures 4K,L).
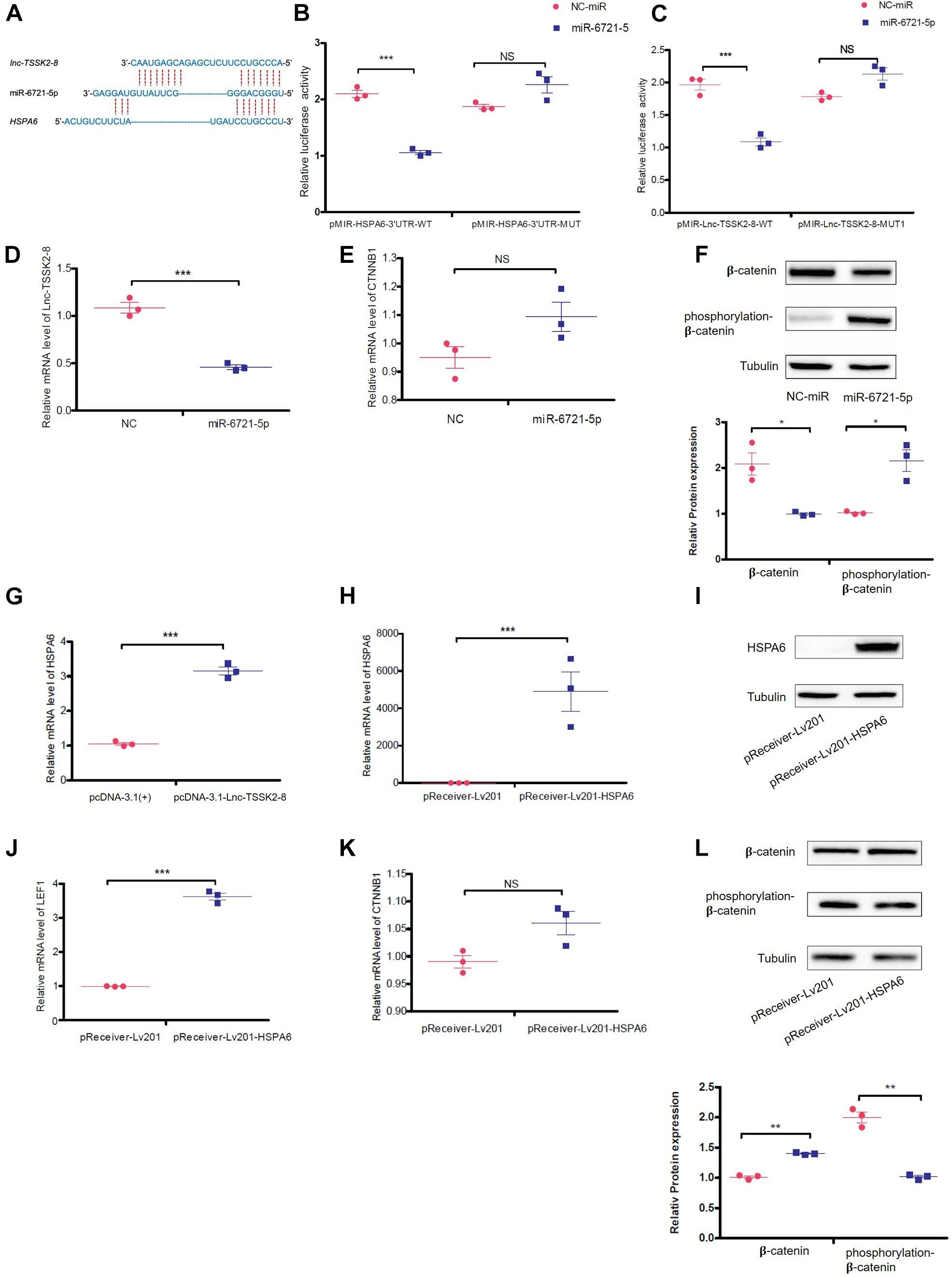
Figure 4. lnc-TSSK2-8 activation promotes β-catenin stabilization through miR-6721-5p/heat shock protein (HSP)A6 axis. (A) The predicted miR-6721-5p binding sites of lnc-TSSK2-8 and HSPA6 3′ UTR. (B) The luciferase reporter assay we performed to confirm the interaction between miR-6721-5p and HSPA6 3′ UTR (B)/lnc-TSSK2-8 (C). (D) RT-qPCR analysis of lnc-TSSK2-8 level in response to miR-6721-5p overexpression. (E) RT-qPCR analysis of β-catenin level in response to miR-6721-5p overexpression. (F) Western blot (WB) analysis of protein expression and phosphorylation of β-catenin in response to miR-6721-5p overexpression. (G) RT-qPCR analysis of HSPA6 level in response to lnc-TSSK2-8 overexpression. (H) Confirmation of HSPA6 overexpression with RT-qPCR. (I) Confirmation of HSPA6 overexpression with WB. (J) RT-qPCR analysis of LEF1 level in response to HSPA6 overexpression. (K) RT-qPCR analysis of β-catenin level in response to HSPA6 overexpression. (L) WB analysis of β-catenin protein expression and phosphorylation in response to HSPA6 overexpression. *p < 0.05, **p < 0.001, **p < 0.0001.
Recently, Mitra et al. (2013) reported that α-B-crystallin (CRYAB) could stabilize β-catenin and promote Wnt signaling in osteogenic differentiation (Zhu et al., 2020). We noticed lnc-TSSK2-8 and CRYAB 3′ UTR also share miRNA target sequences (Figure 5A) and further investigated whether lnc-TSSK2-8 could regulate Wnt/β-catenin signaling by modulating CRYAB expression. Through dual-luciferase reporter assay, we showed that miR-491-5p could interact with both CRYAB and lnc-TSSK2-8 (Figures 5B,C). Overexpression of miR-491-5p significantly downregulated the transcriptional level of CRYAB and LEF1 (Figures 5D,E) and only inhibits β-catenin expression posttranscriptionally (Figures 5F,G). As expected, overexpression of lnc-TSSK2-8 greatly increased the transcription of CRYAB (Figures 5H,I). At last, we overexpressed CRYAB (Figures 5J,K) and verified its posttranscriptional effect on β-catenin expression (Figures 5L,M).
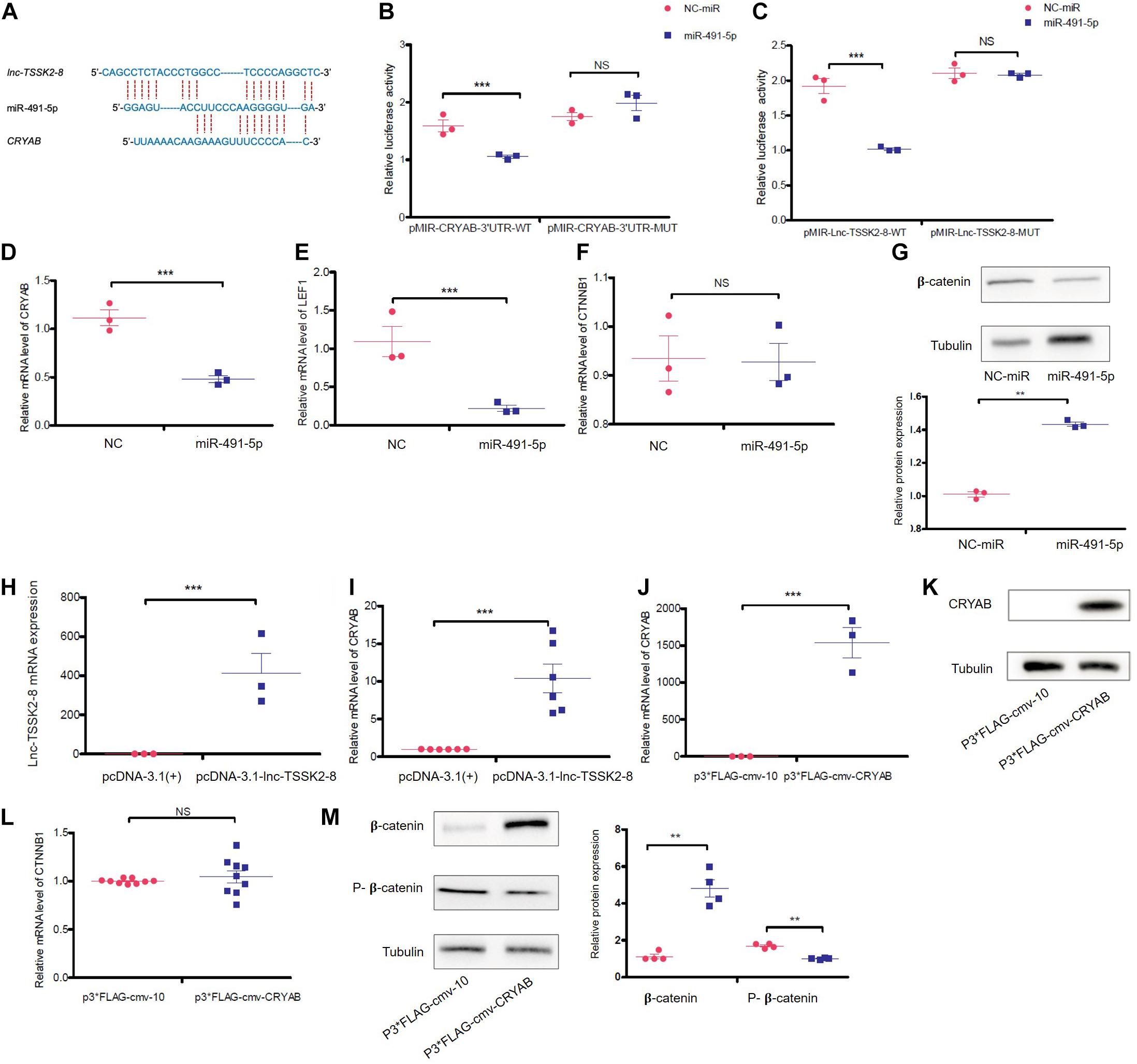
Figure 5. lnc-TSSK2-8 activation promotes β-catenin stabilization through the miR-491-5p/α-B-crystallin (CRYAB) axis. (A) The predicted miR-491-5p binding sites of lnc-TSSK2-8 and CRYAB 3′ UTR. (B) The luciferase reporter assay we performed to confirm the interaction between miR-6721-5p and CRYAB 3′ UTR (B)/lnc-TSSK2-8 (C). (D) RT-qPCR analysis of CRYAB level in response to miR-491-5p overexpression. (E) RT-qPCR analysis of lymphoid enhancer-binding factor 1 (LEF1) level in response to miR-491-5p overexpression. (F) RT-qPCR analysis of β-catenin level in response to miR-491-5p overexpression. (G) Western blot (WB) analysis of protein expression of β-catenin in response to miR-491-5p overexpression. (H) Confirmation of lnc-TSSK2-8 overexpression with RT-qPCR. (I) RT-qPCR analysis of CRYAB level in response to lnc-TSSK2-8 overexpression. (J) Confirmation of CRYAB overexpression with RT-qPCR. (K) Confirmation of CRYAB overexpression with WB. (L) RT-qPCR analysis of β-catenin level in response to CRYAB overexpression. (M) WB analysis of β-catenin protein expression and phosphorylation in response to CRYAB overexpression. *p < 0.05, **p < 0.001, **p < 0.0001.
Discussion
Wnt proteins are a protein family composed of secreted glycoproteins acting as signaling molecules. They bind to Frizzled and several other coreceptors such as lipoprotein receptor-related protein (LRP)-5/6 to trigger intracellular signal transduction (Logan and Nusse, 2004; Kestler and Kuhl, 2008). In the canonical Wnt/β-catenin signaling pathway, when Wnt signal is absent, β-catenin is phosphorylated by the destruction complex. Such processes include CK1-mediated phosphorylation at Ser45, and GSK3β-mediated phosphorylation at Ser33, Ser37, and Thr41. The phosphorylated β-catenin is degraded by the proteasome through the ubiquitin pathway. When Wnt ligands are available, a cascade initiated by the binding of Wnt to the Frzs is activated. The destruction complex consisting of axin, adenomatous polyposis coli (APC), and GSK3β is disassembled, resulting in the stabilization of β-catenin. The β-catenin molecules subsequently accumulate in the cytoplasm and are imported into the nucleus, where they serve as transcriptional coactivators of the TCF/LEF family transcription factors to regulate gene expression (Rao and Kuhl, 2010). The canonical Wnt/β-catenin signaling pathway has been proven to play important roles in multiple aspects of vertebrate heart development such as cardiac differentiation and right ventricular growth (Ai et al., 2007; Gessert and Kuhl, 2010).
The stability of cytosolic β-catenin is controlled by the destruction complex. As the member of the destruction complex, GSK3β acts as a negative regulatory factor. We found that overexpression of lnc-TSSK2-8 promoted the phosphorylation of GSK3β. Subsequently, the phosphorylation of β-catenin was decreased, which might inhibit its ubiquitination. We showed that lnc-TSSK2-8 promotes the stabilization of β-catenin through regulating the expression of HSPA6 and CRYAB. CRYAB is the most abundant small HSP constitutively expressed in cardiomyocytes. It protects against reperfusion injury/myocardial ischemia and suppresses cardiac hypertrophic responses by attenuating nuclear factor of activated T cells (NFAT) signaling (Kumarapeli et al., 2008). CRYAB was also identified as a downstream effector of calcineurin-induced protection against cardiomyocyte apoptosis (Bousette et al., 2010) and a molecular switch in bypassing mitochondrial pathway of apoptosis during myocardial infarction (Mitra et al., 2013). Zhu et al. (2020) recently revealed that CRYAB physically interacts with β-catenin; it promotes osteogenic differentiation of bone marrow stem cells by protecting β-catenin from ubiquitination and degradation (Zhu et al., 2020). HSPA6 has not been reported as associated with Wnt/β-catenin signaling. Our study showed that HSPA6 and CRYAB not only stabilized β-catenin but also activated the downstream transcription factor LEF1. Since dosage of β-catenin significantly affects OFT development upstream of Tbx1 (Racedo et al., 2017), we propose that the small HSP-mediated stabilization of β-catenin should be a common mechanism involved in heart development. The expression of HSPA6 and CRYAB could be modulated by lnc-TSSK2-8 through sponging miR-6721-5p and miR-491-5p, respectively. Wnt/β-catenin signaling could potentiate neonatal mouse cardiomyocyte proliferation and human induced pluripotent stem (iPS) cell-derived cardiomyocyte. A Wnt/β-catenin-dependent transcriptional network governing cardiomyocyte proliferation was discovered (Quaife-Ryan et al., 2020). We showed that lnc-TSSK2-8 promoted cell proliferation of HEK293, which might result from enhancement of Wnt/β-catenin signaling.
Significant achievements have been made in revealing the roles of miRNAs and lncRNAs in heart development (Alexanian and Ounzain, 2020; Das et al., 2020). Cytoplasmic lncRNA could act as ceRNAs to sequester miRNAs from their natural mRNA targets; we established that lnc-TSSK2-8 regulates HSPA6 and CRYAB in this way. Whereas lnc-TSSK2-8 also has substantial nuclear expression, the transcriptomic analysis indicated that lnc-TSSK2-8 might be associated with cilia functions. Cilia have an essential role in the pathogenesis of congenital heart disease (Li et al., 2015; Gabriel et al., 2020). We noted that the DEGs were enriched in dynein binding, which is critical for ciliary processes. The MF of lnc-TSSK2-8 in cilia process and heart development needs to be further investigated. Conotruncal defects such as TOF, truncus arteriosus, conoventricular ventricle septum defect, type B interruption of the aortic arch account for about 70% of heart malformations associated with 22q11.2DS (Agergaard et al., 2012). Although several genes including TBX1 have been identified responsible for heart defects resulting from 22q11.2 deletion, noncoding functions of this common CNV in cardiac development have not received enough attention. We demonstrated that the 22q11.2 lnc-TSSK2-8 is involved in the canonical Wnt signaling pathway by stabilizing β-catenin. There were two main limitations of our study. Firstly, our experiments were based on the HEK293 cell line, which is derived from human embryonic kidney. Additional validation using primary cardiac cells derived from model animals or in vitro cardiomyocyte differentiation from human iPS cells would help reveal the cardiac-specific effect of lnc-TSSK2-8. Secondly, loss-of-function validation was not performed for lnc-TSSK2-8, HSPA6, and CRYAB. It would be meaningful to carry out si/shRNA-mediated knockdown of lnc-TSSK2-8, HSPA6, and CRYAB to investigate their roles in the context of heart development, which would help elucidate the contribution of lnc-TSSK2-8 to cardiac phenotypes of 22q11.2DS.
In summary, we established that the 22q11.2 lncRNA lnc-TSSK2-8 acts as an ceRNA to regulate the expression of HSPA6 and CRYAB through sponging miR-6721-5p and miR-491-5p; subsequently, HSPA6 and CRYAB activates canonical Wnt signaling by promoting the stabilization of β-catenin and activating LEF1 expression. Our study provided evidence of a lncRNA that regulates the dosage of β-catenin, which contributes to the OFT anomalies in 22q11.2DS.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/geo/, GSE165927.
Author Contributions
JF, YG, XpZ, and XqZ performed the lab experiments. BW, MQ, and XyZ performed bioinformatic data analysis. BW wrote the manuscript. QF, YG, ZX, and BW conceived the study and contributed to editing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 81601847, 81672090, 81871717, and 81801460) and grants from Shanghai Municipal Science and Technology Major Project (20JC141850 and 2017SHZDZX01) and Shanghai Key Laboratory of Clinical Molecular Diagnostics for Pediatrics (20dz2260900).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Agergaard, P., Olesen, C., Ostergaard, J. R., Christiansen, M., and Sorensen, K. M. (2012). The prevalence of chromosome 22q11.2 deletions in 2,478 children with cardiovascular malformations. a population based study. Am. J. Med. Genet. A 158A, 498–508. doi: 10.1002/ajmg.a.34250
Ai, D., Fu, X., Wang, J., Lu, M. F., Chen, L., Baldini, A., et al. (2007). Canonical Wnt signaling functions in second heart field to promote right ventricular growth. Proc. Natl. Acad. Sci. U.S.A. 104, 9319–9324. doi: 10.1073/pnas.0701212104
Alexanian, M., and Ounzain, S. (2020). Long noncoding RNAs in cardiac development. Cold Spring Harb. Perspect. Biol. 12, 415–425. doi: 10.1038/nrcardio.2015.55
Anderson, K. M., Anderson, D. M., Mcanally, J. R., Shelton, J. M., Bassel-Duby, R., and Olson, E. N. (2016). Transcription of the non-coding RNA upperhand controls Hand2 expression and heart development. Nature 539, 433–436. doi: 10.1038/nature20128
Blankesteijn, W. M., Van De Schans, V. A., Ter Horst, P., and Smits, J. F. (2008). The Wnt/frizzled/GSK-3 beta pathway: a novel therapeutic target for cardiac hypertrophy. Trends Pharmacol. Sci. 29, 175–180. doi: 10.1016/j.tips.2008.01.003
Botto, L. D., May, K., Fernhoff, P. M., Correa, A., Coleman, K., Rasmussen, S. A., et al. (2003). A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics 112, 101–107. doi: 10.1542/peds.112.1.101
Bousette, N., Chugh, S., Fong, V., Isserlin, R., Kim, K. H., Volchuk, A., et al. (2010). Constitutively active calcineurin induces cardiac endoplasmic reticulum stress and protects against apoptosis that is mediated by alpha-crystallin-B. Proc. Natl. Acad. Sci. U.S.A. 107, 18481–18486. doi: 10.1073/pnas.1013555107
Chapnik, E., Sasson, V., Blelloch, R., and Hornstein, E. (2012). Dgcr8 controls neural crest cells survival in cardiovascular development. Dev. Biol. 362, 50–56. doi: 10.1016/j.ydbio.2011.11.008
Chen, Y., and Wang, X. (2020). miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 48, D127–D131.
Cowan, J. R., and Ware, S. M. (2015). Genetics and genetic testing in congenital heart disease. Clin. Perinatol. 42, 373–393,ix.
Das, S., Shah, R., Dimmeler, S., Freedman, J. E., Holley, C., Lee, J. M., et al. (2020). Noncoding RNAs in cardiovascular disease: current knowledge, tools and technologies for investigation, and future directions: a scientific statement from the american heart association. Circ. Genom. Precis. Med. 13:e000062.
Devaux, Y., Zangrando, J., Schroen, B., Creemers, E. E., Pedrazzini, T., Chang, C. P., et al. (2015). Long noncoding RNAs in cardiac development and ageing. Nat. Rev. Cardiol. 12, 415–425. doi: 10.1038/nrcardio.2015.55
Devriendt, K., Fryns, J. P., Mortier, G., Van Thienen, M. N., and Keymolen, K. (1998). The annual incidence of DiGeorge/velocardiofacial syndrome. J. Med. Genet. 35, 789–790. doi: 10.1136/jmg.35.9.789-a
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Gabriel, G. C., Young, C. B., and Lo, C. W. (2020). Role of cilia in the pathogenesis of congenital heart disease. Semin. Cell Dev. Biol. 110, 2–10. doi: 10.1016/j.semcdb.2020.04.017
Gessert, S., and Kuhl, M. (2010). The multiple phases and faces of wnt signaling during cardiac differentiation and development. Circ. Res. 107, 186–199. doi: 10.1161/circresaha.110.221531
Goodship, J., Cross, I., Liling, J., and Wren, C. (1998). A population study of chromosome 22q11 deletions in infancy. Arch. Dis. Child 79, 348–351. doi: 10.1136/adc.79.4.348
Grote, P., and Herrmann, B. G. (2013). The long non-coding RNA Fendrr links epigenetic control mechanisms to gene regulatory networks in mammalian embryogenesis. RNA Biol. 10, 1579–1585. doi: 10.4161/rna.26165
Gu, M., Zheng, A., Tu, W., Zhao, J., Li, L., Li, M., et al. (2016). Circulating LncRNAs as novel, non-invasive biomarkers for prenatal detection of fetal congenital heart defects. Cell. Physiol. Biochem. 38, 1459–1471. doi: 10.1159/000443088
Hoffman, J. I., and Kaplan, S. (2002). The incidence of congenital heart disease. J. Am. Coll. Cardiol. 39, 1890–1900.
Jiang, Y., Mo, H., Luo, J., Zhao, S., Liang, S., Zhang, M., et al. (2018). HOTAIR is a potential novel biomarker in patients with congenital heart diseases. Biomed. Res. Int. 2018:2850657.
Jiang, Y., Zhuang, J., Lin, Y., Wang, X., Chen, J., and Han, F. (2019). Long noncoding RNA SNHG6 contributes to ventricular septal defect formation via negative regulation of miR-101 and activation of Wnt/beta-catenin pathway. Pharmazie 74, 23–28.
Kestler, H. A., and Kuhl, M. (2008). From individual Wnt pathways towards a Wnt signalling network. Philos. Trans, Soc. Lond B Biol. Sci. 363, 1333–1347. doi: 10.1098/rstb.2007.2251
Klattenhoff, C. A., Scheuermann, J. C., Surface, L. E., Bradley, R. K., Fields, P. A., Steinhauser, M. L., et al. (2013). Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell 152, 570–583. doi: 10.1016/j.cell.2013.01.003
Kumarapeli, A. R., Su, H., Huang, W., Tang, M., Zheng, H., Horak, K. M., et al. (2008). Alpha B-crystallin suppresses pressure overload cardiac hypertrophy. Circ. Res. 103, 1473–1482. doi: 10.1161/circresaha.108.180117
Li, Y., Klena, N. T., Gabriel, G. C., Liu, X., Kim, A. J., Lemke, K., et al. (2015). Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 521, 520–524. doi: 10.1038/nature14269
Liao, Y., Smyth, G. K., and Shi, W. (2014). feature counts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi: 10.1093/bioinformatics/btt656
Lindsay, E. A., Vitelli, F., Su, H., Morishima, M., Huynh, T., Pramparo, T., et al. (2001). Tbx1 haploinsufficieny in the di george syndrome region causes aortic arch defects in mice. Nature 410, 97–101. doi: 10.1038/35065105
Liu, W., and Wang, X. (2019). Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 20:18.
Logan, C. Y., and Nusse, R. (2004). The Wnt signaling pathway in development and disease. Annu. Rev. Cell. Dev. Biol. 20, 781–810.
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550.
Ma, J., Chen, S., Hao, L., Sheng, W., Chen, W., Ma, X., et al. (2020). Hypermethylation-mediated down-regulation of lncRNA TBX5-AS1:2 in tetralogy of fallot inhibits cell proliferation by reducing TBX5 expression. J. Cell. Mol. Med. 24, 6472–6484. doi: 10.1111/jcmm.15298
McDonald-McGinn, D. M., Tonnesen, M. K., Laufer-Cahana, A., Finucane, B., Driscoll, D. A., Emanuel, B. S., et al. (2001). Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: cast a wide FISHing net! Genet. Med. 3, 23–29. doi: 10.1097/00125817-200101000-00006
Mitra, A., Basak, T., Datta, K., Naskar, S., Sengupta, S., and Sarkar, S. (2013). Role of alpha-crystallin B as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction. Cell Death Dis. 4:e582. doi: 10.1038/cddis.2013.114
Naito, A. T., Shiojima, I., Akazawa, H., Hidaka, K., Morisaki, T., Kikuchi, A., et al. (2006). Developmental stage-specific biphasic roles of Wnt/beta-catenin signaling in cardiomyogenesis and hematopoiesis. Proc. Natl. Acad. Sci. U.S.A. 103, 19812–19817. doi: 10.1073/pnas.0605768103
Oskarsdottir, S., Vujic, M., and Fasth, A. (2004). Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden. Arch. Dis. Child 89, 148–151. doi: 10.1136/adc.2003.026880
Pierpont, M. E., Brueckner, M., Chung, W. K., Garg, V., Lacro, R. V., Mcguire, A. L., et al. (2018). Genetic basis for congenital heart disease: revisited: a scientific statement from the american heart association. Circulation 138, e653–e711.
Quaife-Ryan, G. A., Mills, R. J., Lavers, G., Voges, H. K., Vivien, C. J., Elliott, D. A., et al. (2020). beta-Catenin drives distinct transcriptional networks in proliferative and nonproliferative cardiomyocytes. Development 147:dev193417. doi: 10.1242/dev.193417
Racedo, S. E., Hasten, E., Lin, M., Devakanmalai, G. S., Guo, T., Ozbudak, E. M., et al. (2017). Reduced dosage of beta-catenin provides significant rescue of cardiac outflow tract anomalies in a Tbx1 conditional null mouse model of 22q11.2 deletion syndrome. PLoS Genet 13:e1006687. doi: 10.1371/journal.pgen.1006687
Rao, T. P., and Kuhl, M. (2010). An updated overview on Wnt signaling pathways: a prelude for more. Circ. Res. 106, 1798–1806. doi: 10.1161/circresaha.110.219840
Repetto, G. M., Guzman, M. L., Delgado, I., Loyola, H., Palomares, M., Lay-Son, G., et al. (2014). Case fatality rate and associated factors in patients with 22q11 microdeletion syndrome: a retrospective cohort study. BMJ Open 4:e005041. doi: 10.1136/bmjopen-2014-005041
Stark, K. L., Xu, B., Bagchi, A., Lai, W. S., Liu, H., Hsu, R., et al. (2008). Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat. Genet. 40, 751–760. doi: 10.1038/ng.138
Tezenas Du Montcel, S., Mendizabai, H., Ayme, S., Levy, A., and Philip, N. (1996). Prevalence of 22q11 microdeletion. J. Med. Genet. 33:719. doi: 10.1136/jmg.33.8.719
Ueno, S., Weidinger, G., Osugi, T., Kohn, A. D., Golob, J. L., Pabon, L., et al. (2007). Biphasic role for Wnt/beta-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 104, 9685–9690. doi: 10.1073/pnas.0702859104
Van Der Linde, D., Konings, E. E., Slager, M. A., Witsenburg, M., Helbing, W. A., Takkenberg, J. J., et al. (2011). Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J. Am. Coll. Cardiol. 58, 2241–2247.
Wang, B., Shi, G., Zhu, Z., Chen, H., and Fu, Q. (2018). Sexual difference of small RNA expression in tetralogy of fallot. Sci. Rep. 8:12847.
Wilson, K. D., Ameen, M., Guo, H., Abilez, O. J., Tian, L., Mumbach, M. R., et al. (2020). Endogenous retrovirus-derived lncRNA BANCR promotes cardiomyocyte migration in humans and non-human primates. Dev. Cell. 54:e699.
Yu, G., Wang, L. G., Han, Y., and He, Q. Y. (2012). cluster profiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. doi: 10.1089/omi.2011.0118
Zheng, P., Noroski, L. M., Hanson, I. C., Chen, Y., Lee, M. E., Huang, Y., et al. (2015). Molecular mechanisms of functional natural killer deficiency in patients with partial Di George syndrome. J. Allergy Clin. Immunol. 135, 1293–1302. doi: 10.1016/j.jaci.2015.01.011
Zhu, B., Xue, F., Li, G., and Zhang, C. (2020). CRYAB promotes osteogenic differentiation of human bone marrow stem cells via stabilizing beta-catenin and promoting the Wnt signalling. Cell. Prolif. 53:e12709.
Keywords: congenital heart disease, long noncoding RNA, 22q11.2 deletion, lnc-TSSK2-8, Wnt/β-catenin signaling
Citation: Fa J, Zhang X, Zhang X, Qi M, Zhang X, Fu Q, Xu Z, Gao Y and Wang B (2021) Long Noncoding RNA lnc-TSSK2-8 Activates Canonical Wnt/β-Catenin Signaling Through Small Heat Shock Proteins HSPA6 and CRYAB. Front. Cell Dev. Biol. 9:660576. doi: 10.3389/fcell.2021.660576
Received: 29 January 2021; Accepted: 29 March 2021;
Published: 10 May 2021.
Edited by:
Duan Ma, Fudan University, ChinaReviewed by:
Lingqian Wu, Central South University, ChinaDavid D. Eisenstat, Royal Children’s Hospital, Australia
Zhengwei Yuan, ShengJing Hospital of China Medical University, China
Copyright © 2021 Fa, Zhang, Zhang, Qi, Zhang, Fu, Xu, Gao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhuoming Xu, em14Y2ljdUAxNjMuY29t; Yunqian Gao, Z2FveXVucWlhbjJAMTYzLmNvbQ==; Bo Wang, Ym9vZXdAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship