 Bipul Ray1,2
Bipul Ray1,2 Arehally M. Mahalakshmi1Sunanda Tuladhar1,2
Arehally M. Mahalakshmi1Sunanda Tuladhar1,2 Abid Bhat1,2
Abid Bhat1,2 Asha Srinivasan3
Asha Srinivasan3 Christophe Pellegrino4Anbarasu Kannan5Srinivasa Rao Bolla6
Christophe Pellegrino4Anbarasu Kannan5Srinivasa Rao Bolla6 Saravana Babu Chidambaram1,2,7*
Saravana Babu Chidambaram1,2,7* Meena Kishore Sakharkar8*
Meena Kishore Sakharkar8*- 1Department of Pharmacology, JSS College of Pharmacy, JSS Academy of Higher Education & Research, Mysuru, India
- 2Centre for Experimental Pharmacology and Toxicology, Central Animal Facility, JSS Academy of Higher Education & Research, Mysuru, India
- 3Division of Nanoscience & Technology, Faculty of Life Sciences, JSS Academy of Higher Education & Research, Mysuru, India
- 4Institut National de la Santé et de la Recherche Médicale, Institute of Mediterranean Neurobiology, Aix-Marseille University, Marseille, France
- 5Department of Protein Chemistry and Technology, CSIR-Central Food Technological Research Institute, Mysuru, India
- 6Department of Biomedical Sciences, School of Medicine, Nazarbayev University, Nur-Sultan City, Kazakhstan
- 7Special Interest Group – Brain, Behaviour, and Cognitive Neurosciences Research, JSS Academy of Higher Education & Research, Mysuru, India
- 8College of Pharmacy and Nutrition, University of Saskatchewan, Saskatoon, SK, Canada
Parkinson’s disease (PD) is a pathological condition characterized by the aggregation and the resultant presence of intraneuronal inclusions termed Lewy bodies (LBs) and Lewy neurites which are mainly composed of fibrillar α-synuclein (α-syn) protein. Pathogenic aggregation of α-syn is identified as the major cause of LBs deposition. Several mutations in α-syn showing varied aggregation kinetics in comparison to the wild type (WT) α-syn are reported in PD (A30P, E46K, H 50Q, G51D, A53E, and A53T). Also, the cell-to-cell spread of pathological α-syn plays a significant role in PD development. Interestingly, it has also been suggested that the pathology of PD may begin in the gastrointestinal tract and spread via the vagus nerve (VN) to brain proposing the gut–brain axis of α-syn pathology in PD. Despite multiple efforts, the behavior and functions of this protein in normal and pathological states (specifically in PD) is far from understood. Furthermore, the etiological factors responsible for triggering aggregation of this protein remain elusive. This review is an attempt to collate and present latest information on α-syn in relation to its structure, biochemistry and biophysics of aggregation in PD. Current advances in therapeutic efforts toward clearing the pathogenic α-syn via autophagy/lysosomal flux are also reviewed and reported.
Highlights
- Updated information on the structure and biochemistry of α-syn aggregation is discussed and explained with suitable examples.
- Recent evidence on mutations in α-syn (A30P, E46K, H 50Q, G51D, A53E, and A53T) and its cell-to-cell transmission and the consequent impact on PD progression is compiled and discussed in detail.
- Mechanism of transport of pathogenic α-syn from gastrointestinal tract via the vagus nerve (VN) to brain and information on various clinical trials that validate the function of gut–brain axis in Parkinson’s disease (PD) is provided.
- Mechanism of cellular clearance of pathogenic α-syn via autophagy and clinical trials focusing on autophagy facilitation are discussed.
- Various reports indicate that native α-syn has physiological functions, but the mutated and aggregated forms are neurotoxic which play critical role in PD. Hence, understanding the mechanism of its origin, aggregation and cellular clearance will provide new leads in PD drug discovery.
Introduction
Parkinson’s disease is a heterogeneous neurological disorder with progressive loss of dopaminergic neurons in the SNpc region in the brain (Nepal et al., 2019). One of the major hallmarks of PD is the accumulation and aggregation of misfolded α-synuclein (α-syn) protein to form LBs and Lewy neurites that cause disruption of cellular homeostasis, and degeneration of neurons (Mahul-Mellier et al., 2020). α-syn is a presynaptic neuronal protein encoded by SNCA gene and is expressed in several regions of the brain (Polymeropoulos et al., 1997; Meade et al., 2019). The presence of α-syn was observed for the first time by Maroteaux et al. (1988) in the presynaptic nerve terminals and in the neuronal nuclei, hence it was called syn (synapse) and nuclein (nucleus) (Maroteaux et al., 1988). Later, the presence of native and pathogenic α-syn was identified in organelles such as the GA (Gosavi et al., 2002; Mori et al., 2002), endolysosomal system (Lee et al., 2004), and mitochondrial surface (Li et al., 2007; Cole et al., 2008; Parihar et al., 2008). α-syn was also found to be associated with the inner membrane of mitochondria (Devi et al., 2008; Shavali et al., 2008; Ludtmann et al., 2018; Ganjam et al., 2019), ER (Cooper et al., 2006; Hoozemans et al., 2007; Colla et al., 2012a, b) and the mitochondria-associated ER membranes (MAM) (Guardia-Laguarta et al., 2014). Recently, several reports have confirmed the involvement of gut–brain axis in PD (Ghaisas et al., 2019). Gut dysbiosis affects the ENS and the contribute for aggregation of α-syn which is then transported to brain by cell-to-cell contacts exosomes (Danzer et al., 2012; Freundt et al., 2012; Kim et al., 2019; Jang et al., 2020; Siddu et al., 2020). Alternative theories also propose on the involvement of exosomal vesicles from gut microbiome in regulating host gene expression and the in aggregation of α-syn (Danzer et al., 2012; Rokad et al., 2019).
The synuclein family consist of three protein members - α, β, and γ (Lashuel et al., 2013). α-syn and β-syn are primarily found in brain, whereas, γ-syn in the neoplastic tissues (Zhang et al., 2011).
Although the physiological function of α-syn is still not clearly understood, reports suggest that it plays a significant role in neuronal plasticity (Uversky, 2008; Lashuel et al., 2013; Wu J.-Z. et al., 2019) and in dopamine synthesis by regulating TH (Peng et al., 2005). α-syn activates protein phosphatase 2A (PP2A), a serine/threonine phosphatase, that dephosphorylates TH (Leal et al., 2002; Peng et al., 2005; Hua et al., 2015; Qu et al., 2018). Additionally, α-syn is reported to modulate the release of the neurotransmitters in association with the SVs. Overexpression of native α-syn inhibits exocytosis (Logan et al., 2017) and mutations in the SNCA gene that encodes native α-syn are associated with PD with autosomal dominant inheritance pattern with a relatively early onset age than sporadic PD patients (Polymeropoulos et al., 1996; Ikeuchi et al., 2008). Interestingly, α-synucleinopathies are reported increase in the propensity of many neurodegenerative diseases including multiple system atrophy (MSA), Lewy body dementia (LBD) and NBIA Type 1 (formerly known as Hallervorden-Spatz disease) (Jellinger, 2003). Higher Aβ and tau expressions are reported in cortex and striatum in dementia with Lewy bodies (DLB) compared to PD (Jellinger and Korczyn, 2018). On the other hand, the hallmark histopathology feature of MSA is accumulation of α-syn in the cytoplasm of oligodendroglial cells (Dickson, 2012; Kim et al., 2014). Also, MSA has aggregated α-syn inclusions in the nuclei, unlike PD (Lin et al., 2004). Aggregation of α-syn is often observed with hyperphosphorylated Tau, transactive response DNA binding protein 43 kDa (TDP-43), Aβ, and prion protein accumulation in brain (Visanji et al., 2019). The missense mutations in SNCA cause the substitutions of G51D and A53E that result in atypical synucleinopathies (mixture of PD and MSA pathologies) (Schweighauser et al., 2020). Due to the conflicting reports, the physiological and pathophysiological role of α-syn aggregation remains elusive.
Fluorescent labeling report showed the presence of α-syn in several brain regions such as OB, dorsal nucleus of the VN, amygdala, hippocampus, and neocortex, besides SNpc (Braak and Del Tredici, 2009). PD was reported to spread to the connected regions (Braak et al., 2003b). The role of α-syn misfolding in the initiation of PD is well established (Mahul-Mellier et al., 2020). Several studies report on targeting α-syn aggregation and synthesis as a potential therapeutic option in PD (Fields et al., 2019; Chakroun et al., 2020; Gabr and Peccati, 2020; Mahul-Mellier et al., 2020; Ryan et al., 2020). Here, it is important to mention that native α-syn plays a crucial role in releasing the neurotransmitter associated with SV due to its greater curvature, but its over-expression is reported to inhibit the release of neurotransmitters (Sulzer and Edwards, 2019; Cai et al., 2020). Furthermore, mitochondrial localized monomeric α-syn is reported to enhance the bioenergetics of mitochondria (Ludtmann et al., 2016), but the oligomeric form causes detrimental effects and results in mitochondrial dysfunction, particularly in PD (Tripathi and Chattopadhyay, 2019). Here, it must be noted that although misfolded α-syn is reported as a major hallmark of PD, several physiologies associated with native α-syn remain elusive. Hence, there is a need to collate information about this protein from literature and revisit its physiological functions, mechanism of aggregation and its autophagic clearance in view of the latest reports. In this review, we have attempted to summarize the functions of α-syn and various therapeutic approaches that target this protein in PD.
α-Synuclein – Structure and Biochemistry
Structure
The discovery of α-syn using an antibody against the purified cholinergic vesicles from the Torpedo electric organ (Torpedo Californica) gave the first evidence of its existence in the presynaptic nerve terminal (Maroteaux et al., 1988). Native α-syn is normally released α-syn into the extracellular space as exosome via exocytosis (Lee et al., 2005). It is a 14 kDa protein with 140 amino acids and three domains: a N-terminal domain (amino acid 1–60) with incomplete KXKEGV motifs, a non-amyloid-β component of plaques (NAC) domain (amino acid 61–95), and a C-terminal domain (amino acid 96–140) (pKa of 4.7) (Jakes et al., 1994; Hashimoto and Masliah, 1999; Lashuel et al., 2013). Protein containing the NAC (non-amyloid component) domain undergoes three state transition from original structure to β-sheet and further to α-helical structure (Figure 1). Other two domains undergo transformation from native to α-helical structure. Being highly hydrophobic, β-sheet reacts with 1-anilinonaphthalene-8-sulfonic acid to undergo self-aggregation (Li et al., 2002). Native α-syn exists in high concentration in soluble and membrane associated fraction in the brain and makes for as much as 1% of the total protein in the soluble brain cytosolic protein (Iwai et al., 1995). It is predominantly present in the presynaptic brain and cerebrospinal fluid (CSF) (Rokad et al., 2017). The native form of α-syn is a monomer. However, there is a possibility that the protein could form oligomers upon interaction with other proteins.
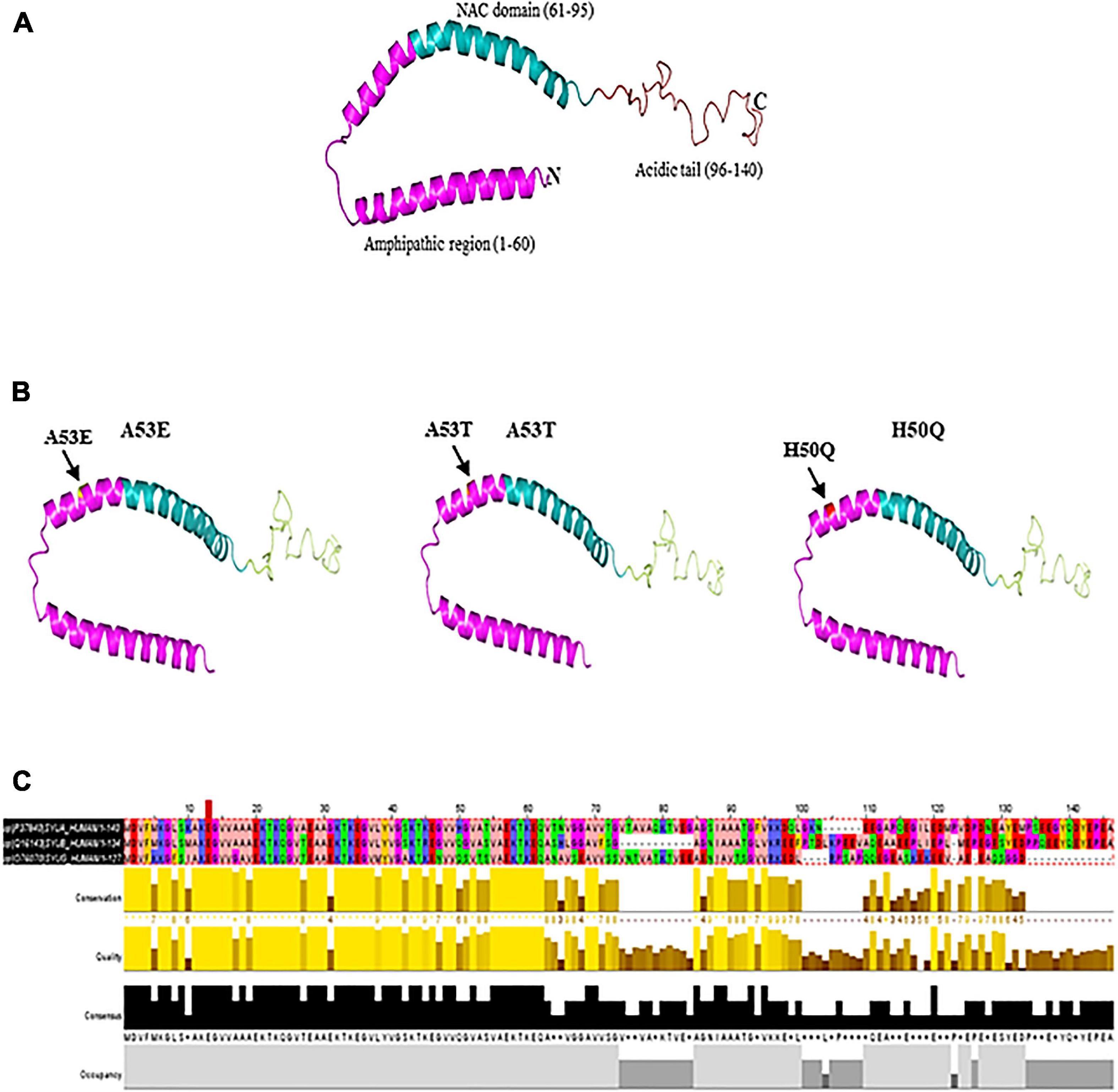
Figure 1. (A) Native α-syn structure (PDB:1XQ8). (B) Mutation regions (A53E, A53T, and H50Q) of α-syn. (C) Multiple sequence alignment of synuclein (α, β, and γ). Residues are colored according to Zappo color scheme.
Oligomerization of α-Synuclein
Depending on the aggregation conditions of α-syn, heterogenous and diverse oligomers are formed, which can be identified by their biophysical and cellular properties. Danzer et al. (2007) had studied three different aggregation protocols of oligomerization of α-syn: where type A had high membrane permeability initiating an elevation of intracellular calcium (Adamczyk and Strosznajder, 2006; Lashuel and Lansbury, 2006) and leading to cell death, whereas, type B and C are able to enter cells directly and seed intracellular α-syn aggregation (Danzer et al., 2007). The report also confirmed that, type B and C do not induce caspase activation or cell loss. It is suggested that this loss might be because of bypassing the toxic oligomeric intermediates aggregation, which might have formed due to α-syn overexpression (Danzer et al., 2007). Along with the ability of self-assemble of α-syn into a variety of oligomeric species, α-syn has been further reported to interact with other proteins undergoing co-oligomerization, including Aβ and tau (Mandal et al., 2006; Qureshi and Paudel, 2011; Sengupta et al., 2015; Chia et al., 2017). In another study, β-sheet geometry between different oligomeric species was reported (Chen et al., 2015) despite mutant variants (A53T, A30P, and E46K) producing similar concentrations and types of oligomeric species to WT protein (Tosatto et al., 2015). High degree of heterogeneity of β-sheet oligomers with the same type of core architecture with different number of β-strands and arrangements and permutations of inter-strand hydrogen bonding interactions is expected as has been observed to occur in fibrillar structures (Qiang et al., 2012; Fitzpatrick et al., 2013). Indeed, the same protein subunits within the same oligomeric species might have different numbers and lengths of β-strands, in the packing of the oligomers and the rearrangement of the β-strands from an antiparallel to a parallel configuration, which might be important for the efficient elongation of these α-syn oligomers to generate the fibrillar architecture (Chen et al., 2015). Recently, Kiechle et al. (2019) had reported that, despite the availability of oligomerization of α-syn throughout the neuronal cell, the oligomerization takes place at the pre-synapse in an animal model of PD (Kiechle et al., 2019). Further in a similar study it was reported that α-syn oligomers accumulate within synaptic terminals of autonomic fibers of the skin in PD patients, which could potentially be a reliable biomarker for detecting the disease (Mazzetti et al., 2020).
Biochemistry
Despite decades of research, the structure and functional relationship of endogenous physiological forms of native α-syn are not elucidated. Anderson et al. (2006) investigated the property of misfolded α-syn in LBs isolated from DLB brains. The phosphorylation of α-syn remains elusive, although kinases including polo-like kinase (PLK), casein kinase (CK)1, CK2, G protein coupled receptor kinase (GRK) families were identified to mediate this event (Dzamko et al., 2014). 90% of α-syn from PD brains is reported to be phosphorylated while 4% phosphorylation of α-syn is observed in normal brains (Dzamko et al., 2014). Phosphorylation of α-syn at Ser 129 has gained a significant importance in the pathogenic aggregation of α-syn (Fujiwara et al., 2002; Paleologou et al., 2008; Ma et al., 2016; Wang et al., 2019). Minor alterations in ubiquitination at Lys residues 12, 21, and 23 and specific truncations at Asp 115, Asp 119, Asn 122, Tyr 133, and Asp 135 are also seen (Anderson et al., 2006). Some studies have suggested that phosphorylation at Ser 129 triggers α-syn-mediated cellular toxicity (Chen and Feany, 2005; Sato et al., 2011; Karampetsou et al., 2017). However, conflicting reports suggest that phosphorylation at Ser 129 promotes proteasomal or autophagic clearance of aggregated α-syn (Gorbatyuk et al., 2008; Machiya et al., 2010; Kuwahara et al., 2012; Nübling et al., 2014; Arawaka et al., 2017). Hence, the exact role Ser 129 phosphorylation in synucleinopathies needs to be investigated.
α-Synuclein undergoes partial folding in the early stages of fibril formation (Uversky et al., 2001). Due to its structure, interactions, and sensitivity to the environment, α-syn is prone to misfolding. Amyloid fibrils can be formed from α-syn upon alterations in pH (Buell et al., 2014), temperature (Uversky et al., 2001), salt concentrations (Munishkina et al., 2004), air-water interference (Campioni et al., 2014), and contact with negatively charged lipid membranes (Galvagnion et al., 2015). Recently, Kurochka et al. (2021) had reported that formation of fibrils from α-syn monomers is significantly decreased in presence of lipids (Kurochka et al., 2021). Anions are also reported to induce partial folding of α-syn at neutral pH (Munishkina et al., 2004). Molecular crowding, i.e., intracellular increase in the concentration of macromolecules (proteins, nucleic acids, and carbohydrates) beyond 400 gram/liter in a cell has been reported to lead to the (Munishkina et al., 2004) production of intrinsically disordered proteins which tend to aggregate. To understand the impact of molecular crowding on α-syn aggregation, Bai et al. (2017) used Ficoll70TM and Sucrose as crowding agents. However, the data did not provide sufficient evidence supporting the role of molecular crowding in α-syn aggregation in PD (Bai et al., 2017).
α-Synuclein Aggregation, Transport and Propagation
The exact process of α-syn aggregation is not elucidated. Reports on random and instant transformation of the α-syn structure to the unfolded or partially folded state are available (Uversky, 2007). Under physiological conditions, native α-syn has a tendency to remain folded (Bartels et al., 2011; Wang et al., 2011). However, there is other report also, that says native α-syn is large unstructured monomer and they are aggregation prone (Burré et al., 2013). Further research in this filed will give the clear turn of the debate. The first evidence on the role of misfolded α-syn and amyloid β (Aβ) in neurodegenerative diseases was in the brains of AD patients (Uéda et al., 1993). The cleaved peptide from the plaques in AD brains was reported to be the central hydrophobic core of α-syn and was given the description of ‘non-amyloidogenic component,’ or NAC region (Uéda et al., 1993). Native α-syn requires the presence of interacting partner for aggregation in response to environmental stress, as this involves changes in the structural configuration of the protein (Weinreb et al., 1996; Uversky, 2002; Dyson and Wright, 2005; Villar-Piqué et al., 2016; Candelise et al., 2020).
Apart from interacting with other proteins, α-syn forms homo-multimers (Bartels et al., 2011; Dettmer et al., 2013, 2015). Native α-syn also self-assembles and forms β-pleated sheets (Serpell et al., 2000) which further lead to the formation of insoluble aggregations (Conway et al., 1998). NMR spectroscopy, AFM and circular dichroism have revealed the presence of increased β-sheet structure during α-syn aggregations (Pfefferkorn et al., 2012; Lashuel et al., 2013). The factors that are involved in the process of forming insoluble α-syn aggregation include genetic mutation (Conway et al., 1998; Fredenburg et al., 2007), molecular crowding induced by high concentration of macromolecules (Conway et al., 1998; Shtilerman et al., 2002; Uversky, 2002, 2007), alterations in temperature and low pH (Ahmad et al., 2012). Additionally, minute variations in ionic strength of α-syn (25 instead of 50 mM NaCl) (Sandal et al., 2008; Roeters et al., 2017), oxidative stress (Hashimoto et al., 1999), proteins with lipid bilayer surface (Burke et al., 2013) or phospholipids (Bodner et al., 2009) also contribute to the formation of aggregates. Under these alterations in the environment, α-syn forms oligomers (Figure 2), then proto fibrils and finally the insoluble fibrils (Uversky et al., 2001; Kanaan and Manfredsson, 2012). The reaction is reported to proceed in first-order kinetics (Uversky et al., 2001; Kanaan and Manfredsson, 2012), where, in each stage the product is more stable compared to the reactants, signifying the irreversible process (Uversky and Eliezer, 2009). The hydrophobic core of various α-syn point mutations (like A30P, E46K, and A53T) reduces the α-helical content prompting the aggregation of the protein (Waxman et al., 2009; Burré et al., 2012). Srivastava et al. (2020) reported the rotenone induced alterations in the environment. Rotenone increases the hydrophobicity favoring misfolding of α-syn by reducing the lag phase and triggering aggregations (Srivastava et al., 2020).
Figure 2. α-Synuclein aggregation process (α-syn, principal constituent of Lewy’s body). This figure was drawn using Motifolio.
Alerte et al. (2008) reported that the aggregation of α-syn is associated with the hyperphosphorylation of PP2A in dopaminergic neurons (Alerte et al., 2008). A small amount of intracellular α-syn translocates into the lumen of vesicles which are facilitate aggregation (Lee et al., 2005). Misfolded α-syn is removed by extracellular proteolytic enzymes or is taken up by the neighboring cells especially by microglia and astrocytes, and degraded inside lysosomes (Ahn et al., 2006; Stefanis et al., 2019). Mitochondrial and proteasomal dysfunctions triggers α-syn aggregation and further activate microglial neuroinflammation associated with PD (Ciechanover and Brundin, 2003; Lee, 2003; Zhang et al., 2005). Recent studies also propose that the aggregated α-syn gets transported like prions to other neurons (Desplats et al., 2009; Luk et al., 2012). Supporting the data, George et al. (2019) have reported microglia modulated cell to cell transfer of α-syn in PD in non-inflammatory conditions (George et al., 2019). A study has reported that lipid peroxidation by-product 4-hydroxy-2-non-enal (HNE) plays a crucial role in oligomerization and cell to cell transmission of misfolded α-syn (Bae et al., 2013).
The exact mechanism of α-syn spread is yet to be clearly understood. However, secretion of exosome like vesicles has been reported to be involved in the spread of intracellular α-syn to the extracellular space of another cell (Emmanouilidou et al., 2010; Alvarez-Erviti et al., 2011). Furthermore, exosomes are also reported to be associated the formation of α-syn oligomers which are easily uptaken by the neighboring cells (Danzer et al., 2012). Microglia was reported to uptake the α-syn via receptor mediated endocytosis (Lee et al., 2008). Along with the exosomal α-syn, free oligomeric α-syn was also reported to be up taken by neighboring cells (Danzer et al., 2012). There are several reported mechanism of cell-to-cell transfer of α-syn in PD including tunneling nanotubes, trans-synaptic junctions (Lee et al., 2005; Jao et al., 2008; Jang et al., 2010; Freundt et al., 2012; Masuda-Suzukake et al., 2014; Abounit et al., 2016; Dieriks et al., 2017). Recent studies also reported that the introduction of α-syn exosomes derived from patients with synucleinopathies to cell culture and mice model leads to propagating of α-syn aggregation (Stuendl et al., 2016; Ngolab et al., 2017). The pathological α-syn is also reported to be taken up by the surrounding microglia, which causes neuroinflammation (Chang et al., 2013; Bliederhaeuser et al., 2016) and inhibits autophagy and promotes the transmission α-syn (Xia et al., 2019). On the other hand, tunneling nanotubes (TnTs), the non-adherent actin-based cytoplasmic extensions act as a membrane bridges for intercellular transport of α-syn between two cells within a short time span of 30 sec (Gousset and Zurzolo, 2009; Abounit et al., 2016; Dieriks et al., 2017; Rostami et al., 2017). The TnTs mediated transport is not restricted to α-syn and may have a general role in transportation (Dieriks et al., 2017). The spread of α-syn over long distances via axon could possibly be involved in transferring misfolded α-syn to different regions of the brain (Jensen et al., 1999; Utton et al., 2005). Neuronal cell-to-cell transmission of α-syn fibrils is reported through axonal transport (Freundt et al., 2012). Beyond the classical exocytosis of exosome in, the transportation of α-syn from cell-to-cell (Lee et al., 2005; Danzer et al., 2012) has also been suggested to happen by trans-synaptic spreading (Danzer et al., 2011; Pan-Montojo et al., 2012; Van Den Berge et al., 2019; Mezias et al., 2020).
Several drugs inhibit cell to cell transportation of aggregated α-syn (Schofield et al., 2019; Weihofen et al., 2019; Hijaz and Volpicelli-Daley, 2020). 14-3-3θ protein was reported to inhibit the cell to cell transmission of α-syn and its toxicity by reducing the oligomerization in PD (Wang et al., 2018). Liu et al. (2021) suggest on the protective role of biocompatible antioxidant nanozyme, PtCu nanoalloys (NAs) that inhibits the prion-like spreading of α-syn in PD (Liu et al., 2021).
Mutant α-Synuclein and PD
The SNCA gene is located on chromosome 4. Several mutations and polymorphisms have been observed in SNCA gene. Alterations in SNCA expression levels due to mutations have been associated to PD (Rutherford et al., 2014). The link between mutant α-syn and PD was established in an autosomal dominant form of PD with the missense mutation in the chromosome 4q21-q23 (Polymeropoulos et al., 1996). The change in a single base pair in the chromosome from guanine to adenosine (G to A transition) at the position 209 of exon number 4 in SNCA gene results in alteration of alanine to threonine at position 53 of the α-syn protein (A53T) (Golbe et al., 1990; Polymeropoulos et al., 1997). Furthermore, A30P (Krüger et al., 1998), E46K (Zarranz et al., 2004; Sakai et al., 2019), H50Q (Appel-Cresswell et al., 2013), G51D (Lesage et al., 2013), A53E (Pasanen et al., 2014), A53V (Yoshino et al., 2017), A18T and A29S (Hoffman-Zacharska et al., 2013) mutations are also reported. Remarkably, all the recognized mutations in the SNCA gene occur at the N-terminus of the protein which either disrupt the membrane binding property or result in increase in the aggregation of α-syn, thereby impairing the native α-syn functions at the pre-synaptic terminal (Conway et al., 1998; Fredenburg et al., 2007; Burré et al., 2012; Lesage et al., 2013; Fares et al., 2014). Although single base changes and small indels have been reported as the most widely studies DNA variations in PD, Copy Number Variations are also emerging as a prevalent source of genetic variations in PD (La Cognata et al., 2017). For example, Singleton et al. (2003) determined that triplication of the SNCA genomic locus on chromosome 4q21 is associated with PD (Singleton et al., 2003).
Majority of the identified PD mutations are located within the lipid-binding domain of α-syn suggesting that alterations in lipid binding might be associated with α-syn pathology (Pineda and Burré, 2017). SNCA variants have been shown to have differential affinity in binding to the phospholipid membranes. SNCA WT and A53T were reported to bind to rat brain vesicles whereas A30P was reported not to bind to phospholipid membranes. It was proposed that mutant α-syn potentially accumulates in the cells and assembles into Lewy body filaments (Jensen et al., 1998). Later on, it was reported that familial mutant A30P had a lesser affinity and A53T had no affinity to bind lipid membranes (Perrin et al., 2000). However, subsequently, it was confirmed that A30P, but not A53T shows decreased lipid binding affinity (Bussell and Eliezer, 2004). This was suggested to be due to the disruption of local helix formation as a result of A30P mutation (Fares et al., 2014; Ysselstein et al., 2015). In other reports, A53T mutant has been shown to have reduced (Samuel et al., 2016; Robotta et al., 2017) or similar (Middleton and Rhoades, 2010) binding affinities when compared to WT α-syn. Mutations specifically in G51D (Fares et al., 2014) and A53E (Ghosh et al., 2014) have reduced phospholipid binding. E46K variant of α-syn, binds more efficiently to anionic phospholipids, while the A30P variant shows less binding, suggesting the alterations in lipid membrane binding in PD for this variant (Stöckl et al., 2008). However, H50Q mutation does not alter lipid binding affinity (Ruf et al., 2019). These observations suggest that lipid-induced generation of fibrils is highly sensitive to the specific sequence of the SNCA protein, in particular, the region encompassing residues 46–51 (Flagmeier et al., 2016). However, the question on whether α-syn aggregation occurs in lipid bound or unbound state is under investigation (Narayanan and Scarlata, 2001; Cole et al., 2002; Lee et al., 2002a; Zhu and Fink, 2003; Burré et al., 2015; Perni et al., 2017; Mori et al., 2019).
The E46K, G51D, and the H50Q mutants of α-syn protein have significantly delayed degradation compared to WT α-syn, concurring to the data on higher resistance to degradation of these mutants in fly model of PD (Mohite et al., 2018; Sakai et al., 2019). However, H50Q variant of α-syn does not affect the structure or subcellular localization of α-syn (Khalaf et al., 2014). α-syn overexpressing SH-SY5Y cells show increased toxicity and are resistant to degradation and these aggregates are enriched in A53T α-syn (Paleologou et al., 2008; Sugeno et al., 2008; Karampetsou et al., 2017). Here it is important to mention that α-syn mutant A30P has also been reported to have slower degradation rate compared to WT α-syn (Bennett et al., 1999; Kasai et al., 2008). Unlike WT α-syn, A53T mutant has tendency for early-stage aggregation by acquiring β-sheet structure (Bertoncini et al., 2005; Camilloni and Vendruscolo, 2013) during protofibril growth, explaining the early onset of familial PD (Kang et al., 2011; de Oliveira and Silva, 2019). E46K mutations also show the propensity to acquiring β-sheet structure. However, increased N-terminal and C-terminal contacts with proteins (Rospigliosi et al., 2009; Wise-Scira et al., 2013) result in more complex and compact structure compared to WT α-syn (Fredenburg et al., 2007; Wise-Scira et al., 2013; Boyer et al., 2020). In a systematic analysis, A30P mutant was shown to have reduced tendency to form inclusions in comparison to E46K and G51D mutants. This is probably due to long-range contacts between the N and C-termini that shield the central domain, which is reported to promote aggregation (Lázaro et al., 2014). Further investigations will help to understand the role of point mutations in the pathogenic aggregation of α-syn.
Post Translational Modifications (PTMs) of α-Syn
α-Synuclein undergoes various post-translational modifications (PTMs) and plays a crucial role in PD pathology. Until now, acetylation, phosphorylation, and nitration are the key PTMs. Phosphorylation and ubiquitination have emerged as consistent markers of α-syn pathology. Apart from Ser 129, phosphorylation at Ser 87 (Paleologou et al., 2010) is also reported in α-syn aggregation. Despite strong evidences of phosphorylation, synucleinopathic lesions also contain monoubiquitinated α-syn (Tofaris et al., 2011). However, the ubiquitination mechanism needs to be understood clearly. A small proportion of aggregated α-syn is ubiquitinated, despite the presence of ubiquitin chains in LBs inclusions (Hasegawa et al., 2002). Supporting the data, transgenic mice expressing a form of α-syn unable to undergo developmentally down-regulated gene 4 (Nedd4) associated ubiquitination showed increased α-synuclein aggregation and un-ubiquitinated synucleinopathy lesions (Periquet et al., 2007).
Nitrative stress plays a critical role in α-syn aggregation. α-syn has four tyrosine residues Y39, Y125, Y133, and Y136 which are susceptible to nitration (Chavarría and Souza, 2013; Barrett and Timothy Greenamyre, 2015). Nitration of α-syn is reported as a biomarker that is the indicative of nitrative damage in the PD patients in human and animal models (Good et al., 1998; Giasson et al., 2000; Sathiya et al., 2013; Ma et al., 2019a). Stone et al. (2012) reported that overexpression of NO synthase and NO levels triggers nitration of α-syn followed by its oligomerization in neurons (Stone et al., 2012).
Oxidation of the four methionine residues: N-terminal (M1 and 5) and the C-terminal (M116 and 127) of α-syn produce methionine sulfoxides which inhibit fibrillization (Hokenson et al., 2004). Oxidative modifications of the tyrosine’s via nitration leads to the partial folded conformation that stabilizes soluble oligomers and stops elongation into fibrils (Uversky et al., 2002b, 2005; Yamin et al., 2003). In presence of H2O2 4 methionines converts to sulfoxides (Glaser et al., 2005) and rotenone leads to methionine oxidation and subsequent intracellular aggregation (Sanders and Greenamyre, 2013).α-syn forms either antiparallel α-helices or one contiguous α-helix when interacting with acidic lipid membrane. α-syn is believed to be present as an unfolded protein in native form, which undergoes conformational change (Weinreb et al., 1996) when interacting with other molecular partners (Uversky, 2002; Dyson and Wright, 2005). Upon interaction with acidic lipid or with high curvature membrane, the N-terminus of α-syn folds into an α-helix that interacts with membranes for physiological functions (Benskey et al., 2016). Studies have reported that α-syn directly interacts with SV, SNARE complex proteins, proteins involved in calcium regulation, and the catalytic subunit of PP2A (Burré et al., 2010, 2012). Parallelly, Ruf et al. (2019) have investigated the potential of various mutant α-syn to form fibrils from monomeric α-syn (Figure 3) (Ruf et al., 2019). In conditions like genetic mutation, increased α-syn protein concentrations, post translational modifications and oxidative stress promote α-syn aggregation (Benskey et al., 2016). α-syn fibrogenesis impairs mitochondria, disrupts synapses and is toxic to the lysosome-autophagy axis and results in neurodegeneration (Lashuel et al., 2013).
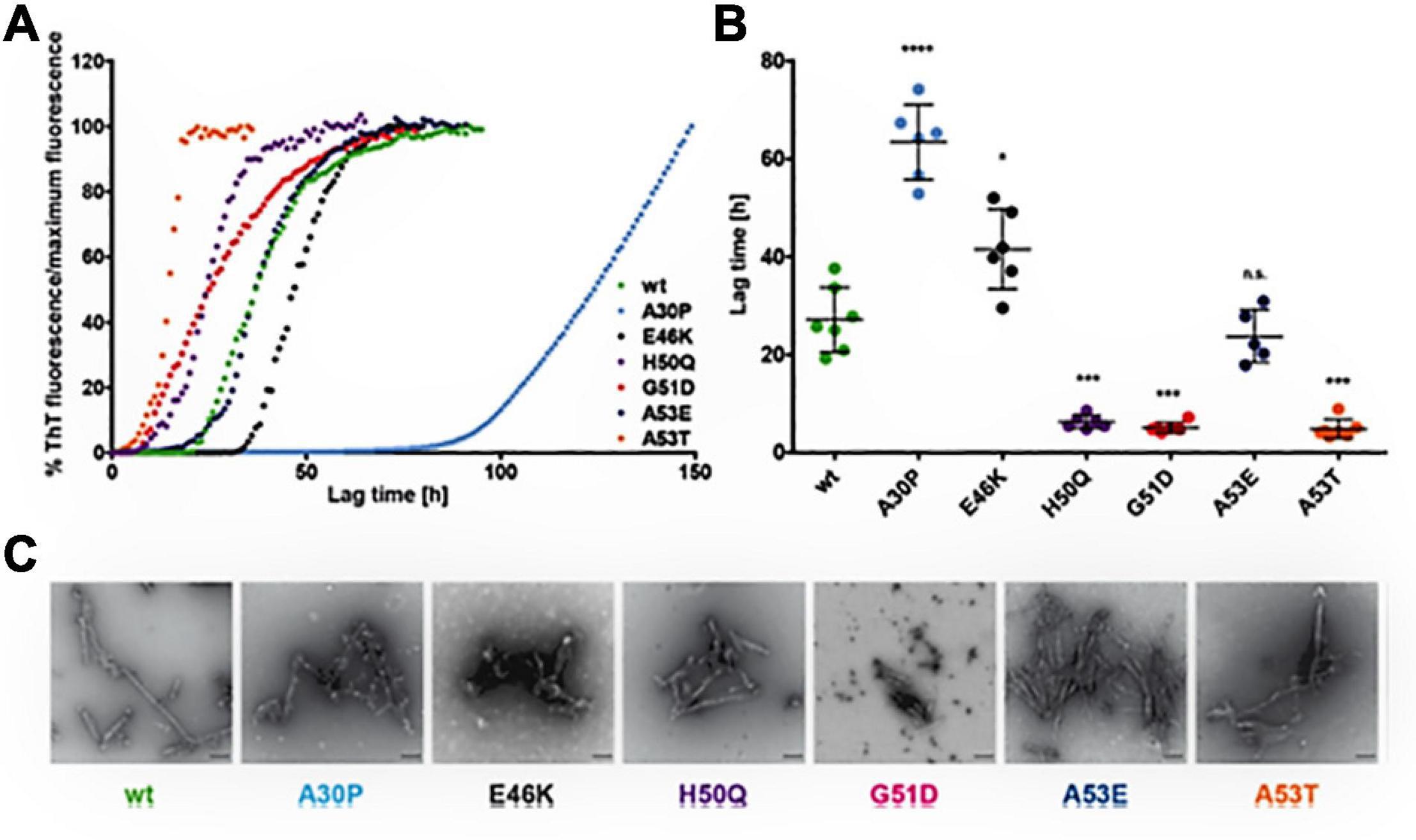
Figure 3. Effect of α-syn mutants on the kinetics of fibril development. (A) Curves indicate the aggregation kinetics of mutant α-syn, observed by ThT fluorescence over time up to 150 h. (B) Denotes the quantification of the lag times (time taken to show ThT fluorescence from the reference line) represent the slow fibril development of A30P and rapid fibrillation of H50Q, G51D, and A53T. E46K α-syn shows minor increase of lag times and of A53E α-syn shows no variance in fibrillation in comparison to WTα-syn. The Single dot indicates independent value (n ≥ 5) and the Error bars specify standard deviation, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. (C) The electron microscopy images representing fibril development of all α-syn mutants after incubation of 50 μM monomeric α-syn for 96 h at 37°C under 1400 rpm (scale bar: 200 nm). Reprinted (adapted) with permission from Ruf et al. (2019). Copyright (2019) American Chemical Society.
It is widely acknowledged that native α-syn exists as an intrinsically disordered monomeric protein (Conway et al., 1998). However, physiologically, α-syn exists as a steady tetramer with rich α-helical structure which is immune to aggregation (Bartels et al., 2011). α-syn contains a highly amyloidogenic hydrophobic domain in the N-terminus region (amino acid 61–95), that is partly absent in β-syn (Hashimoto et al., 2001; Uversky et al., 2002a). α-syn oligomerization occurs with hydrophobic residue of the amphipathic helices to form tetrameric structures (Zhu et al., 2003; Ullman et al., 2011). Anderson et al. (2006) isolated insoluble α-syn from synucleinopathy patients to investigate changes in its primary structure in a diseased state. Adding to this data, Wang et al. (2011) reported that α-syn produced in Escherichia coli exists as a stable form in absence of lipids or micelles (Wang et al., 2011). However, the factors that are responsible for promoting and/or inhibiting the pathogenic α-syn accumulation are not clearly understood.
O-GlcNAcylation is a dynamic biochemical process, in which N-acetylglucosamine (GlcNAc) from uridine 5′-diphospho-N-acetylglucosamine (UDP-GlcNAc) is transferred to the serine and threonine residues of proteins by O-GlcNAc transferase (OGT) and removed by O-GlcNAcase (OGA) (Hart et al., 2007). O-GlcNAcylation identifies threonine (T) residues of α-syn isolated from mouse and human samples (Wang et al., 2010; Alfaro et al., 2012; Morris et al., 2015). O-GlcNAcylation at T72 completely blocks the formation of both fiber and oligomer aggregates in vitro (Marotta et al., 2015). The full-length α-syn with O-GlcNAcylation at Ser 87, aggregates with slower kinetics than the unmodified protein (Lewis et al., 2017). Several O-GlcNAcylated sites inhibit the toxicity of extracellular α-syn fibers that are the likely culprits in the spread of PD (Levine et al., 2019).
α-Synuclein Pathology and Cellular Organelles in PD
It is known that α-syn aggregation is linked to various pathological cascades such as down regulation of mitochondrial complex I activity, ER stress, neuro-inflammation, disrupted cell membrane integrity, inhibition of ubiquitin proteasome system (UPS) and impaired autophagy-lysosomal pathway (ALP) in PD (Lin et al., 2019; Mahul-Mellier et al., 2020). Also, mitochondrial dysfunction plays a crucial role in the PD pathogenesis (Park et al., 2018). Interestingly, α-syn has a high affinity for mitochondrial membrane compared to other organelles (Nakamura et al., 2008; Kamp et al., 2010). Colocalization of α-syn in the mitochondrial and cytosolic fraction of rat brain tissues (Li et al., 2007) and in the SNpc and the striatum of PD patients brain is well established (Devi et al., 2008). α-syn is shown to be present in the inner mitochondrial membrane (IMM), outer mitochondrial membrane (OMM) and mitochondrial matrix (Cole et al., 2008; Liu et al., 2009; Kamp et al., 2010; Robotta et al., 2014). Its translocation to mitochondrial matrix causes alterations in complex I and increases the oxidative stress (Martínez et al., 2018). Taken altogether these data indicate that α-syn is of importance in the mitochondrial function in PD. Nevertheless, the causal link between secondary effect of PD and the associated pathogenesis is not clearly understood and needs investigation. ER is responsible for protein synthesis, folding, lipid synthesis and trafficking to Golgi. ER activates UPR when degradation of misfolded proteins is required (Walter and Ron, 2011). Proteins which fail to fold properly are degraded by proteasomes (Colla, 2019). Aggregation of α-syn triggers UPR which causes cell death (Cooper et al., 2006; Sugeno et al., 2008). Glucose regulated protein 78 (GRP78)/BiP, is the key mediator of the UPR and also senses of ER stress. Oligomeric α-syn is reported to accumulate in ER thereby triggering PD (Colla et al., 2012b; Liu et al., 2018a; Yan et al., 2019). Furthermore, the UPR activation caused by ER stress was also reported in the histopathological studies of brains of PD patients (Conn et al., 2004; Hoozemans et al., 2007; Colla, 2019).
Recently, lysosomal dysfunction, oxidative stress, and apoptosis were reported to trigger the nuclear translocations of α-syn (Ryu et al., 2019). Accumulation of α-syn in nucleus is shown to interfere with cell cycle process in PC12 cells and cause PD like motor symptoms in C57 mice (Ma et al., 2014). Mutations in GBA gene, which encodes for lysosomal enzyme glucocerebrosidase (GCase) is also associated with PD (Tayebi et al., 2003; Lwin et al., 2004; Gegg et al., 2020). Mutations such as N370S and L444P in GBA protein are reported in various PD patient based clinical studies (Toft et al., 2006; Marco et al., 2008; Mata et al., 2008; Hu et al., 2010; Emelyanov et al., 2012) as well as in in vitro (Maor et al., 2019) and in vivo (Taguchi et al., 2017; Yun et al., 2018) models of PD. The endo-lysosomal system regulates vesicle traffic and comprises a unique environment for proteolysis. Mutations in the endo-lysosomal protein ATP13A2 are reported to increase the aggregation of α-syn (Lopes da Fonseca et al., 2016). Recently, Tsunemi et al. (2020) reported the impaired astrocyte mediated α-syn clearance due to the mutation in ATP13A2 gene (Tsunemi et al., 2020). Overexpression of α-syn inhibits Ras-related protein Rab-1A (RAb1A), a GTPase, which in turn causes mis-localization of Atg9 in the TGN, an important process in autophagosome formation (Winslow et al., 2010). Mutant (A30P) α-syn suppresses c-Jun N-terminal kinase activity and inhibits autophagy in dopaminergic neurons which further increases the intracellular burden of α-syn accumulation in PD (Lei et al., 2019).
Gut–Brain Axis and Synucleinopathy: Does PD Starts From Gut?
Enteric nervous system and parasympathetic nerves get affected due to α-synucleinopathies (Edwards et al., 1992; O’Donovan et al., 2020). The VN is reported to be involved in spreading the neurogenerative process to the lower brainstem and the dopaminergic nigrostriatal system (Braak and Del Tredici, 2017; Kujawska and Jodynis-Liebert, 2018). Constipation is a common non-motor symptoms observed in the early onset of PD (Yu et al., 2018). GI dysfunction, in particular constipation, affects up to 80% of PD patients (Poewe, 2008; Cersosimo and Benarroch, 2012; Noyce et al., 2012; Müller et al., 2013). Dental deterioration, gastroparesis, delayed intestinal transit time and constipation are other symptoms associated with ENS neurodegenerative diseases (Pfeiffer, 2011; Cersosimo and Benarroch, 2012). These symptoms may appear even before the loss in motor functions and become established as early diagnostic information on PD (Braak et al., 2006; Shannon et al., 2012). The intestinal environmental factors such as the gut microbiota and the metabolites also exert their influences primarily via the gut in PD (Braak et al., 2006; Kieburtz and Wunderle, 2013; Vascellari et al., 2020). Intestinal microbiota interacts with CNS including ENS and vagal nerve (Carabotti et al., 2015; Ma et al., 2019b). Pyrosequencing of the V1–V3 regions of the bacterial 16S ribosomal RNA gene from the fecal microbiome of PD patients suggested that there are alterations in intestinal microbiome (Scheperjans et al., 2015). Extreme stimulus of innate immunity by gut dysbiosis and/or intestinal pathobionts overgrowth and the consequent increase in intestinal penetrability triggers systemic inflammation (Figure 4). Simultaneously, enteric neurons and enteric glial cells activation contribute to the aggregation of α-syn pathology (Holmqvist et al., 2014; Sampson et al., 2016). Accumulation of α-syn in PNS is reported to be associated with impairment of enteric neurons which in turn is linked to GI dysfunctions (Gold et al., 2013; Sánchez-Ferro et al., 2015). Impaired intestinal barrier integrity in PD patients increases the susceptibility of patients to microbial infections (Forsyth et al., 2011). Increased intestinal accumulation of α-syn is referred to as “leaky gut” and is common in PD patients (Chiang and Lin, 2019). Leaky gut promotes translocation of bacteria and endotoxins (bacterial products) from the gut to the brain triggering pro-inflammatory conditions and oxidative stress in the ENS (Forsythe et al., 2014). Pro-inflammatory factors associated with chronic GI disease lead to PNS inflammation which is one of the major risk factors for the observed neuroinflammation in PD (Dobbs et al., 1999; Villarán et al., 2010).
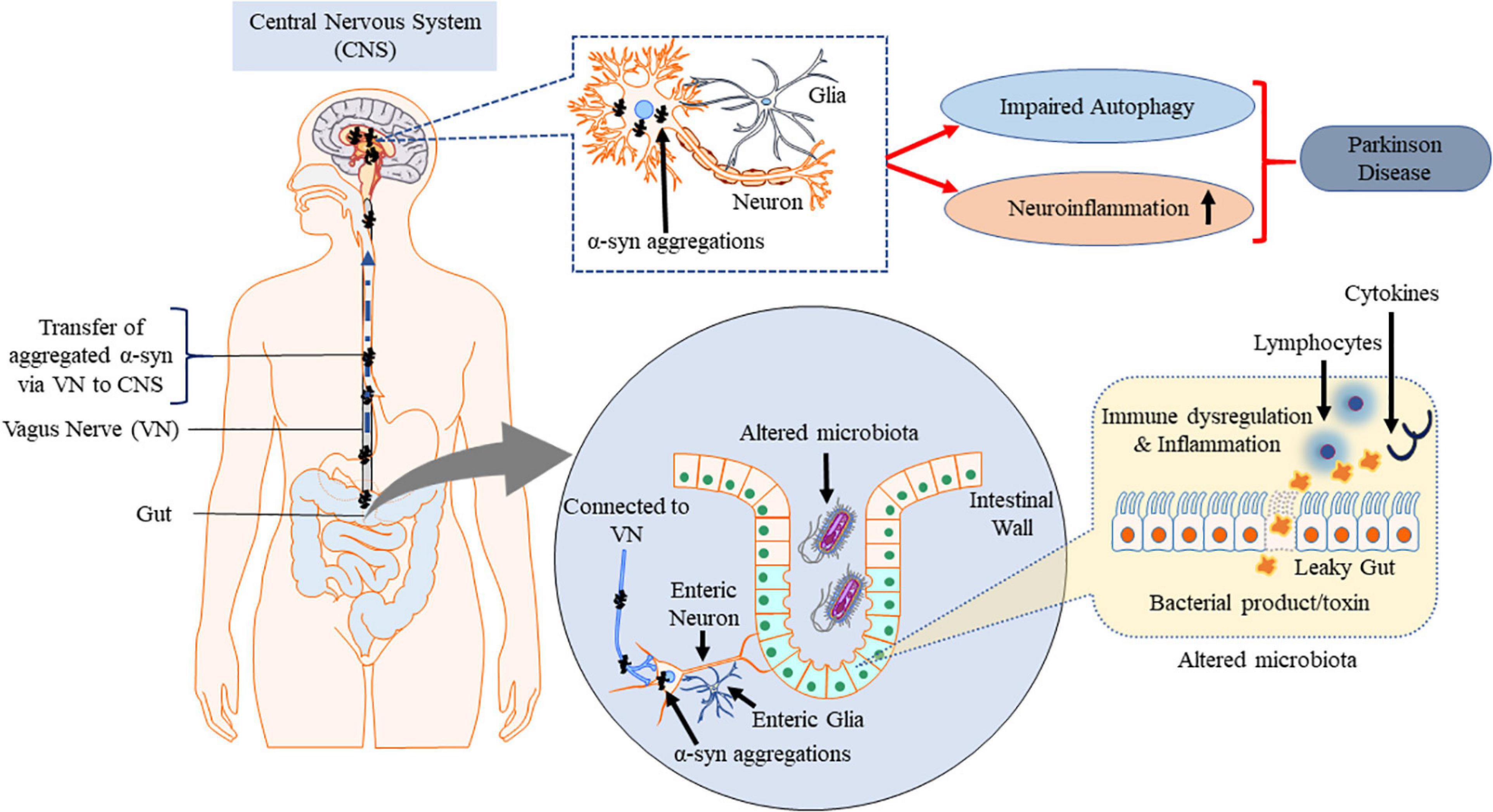
Figure 4. Misfolded α-syn and gut–brain axis. Gut dysbiosis, cytokines and endotoxins potentially cause inflammation at ENS which spreads to CNS via VN along with the aggregated α-syn triggered by ENS, contributing to PD pathogenesis. This figure was drawn using Motifolio.
The gut bacteria synthesize many neurotransmitters as well as neuromodulators such as γ-aminobutyric acid, serotonin, dopamine, short-chain fatty acids, etc. (Lyte, 2014; Mayer et al., 2015). The gut upholds a neuronal connection via VN, as bacteria can trigger afferent neurons of the ENS (Forsythe et al., 2014). The local reflexes (migrating motor complex and peristaltic reflex) are managed by ENS via IPANs (Nezami and Srinivasan, 2010). Enteric dopaminergic neurons are present in GI and inhibit intestinal motility (Anlauf et al., 2003). The spinal cord with DMVN accepts and gives rise to the afferent and efferent fibers of the VN and influences the GI tract (Chang et al., 2003).
Electrogastrography examination of patients in early and advanced PD state confirmed the persistent gastric motility irregularities (Soykan et al., 1999). Reduced amplitude of stomach contractions in PD is reported in real-time magnetic resonance imaging (Ajaj et al., 2004). The lesions in the medullar, spinal and peripheral autonomic nervous system in PD are the reasons for GI disturbances (Wakabayashi and Takahashi, 1997; Benarroch et al., 2005). In normal physiological circumstances, native α-syn is highly expressed in the CNS and is associated with regulating neurotransmission. The α-syn pathology begins in submucosal plexus of the ENS and spreads retrogradely to the CNS through vagal preganglionic axons of the DMVN (Figure 5) (Braak et al., 2006). From the DMVN a predictable caudo-rostral spread of α-syn associated pathology to other parts of the brain α-syn associated (SNpc, basal forebrain and finally neocortex region (Del Tredici et al., 2002; Braak et al., 2003b; Hawkes et al., 2007; Reichmann, 2011). This α-syn pathology spread has recently also been observed in non-human primates (Arotcarena et al., 2020). Recently, Musgrove et al. (2019) reported that oxidative stress increases at VN increasing cell to cell transmission of α-syn and promotes PD (Musgrove et al., 2019).
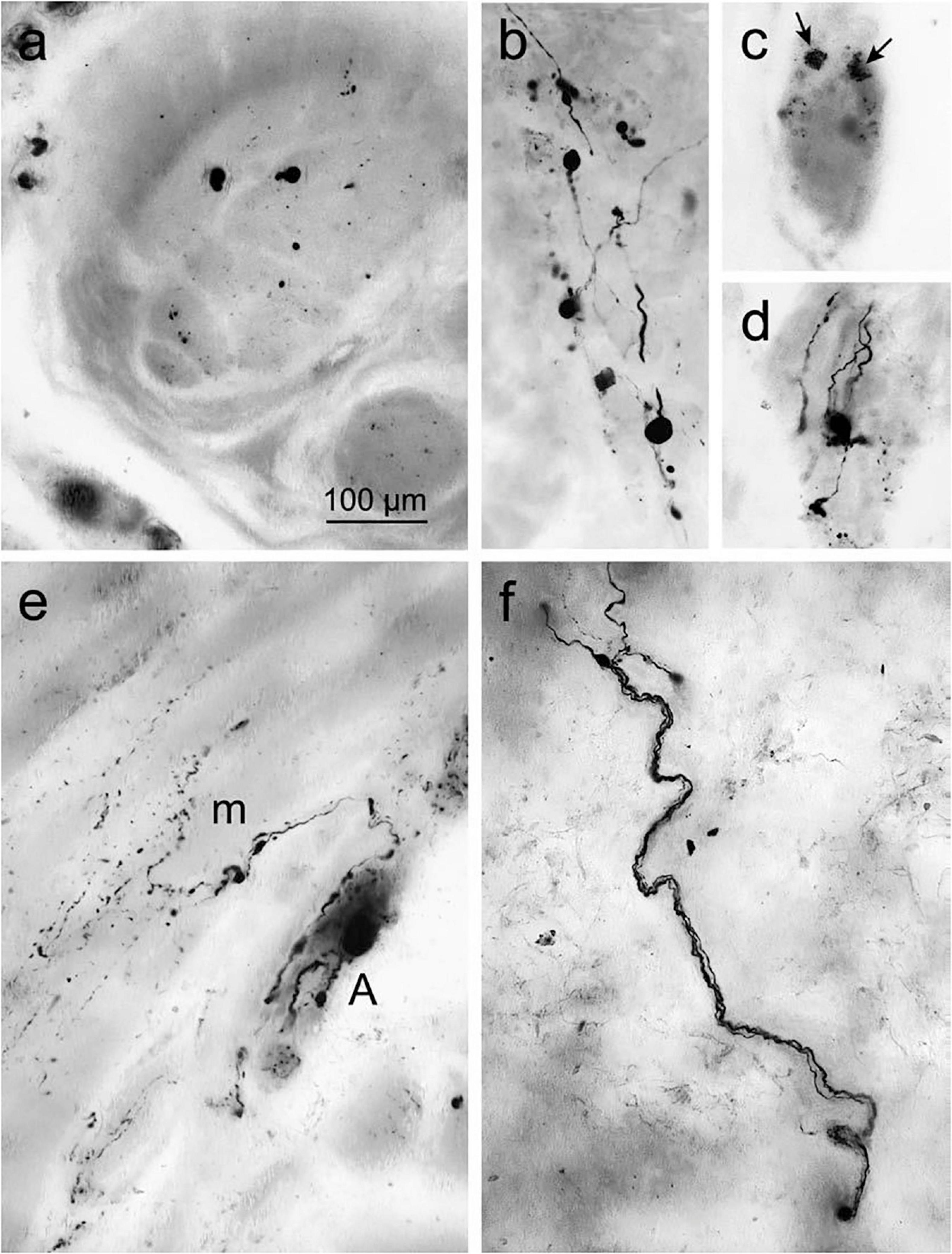
Figure 5. Accumulated α-syn in the surface of gastric wall. (a) Denotes immunoreactive inclusions in axons within the peripheral nerve. (b–d) Lewy neurites and LBs in the Auerbach plexus. (b) Presence of Lewy body pathology. (c) Punctate α-syn aggregations (arrows) in ENS neurons in the fundus, indicates the early signs of LBs. (d) Fiber-like Lewy neurites associated with the ganglia of the Auerbach plexus. (e) Immunoreactive fibers produced from the Auerbach plexus (A) bifurcate recurrently and fragmented into terminal ramifications along with the smooth muscle cells next to muscle layer (m). (f) The nerve fiber bundle of Meissner’s plexus coursing via the gastric submucosa. Reused (adapted) with the permission from Copyright Clearance Centre (License Number 5057740321467) (Braak et al., 2006).
In early PD, phosphorylated and aggregated α-syn is identified in the ENS neurons and OB (Braak et al., 2006; Shannon et al., 2011). α-syn deposition in the neurons might start from ENS and OBs, through VN and olfactory tract, respectively (Braak et al., 2003a; Hawkes et al., 2009, 2010; Klingelhoefer and Reichmann, 2015). Interestingly, the evidence for α-syn pathology spread from the GI tract to the brain in a rat model is available (Holmqvist et al., 2014; Kim et al., 2019). Interestingly, decreased gastric motility observed in 6-hydroxydopamine-model of PD lesion in rats is also reported (Zheng et al., 2014). Also, decrease in the levels of short chain fatty acids (SCFA), the prime metabolic product of certain gut bacteria, causes alterations in the ENS and contributes to GI dysmotility in the PD (Unger et al., 2016). Lai et al. (2018) reported that a chronic low-dose MPTP may be used to assess the development of intestinal pathology as well as gut microbiota dysbiosis. This may provide new insights into the pathogenesis of PD (Lai et al., 2018; Kim et al., 2019). Many chemical signals from the gut to specific regions of the brain are also speculated to affect blood brain barrier integrity through formation of endothelial clusters, which is often recorded in PD (Guan et al., 2013). Further investigations are warranted to elucidate the exact role of the gut–brain axis in PD (Table 1).
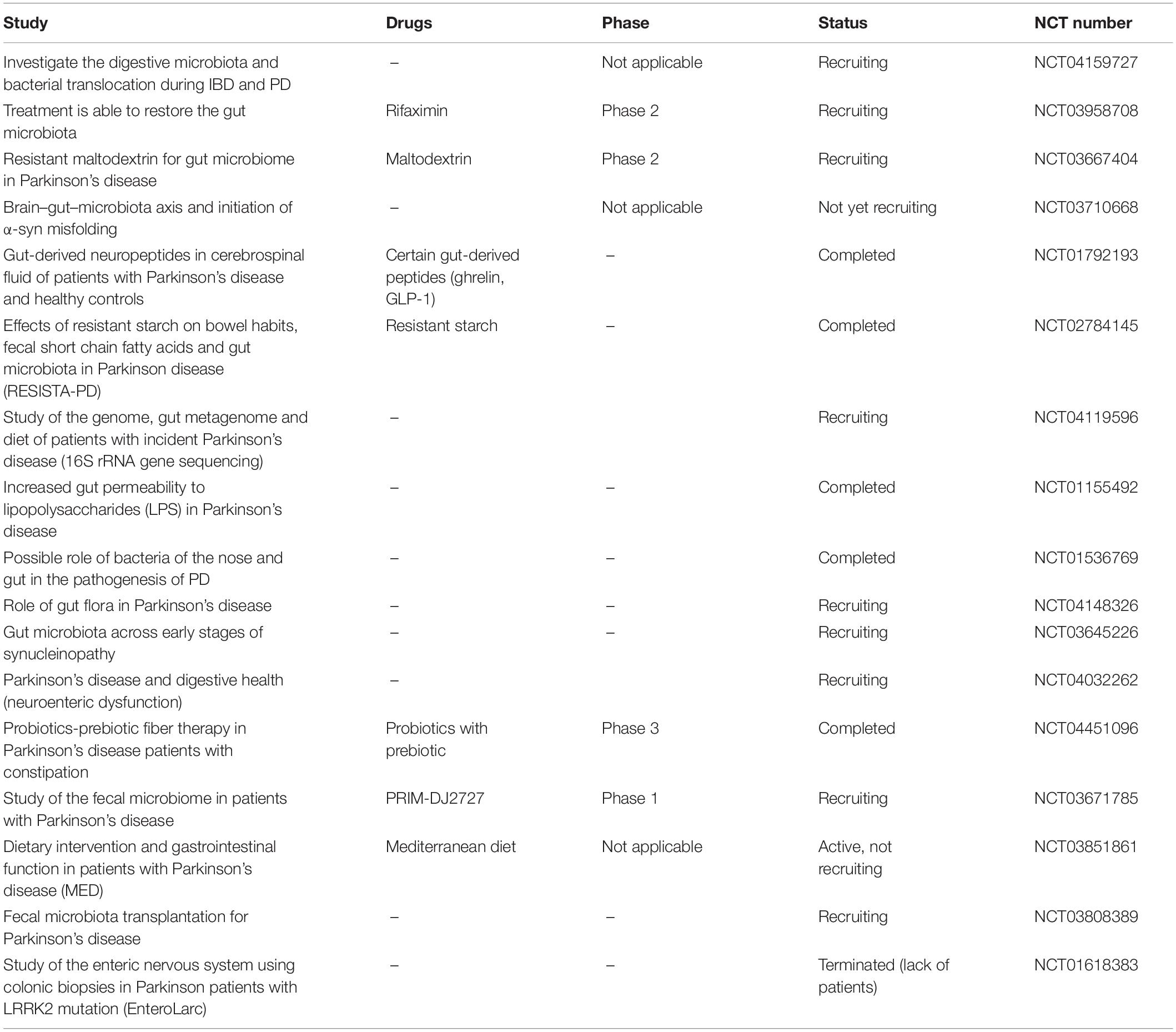
Table 1. Clinical trials on gut–brain axis in PD.
Mechanism of Neuronal Clearance of Misfolded α-Synuclein by Autophagy
Cellular aggregation and impaired clearance of α-syn are the major pathological hallmarks of PD. Cellular clearance of misfolded proteins including α-syn is regulated by the ALP and the UPS (Lopes da Fonseca et al., 2015). Monomeric α-syn is degraded by both ALP and UPS (Liu et al., 2003; Cuervo et al., 2004) by compensatory mechanisms, i.e., when one fails the other will execute (Yang et al., 2013).
In an compensatory mechanism of ALP, a peptide based therapy protects α-syn neurotoxicity by activating proteasome pathway (Betarbet et al., 2006; Qu et al., 2020). Purified human 20S proteasomes are also reported to degrade accumulated α-syn in an ubiquitin-independent manner in PD (Tofaris et al., 2001; McKinnon et al., 2020). Supporting the data further, activation of UPS by natural alkaloid (Cai et al., 2019) and Orobol derivatives (ethanolic extracts of Cudrania tricuspidata fruits) is reported to decrease α-syn accumulation in PD. Additionally, phosphorylated α-syn (Ser 129) aggregates are reported to degrade via proteasome pathway (Machiya et al., 2010). Interestingly, α-syn oligomers and fibril are reported to inhibit the activity of 20S/26S proteasome subunits (Snyder et al., 2003; Zhang et al., 2008; McKinnon et al., 2020; Suzuki et al., 2020).
In the case of removal of high molecular weight proteins, including oligomers and aggregates, the disposal mechanism shifts to autophagy (Lee et al., 2004). Based on its cargo delivery process, it is divided into CMA, macroautophagy and microautophagy. As of today, and to the best of our knowledge, there is no report on microautophagy clearing α-syn aggregation. The other two types of autophagic processes are discussed below (Figure 6).
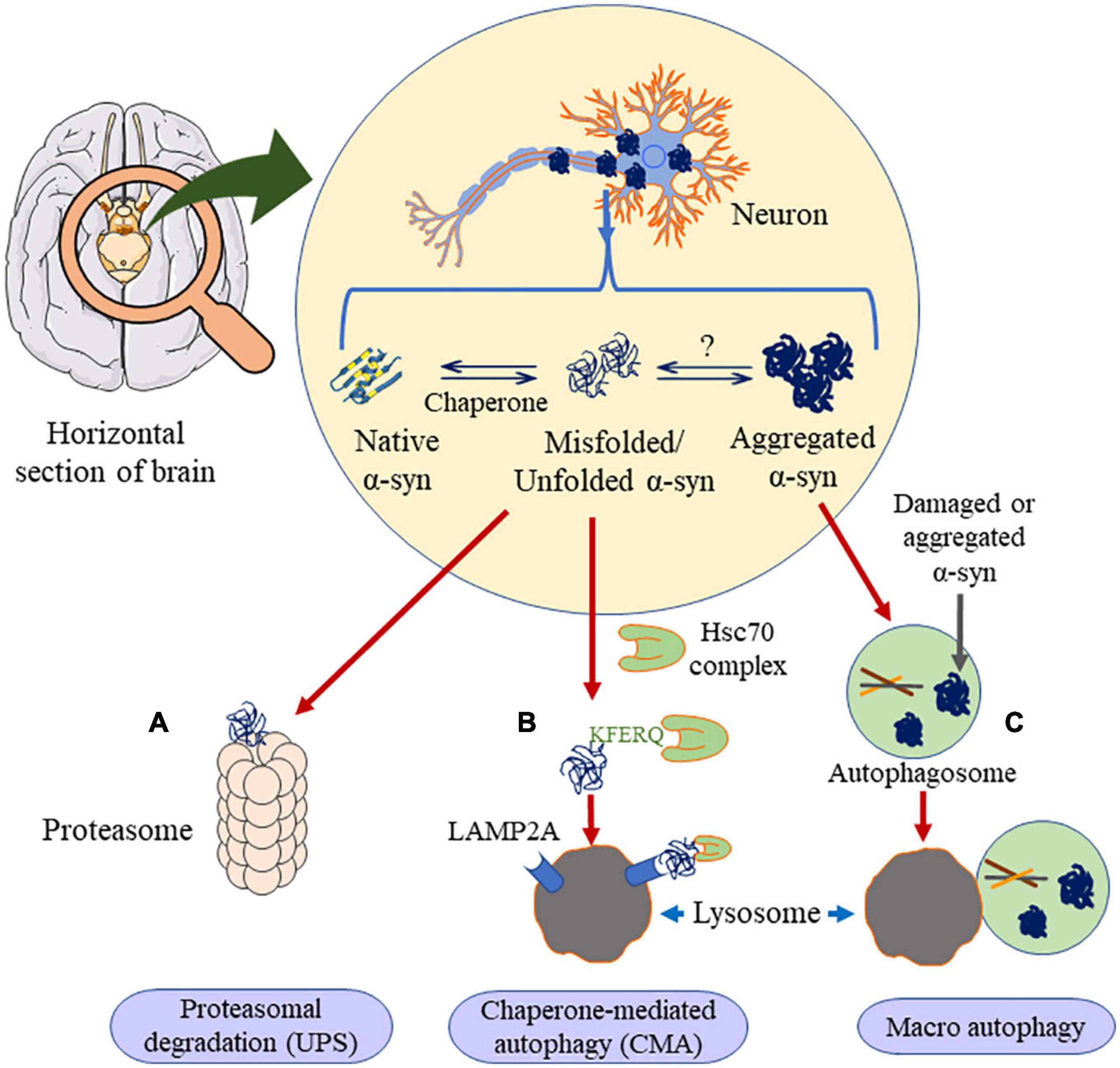
Figure 6. Clearance of α-syn occurs through three major ways. If the α-syn is in the unfolded dimer or the small oligomer state it undergoes proteasomal degradation (A) or chaperone mediated autophagy (B), in presence of HSc70, α-syn is engulfed by lysosome through LAMP2A receptor. Else, if the aggregation is higher than oligomer state, it prefers macro autophagy (C), where it forms autophagosome, which is engulfed by the lysosome. This figure was drawn using Motifolio.
Wild-type soluble α-syn is efficiently degraded in lysosomes by CMA, but the mutant α-syn is poorly degraded by CMA despite having an affinity for the CMA receptor. The lysosome-associated Hsc70 (lHsc70) protein helps in translocation of the targeted substrates for degradation (Chiang et al., 1989; Agarraberes et al., 1997). Mutant α-syn (A53T and A30P) also inhibits CMA substrates and lysosomal uptake that results in compensatory activation of macroautophagy (Cuervo et al., 2004). α-syn monomers and dimers, but not oligomers, are degraded via CMA (Martinez-Vicente et al., 2008; Xilouri et al., 2009). Alvarez-Erviti et al. (2010) had reported that decreased expression of LAMP2A, slows down the degradation of wild-type α-syn (Alvarez-Erviti et al., 2010). Hence, it was concluded that CMA is not involved in the degradation of misfolded α-syn directly. In contrast, Wu J.-Z. et al. (2019) reported that two bioactive ingredients dihydromyricetin and salvianolic acid B extracted from natural medicinal plants downregulate α-syn aggregation by activating both CMA and macroautophagy processes (Wu J.-Z. et al., 2019).
Endoplasmic reticulum stress is mainly an outcome of accumulated misfolded proteins, for example, α-syn, that undergoes ER associated degradation (ERAD) (McCracken and Brodsky, 2003). Misfolded/mutated proteins impair the ERAD system and contribute to PD pathogenesis (Lehtonen et al., 2019). GA regulates post-translational protein modifications, for example, glycosylation and proteolytic cleavage that occurs in the ER (Rabouille and Haase, 2016). Misfolded α-syn inhibits ER-Golgi transportation and leads to the aggregation of proteins in ER and triggers cell death in PD (Cooper et al., 2006; Wang and Hay, 2015). Furthermore, α-syn inhibits Rab1a which not only alters the ER-Golgi transportation, but also causes mislocalization of Atg9 trafficking, thereby, inhibiting autophagy (Winslow et al., 2010; Xilouri et al., 2016; Tomás et al., 2020).
Inositol-requiring enzyme 1 (IRE1), a key UPR signal activator, under ER stress clears protein aggregation via autophagy IRE1-X-box–binding protein 1 (XBP1) (Sado et al., 2009, 1; Fouillet et al., 2012; Valdés et al., 2014, 1; Ghavidel et al., 2015; Adams et al., 2019). On the other hand, Yan et al. (2019) reported that α-syn accumulation promotes neuronal death in Drosophila model of PD through the hyperactivation of IRE1 via the c-Jun N-terminal kinase (JNK)-dependent manner (Yan et al., 2019). Further research on the exact mechanisms of α-syn clearance will help understand the neuroprotective role of IRE1. Mesencephalic astrocyte-derived neurotrophic factor (MANF), also known as ARMET (arginine-rich mutated in early-stage tumors) is present in ER and promotes neuronal cell survival through UPR regulation (Apostolou et al., 2008; Renko et al., 2018; Wang et al., 2021). ER stress triggers the accumulation of misfolded α-syn (Colla et al., 2012a). MANF is shown to have neuroprotective activity in in vitro and in vivo models of PD (Voutilainen et al., 2009; Liu et al., 2018b). MANF is also reported to facilitate the cellular clearance of misfolded α-syn in a Caenorhabditis elegans model of PD. Inhibition of autophagy related genes by RNAi approach has been shown to decrease the expression of MANF suggesting its potential therapeutic role in PD (Zhang et al., 2018). In a recent clinical study, MANF level was also reported to be higher in the blood of PD patients. However further studies are required to reveal if MANF is a clinical marker for PD (Galli et al., 2019).
Zinc finger with KRAB and SCAN domains 3 (ZKSCAN3), a zinc-finger family DNA-binding protein initiates autophagosome biogenesis. However, it works antiparallel to TFEB (Chauhan et al., 2013). A30P mutant α-syn inhibits ZKSCAN3 and impairs autophagy in dopaminergic neuron (Lei et al., 2019).
Recent evidence proposes that AMPK signaling plays a crucial role in neurodegeneration. Rapamycin-induced initiation of autophagy, or AMPK agonists, promote the clearance of fibril-mediated α-syn pathology (Gao et al., 2019). Overexpressed AMPKα1 or α2 subunits integrate into the AMPK complex and protect dopamine neurons against human α-syn accumulation toxic effects (Bobela et al., 2017). AMPK also regulates PGC-1α, which is a transcriptional co-activator and master regulator of mitochondrial biogenesis (Wan et al., 2014). α-syn binds to the promoter sequence of PGC-1α and causes promoter methylation, a sporadic PD associated phenomenon which ultimately decreases PGC-1α expression (Su et al., 2015). AMPK inhibits mTORC1 by phosphorylating Raptor (Gwinn et al., 2008), along with indirect phosphorylation and activation of TSC2 (Inoki et al., 2003). unc-51-like autophagy activating kinase 1 (ULK1) drives autophagosome formation whilst mTORC1 suppresses (under nutrient condition) autophagy by phosphorylating ULK1 at Ser 757. In contrast, phosphorylation of ULK1 at Ser 317, Ser 777 or Ser 555 by AMPK promotes autophagy (Egan et al., 2011; Kim et al., 2011). Various reports suggest on the protective role of AMPK against the toxicity of both intracellular and extracellular α-syn (Choi et al., 2010; Wu et al., 2011; Dulovic et al., 2014; Anandhan et al., 2017; Bobela et al., 2017; Gao et al., 2019). α-syn is suggested to reduce AMPK phosphorylation and downstream target Raptor in SH-SY5Y neuroblastoma cells (Dulovic et al., 2014). However, several conflicting reports on AMPK signaling data are available. For example, Kim et al. (2013) and Xu et al. (2014) reported that activation of AMPK and inactivation of Akt causes neuronal cell death via inhibition of the mTOR pathway (Kim et al., 2013; Xu et al., 2014). Along similar lines, AMPK activation is also reported to trigger the aggregation of α-syn in primary neurons (Jiang et al., 2013). Future molecular investigations will help understand the role of AMPK in PD.
Cellular clearance of expired or damaged organelles is processed by autophagy, including the selective autophagy processes such as, (mitochondria) mitophagy, (peroxisomes) pexophagy, (ribosomes) ribophagy and parts of the nucleus involved in nucleophagy (Martinez-Vicente and Cuervo, 2007; Senkevich and Gan-Or, 2019). α-syn has been reported to impair mitophagy in PD (Shaltouki et al., 2018). α-syn exosome/extracellular vesicle (EV) fractions range from 60 to 160 nm in diameter, and are cleared by ALP. Inhibition of ALP increases α-syn levels (Alvarez-Erviti et al., 2011; Danzer et al., 2012; Minakaki et al., 2018). α-syn fibrils are transported to an endosomal compartment and lysosomes. The lysosomal inhibition is shown to accumulate α-syn aggregations, supporting the autophagy/lysosomal clearance pathway.
Strategies That Facilitate Neuronal Clearance of α-Synuclein
Reducing α-Synuclein Production
Many research efforts are focused on protecting neuronal cells from α-syn toxicity, for example, reducing the synthesis of α-syn (Figure 7) by the infusion of siRNA in the hippocampal and cortical regions of mice (Lewis et al., 2008). In another study, injecting siRNA-containing exosomes is shown to lead to decrease in α-syn in the SNpc of Ser 129D α-syn transgenic mice (Cooper et al., 2014). α-syn propagation is also shown to participate in the neurotoxicity process and here the C-terminus (CT) of the protein plays a significant role (Games et al., 2014). Hence, monoclonal antibodies 1H7, 5C1, or 5D12 that target the CT, decrease α-syn in neurons and rescue TH in striatum which have been reported to improve motor ability and memory deficits (Games et al., 2014). Selective silencing of mutant SNCA gene has been shown to reverse the pathogenic characteristics of mutated α-syn while preserving the physiological functions of the native α-syn. These effects are consistently observed both in in vitro and in vivo studies using lentivirus mediated RNA interference (Sapru et al., 2006; Takahashi et al., 2015). Naked small interfering RNA (siRNA) is shown to reduce endogenous SNCA in hippocampus region, in vitro and in vivo models of PD. This has been translated to potential neuroprotective effect in α-synucleinopathies (Lewis et al., 2008). Also, Zharikov et al. (2015) have reported that knockdown of α-syn exerts neuroprotective role in a rotenone model of PD (Zharikov et al., 2015). Also, AAV vectors expressing miSyn4 siRNAs are reported to downregulate the α-syn (overexpressed) in mice (Kim et al., 2017). However, long-term RNAi knockdown of α-syn did not show any beneficial effects on dopaminergic functions in the adult rats (Zharikov et al., 2019).
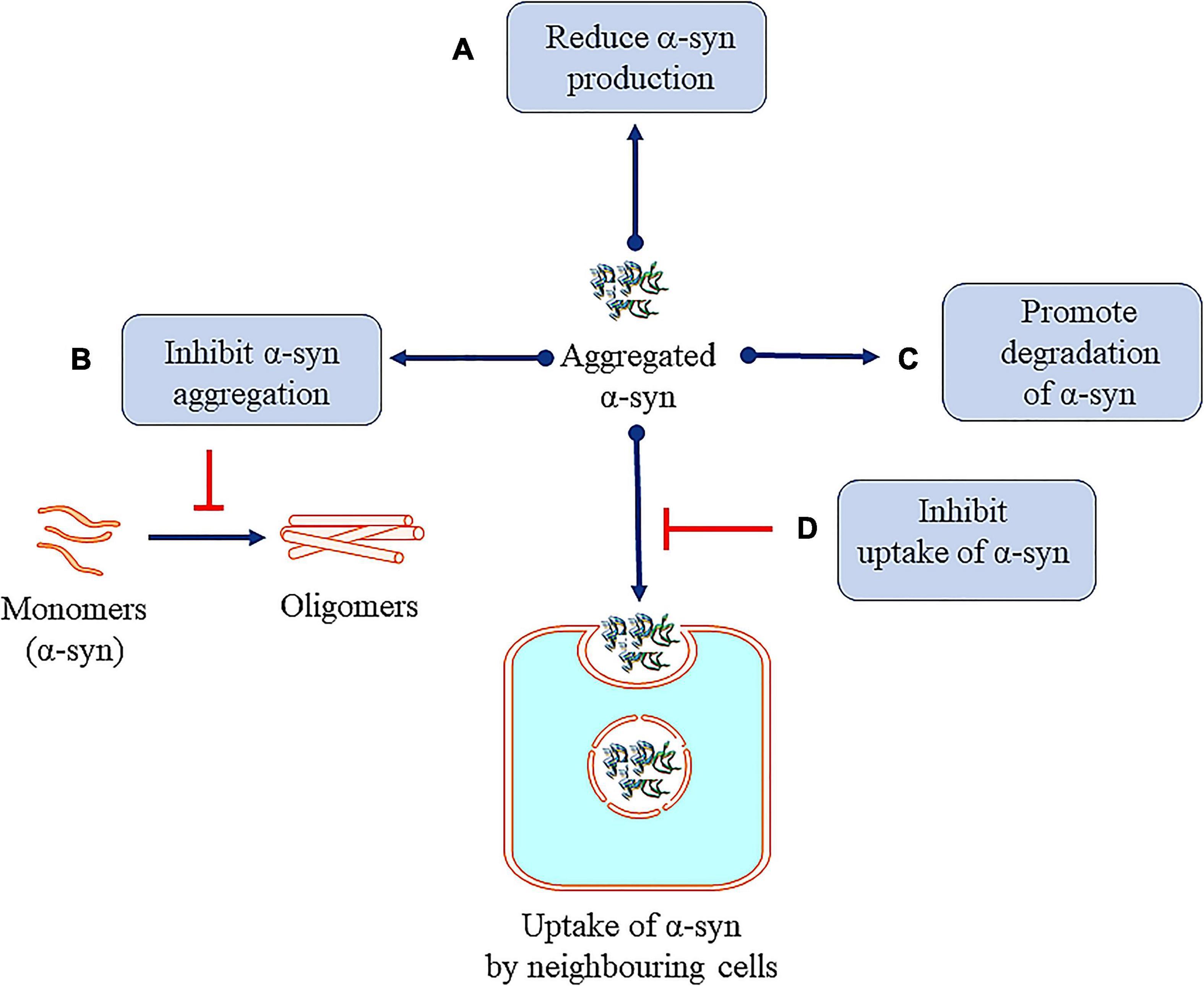
Figure 7. Therapeutic approaches against toxic α-syn. (A) Reducing α-syn production. (B) Inhibiting α-syn aggregation. (C) Promoting degradation of α-syn. (D) Inhibition of uptake of α-syn by neighboring cells.
Inhibiting α-Synuclein Aggregation
Some reports have focused on inhibiting the aggregation of α-syn. Bae et al. (2012) used antibodies targeting the Fcγ receptors present on the surface of microglia to inhibit microglial triggered α-syn aggregation. Fonseca-Ornelas et al. (2014) reported that porphyrin phtalocyanine tetrasulfonate delays the aggregation of vesicle bound α-syn in H4 neuroglioma cells. Similarly, an α-syn protofibril-selective monoclonal antibody (mAb47) is shown to decrease its aggregation in A30P α-syn mutant mouse model (Lindström et al., 2014). Novel compounds NPT200-11 (Price et al., 2018) and NPT100-18A (Wrasidlo et al., 2016) are also reported to inhibit the aggregation of α-syn in preclinical models. It must be mentioned that compound NPT200-11 has cleared phase 1 of clinical trials (Table 2). A novel compound PBT434, is reported to slow down the progression of PD in hA53T α-syn transgenic mouse (Finkelstein et al., 2017). First generation epitope vaccines targeting the aggregated α -syn, are reported to be immunogenic in B6SJL mice (Ghochikyan et al., 2014). Heat shock proteins (HSP), especially small HSPs are molecular chaperones which have also been reported to inhibit α-syn aggregation in both in vitro and in vivo (McLean et al., 2002; Klucken et al., 2004; Gorenberg and Chandra, 2017).
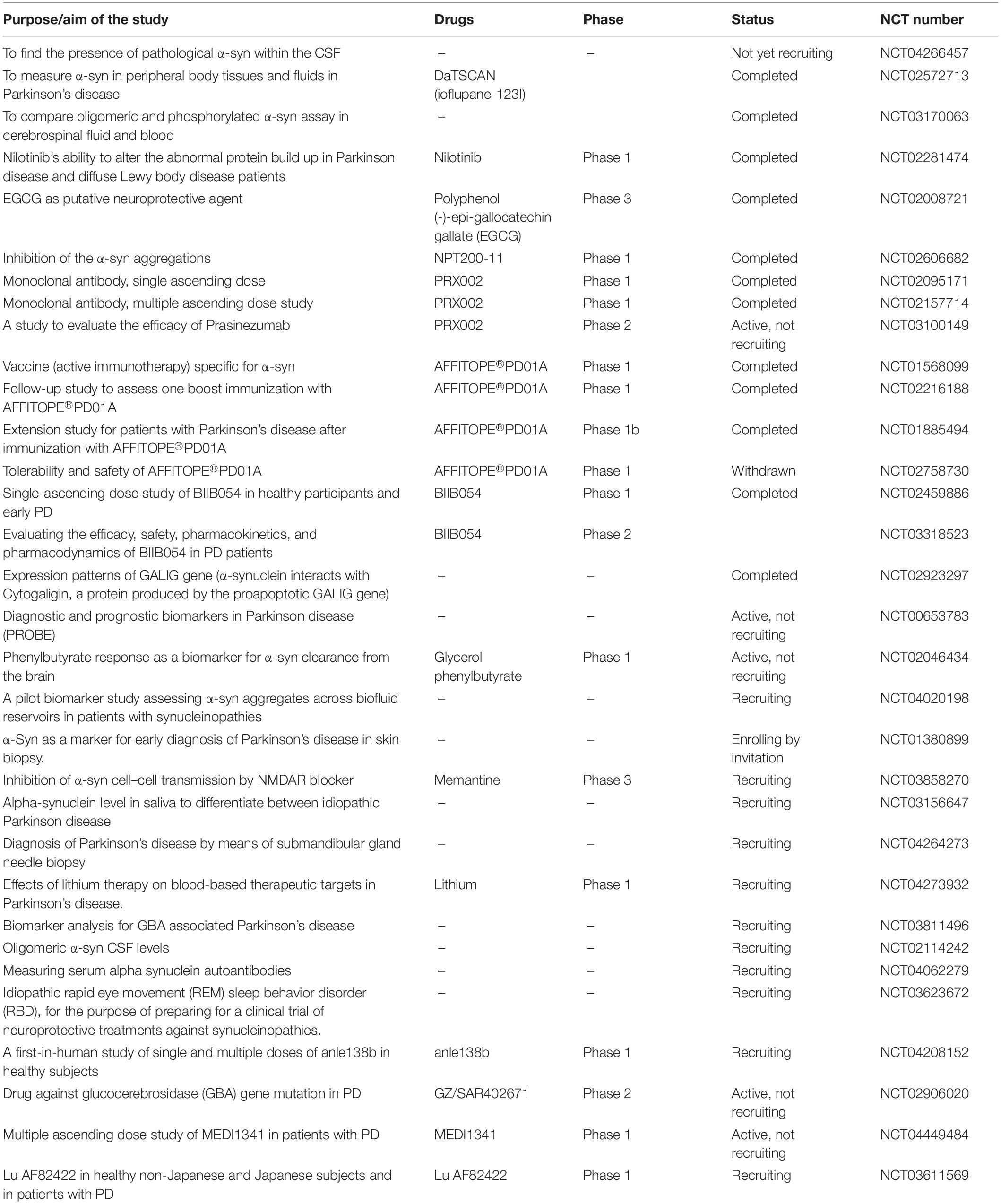
Table 2. List the clinical trials targeting α-syn.
Promoting Degradation of α-Synuclein
Increasing α-syn clearance through lysosomal/or autophagic process also leads to decrease in the cellular levels. Decressac et al. (2013) reported that stimulation of TFEB function or blocking of mTOR prevents the degeneration of dopaminergic neurons caused by α-syn toxicity (Decressac et al., 2013). Passive immunization with monoclonal α-syn antibodies (9E4) is also shown to clear α-syn aggregation via a lysosomal pathway (Masliah et al., 2011; Bae et al., 2012).
Additionally, deficiency of GD-linked glucocerebrosidase (GCase) is also reported to impair the lysosomal proteolytic enzyme in primary cultures or induce hiPSC neurons, triggering aggregation of α-syn (Mazzulli et al., 2011). Toward this end, increasing the GCase activity by AAV-GBA1 (gene encoding glucocerebrosidase) intra-cerebral gene delivery has also been shown to protect against α-syn toxicity in rodents (Rocha et al., 2015). Furthermore, NCGC607 (Aflaki et al., 2016) and NCGC00188758 (Mazzulli et al., 2016) (new leads against α-syn) are shown to improve the GCase activity and decrease α-syn accumulation in human neurons. Interestingly, Kalekrein 6 (KLK6) is a serine protease in PD whose expression levels are inversely correlated toα-syn and recombinant KLK6 is reported to degrade of extracellular α-syn directly (Pampalakis et al., 2016).
Several other studies of active and passive immunization against α-syn aggregation are reported and are shown to be neuroprotective (Masliah et al., 2005, 2011; Sanchez-Guajardo et al., 2013; Christiansen et al., 2016). Here, drugs like PRX002 (Table 2) (a humanized IgG1 monoclonal antibody), that has successfully entered in Phase 2 (Jankovic et al., 2018) clinical trials (NCT02157714) needs a special mention. Further, safety and tolerability is being tested in the PD patients (Schenk et al., 2017).
Inhibition of Uptake of α-Synuclein by Neighboring Cells
α-Synuclein monoclonal antibodies (mAbs) are reported to inhibit propagation and uptake of α-syn and prevent the aggregation of α-syn in a mouse model (Tran et al., 2014). Mao et al. (2016) have demonstrated that α-syn fibrils bind to lymphocyte-activation-gene 3 (LAG3) protein and initiate endocytosis into neuronal cells (Mao et al., 2016). The involvement of other proteins in the initiating endocytosis is suggested (Shrivastava et al., 2015). Further research in this area is ongoing and will help in understanding the role of endocytosis in removal of pathogenic α-syn. In line with this, it is important to mention that Gustafsson et al. (2017) had also reported that, inhibiting Fcγ receptors (FcγRI and FcγRIIB/C) results in reduced uptake of α-synu oligomer/protofibril (Gustafsson et al., 2017). Here, astrocytes are reported to take up α-syn preformed fibrils (pffs) via endocytosis process. Clusterin interacts with α-syn pffs in the extracellular compartment and the clusterin/α-syn complexes are internalized by astrocytes. To this end, clusterin knock-out primary astrocytes and clusterin knock-down hiPSC-derived astrocytes are also reported that limits the uptake of α-syn pffs by the cells (Filippini et al., 2021).
“Janus-Faced” α-Synuclein
α-Synuclein and cysteine-string protein-alpha (CSPalpha) are present abundantly in SV. CSPalpha plays a vital role in neuronal growth and its deletion is shown to cause progressive neurodegeneration in mouse model. Interestingly, abnormal expression of α-syn causes neurodegeneration and motor impairment due to the deletion of CSPalpha. Also, α-syn is shown to inverse the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE)-complex assembly, a pathological impediment observed in the CSPα knockout mice (Chandra et al., 2005). α-syn binds at the N-terminus of SNARE protein synaptobrevin-2 by its C-terminus (Burré et al., 2010). Greten-Harrison et al. (2010) demonstrated that deletion of α-syn causes alterations in the synaptic structure and leads to transferable and age dependent neuronal dysfunction. It further causes decrease in synapse size by ∼30% both in vivo and in vitro (Greten-Harrison et al., 2010). These data indicate the neuroprotective roles of α-syn at the synapse. However, Darios et al. (2010) reported indirect inhibitory effect of α-syn on SNARE-complex assembly by inhibition of arachidonic acid. Arachidonic acid is reported to stimulate SNARE-complex formation and exocytosis (Darios et al., 2010). It is widely known that the mutated form of α-syn is linked to PD pathology (Chandra et al., 2005; Stefanis, 2012).
Furthermore, α-syn is also reported to exert protection of neurons against various apoptotic stimuli (da Costa et al., 2000). Additionally, the involvement of α-syn in various biological functions such as synaptic transmission, calcium regulation, mitochondrial homeostasis, gene expression, protein phosphorylation cannot be ignored (Sharon et al., 2001; Ellis et al., 2005). α-syn interacts in SV and SNARE proteins, mediating the vesicular transport to presynaptic membrane (Maroteaux et al., 1988; Burré et al., 2010, 2012). α-syn inhibits TH (Perez et al., 2002; Perez and Hastings, 2004) and its phosphorylation either by increasing PP2A activity or by altering the binding sites of TH for phosphorylation (Peng et al., 2005; Wang et al., 2009). Supporting the phenomenon of DA release from presynaptic membrane of neuronal cells, α-syn also inhibits AADC, an inhibitor of DA synthesis (Tehranian et al., 2006). Moreover, α-syn is shown to interact with the DAT, modulate DAT activity (increasing or decreasing) and increase the amount of VMAT on vesicles (Lee et al., 2001; Dauer et al., 2002; Wersinger and Sidhu, 2003; Wersinger et al., 2003; Fountaine and Wade-Martins, 2007; Fountaine et al., 2008).
The SNCA expression in terms of PD pathogenesis is delicately balanced. Supporting this concept, a clinical study on well characterized PD patients described that the low repeat REP1 allele, a complex microsatellite (259 base pairs; resulting in decreased SNCA expression) is associated with motor and cognitive dysfunctions, whereas the high-repeat REP1 allele (263 base pairs; increases SNCA expression) is associated with improving the motor and non-motor symptoms like cognition (Markopoulou et al., 2014). Contradictory data by Corrado et al. (2018) reported that, REP1 allele (263 base pairs) is associated with inferior cognitive outcome (Corrado et al., 2018) in PD. In a clinical patients based study it was reported that, long REP1 alleles are associated with motor and non-motor functions in PD (Ng et al., 2019). Adding to this study, the role of SNCA Rep1 allele length in non-motor functions as well as depression in the early PD patients was also reported (Yong et al., 2020). It was argued that Markopoulou et al. (2014) collected the data through telephonic interviews and hence there could be possibility of miscommunication. Secondly, the biological effect of REP1 allele could also vary with the patients, especially in different ethnic groups. Hence, it was concluded that future studies with more patients are required to resolve these contradictory findings.
Interestingly, in another report both aggregation and knockdown of α-syn were reported to impair mitochondrial Ca2+ homeostasis and induce toxicity (Calì et al., 2012). Ludtmann et al. (2016) had also reported that monomeric α-syn enters the mitochondria and enhances ATP synthase function (Ludtmann et al., 2016). α-syn was also reported to participate in the physiological functions of mitochondria like fusion, ETC, and VDAC permeability (Ellis et al., 2005; Kamp et al., 2010; Rostovtseva et al., 2015). Supporting the protective role of α-syn, Seo et al. (2002) reported that at nanomolecular concentrations, α-syn is shown to protect the primary neurons against oxidative stress (Seo et al., 2002). Recently, Carmo-Gonçalves et al. (2020) had studied the role of monomeric and fibrillar α-syn on mesencephalic dopaminergic neurons in primary cultures using neurotoxic salsolinol and 3,4-dihydroxyphenylacetaldehyde (DOPAL). They reported that the protective properties of monomeric α-syn involve the inhibition of caspase 3 mediated apoptosis (Carmo-Gonçalves et al., 2020).
Furthermore, increased oxidative stress is shown to trigger the aggregation of α-syn in PD (Scudamore and Ciossek, 2018). Accumulations and overexpression of α-syn further triggers α-syn misfolding (Hsu et al., 2000; Lee et al., 2002b; Chinta et al., 2010) leading to mitochondrial fragmentation and dopaminergic cell death (Menges et al., 2017). Here, it must be mentioned that overexpression and aggregation of α-syn is linked to decrease in the neurotransmitters and consequent motor dysfunctions (Larsen et al., 2006; Gaugler et al., 2012; Scott and Roy, 2012).
It was observed that, in α-synucleinopathy, some neurons express abundant Lewy pathology than other neuronal types. For example, dopaminergic, noradrenergic, cholinergic and the glutamatergic neurons express abundant α-syn aggregation, whereas, most of the GABAergic neurons are spared (Wakabayashi et al., 1995; Spillantini et al., 1998; Gómez-Tortosa et al., 2001; Del Tredici and Braak, 2013; Hall et al., 2014; Kay et al., 2015; Taguchi et al., 2019). Expression of α-syn protein is positively correlated with susceptibility to aggregate (Figure 8) (Wakabayashi et al., 1995; Erskine et al., 2018; Taguchi et al., 2019). Thus, conflicting reports on α-syn in literature need to be resolved.
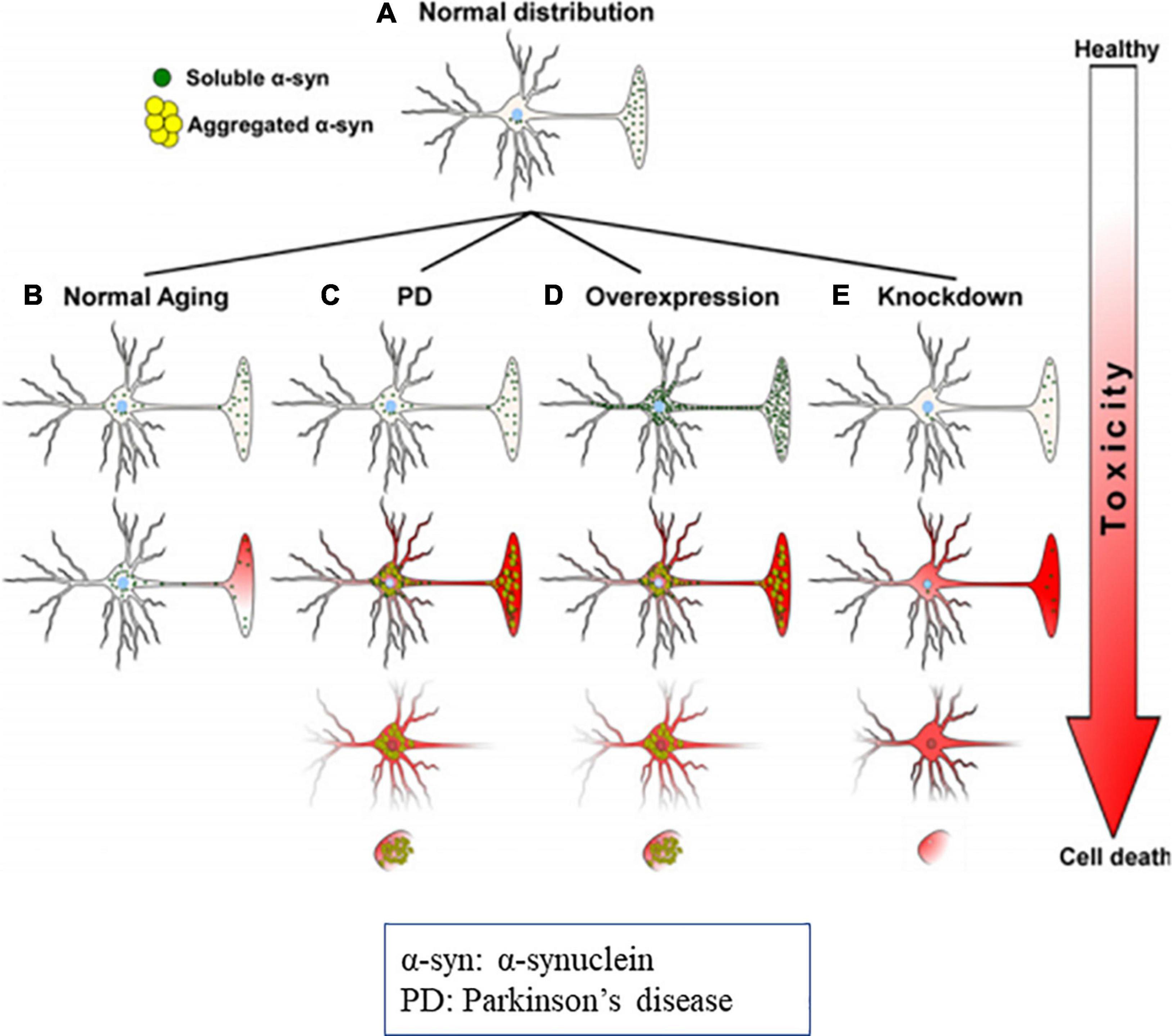
Figure 8. Diagrammatic representation of α-syn distribution and its associated toxicities (Cell death). (A) Physiological distribution of α-syn in healthy neurons mainly in presynaptic terminal. (B) During the aging distribution of α-syn spreads from the presynaptic terminal to the soma, that causes subsequent toxicity. In PD, due to genetic mutations, oxidative stress, α-syn aggregates and produce toxicities and cell death. (C) Overexpression or molecular crowding of α-syn causes toxicities. (D) Knockdown of α-syn below threshold (protein concentration) results in cell death. Image reused as per Creative Commons Attribution-Non-commercial-NoDerivs License (Benskey et al., 2016).
Conclusion
α-Synuclein is reported to be involved in the DA release in the synapse and also has a neuroprotective roles in apoptotic stimuli. There are many scientific reports which establish the physiological role of α-syn in healthy individuals. α-syn is a vital component of LBs which are the major pathological hallmarks in PD. Reports also suggest that misfolded α-syn can travel from cell-to-cell (Freundt et al., 2012; Domert et al., 2016; Tyson et al., 2017; George et al., 2019; Rey et al., 2019). The point mutations that result in change of amino acid in α-syn (A30P, E46K, H50Q, G51D, A53E, and A53T) are studied along with their aggregation kinetics in PD. Further studies confirming the pathogenic mutations and aggregation could help to target α-syn and understand its role in disease pathogenesis. Several studies are being conducted that target pathogenic α-syn and cause impairing of the autophagy and proteasomal processes. Furthermore, the pathogenic origin of α-syn is being explored in relation to gut dysbiosis. Early diagnosis of PD is a major field of interest in modern science. The identification of a biomarker which can detect α-syn toxicity could potentially lead to novel strategies for effective PD diagnosis and treatment. There is a need to collate and present latest data on α-syn and provide a unified view of the protein. This review is an attempt in this direction and aims to help understand the pathophysiological role of α-syn and its aggregation in PD.
Author Contributions
BR contributed to the investigation, writing – original draft, writing – review and editing and visualization. AM and ST contributed to the writing – review and editing. AB contributed to the writing – review and editing, and visualization. AS, CP, AK, and SB contributed to the validation. MS contributed to formal analysis and writing. SC contributed to the conceptualization, methodology, validation, and formal analysis. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
BR acknowledges the Indian Council of Medical Research (ICMR), New Delhi, Government of India, for the Senior Research Fellowship. The authors thank JSS AHER for providing facilities. MS would like to acknowledge the support from the College of Pharmacy and Nutrition, University of Saskatchewan, Canada (Grant No. 415802).
Abbreviations
α -syn, α -synuclein; A β, amyloid- β; AMPK, AMP-activated protein kinase; AADC, aromatic amino acid decarboxylase; AFM, atomic force microscopy; CMA, chaperone-mediated autophagy; DMVN, dorsal motor nucleus of the vagus nerve; DAT, dopamine transporter; ER, endoplasmic reticulum; ETC, electron transport chain; ENS, enteric nervous system; ERAD, endoplasmic reticulum associated degradation; GI, gastrointestinal; GA, Golgi apparatus; hiPSC, human pluripotent stem cells; IPANs, intrinsic primary afferent neurons; IRE1, inositol-requiring enzyme 1; lHsc70, lysosome-associated Hsc70; LAMP2A, lysosome-associated membrane protein type 2a; LBs, Lewy bodies; LBD, Lewy body disorders; mTOR, mammalian target of rapamycin; NO, nitric oxide; NMR, nuclear magnetic resonance; OB, olfactory bulb; PP2A, protein phosphatase 2A; PD, Parkinson’s disease; RAb1A, Ras-related protein Rab-1A; SNpc, substantia nigra pars compacta; SV, synaptic vesicles; TFEB, transcription factor EB; TH, tyrosine hydroxylase; TGN, trans-Golgi network; UPR, unfolded protein response; ULK1, Unc-51-like autophagy activating kinase 1; VMAT, vesicular monoamine transporter; XBP, X-box –binding protein 1; ZKSCAN3, zinc finger with KRAB and SCAN domains 3.
References
Abounit, S., Bousset, L., Loria, F., Zhu, S., de Chaumont, F., Pieri, L., et al. (2016). Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J. 35, 2120–2138. doi: 10.15252/embj.201593411
Adamczyk, A., and Strosznajder, J. B. (2006). Alpha-synuclein potentiates Ca2+ influx through voltage-dependent Ca2+ channels. Neuroreport 17, 1883–1886. doi: 10.1097/WNR.0b013e3280115185
Adams, C. J., Kopp, M. C., Larburu, N., Nowak, P. R., and Ali, M. M. U. (2019). Structure and Molecular Mechanism of ER Stress Signaling by the Unfolded Protein Response Signal Activator IRE1. Front. Mol. Biosci. 6:11. doi: 10.3389/fmolb.2019.00011
Aflaki, E., Borger, D. K., Moaven, N., Stubblefield, B. K., Rogers, S. A., Patnaik, S., et al. (2016). A New Glucocerebrosidase Chaperone Reduces α-Synuclein and Glycolipid Levels in iPSC-Derived Dopaminergic Neurons from Patients with Gaucher Disease and Parkinsonism. J. Neurosci. 36, 7441–7452. doi: 10.1523/JNEUROSCI.0636-16.2016
Agarraberes, F. A., Terlecky, S. R., and Dice, J. F. (1997). An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J. Cell Biol. 137, 825–834. doi: 10.1083/jcb.137.4.825
Ahmad, B., Chen, Y., and Lapidus, L. J. (2012). Aggregation of α-synuclein is kinetically controlled by intramolecular diffusion. Proc. Natl. Acad. Sci. U. S. A. 109, 2336–2341. doi: 10.1073/pnas.1109526109
Ahn, K. J., Paik, S. R., Chung, K. C., and Kim, J. (2006). Amino acid sequence motifs and mechanistic features of the membrane translocation of alpha-synuclein. J. Neurochem. 97, 265–279. doi: 10.1111/j.1471-4159.2006.03731.x
Ajaj, W., Goehde, S. C., Papanikolaou, N., Holtmann, G., Ruehm, S. G., Debatin, J. F., et al. (2004). Real time high resolution magnetic resonance imaging for the assessment of gastric motility disorders. Gut 53, 1256–1261. doi: 10.1136/gut.2003.038588
Alerte, T. N. M., Akinfolarin, A. A., Friedrich, E. E., Mader, S. A., Hong, C.-S., and Perez, R. G. (2008). Alpha-synuclein aggregation alters tyrosine hydroxylase phosphorylation and immunoreactivity: lessons from viral transduction of knockout mice. Neurosci. Lett. 435, 24–29. doi: 10.1016/j.neulet.2008.02.014
Alfaro, J. F., Gong, C.-X., Monroe, M. E., Aldrich, J. T., Clauss, T. R. W., Purvine, S. O., et al. (2012). Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc. Natl. Acad. Sci. U. S. A. 109, 7280–7285. doi: 10.1073/pnas.1200425109
Alvarez-Erviti, L., Rodriguez-Oroz, M. C., Cooper, J. M., Caballero, C., Ferrer, I., Obeso, J. A., et al. (2010). Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 67, 1464–1472. doi: 10.1001/archneurol.2010.198
Alvarez-Erviti, L., Seow, Y., Schapira, A. H., Gardiner, C., Sargent, I. L., Wood, M. J. A., et al. (2011). Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 42, 360–367. doi: 10.1016/j.nbd.2011.01.029
Anandhan, A., Lei, S., Levytskyy, R., Pappa, A., Panayiotidis, M. I., Cerny, R. L., et al. (2017). Glucose metabolism and AMPK signaling regulate dopaminergic cell death induced by gene (α-synuclein)-environment (paraquat) interactions. Mol. Neurobiol. 54, 3825–3842. doi: 10.1007/s12035-016-9906-2
Anderson, J. P., Walker, D. E., Goldstein, J. M., Laat, R., Banducci, K., Caccavello, R. J., et al. (2006). Phosphorylation of Ser-129 Is the Dominant Pathological Modification of α-Synuclein in Familial and Sporadic Lewy Body Disease. J. Biol. Chem. 281, 29739–29752. doi: 10.1074/jbc.M600933200
Anlauf, M., Schäfer, M. K.-H., Eiden, L., and Weihe, E. (2003). Chemical coding of the human gastrointestinal nervous system: cholinergic, VIPergic, and catecholaminergic phenotypes. J. Comp. Neurol. 459, 90–111. doi: 10.1002/cne.10599
Apostolou, A., Shen, Y., Liang, Y., Luo, J., and Fang, S. (2008). Armet, a UPR-upregulated protein, inhibits cell proliferation and ER stress-induced cell death. Exp. Cell Res. 314, 2454–2467. doi: 10.1016/j.yexcr.2008.05.001
Appel-Cresswell, S., Vilarino-Guell, C., Encarnacion, M., Sherman, H., Yu, I., Shah, B., et al. (2013). Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 28, 811–813. doi: 10.1002/mds.25421
Arawaka, S., Sato, H., Sasaki, A., Koyama, S., and Kato, T. (2017). Mechanisms underlying extensive Ser129-phosphorylation in α-synuclein aggregates. Acta Neuropathol. Commun. 5:48. doi: 10.1186/s40478-017-0452-6
Arotcarena, M.-L., Dovero, S., Prigent, A., Bourdenx, M., Camus, S., Porras, G., et al. (2020). Bidirectional gut-to-brain and brain-to-gut propagation of synucleinopathy in non-human primates. Brain J. Neurol. 143, 1462–1475. doi: 10.1093/brain/awaa096
Bae, E.-J., Ho, D.-H., Park, E., Jung, J. W., Cho, K., Hong, J. H., et al. (2013). Lipid Peroxidation Product 4-Hydroxy-2-Nonenal Promotes Seeding-Capable Oligomer Formation and Cell-to-Cell Transfer of α-Synuclein. Antioxid. Redox Signal. 18, 770–783. doi: 10.1089/ars.2011.4429
Bae, E.-J., Lee, H.-J., Rockenstein, E., Ho, D.-H., Park, E.-B., Yang, N.-Y., et al. (2012). Antibody-Aided Clearance of Extracellular α-Synuclein Prevents Cell-to-Cell Aggregate Transmission. J. Neurosci. 32, 13454–13469. doi: 10.1523/JNEUROSCI.1292-12.2012
Bai, J., Liu, M., Pielak, G. J., and Li, C. (2017). Macromolecular and Small Molecular Crowding Have Similar Effects on α-Synuclein Structure. ChemPhysChem 18, 55–58. doi: 10.1002/cphc.201601097
Barrett, P. J., and Timothy Greenamyre, J. (2015). Post-translational modification of α-synuclein in Parkinson’s disease. Brain Res. 1628, 247–253. doi: 10.1016/j.brainres.2015.06.002
Bartels, T., Choi, J. G., and Selkoe, D. J. (2011). α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110. doi: 10.1038/nature10324
Benarroch, E. E., Schmeichel, A. M., Low, P. A., Boeve, B. F., Sandroni, P., and Parisi, J. E. (2005). Involvement of medullary regions controlling sympathetic output in Lewy body disease. Brain J. Neurol. 128, 338–344. doi: 10.1093/brain/awh376
Bennett, M. C., Bishop, J. F., Leng, Y., Chock, P. B., Chase, T. N., and Mouradian, M. M. (1999). Degradation of alpha-synuclein by proteasome. J. Biol. Chem. 274, 33855–33858. doi: 10.1074/jbc.274.48.33855
Benskey, M. J., Perez, R. G., and Manfredsson, F. P. (2016). The contribution of alpha synuclein to neuronal survival and function - Implications for Parkinson’s disease. J. Neurochem. 137, 331–359. doi: 10.1111/jnc.13570
Bertoncini, C. W., Fernandez, C. O., Griesinger, C., Jovin, T. M., and Zweckstetter, M. (2005). Familial Mutants of α-Synuclein with Increased Neurotoxicity Have a Destabilized Conformation. J. Biol. Chem. 280, 30649–30652. doi: 10.1074/jbc.C500288200
Betarbet, R., Canet-Aviles, R. M., Sherer, T. B., Mastroberardino, P. G., McLendon, C., Kim, J.-H., et al. (2006). Intersecting pathways to neurodegeneration in Parkinson’s disease: effects of the pesticide rotenone on DJ-1, alpha-synuclein, and the ubiquitin-proteasome system. Neurobiol. Dis. 22, 404–420. doi: 10.1016/j.nbd.2005.12.003
Bliederhaeuser, C., Grozdanov, V., Speidel, A., Zondler, L., Ruf, W. P., Bayer, H., et al. (2016). Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol. 131, 379–391. doi: 10.1007/s00401-015-1504-2
Bobela, W., Nazeeruddin, S., Knott, G., Aebischer, P., and Schneider, B. L. (2017). Modulating the catalytic activity of AMPK has neuroprotective effects against α-synuclein toxicity. Mol. Neurodegener. 12:80. doi: 10.1186/s13024-017-0220-x
Bodner, C. R., Dobson, C. M., and Bax, A. (2009). Multiple tight phospholipid-binding modes of alpha-synuclein revealed by solution NMR spectroscopy. J. Mol. Biol. 390, 775–790. doi: 10.1016/j.jmb.2009.05.066
Boyer, D. R., Li, B., Sun, C., Fan, W., Zhou, K., Hughes, M. P., et al. (2020). The α-synuclein hereditary mutation E46K unlocks a more stable, pathogenic fibril structure. Proc. Natl. Acad. Sci. U. S. A. 117, 3592–3602. doi: 10.1073/pnas.1917914117
Braak, H., and Del Tredici, K. (2009). Neuroanatomy and pathology of sporadic Parkinson’s disease. Adv. Anat. Embryol. Cell Biol. 201, 1–119. doi: 10.1007/978-3-540-79850-7_1
Braak, H., and Del Tredici, K. (2017). Neuropathological Staging of Brain Pathology in Sporadic Parkinson’s disease: separating the Wheat from the Chaff. J. Park. Dis. 7, S71–S85. doi: 10.3233/JPD-179001
Braak, H., de Vos, R. A. I., Bohl, J., and Del Tredici, K. (2006). Gastric α-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 396, 67–72. doi: 10.1016/j.neulet.2005.11.012
Braak, H., Rüb, U., Gai, W. P., and Del Tredici, K. (2003a). Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. (Vienna) 110, 517–536. doi: 10.1007/s00702-002-0808-2
Braak, H., Tredici, K. D., Rüb, U., de Vos, R. A. I., Jansen Steur, E. N. H., and Braak, E. (2003b). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. doi: 10.1016/S0197-4580(02)00065-9
Buell, A. K., Galvagnion, C., Gaspar, R., Sparr, E., Vendruscolo, M., Knowles, T. P. J., et al. (2014). Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. U. S. A. 111, 7671–7676. doi: 10.1073/pnas.1315346111
Burke, K. A., Yates, E. A., and Legleiter, J. (2013). Biophysical insights into how surfaces, including lipid membranes, modulate protein aggregation related to neurodegeneration. Front. Neurol. 4:17. doi: 10.3389/fneur.2013.00017
Burré, J., Sharma, M., and Südhof, T. C. (2012). Systematic mutagenesis of α-synuclein reveals distinct sequence requirements for physiological and pathological activities. J. Neurosci. 32, 15227–15242. doi: 10.1523/JNEUROSCI.3545-12.2012
Burré, J., Sharma, M., and Südhof, T. C. (2015). Definition of a molecular pathway mediating α-synuclein neurotoxicity. J. Neurosci. 35, 5221–5232. doi: 10.1523/JNEUROSCI.4650-14.2015
Burré, J., Sharma, M., Tsetsenis, T., Buchman, V., Etherton, M. R., and Südhof, T. C. (2010). Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667. doi: 10.1126/science.1195227
Burré, J., Vivona, S., Diao, J., Sharma, M., Brunger, A. T., and Südhof, T. C. (2013). Properties of native brain α-synuclein. Nature 498, E4–E6. doi: 10.1038/nature12125
Bussell, R., and Eliezer, D. (2004). Effects of Parkinson’s Disease-Linked Mutations on the Structure of Lipid-Associated α-Synuclein. Biochemistry 43, 4810–4818. doi: 10.1021/bi036135+
Cai, B., Liu, J., Zhao, Y., Xu, X., Bu, B., Li, D., et al. (2020). Single-vesicle imaging quantifies calcium’s regulation of nanoscale vesicle clustering mediated by α-synuclein. Microsyst. Nanoeng. 6, 1–10. doi: 10.1038/s41378-020-0147-1
Cai, C.-Z., Zhou, H.-F., Yuan, N.-N., Wu, M.-Y., Lee, S. M.-Y., Ren, J.-Y., et al. (2019). Natural alkaloid harmine promotes degradation of alpha-synuclein via PKA-mediated ubiquitin-proteasome system activation. Phytomedicine 61:152842. doi: 10.1016/j.phymed.2019.152842
Calì, T., Ottolini, D., Negro, A., and Brini, M. (2012). α-Synuclein Controls Mitochondrial Calcium Homeostasis by Enhancing Endoplasmic Reticulum-Mitochondria Interactions. J. Biol. Chem. 287, 17914–17929. doi: 10.1074/jbc.M111.302794
Camilloni, C., and Vendruscolo, M. (2013). A relationship between the aggregation rates of α-synuclein variants and the β-sheet populations in their monomeric forms. J. Phys. Chem. B 117, 10737–10741. doi: 10.1021/jp405614j
Campioni, S., Carret, G., Jordens, S., Nicoud, L., Mezzenga, R., and Riek, R. (2014). The presence of an air-water interface affects formation and elongation of α-Synuclein fibrils. J. Am. Chem. Soc. 136, 2866–2875. doi: 10.1021/ja412105t
Candelise, N., Schmitz, M., Thüne, K., Cramm, M., Rabano, A., Zafar, S., et al. (2020). Effect of the micro-environment on α-synuclein conversion and implication in seeded conversion assays. Transl. Neurodegener. 9:5. doi: 10.1186/s40035-019-0181-9
Carabotti, M., Scirocco, A., Maselli, M. A., and Severi, C. (2015). The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 28, 203–209.
Carmo-Gonçalves, P., Romão, L., and Follmer, C. (2020). In Vitro Protective Action of Monomeric and Fibrillar α-Synuclein on Neuronal Cells Exposed to the Dopaminergic Toxins Salsolinol and DOPAL. ACS Chem. Neurosci. 11, 3541–3548. doi: 10.1021/acschemneuro.0c00527
Cersosimo, M. G., and Benarroch, E. E. (2012). Pathological correlates of gastrointestinal dysfunction in Parkinson’s disease. Neurobiol. Dis. 46, 559–564. doi: 10.1016/j.nbd.2011.10.014
Chakroun, T., Evsyukov, V., Nykänen, N.-P., Höllerhage, M., Schmidt, A., Kamp, F., et al. (2020). Alpha-synuclein fragments trigger distinct aggregation pathways. Cell Death Dis. 11, 1–16. doi: 10.1038/s41419-020-2285-7
Chandra, S., Gallardo, G., Fernández-Chacón, R., Schlüter, O. M., and Südhof, T. C. (2005). Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123, 383–396. doi: 10.1016/j.cell.2005.09.028
Chang, C., Lang, H., Geng, N., Wang, J., Li, N., and Wang, X. (2013). Exosomes of BV-2 cells induced by alpha-synuclein: important mediator of neurodegeneration in PD. Neurosci. Lett. 548, 190–195. doi: 10.1016/j.neulet.2013.06.009
Chang, H. Y., Mashimo, H., and Goyal, R. K. (2003). Musings on the wanderer: what’s new in our understanding of vago-vagal reflex? IV. Current concepts of vagal efferent projections to the gut. Am. J. Physiol. Gastrointest. Liver Physiol. 284, G357–G366. doi: 10.1152/ajpgi.00478.2002
Chauhan, S., Goodwin, J. G., Chauhan, S., Manyam, G., Wang, J., Kamat, A. M., et al. (2013). ZKSCAN3 (ZNF306) is a Master Transcriptional Repressor of Autophagy. Mol. Cell 50, 16–28. doi: 10.1016/j.molcel.2013.01.024
Chavarría, C., and Souza, J. M. (2013). Oxidation and nitration of α-synuclein and their implications in neurodegenerative diseases. Arch. Biochem. Biophys. 533, 25–32. doi: 10.1016/j.abb.2013.02.009
Chen, L., and Feany, M. B. (2005). α-Synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 8, 657–663. doi: 10.1038/nn1443
Chen, S. W., Drakulic, S., Deas, E., Ouberai, M., Aprile, F. A., Arranz, R., et al. (2015). Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc. Natl. Acad. Sci. U. S. A. 112, E1994–E2003. doi: 10.1073/pnas.1421204112
Chia, S., Flagmeier, P., Habchi, J., Lattanzi, V., Linse, S., Dobson, C. M., et al. (2017). Monomeric and fibrillar α-synuclein exert opposite effects on the catalytic cycle that promotes the proliferation of Aβ42 aggregates. Proc. Natl. Acad. Sci. U. S. A. 114, 8005–8010. doi: 10.1073/pnas.1700239114
Chiang, H. L., Terlecky, S. R., Plant, C. P., and Dice, J. F. (1989). A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246, 382–385. doi: 10.1126/science.2799391
Chiang, H.-L., and Lin, C.-H. (2019). Altered gut microbiome and intestinal pathology in Parkinson’s disease. J. Mov. Disord. 12:67. doi: 10.14802/jmd.18067
Chinta, S. J., Mallajosyula, J. K., Rane, A., and Andersen, J. K. (2010). Mitochondrial α-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 486, 235–239. doi: 10.1016/j.neulet.2010.09.061
Choi, J.-S., Park, C., and Jeong, J.-W. (2010). AMP-activated protein kinase is activated in Parkinson’s disease models mediated by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem. Biophys. Res. Commun. 391, 147–151. doi: 10.1016/j.bbrc.2009.11.022
Christiansen, J. R., Olesen, M. N., Otzen, D. E., Romero-Ramos, M., and Sanchez-Guajardo, V. (2016). α-Synuclein vaccination modulates regulatory T cell activation and microglia in the absence of brain pathology. J. Neuroinflammation 13:74. doi: 10.1186/s12974-016-0532-8
Ciechanover, A., and Brundin, P. (2003). The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40, 427–446. doi: 10.1016/s0896-6273(03)00606-8
Cole, N. B., Dieuliis, D., Leo, P., Mitchell, D. C., and Nussbaum, R. L. (2008). Mitochondrial translocation of alpha-synuclein is promoted by intracellular acidification. Exp. Cell Res. 314, 2076–2089. doi: 10.1016/j.yexcr.2008.03.012
Cole, N. B., Murphy, D. D., Grider, T., Rueter, S., Brasaemle, D., and Nussbaum, R. L. (2002). Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein alpha-synuclein. J. Biol. Chem. 277, 6344–6352. doi: 10.1074/jbc.M108414200
Colla, E. (2019). Linking the Endoplasmic Reticulum to Parkinson’s Disease and Alpha-Synucleinopathy. Front. Neurosci. 13:560. doi: 10.3389/fnins.2019.00560
Colla, E., Coune, P., Liu, Y., Pletnikova, O., Troncoso, J. C., Iwatsubo, T., et al. (2012a). Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J. Neurosci. 32, 3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012
Colla, E., Jensen, P. H., Pletnikova, O., Troncoso, J. C., Glabe, C., and Lee, M. K. (2012b). Accumulation of toxic α-synuclein oligomer within endoplasmic reticulum occurs in α-synucleinopathy in vivo. J. Neurosci. 32, 3301–3305. doi: 10.1523/JNEUROSCI.5368-11.2012
Conn, K. J., Gao, W., McKee, A., Lan, M. S., Ullman, M. D., Eisenhauer, P. B., et al. (2004). Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson’s disease and Lewy body pathology. Brain Res. 1022, 164–172. doi: 10.1016/j.brainres.2004.07.026
Conway, K. A., Harper, J. D., and Lansbury, P. T. (1998). Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat. Med. 4, 1318–1320. doi: 10.1038/3311
Cooper, A. A., Gitler, A. D., Cashikar, A., Haynes, C. M., Hill, K. J., Bhullar, B., et al. (2006). α-Synuclein Blocks ER-Golgi Traffic and Rab1 Rescues Neuron Loss in Parkinson’s Models. Science 313, 324–328. doi: 10.1126/science.1129462
Cooper, J. M., Wiklander, P. O., Nordin, J. Z., Al-Shawi, R., Wood, M. J., Vithlani, M., et al. (2014). Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov. Disord. 29, 1476–1485. doi: 10.1002/mds.25978
Corrado, L., De Marchi, F., Tunesi, S., Oggioni, G. D., Carecchio, M., Magistrelli, L., et al. (2018). The Length of SNCA Rep1 Microsatellite May Influence Cognitive Evolution in Parkinson’s Disease. Front. Neurol. 9:213. doi: 10.3389/fneur.2018.00213
Cuervo, A. M., Stefanis, L., Fredenburg, R., Lansbury, P. T., and Sulzer, D. (2004). Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295. doi: 10.1126/science.1101738
da Costa, C. A., Ancolio, K., and Checler, F. (2000). Wild-type but not Parkinson’s disease-related ala-53 –> Thr mutant alpha -synuclein protects neuronal cells from apoptotic stimuli. J. Biol. Chem. 275, 24065–24069. doi: 10.1074/jbc.M002413200
Danzer, K. M., Haasen, D., Karow, A. R., Moussaud, S., Habeck, M., Giese, A., et al. (2007). Different Species of α-Synuclein Oligomers Induce Calcium Influx and Seeding. J. Neurosci. 27, 9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007
Danzer, K. M., Kranich, L. R., Ruf, W. P., Cagsal-Getkin, O., Winslow, A. R., Zhu, L., et al. (2012). Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 7:42. doi: 10.1186/1750-1326-7-42
Danzer, K. M., Ruf, W. P., Putcha, P., Joyner, D., Hashimoto, T., Glabe, C., et al. (2011). Heat-shock protein 70 modulates toxic extracellular α-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 25, 326–336. doi: 10.1096/fj.10-164624
Darios, F., Ruipérez, V., López, I., Villanueva, J., Gutierrez, L. M., and Davletov, B. (2010). Alpha-synuclein sequesters arachidonic acid to modulate SNARE-mediated exocytosis. EMBO Rep. 11, 528–533. doi: 10.1038/embor.2010.66
Dauer, W., Kholodilov, N., Vila, M., Trillat, A.-C., Goodchild, R., Larsen, K. E., et al. (2002). Resistance of α-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. U. S. A. 99, 14524–14529. doi: 10.1073/pnas.172514599
de Oliveira, G. A. P., and Silva, J. L. (2019). Alpha-synuclein stepwise aggregation reveals features of an early onset mutation in Parkinson’s disease. Commun. Biol. 2, 1–13. doi: 10.1038/s42003-019-0598-9
Decressac, M., Mattsson, B., Weikop, P., Lundblad, M., Jakobsson, J., and Björklund, A. (2013). TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl. Acad. Sci. U. S. A. 110, E1817–E1826. doi: 10.1073/pnas.1305623110
Del Tredici, K., and Braak, H. (2013). Dysfunction of the locus coeruleus-norepinephrine system and related circuitry in Parkinson’s disease-related dementia. J. Neurol. Neurosurg. Psychiatry 84, 774–783. doi: 10.1136/jnnp-2011-301817
Del Tredici, K., Rüb, U., De Vos, R. A. I., Bohl, J. R. E., and Braak, H. (2002). Where does parkinson disease pathology begin in the brain? J. Neuropathol. Exp. Neurol. 61, 413–426. doi: 10.1093/jnen/61.5.413
Desplats, P., Lee, H.-J., Bae, E.-J., Patrick, C., Rockenstein, E., Crews, L., et al. (2009). Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. U. S. A. 106, 13010–13015. doi: 10.1073/pnas.0903691106
Dettmer, U., Newman, A. J., Luth, E. S., Bartels, T., and Selkoe, D. (2013). In vivo cross-linking reveals principally oligomeric forms of α-synuclein and β-synuclein in neurons and non-neural cells. J. Biol. Chem. 288, 6371–6385. doi: 10.1074/jbc.M112.403311
Dettmer, U., Newman, A. J., von Saucken, V. E., Bartels, T., and Selkoe, D. (2015). KTKEGV repeat motifs are key mediators of normal α-synuclein tetramerization: their mutation causes excess monomers and neurotoxicity. Proc. Natl. Acad. Sci. U. S. A. 112, 9596–9601. doi: 10.1073/pnas.1505953112
Devi, L., Raghavendran, V., Prabhu, B. M., Avadhani, N. G., and Anandatheerthavarada, H. K. (2008). Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283, 9089–9100. doi: 10.1074/jbc.M710012200
Dickson, D. W. (2012). Parkinson’s Disease and Parkinsonism: neuropathology. Cold Spring Harb. Perspect. Med. 2:a009258. doi: 10.1101/cshperspect.a009258
Dieriks, B. V., Park, T. I.-H., Fourie, C., Faull, R. L. M., Dragunow, M., and Curtis, M. A. (2017). α-synuclein transfer through tunneling nanotubes occurs in SH-SY5Y cells and primary brain pericytes from Parkinson’s disease patients. Sci. Rep. 7:42984. doi: 10.1038/srep42984
Dobbs, R. J., Charlett, A., Purkiss, A. G., Dobbs, S. M., Weller, C., and Peterson, D. W. (1999). Association of circulating TNF-alpha and IL-6 with ageing and parkinsonism. Acta Neurol. Scand. 100, 34–41. doi: 10.1111/j.1600-0404.1999.tb00721.x
Domert, J., Sackmann, C., Severinsson, E., Agholme, L., Bergström, J., Ingelsson, M., et al. (2016). Aggregated Alpha-Synuclein Transfer Efficiently between Cultured Human Neuron-Like Cells and Localize to Lysosomes. PLoS One 11:e0168700. doi: 10.1371/journal.pone.0168700
Dulovic, M., Jovanovic, M., Xilouri, M., Stefanis, L., Harhaji-Trajkovic, L., Kravic-Stevovic, T., et al. (2014). The protective role of AMP-activated protein kinase in alpha-synuclein neurotoxicity in vitro. Neurobiol. Dis. 63, 1–11. doi: 10.1016/j.nbd.2013.11.002
Dyson, H. J., and Wright, P. E. (2005). Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208. doi: 10.1038/nrm1589
Dzamko, N., Zhou, J., Huang, Y., and Halliday, G. M. (2014). Parkinson’s disease-implicated kinases in the brain; insights into disease pathogenesis. Front. Mol. Neurosci. 7:57. doi: 10.3389/fnmol.2014.00057
Edwards, L. L., Quigley, E. M., and Pfeiffer, R. F. (1992). Gastrointestinal dysfunction in Parkinson’s disease: frequency and pathophysiology. Neurology 42, 726–732. doi: 10.1212/wnl.42.4.726
Egan, D. F., Shackelford, D. B., Mihaylova, M. M., Gelino, S., Kohnz, R. A., Mair, W., et al. (2011). Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461. doi: 10.1126/science.1196371
Ellis, C. E., Murphy, E. J., Mitchell, D. C., Golovko, M. Y., Scaglia, F., Barceló-Coblijn, G. C., et al. (2005). Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein. Mol. Cell. Biol. 25, 10190–10201. doi: 10.1128/MCB.25.22.10190-10201.2005
Emelyanov, A., Boukina, T., Yakimovskii, A., Usenko, T., Drosdova, A., Zakharchuk, A., et al. (2012). Glucocerebrosidase gene mutations are associated with Parkinson’s disease in Russia. Mov. Disord. 27, 158–159. doi: 10.1002/mds.23950
Emmanouilidou, E., Melachroinou, K., Roumeliotis, T., Garbis, S. D., Ntzouni, M., Margaritis, L. H., et al. (2010). Cell-Produced α-Synuclein Is Secreted in a Calcium-Dependent Manner by Exosomes and Impacts Neuronal Survival. J. Neurosci. 30, 6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010
Erskine, D., Patterson, L., Alexandris, A., Hanson, P. S., McKeith, I. G., Attems, J., et al. (2018). Regional levels of physiological α-synuclein are directly associated with Lewy body pathology. Acta Neuropathol. 135, 153–154. doi: 10.1007/s00401-017-1787-6
Fares, M.-B., Ait-Bouziad, N., Dikiy, I., Mbefo, M. K., Jovièiæ, A., Kiely, A., et al. (2014). The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet. 23, 4491–4509. doi: 10.1093/hmg/ddu165
Fields, C. R., Bengoa-Vergniory, N., and Wade-Martins, R. (2019). Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 12:299. doi: 10.3389/fnmol.2019.00299
Filippini, A., Mutti, V., Faustini, G., Longhena, F., Ramazzina, I., Rizzi, F., et al. (2021). Extracellular clusterin limits the uptake of α−synuclein fibrils by murine and human astrocytes. Glia 69, 681–696. doi: 10.1002/glia.23920
Finkelstein, D. I., Billings, J. L., Adlard, P. A., Ayton, S., Sedjahtera, A., Masters, C. L., et al. (2017). The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol. Commun. 5:53. doi: 10.1186/s40478-017-0456-2
Fitzpatrick, A. W. P., Debelouchina, G. T., Bayro, M. J., Clare, D. K., Caporini, M. A., Bajaj, V. S., et al. (2013). Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc. Natl. Acad. Sci. U. S. A. 110, 5468–5473. doi: 10.1073/pnas.1219476110
Flagmeier, P., Meisl, G., Vendruscolo, M., Knowles, T. P. J., Dobson, C. M., Buell, A. K., et al. (2016). Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of α-synuclein aggregation. Proc. Natl. Acad. Sci. U. S. A. 113, 10328–10333. doi: 10.1073/pnas.1604645113
Fonseca-Ornelas, L., Eisbach, S. E., Paulat, M., Giller, K., Fernández, C. O., Outeiro, T. F., et al. (2014). Small molecule-mediated stabilization of vesicle-associated helical α-synuclein inhibits pathogenic misfolding and aggregation. Nat. Commun. 5:5857. doi: 10.1038/ncomms6857
Forsyth, C. B., Shannon, K. M., Kordower, J. H., Voigt, R. M., Shaikh, M., Jaglin, J. A., et al. (2011). Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS One 6:e28032. doi: 10.1371/journal.pone.0028032
Forsythe, P., Bienenstock, J., and Kunze, W. A. (2014). Vagal pathways for microbiome-brain-gut axis communication. Adv. Exp. Med. Biol. 817, 115–133. doi: 10.1007/978-1-4939-0897-4_5
Fouillet, A., Levet, C., Virgone, A., Robin, M., Dourlen, P., Rieusset, J., et al. (2012). ER stress inhibits neuronal death by promoting autophagy. Autophagy 8, 915–926. doi: 10.4161/auto.19716
Fountaine, T. M., and Wade-Martins, R. (2007). RNA interference-mediated knockdown of alpha-synuclein protects human dopaminergic neuroblastoma cells from MPP(+) toxicity and reduces dopamine transport. J. Neurosci. Res. 85, 351–363. doi: 10.1002/jnr.21125
Fountaine, T. M., Venda, L. L., Warrick, N., Christian, H. C., Brundin, P., Channon, K. M., et al. (2008). The effect of alpha-synuclein knockdown on MPP+ toxicity in models of human neurons. Eur. J. Neurosci. 28, 2459–2473. doi: 10.1111/j.1460-9568.2008.06527.x
Fredenburg, R. A., Rospigliosi, C., Meray, R. K., Kessler, J. C., Lashuel, H. A., Eliezer, D., et al. (2007). The impact of the E46K mutation on the properties of alpha-synuclein in its monomeric and oligomeric states. Biochemistry 46, 7107–7118. doi: 10.1021/bi7000246
Freundt, E. C., Maynard, N., Clancy, E. K., Roy, S., Bousset, L., Sourigues, Y., et al. (2012). Neuron-to-neuron transmission of α-synuclein fibrils through axonal transport. Ann. Neurol. 72, 517–524. doi: 10.1002/ana.23747
Fujiwara, H., Hasegawa, M., Dohmae, N., Kawashima, A., Masliah, E., Goldberg, M. S., et al. (2002). alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160–164. doi: 10.1038/ncb748
Gabr, M. T., and Peccati, F. (2020). Dual Targeting of Monomeric Tau and α-Synuclein Aggregation: a New Multitarget Therapeutic Strategy for Neurodegeneration. ACS Chem. Neurosci. 11, 2051–2057. doi: 10.1021/acschemneuro.0c00281
Galli, E., Planken, A., Kadastik-Eerme, L., Saarma, M., Taba, P., and Lindholm, P. (2019). Increased Serum Levels of Mesencephalic Astrocyte-Derived Neurotrophic Factor in Subjects With Parkinson’s Disease. Front. Neurosci. 13:929. doi: 10.3389/fnins.2019.00929
Galvagnion, C., Buell, A. K., Meisl, G., Michaels, T. C. T., Vendruscolo, M., Knowles, T. P. J., et al. (2015). Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol. 11, 229–234. doi: 10.1038/nchembio.1750
Games, D., Valera, E., Spencer, B., Rockenstein, E., Mante, M., Adame, A., et al. (2014). Reducing C-Terminal-Truncated Alpha-Synuclein by Immunotherapy Attenuates Neurodegeneration and Propagation in Parkinson’s Disease-Like Models. J. Neurosci. 34, 9441–9454. doi: 10.1523/JNEUROSCI.5314-13.2014
Ganjam, G. K., Bolte, K., Matschke, L. A., Neitemeier, S., Dolga, A. M., Höllerhage, M., et al. (2019). Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis. 10, 1–16. doi: 10.1038/s41419-019-2091-2
Gao, J., Perera, G., Bhadbhade, M., Halliday, G. M., and Dzamko, N. (2019). Autophagy activation promotes clearance of α-synuclein inclusions in fibril-seeded human neural cells. J. Biol. Chem. 294, 14241–14256. doi: 10.1074/jbc.RA119.008733
Gaugler, M. N., Genc, O., Bobela, W., Mohanna, S., Ardah, M. T., El-Agnaf, O. M., et al. (2012). Nigrostriatal overabundance of α-synuclein leads to decreased vesicle density and deficits in dopamine release that correlate with reduced motor activity. Acta Neuropathol. 123, 653–669. doi: 10.1007/s00401-012-0963-y
Gegg, M. E., Verona, G., and Schapira, A. H. V. (2020). Glucocerebrosidase deficiency promotes release of α-synuclein fibrils from cultured neurons. Hum. Mol. Genet. 29, 1716–1728. doi: 10.1093/hmg/ddaa085
George, S., Rey, N. L., Tyson, T., Esquibel, C., Meyerdirk, L., Schulz, E., et al. (2019). Microglia affect α-synuclein cell-to-cell transfer in a mouse model of Parkinson’s disease. Mol. Neurodegener. 14:34. doi: 10.1186/s13024-019-0335-3
Ghaisas, S., Langley, M. R., Palanisamy, B. N., Dutta, S., Narayanaswamy, K., Plummer, P. J., et al. (2019). MitoPark transgenic mouse model recapitulates the gastrointestinal dysfunction and gut-microbiome changes of Parkinson’s disease. NeuroToxicology 75, 186–199. doi: 10.1016/j.neuro.2019.09.004
Ghavidel, A., Baxi, K., Ignatchenko, V., Prusinkiewicz, M., Arnason, T. G., Kislinger, T., et al. (2015). A Genome Scale Screen for Mutants with Delayed Exit from Mitosis: ire1-Independent Induction of Autophagy Integrates ER Homeostasis into Mitotic Lifespan. PLoS Genet. 11:e1005429. doi: 10.1371/journal.pgen.1005429
Ghochikyan, A., Petrushina, I., Davtyan, H., Hovakimyan, A., Saing, T., Davtyan, A., et al. (2014). Immunogenicity of epitope vaccines targeting different B cell antigenic determinants of human α-Synuclein: feasibility study. Neurosci. Lett. 560, 86–91. doi: 10.1016/j.neulet.2013.12.028
Ghosh, D., Sahay, S., Ranjan, P., Salot, S., Mohite, G. M., Singh, P. K., et al. (2014). The Newly Discovered Parkinson’s Disease Associated Finnish Mutation (A53E) Attenuates α-Synuclein Aggregation and Membrane Binding. Biochemistry 53, 6419–6421. doi: 10.1021/bi5010365
Giasson, B. I., Duda, J. E., Murray, I. V., Chen, Q., Souza, J. M., Hurtig, H. I., et al. (2000). Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290, 985–989. doi: 10.1126/science.290.5493.985
Glaser, C. B., Yamin, G., Uversky, V. N., and Fink, A. L. (2005). Methionine oxidation, alpha-synuclein and Parkinson’s disease. Biochim. Biophys. Acta 1703, 157–169. doi: 10.1016/j.bbapap.2004.10.008
Golbe, L. I., Di Iorio, G., Bonavita, V., Miller, D. C., and Duvoisin, R. C. (1990). A large kindred with autosomal dominant Parkinson’s disease. Ann. Neurol. 27, 276–282. doi: 10.1002/ana.410270309
Gold, A., Turkalp, Z. T., and Munoz, D. G. (2013). Enteric alpha-synuclein expression is increased in Parkinson’s disease but not Alzheimer’s disease. Mov. Disord. 28, 237–241. doi: 10.1002/mds.25298
Gómez-Tortosa, E., Sanders, J. L., Newell, K., and Hyman, B. T. (2001). Cortical neurons expressing calcium binding proteins are spared in dementia with Lewy bodies. Acta Neuropathol. 101, 36–42. doi: 10.1007/s004010000270
Good, P. F., Hsu, A., Werner, P., Perl, D. P., and Olanow, C. W. (1998). Protein nitration in Parkinson’s disease. J. Neuropathol. Exp. Neurol. 57, 338–342. doi: 10.1097/00005072-199804000-00006
Gorbatyuk, O. S., Li, S., Sullivan, L. F., Chen, W., Kondrikova, G., Manfredsson, F. P., et al. (2008). The phosphorylation state of Ser-129 in human α-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc. Natl. Acad. Sci. U. S. A. 105, 763–768. doi: 10.1073/pnas.0711053105
Gorenberg, E. L., and Chandra, S. S. (2017). The Role of Co-chaperones in Synaptic Proteostasis and Neurodegenerative Disease. Front. Neurosci. 11:248. doi: 10.3389/fnins.2017.00248
Gosavi, N., Lee, H.-J., Lee, J. S., Patel, S., and Lee, S.-J. (2002). Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J. Biol. Chem. 277, 48984–48992. doi: 10.1074/jbc.M208194200
Gousset, K., and Zurzolo, C. (2009). Tunnelling nanotubes. Prion 3, 94–98. doi: 10.4161/pri.3.2.8917
Greten-Harrison, B., Polydoro, M., Morimoto-Tomita, M., Diao, L., Williams, A. M., Nie, E. H., et al. (2010). αβγ-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. U. S. A. 107, 19573–19578. doi: 10.1073/pnas.1005005107
Guan, J., Pavlovic, D., Dalkie, N., Waldvogel, H. J., O’Carroll, S. J., Green, C. R., et al. (2013). Vascular Degeneration in Parkinson’s Disease. Brain Pathol. 23, 154–164. doi: 10.1111/j.1750-3639.2012.00628.x
Guardia-Laguarta, C., Area-Gomez, E., Rüb, C., Liu, Y., Magrané, J., Becker, D., et al. (2014). α-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 34, 249–259. doi: 10.1523/JNEUROSCI.2507-13.2014
Gustafsson, G., Eriksson, F., Möller, C., da Fonseca, T. L., Outeiro, T. F., Lannfelt, L., et al. (2017). Cellular Uptake of α-Synuclein Oligomer-Selective Antibodies is Enhanced by the Extracellular Presence of α-Synuclein and Mediated via Fcγ Receptors. Cell. Mol. Neurobiol. 37, 121–131. doi: 10.1007/s10571-016-0352-5
Gwinn, D. M., Shackelford, D. B., Egan, D. F., Mihaylova, M. M., Mery, A., Vasquez, D. S., et al. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226. doi: 10.1016/j.molcel.2008.03.003
Hall, H., Reyes, S., Landeck, N., Bye, C., Leanza, G., Double, K., et al. (2014). Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson’s disease. Brain J. Neurol. 137, 2493–2508. doi: 10.1093/brain/awu193
Hart, G. W., Housley, M. P., and Slawson, C. (2007). Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 446, 1017–1022. doi: 10.1038/nature05815
Hasegawa, M., Fujiwara, H., Nonaka, T., Wakabayashi, K., Takahashi, H., Lee, V. M.-Y., et al. (2002). Phosphorylated α-Synuclein Is Ubiquitinated in α-Synucleinopathy Lesions. J. Biol. Chem. 277, 49071–49076. doi: 10.1074/jbc.M208046200
Hashimoto, M., and Masliah, E. (1999). Alpha-synuclein in Lewy body disease and Alzheimer’s disease. Brain Pathol. 9, 707–720. doi: 10.1111/j.1750-3639.1999.tb00552.x
Hashimoto, M., Hsu, L. J., Xia, Y., Takeda, A., Sisk, A., Sundsmo, M., et al. (1999). Oxidative stress induces amyloid-like aggregate formation of NACP/alpha-synuclein in vitro. Neuroreport 10, 717–721. doi: 10.1097/00001756-199903170-00011
Hashimoto, M., Rockenstein, E., Mante, M., Mallory, M., and Masliah, E. (2001). beta-Synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron 32, 213–223. doi: 10.1016/s0896-6273(01)00462-7
Hawkes, C. H., Del Tredici, K., and Braak, H. (2007). Parkinson’s disease: a dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 33, 599–614. doi: 10.1111/j.1365-2990.2007.00874.x
Hawkes, C. H., Del Tredici, K., and Braak, H. (2009). Parkinson’s disease: the dual hit theory revisited. Ann. N. Y. Acad. Sci. 1170, 615–622. doi: 10.1111/j.1749-6632.2009.04365.x
Hawkes, C. H., Del Tredici, K., and Braak, H. (2010). A timeline for Parkinson’s disease. Parkinsonism Relat. Disord. 16, 79–84. doi: 10.1016/j.parkreldis.2009.08.007
Hijaz, B. A., and Volpicelli-Daley, L. A. (2020). Initiation and propagation of α-synuclein aggregation in the nervous system. Mol. Neurodegener. 15:19. doi: 10.1186/s13024-020-00368-6
Hoffman-Zacharska, D., Koziorowski, D., Ross, O. A., Milewski, M., Poznanski, J. A., Jurek, M., et al. (2013). Novel A18T and pA29S substitutions in α-synuclein may be associated with sporadic Parkinson’s disease. Parkinsonism Relat. Disord. 19, 1057–1060. doi: 10.1016/j.parkreldis.2013.07.011
Hokenson, M. J., Uversky, V. N., Goers, J., Yamin, G., Munishkina, L. A., and Fink, A. L. (2004). Role of individual methionines in the fibrillation of methionine-oxidized alpha-synuclein. Biochemistry 43, 4621–4633. doi: 10.1021/bi049979h
Holmqvist, S., Chutna, O., Bousset, L., Aldrin-Kirk, P., Li, W., Björklund, T., et al. (2014). Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 128, 805–820. doi: 10.1007/s00401-014-1343-6
Hoozemans, J. J. M., van Haastert, E. S., Eikelenboom, P., de Vos, R. A. I., Rozemuller, J. M., and Scheper, W. (2007). Activation of the unfolded protein response in Parkinson’s disease. Biochem. Biophys. Res. Commun. 354, 707–711. doi: 10.1016/j.bbrc.2007.01.043
Hsu, L. J., Sagara, Y., Arroyo, A., Rockenstein, E., Sisk, A., Mallory, M., et al. (2000). alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 157, 401–410. doi: 10.1016/s0002-9440(10)64553-1
Hu, F.-Y., Xi, J., Guo, J., Yu, L.-H., Liu, L., He, X.-H., et al. (2010). Association of the glucocerebrosidase N370S allele with Parkinson’s disease in two separate Chinese Han populations of mainland China. Eur. J. Neurol. 17, 1476–1478. doi: 10.1111/j.1468-1331.2010.03097.x
Hua, G., Xiaolei, L., Weiwei, Y., Hao, W., Yuangang, Z., Dongmei, L., et al. (2015). Protein Phosphatase 2A is Involved in the Tyrosine Hydroxylase Phosphorylation Regulated by α-Synuclein. Neurochem. Res. 40, 428–437. doi: 10.1007/s11064-014-1477-x
Ikeuchi, T., Kakita, A., Shiga, A., Kasuga, K., Kaneko, H., Tan, C.-F., et al. (2008). Patients homozygous and heterozygous for SNCA duplication in a family with parkinsonism and dementia. Arch. Neurol. 65, 514–519. doi: 10.1001/archneur.65.4.514
Inoki, K., Zhu, T., and Guan, K.-L. (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. doi: 10.1016/s0092-8674(03)00929-2
Iwai, A., Masliah, E., Yoshimoto, M., Ge, N., Flanagan, L., de Silva, H. A., et al. (1995). The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 14, 467–475. doi: 10.1016/0896-6273(95)90302-x
Jakes, R., Spillantini, M. G., and Goedert, M. (1994). Identification of two distinct synucleins from human brain. FEBS Lett. 345, 27–32. doi: 10.1016/0014-5793(94)00395-5
Jang, A., Lee, H.-J., Suk, J.-E., Jung, J.-W., Kim, K.-P., and Lee, S.-J. (2010). Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 113, 1263–1274. doi: 10.1111/j.1471-4159.2010.06695.x
Jang, J.-H., Yeom, M.-J., Ahn, S., Oh, J.-Y., Ji, S., Kim, T.-H., et al. (2020). Acupuncture inhibits neuroinflammation and gut microbial dysbiosis in a mouse model of Parkinson’s disease. Brain. Behav. Immun. 89, 641–655. doi: 10.1016/j.bbi.2020.08.015
Jankovic, J., Goodman, I., Safirstein, B., Marmon, T. K., Schenk, D. B., Koller, M., et al. (2018). Safety and Tolerability of Multiple Ascending Doses of PRX002/RG7935, an Anti-α-Synuclein Monoclonal Antibody, in Patients With Parkinson Disease: a Randomized Clinical Trial. JAMA Neurol. 75, 1206–1214. doi: 10.1001/jamaneurol.2018.1487
Jao, C. C., Hegde, B. G., Chen, J., Haworth, I. S., and Langen, R. (2008). Structure of membrane-bound α-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. U. S. A. 105, 19666–19671. doi: 10.1073/pnas.0807826105
Jellinger, K. A. (2003). Neuropathological spectrum of synucleinopathies. Mov. Disord. 18, S2–S12. doi: 10.1002/mds.10557
Jellinger, K. A., and Korczyn, A. D. (2018). Are dementia with Lewy bodies and Parkinson’s disease dementia the same disease? BMC Med. 16:34. doi: 10.1186/s12916-018-1016-8
Jensen, P. H., Li, J. Y., Dahlström, A., and Dotti, C. G. (1999). Axonal transport of synucleins is mediated by all rate components. Eur. J. Neurosci. 11, 3369–3376. doi: 10.1046/j.1460-9568.1999.00754.x
Jensen, P. H., Nielsen, M. S., Jakes, R., Dotti, C. G., and Goedert, M. (1998). Binding of α-Synuclein to Brain Vesicles Is Abolished by Familial Parkinson’s Disease Mutation. J. Biol. Chem. 273, 26292–26294. doi: 10.1074/jbc.273.41.26292
Jiang, P., Gan, M., Ebrahim, A. S., Castanedes-Casey, M., Dickson, D. W., and Yen, S.-H. C. (2013). Adenosine monophosphate-activated protein kinase overactivation leads to accumulation of α-synuclein oligomers and decrease of neurites. Neurobiol. Aging 34, 1504–1515. doi: 10.1016/j.neurobiolaging.2012.11.001
Kamp, F., Exner, N., Lutz, A. K., Wender, N., Hegermann, J., Brunner, B., et al. (2010). Inhibition of mitochondrial fusion by α−synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 29, 3571–3589. doi: 10.1038/emboj.2010.223
Kanaan, N. M., and Manfredsson, F. P. (2012). Loss of functional alpha-synuclein: a toxic event in Parkinson’s disease? J. Park. Dis. 2, 249–267. doi: 10.3233/JPD-012138
Kang, L., Wu, K.-P., Vendruscolo, M., and Baum, J. (2011). The A53T mutation is key in defining the differences in the aggregation kinetics of human and mouse α-synuclein. J. Am. Chem. Soc. 133, 13465–13470. doi: 10.1021/ja203979j
Karampetsou, M., Ardah, M. T., Semitekolou, M., Polissidis, A., Samiotaki, M., Kalomoiri, M., et al. (2017). Phosphorylated exogenous alpha-synuclein fibrils exacerbate pathology and induce neuronal dysfunction in mice. Sci. Rep. 7:16533. doi: 10.1038/s41598-017-15813-8
Kasai, T., Tokuda, T., Yamaguchi, N., Watanabe, Y., Kametani, F., Nakagawa, M., et al. (2008). Cleavage of normal and pathological forms of alpha-synuclein by neurosin in vitro. Neurosci. Lett. 436, 52–56. doi: 10.1016/j.neulet.2008.02.057
Kay, S., Josefine, M., Siswanto, S., Reijko, K., Helmut, H., Georg, A., et al. (2015). The brainstem pathologies of Parkinson’s disease and dementia with Lewy bodies. Brain Pathol. 25, 121–135. doi: 10.1111/bpa.12168
Khalaf, O., Fauvet, B., Oueslati, A., Dikiy, I., Mahul-Mellier, A.-L., Ruggeri, F. S., et al. (2014). The H50Q mutation enhances α-synuclein aggregation, secretion, and toxicity. J. Biol. Chem. 289, 21856–21876. doi: 10.1074/jbc.M114.553297
Kieburtz, K., and Wunderle, K. B. (2013). Parkinson’s disease: evidence for environmental risk factors. Mov. Disord. 28, 8–13. doi: 10.1002/mds.25150
Kiechle, M., von Einem, B., Höfs, L., Voehringer, P., Grozdanov, V., Markx, D., et al. (2019). In Vivo Protein Complementation Demonstrates Presynaptic α-Synuclein Oligomerization and Age-Dependent Accumulation of 8–16-mer Oligomer Species. Cell Rep. 29, 2862–2874.e9. doi: 10.1016/j.celrep.2019.10.089
Kim, J., Kundu, M., Viollet, B., and Guan, K.-L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Kim, S., Kwon, S.-H., Kam, T.-I., Panicker, N., Karuppagounder, S. S., Lee, S., et al. (2019). Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 103, 627–641.e7. doi: 10.1016/j.neuron.2019.05.035
Kim, T. W., Cho, H. M., Choi, S. Y., Suguira, Y., Hayasaka, T., Setou, M., et al. (2013). (ADP-ribose) polymerase 1 and AMP-activated protein kinase mediate progressive dopaminergic neuronal degeneration in a mouse model of Parkinson’s disease. Cell Death Dis. 4:e919. doi: 10.1038/cddis.2013.447
Kim, W. S., Kågedal, K., and Halliday, G. M. (2014). Alpha-synuclein biology in Lewy body diseases. Alzheimers Res. Ther. 6:73. doi: 10.1186/s13195-014-0073-2
Kim, Y.-C., Miller, A., Lins, L. C. R. F., Han, S.-W., Keiser, M. S., Boudreau, R. L., et al. (2017). RNA Interference of Human α-Synuclein in Mouse. Front. Neurol. 8:13. doi: 10.3389/fneur.2017.00013
Klingelhoefer, L., and Reichmann, H. (2015). Pathogenesis of Parkinson disease–the gut-brain axis and environmental factors. Nat. Rev. Neurol. 11, 625–636. doi: 10.1038/nrneurol.2015.197
Klucken, J., Shin, Y., Masliah, E., Hyman, B. T., and McLean, P. J. (2004). Hsp70 Reduces alpha-Synuclein Aggregation and Toxicity. J. Biol. Chem. 279, 25497–25502. doi: 10.1074/jbc.M400255200
Krüger, R., Kuhn, W., Müller, T., Woitalla, D., Graeber, M., Kösel, S., et al. (1998). Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108. doi: 10.1038/ng0298-106
Kujawska, M., and Jodynis-Liebert, J. (2018). What Is the Evidence that Parkinson’s Disease Is a Prion Disorder, Which Originates in the Gut? Int. J. Mol. Sci. 19:3573. doi: 10.3390/ijms19113573
Kurochka, A. S., Yushchenko, D. A., Bouř, P., and Shvadchak, V. V. (2021). Influence of Lipid Membranes on α-Synuclein Aggregation. ACS Chem. Neurosci. 12, 825–830. doi: 10.1021/acschemneuro.0c00819
Kuwahara, T., Tonegawa, R., Ito, G., Mitani, S., and Iwatsubo, T. (2012). Phosphorylation of α-synuclein protein at Ser-129 reduces neuronal dysfunction by lowering its membrane binding property in Caenorhabditis elegans. J. Biol. Chem. 287, 7098–7109. doi: 10.1074/jbc.M111.237131
La Cognata, V., Morello, G., D’Agata, V., and Cavallaro, S. (2017). Copy number variability in Parkinson’s disease: assembling the puzzle through a systems biology approach. Hum. Genet. 136, 13–37. doi: 10.1007/s00439-016-1749-4
Lai, F., Jiang, R., Xie, W., Liu, X., Tang, Y., Xiao, H., et al. (2018). Intestinal Pathology and Gut Microbiota Alterations in a Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Mouse Model of Parkinson’s Disease. Neurochem. Res. 43, 1986–1999. doi: 10.1007/s11064-018-2620-x
Larsen, K. E., Schmitz, Y., Troyer, M. D., Mosharov, E., Dietrich, P., Quazi, A. Z., et al. (2006). Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci. 26, 11915–11922. doi: 10.1523/JNEUROSCI.3821-06.2006
Lashuel, H. A., and Lansbury, P. T. (2006). Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Q. Rev. Biophys. 39, 167–201. doi: 10.1017/S0033583506004422
Lashuel, H. A., Overk, C. R., Oueslati, A., and Masliah, E. (2013). The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 14, 38–48. doi: 10.1038/nrn3406
Lázaro, D. F., Rodrigues, E. F., Langohr, R., Shahpasandzadeh, H., Ribeiro, T., Guerreiro, P., et al. (2014). Systematic Comparison of the Effects of Alpha-synuclein Mutations on Its Oligomerization and Aggregation. PLoS Genet. 10:e1004741. doi: 10.1371/journal.pgen.1004741
Leal, R. B., Sim, A. T., Gonçalves, C. A., and Dunkley, P. R. (2002). Tyrosine hydroxylase dephosphorylation by protein phosphatase 2A in bovine adrenal chromaffin cells. Neurochem. Res. 27, 207–213.
Lee, F. J., Liu, F., Pristupa, Z. B., and Niznik, H. B. (2001). Direct binding and functional coupling of alpha-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J. 15, 916–926. doi: 10.1096/fj.00-0334com
Lee, H.-J., Choi, C., and Lee, S.-J. (2002a). Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J. Biol. Chem. 277, 671–678. doi: 10.1074/jbc.M107045200
Lee, H.-J., Khoshaghideh, F., Patel, S., and Lee, S.-J. (2004). Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J. Neurosci. 24, 1888–1896. doi: 10.1523/JNEUROSCI.3809-03.2004
Lee, H.-J., Patel, S., and Lee, S.-J. (2005). Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 25, 6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005
Lee, H.-J., Shin, S. Y., Choi, C., Lee, Y. H., and Lee, S.-J. (2002b). Formation and removal of alpha-synuclein aggregates in cells exposed to mitochondrial inhibitors. J. Biol. Chem. 277, 5411–5417. doi: 10.1074/jbc.M105326200
Lee, H.-J., Suk, J.-E., Bae, E.-J., and Lee, S.-J. (2008). Clearance and deposition of extracellular α-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 372, 423–428. doi: 10.1016/j.bbrc.2008.05.045
Lee, S.-J. (2003). alpha-synuclein aggregation: a link between mitochondrial defects and Parkinson’s disease? Antioxid. Redox Signal. 5, 337–348. doi: 10.1089/152308603322110904
Lehtonen, Š, Sonninen, T.-M., Wojciechowski, S., Goldsteins, G., and Koistinaho, J. (2019). Dysfunction of Cellular Proteostasis in Parkinson’s Disease. Front. Neurosci. 13:457. doi: 10.3389/fnins.2019.00457
Lei, Z., Cao, G., and Wei, G. (2019). A30P mutant α-synuclein impairs autophagic flux by inactivating JNK signaling to enhance ZKSCAN3 activity in midbrain dopaminergic neurons. Cell Death Dis. 10:133. doi: 10.1038/s41419-019-1364-0
Lesage, S., Anheim, M., Letournel, F., Bousset, L., Honoré, A., Rozas, N., et al. (2013). G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 73, 459–471. doi: 10.1002/ana.23894
Levine, P. M., Galesic, A., Balana, A. T., Mahul-Mellier, A.-L., Navarro, M. X., De Leon, C. A., et al. (2019). α-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease. Proc. Natl. Acad. Sci. U. S. A. 116, 1511–1519. doi: 10.1073/pnas.1808845116
Lewis, J., Melrose, H., Bumcrot, D., Hope, A., Zehr, C., Lincoln, S., et al. (2008). In vivo silencing of alpha-synuclein using naked siRNA. Mol. Neurodegener. 3:19. doi: 10.1186/1750-1326-3-19
Lewis, Y. E., Galesic, A., Levine, P. M., De Leon, C. A., Lamiri, N., Brennan, C. K., et al. (2017). O-GlcNAcylation of α-Synuclein at Serine 87 Reduces Aggregation without Affecting Membrane Binding. ACS Chem. Biol. 12, 1020–1027. doi: 10.1021/acschembio.7b00113
Li, H.-T., Du, H.-N., Tang, L., Hu, J., and Hu, H.-Y. (2002). Structural transformation and aggregation of human alpha-synuclein in trifluoroethanol: non-amyloid component sequence is essential and beta-sheet formation is prerequisite to aggregation. Biopolymers 64, 221–226. doi: 10.1002/bip.10179
Li, W.-W., Yang, R., Guo, J.-C., Ren, H.-M., Zha, X.-L., Cheng, J.-S., et al. (2007). Localization of alpha-synuclein to mitochondria within midbrain of mice. Neuroreport 18, 1543–1546. doi: 10.1097/WNR.0b013e3282f03db4
Lin, K.-J., Lin, K.-L., Chen, S.-D., Liou, C.-W., Chuang, Y.-C., Lin, H.-Y., et al. (2019). The Overcrowded Crossroads: mitochondria, Alpha-Synuclein, and the Endo-Lysosomal System Interaction in Parkinson’s Disease. Int. J. Mol. Sci. 20:5312. doi: 10.3390/ijms20215312
Lin, W.-L., DeLucia, M. W., and Dickson, D. W. (2004). Alpha-synuclein immunoreactivity in neuronal nuclear inclusions and neurites in multiple system atrophy. Neurosci. Lett. 354, 99–102. doi: 10.1016/j.neulet.2003.09.075
Lindström, V., Fagerqvist, T., Nordström, E., Eriksson, F., Lord, A., Tucker, S., et al. (2014). Immunotherapy targeting α-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] α-synuclein mice. Neurobiol. Dis. 69, 134–143. doi: 10.1016/j.nbd.2014.05.009
Liu, C.-W., Corboy, M. J., DeMartino, G. N., and Thomas, P. J. (2003). Endoproteolytic activity of the proteasome. Science 299, 408–411. doi: 10.1126/science.1079293
Liu, G., Zhang, C., Yin, J., Li, X., Cheng, F., Li, Y., et al. (2009). alpha-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci. Lett. 454, 187–192. doi: 10.1016/j.neulet.2009.02.056
Liu, M., Qin, L., Wang, L., Tan, J., Zhang, H., Tang, J., et al. (2018a). α−synuclein induces apoptosis of astrocytes by causing dysfunction of the endoplasmic reticulum-Golgi compartment. Mol. Med. Rep. 18, 322–332. doi: 10.3892/mmr.2018.9002
Liu, Y., Zhang, J., Jiang, M., Cai, Q., Fang, J., and Jin, L. (2018b). MANF improves the MPP+/MPTP-induced Parkinson’s disease via improvement of mitochondrial function and inhibition of oxidative stress. Am. J. Transl. Res. 10, 1284–1294.
Liu, Y.-Q., Mao, Y., Xu, E., Jia, H., Zhang, S., Dawson, V. L., et al. (2021). Nanozyme scavenging ROS for prevention of pathologic α-synuclein transmission in Parkinson’s disease. Nano Today 36:101027. doi: 10.1016/j.nantod.2020.101027
Logan, T., Bendor, J., Toupin, C., Thorn, K., and Edwards, R. H. (2017). α-Synuclein promotes dilation of the exocytotic fusion pore. Nat. Neurosci. 20, 681–689. doi: 10.1038/nn.4529
Lopes da Fonseca, T., Pinho, R., and Outeiro, T. F. (2016). A familial ATP13A2 mutation enhances alpha-synuclein aggregation and promotes cell death. Hum. Mol. Genet. 25, 2959–2971. doi: 10.1093/hmg/ddw147
Lopes da Fonseca, T., Villar-Piqué, A., and Outeiro, T. F. (2015). The Interplay between Alpha-Synuclein Clearance and Spreading. Biomolecules 5, 435–471. doi: 10.3390/biom5020435
Ludtmann, M. H. R., Angelova, P. R., Horrocks, M. H., Choi, M. L., Rodrigues, M., Baev, A. Y., et al. (2018). α-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 9:2293. doi: 10.1038/s41467-018-04422-2
Ludtmann, M. H. R., Angelova, P. R., Ninkina, N. N., Gandhi, S., Buchman, V. L., and Abramov, A. Y. (2016). Monomeric Alpha-Synuclein Exerts a Physiological Role on Brain ATP Synthase. J. Neurosci. 36, 10510–10521. doi: 10.1523/JNEUROSCI.1659-16.2016
Luk, K. C., Kehm, V., Carroll, J., Zhang, B., O’Brien, P., Trojanowski, J. Q., et al. (2012). Pathological α-Synuclein Transmission Initiates Parkinson-like Neurodegeneration in Non-transgenic Mice. Science 338, 949–953. doi: 10.1126/science.1227157
Lwin, A., Orvisky, E., Goker-Alpan, O., LaMarca, M. E., and Sidransky, E. (2004). Glucocerebrosidase mutations in subjects with parkinsonism. Mol. Genet. Metab. 81, 70–73. doi: 10.1016/j.ymgme.2003.11.004
Lyte, M. (2014). Microbial endocrinology: host-microbiota neuroendocrine interactions influencing brain and behavior. Gut Microbes 5, 381–389. doi: 10.4161/gmic.28682
Ma, K.-L., Song, L.-K., Yuan, Y.-H., Zhang, Y., Han, N., Gao, K., et al. (2014). The nuclear accumulation of alpha-synuclein is mediated by importin alpha and promotes neurotoxicity by accelerating the cell cycle. Neuropharmacology 82, 132–142. doi: 10.1016/j.neuropharm.2013.07.035
Ma, L.-Y., Gao, L., Li, X., Ma, H.-Z., and Feng, T. (2019a). Nitrated alpha-synuclein in minor salivary gland biopsies in Parkinson’s disease. Neurosci. Lett. 704, 45–49. doi: 10.1016/j.neulet.2019.03.054
Ma, M.-R., Hu, Z.-W., Zhao, Y.-F., Chen, Y.-X., and Li, Y.-M. (2016). Phosphorylation induces distinct alpha-synuclein strain formation. Sci. Rep. 6:37130. doi: 10.1038/srep37130
Ma, Q., Xing, C., Long, W., Wang, H. Y., Liu, Q., and Wang, R.-F. (2019b). Impact of microbiota on central nervous system and neurological diseases: the gut-brain axis. J. Neuroinflammation 16:53. doi: 10.1186/s12974-019-1434-3
Machiya, Y., Hara, S., Arawaka, S., Fukushima, S., Sato, H., Sakamoto, M., et al. (2010). Phosphorylated α-Synuclein at Ser-129 Is Targeted to the Proteasome Pathway in a Ubiquitin-independent Manner. J. Biol. Chem. 285, 40732–40744. doi: 10.1074/jbc.M110.141952
Mahul-Mellier, A.-L., Burtscher, J., Maharjan, N., Weerens, L., Croisier, M., Kuttler, F., et al. (2020). The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. U. S. A. 117, 4971–4982. doi: 10.1073/pnas.1913904117
Mandal, P. K., Pettegrew, J. W., Masliah, E., Hamilton, R. L., and Mandal, R. (2006). Interaction between Abeta peptide and alpha synuclein: molecular mechanisms in overlapping pathology of Alzheimer’s and Parkinson’s in dementia with Lewy body disease. Neurochem. Res. 31, 1153–1162. doi: 10.1007/s11064-006-9140-9
Mao, X., Ou, M. T., Karuppagounder, S. S., Kam, T.-I., Yin, X., Xiong, Y., et al. (2016). Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353:aah3374. doi: 10.1126/science.aah3374
Maor, G., Rapaport, D., and Horowitz, M. (2019). The effect of mutant GBA1 on accumulation and aggregation of α-synuclein. Hum. Mol. Genet. 28, 1768–1781. doi: 10.1093/hmg/ddz005
Marco, E. V. D., Annesi, G., Tarantino, P., Rocca, F. E., Provenzano, G., Civitelli, D., et al. (2008). Glucocerebrosidase gene mutations are associated with Parkinson’s disease in southern Italy. Mov. Disord. 23, 460–463. doi: 10.1002/mds.21892
Markopoulou, K., Biernacka, J. M., Armasu, S. M., Anderson, K. J., Ahlskog, J. E., Chase, B. A., et al. (2014). Does α-synuclein have a dual and opposing effect in preclinical vs. clinical Parkinson’s disease? Parkinsonism Relat. Disord 20, 584–589. doi: 10.1016/j.parkreldis.2014.02.021
Maroteaux, L., Campanelli, J. T., and Scheller, R. H. (1988). Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 8, 2804–2815. doi: 10.1523/jneurosci.08-08-02804.1988
Marotta, N. P., Lin, Y. H., Lewis, Y. E., Ambroso, M. R., Zaro, B. W., Roth, M. T., et al. (2015). O-GlcNAc modification blocks the aggregation and toxicity of the Parkinson’s disease associated protein α-synuclein. Nat. Chem. 7, 913–920. doi: 10.1038/nchem.2361
Martínez, J. H., Fuentes, F., Vanasco, V., Alvarez, S., Alaimo, A., Cassina, A., et al. (2018). Alpha-synuclein mitochondrial interaction leads to irreversible translocation and complex I impairment. Arch. Biochem. Biophys. 651, 1–12. doi: 10.1016/j.abb.2018.04.018
Martinez-Vicente, M., and Cuervo, A. M. (2007). Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 6, 352–361. doi: 10.1016/S1474-4422(07)70076-5
Martinez-Vicente, M., Talloczy, Z., Kaushik, S., Massey, A. C., Mazzulli, J., Mosharov, E. V., et al. (2008). Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest. 118, 777–788. doi: 10.1172/JCI32806
Masliah, E., Rockenstein, E., Adame, A., Alford, M., Crews, L., Hashimoto, M., et al. (2005). Effects of α-Synuclein Immunization in a Mouse Model of Parkinson’s Disease. Neuron 46, 857–868. doi: 10.1016/j.neuron.2005.05.010
Masliah, E., Rockenstein, E., Mante, M., Crews, L., Spencer, B., Adame, A., et al. (2011). Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One 6:e19338. doi: 10.1371/journal.pone.0019338
Masuda-Suzukake, M., Nonaka, T., Hosokawa, M., Kubo, M., Shimozawa, A., Akiyama, H., et al. (2014). Pathological alpha-synuclein propagates through neural networks. Acta Neuropathol. Commun. 2, 1–12.
Mata, I. F., Samii, A., Schneer, S. H., Roberts, J. W., Griffith, A., Leis, B. C., et al. (2008). Glucocerebrosidase Gene Mutations. Arch. Neurol. 65, 379–382. doi: 10.1001/archneurol.2007.68
Mayer, E. A., Tillisch, K., and Gupta, A. (2015). Gut/brain axis and the microbiota. J. Clin. Invest. 125, 926–938. doi: 10.1172/JCI76304
Mazzetti, S., Basellini, M. J., Ferri, V., Cassani, E., Cereda, E., Paolini, M., et al. (2020). α-Synuclein oligomers in skin biopsy of idiopathic and monozygotic twin patients with Parkinson’s disease. Brain 143, 920–931. doi: 10.1093/brain/awaa008
Mazzulli, J. R., Xu, Y.-H., Sun, Y., Knight, A. L., McLean, P. J., Caldwell, G. A., et al. (2011). Gaucher’s Disease Glucocerebrosidase and <alpha>-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52. doi: 10.1016/j.cell.2011.06.001
Mazzulli, J. R., Zunke, F., Tsunemi, T., Toker, N. J., Jeon, S., Burbulla, L. F., et al. (2016). Activation of β-Glucocerebrosidase Reduces Pathological α-Synuclein and Restores Lysosomal Function in Parkinson’s Patient Midbrain Neurons. J. Neurosci. 36, 7693–7706. doi: 10.1523/JNEUROSCI.0628-16.2016
McCracken, A. A., and Brodsky, J. L. (2003). Evolving questions and paradigm shifts in endoplasmic-reticulum-associated degradation (ERAD). Bioessays 25, 868–877. doi: 10.1002/bies.10320
McKinnon, C., De Snoo, M. L., Gondard, E., Neudorfer, C., Chau, H., Ngana, S. G., et al. (2020). Early-onset impairment of the ubiquitin-proteasome system in dopaminergic neurons caused by α-synuclein. Acta Neuropathol. Commun. 8:17. doi: 10.1186/s40478-020-0894-0
McLean, P. J., Kawamata, H., Shariff, S., Hewett, J., Sharma, N., Ueda, K., et al. (2002). TorsinA and heat shock proteins act as molecular chaperones: suppression of alpha-synuclein aggregation. J. Neurochem. 83, 846–854. doi: 10.1046/j.1471-4159.2002.01190.x
Meade, R. M., Fairlie, D. P., and Mason, J. M. (2019). Alpha-synuclein structure and Parkinson’s disease – lessons and emerging principles. Mol. Neurodegener. 14:29. doi: 10.1186/s13024-019-0329-1
Menges, S., Minakaki, G., Schaefer, P. M., Meixner, H., Prots, I., Schlötzer-Schrehardt, U., et al. (2017). Alpha-synuclein prevents the formation of spherical mitochondria and apoptosis under oxidative stress. Sci. Rep. 7:42942. doi: 10.1038/srep42942
Mezias, C., Rey, N., Brundin, P., and Raj, A. (2020). Neural connectivity predicts spreading of alpha-synuclein pathology in fibril-injected mouse models: involvement of retrograde and anterograde axonal propagation. Neurobiol. Dis. 134:104623. doi: 10.1016/j.nbd.2019.104623
Middleton, E. R., and Rhoades, E. (2010). Effects of Curvature and Composition on α-Synuclein Binding to Lipid Vesicles. Biophys. J. 99, 2279–2288. doi: 10.1016/j.bpj.2010.07.056
Minakaki, G., Menges, S., Kittel, A., Emmanouilidou, E., Schaeffner, I., Barkovits, K., et al. (2018). Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 14, 98–119. doi: 10.1080/15548627.2017.1395992
Mohite, G. M., Dwivedi, S., Das, S., Kumar, R., Paluri, S., Mehra, S., et al. (2018). Parkinson’s Disease Associated α-Synuclein Familial Mutants Promote Dopaminergic Neuronal Death in Drosophila melanogaster. ACS Chem. Neurosci. 9, 2628–2638. doi: 10.1021/acschemneuro.8b00107
Mori, A., Hatano, T., Inoshita, T., Shiba-Fukushima, K., Koinuma, T., Meng, H., et al. (2019). Parkinson’s disease-associated iPLA2-VIA/PLA2G6 regulates neuronal functions and α-synuclein stability through membrane remodeling. Proc. Natl. Acad. Sci. U. S. A. 116, 20689–20699. doi: 10.1073/pnas.1902958116
Mori, F., Inenaga, C., Yoshimoto, M., Umezu, H., Tanaka, R., Takahashi, H., et al. (2002). Alpha-synuclein immunoreactivity in normal and neoplastic Schwann cells. Acta Neuropathol. 103, 145–151. doi: 10.1007/s004010100443
Morris, M., Knudsen, G. M., Maeda, S., Trinidad, J. C., Ioanoviciu, A., Burlingame, A. L., et al. (2015). Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 18, 1183–1189. doi: 10.1038/nn.4067
Müller, B., Assmus, J., Herlofson, K., Larsen, J. P., and Tysnes, O.-B. (2013). Importance of motor vs. non-motor symptoms for health-related quality of life in early Parkinson’s disease. Parkinsonism Relat. Disord. 19, 1027–1032. doi: 10.1016/j.parkreldis.2013.07.010
Munishkina, L. A., Cooper, E. M., Uversky, V. N., and Fink, A. L. (2004). The effect of macromolecular crowding on protein aggregation and amyloid fibril formation. J. Mol. Recognit. 17, 456–464. doi: 10.1002/jmr.699
Musgrove, R. E., Helwig, M., Bae, E.-J., Aboutalebi, H., Lee, S.-J., Ulusoy, A., et al. (2019). Oxidative stress in vagal neurons promotes parkinsonian pathology and intercellular α-synuclein transfer. J. Clin. Invest. 129, 3738–3753. doi: 10.1172/jci127330
Nakamura, K., Nemani, V. M., Wallender, E. K., Kaehlcke, K., Ott, M., and Edwards, R. H. (2008). Optical reporters for the conformation of alpha-synuclein reveal a specific interaction with mitochondria. J. Neurosci. 28, 12305–12317. doi: 10.1523/JNEUROSCI.3088-08.2008
Narayanan, V., and Scarlata, S. (2001). Membrane binding and self-association of alpha-synucleins. Biochemistry 40, 9927–9934. doi: 10.1021/bi002952n
Nepal, G., Rehrig, J. H., and Ojha, R. (2019). Glutamate ionotropic receptor NMDA type subunit 2A (GRIN2A) gene polymorphism (rs4998386) and Parkinson’s disease susceptibility: a meta-analysis. Aging Med. (Milton) 2, 174–183. doi: 10.1002/agm2.12075
Nezami, B. G., and Srinivasan, S. (2010). Enteric Nervous System in the Small Intestine: pathophysiology and Clinical Implications. Curr. Gastroenterol. Rep. 12, 358–365. doi: 10.1007/s11894-010-0129-9
Ng, A. S. L., Tan, Y. J., Zhao, Y., Saffari, S. E., Lu, Z., Ng, E. Y. L., et al. (2019). SNCA Rep1 promoter variability influences cognition in Parkinson’s disease. Mov. Disord. 34, 1232–1236. doi: 10.1002/mds.27768
Ngolab, J., Trinh, I., Rockenstein, E., Mante, M., Florio, J., Trejo, M., et al. (2017). Brain-derived exosomes from dementia with Lewy bodies propagate α-synuclein pathology. Acta Neuropathol. Commun. 5:46. doi: 10.1186/s40478-017-0445-5
Noyce, A. J., Bestwick, J. P., Silveira-Moriyama, L., Hawkes, C. H., Giovannoni, G., Lees, A. J., et al. (2012). Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann. Neurol. 72, 893–901. doi: 10.1002/ana.23687
Nübling, G. S., Levin, J., Bader, B., Lorenzl, S., Hillmer, A., Högen, T., et al. (2014). Modelling Ser129 phosphorylation inhibits membrane binding of pore-forming alpha-synuclein oligomers. PLoS One 9:e98906. doi: 10.1371/journal.pone.0098906
O’Donovan, S. M., Crowley, E. K., Brown, J. R.-M., O’Sullivan, O., O’Leary, O. F., Timmons, S., et al. (2020). Nigral overexpression of α-synuclein in a rat Parkinson’s disease model indicates alterations in the enteric nervous system and the gut microbiome. Neurogastroenterol. Motil. 32:e13726. doi: 10.1111/nmo.13726
Paleologou, K. E., Oueslati, A., Shakked, G., Rospigliosi, C. C., Kim, H.-Y., Lamberto, G. R., et al. (2010). Phosphorylation at S87 Is Enhanced in Synucleinopathies, Inhibits α-Synuclein Oligomerization, and Influences Synuclein-Membrane Interactions. J. Neurosci. 30, 3184–3198. doi: 10.1523/JNEUROSCI.5922-09.2010
Paleologou, K. E., Schmid, A. W., Rospigliosi, C. C., Kim, H. Y., Lamberto, G. R., Fredenburg, R. A., et al. (2008). Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of alpha-synuclein. J. Biol. Chem. 283, 16895–16905. doi: 10.1074/jbc.m800747200
Pampalakis, G., Sykioti, V.-S., Ximerakis, M., Stefanakou-Kalakou, I., Melki, R., Vekrellis, K., et al. (2016). KLK6 proteolysis is implicated in the turnover and uptake of extracellular alpha-synuclein species. Oncotarget 8, 14502–14515. doi: 10.18632/oncotarget.13264
Pan-Montojo, F., Schwarz, M., Winkler, C., Arnhold, M., O’Sullivan, G. A., Pal, A., et al. (2012). Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci. Rep. 2:898. doi: 10.1038/srep00898
Parihar, M., Parihar, A., Fujita, M., Hashimoto, M., and Ghafourifar, P. (2008). Mitochondrial association of alpha-synuclein causes oxidative stress. Cell. Mol. Life Sci. 65, 1272–1284. doi: 10.1007/s00018-008-7589-1
Park, J.-S., Davis, R. L., and Sue, C. M. (2018). Mitochondrial Dysfunction in Parkinson’s Disease: new Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 18:21. doi: 10.1007/s11910-018-0829-3
Pasanen, P., Myllykangas, L., Siitonen, M., Raunio, A., Kaakkola, S., Lyytinen, J., et al. (2014). Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 35:2180.e1–5. doi: 10.1016/j.neurobiolaging.2014.03.024
Peng, X., Peng, X. M., Tehranian, R., Dietrich, P., Stefanis, L., and Perez, R. G. (2005). Alpha-synuclein activation of protein phosphatase 2A reduces tyrosine hydroxylase phosphorylation in dopaminergic cells. J. Cell Sci. 118, 3523–3530. doi: 10.1242/jcs.02481
Perez, R. G., and Hastings, T. G. (2004). Could a loss of alpha-synuclein function put dopaminergic neurons at risk? J. Neurochem. 89, 1318–1324. doi: 10.1111/j.1471-4159.2004.02423.x
Perez, R. G., Waymire, J. C., Lin, E., Liu, J. J., Guo, F., and Zigmond, M. J. (2002). A role for alpha-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 22, 3090–3099. doi: 10.1523/JNEUROSCI.22-08-03090.2002
Periquet, M., Fulga, T., Myllykangas, L., Schlossmacher, M. G., and Feany, M. B. (2007). Aggregated α-Synuclein Mediates Dopaminergic Neurotoxicity In Vivo. J. Neurosci. 27, 3338–3346. doi: 10.1523/JNEUROSCI.0285-07.2007
Perni, M., Galvagnion, C., Maltsev, A., Meisl, G., Müller, M. B. D., Challa, P. K., et al. (2017). A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity. Proc. Natl. Acad. Sci. U. S. A. 114, E1009–E1017. doi: 10.1073/pnas.1610586114
Perrin, R. J., Woods, W. S., Clayton, D. F., and George, J. M. (2000). Interaction of human α-Synuclein and Parkinson’s disease variants with phospholipids: structural analysis using site-directed mutagenesis. J. Biol. Chem. 275, 34393–34398. doi: 10.1074/jbc.M004851200
Pfefferkorn, C. M., Jiang, Z., and Lee, J. C. (2012). Biophysics of α-synuclein membrane interactions. Biochim. Biophys. Acta 1818, 162–171. doi: 10.1016/j.bbamem.2011.07.032
Pfeiffer, R. F. (2011). Gastrointestinal dysfunction in Parkinson’s disease. Parkinsonism Relat. Disord. 17, 10–15. doi: 10.1016/j.parkreldis.2010.08.003
Pineda, A., and Burré, J. (2017). Modulating membrane binding of α-synuclein as a therapeutic strategy. Proc. Natl. Acad. Sci. U. S. A. 114, 1223–1225. doi: 10.1073/pnas.1620159114
Poewe, W. (2008). Non-motor symptoms in Parkinson’s disease. Eur. J. Neurol. 15, 14–20. doi: 10.1111/j.1468-1331.2008.02056.x
Polymeropoulos, M. H., Higgins, J. J., Golbe, L. I., Johnson, W. G., Ide, S. E., Di Iorio, G., et al. (1996). Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science 274, 1197–1199. doi: 10.1126/science.274.5290.1197
Polymeropoulos, M. H., Lavedan, C., Leroy, E., Ide, S. E., Dehejia, A., Dutra, A., et al. (1997). Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 276, 2045–2047. doi: 10.1126/science.276.5321.2045
Price, D. L., Koike, M. A., Khan, A., Wrasidlo, W., Rockenstein, E., Masliah, E., et al. (2018). The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci. Rep. 8:16165. doi: 10.1038/s41598-018-34490-9
Qiang, W., Yau, W.-M., Luo, Y., Mattson, M. P., and Tycko, R. (2012). Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc. Natl. Acad. Sci. U. S. A. 109, 4443–4448. doi: 10.1073/pnas.1111305109
Qu, J., Ren, X., Xue, F., He, Y., Zhang, R., Zheng, Y., et al. (2020). Specific Knockdown of α-Synuclein by Peptide-Directed Proteasome Degradation Rescued Its Associated Neurotoxicity. Cell Chem. Biol. 27, 751–762.e4. doi: 10.1016/j.chembiol.2020.03.010
Qu, J., Yan, H., Zheng, Y., Xue, F., Zheng, Y., Fang, H., et al. (2018). The Molecular Mechanism of Alpha-Synuclein Dependent Regulation of Protein Phosphatase 2A Activity. Cell. Physiol. Biochem. 47, 2613–2625. doi: 10.1159/000491657
Qureshi, H. Y., and Paudel, H. K. (2011). Parkinsonian neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and alpha-synuclein mutations promote Tau protein phosphorylation at Ser262 and destabilize microtubule cytoskeleton in vitro. J. Biol. Chem. 286, 5055–5068. doi: 10.1074/jbc.M110.178905
Rabouille, C., and Haase, G. (2016). Editorial: golgi Pathology in Neurodegenerative Diseases. Front. Neurosci. 9:489. doi: 10.3389/fnins.2015.00489
Reichmann, H. (2011). View point: etiology in Parkinson’s disease. Dual hit or spreading intoxication. J. Neurol. Sci. 310, 9–11. doi: 10.1016/j.jns.2011.04.016
Renko, J.-M., Bäck, S., Voutilainen, M. H., Piepponen, T. P., Reenilä, I., Saarma, M., et al. (2018). Mesencephalic Astrocyte-Derived Neurotrophic Factor (MANF) Elevates Stimulus-Evoked Release of Dopamine in Freely-Moving Rats. Mol. Neurobiol. 55, 6755–6768. doi: 10.1007/s12035-018-0872-8
Rey, N. L., Bousset, L., George, S., Madaj, Z., Meyerdirk, L., Schulz, E., et al. (2019). α-Synuclein conformational strains spread, seed and target neuronal cells differentially after injection into the olfactory bulb. Acta Neuropathol. Commun. 7:221. doi: 10.1186/s40478-019-0859-3
Robotta, M., Cattani, J., Martins, J. C., Subramaniam, V., and Drescher, M. (2017). Alpha-Synuclein Disease Mutations Are Structurally Defective and Locally Affect Membrane Binding. J. Am. Chem. Soc. 139, 4254–4257. doi: 10.1021/jacs.6b05335
Robotta, M., Gerding, H. R., Vogel, A., Hauser, K., Schildknecht, S., Karreman, C., et al. (2014). Alpha-synuclein binds to the inner membrane of mitochondria in an α-helical conformation. Chembiochem 15, 2499–2502. doi: 10.1002/cbic.201402281
Rocha, E. M., Smith, G. A., Park, E., Cao, H., Brown, E., Hayes, M. A., et al. (2015). Glucocerebrosidase gene therapy prevents α-synucleinopathy of midbrain dopamine neurons. Neurobiol. Dis. 82, 495–503. doi: 10.1016/j.nbd.2015.09.009
Roeters, S. J., Iyer, A., Pletikapiæ, G., Kogan, V., Subramaniam, V., and Woutersen, S. (2017). Evidence for Intramolecular Antiparallel Beta-Sheet Structure in Alpha-Synuclein Fibrils from a Combination of Two-Dimensional Infrared Spectroscopy and Atomic Force Microscopy. Sci. Rep. 7:41051. doi: 10.1038/srep41051
Rokad, D., Ghaisas, S., Harischandra, D. S., Jin, H., Anantharam, V., Kanthasamy, A., et al. (2017). Role of neurotoxicants and traumatic brain injury in α-synuclein protein misfolding and aggregation. Brain Res. Bull. 133, 60–70. doi: 10.1016/j.brainresbull.2016.12.003
Rokad, D., Jin, H., Anantharam, V., Kanthasamy, A., and Kanthasamy, A. G. (2019). Exosomes as Mediators of Chemical-induced Toxicity. Curr. Environ. Health Rep. 6, 73–79. doi: 10.1007/s40572-019-00233-9
Rospigliosi, C. C., McClendon, S., Schmid, A. W., Ramlall, T. F., Barré, P., Lashuel, H. A., et al. (2009). E46K Parkinson’s-Linked Mutation Enhances C-Terminal-to-N-Terminal Contacts in α-Synuclein. J. Mol. Biol. 388, 1022–1032. doi: 10.1016/j.jmb.2009.03.065
Rostami, J., Holmqvist, S., Lindström, V., Sigvardson, J., Westermark, G. T., Ingelsson, M., et al. (2017). Human Astrocytes Transfer Aggregated Alpha-Synuclein via Tunneling Nanotubes. J. Neurosci. 37, 11835–11853. doi: 10.1523/JNEUROSCI.0983-17.2017
Rostovtseva, T. K., Gurnev, P. A., Protchenko, O., Hoogerheide, D. P., Yap, T. L., Philpott, C. C., et al. (2015). α-Synuclein Shows High Affinity Interaction with Voltage-dependent Anion Channel, Suggesting Mechanisms of Mitochondrial Regulation and Toxicity in Parkinson Disease. J. Biol. Chem. 290, 18467–18477. doi: 10.1074/jbc.M115.641746
Ruf, V. C., Nübling, G. S., Willikens, S., Shi, S., Schmidt, F., Levin, J., et al. (2019). Different Effects of α-Synuclein Mutants on Lipid Binding and Aggregation Detected by Single Molecule Fluorescence Spectroscopy and ThT Fluorescence-Based Measurements. ACS Chem. Neurosci. 10, 1649–1659. doi: 10.1021/acschemneuro.8b00579
Rutherford, N. J., Moore, B. D., Golde, T. E., and Giasson, B. I. (2014). Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of α-synuclein. J. Neurochem. 131, 859–867. doi: 10.1111/jnc.12806
Ryan, P., Xu, M., Jahan, K., Davey, A. K., Bharatam, P. V., Anoopkumar-Dukie, S., et al. (2020). Novel Furan-2-yl-1H-pyrazoles Possess Inhibitory Activity against α-Synuclein Aggregation. ACS Chem. Neurosci. 11, 2303–2315. doi: 10.1021/acschemneuro.0c00252
Ryu, S., Baek, I., and Liew, H. (2019). Sumoylated α-synuclein translocates into the nucleus by karyopherin α6. Mol. Cell. Toxicol. 15, 103–109. doi: 10.1007/s13273-019-0012-1
Sado, M., Yamasaki, Y., Iwanaga, T., Onaka, Y., Ibuki, T., Nishihara, S., et al. (2009). Protective effect against Parkinson’s disease-related insults through the activation of XBP1. Brain Res. 1257, 16–24. doi: 10.1016/j.brainres.2008.11.104
Sakai, R., Suzuki, M., Ueyama, M., Takeuchi, T., Minakawa, E. N., Hayakawa, H., et al. (2019). E46K mutant α-synuclein is more degradation resistant and exhibits greater toxic effects than wild-type α-synuclein in Drosophila models of Parkinson’s disease. PLoS One 14:e0218261. doi: 10.1371/journal.pone.0218261
Sampson, T. R., Debelius, J. W., Thron, T., Janssen, S., Shastri, G. G., Ilhan, Z. E., et al. (2016). Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 167, 1469–1480.e12. doi: 10.1016/j.cell.2016.11.018
Samuel, F., Flavin, W. P., Iqbal, S., Pacelli, C., Sri Renganathan, S. D., Trudeau, L.-E., et al. (2016). Effects of Serine 129 Phosphorylation on α-Synuclein Aggregation, Membrane Association, and Internalization. J. Biol. Chem. 291, 4374–4385. doi: 10.1074/jbc.M115.705095
Sánchez-Ferro, Á, Rábano, A., Catalán, M. J., Rodríguez-Valcárcel, F. C., Fernández Díez, S., Herreros-Rodríguez, J., et al. (2015). In vivo gastric detection of α-synuclein inclusions in Parkinson’s disease. Mov. Disord. 30, 517–524. doi: 10.1002/mds.25988
Sanchez-Guajardo, V., Annibali, A., Jensen, P. H., and Romero-Ramos, M. (2013). α-Synuclein vaccination prevents the accumulation of parkinson disease-like pathologic inclusions in striatum in association with regulatory T cell recruitment in a rat model. J. Neuropathol. Exp. Neurol. 72, 624–645. doi: 10.1097/NEN.0b013e31829768d2
Sandal, M., Valle, F., Tessari, I., Mammi, S., Bergantino, E., Musiani, F., et al. (2008). Conformational equilibria in monomeric α-synuclein at the single-molecule level. PLoS Biol. 6:e6. doi: 10.1371/journal.pbio.0060006
Sanders, L. H., and Greenamyre, J. T. (2013). Oxidative damage to macromolecules in human Parkinson’s disease and the rotenone model. Free Radic. Biol. Med. 62, 111–120. doi: 10.1016/j.freeradbiomed.2013.01.003
Sapru, M. K., Yates, J. W., Hogan, S., Jiang, L., Halter, J., and Bohn, M. C. (2006). Silencing of human alpha-synuclein in vitro and in rat brain using lentiviral-mediated RNAi. Exp. Neurol. 198, 382–390. doi: 10.1016/j.expneurol.2005.12.024
Sathiya, S., Ranju, V., Kalaivani, P., Priya, R. J., Sumathy, H., Sunil, A. G., et al. (2013). Telmisartan attenuates MPTP induced dopaminergic degeneration and motor dysfunction through regulation of α-synuclein and neurotrophic factors (BDNF and GDNF) expression in C57BL/6J mice. Neuropharmacology 73, 98–110. doi: 10.1016/j.neuropharm.2013.05.025
Sato, H., Arawaka, S., Hara, S., Fukushima, S., Koga, K., Koyama, S., et al. (2011). Authentically phosphorylated α-synuclein at Ser129 accelerates neurodegeneration in a rat model of familial Parkinson’s disease. J. Neurosci. 31, 16884–16894. doi: 10.1523/JNEUROSCI.3967-11.2011
Schenk, D. B., Koller, M., Ness, D. K., Griffith, S. G., Grundman, M., Zago, W., et al. (2017). First-in-human assessment of PRX002, an anti–α−synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. 32, 211–218. doi: 10.1002/mds.26878
Scheperjans, F., Aho, V., Pereira, P. A. B., Koskinen, K., Paulin, L., Pekkonen, E., et al. (2015). Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 30, 350–358. doi: 10.1002/mds.26069
Schofield, D. J., Irving, L., Calo, L., Bogstedt, A., Rees, G., Nuccitelli, A., et al. (2019). Preclinical development of a high affinity α-synuclein antibody, MEDI1341, that can enter the brain, sequester extracellular α-synuclein and attenuate α-synuclein spreading in vivo. Neurobiol. Dis. 132:104582. doi: 10.1016/j.nbd.2019.104582
Schweighauser, M., Shi, Y., Tarutani, A., Kametani, F., Murzin, A. G., Ghetti, B., et al. (2020). Structures of α-synuclein filaments from multiple system atrophy. Nature 585, 464–469. doi: 10.1038/s41586-020-2317-6
Scott, D., and Roy, S. (2012). α-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J. Neurosci. 32, 10129–10135. doi: 10.1523/JNEUROSCI.0535-12.2012
Scudamore, O., and Ciossek, T. (2018). Increased Oxidative Stress Exacerbates α-Synuclein Aggregation In Vivo. J. Neuropathol. Exp. Neurol. 77, 443–453. doi: 10.1093/jnen/nly024
Sengupta, U., Guerrero-Muñoz, M. J., Castillo-Carranza, D. L., Lasagna-Reeves, C. A., Gerson, J. E., Paulucci-Holthauzen, A. A., et al. (2015). Pathological interface between oligomeric alpha-synuclein and tau in synucleinopathies. Biol. Psychiatry 78, 672–683. doi: 10.1016/j.biopsych.2014.12.019
Senkevich, K., and Gan-Or, Z. (2019). Autophagy lysosomal pathway dysfunction in Parkinson’s disease; evidence from human genetics. Parkinsonism Relat. Disord. 73, 60–71. doi: 10.1016/j.parkreldis.2019.11.015
Seo, J.-H., Rah, J.-C., Choi, S. H., Shin, J. K., Min, K., Kim, H.-S., et al. (2002). Alpha-synuclein regulates neuronal survival via Bcl-2 family expression and PI3/Akt kinase pathway. FASEB J. 16, 1826–1828. doi: 10.1096/fj.02-0041fje
Serpell, L. C., Berriman, J., Jakes, R., Goedert, M., and Crowther, R. A. (2000). Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. U. S. A. 97, 4897–4902. doi: 10.1073/pnas.97.9.4897
Shaltouki, A., Hsieh, C.-H., Kim, M. J., and Wang, X. (2018). Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson’s models. Acta Neuropathol. 136, 607–620. doi: 10.1007/s00401-018-1873-4
Shannon, K. M., Keshavarzian, A., Dodiya, H. B., Jakate, S., and Kordower, J. H. (2012). Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov. Disord. 27, 716–719. doi: 10.1002/mds.25020
Shannon, K. M., Keshavarzian, A., Mutlu, E., Dodiya, H. B., Daian, D., Jaglin, J. A., et al. (2011). Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov. Disord. 27, 709–715. doi: 10.1002/mds.23838
Sharon, R., Goldberg, M. S., Bar-Josef, I., Betensky, R. A., Shen, J., and Selkoe, D. J. (2001). alpha-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc. Natl. Acad. Sci. U. S. A. 98, 9110–9115. doi: 10.1073/pnas.171300598
Shavali, S., Brown-Borg, H. M., Ebadi, M., and Porter, J. (2008). Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci. Lett. 439, 125–128. doi: 10.1016/j.neulet.2008.05.005
Shrivastava, A. N., Redeker, V., Fritz, N., Pieri, L., Almeida, L. G., Spolidoro, M., et al. (2015). α-synuclein assemblies sequester neuronal α3-Na+/K+-ATPase and impair Na+ gradient. EMBO J. 34, 2408–2423. doi: 10.15252/embj.201591397
Shtilerman, M. D., Ding, T. T., and Lansbury, P. T. (2002). Molecular crowding accelerates fibrillization of alpha-synuclein: could an increase in the cytoplasmic protein concentration induce Parkinson’s disease? Biochemistry 41, 3855–3860. doi: 10.1021/bi0120906
Siddu, A., David, L. S., Lauinger, N., Chen, X., Saint-Pierre, M., Alpaugh, M., et al. (2020). Beneficial effects of cysteamine in Thy1-α-Syn mice and induced pluripotent stem cells with a SNCA gene triplication. Neurobiol. Dis. 145:105042. doi: 10.1016/j.nbd.2020.105042
Singleton, A. B., Farrer, M., Johnson, J., Singleton, A., Hague, S., Kachergus, J., et al. (2003). alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302:841. doi: 10.1126/science.1090278
Snyder, H., Mensah, K., Theisler, C., Lee, J., Matouschek, A., and Wolozin, B. (2003). Aggregated and monomeric α-synuclein bind to the S6’ proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 278, 11753–11759. doi: 10.1074/jbc.M208641200
Soykan, I., Lin, Z., Bennett, J. P., and McCallum, R. W. (1999). Gastric myoelectrical activity in patients with Parkinson’s disease: evidence of a primary gastric abnormality. Dig. Dis. Sci. 44, 927–931. doi: 10.1023/a:1026648311646
Spillantini, M. G., Crowther, R. A., Jakes, R., Hasegawa, M., and Goedert, M. (1998). α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. U. S. A. 95, 6469–6473. doi: 10.1073/pnas.95.11.6469
Srivastava, T., Raj, R., Dubey, A., Kumar, D., Chaturvedi, R. K., Sharma, S. K., et al. (2020). Fast kinetics of environmentally induced α-synuclein aggregation mediated by structural alteration in NAC region and result in structure dependent cytotoxicity. Sci. Rep. 10:18412. doi: 10.1038/s41598-020-75361-6
Stefanis, L. (2012). α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2:a009399. doi: 10.1101/cshperspect.a009399
Stefanis, L., Emmanouilidou, E., Pantazopoulou, M., Kirik, D., Vekrellis, K., and Tofaris, G. K. (2019). How is alpha-synuclein cleared from the cell? J. Neurochem. 150, 577–590. doi: 10.1111/jnc.14704
Stöckl, M., Fischer, P., Wanker, E., and Herrmann, A. (2008). Alpha-synuclein selectively binds to anionic phospholipids embedded in liquid-disordered domains. J. Mol. Biol. 375, 1394–1404. doi: 10.1016/j.jmb.2007.11.051
Stone, D. K., Kiyota, T., Mosley, R. L., and Gendelman, H. E. (2012). A Model of Nitric Oxide Induced α-Synuclein Misfolding in Parkinson’s Disease. Neurosci. Lett. 523, 167–173. doi: 10.1016/j.neulet.2012.06.070
Stuendl, A., Kunadt, M., Kruse, N., Bartels, C., Moebius, W., Danzer, K. M., et al. (2016). Induction of α-synuclein aggregate formation by CSF exosomes from patients with Parkinson’s disease and dementia with Lewy bodies. Brain 139, 481–494. doi: 10.1093/brain/awv346
Su, X., Chu, Y., Kordower, J. H., Li, B., Cao, H., Huang, L., et al. (2015). PGC-1α Promoter Methylation in Parkinson’s Disease. PLoS One 10:e0134087. doi: 10.1371/journal.pone.0134087
Sugeno, N., Takeda, A., Hasegawa, T., Kobayashi, M., Kikuchi, A., Mori, F., et al. (2008). Serine 129 phosphorylation of alpha-synuclein induces unfolded protein response-mediated cell death. J. Biol. Chem. 283, 23179–23188. doi: 10.1074/jbc.M802223200
Sulzer, D., and Edwards, R. H. (2019). The physiological role of α-synuclein and its relationship to Parkinson’s Disease. J. Neurochem. 150, 475–486. doi: 10.1111/jnc.14810
Suzuki, G., Imura, S., Hosokawa, M., Katsumata, R., Nonaka, T., Hisanaga, S.-I., et al. (2020). α-synuclein strains that cause distinct pathologies differentially inhibit proteasome. Elife 9:e56825. doi: 10.7554/eLife.56825
Taguchi, K., Watanabe, Y., Tsujimura, A., and Tanaka, M. (2019). Expression of α-synuclein is regulated in a neuronal cell type-dependent manner. Anat. Sci. Int. 94, 11–22. doi: 10.1007/s12565-018-0464-8
Taguchi, Y. V., Liu, J., Ruan, J., Pacheco, J., Zhang, X., Abbasi, J., et al. (2017). Glucosylsphingosine Promotes α-Synuclein Pathology in Mutant GBA-Associated Parkinson’s Disease. J. Neurosci. 37, 9617–9631. doi: 10.1523/JNEUROSCI.1525-17.2017
Takahashi, M., Suzuki, M., Fukuoka, M., Fujikake, N., Watanabe, S., Murata, M., et al. (2015). Normalization of Overexpressed α-Synuclein Causing Parkinson’s Disease By a Moderate Gene Silencing With RNA Interference. Mol. Ther. Nucleic Acids 4:e241. doi: 10.1038/mtna.2015.14
Tayebi, N., Walker, J., Stubblefield, B., Orvisky, E., LaMarca, M. E., Wong, K., et al. (2003). Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol. Genet. Metab. 79, 104–109. doi: 10.1016/s1096-7192(03)00071-4
Tehranian, R., Montoya, S. E., Van Laar, A. D., Hastings, T. G., and Perez, R. G. (2006). Alpha-synuclein inhibits aromatic amino acid decarboxylase activity in dopaminergic cells. J. Neurochem. 99, 1188–1196. doi: 10.1111/j.1471-4159.2006.04146.x
Tofaris, G. K., Kim, H. T., Hourez, R., Jung, J.-W., Kim, K. P., and Goldberg, A. L. (2011). Ubiquitin ligase Nedd4 promotes α-synuclein degradation by the endosomal–lysosomal pathway. Proc. Natl. Acad. Sci. U. S. A. 108, 17004–17009. doi: 10.1073/pnas.1109356108
Tofaris, G. K., Layfield, R., and Spillantini, M. G. (2001). α-Synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 509, 22–26. doi: 10.1016/S0014-5793(01)03115-5
Toft, M., Pielsticker, L., Ross, O. A., Aasly, J., and Farrer, M. (2006). Glucocerebrosidase gene mutations and Parkinson disease in the Norwegian population. Neurology 66, 415–417. doi: 10.1212/01.wnl.0000196492.80676.7c
Tomás, M., Martínez-Alonso, E., Martínez-Martínez, N., Cara-Esteban, M., and Martínez-Menárguez, J. A. (2020). Fragmentation of the Golgi complex of dopaminergic neurons in human substantia nigra: new cytopathological findings in Parkinson’s disease. Histol. Histopathol. 36, 47–60. doi: 10.14670/HH-18-270
Tosatto, L., Horrocks, M. H., Dear, A. J., Knowles, T. P. J., Dalla Serra, M., Cremades, N., et al. (2015). Single-molecule FRET studies on alpha-synuclein oligomerization of Parkinson’s disease genetically related mutants. Sci. Rep. 5:16696. doi: 10.1038/srep16696
Tran, H. T., Chung, C. H.-Y., Iba, M., Zhang, B., Trojanowski, J. Q., Luk, K. C., et al. (2014). α-Synuclein Immunotherapy Blocks Uptake and Templated Propagation of Misfolded α-Synuclein and Neurodegeneration. Cell Rep. 7, 2054–2065. doi: 10.1016/j.celrep.2014.05.033
Tripathi, T., and Chattopadhyay, K. (2019). Interaction of α-Synuclein with ATP Synthase: switching Role from Physiological to Pathological. ACS Chem. Neurosci. 10, 16–17. doi: 10.1021/acschemneuro.8b00407
Tsunemi, T., Ishiguro, Y., Yoroisaka, A., Valdez, C., Miyamoto, K., Ishikawa, K., et al. (2020). Astrocytes Protect Human Dopaminergic Neurons from α-Synuclein Accumulation and Propagation. J. Neurosci. 40, 8618–8628. doi: 10.1523/JNEUROSCI.0954-20.2020
Tyson, T., Senchuk, M., Cooper, J. F., George, S., Van Raamsdonk, J. M., and Brundin, P. (2017). Novel animal model defines genetic contributions for neuron-to-neuron transfer of α-synuclein. Sci. Rep. 7:7506. doi: 10.1038/s41598-017-07383-6
Uéda, K., Fukushima, H., Masliah, E., Xia, Y., Iwai, A., Yoshimoto, M., et al. (1993). Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A. 90, 11282–11286. doi: 10.1073/pnas.90.23.11282
Ullman, O., Fisher, C. K., and Stultz, C. M. (2011). Explaining the Structural Plasticity of α-Synuclein. J. Am. Chem. Soc. 133, 19536–19546. doi: 10.1021/ja208657z
Unger, M. M., Spiegel, J., Dillmann, K.-U., Grundmann, D., Philippeit, H., Bürmann, J., et al. (2016). Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat. Disord. 32, 66–72. doi: 10.1016/j.parkreldis.2016.08.019
Utton, M. A., Noble, W. J., Hill, J. E., Anderton, B. H., and Hanger, D. P. (2005). Molecular motors implicated in the axonal transport of tau and alpha-synuclein. J. Cell Sci. 118, 4645–4654. doi: 10.1242/jcs.02558
Uversky, V. N. (2002). What does it mean to be natively unfolded? Eur. J. Biochem. 269, 2–12. doi: 10.1046/j.0014-2956.2001.02649.x
Uversky, V. N. (2007). Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J. Neurochem. 103, 17–37. doi: 10.1111/j.1471-4159.2007.04764.x
Uversky, V. N. (2008). α-synuclein misfolding and neurodegenerative diseases. Curr. Protein Pept. Sci. 9, 507–540. doi: 10.2174/138920308785915218
Uversky, V. N., and Eliezer, D. (2009). Biophysics of Parkinson’s disease: structure and aggregation of alpha-synuclein. Curr. Protein Pept. Sci. 10, 483–499. doi: 10.2174/138920309789351921
Uversky, V. N., Li, J., and Fink, A. L. (2001). Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J. Biol. Chem. 276, 10737–10744. doi: 10.1074/jbc.M010907200
Uversky, V. N., Li, J., Souillac, P., Millett, I. S., Doniach, S., Jakes, R., et al. (2002a). Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J. Biol. Chem. 277, 11970–11978. doi: 10.1074/jbc.M109541200
Uversky, V. N., Yamin, G., Munishkina, L. A., Karymov, M. A., Millett, I. S., Doniach, S., et al. (2005). Effects of nitration on the structure and aggregation of alpha-synuclein. Brain Res. Mol. Brain Res. 134, 84–102. doi: 10.1016/j.molbrainres.2004.11.014
Uversky, V. N., Yamin, G., Souillac, P. O., Goers, J., Glaser, C. B., and Fink, A. L. (2002b). Methionine oxidation inhibits fibrillation of human alpha-synuclein in vitro. FEBS Lett. 517, 239–244. doi: 10.1016/s0014-5793(02)02638-8
Valdés, P., Mercado, G., Vidal, R. L., Molina, C., Parsons, G., Court, F. A., et al. (2014). Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc. Natl. Acad. Sci. U. S. A. 111, 6804–6809. doi: 10.1073/pnas.1321845111
Van Den Berge, N., Ferreira, N., Gram, H., Mikkelsen, T. W., Alstrup, A. K. O., Casadei, N., et al. (2019). Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 138, 535–550. doi: 10.1007/s00401-019-02040-w
Vascellari, S., Palmas, V., Melis, M., Pisanu, S., Cusano, R., Uva, P., et al. (2020). Gut Microbiota and Metabolome Alterations Associated with Parkinson’s Disease. mSystems 5:2020. doi: 10.1128/mSystems.00561-20
Villarán, R. F., Espinosa-Oliva, A. M., Sarmiento, M., De Pablos, R. M., Argüelles, S., Delgado-Cortés, M. J., et al. (2010). Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: potential risk factor in Parkinson‘s disease. J. Neurochem. 114, 1687–1700. doi: 10.1111/j.1471-4159.2010.06879.x
Villar-Piqué, A., Lopes da Fonseca, T., Sant’Anna, R., Szegö, ÉM., Fonseca-Ornelas, L., Pinho, R., et al. (2016). Environmental and genetic factors support the dissociation between α-synuclein aggregation and toxicity. Proc. Natl. Acad. Sci. U. S. A. 113, E6506–E6515. doi: 10.1073/pnas.1606791113
Visanji, N. P., Lang, A. E., and Kovacs, G. G. (2019). Beyond the synucleinopathies: alpha synuclein as a driving force in neurodegenerative comorbidities. Transl. Neurodegener. 8:28. doi: 10.1186/s40035-019-0172-x
Voutilainen, M. H., Bäck, S., Pörsti, E., Toppinen, L., Lindgren, L., Lindholm, P., et al. (2009). Mesencephalic Astrocyte-Derived Neurotrophic Factor Is Neurorestorative in Rat Model of Parkinson’s Disease. J. Neurosci. 29, 9651–9659. doi: 10.1523/JNEUROSCI.0833-09.2009
Wakabayashi, K., and Takahashi, H. (1997). Neuropathology of autonomic nervous system in Parkinson’s disease. Eur. Neurol. 38, 2–7. doi: 10.1159/000113469
Wakabayashi, K., Hansen, L. A., and Masliah, E. (1995). Cortical Lewy body-containing neurons are pyramidal cells: laser confocal imaging of double-immunolabeled sections with anti-ubiquitin and SMI32. Acta Neuropathol. 89, 404–408. doi: 10.1007/BF00307643
Walter, P., and Ron, D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. doi: 10.1126/science.1209038
Wan, Z., Root-McCaig, J., Castellani, L., Kemp, B. E., Steinberg, G. R., and Wright, D. C. (2014). Evidence for the role of AMPK in regulating PGC-1 alpha expression and mitochondrial proteins in mouse epididymal adipose tissue. Obesity (Silver Spring) 22, 730–738. doi: 10.1002/oby.20605
Wang, B., Underwood, R., Kamath, A., Britain, C., McFerrin, M. B., McLean, P. J., et al. (2018). 14-3-3 Proteins Reduce Cell-to-Cell Transfer and Propagation of Pathogenic α-Synuclein. J. Neurosci. 38, 8211–8232. doi: 10.1523/JNEUROSCI.1134-18.2018
Wang, J., Lou, H., Pedersen, C. J., Smith, A. D., and Perez, R. G. (2009). 14-3-3zeta contributes to tyrosine hydroxylase activity in MN9D cells: localization of dopamine regulatory proteins to mitochondria. J. Biol. Chem. 284, 14011–14019. doi: 10.1074/jbc.M901310200
Wang, T., and Hay, J. C. (2015). Alpha-synuclein Toxicity in the Early Secretory Pathway: how It Drives Neurodegeneration in Parkinsons Disease. Front. Neurosci. 9:433. doi: 10.3389/fnins.2015.00433
Wang, W., Perovic, I., Chittuluru, J., Kaganovich, A., Nguyen, L. T. T., Liao, J., et al. (2011). A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. U. S. A. 108, 17797–17802. doi: 10.1073/pnas.1113260108
Wang, X., Becker, K., Levine, N., Zhang, M., Lieberman, A. P., Moore, D. J., et al. (2019). Pathogenic alpha-synuclein aggregates preferentially bind to mitochondria and affect cellular respiration. Acta Neuropathol. Commun. 7:41. doi: 10.1186/s40478-019-0696-4
Wang, Y., Wen, W., Li, H., Clementino, M., Xu, H., Xu, M., et al. (2021). MANF is neuroprotective against ethanol-induced neurodegeneration through ameliorating ER stress. Neurobiol. Dis. 148:105216. doi: 10.1016/j.nbd.2020.105216
Wang, Z., Udeshi, N. D., O’Malley, M., Shabanowitz, J., Hunt, D. F., and Hart, G. W. (2010). Enrichment and site mapping of O-linked N-acetylglucosamine by a combination of chemical/enzymatic tagging, photochemical cleavage, and electron transfer dissociation mass spectrometry. Mol. Cell. Proteomics 9, 153–160. doi: 10.1074/mcp.M900268-MCP200
Waxman, E. A., Mazzulli, J. R., and Giasson, B. I. (2009). Characterization of Hydrophobic Residue Requirements for α-Synuclein Fibrillization. Biochemistry 48, 9427–9436. doi: 10.1021/bi900539p
Weihofen, A., Liu, Y., Arndt, J. W., Huy, C., Quan, C., Smith, B. A., et al. (2019). Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol. Dis. 124, 276–288. doi: 10.1016/j.nbd.2018.10.016
Weinreb, P. H., Zhen, W., Poon, A. W., Conway, K. A., and Lansbury, P. T. (1996). NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 35, 13709–13715. doi: 10.1021/bi961799n
Wersinger, C., and Sidhu, A. (2003). Attenuation of dopamine transporter activity by alpha-synuclein. Neurosci. Lett. 340, 189–192. doi: 10.1016/s0304-3940(03)00097-1
Wersinger, C., Prou, D., Vernier, P., and Sidhu, A. (2003). Modulation of dopamine transporter function by alpha-synuclein is altered by impairment of cell adhesion and by induction of oxidative stress. FASEB J. 17, 2151–2153. doi: 10.1096/fj.03-0152fje
Winslow, A. R., Chen, C.-W., Corrochano, S., Acevedo-Arozena, A., Gordon, D. E., Peden, A. A., et al. (2010). α-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J. Cell Biol. 190, 1023–1037. doi: 10.1083/jcb.201003122
Wise-Scira, O., Dunn, A., Aloglu, A. K., Sakallioglu, I. T., and Coskuner, O. (2013). Structures of the E46K Mutant-Type α-Synuclein Protein and Impact of E46K Mutation on the Structures of the Wild-Type α-Synuclein Protein. ACS Chem. Neurosci. 4, 498–508. doi: 10.1021/cn3002027
Wrasidlo, W., Tsigelny, I. F., Price, D. L., Dutta, G., Rockenstein, E., Schwarz, T. C., et al. (2016). A de novo compound targeting α-synuclein improves deficits in models of Parkinson’s disease. Brain 139, 3217–3236. doi: 10.1093/brain/aww238
Wu, J.-Z., Ardah, M., Haikal, C., Svanbergsson, A., Diepenbroek, M., Vaikath, N. N., et al. (2019). Dihydromyricetin and Salvianolic acid B inhibit alpha-synuclein aggregation and enhance chaperone-mediated autophagy. Transl. Neurodegener. 8:18. doi: 10.1186/s40035-019-0159-7
Wu, Q., Takano, H., Riddle, D. M., Trojanowski, J. Q., Coulter, D. A., and Lee, V. M.-Y. (2019). α-Synuclein (αSyn) Preformed Fibrils Induce Endogenous αSyn Aggregation, Compromise Synaptic Activity and Enhance Synapse Loss in Cultured Excitatory Hippocampal Neurons. J. Neurosci. 39, 5080–5094. doi: 10.1523/JNEUROSCI.0060-19.2019
Wu, Y., Li, X., Zhu, J. X., Xie, W., Le, W., Fan, Z., et al. (2011). Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neurosignals 19, 163–174. doi: 10.1159/000328516
Xia, Y., Zhang, G., Han, C., Ma, K., Guo, X., Wan, F., et al. (2019). Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis. 10, 1–15. doi: 10.1038/s41419-019-1404-9
Xilouri, M., Brekk, O. R., and Stefanis, L. (2016). Autophagy and Alpha-Synuclein: relevance to Parkinson’s Disease and Related Synucleopathies. Mov. Disord. 31, 178–192. doi: 10.1002/mds.26477
Xilouri, M., Vogiatzi, T., Vekrellis, K., Park, D., and Stefanis, L. (2009). Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One 4:e5515. doi: 10.1371/journal.pone.0005515
Xu, Y., Liu, C., Chen, S., Ye, Y., Guo, M., Ren, Q., et al. (2014). Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson’s disease. Cell. Signal. 26, 1680–1689. doi: 10.1016/j.cellsig.2014.04.009
Yamin, G., Uversky, V. N., and Fink, A. L. (2003). Nitration inhibits fibrillation of human alpha-synuclein in vitro by formation of soluble oligomers. FEBS Lett. 542, 147–152. doi: 10.1016/s0014-5793(03)00367-3
Yan, C., Liu, J., Gao, J., Sun, Y., Zhang, L., Song, H., et al. (2019). IRE1 promotes neurodegeneration through autophagy-dependent neuron death in the Drosophila model of Parkinson’s disease. Cell Death Dis. 10, 1–15. doi: 10.1038/s41419-019-2039-6
Yang, F., Yang, Y., Mao, C., Liu, L., Zheng, H., Hu, L., et al. (2013). Crosstalk between the proteasome system and autophagy in the clearance of α-synuclein. Acta Pharmacol. Sin. 34, 674–680. doi: 10.1038/aps.2013.29
Yong, A. C., Tan, Y. J., Zhao, Y., Lu, Z., Ng, E. Y., Ng, S. Y., et al. (2020). SNCA Rep1 microsatellite length influences non-motor symptoms in early Parkinson’s disease. Aging 12, 20880–20887. doi: 10.18632/aging.104111
Yoshino, H., Hirano, M., Stoessl, A. J., Imamichi, Y., Ikeda, A., Li, Y., et al. (2017). Homozygous alpha-synuclein p. A53V in familial Parkinson’s disease. Neurobiol. Aging 57, 248.e7–248.e12.
Ysselstein, D., Joshi, M., Mishra, V., Griggs, A. M., Asiago, J. M., McCabe, G. P., et al. (2015). Effects of impaired membrane interactions on α-synuclein aggregation and neurotoxicity. Neurobiol. Dis. 79, 150–163. doi: 10.1016/j.nbd.2015.04.007
Yu, Q.-J., Yu, S.-Y., Zuo, L.-J., Lian, T.-H., Hu, Y., Wang, R.-D., et al. (2018). Parkinson disease with constipation: clinical features and relevant factors. Sci. Rep. 8:567. doi: 10.1038/s41598-017-16790-8
Yun, S. P., Kim, D., Kim, S., Kim, S., Karuppagounder, S. S., Kwon, S.-H., et al. (2018). α-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol. Neurodegener. 13:1. doi: 10.1186/s13024-017-0233-5
Zarranz, J. J., Alegre, J., Gómez-Esteban, J. C., Lezcano, E., Ros, R., Ampuero, I., et al. (2004). The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173. doi: 10.1002/ana.10795
Zhang, H., Maitta, R. W., Bhattacharyya, P. K., Florea, A. D., Sen, F., Wang, Q., et al. (2011). γ-Synuclein is a promising new marker for staining reactive follicular dendritic cells, follicular dendritic cell sarcoma, Kaposi sarcoma, and benign and malignant vascular tumors. Am. J. Surg. Pathol. 35, 1857–1865. doi: 10.1097/PAS.0b013e3182297c2e
Zhang, N.-Y., Tang, Z., and Liu, C.-W. (2008). alpha-Synuclein protofibrils inhibit 26 S proteasome-mediated protein degradation: understanding the cytotoxicity of protein protofibrils in neurodegenerative disease pathogenesis. J. Biol. Chem. 283, 20288–20298. doi: 10.1074/jbc.M710560200
Zhang, W., Wang, T., Pei, Z., Miller, D. S., Wu, X., Block, M. L., et al. (2005). Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J. 19, 533–542. doi: 10.1096/fj.04-2751com
Zhang, Z., Shen, Y., Luo, H., Zhang, F., Peng, D., Jing, L., et al. (2018). MANF protects dopamine neurons and locomotion defects from a human α-synuclein induced Parkinson’s disease model in C. elegans by regulating ER stress and autophagy pathways. Exp. Neurol. 308, 59–71. doi: 10.1016/j.expneurol.2018.06.016
Zharikov, A. D., Cannon, J. R., Tapias, V., Bai, Q., Horowitz, M. P., Shah, V., et al. (2015). shRNA targeting α-synuclein prevents neurodegeneration in a Parkinson’s disease model. J. Clin. Invest. 125, 2721–2735. doi: 10.1172/JCI64502
Zharikov, A., Bai, Q., De Miranda, B. R., Van Laar, A., Greenamyre, J. T., and Burton, E. A. (2019). Long-term RNAi knockdown of α-synuclein in the adult rat substantia nigra without neurodegeneration. Neurobiol. Dis. 125, 146–153. doi: 10.1016/j.nbd.2019.01.004
Zheng, L.-F., Song, J., Fan, R.-F., Chen, C.-L., Ren, Q.-Z., Zhang, X.-L., et al. (2014). The role of the vagal pathway and gastric dopamine in the gastroparesis of rats after a 6-hydroxydopamine microinjection in the substantia nigra. Acta Physiol. (Oxf) 211, 434–446. doi: 10.1111/apha.12229
Zhu, M., and Fink, A. L. (2003). Lipid binding inhibits alpha-synuclein fibril formation. J. Biol. Chem. 278, 16873–16877. doi: 10.1074/jbc.M210136200
Keywords: α-synuclein, autophagy, neurotoxicity, gut–brain axis, Parkinson’s disease
Citation: Ray B, Mahalakshmi AM, Tuladhar S, Bhat A, Srinivasan A, Pellegrino C, Kannan A, Bolla SR, Chidambaram SB and Sakharkar MK (2021) “Janus-Faced” α-Synuclein: Role in Parkinson’s Disease. Front. Cell Dev. Biol. 9:673395. doi: 10.3389/fcell.2021.673395
Received: 27 February 2021; Accepted: 15 April 2021;
Published: 28 May 2021.
Edited by:
Franziska Richter, University of Veterinary Medicine Hannover, GermanyReviewed by:
Raman Chandrasekar, Kansas State University, United StatesMathieu Bourdenx, Albert Einstein College of Medicine, United States
Matthew Robert Pratt, University of Southern California, Los Angeles, United States
Copyright © 2021 Ray, Mahalakshmi, Tuladhar, Bhat, Srinivasan, Pellegrino, Kannan, Bolla, Chidambaram and Sakharkar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saravana Babu Chidambaram, YmFidXB1YmxpY2F0aW9uc0BnbWFpbC5jb20=; Meena Kishore Sakharkar, bWVlbmEuc2FraGFya2FyQHVzYXNrLmNh