 Shun Yan
Shun Yan Jin Lu
Jin Lu Kai Jiao
Kai Jiao- Department of Genetics, The University of Alabama at Birmingham, Birmingham, AL, United States
The cardiac neural crest cells (cNCCs) is a transient, migratory cell population that contribute to the formation of major arteries and the septa and valves of the heart. Abnormal development of cNCCs leads to a spectrum of congenital heart defects that mainly affect the outflow region of the hearts. Signaling molecules and transcription factors are the best studied regulatory events controlling cNCC development. In recent years, however, accumulated evidence supports that epigenetic regulation also plays an important role in cNCC development. Here, we summarize the functions of epigenetic regulators during cNCC development as well as cNCC related cardiovascular defects. These factors include ATP-dependent chromatin remodeling factors, histone modifiers and DNA methylation modulators. In many cases, mutations in the genes encoding these factors are known to cause inborn heart diseases. A better understanding of epigenetic regulators, their activities and their roles during heart development will ultimately contribute to the development of new clinical applications for patients with congenital heart disease.
cNCCs Contribute to Cardiovascular Development
Neural crest cells are multipotent stem-like cells that are exclusive to vertebrate embryos. They are formed at the ectoderm-neural ectoderm border through epithelial-to-mesenchymal-transition during embryonic development. After formation, they quickly migrate ventrally to various destinations where they differentiate into different cell types based on local instructive cues (Hutson and Kirby, 2003; Snider et al., 2007; Keyte and Hutson, 2012; Lin et al., 2012; Keyte et al., 2014; Plein et al., 2015). The cardiac neural crest cells (cNCCs) is a subpopulation of NCCs that is derived from the dorsal neural tube between the otic placode and the posterior border of the third somite (Hutson and Kirby, 2003; Snider et al., 2007; Keyte and Hutson, 2012; Lin et al., 2012; Keyte et al., 2014; Plein et al., 2015). These cells migrate to the circumpharyngeal ridge, briefly pause there, and then invade into the newly formed pharyngeal arches (PAs) 3, 4, and 6. In PAs, they proliferate and become the major source of PA mesenchyme. Some cNCCs in PAs differentiate into vessel smooth muscle cells (SMCs) to support the formation of pharyngeal arch arteries (PAAs) and further remodeling of PAAs into aortic arch arteries. A subgroup of cNCCs continue to migrate into cardiac outflow tract (OFT) cushions and act together with endocardial-derived mesenchymal cells to separate the common OFT into the aorta and pulmonary trunk. In addition, OFT cNCCs participate in valvulogenesis, and some of them eventually become interstitial cells of semilunar valves (Hutson and Kirby, 2003; Snider et al., 2007; Keyte and Hutson, 2012; Lin et al., 2012; Keyte et al., 2014; Plein et al., 2015). Some recent studies have shown that cNCCs can also differentiate into cardiomyocytes in zebrafish, chicken and mice (George et al., 2020).
The critical roles of cNCCs in PAA development and OFT septation are well established in mammalian development (Hutson and Kirby, 2003; Snider et al., 2007; Keyte and Hutson, 2012; Lin et al., 2012; Keyte et al., 2014; Plein et al., 2015). Abnormalities in cNCCs may lead to a spectrum of congenital heart defects, including ventricular septal defect (VSD), overriding aorta, double outlet right ventricle (DORV), persistent truncus arteriosus (PTA), transposition of the great arteries, tetralogy of Fallot (TOF) and interruption of the aortic arch (IAA; Hutson and Kirby, 2003; Snider et al., 2007; Keyte and Hutson, 2012; Lin et al., 2012; Keyte et al., 2014; Plein et al., 2015). PAA and OFT defects account for 20 to 30% of congenital heart disease (CHD), one of the most common birth defects that affects about 1–5% of newborns every year (Bruneau, 2008). While signaling molecules and transcription factors are the best studied regulatory events controlling cNCC development, growing evidence in recent years supports the idea that epigenetic regulating factors also play crucial roles for normal development of cNCCs during mammalian embryogenesis.
Epigenetic Regulators of cNCCs
In this section, we will summarize the known functions of epigenetic regulators during cNCC development that have been reported in recent years. These factors are classified into three groups: ATP-dependent chromatin remodeling factors, histone modifiers and DNA methylation modulators.
ATP-Dependent Chromatin Remodeling Factors
The ATP-dependent chromatin remodeling complexes utilize energy from ATP hydrolysis to alter nucleosome structure or conformation, thereby regulating the accessibility of DNA to transcription factors and other regulators (Wu et al., 2009; Hota and Bruneau, 2016; Runge et al., 2016; Clapier et al., 2017). There are four major families of SWI-like ATP-dependent chromatin remodeling complexes: SWI/SNF (switch/sucrose non-fermentable), ISWI (imitation SWI), INO80 (inositol requiring 80), and CHD (chromodomain helicase DNA-binding).
(i) Coffin-Siris syndrome is a rare genetic disease affecting multiple body systems such as the head, face and heart. The associated cardiac anomalies include VSD, atrial septal defect (ASD), TOF and patent ductus arteriosus (PDA; Schrier et al., 2012; Kosho et al., 2013). More than 80% of Coffin-Siris syndrome cases are caused by mutations in one of the following genes: Arid1A, Arid1B, Brg1, Brm, Snf5/Ini1, or BAF57, which encode the subunits of the BRG1-associated factors (BAF) chromatin-remodeling complex (vertebrate homolog of the SWI/SNF complex; Schrier et al., 2012; Kosho et al., 2013, 2014). Brg1 (brahma-relate gene 1), which encodes an ATPase subunit of BAF complex, plays crucial roles in cardiovascular development. In zebrafish, Brg1 is required for neural crest induction and differentiation (Eroglu et al., 2006). Brg1-null mutant mice are lethal prior to the initiation of decidualization, preventing the analysis of the role of BRG1 in the neural crest (Bultman et al., 2000). Brg1 haploinsufficient mice have congenital heart defects such as ASD, VSD, and dilated hearts at birth (Takeuchi et al., 2011). While these defects were thought to be due to combined abnormalities in different cell types, including cNCCs, no further study was performed on NCCs in Brg1± mice (Takeuchi et al., 2011). To address its specific role in NCCs, Brg1 was specifically inactivated in NCCs using a conditional gene inactivation approach (Li et al., 2013). NCC specific deletion of Brg1 results in aberrant patterning of PAAs (loss of PAA3 and PAA4) and shortened OFT at E10.5. In NCCs, BRG1 suppresses the expression of Ask1 (an apoptosis factor) and p21cip1 (a cell cycle inhibitor) to decrease cell death and promote cell proliferation. Furthermore, BRG1 also promotes Myh11 expression to support differentiation of NCCs into SMCs. These results suggest that Brg1 maintains cNCC progenitor pool by inhibiting apoptosis and promoting proliferation, regulating cNCC migration to the OFT and helping cNCC differentiation into vascular SMCs (Li et al., 2013). In addition to the studies on Brg1, NCC-specific Arid1A mutant mice also display OFT and posterior PAA patterning defects (incomplete PAA6 formation) at E10.5 (Chandler and Magnuson, 2016). Collectively, these data suggest that SWI/SNF remodelers play essential roles at various stages of cNCC development.
(ii) Haploinsufficiency of CHD7 gene, a member of CHD family, is the major cause of CHARGE syndrome (Vissers et al., 2004; Zentner et al., 2010; Bergman et al., 2011; Corsten-Janssen et al., 2013; Basson and van Ravenswaaij-Arts, 2015; Micucci et al., 2015; Corsten-Janssen and Scambler, 2017; Pauli et al., 2017; van Ravenswaaij-Arts and Martin, 2017). CHARGE syndrome is a genetic disorder characterized by abnormalities in multiple NCC-derived organs including the eyes, ears and heart. The associated congenital heart defects in CHARGE patients include TOF, DORV, VSD, atrioventricular canal defect, PDA, pulmonary stenosis and interrupted aortic arch type B (IAA-B; Cyran et al., 1987; Blake et al., 1990; Lin et al., 1990; Tellier et al., 1998; Oswal et al., 2014). In mouse and Xenopus, CHD7 expression is detected in pre-migratory and migratory neural crest cells (Bajpai et al., 2010; Fujita et al., 2014). While CHD7 null mutant mice are embryonic lethal, CHD7 heterozygous mutant mice display variable cardiovascular defects including IAA-B and VSD, similar to that observed in CHARGE patients (Bosman et al., 2005; Randall et al., 2009). The deletion of CHD7 in mouse NCCs using Wnt1-Cre2 driver leads to severe conotruncal defects and perinatal lethality, resulting from increased NCC apoptosis along with impaired NCC migration and differentiation (Yan et al., 2020). Multiple evidence suggests that CHD7 functionally interacts with other chromatin remodelers like PBAF (polybromo- and BAF) and CHD8 (chromodomain helicase DNA-binding 8). In human NC-like cells, CHD7 cooperates with PBAF to control the activation of genes (including Sox9 and Twist1) important for NCC development (Bajpai et al., 2010). In addition, CHD7 functions with BRG1 to activate PlexinA2, a SEMA3-family receptor that is required for guiding cNCCs into the OFT (Li et al., 2013). Our recent study (Yan et al., 2020) revealed that CHD7 interacts with WDR5, which is a core component of H3K4 methyltransferase complexes (Eissenberg and Shilatifard, 2010; Justin et al., 2010; Black et al., 2012; Zhang and Liu, 2015). Our data showed for the first time that CHD7 regulates cNCC development through both nucleosome remodeling and recruitment of histone modifying enzymes to target loci.
(iii) Williams Syndrome (WS) is a congenital disorder characterized by developmental delays, learning challenges and cardiovascular disease. It occurs in approximately one per 7,500 births, and associated heart defects include supravalvular aortic stenosis and pulmonary stenosis. Most cases of WS are caused by the spontaneous deletion of a specific region on chromosome 7. One gene within that region is Baz1B (bromodomain adjacent to zinc finger domain 1B), which encodes WSTF (Williams Syndrome transcription factor). WSTF is a major subunit of two distinct ATP-dependent chromatin remodeling complexes: WICH (WSTF-ISWI chromatin remodeling complex), a subclass of the SWI/SNF class and WINAC (WSTF including the nucleosome assembly complex), a subclass of the ISWI class (Barnett and Krebs, 2011). While Baz1B null mutant mice are neonatal lethal, at E10.5 the mutant embryos have hypoplastic fourth pharyngeal arch artery. Additionally, around 10% of Baz1B haploinsufficient neonates exhibit cardiovascular abnormalities resembling those observed in autosomal-dominant WS patients (Yoshimura et al., 2009). In Xenopus embryos, WSTF is expressed in the migratory neural crest cells, and the knockout of Baz1B results in severe defects in neural crest migration and maintenance (Barnett et al., 2012). Thus, these data indicate that WSTF malfunction contributes to WS possibly via improper epigenetic regulation of cNCCs.
Histone Modifiers
Two of each core histones (H2A, H2B, H3, and H4) form an octameric structure called the nucleosome core, which associates with wrapped DNA to organize into a nucleosome, the basic building block of chromatin. The core histones are highly evolutionary conserved, and they can be modified in a variety of ways, including methylation, acetylation, phosphorylation and ubiquitination (Kouzarides, 2007). These post-translational modifications of histone proteins influence gene expression by altering the histone-DNA interaction or by acting as markers that recruit specific histone modifiers (Kouzarides, 2007).
Histone Methylation Mainly Occurs on Lysine and Arginine Residues on Histone Side Chains
Lysine can be mono-, di-, or tri-methylated, whereas arginine can be mono- or di-methylated. Histone methylation is associated with both transcriptional activation and repression (Kouzarides, 2007). We will review the most characterized histone methylations whose dysregulation may be associated with CHDs.
(i) H3K4 methylation and H3K27 demethylation
Kabuki syndrome is a rare congenital disorder that affects multiple parts of the body. It is characterized by a distinctive set of facial features, short stature, skeletal abnormalities, intellectual disability and heart defects including coarctation of the aorta, ASD or VSD (Digilio et al., 2001, 2017). Kabuki syndrome is usually caused by mutations in two genes: KMT2D (histone-lysine N-methyltransferase 2D) and KDM6A (lysine-specific demethylase 6A; Ng et al., 2010; Hannibal et al., 2011; Lederer et al., 2012; Miyake et al., 2013). The human KMT2D gene (also known as MLL2 or MLL4) encodes for a ubiquitously expressed H3K4 methylase, which is predominantly associated with active gene transcription (Shpargel et al., 2017). Heterozygous mutations in KMT2D are identified in more than 50% of Kabuki patients, most of which lead to the premature termination of the protein product, likely resulting in reduced activity of the KMT2D protein (Ng et al., 2010; Lederer et al., 2012; Bögershausen and Wollnik, 2013; Micale et al., 2014). A smaller percentage of Kabuki individuals (less than 10%) carry mutations in KDM6A, a gene that encodes a H3K27 demethylase, which removes repressive chromatin modifications and enables gene transcription (Shpargel et al., 2017).
Knockdown of Kmt2d in zebrafish results in craniofacial, brain and heart abnormalities close to the Kabuki syndromic features. The Kmt2d morphants exhibit defects in heart looping, which lead to abnormal development of the atria and/or ventricle, as well as prominent bulging of the myocardial wall (Van Laarhoven et al., 2015). A Kabuki Xenopus model reveals that Kmt2d is required for neural crest cell formation and migration. The loss-of-function of Kmt2d correlates with reduced H3K4me1 and H3K27Ac modifications (Schwenty-Lara et al., 2020). Additionally, KMT2D haploinsufficiency impairs differentiation of the cultured human neural crest cells through dysregulation of H3K4me3 and H3K27Ac at the TFAP2A locus, which is a master regulator of NCC lineage progression (Lavery et al., 2020). In mouse, deletion of Kmt2d in cardiac precursors and myocardium results in embryonic lethality and cardiac defects including disorganized interventricular septum, thin compact myocardium and OFT septation defects. The level of H3K4me1 and H3K4me2 at enhancers and promoters is decreased in mutant embryos (Ang et al., 2016). While the deletion of Kmt2d in NCCs in mice leads to severe cranial facial defects, no cardiovascular phenotype has been reported (Shpargel et al., 2020). Therefore, it remains unclear if Kmt2d in NCCs is important for cardiovascular development. It is possible that the loss of KMT2D in cNCCs may be compensated by other histone lysine methyltransferases.
Neural crest cell-specific knockout of Kdm6a in mice leads to clinical features of Kabuki syndrome including heart defects (aorta coarctation, PDA and VSD; Shpargel et al., 2017). Kdm6a NCC loss-of-function mutants exhibit reduced post-migratory embryonic neural crest cells. In mouse Kdm6a-knockout NCCs, a subset of the H3K27me3 peaks is elevated compared with wild type cells, supporting its role of H3K27me3 demethylation in NCCs. However, multiple lines of evidence presented in this paper suggest that KDM6A also possess histone demethylase-independent activity critical for NCC development (Shpargel et al., 2017). First, the male Kdm6a knockout mice displayed weaker phenotypes than female mutant mice due to the partial redundancy of Uty, which encodes a Y-chromosome demethylase-dead homolog of KDM6A. Second, the methylation status of the majority of KDM6A target genes is not altered in NCCs with Kdm6a deleted. Third, inactivation of the demethylase activity of KDM6A via a homozygous knockin allele of Kdm6a does not lead to aberrant craniofacial development in mice embryos. Fourth, while most Kabuki causative mutations in KDM6A impair the demethylase activity of KDM6A, several mutations do not. Collectively, all these data support the idea that KDM6A promotes normal NCC development through both demethylase -dependent and –independent activities.
(ii) H3K9 methylation and demethylation
Kleefstra Syndrome (KS) is a rare genetic disorder characterized by childhood hypotonia, intellectual disability, and distinctive facial features. Approximately 40% of KS patients have CHDs, including ASD/VSD, TOF, aortic coarctation, bicuspid aortic valve, and pulmonic stenosis. Mutations in EHMT1 (euchromatic histone-lysine N-methyltransferase 1), a gene encoding H3K9 methyltransferase, is the major cause of KS (Kleefstra et al., 2005, 2006). Ehmt1± mice recapitulate the core developmental features of KS phenotype; however, no heart abnormalities have been reported (Balemans et al., 2014). Therefore, an experimental animal model for cardiac manifestations in KS should be developed.
Jumonji, encoded by Jmj (jumonji) gene, functions as histone demethylase. Jmj expression is detected in the heart throughout all the cardiac developmental stages. Homozygous Jmj mutant mice die soon after birth with various cardiovascular defects such as VSD and DORV (Lee et al., 2000). Knockdown of JmjD2A (Jumonji domain containing 2A) in chick embryos leads to down-regulated expression of neural crest specifier genes (Sox10, Snail2, and FoxD3), resulting in abnormal neural crest derivatives at later embryonic development stages (Strobl-Mazzulla et al., 2010). Moreover, JmjD2A is localized at the promoter regions of Sox10 and Sn6ail2, which are occupied by H3K9me3 (a heterochromatin epigenetic mark), and JmjD2A knock-down represses Sox10 transcription by inhibiting H3K9me3 demethylation (Strobl-Mazzulla et al., 2010). Thus, JmjD2A is required in neural crest cell development by carrying out its demethylase activity.
(iii) H3K36 methylation
Wolf-Hirschhorn Syndrome (WHS) is a rare congenital neurodevelopmental disorder characterized by prenatal and postnatal growth deficiency, intellectual disability, seizure, craniofacial malformation and heart defects (Cooper, 1961; Wolf et al., 1965). The associated cardiac defects include ASD. The putative candidate genes responsible for WHS are WHS Candidates 1 and 2 (WHSC1 and WHSC2) and LETM1 (leucine zipper and EF-hand containing transmembrane protein 1). The most established role of WHSC1 is that of an H3K36me3-specific histone methyltransferase, which is associated with both transcriptional activation and inhibition. In mouse embryonic hearts, loss of WHSC1 leads to reduced H3K36me3 at the Pdgfra locus, up-regulated transcription of Pdgfra and WHS-like defects including congenital cardiovascular anomalies (Nimura et al., 2009; Yu et al., 2017). Therefore, WHSC1 contributes to WHS by carrying out its H3K36 methyltransferase function. The evidence supporting the role of WHSC1 in regulating cNCC development mainly comes from a Xenopus study. WHSC1 expression is enriched in PAs of X. laevis embryos, and Whsc1 deletion in vivo impairs neural crest cell migration into the PAs (Mills et al., 2019).
Histone Acetylation/Deacetylation Is Mediated by Histone Acetyltransferases/Histone Deacetylases, Respectively
Histone acetylation is associated with active transcription, while histone deacetylation corresponds to transcriptional repression (Jenuwein and Allis, 2001). In mice, NCC-deletion of Hdac3, which encodes histone deacetylase 3, results in perinatal lethality and severe cardiovascular defects including IAA-B, PTA, VSD, DORV, and aortic arch hypoplasia. The cardiovascular abnormalities are probably due to deficiency in smooth muscle differentiation, indicating that HDAC3 is required for differentiation of cNCCs into SMCs that are involved in septation of the distal OFT in mice (Singh et al., 2011). Although other Hdacs (Hdac1, Hdac4, and Hdac8) also play important roles in neural crest development, there is no evidence for their requirement in cNCCs (Haberland et al., 2009; DeLaurier et al., 2012; Ignatius et al., 2013).
DNA Methylation Modulators
DNA methylation is a common epigenetic mechanism that typically acts to repress gene expression (Moore et al., 2013). In mammals, it is associated with numerous key processes including genomic imprinting, inactivation of the silent X chromosome, and normal prenatal development (Paulsen and Ferguson-Smith, 2001; Smith and Meissner, 2013; Elhamamsy, 2017). The mechanism of DNA methylation involves the covalent transfer of a methyl group to C-5 position of cytosine, and generally occurs in the context of CpG dinucleotides (Hu et al., 2014a). This process is mediated by the family of DNA methyltransferases, which includes DNMT1, DNMT3A, 3B, and 3L (Hu et al., 2014a). DNMT1 is considered to be a key maintenance methyltransferase, while DNMT3A and 3B act as de novo methyltransferases of either unmethylated or hemi-methylated DNA (Hu et al., 2014a). DNMT3L is inactive on its own, instead acting as a general stimulating factor for DNMT3A and 3B (Wienholz et al., 2010).
(i) DNMT3A and 3B
Both DNMT3A and 3B have been shown to be crucial for normal mammalian development (Hu et al., 2014a). For example, Dnmt3A homozygous null mice typically die 4 weeks after birth, and Dnmt3B homozygous null mice die in utero (Okano et al., 1999). In the chicken embryo, DNMT3A is primarily expressed at sites of neural crest formation, and knockdown of Dnmt3A blocks neural crest specification (Hu et al., 2012). In particular, knockdown of Dnmt3A results in expansion of neural tube genes Sox2 and Sox3 into the neural crest region and down-regulation of neural crest specifier genes (Snail2, Sox9, TNIP1, FoxD3, Sox8, and Sox10). Furthermore, DNMT3A directly represses the transcription of Sox2 and Sox3 through promoter DNA methylation (Hu et al., 2012). Together, these results indicate that DNMT3A functions as a molecular switch that promotes neural crest cell fate through repression of neural tube genes (Hu et al., 2012).
DNMT3B is broadly expressed throughout the neural plate during the gastrula stages, but its expression becomes subsequently restricted to the dorsal neural tube and migratory neural crest cells (Hu et al., 2014b). Knockdown of DNMT3B in human embryonic stem cells results in upregulation of neural crest specifier genes (Pax3/7, NGFR, FoxD3, Sox10, and Snail2; Martins-Taylor et al., 2012). In chicken embryos, knockdown of Dnmt3B results in prolonging of neural crest production by the neural tubes and excess migratory neural crest cells (Hu et al., 2014a). DNMT3B has also been shown to repress Sox10 directly through promoter methylation (Hu et al., 2014a). These results suggest that DNMT3B restricts the temporal window in which the dorsal neural tube undergoes epithelial-to-mesenchymal transition to produce neural crest cells (Hu et al., 2014a).
Immunodeficiency with centromeric instability and facial anomalies (ICF) syndrome is a rare autosomal recessive disease with just less than 70 known cases at present (Weemaes et al., 2013). Approximately 50% of ICF cases have mutations in DNMT3B, and are designated as ICF1. Nearly all patients with ICF1 exhibit craniofacial abnormalities such as hypertelorism and flat nasal bridge as well as neurological dysfunction (Weemaes et al., 2013). Furthermore, three patients with ICF1 were reported to have congenital heart defects: two with VSD and one with ASD (Weemaes et al., 2013). These abnormalities are consistent with the crucial role of DNMT3B in neural crest development. Mouse models for ICF syndrome show similar congenital defects. Most of the Dnmt3B null mice exhibit embryonic lethality between E13.5 to E16.5 (Ueda et al., 2006). Sectioning of the recovered Dnmt3B null mutants show that the ventricular septum is not closed at E14.5 and E15.5, although the ventricular septum is normally closed by E13.5. Furthermore, mice with ICF1 mutations, although alive at birth, exhibit craniofacial abnormalities similar to those found in ICF1 patients (Ueda et al., 2006). In contrast to the above studies, condition knockout of Dnmt3B in mice using Wnt1-cre and Sox10-cre only results in subtle migration defects, and does not result in any abnormalities in cardiac or craniofacial structures (Jacques-Fricke et al., 2012). This may indicate that the defects in neural crest derivatives are due to the requirement of DNMT3B in neighboring cell types (Jacques-Fricke et al., 2012).
(ii) Folic acid
Folic acid is a B-vitamin crucial for vertebrate development (Jimenez et al., 2018). Folate is converted into tetrahydrofolate, which is then used as a cofactor for nucleotide synthesis and generation of S-adenosylmethionine (SAM; Rosenquist, 2013). SAM is the primary source of methyl groups for DNA and histone methylation (Jimenez et al., 2018). The relationship between folate and CHD has been well studied, and numerous studies have shown that maternal folate supplementation reduces the risk of CHD, especially conotruncal defects (Rosenquist, 2013). Folate deficiency results in severe neural tube defects (Beaudin and Stover, 2007), as well as neural-crest associated defects such as orofacial abnormalities, VSD, as well as enlarged pericardial cavity (Rosenquist et al., 1996; Li et al., 2011; Wahl et al., 2015; Jimenez et al., 2018). Currently, there is evidence that folate regulation of DNA methylation may play a role in cNCC development. Studies have shown that folate levels directly correlate with DNA methylation (Choi et al., 2005; Pufulete et al., 2005), and maternal hypomethylation is associated with an increased risk of CHD (Chowdhury et al., 2011). Folate transporters FolR1 and Rfc1 are required for Sox2 methylation on neural crest region in mouse embryos, and knockdown of FolR1 and Rfc1 results in severe reduction of Sox10 expression and associated orofacial defects (Jimenez et al., 2018). Finally, in one human fetus with DORV, VSD, patent foramen ovale and coarctation of the preductal aorta, hypermethylation with associated decrease in expression of the folate metabolism gene MTHFS is detected (Serra-Juhé et al., 2015). Ultimately, more studies are needed to further establish the relationship between folate, DNA methylation and cNCC development.
Summary and Perspective
We summarize the major genes discussed above in Table 1. Different types of epigenetic regulators, including chromatin remodeling factors, histone modifiers and DNA methylation modulators, are all involved in cNCC development (Figure 1). Significantly, mutations in many of these genes lead to inborn heart defects in human patients, and functional studies using various model systems have provided strong evidence supporting their critical roles in cNCCs.
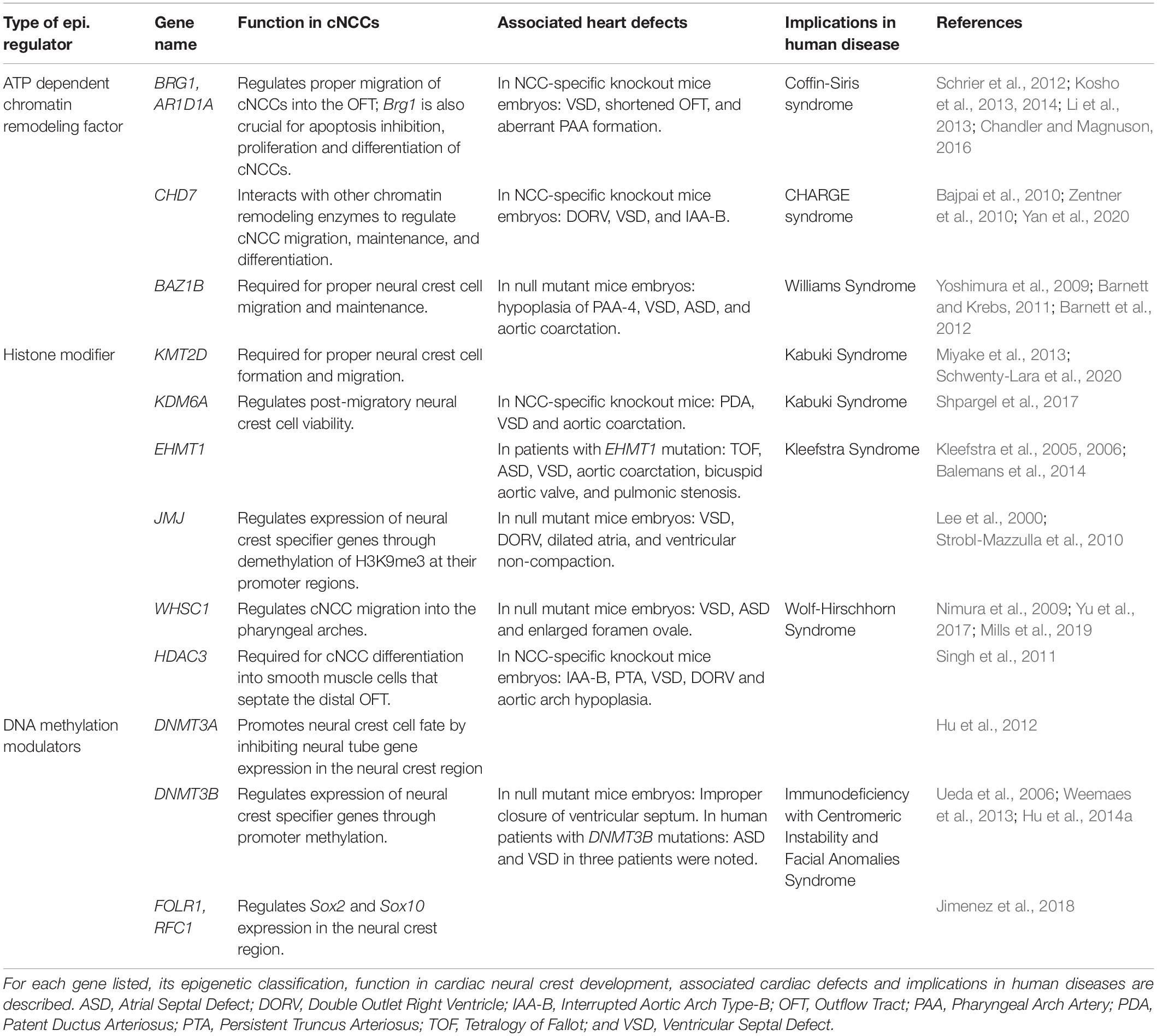
Table 1. A summary of epigenetic regulators of cardiac neural crest cells (cNCCs).
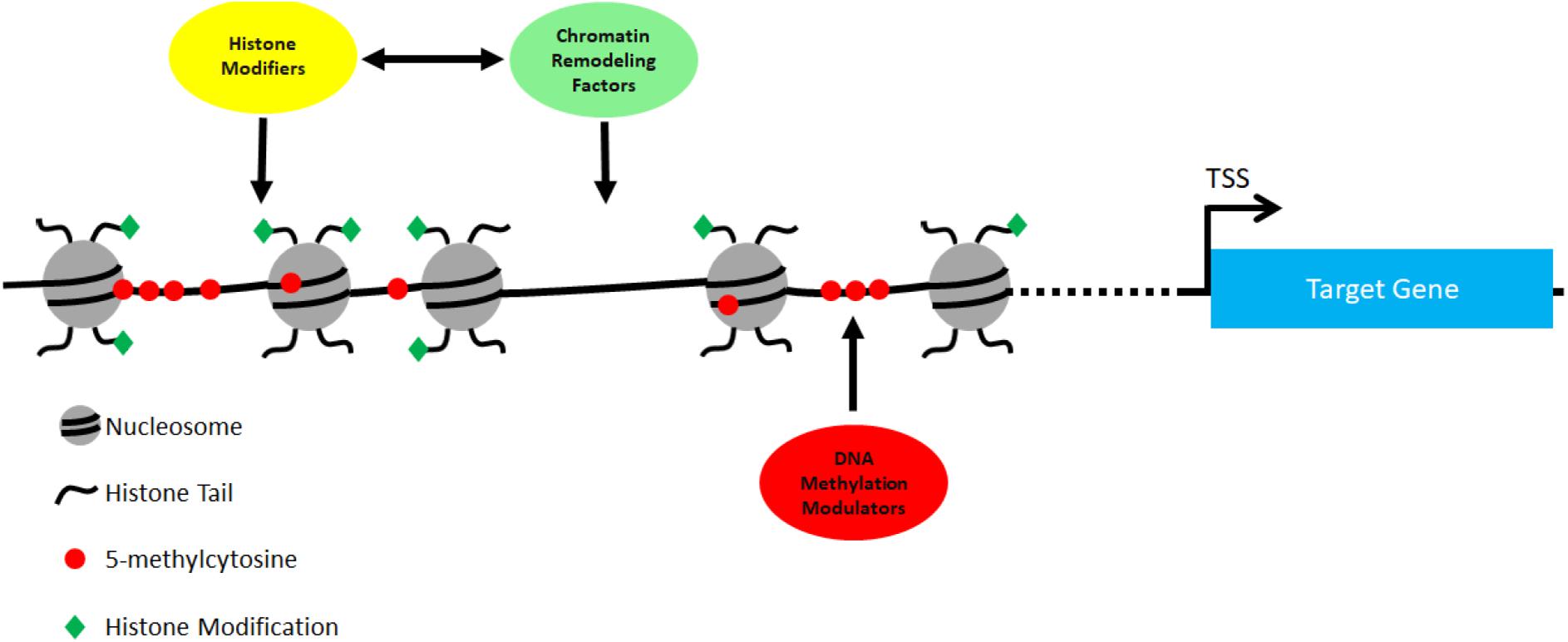
Figure 1. All three types of epigenetic regulators act together to regulate gene expression in cNCCs. ATP-dependent chromatin regulators regulate the density and positions of nucleosomes in target sites, while histone modifiers regulates post-translational modifications in the histone tails. There are many examples showing the interaction, physical and functional, between chromatin regulators and histone modifiers (see main text). In addition, DNA methylation modulators regulate the status of DNA methylation at target sites to control gene expression in cNCCs.
Despite the significant progress made on epigenetic regulation of cNCC development, there remain some particularly challenging questions warranting further investigation. First, many epigenetic regulators are expressed in different cell types, and how these factors execute their specific functions in cNCCs and other cardiac cell types remain highly elusive. One likely explanation is that these regulators interact with cell-type-specific transcriptional regulators to control expression of cell-type specific genes. In this case, understanding their interaction is particularly important, as it holds the key to designing specific therapeutic strategies for a particular cell type. Second, numerous examples have shown that epigenetic regulators from different classes can act in concert to regulate gene expression. How such harmony is achieved in cNCCs and other cell types remains largely unknown. This question is particularly challenging, as it requires a good understanding on how regulatory mechanisms act individually and in tandem. Third, a single epigenetic regulator may have distinct functions. For example, we recently showed that in addition to its well-known function in nucleosome remodeling, CHD7 can also recruit histone modifying enzymes to target loci (Yan et al., 2020). This latter activity of CHD7 does not reply on nucleosome remodeling. Another example would be WDR5, which not only acts in the nucleus as a core component of H3K4 methyltransferase complexes (Eissenberg and Shilatifard, 2010; Justin et al., 2010; Black et al., 2012; Zhang and Liu, 2015), but also serves as scaffolding protein between the basal body and F-actin in cilia (Kulkarni and Khokha, 2018; Kulkarni et al., 2018). Therefore, the inborn cardiac phenotypes observed in human patients with different mutations in the same gene can be due to different molecular alterations. Such information is particularly important for personalized therapeutic interventions. Overall, a better understanding of the functions of epigenetic regulators in cNCCs and their underlying mechanisms will ultimately shed light on developing new therapies for CHD.
Author Contributions
SY, JL, and KJ wrote the review manuscript. All authors contributed to the article and approved the submitted version.
Funding
Research in the authors’ laboratory is supported by R56 (HL130711), R01 (HL095783), AHA (17GRNT33410623), and CHARGE Scientific Research grants (awarded to KJ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge the many valuable publications of our colleagues, but regret that all studies could not be cited.
References
Ang, S.-Y., Uebersohn, A., Spencer, C. I., Huang, Y., Lee, J.-E., Ge, K., et al. (2016). KMT2D regulates specific programs in heart development via histone H3 lysine 4 di-methylation. Development 143, 810–821. doi: 10.1242/dev.132688
Bajpai, R., Chen, D. A., Rada-Iglesias, A., Zhang, J., Xiong, Y., Helms, J., et al. (2010). CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463, 958–962. doi: 10.1038/nature08733
Balemans, M. C., Ansar, M., Oudakker, A. R., van Caam, A. P., Bakker, B., Vitters, E. L., et al. (2014). Reduced euchromatin histone methyltransferase 1 causes developmental delay, hypotonia, and cranial abnormalities associated with increased bone gene expression in Kleefstra syndrome mice. Dev. Biol. 386, 395–407. doi: 10.1016/j.ydbio.2013.12.016
Barnett, C., and Krebs, J. E. (2011). WSTF does it all: a multifunctional protein in transcription, repair, and replication. Biochem. Cell Biol. 89, 12–23. doi: 10.1139/o10-114
Barnett, C., Yazgan, O., Kuo, H.-C., Malakar, S., Thomas, T., Fitzgerald, A., et al. (2012). Williams syndrome transcription factor is critical for neural crest cell function in Xenopus laevis. Mech. Dev. 129, 324–338. doi: 10.1016/j.mod.2012.06.001
Basson, M. A., and van Ravenswaaij-Arts, C. (2015). Functional insights into chromatin remodelling from studies on CHARGE syndrome. Trends Genet. 31, 600–611. doi: 10.1016/j.tig.2015.05.009
Beaudin, A. E., and Stover, P. J. (2007). Folate-mediated one-carbon metabolism and neural tube defects: balancing genome synthesis and gene expression. Birth Defects Res. Part C: Embryo Today: Rev. 81, 183–203. doi: 10.1002/bdrc.20100
Bergman, J. E., Janssen, N., Hoefsloot, L. H., Jongmans, M. C., Hofstra, R. M., and van Ravenswaaij-Arts, C. M. (2011). CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J. Med. Genet. 48, 334–342. doi: 10.1136/jmg.2010.087106
Black, J. C., Van Rechem, C., and Whetstine, J. R. (2012). Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol. Cell 48, 491–507. doi: 10.1016/j.molcel.2012.11.006
Blake, K., Russell-Eggitt, I., Morgan, D., Ratcliffe, J., and Wyse, R. (1990). Who’s in CHARGE? multidisciplinary management of patients with charge association. Arch. Dis. Child. 65, 217–223. doi: 10.1136/adc.65.2.217
Bögershausen, N., and Wollnik, B. (2013). Unmasking kabuki syndrome. Clin. Genet. 83, 201–211. doi: 10.1111/cge.12051
Bosman, E. A., Penn, A. C., Ambrose, J. C., Kettleborough, R., Stemple, D. L., and Steel, K. P. (2005). Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum. Mol. Genet. 14, 3463–3476. doi: 10.1093/hmg/ddi375
Bruneau, B. G. (2008). The developmental genetics of congenital heart disease. Nature 451, 943–948. doi: 10.1038/nature06801
Bultman, S., Gebuhr, T., Yee, D., La Mantia, C., Nicholson, J., Gilliam, A., et al. (2000). A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol. Cell. 6, 1287–1295. doi: 10.1016/s1097-2765(00)00127-1
Chandler, R. L., and Magnuson, T. (2016). The SWI/SNF BAF-A complex is essential for neural crest development. Dev. Biol. 411, 15–24. doi: 10.1016/j.ydbio.2016.01.015
Choi, S.-W., Friso, S., Keyes, M. K., and Mason, J. B. (2005). Folate supplementation increases genomic DNA methylation in the liver of elder rats. Br. J. Nutrition 93, 31–35. doi: 10.1079/bjn20041283
Chowdhury, S., Cleves, M. A., MacLeod, S. L., James, S. J., Zhao, W., and Hobbs, C. A. (2011). Maternal DNA hypomethylation and congenital heart defects. Birth Defects Res. Part A: Clin. Mol. Teratol. 91, 69–76. doi: 10.1002/bdra.20761
Clapier, C. R., Iwasa, J., Cairns, B. R., and Peterson, C. L. (2017). Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol. 18, 407–422. doi: 10.1038/nrm.2017.26
Cooper, H. (1961). Apparent deletion of short arms of one chromosome (4 or 5) in a child with defects of midline fusion. Mamm. Chrom. Newslett. 4:14.
Corsten-Janssen, N., Kerstjens-Frederikse, W. S., du Marchie, Sarvaas, G. J., Baardman, M. E., Bakker, M. K., et al. (2013). The cardiac phenotype in patients with a CHD7 mutation. Circulation. Cardiovascular Genet. 6, 248–254. doi: 10.1161/circgenetics.113.000054
Corsten-Janssen, N., and Scambler, P. J. (2017). Clinical and molecular effects of CHD7 in the heart. Am. J. Med. Genet. C Semin. Med. Genet. 175, 487–495.
Cyran, S. E., Martinez, R., Daniels, S., Dignan, P. S. J., and Kaplan, S. (1987). Spectrum of congenital heart disease in CHARGE association. J. Pediatr. 110, 576–578. doi: 10.1016/s0022-3476(87)80555-3
DeLaurier, A., Nakamura, Y., Braasch, I., Khanna, V., Kato, H., Wakitani, S., et al. (2012). Histone deacetylase-4 is required during early cranial neural crest development for generation of the zebrafish palatal skeleton. BMC Dev. Biol. 12:16. doi: 10.1186/1471-213X-12-16
Digilio, M. C., Gnazzo, M., Lepri, F., Dentici, M. L., Pisaneschi, E., Baban, A., et al. (2017). Congenital heart defects in molecularly proven Kabuki syndrome patients. Am. J. Med. Genet. Part A 173, 2912–2922. doi: 10.1002/ajmg.a.38417
Digilio, M. C., Marino, B., Toscano, A., Giannotti, A., and Dallapiccola, B. (2001). Congenital heart defects in Kabuki syndrome. Am. J. Med. Genet. 100, 269–274. doi: 10.1002/ajmg.1265
Eissenberg, J. C., and Shilatifard, A. (2010). Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev. Biol. 339, 240–249. doi: 10.1016/j.ydbio.2009.08.017
Elhamamsy, A. R. (2017). Role of DNA methylation in imprinting disorders: an updated review. J. Assist. Reprod. Genet. 34, 549–562. doi: 10.1007/s10815-017-0895-5
Eroglu, B., Wang, G., Tu, N., Sun, X., and Mivechi, N. F. (2006). Critical role of Brg1 member of the SWI/SNF chromatin remodeling complex during neurogenesis and neural crest induction in zebrafish. Dev. Dyn. 235, 2722–2735. doi: 10.1002/dvdy.20911
Fujita, K., Ogawa, R., Kawawaki, S., and Ito, K. (2014). Roles of chromatin remodelers in maintenance mechanisms of multipotency of mouse trunk neural crest cells in the formation of neural crest-derived stem cells. Mech. Dev. 133, 126–145. doi: 10.1016/j.mod.2014.05.001
George, R. M., Maldonado-Velez, G., and Firulli, A. B. (2020). The heart of the neural crest: cardiac neural crest cells in development and regeneration. Development 147:dev188706. doi: 10.1242/dev.188706
Haberland, M., Mokalled, M. H., Montgomery, R. L., and Olson, E. N. (2009). Epigenetic control of skull morphogenesis by histone deacetylase 8. Genes Dev. 23, 1625–1630. doi: 10.1101/gad.1809209
Hannibal, M. C., Buckingham, K. J., Ng, S. B., Ming, J. E., Beck, A. E., McMillin, M. J., et al. (2011). Spectrum of MLL2 (ALR) mutations in 110 cases of Kabuki syndrome. Am. J. Med. Genet. Part A 155, 1511–1516.
Hota, S. K., and Bruneau, B. G. (2016). ATP-dependent chromatin remodeling during mammalian development. Development 143, 2882–2897. doi: 10.1242/dev.128892
Hu, N., Strobl-Mazzulla, P., Sauka-Spengler, T., and Bronner, M. E. (2012). DNA methyltransferase3A as a molecular switch mediating the neural tube-to-neural crest fate transition. Genes Dev. 26, 2380–2385. doi: 10.1101/gad.198747.112
Hu, N., Strobl-Mazzulla, P. H., and Bronner, M. E. (2014a). Epigenetic regulation in neural crest development. Dev. Biol. 396, 159–168. doi: 10.1016/j.ydbio.2014.09.034
Hu, N., Strobl-Mazzulla, P. H., Simoes-Costa, M., Sánchez-Vásquez, E., and Bronner, M. E. (2014b). DNA methyltransferase 3B regulates duration of neural crest production via repression of Sox10. Proc. Natl. Acad. Sci. U S A. 111, 17911–17916. doi: 10.1073/pnas.1318408111
Hutson, M. R., and Kirby, M. L. (2003). Neural crest and cardiovascular development: a 20-year perspective. Birth Defects Res. C Embryo Today 69, 2–13. doi: 10.1002/bdrc.10002
Ignatius, M. S., Eroglu, A. U., Malireddy, S., Gallagher, G., Nambiar, R. M., and Henion, P. D. (2013). Distinct functional and temporal requirements for zebrafish Hdac1 during neural crest-derived craniofacial and peripheral neuron development. PLoS One 8:e63218. doi: 10.1371/journal.pone.0063218
Jacques-Fricke, B. T., Roffers-Agarwal, J., and Gammill, L. S. (2012). DNA methyltransferase 3b is dispensable for mouse neural crest development. PLoS One 7:e47794. doi: 10.1371/journal.pone.0047794
Jenuwein, T., and Allis, C. D. (2001). Translating the histone code. Science 293, 1074–1080. doi: 10.1126/science.1063127
Jimenez, N. A., Pérez, S. A. T., Sánchez-Vásquez, E., Fernandino, J. I., and Strobl-Mazzulla, P. H. (2018). Folate deficiency prevents neural crest fate by disturbing the epigenetic Sox2 repression on the dorsal neural tube. Dev. Biol. 444, S193–S201.
Justin, N., De Marco, V., Aasland, R., and Gamblin, S. J. (2010). Reading, writing and editing methylated lysines on histone tails: new insights from recent structural studies. Curr. Opin. Struct. Biol. 20, 730–738. doi: 10.1016/j.sbi.2010.09.012
Keyte, A., and Hutson, M. R. (2012). The neural crest in cardiac congenital anomalies. Differentiation 84, 25–40. doi: 10.1016/j.diff.2012.04.005
Keyte, A. L., Alonzo-Johnsen, M., and Hutson, M. R. (2014). Evolutionary and developmental origins of the cardiac neural crest: building a divided outflow tract. Birth Defects Res. C Embryo Today 102, 309–323. doi: 10.1002/bdrc.21076
Kleefstra, T., Brunner, H. G., Amiel, J., Oudakker, A. R., Nillesen, W. M., Magee, A., et al. (2006). Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am. J. Hum. Genet. 79, 370–377. doi: 10.1086/505693
Kleefstra, T., Smidt, M., Banning, M., Oudakker, A., Van Esch, H., De Brouwer, A., et al. (2005). Disruption of the gene Euchromatin Histone Methyl Transferase1 (Eu-HMTase1) is associated with the 9q34 subtelomeric deletion syndrome. J. Med. Genet. 42, 299–306. doi: 10.1136/jmg.2004.028464
Kosho, T., Okamoto, N., and Coffin-Siris Syndrome, and International Collaborators. (2014). Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. Am. J. Med. Genet. Part C: Sem. Med. Genet. 166C, 262–275. doi: 10.1002/ajmg.c.31407
Kosho, T., Okamoto, N., Ohashi, H., Tsurusaki, Y., Imai, Y., Hibi-Ko, Y., et al. (2013). Clinical correlations of mutations affecting six components of the SWI/SNF complex: detailed description of 21 patients and a review of the literature. Am. J. Med. Genet. Part A 161, 1221–1237. doi: 10.1002/ajmg.a.35933
Kouzarides, T. (2007). Chromatin modifications and their function. Cell 128, 693–705. doi: 10.1016/j.cell.2007.02.005
Kulkarni, S. S., Griffin, J. N., Date, P. P., Liem, K. F. Jr., and Khokha, M. K. (2018). WDR5 Stabilizes Actin Architecture to Promote Multiciliated Cell Formation. Dev. Cell 46, 595–610.e3.
Kulkarni, S. S., and Khokha, M. K. (2018). WDR5 regulates left-right patterning via chromatin-dependent and -independent functions. Development 145.
Lavery, W. J., Kotliar, M., Chang, C. F., Brugmann, S. A., Barski, A., and Lindsley, A. W. (2020). KMT2D Haploinsufficiency in Kabuki Syndrome Impairs Differentiation of Neural Crest Cells. FASEB J. 34, 1–1. doi: 10.1096/fasebj.2020.34.s1.09189
Lederer, D., Grisart, B., Digilio, M. C., Benoit, V., Crespin, M., Ghariani, S. C., et al. (2012). Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am. J. Hum. Genet. 90, 119–124. doi: 10.1016/j.ajhg.2011.11.021
Lee, Y., Song, A. J., Baker, R., Micales, B., Conway, S. J., and Lyons, G. E. (2000). Jumonji, a nuclear protein that is necessary for normal heart development. Circ. Res. 86, 932–938. doi: 10.1161/01.res.86.9.932
Li, J., Shi, Y., Sun, J., Zhang, Y., and Mao, B. (2011). Xenopus reduced folate carrier regulates neural crest development epigenetically. PLoS One 6:e27198. doi: 10.1371/journal.pone.0027198
Li, W., Xiong, Y., Shang, C., Twu, K. Y., Hang, C. T., Yang, J., et al. (2013). Brg1 governs distinct pathways to direct multiple aspects of mammalian neural crest cell development. Proc. Natl. Acad. Sci. U S A. 110, 1738–1743. doi: 10.1073/pnas.1218072110
Lin, A. E., Siebert, J. R., and Graham, J. M. Jr. (1990). Central nervous system malformations in the CHARGE association. Am. J. Med. Genet. 37, 304–310. doi: 10.1002/ajmg.1320370303
Lin, C. J., Lin, C. Y., Chen, C. H., Zhou, B., and Chang, C. P. (2012). Partitioning the heart: mechanisms of cardiac septation and valve development. Development 139, 3277–3299. doi: 10.1242/dev.063495
Martins-Taylor, K., Schroeder, D. I., LaSalle, J. M., Lalande, M., and Xu, R.-H. (2012). Role of DNMT3B in the regulation of early neural and neural crest specifiers. Epigenetics 7, 71–82. doi: 10.4161/epi.7.1.18750
Micale, L., Augello, B., Maffeo, C., Selicorni, A., Zucchetti, F., Fusco, C., et al. (2014). Molecular analysis, pathogenic mechanisms, and readthrough therapy on a large cohort of K abuki syndrome patients. Hum. Mutat. 35, 841–850. doi: 10.1002/humu.22547
Micucci, J. A., Sperry, E. D., and Martin, D. M. (2015). Chromodomain helicase DNA-binding proteins in stem cells and human developmental diseases. Stem Cells Dev. 24, 917–926. doi: 10.1089/scd.2014.0544
Mills, A., Bearce, E. A., Cella, R., Kim, S. W., Selig, M., Lee, S., et al. (2019). Wolf-Hirschhorn Syndrome-associated genes are enriched in motile neural crest cells and affect craniofacial development in Xenopus laevis. Front. Physiol. 10:431. doi: 10.3389/fphys.2019.00431
Miyake, N., Koshimizu, E., Okamoto, N., Mizuno, S., Ogata, T., Nagai, T., et al. (2013). MLL2 and KDM6A mutations in patients with Kabuki syndrome. Am. J. Med. Genet. Part A 161, 2234–2243.
Moore, L. D., Le, T., and Fan, G. (2013). DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38. doi: 10.1038/npp.2012.112
Ng, S. B., Bigham, A. W., Buckingham, K. J., Hannibal, M. C., McMillin, M. J., Gildersleeve, H. I., et al. (2010). Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 42, 790–793. doi: 10.1038/ng.646
Nimura, K., Ura, K., Shiratori, H., Ikawa, M., Okabe, M., Schwartz, R. J., et al. (2009). A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf–Hirschhorn syndrome. Nature 460, 287–291. doi: 10.1038/nature08086
Okano, M., Bell, D. W., Haber, D. A., and Li, E. (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257. doi: 10.1016/s0092-8674(00)81656-6
Oswal, N., Christov, G., Sridharan, S., Khambadkone, S., Bull, C., and Sullivan, I. (2014). Aberrant subclavian artery origin in tetralogy of Fallot with pulmonary stenosis is associated with chromosomal or genetic abnormality. Cardiol. Young 24, 478–484. doi: 10.1017/s1047951113000644
Pauli, S., Bajpai, R., and Borchers, A. (2017). CHARGEd with neural crest defects. Am. J. Med. Genet. C Semin. Med. Genet. 175, 478–486. doi: 10.1002/ajmg.c.31584
Paulsen, M., and Ferguson-Smith, A. C. (2001). DNA methylation in genomic imprinting, development, and disease. J. Pathol. 195, 97–110. doi: 10.1002/path.890
Plein, A., Fantin, A., and Ruhrberg, C. (2015). Neural crest cells in cardiovascular development. Curr. Top. Dev. Biol. 111, 183–200. doi: 10.1016/bs.ctdb.2014.11.006
Pufulete, M., Al-Ghnaniem, R., Rennie, J., Appleby, P., Harris, N., Gout, S., et al. (2005). Influence of folate status on genomic DNA methylation in colonic mucosa of subjects without colorectal adenoma or cancer. Br. J. Cancer 92, 838–842. doi: 10.1038/sj.bjc.6602439
Randall, V., McCue, K., Roberts, C., Kyriakopoulou, V., Beddow, S., Barrett, A. N., et al. (2009). Great vessel development requires biallelic expression of Chd7 and Tbx1 in pharyngeal ectoderm in mice. J. Clin. Invest. 119, 3301–3310.
Rosenquist, T. H. (2013). Folate, homocysteine and the cardiac neural crest. Dev. Dyn. 242, 201–218. doi: 10.1002/dvdy.23922
Rosenquist, T. H., Ratashak, S. A., and Selhub, J. (1996). Homocysteine induces congenital defects of the heart and neural tube: effect of folic acid. Proc. Natl. Acad. Sci. U S A. 93, 15227–15232. doi: 10.1073/pnas.93.26.15227
Runge, J. S., Raab, J. R., and Magnuson, T. (2016). Epigenetic regulation by ATP-Dependent chromatin-remodeling enzymes: SNF-ing out crosstalk. Curr. Top. Dev. Biol. 117, 1–13. doi: 10.1016/bs.ctdb.2015.10.009
Schrier, S. A., Bodurtha, J. N., Burton, B., Chudley, A. E., Chiong, M. A. D., D’avanzo, M. G., et al. (2012). The coffin–siris syndrome: a proposed diagnostic approach and assessment of 15 overlapping cases. Am. J. Med. Genet. Part A 158, 1865–1876. doi: 10.1002/ajmg.a.35415
Schwenty-Lara, J., Nehl, D., and Borchers, A. (2020). The histone methyltransferase KMT2D, mutated in Kabuki syndrome patients, is required for neural crest cell formation and migration. Hum. Mol. Genet. 29, 305–319. doi: 10.1093/hmg/ddz284
Serra-Juhé, C., Cuscó, I., Homs, A., Flores, R., Torán, N., and Pérez-Jurado, L. A. (2015). DNA methylation abnormalities in congenital heart disease. Epigenetics 10, 167–177. doi: 10.1080/15592294.2014.998536
Shpargel, K. B., Mangini, C. L., Xie, G., Ge, K., and Magnuson, T. (2020). The KMT2D Kabuki syndrome histone methylase controls neural crest cell differentiation and facial morphology. Development 147:dev187997. doi: 10.1242/dev.187997
Shpargel, K. B., Starmer, J., Wang, C., Ge, K., and Magnuson, T. (2017). UTX-guided neural crest function underlies craniofacial features of Kabuki syndrome. Proc. Natl. Acad. Sci. U S A. 114, E9046–E9055.
Singh, N., Trivedi, C. M., Lu, M., Mullican, S. E., Lazar, M. A., and Epstein, J. A. (2011). Histone deacetylase 3 regulates smooth muscle differentiation in neural crest cells and development of the cardiac outflow tract. Circ. Res. 109, 1240–1249. doi: 10.1161/circresaha.111.255067
Smith, Z. D., and Meissner, A. (2013). DNA methylation: roles in mammalian development. Nat. Rev. Genet. 14, 204–220. doi: 10.1038/nrg3354
Snider, P., Olaopa, M., Firulli, A. B., and Conway, S. J. (2007). Cardiovascular development and the colonizing cardiac neural crest lineage. TheScientificWorldJournal 7, 1090–1113. doi: 10.1100/tsw.2007.189
Strobl-Mazzulla, P. H., Sauka-Spengler, T., and Bronner-Fraser, M. (2010). Histone demethylase JmjD2A regulates neural crest specification. Dev. Cell 19, 460–468. doi: 10.1016/j.devcel.2010.08.009
Takeuchi, J. K., Lou, X., Alexander, J. M., Sugizaki, H., Delgado-Olguin, P., Holloway, A. K., et al. (2011). Chromatin remodelling complex dosage modulates transcription factor function in heart development. Nat. Commun. 2:187.
Tellier, A., Cormier-Daire, V., Abadie, V., Amiel, J., Sigaudy, S., Bonnet, D., et al. (1998). CHARGE syndrome: report of 47 cases and review. Am. J. Med. Genet. 76, 402–409.
Ueda, Y., Okano, M., Williams, C., Chen, T., Georgopoulos, K., and Li, E. (2006). Roles for Dnmt3b in mammalian development: a mouse model for the ICF syndrome. Development 133, 1183–1192. doi: 10.1242/dev.02293
Van Laarhoven, P. M., Neitzel, L. R., Quintana, A. M., Geiger, E. A., Zackai, E. H., Clouthier, D. E., et al. (2015). Kabuki syndrome genes KMT2D and KDM6A: functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum. Mol. Genet. 24, 4443–4453. doi: 10.1093/hmg/ddv180
van Ravenswaaij-Arts, C., and Martin, D. M. (2017). New insights and advances in CHARGE syndrome: diagnosis, etiologies, treatments, and research discoveries. Am. J. Med. Genet. C Semin. Med. Genet. 175, 397–406. doi: 10.1002/ajmg.c.31592
Vissers, L. E., van Ravenswaaij, C. M., Admiraal, R., Hurst, J. A., de Vries, B. B., Janssen, I. M., et al. (2004). Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 36, 955–957. doi: 10.1038/ng1407
Wahl, S. E., Kennedy, A. E., Wyatt, B. H., Moore, A. D., Pridgen, D. E., Cherry, A. M., et al. (2015). The role of folate metabolism in orofacial development and clefting. Dev. Biol. 405, 108–122. doi: 10.1016/j.ydbio.2015.07.001
Weemaes, C. M., van Tol, M. J., Wang, J., van Ostaijen-ten Dam, M. M., van Eggermond, M. C., Thijssen, P. E., et al. (2013). Heterogeneous clinical presentation in ICF syndrome: correlation with underlying gene defects. Eur. J. Hum. Genet. 21, 1219–1225. doi: 10.1038/ejhg.2013.40
Wienholz, B. L., Kareta, M. S., Moarefi, A. H., Gordon, C. A., Ginno, P. A., and Chédin, F. (2010). DNMT3L modulates significant and distinct flanking sequence preference for DNA methylation by DNMT3A and DNMT3B in vivo. PLoS Genet. 6:e1001106. doi: 10.1371/journal.pgen.1001106
Wolf, U., Reinwein, H., Porsch, R., Schröter, R., and Baitsch, H. (1965). Defizienz an den kurzen Armen eines Chromosomes Nr. 4. Humangenetik 1, 397–413.
Wu, J. I., Lessard, J., and Crabtree, G. R. (2009). Understanding the words of chromatin regulation. Cell 136, 200–206. doi: 10.1016/j.cell.2009.01.009
Yan, S., Thienthanasit, R., Chen, D., Engelen, E., Brühl, J., Crossman, D. K., et al. (2020). CHD7 regulates cardiovascular development through ATP-dependent and-independent activities. Proc. Natl. Acad. Sci. U S A. 117, 28847–28858. doi: 10.1073/pnas.2005222117
Yoshimura, K., Kitagawa, H., Fujiki, R., Tanabe, M., Takezawa, S., Takada, I., et al. (2009). Distinct function of 2 chromatin remodeling complexes that share a common subunit, Williams syndrome transcription factor (WSTF). Proc. Natl. Acad. Sci. U S A. 106, 9280–9285. doi: 10.1073/pnas.0901184106
Yu, C., Yao, X., Zhao, L., Wang, P., Zhang, Q., Zhao, C., et al. (2017). Wolf–Hirschhorn Syndrome Candidate 1 (whsc1) functions as a tumor suppressor by governing cell differentiation. Neoplasia 19, 606–616. doi: 10.1016/j.neo.2017.05.001
Zentner, G. E., Layman, W. S., Martin, D. M., and Scacheri, P. C. (2010). Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am. J. Med. Genet. A 152A, 674–686.
Keywords: cardiac neural crest cell, epigenetic regulation, heart development, cardiovascular development, congenital heart diasease
Citation: Yan S, Lu J and Jiao K (2021) Epigenetic Regulation of Cardiac Neural Crest Cells. Front. Cell Dev. Biol. 9:678954. doi: 10.3389/fcell.2021.678954
Received: 10 March 2021; Accepted: 29 March 2021;
Published: 21 April 2021.
Edited by:
Weinian Shou, Indiana University Bloomington, United StatesReviewed by:
Martin Chi Hang Cheung, The University of Hong Kong, Hong KongShuyi Nie, Georgia Institute of Technology, United States
Copyright © 2021 Yan, Lu and Jiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai Jiao, a2ppYW9AdWFiLmVkdQ==
†These authors have contributed equally to this work