 Joseph H. Garcia
Joseph H. Garcia Saket Jain
Saket Jain Manish K. Aghi
Manish K. Aghi- Department of Neurological Surgery, University of California, San Francisco, San Francisco, CA, United States
Glioblastoma is a primary malignant brain tumor with a median survival under 2 years. The poor prognosis glioblastoma caries is largely due to cellular invasion, which enables escape from resection, and drives inevitable recurrence. While most studies to date have focused on pathways that enhance the invasiveness of tumor cells in the brain microenvironment as the primary driving forces behind GBM’s ability to invade adjacent tissues, more recent studies have identified a role for adaptations in cellular metabolism in GBM invasion. Metabolic reprogramming allows invasive cells to generate the energy necessary for colonizing surrounding brain tissue and adapt to new microenvironments with unique nutrient and oxygen availability. Historically, enhanced glycolysis, even in the presence of oxygen (the Warburg effect) has dominated glioblastoma research with respect to tumor metabolism. More recent global profiling experiments, however, have identified roles for lipid, amino acid, and nucleotide metabolism in tumor growth and invasion. A thorough understanding of the metabolic traits that define invasive GBM cells may provide novel therapeutic targets for this devastating disease. In this review, we focus on metabolic alterations that have been characterized in glioblastoma, the dynamic nature of tumor metabolism and how it is shaped by interaction with the brain microenvironment, and how metabolic reprogramming generates vulnerabilities that may be ripe for exploitation.
Introduction
Glioblastoma (GBM) is a primary malignant brain tumor with a median survival under 2 years (Wen and Kesari, 2008; Stupp et al., 2019). The poor prognosis GBM caries is largely due to cellular invasion, which enables escape from resection and drives inevitable recurrence (de Gooijer et al., 2018; Wolf et al., 2019). To successfully invade, GBM cells must be motile and penetrate adjacent brain tissue (de Gooijer et al., 2018). Though it is largely agreed upon that specific molecular factors are the primary drivers of invasion, attempts to identify and target these drivers have been unsuccessful thus far (de Gooijer et al., 2018).
Clinical observations and experimental studies have led to numerous hypotheses attempting to define the factors which drive tumor invasion, but whether these changes are causative is widely contested (Hatoum et al., 2019; Vollmann-Zwerenz et al., 2020). Regardless, whether the sequential acquisition of random mutations in GBM might provide and select for invasive traits in individual cells, or the invasive predisposition of a tumor is already imprinted in the majority of cancer cells, invading cells need to be able to continuously adapt to changing environments during the invasive process (de Gooijer et al., 2018).
Subsequent studies have revealed differentially expressed genes at individual steps in the process of cellular invasion (Paw et al., 2015). The expression of gene clusters associated with rapid growth and proliferation have been proven to confer a selective advantage to cells invading into the adjacent brain microenvironment (Cuddapah et al., 2014). Invasion is facilitated by multiple factors including tumor-intrinsic factors, central nervous system-specific niches and the interaction between tumor cells and the cerebral microenvironment (Paw et al., 2015; de Gooijer et al., 2018). Furthermore, through the secretion of specific factors and extracellular vesicles, tumors can degrade structural matrices and avoid immune detection (Goldbrunner et al., 1998; Cuddapah et al., 2014).
More recently, attention has been given to the notion that invasive cells also require specific metabolic traits to survive and grow in new environments which substantially vary in their nutrient and oxygen availability from the tumor core (Marin-Valencia et al., 2012; Strickland and Stoll, 2017). Such metabolic rewiring can be controlled transcriptionally (i.e., epigenetic alterations), but also post-translationally or through metabolite availability (Strickland and Stoll, 2017). In this Review, we discuss the pathways implicated in glioblastoma’s metabolism and how they potentially contribute to invasive cells, the growing evidence for the dynamic metabolic shifts these cells display to survive and propagate during the different steps of the invasive transition, and how targeting metabolism can potentially be used to inhibit invasion in glioblastoma.
Metabolic Pathways in Glioblastoma
Carbohydrate Metabolism
Cellular growth and division depend on the uptake of nutrients from the surrounding environment, which are in turn metabolized to produce energy and maintain cellular homeostasis (Phan et al., 2014). The brain is highly metabolic and utilizes approximately 20% of the body’s total oxygen consumption as well as 60% of the body’s daily glucose intake (Mergenthaler et al., 2013). Additionally, the brain requires a constant supply of glucose because it lacks the ability to store glucose as glycogen. This high metabolic demand and the inability of the brain to store glucose effectively lead to glucose being the most widely available nutrient in the brain microenvironment (Thorens, 2012; Mergenthaler et al., 2013).
High glucose levels have been linked to increased tumor invasion and poor patient survival (Labak et al., 2016; Martin-McGill et al., 2018). To gain insight into the mechanistic pathways that link high glucose levels and GBM invasion, studies in other cancer types point to reasonable hypotheses. A study in Lung adenocarcinoma cells revealed high glucose levels induced increased heme oxygenase-1 (HO-1), and subsequent increases in tumor cell invasiveness via increased PI3K/akt signaling (Han et al., 2013; Sferrazzo et al., 2020). Moreover, silencing of HO-1 led to an attenuation of high glucose induced invasion. The expression of HO-1 is increased in GBM in comparison to both normal brain tissue and compared to lower grade tumors (Sferrazzo et al., 2020). While a promising lead, future studies will determine whether glucose drives invasion in GBM through direct mechanistic effects, or subsequent effects of its downstream metabolites.
In neurons and glia alike, glucose is metabolized via glycolysis into pyruvate, which then enter the tricarboxylic acid (TCA) cycle to generate ATP through oxidative phosphorylation (OXPHOS) (Thorens, 2012). In contrast to non-cancerous glial cells, GBM cells preferentially metabolize glucose into lactate despite the presence of ample oxygen (known as the “Warburg” effect) (Strickland and Stoll, 2017; Pirmoradi et al., 2019). It is hypothesized that this metabolic shift enables tumor cells to use glucose-derived carbons for the synthesis of essential cellular ingredients while generating sufficient ATP to fuel cellular reactions (Phan et al., 2014; Kalyanaraman, 2017).
Aside from the benefits glycolysis brings to invading tumor cells by providing carbon skeletons for cellular building blocks several glycolytic enzymes play a more direct role in invasion. Phosphoglucose isomerase (PGI) is a cytosolic enzyme that catalyzes the conversion of glucose-6-phosphate into fructose-6-phosphate in the second step of glycolysis (Zhu et al., 2014). PGI is a secreted protein that behaves as a potent cytokine in extracellular environment. It has been hypothesized that PGI is an autocrine motility factor (AMF), and a tumor-secreted cytokine that stimulates cell migration in vitro and metastasis in vivo (Zhu et al., 2014; Kathagen-Buhmann et al., 2016). Fructose-1,6-bisphosphatase (FBP1), a gluconeogenesis enzyme, which catalyzes the splitting of fructose-1,6-bisphosphate (F-1,6-BP) into fructose 6-phosphate, also plays an important role in the epithelial to mesenchymal transition EMT (Han et al., 2013; Shi et al., 2017). Loss of FBP1 in breast cancer cells induces EMT and increases invasiveness, the enzyme may perform a similar role in GBM (Shi et al., 2017). Pyruvate kinase (PK) mediates the final rate-limiting step of glycolysis by catalyzing the dephosphorylation of phosphoenolpyruvate (PEP) to pyruvate (Mukherjee et al., 2013). In colon adenocarcinoma cells, a PK subtype knockdown suppressed invasion through reduced EGFR signaling, perhaps shedding light on this glycolytic enzyme’s invasive role across cancer types (Han et al., 2013).
The Warburg effect is well studied in glioblastoma (Strickland and Stoll, 2017). Like many other aggressive tumors, GBM cells ultimately gain oncogenic signaling pathways that regulate cell survival, cell proliferation, and aerobic glycolysis (Kalyanaraman, 2017). This metabolic adaptation, while a promising target for genetic and pharmacologic interventions, is only scratching the surface concerning metabolism in GBM invasion.
When metabolizing glucose, invading tumor cells must also divert carbon from glycolysis into the pentose phosphate pathway (PPP) for nucleotide synthesis and to combat oxidative stress (Jin and Zhou, 2019). The PPP has two primary roles: the generation of reducing equivalents (oxidative phase), and the production of ribose 5-phosphate for nucleotide generation (non-oxidative phase) (Loreck et al., 1987).
The oxidative arm of the PPP utilizes G6P as its substrate and leads to the generation of NADPH. The non-oxidative reactions of the PPP lead to the generation of ribose-5-phosphate (R5P) for nucleotide biosynthesis (Loreck et al., 1987; Jin and Zhou, 2019). Many glycolytic enzymes including phosphofructokinase 1, phosphoglycerate mutase, and pyruvate kinase M2 (PKM2) are tightly controlled by tumor cells (Kowalik et al., 2017). Regulation of these glycolytic enzymes can result in accumulation of substrates leading into diversion of carbon toward R5P for nucleotide synthesis (Strickland and Stoll, 2017; Jin and Zhou, 2019). The non-oxidative arm of PPP is also important for tumor cells, based on higher expression and activity of transketolase, which correlates with the rate of tumor growth in some cancers, including GBMs (Jiang et al., 2014; Jin and Zhou, 2019). A further level of regulation for R5P synthesis is the ratio of NADP + /NADPH in cells through the oxidative arm of PPP. The reversible reduction of glucose-6-phosphase (G6P) by G6P dehydrogenase is associated with reduction of NADP to NADPH, a critical reducing agent for several reactions including fatty acid and glutathione synthesis, for building biomass and controlling oxidative stress (Loreck et al., 1987; Kathagen-Buhmann et al., 2016).
To meet GBM’s high metabolic demands, glucose uptake is increased through the upregulation of glucose transport proteins (Strickland and Stoll, 2017). Expression of GLUT1, and to a lesser extent GLUT3 and GLUT4, have been shown to be increased in GBM cells both in conjunction with the relative glucose concentrations in the tumor microenvironment (Labak et al., 2016). Additionally, signaling pathways upstream of the metabolic shifts seen at the tumor invasive edge have been tied to increased glucose transporter expression (Labak et al., 2016; Strickland and Stoll, 2017). These results in GBM as well as other cancer types suggest that GLUT proteins not only play a role as glucose transporters, but also acts as a regulator of signaling cascades in the invasive phenotype of GBM (Labak et al., 2016).
Lipid Metabolism
Lipids are a diverse group of water-insoluble molecules essential for numerous biological processes (Snaebjornsson et al., 2020). Their functions at the cellular level include energy storage and homeostasis, maintaining structural integrity as components of cellular membranes, and acting as messengers in cellular signaling. There is increasing evidence that cancer cells develop specific alterations in several aspects of lipid metabolism to supply their high bioenergetic demand (Guo et al., 2013; Shakya et al., 2021). By reprogramming lipid metabolism tumor cells can effectively enhance processes central to invasion, including cell growth, proliferation, and motility (Guo et al., 2013; Shakya et al., 2021). Lipids are abundant in the cerebral microenvironment and play a fundamental role in normal astrocyte function (van Deijk et al., 2017; Barber and Raben, 2019). Given this evidence, lipid metabolism has become an attractive target for investigation in metabolic drivers of glioblastoma invasion.
Among lipids in the brain cholesterol is among the highest in its relative abundance and represents 20–25% of total body cholesterol (Pirmoradi et al., 2019). Due to the inability of peripheral cholesterol to cross the blood–brain barrier (BBB), the majority of cholesterol in the brain is generated via de novo biosynthesis by astrocytes and delivered to neurons within high-density lipoproteins containing apolipoprotein E (Apo-E) (Barber and Raben, 2019; Pirmoradi et al., 2019). Unlike benign non-cancerous astrocytes (Ahmad et al., 2019; Pirmoradi et al., 2019), the metabolic needs of GBM cells are supplied mainly by exogenously rather than endogenously synthesized cholesterol, and cholesterol uptake is a crucial step for growth and survival for GBM cells (Geng et al., 2016). Studies have indicated that this uptake largely is driven via upregulation of sterol regulatory element-binding protein (SREBP-1) (Geng et al., 2016).
In addition to increased extracellular cholesterol uptake, GBM cells exhibit dysregulated cholesterol efflux and synthesis (An and Weiss, 2016; Ahmad et al., 2019). At high cell density, benign astrocytes reduce intracellular cholesterol by both upregulating the cholesterol efflux transporter ABCA1, and by reducing expression of genes in the mevalonate pathway (Patel et al., 2019). Glioblastoma cells conversely do not display this density-dependent regulation and maintain cholesterol synthesis (Ahmad et al., 2019; Patel et al., 2019). This dysregulation combined with the enhanced cholesterol uptake exhibited by GBM cells leads to the increased cholesterol required by these invasive cells (An and Weiss, 2016).
Lipidomics studies have shown the presence of triglycerides in two biopsies from glioblastomas (GBM) that had no treatment, but absent in healthy adult brain. Given that triglycerides contain three fatty acids and act as energy storage in addition to unsaturated fatty acids prominent in high-grade intracranial tumors, it appears that gliomas have developed an altered metabolism for fatty acids (Guo et al., 2013). Triglyceride accumulation has been linked directly to invasion in prostate cancer, and it is possible a similar mechanism occurs in GBM (Schlaepfer et al., 2015). Given these high levels of polyunsaturated fatty acids are present in gliomas, it is worth investigating whether the conversion of saturated fatty acids to unsaturated acids is a targetable driver of tumor invasion.
Fatty acid (FA) synthesis and catabolism are also among the lipid metabolic pathways with demonstrated alteration in Glioblastoma, with paradoxical elevation observed in both pathways. Fatty acids are able to cross the BBB and are readily available at the tumor’s invasive front (Barber and Raben, 2019; Taïb et al., 2019). The Fatty acid synthesis genes ACC and FAS have both been demonstrated to be highly expressed in glioblastoma and have been associated with poor patient outcomes (Zhao et al., 2006; Taïb et al., 2019). Genetic inhibition of both genes has been demonstrated to significantly suppress tumor growth in vitro and in xenograft mouse models (Guo et al., 2013; Yasumoto et al., 2016). Global metabolic profiling has suggested fatty acid oxidation is also upregulated in high grade glioma cells (Taïb et al., 2019). The simultaneous upregulation of these pathways could potentially supply GBM cells with ample lipids for cellular components while also allowing metabolic flexibility to invasive cells encountering novel microenvironments.
On a mechanistic level, expression of the FA uptake channel CD36, is upregulated under hypoxia in several types of cancer (Zhao et al., 2006; Liang et al., 2018, 36). CD36 has been previously implicated in tumor invasion and progression. Metastasis-initiating cells in human oral carcinomas display high levels of the CD36 (Liang et al., 2018, 3). Clinical data implies that the presence of CD36 + cells correlates with a poor prognosis and greater invasion numerous types of carcinomas, and inhibition of CD36 also impairs metastasis, in human melanoma and breast cancer-derived tumors (Ben-Shoshan et al., 2008; Liang et al., 2018, 3). In GBM, CD36 is expressed in tumorigenic cancer stem cells, and is associated with a pro-invasion phenotype (Hale et al., 2014). Further studies are required to study mechanism by which this channel protein drives invasion.
Amino Acid Metabolism
Amino acids are increasingly recognized as important fuels for supporting cancer growth and division (Lieu et al., 2020). Biosynthetic and bioenergetic pathways alike rely on various amino acid contributors (Wu, 2009). Additionally, the breakdown of amino acids produces derivatives that can support tumor growth and invasive potential (Panosyan et al., 2017; Lieu et al., 2020). The diversity of roles amino acid metabolism plays in bioenergetic regulation, the synthesis of essential biomolecules, and in homeostatic maintenance, have made amino acid metabolism increasingly popular in the research of many cancer types including glioblastoma (Reeds et al., 1998; Bianchi et al., 2004; Wu, 2009).
Among the most intriguing amino acids in the study of glioblastoma invasion is glutamine (Obara-Michlewska and Szeliga, 2020). Glutamine is abundant in the cerebral microenvironment and serves as the primary precursor to the excitatory neurotransmitter glutamate (Smith, 1990; Tardito et al., 2015). Astrocytes play a key role in maintaining glutamine homeostasis in the brain by regulating its synthesis via glutamate recycling (Smith, 1990; Schousboe et al., 2014). Following this conversion, glutamine is released into the extracellular space through N or sodium-coupled amino acid transporters (SNATs) (Natarajan and Venneti, 2019). Gliomas take in the recycled glutamine through the upregulation of glutamine and glutamate transporters (Obara-Michlewska and Szeliga, 2020). Gene expression profiling has shown an upregulation of the glutamine importer ASCT2 (SLC1A5) compared to low grade gliomas, and glutamine deprivation has slowed GBM tumor growth in some in vitro studies (Hassanein et al., 2013; Obara-Michlewska and Szeliga, 2020).
There are a variety of potential roles played by glutamine as a driver of invasion in glioblastoma. Glutamine is the obligate nitrogen donor in several enzymatic reactions in the formation of both purines and pyrimidines (Natarajan and Venneti, 2019). The nucleotides formed by these reactions are essential to the growth and division required for tumor invasion. Glutamine also contributes to the pool of available non-essential amino acids (NEAAs) mitochondrial substrates in rapidly proliferating tumor cells (Obara-Michlewska and Szeliga, 2020). Finally, glutamine has been shown to stimulate mechanistic target of rapamycin (mTOR) complex 1 (mTORC1) signaling via translocation to the cellular lysosome. This pathway has been indicated as a potent driver of GBM growth and progression (Jewell et al., 2015; Natarajan and Venneti, 2019).
Glutamine starvation has also been recently been associated with tumor invasion, through the amino acid’s association with Cancer Associated Fibroblasts (CAFs), which facilitate epithelial tumor cell invasion (Schousboe et al., 2014; Mestre-Farrera et al., 2021). CAFs and other mesenchymal cells appear to rely on glutamine metabolism to a much greater degree than their epithelial counterparts. Deprivation of glutamine caused CAF and subsequent tumor invasion toward glutamine sources (Mestre-Farrera et al., 2021). Given the importance of CAFs in glioblastoma’s invasive capacity, it may be worthwhile to investigate the role of glutamine seeking behavior as a driver of tumor invasion.
In addition to glutamine, several other amino acids are utilized to fuel bioenergetic reactions and the synthesis of macromolecules in GBM (Panosyan et al., 2017; Lieu et al., 2020). Arginine has demonstrated importance in GBM cell adhesion, and it is been speculated that the molecular mechanisms that drive this process are also important for tumor cell migration and invasion (Pavlyk et al., 2015; Agarwal et al., 2017). Aspartate has been shown to be a limiting metabolite for glioblastoma cellular proliferation in hypoxic conditions, which has important implications as these tumors typically outgrow their blood (and therefore oxygen) supplies rapidly (Garcia-Bermudez et al., 2018). In vitro studies have shown restriction of the essential amino acid methionine slows GBM cell in vitro via inhibition of key oncologic signaling proteins including PI3K, p38MAPK, and ERK (Palanichamy et al., 2016; Palanichamy and Chakravarti, 2017).
Lastly, metabolism of the amino acids arginine and tryptophan have been linked to decreased detection by neighboring immune cells, which is crucial to successful tumor invasion (Kesarwani et al., 2018).
Oxidative Phosphorylation and IDH Mutations
The tricarboxylic acid cycle (TCA cycle) is the central point of convergence for intermediates generated in the metabolic pathways altered in GBM (Strickland and Stoll, 2017). Carbohydrate, lipid, and amino acid metabolism generate metabolites that are fed into the TCA cycle in tumors through differential regulation of many of the pathways described above (Marie and Shinjo, 2011; Maus and Peters, 2017). Though the TCA cycle has at times been overlooked due to metabolic research in GBM being primarily focused on aerobic glycolysis, emerging evidence has indicated that GBM cells, especially certain cellular subtypes, utilize the TCA cycle to fuel energy production and biomolecule synthesis crucial for growth (Marin-Valencia et al., 2012; Shi et al., 2019).
Interestingly, it has been primarily genomic, rather than metabolic studies that have shed light on the importance of oxidative phosphorylation in GBM (Özcan and Çakır, 2016; Anderson et al., 2018). Several mutations in TCA Cycle enzymes and enzymes in adjacent metabolic pathways are commonly found in glioblastomas, and none more prevalently than isocitrate dehydrogenase 1 (IDH1) and 2 (IDH2) (Cohen et al., 2013; Quinones and Le, 2018).
IDH mutant tumors constitute roughly 10 percent of all glioblastomas (Cohen et al., 2013; Wu et al., 2019). Tumors with IDH mutations are typically secondary GBMs (Fack et al., 2015; Jahangiri et al., 2017; Chandra et al., 2020) and have a longer mean survival, and different mutation and histopathological profiles than their IDH wild-type counterparts (Cohen et al., 2013; Olar and Aldape, 2014). Additionally, IDH mutant tumors have metabolic profiles entirely distinct from IDH wildtype GBMs including an exaggerated dependance on glutamate as an energy source (Li et al., 2017; Maus and Peters, 2017). IDH1-mutatnt GBMs have a high demand for glutamate and are believed to use this amino acid as a chemotactic signal. As healthy astrocytes excrete glutamate, IDH1-mutated GBM cells tend to lack dense tumor structures, and instead migrate, invade, and disperse into adjacent cerebral tissue where glutamate concentrations are higher (Marin-Valencia et al., 2012; Maus and Peters, 2017). The abundance of IDH mutations in GBM, and the differences found in tumors with the mutation has led to IDH status being the primary mode of classification of GBM tumors (Cohen et al., 2013).
IDH1 and IDH2 are NADP+-dependent enzymes that interconvert isocitrate and α-ketoglutarate (αKG) in cytosol and mitochondria, respectively. Genome-wide exon-sequencing studies of gliomas have revealed IDH1 R132H activating point mutations in as many as 80–90% of low-grade gliomas (LGGs) (Cohen et al., 2013; Li et al., 2017). Mutations affecting IDH2 and additional IDH1 variants have also been reported in gliomas, but at a lower frequency, with the majority conferring similar changes in IDH activity (Cohen et al., 2013). The primary mechanism by which mutant IDH contributes to the pathogenesis of GBM is ascribed to the deregulated enzymatic activity of mutant IDH, which converts αKG into the metabolite D2HG, which in turn inhibits αKG-dependent dioxygenases, such as ten–eleven translocation (TET) family 5-methylcytosine DNA hydroxylases and the Jumonji C domain-containing histone–lysine demethylases (KDMs) (Xiao et al., 2012; Miller et al., 2017). Consequently, mutant IDH1and IDH2 activity cause aberrant DNA and histone methylation, which lead to widespread hypermethylation of cytosine–phosphate–guanine (CpG) islands a phenomenon termed the glioma CpG-island methylator phenotype (G-CIMP) (Malta et al., 2018).
Mutations in additional TCA and TCA adjacent enzymes are found in GBM with lower frequency and comparatively fewer known functional implications compared to IDH (Cohen et al., 2013; Huang et al., 2019). Notably, mutation of fumarate hydratase (FH), and succinate dehydrogenase (SDH) have been reported and further solidify the TCA cycle’s importance in GBM’s metabolic reprogramming (Bardella et al., 2011; Schmidt et al., 2019). Further studies are needed to elucidate the advantages conferred to GBM cells through these mutations, and how they might contribute to GBM’s invasive phenotype. A graphical summary of the enzymes and pathways that have been tied to GBM tumor invasion is displayed in Figure 1.
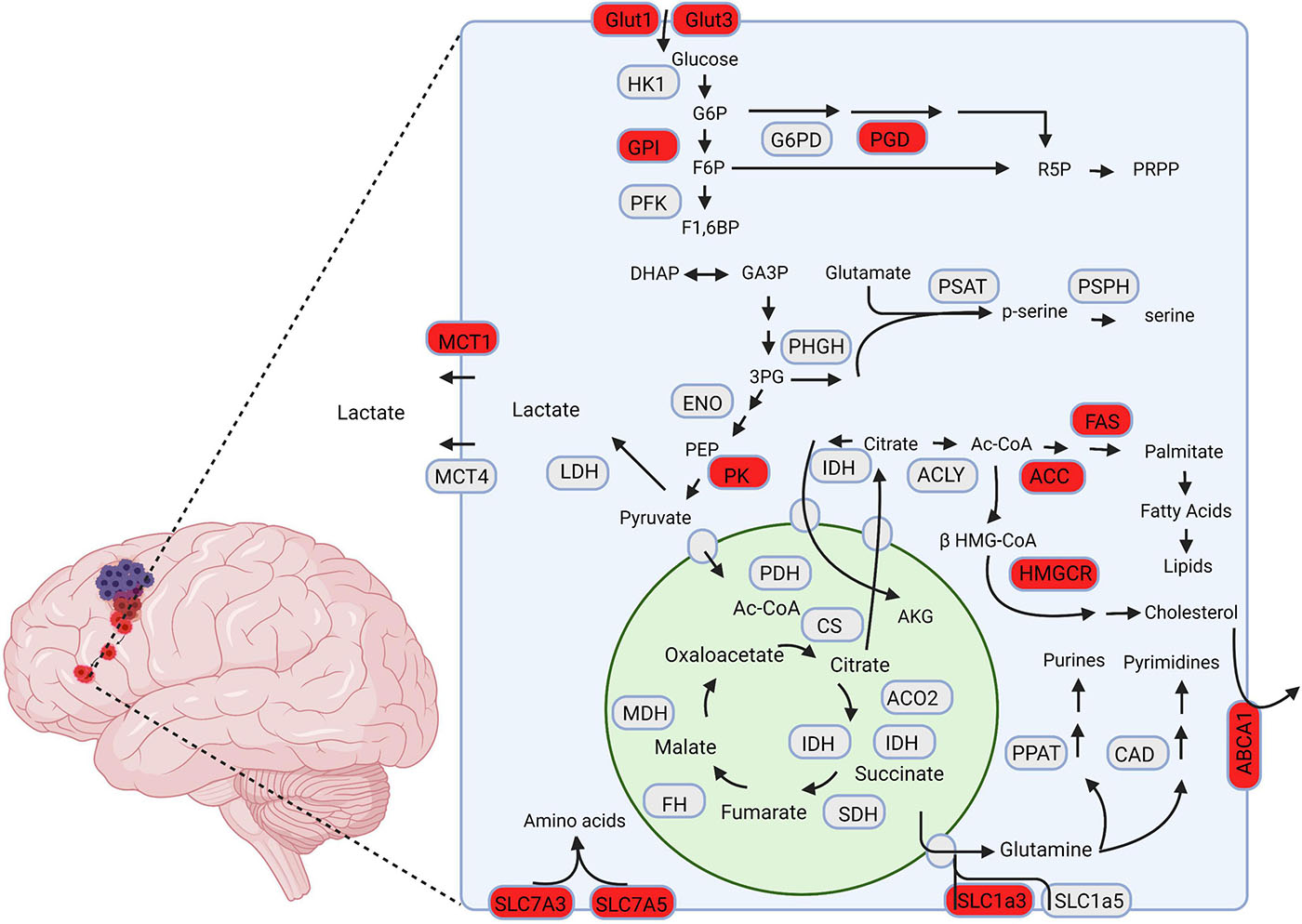
Figure 1. A schematic representation of metabolic pathways associated with GBM invasion. The proteins highlighted in red are hypothesized to drive invasion in GBM cells. Created using Biorender.com.
Effects of the GBM Microenvironment on Metabolism
The Brain Extra Cellular Matrix as a Driver of Altered Metabolism
The brain extracellular matrix (ECM) occupies a notable portion of the CNS and contributes to its normal physiology (Goldbrunner et al., 1998). It is produced intracellularly and secreted to form a network of proteins and glycans, occupying the parenchyma of virtually all CNS cells (Bonneh-Barkay and Wiley, 2009; Lau et al., 2013). Structurally, the ECM provides cerebral cells with anchorage points and facilitates the organization of these cells into distinct CNS regions (Lau et al., 2013). It is a source of important molecular signals that influence cellular growth and survival (Bonneh-Barkay and Wiley, 2009).
Alterations to the ECM occur in many diseases of the CNS, including glioblastoma (Goldbrunner et al., 1998; Bonneh-Barkay and Wiley, 2009). As GBM cells navigate through the ECM, molecular changes allow the tumors to adhere to, detach from, and degrade ECM as needed to facilitate their invasive processes (Goldbrunner et al., 1998; Malric et al., 2017). Changes in cellular metabolism appear to be important players in many of the interactions between tumor cells and the ECM, though it is contested whether these changes are causative or merely consequential.
Effects of GBM Cell Adhesion and Detachment on Tumor Metabolsim
During the invasive process, GBM cells become polarized, and their outer edge undergoes dynamic cytoskeletal rearrangements that facilitate adhesion to the adjacent ECM. This process is generally believed to be regulated by receptors in the integrin protein family (Malric et al., 2017). Integrins, which are characterized by a large extracellular domain, a short transmembrane domain, and a small intracellular non-catalytic cytoplasmic tail, are key components of the crosstalk between GBM cells and the microenvironment (Alberts et al., 2002; Bellail et al., 2004). Expression levels of several integrins are associated with poor prognosis and decreased survival in GBM patients (Paolillo et al., 2016; Malric et al., 2017).
Given the important link integrins provide between GBM cells and the tumor microenvironment, their role in tumor cell metabolism has been explored in several in vitro studies (Alberts et al., 2002; Paolillo et al., 2016). Unsurprisingly, a plethora of metabolism related signaling pathways have been shown to exert regulatory effects on integrins (Ata and Antonescu, 2017). Cellular signaling via the hypoxia related transcription factor HIF1α, mTOR signaling through amino acid relative abundance, and activity of the energy sensing protein AMP-Protein kinase have established regulatory effects on integrin function (Ata and Antonescu, 2017; Georgiadou et al., 2017; Ju et al., 2017). A better understanding of reciprocal integrin-metabolism interactions through targeted mechanistic studies is needed in the future.
In addition to their influence via integrins, several metabolites and metabolic pathways have direct effects on tumor cell adhesion properties. Upregulation of glycolysis and the PPP have both been associated with the functional activity of the adhesion proteins E-cadherin and P- cadherin (Liu et al., 2019; Sousa et al., 2019). Furthermore, increased levels of the metabolite Acetyl-coA directly promote cell-ECM adhesion by donating the necessary acetyl group for lysine acetylation in cross-linking (Lee et al., 2018). Specific metabolic dependencies such as these can potentially be exploited wither through small molecule inhibition or nutrient deprivation.
Effects of GBM Proteases on Cellular Metabolism
While adhering to the ECM allows GBM cells to migrate and disperse throughout the cerebral microenvironment efficiently, invasive tumors must also slice through the matrix to seed tumor cells in adjacent brain tissue. This is primarily accomplished via specific degradation enzymes called proteases (Lakka et al., 2005; Paw et al., 2015). Links between proteases and the cellular metabolism of GBM have been poorly explored relative to other invasive factors (Strickland and Stoll, 2017). Existing studies have highlighted instances of both metabolic factors influencing protease activity and vice versa (Rao, 2003; Lakka et al., 2005). The role these enzymes play in GBM invasion and the paucity of data regarding the extent of their links to tumor metabolism represent an attractive target for research.
Matrix metalloproteinases (MMPs) are proteases believed to play a central role in GBM invasion, owed to their ability to degrade many brain ECM components (Hagemann et al., 2012). This family of zinc ion-dependent enzymes is broadly divided into six classes based on substrate specificity (Klein and Bischoff, 2011). Acting through both the degradation of the ECM and activation of pro-migratory signaling cascades, MMPs are able to promote highly invasive tumor behavior (Goldbrunner et al., 1998; Hagemann et al., 2012). The main MMPs implicated in GBM are the gelatinases MMP-2 and MMP-9, as well as the membrane-type MT1-MMP (MMP-14) (Hagemann et al., 2012; Ulasov et al., 2014; Zhang et al., 2019). Though MMPs are not typically viewed as important metabolic modulators, there is emerging evidence that MMP-2, MMP-9, and MMP-14 are potent modulators of cholesterol metabolism, a crucial pathway in glioblastoma growth and invasion (Ulasov et al., 2014; Hernandez-Anzaldo et al., 2016).
Urokinase-type plasminogen activator (uPA) is a serine protease which, along with its receptor uPAR, plays a role in invasion and neovascularization in gliomas (Lakka et al., 2001). Molecular characterization of high-grade gliomas has revealed increased expression of uPA and uPAR and has correlated upregulation of these genes with a more invasive tumor phenotype (Lakka et al., 2001; Chandrasekar et al., 2003). In addition to its intrinsic protease activity, uPA is able to indirectly activate other pro-form collagenases responsible for the degradation of plasmin-resistant ECM components (Zhou et al., 2000; Bekes et al., 2011). Upregulation of uPA has been shown to inhibit the Phosphoinositide 3-kinase (PI3K)–AKT signaling network which has diverse downstream effects on cellular metabolism, both through direct regulation of nutrient transporters and metabolic enzymes, and the control of transcription factors that regulate the expression of key components of metabolic pathways (Chandrasekar et al., 2003). The number of links found between tumor invasion and cellular metabolism in proteases in a relatively short amount of time warrant a deeper dive into this area of GBM research.
GBM Stem Cells and Metabolism
Glioblastoma Stem Cells (CSCs) constitute a small percentage of GBM tumor cells and demonstrate two principal features of stem cells: self-renewal and differentiation (Lathia et al., 2015). GSCs are enriched in factors responsible for invasive potential and are found at the leading edge of recurrent tumors following surgical resection. These cells migrate along the vasculature and white matter tracts utilizing cadherins and integrins and cleave their way through extracellular matrix using matrix metalloproteinases such as MMP9 and ADAMT2 (Lathia et al., 2015; Prager et al., 2020). Additionally, several signaling pathways that are upregulated in GSCs, including L1CAM and ephrin-B2, have been shown to enhance tumor cell invasioveness. GSCs are non-autonomous cells and are crucial to the interplay between GBM tumors and their surrounding microenvironment (Prager et al., 2020). Thus, GSCs may represent an important link between GBM invasion and metabolism.
Distinct microenvironments in GBM tumors; including the hypoxic core, the perivascular niche, and the invasive tumor edge, add to the heterogeneity and the dynamic behavior of GSCs in these tumors (Lathia et al., 2015). GSCs hold the unique ability to rapidly adapt to the metabolic changes in various tumor microenvironments (Garnier et al., 2019). GSCs residing in the perivascular niche exhibit an entirely different metabolic profile than those residing in the hypoxic niche of the tumor core. GSCs in the perivascular niche show a proneural phenotype while the GSCs in the hypoxic core exhibit a mesenchymal phenotype (Garnier et al., 2019; Prager et al., 2020). While GBM cells predominantly express PKM2 which promotes aerobic glycolysis for their glucose metabolism, GSCs express both PKM2 and PKM1 (Prager et al., 2020). PKM1 promotes mitochondrial metabolism allowing GSCs to shift between aerobic glycolysis and oxidative phosphorylation. While GSCs in the perivascular niche rely mostly on glucose metabolism, glutamine dependency has been seen in the mesenchymal GSCs residing in hypoxic niche (Garnier et al., 2019).
These dynamic metabolic changes in GSCs create a challenge when selecting therapeutic targets, and warrants exploration of targeting multiple pathways simultaneously. For example, radiation targets highly proliferative cells sparing slow cycling cells (Lathia et al., 2015; Garnier et al., 2019). Therefore, a combination of radiation and metabolic inhibition may hold promise. Understanding the role GSCs play in tumor metabolism is likely a crucial step of solving the puzzle that is GBM.
The Immune Systems and Metabolism in GBM Invasion
To invade adjacent brain tissue successfully, GBM cells must avoid detection and defend themselves from the host immune system. The immune composition of GBM’s microenvironment evolves with tumor stage but is predominantly comprised of immune suppressive cells (Pombo Antunes et al., 2020). There are distinct populations of immune-modulating macrophages, regulatory T cells (Tregs), T as well as dendritic cells (DC) present (Pombo Antunes et al., 2020). Recent studies have highlighted the intimate relationship between tumor metabolism and immune modulation, and the influence these processes have on tumor growth and progression (Chinnaiyan et al., 2012). Global metabolic profiling has identified distinct alterations that play key roles in immune modulation and the subsequent facilitation of tumor cell invasion (Chinnaiyan et al., 2012; Pearce and Pearce, 2013).
Following activation, T cells and DC undergo rapid expansion, with an accompanying increase in bioenergetic demand (Everts et al., 2012). These activated immune cells shift their metabolism by increasing glycolysis, increasing glucose uptake, decreasing carbon flux into the mitochondria, and enhancing lactate production (Ghesquière et al., 2014; Kesarwani et al., 2017).
This preference for glycolysis has also been demonstrated in other immune cells including macrophages, neutrophils, B cells and natural killer (NK) cells (Biswas, 2015). However, the elevated glycolysis exhibited by tumors cells leads to a microenvironment devoid of glucose which can significantly impact immune response (Singer et al., 2011; Becker et al., 2013). There is substantial evidence suggesting in the cerebral microenvironment, intense competition for nutrients exists between neural, immune, and tumor cells (Becker et al., 2013). Furthermore, accumulation of lactic acid due to enhanced glycolysis by tumor cells also impacts the immune cell function (Singer et al., 2011). Increased lactic acid can inhibit monocyte differentiation into DCs and increase transcription and secretion of pro-tumorigenic cytokines such as IL-23 and reduction of T-cell response (Correia et al., 2017). Therefore, enhanced GBM glycolysis has both active and passive inhibitory effects on immune cells in the adjacent microenvironment.
Glioma cells also influence their adjacent immune microenvironment through amino acid metabolic pathways via increased uptake of branched chain amino acids (BCAAs) (Silva et al., 2017). Glioblastoma cells overexpress branched chain amino acid transaminase 1 (BCAT1) which enhances excretion of branched chain ketoacids (BCKA) through MCT1 that influx into nearby macrophages and reduce their phagocytic ability (Silva et al., 2017).
Furthermore, GBM tumors promote macrophage and T cell dysfunction through expression of the ectonucleosides CD39 and CD73 (Xu et al., 2013; Yan et al., 2019). These integral membrane proteins induce the production of the immunosuppressive metabolite adenosine (Xu et al., 2013). Another link between amino acid metabolism and GBM immune invasion has been demonstrated in studies exploring the role of indoleamine 2,3-dioxygenase 1 (IDO1) (Zhai et al., 2015; Valtorta et al., 2020). IDO1 is an enzyme in the tryptophan metabolism pathway and converts tryptophan to kynurenine (Zhai et al., 2015). This conversion results in effector T cell energy, while concurrently promoting maturation and activation of Tregs blunting the host immune response to GBM cells (Munn et al., 2004). Pharmacologic inhibition of IDO has successfully enhanced the efficacy of immune checkpoint inhibitors in intracranial GBM mouse models (Wainwright et al., 2014).
Moreover, hypoxic conditions associated with the GBM tumor microenvironment can also contribute to immune system suppression (Wei et al., 2011). Hypoxia stimulates STAT3 phosphorylation which in turn activates immune-suppressive Tregs (Ben-Shoshan et al., 2008). Hypoxia also enhances the production of immune-suppressive cytokines including transforming growth factor (TGF)-β and vascular endothelial growth factor (VEGF) (Wei et al., 2011). Hypoxia also influences the immune cell composition of the adjacent microenvironment (Hussain et al., 2006; Wei et al., 2011). CNS macrophages are primary immune cells infiltrating GBM tumors and play an important role in mediating innate immunity in GBM (Pollard, 2004). These macrophages become tumor-associated macrophages (TAMs) when exposed to hypoxia (Lin et al., 2006). TAMs are polarized toward immune-suppressive and pro-tumorigenic phenotype (M2) via the STAT3 pathway (Pollard, 2004; Lin et al., 2006). In addition, TAMs further reinforce the GBM metabolic switch to aerobic glycolysis by secreting IL-6 which promotes the phosphorylation of phosphoglycerate kinase 1 (PGK1) and enhances its activity (Zhang et al., 2019).
In summary, tumor metabolism plays a crucial role in negating local immune detection and response. This dynamic interplay must be taken into consideration when targeting invasion in GBM.
Opportunities to Target Metabolism in GBM as a Way of Targeting GBM Invasion
The metabolic divergence between GBM cells and normal astrocytes holds potential for the discovery of novel therapeutic targets (Wolf et al., 2010; Nakano, 2014). Metabolomic and gene expression profiling can assist in the quantification of metabolites and metabolic enzymes in invasive cells, which can be combined to examine metabolic pathways of particular importance in these cells (Wolf et al., 2010; Chinnaiyan et al., 2012). Additionally, the tumor’s metabolomic profile can be correlated with the gene expression or proteomic profile of corresponding tissues, potentially yielding novel diagnostic or prognostic markers (Zhou and Wahl, 2019). Understanding how these metabolic networks vary and their importance for rapid invasion and proliferation may identify further targeted therapeutic strategies for GBM patients. A graphical representation of targeted metabolic inhibitors, dietary interventions, and repurposed drugs with potential utilization in invasive GBM is displayed in Figure 2.
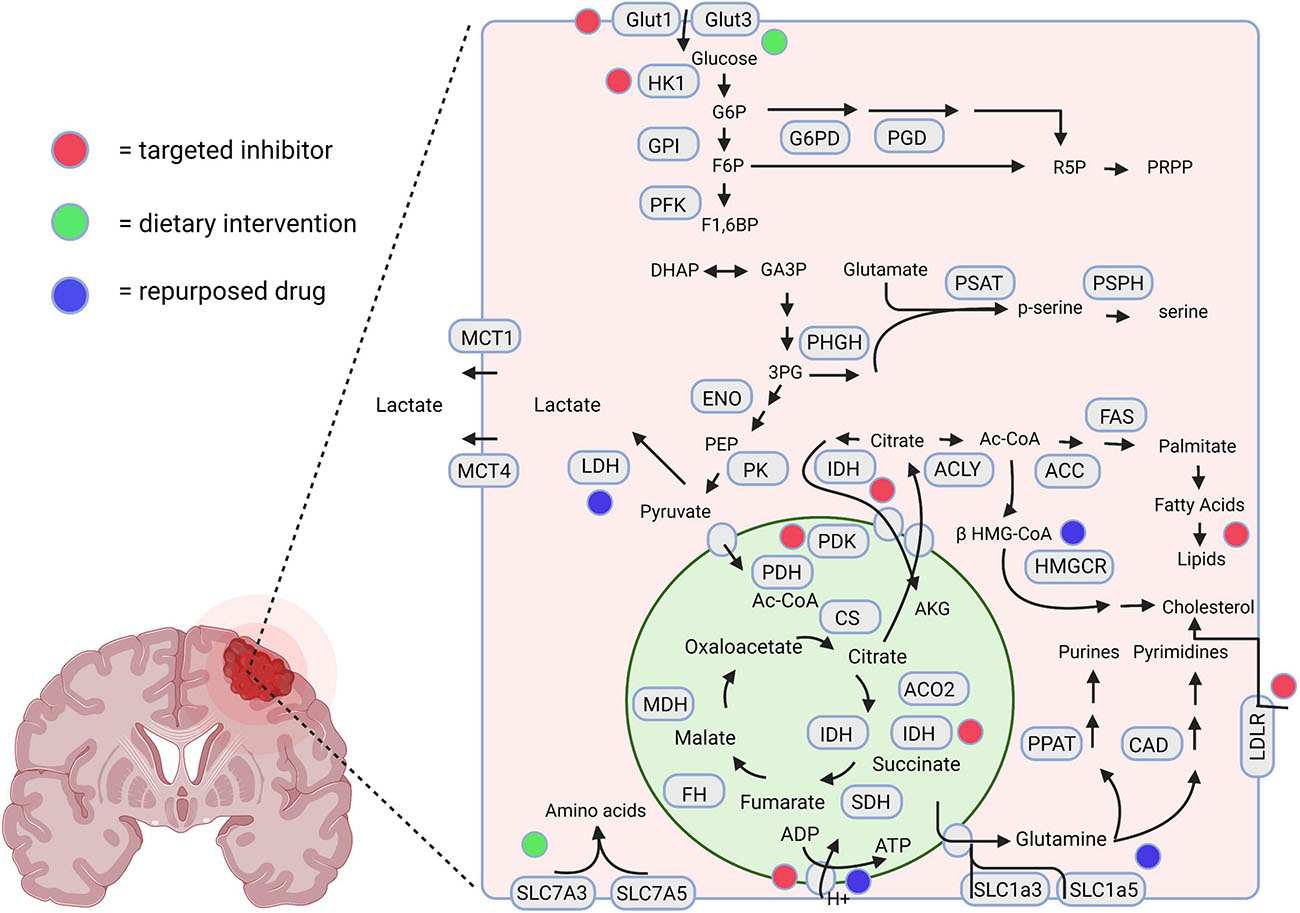
Figure 2. A schematic representation highlighting the therapeutic interventions associated with GBM metabolism. The red, green, and blue dots depict the interventions using novel targeted inhibitors, dietary interventions, and repurposed drugs, respectively. Created using Biorender.com.
Metabolic Targets in GBM Resistant to Standard Therapies
Current standard treatment for GBM involves surgical resection followed by concurrent radio and chemotherapy (Wen and Kesari, 2008). Despite this aggressive treatment regimen, the median survival of GBM patients remains under 2 years (Davis, 2016). The devastating prognosis carried by this tumor is inseparable from its invasive capacity, which makes total surgical resection or treatment with local therapies extremely challenging (de Gooijer et al., 2018). When local therapies fail, systemic treatments are used to attempt to slow tumor growth. However, aspects unique to GBM, including shifts in its cellular metabolism, render these mainstay cancer treatments often ineffective, allowing the tumor cells to continue down the path of uncontrolled growth and invasion (Wolf et al., 2010; Zhou and Wahl, 2019).
Whole brain radiation has previously been shown to induce metabolic changes in GBM cells (Gupta et al., 2020). Untargeted metabolomics experiments have identified significant differences in metabolite levels pre and post radiation in both GBM and adjacent normal brain (Wibom et al., 2010; Gupta et al., 2020). Increased levels of glutamine and alanine and decreased levels of galactose and tyrosine were observed in biopsied tumor tissue post irradiation (Wibom et al., 2010). Additional pathway specific studies in GBM cell lines have shown that radiation induces a transition in metabolic preference from glycolysis to oxidative phosphorylation via mTOR mediated HEK2 inhibition (Lu et al., 2015; Zhou and Wahl, 2019).
Several studies have reported the metabolic changes associated with Temozolomide (TMZ) resistance. GBM treatment with TMZ induces changes in mitochondrial complex activity and glutamate metabolism (Oliva et al., 2010). TMZ resistant cells show increased levels of complexes II/III and CcO (complex IV) (Oliva et al., 2010; Ulasov et al., 2014). Selective inhibition of the OXPHOS components is currently being explored as a strategy to overcome this therapeutic resistance (Shi et al., 2019).
The VEGF inhibitor bevacizumab does not improve patient overall survival and has shown to be associated with increased invasion of GBM cells (Fack et al., 2015; Jahangiri et al., 2017; Chandra et al., 2020). VEGF inhibition results in a hypoxic tumor microenvironment which GBM overcomes through dynamic shifts in glucose uptake and carbohydrate metabolism (Kuang et al., 2017). Tumors treated with bevacizumab show enhanced glucose influx via upregulation of the glucose transporters GLUT1 and GLUT3, and an increased production of lactate compared to untreated cells (Fack et al., 2015; Kuang et al., 2017). While the correlation between these adaptive metabolic changes seen after bevacizumab treatment of GBM and the invasive phenotype seen in bevacizumab-resistant GBM has yet to be definitively established, the metabolic adaptations GBM cells undergo to overcome systemic cancer treatments represent an attractive target to unlock the full potential of anti-angiogenic therapy in GBM.
Targeting Carbohydrate Metabolism
Among the metabolic pathways targeted in GBM by researchers, glycolysis has historically been the most prevalent (Wang et al., 2019). Most metabolic interventions to reach the clinical trial stage in GBM thus far have been dietary interventions rather than targeted inhibitors (Martin-McGill et al., 2017; Schwartz et al., 2018; van der Louw et al., 2019). Through the alteration of their intake composition, it is hypothesized that patients can deprive GBM tumors of the fuels that allow them to grow and proliferate (Schwartz et al., 2018). The ketogenic diet (KD), which aims to deprive GBM cells of their preferred metabolic fuel, glucose, aims to slow GBM growth and increase survival time among patients. The KD has been shown to reduce rodent tumor growth and tumor size and increase survival of animals (Lussier et al., 2016; Martin-McGill et al., 2018). Numerous clinical trials have tested different formulations of the ketogenic diet in GBM patients, and combined use of the KD with traditional chemotherapies (Martin-McGill et al., 2017; van der Louw et al., 2019). A summary of clinical trials using dietary modifications to treat GBM are summarized in Table 1.

Table 1. Clinical trials featuring dietary interventions to treat Glioblastoma multiforme.
The results of the aforementioned trials suggest the ketogenic diet can be safely implemented in patients and can achieve sufficient levels of ketogenesis (Santos et al., 2018; Schwartz et al., 2018). Patient recruitment and retention are two major factors which limit longer-term and larger trials (Martin-McGill et al., 2018; Schwartz et al., 2018; van der Louw et al., 2019). In those that have been conducted so far, a few smaller studies have shown modest effects on either tumor burden or progression (Martin-McGill et al., 2018). Optimization of timing and synergistic therapies are needed to assess whether the affects seen with this diet can match the results found across animal studies.
Carbohydrate metabolism has also been targeted in GBM both in vitro and in vivo with small molecules that disrupt key steps in crucial metabolic pathways. 2-deoxyglucose (2DG), which halts glycolysis following its initial phosphorylation by hexokinase, has been shown to inhibit GBM growth to a greater degree when combined with targeted radiation than radiation alone, and was safely used in a clinical trial in India with modest improvements in survival and reported patient quality of life (Dwarakanath et al., 2009; Shah et al., 2019). ManWZB117, a GLUT1 inhibitor, has been shown to inhibit tumor formation from GSCs but failed to limit progression of existing tumors (Shibuya et al., 2014). Additional GLUT1 inhibitors ritonavir and idinivar have been shown to reduce glucose consumption and GBM cell proliferation in vitro. 3-bromo-2-oxopropionate-1-propyl ester (3-BrOP), an inhibitor of hexokinase (HK) and 3-phospho dehydrogenase (3-PD) targets GSCs and therapeutic resistance acquired by GSCs. Combination treatment of 3-BrOP with carmustine has been shown to have a synergistic effect on decreased tumor formation (Shibuya et al., 2014). Dichloroacetate (DCA), a pyruvate dehydrogenase kinase (PDK) inhibitor has been shown to suppress tumor growth in GBM animal models, though its efficacy in human patients alone or in combination with other therapies remains to be seen (Yuan et al., 2013).
Finally, with advances in synthetic biology and biochemical techniques, molecular therapies focused on protein and gene level regulation are becoming the norm in cancer therapeutic research, and GBM is no exception. MicroRNAs (miRNAs) have come into focus as potential therapeutic targets to mediate the metabolic shifts in GBM due to their ability to regulate levels of specific metabolic genes of interest (Semonche et al., 2019). Examples include miR-106a which regulates GLUT3, miR-143 which regulates HKII, and miR-326 which regulates PKM2 (Dai et al., 2013; Zhao et al., 2013). Other investigators have attempted to inhibit tumor progression through modulation of miRNA-451 which exhibits negative regulatory effects on the LKB1/AMPK pathway (Godlewski et al., 2010). Exploitation of GBM’s downregulation of miRNA-451, which in turn results in increased cell migration through activation of the LKB1-AMPK pathway is currently being investigated with respect to its therapeutic potential (Godlewski et al., 2010; Ahir et al., 2017).
Targeting Lipid Metabolism
Long overlooked in favor of other pathways, lipid metabolism has increasingly come into focus in the targeting of metabolic pathways in GBM. LXR-623 and archazolid B are two promising metabolic therapies and inhibit cholesterol uptake and recycling, respectively. One or both drugs can potentially be used to target cholesterol metabolism which has been linked to GBM’s invasive spread (Hamm et al., 2014; Villa et al., 2016). Arachidonyl trifluoromethyl ketone (AACOCF3) which inhibits cytoplasmic phospholipase A2 in phospholipid metabolism, and A922500, which inhibits diacylglycerol-acyltransferase 1 (DGAT1) in the formation of lipid droplets from FFAs, are two more targets that the inhibition of which has stunted tumor growth in intracranial xenograft mouse models (Anfuso et al., 2014; Cheng et al., 2020, p. 1).
Targeting Oxidative Phosphorylation
Altering mitochondrial activity holds promise for targeting metabolism in GBM. Nigericin, a drug identified in a small molecule screen was able to induce mTOR inactivation, increase autophagy and decrease tumor growth in vivo (Hegazy et al., 2016). Metformin, a mainstay treatment for diabetes, has also shown potential in the treatment of GBM potentially through its effects on mitochondrial bioenergetics (Würth et al., 2013). Furthermore, treatment with Phenformin, an analog of Metformin, has been shown to decrease tumor growth and reduce markers of GSCs in vivo. Combination treatments using Phenformin and TMZ inhibit tumor growth in vitro and in vivo (Jiang et al., 2016). An arsenic based mitochondrial toxin, 4-(N-(S-penicillaminylacetyl)amion) phenylarsonous acid (PENAO) in combination with DCA results in inhibition of GBM proliferation, and induces G2/M cell cycle arrest both in vitro and in vivo (Shen et al., 2015). Recently, researchers identified the compound benzimidazolinium Gboxin. This small molecule, which inhibits ATP synthase activity in a proton gradient dependent manner, is a novel inhibitor of oxidative phosphorylation, and targets the specific mitochondrial alterations found in cancer cells (Shi et al., 2019). Further studies confirmed Gboxin accumulation and growth inhibition in GBM allografts and patient derived xenografts, providing additional validation of OXPHOS as a viable therapeutic target in GBM.
While therapies targeting metabolic pathways to reduce invasion in GBM have shown tremendous promise, there are multifactorial challenges in utilizing these treatments in patients. The suppression of invasion via inhibition of cellular metabolism in GBM is subject to the same difficulties faced by pharmacologic treatments of any disease of the central nervous system, that is, penetration of the BBB (Daneman and Prat, 2015; Strickland and Stoll, 2017). Furthermore, the incredible heterogeneity of GBM’s molecular profile pose the same challenges in choosing specific targets of treating any malignant neoplasm (Hatoum et al., 2019). Finally, therapies focused on metabolic pathways meet the new dilemma of targeting factors that drive the metabolism of normal and cancerous cells alike (Strickland and Stoll, 2017). Successful future therapies will not only have to suppress the metabolism that drives invasion in tumors but do so in a way that non-cancerous cells are not significantly harmed.
Drug Repurposing
Although potential therapies targeting tumor metabolism hold promise in the treatment of invasive GBM, several challenges in the drug development pipeline exist (Seliger and Hau, 2018; Tran and Prasad, 2020). Drugs designed for diseases other than cancer, such as antibiotics and antidiabetic agents, have the potential to be used as anti-tumor agents. Drug repurposing is best defined as the utilization of approved drugs, outside the scope of their original indication (Tran and Prasad, 2020). The advantage of repurposed drugs is that they circumvent many of the laborious and time-consuming challenges in the drug development pipeline, making them an attractive strategy in cancer therapeutic research (Seliger and Hau, 2018). Many pharmaceuticals with metabolic targets exist as strategies to treat other diseases, including heart disease and diabetes. Several of these drugs are currently being researched in animal models and in clinical trials as a method of slowing cancer growth, with GBM being no exception (Jiang et al., 2016; Seliger and Hau, 2018).
Metformin, an example of a repurposed drug with growing interest for its potential to target tumor metabolism, is a first-line treatment for type II diabetes (Kasznicki et al., 2014). Metformin has been reported to possess anticancer properties affecting the survival of cancer stem cells in breast cancer models, likely through the regulation of AMP kinase (AMPk), triggered by reduction in ATP/AMP ratio (Würth et al., 2013; Kasznicki et al., 2014). Treatment with metformin reduced the proliferation rate of tumor-initiating cell-enriched cultures isolated from four human glioblastomas. Metformin has also been shown to impair tumor-initiating cell spherogenesis, indicating a direct effect on self-renewal mechanisms (Würth et al., 2013). Additional studies in cell models have shown metformin can inhibit migration of GBM cells.
Another commonly used drug group, statins, have been implicated for potential repurposing to treat glioma based on promising in vitro studies and observational research in patient cohorts (Fatehi Hassanabad, 2019). Statins inhibit 3-hydroxy-3-methylglutaryl-CoA reductase, the rate-limiting enzyme of the mevalonate pathway. It has been proposed that statins induce tumor-specific apoptosis through mitochondrial apoptotic signaling pathways, which are activated by the suppression of mevalonate or geranylgeranyl pyrophosphate biosynthesis (Gong et al., 2019). These drugs have shown the ability to limit migration in cultured tumor cells, which some researchers have speculated could translate to slowing tumor invasion in vitro (Bababeygy et al., 2009). Existing evidence in humans has been less straightforward. A Meta-analysis showed no progression-free survival benefit or overall survival benefit to those patients taking statins (Fatehi Hassanabad, 2019).
Additional drugs that have been repurposed to stymie invasion in GBM include, NSAIDS, Disulfiram, and Ritonvir. These act through COX-2 inhibition and downstream reduction of c-myc and LDH expression, inhibition if ALDH and oxphos, and disruption of glutamine metabolism, respectively (Seliger and Hau, 2018). The evidence for these drugs benefitting GBM patients comes, again, from primarily basic science work in cell culture models (Maiti, 2014; Xu et al., 2015). Retrospective survival analyses on patients taking these drugs has shown mixed results, with modest survival benefits observed in some cohorts and no effect at all observed in others (Agarwal et al., 2017; Seliger and Hau, 2018). In addition to being largely inconclusive, such studies are commonly criticized for selection bias and immortal-time bias (Jiang et al., 2016; Tran and Prasad, 2020). Future well-designed, multicenter randomized controlled clinical trials will likely be needed to gauge the potential therapeutic benefit of these drugs.
Conclusion
The collective body of knowledge concerning the metabolic profile of invasive glioblastoma is rapidly evolving. The study of tumor metabolism is shifting from a static picture shaped by genetic mutations or environment alone to a dynamic view in which genotype and microenvironment interact to form the metabolic profile of invasive tumor cells, exposing potential vulnerabilities that are ripe for therapeutic exploitation. Research in this area is challenging because it is not amenable tracking data at any particular point in time, but rather requires a nuanced understanding of physiologic and biochemical alterations in flux, especially in the dynamic environments which brain tumors encounter. Fortunately, novel clinical and research tools are providing powerful insights. By integrating the full spectrum of system-wide, unbiased “omics” screens available to researchers, and cutting edge diagnostic clinical tools including radiotracer and flux imaging technologies, a new understanding of the molecular basis of glioblastoma metabolism has begun to emerge, including metabolic vulnerabilities that could be targeted therapeutically. Additionally, the development of integrative models based on tumor molecular markers, microenvironment, or both are continuously revealing additional therapeutic targets. This review has focused primarily on the actionable targets and routes of exploration that will be the most useful for finding them, with the goal of expanding the breadth of pharmacologic interventions through new drug development and repurposing. Glioblastoma research has been at the forefront of cancer research for the past 10 years, including the elucidation of critical mechanisms by which tumors undergo metabolic reprogramming. It is time for this research to translate to the clinical setting for the benefit of patients.
Author Contributions
JG prepared the figures and table. All authors contributed to the conception, drafting, and revising of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Agarwal, P., Pajor, M. J., Anson, D. M., Guda, M. R., Labak, C. M., Tsung, A. J., et al. (2017). Elucidating immunometabolic targets in glioblastoma. Am. J. Cancer Res. 7, 1990–1995.
Ahir, B. K., Ozer, H., Engelhard, H. H., and Lakka, S. S. (2017). MicroRNAs in glioblastoma pathogenesis and therapy: a comprehensive review. Crit. Rev. Oncol. Hematol. 120, 22–33. doi: 10.1016/j.critrevonc.2017.10.003
Ahmad, F., Sun, Q., Patel, D., and Stommel, J. M. (2019). Cholesterol metabolism: a potential therapeutic target in glioblastoma. Cancers (Basel) 11:146. doi: 10.3390/cancers11020146
Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., and Walter, P. (2002). “Integrins,” in Molecular Biology of the Cell, 4 Edn. Available online at: https://www.ncbi.nlm.nih.gov/books/NBK26867/ (accessed March 20, 2021).
An, Z., and Weiss, W. A. (2016). Cholesterol: an achilles’ heel for glioblastoma? Cancer Cell 30, 653–654. doi: 10.1016/j.ccell.2016.10.011
Anderson, N. M., Mucka, P., Kern, J. G., and Feng, H. (2018). The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 9, 216–237. doi: 10.1007/s13238-017-0451-1
Anfuso, C. D., Motta, C., Giurdanella, G., Arena, V., Alberghina, M., and Lupo, G. (2014). Endothelial PKCα-MAPK/ERK-phospholipase A2 pathway activation as a response of glioma in a triple culture model. A new role for pericytes? Biochimie 99, 77–87. doi: 10.1016/j.biochi.2013.11.013
Ata, R., and Antonescu, C. N. (2017). Integrins and cell metabolism: an intimate relationship impacting cancer. Int. J. Mol. Sci. 18:189. doi: 10.3390/ijms18010189
Bababeygy, S. R., Polevaya, N. V., Youssef, S., Sun, A., Xiong, A., Prugpichailers, T., et al. (2009). HMG-CoA reductase inhibition causes increased necrosis and apoptosis in an in vivo mouse glioblastoma multiforme model. Anticancer Res. 29, 4901–4908.
Barber, C. N., and Raben, D. M. (2019). Lipid metabolism crosstalk in the brain: glia and neurons. Front. Cell Neurosci. 13:212. doi: 10.3389/fncel.2019.00212
Bardella, C., Pollard, P. J., and Tomlinson, I. (2011). SDH mutations in cancer. Biochim. Biophys. Acta 1807, 1432–1443. doi: 10.1016/j.bbabio.2011.07.003
Becker, J. C., Andersen, M. H., Schrama, D., and Thor Straten, P. (2013). Immune-suppressive properties of the tumor microenvironment. Cancer Immunol. Immunother. 62, 1137–1148. doi: 10.1007/s00262-013-1434-6
Bekes, E. M., Deryugina, E. I., Kupriyanova, T. A., Zajac, E., Botkjaer, K. A., Andreasen, P. A., et al. (2011). Activation of Pro-uPA is critical for initial escape from the primary tumor and hematogenous dissemination of human carcinoma cells. Neoplasia 13, 806–821.
Bellail, A. C., Hunter, S. B., Brat, D. J., Tan, C., and Van Meir, E. G. (2004). Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int. J. Biochem. Cell Biol. 36, 1046–1069. doi: 10.1016/j.biocel.2004.01.013
Ben-Shoshan, J., Maysel-Auslender, S., Mor, A., Keren, G., and George, J. (2008). Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1alpha. Eur. J. Immunol. 38, 2412–2418. doi: 10.1002/eji.200838318
Bianchi, L., De Micheli, E., Bricolo, A., Ballini, C., Fattori, M., Venturi, C., et al. (2004). Extracellular levels of amino acids and choline in human high grade gliomas: an intraoperative microdialysis study. Neurochem. Res. 29, 325–334. doi: 10.1023/b:nere.0000010462.72557.6d
Biswas, S. K. (2015). Metabolic reprogramming of immune cells in cancer progression. Immunity 43, 435–449. doi: 10.1016/j.immuni.2015.09.001
Bonneh-Barkay, D., and Wiley, C. A. (2009). Brain extracellular matrix in neurodegeneration. Brain Pathol. 19, 573–585. doi: 10.1111/j.1750-3639.2008.00195.x
Chandra, A., Jahangiri, A., Chen, W., Nguyen, A. T., Yagnik, G., Pereira, M. P., et al. (2020). Clonal ZEB1-driven mesenchymal transition promotes targetable oncologic antiangiogenic therapy resistance. Cancer Res. 80, 1498–1511. doi: 10.1158/0008-5472.CAN-19-1305
Chandrasekar, N., Mohanam, S., Gujrati, M., Olivero, W. C., Dinh, D. H., and Rao, J. S. (2003). Downregulation of uPA inhibits migration and PI3k/Akt signaling in glioblastoma cells. Oncogene 22, 392–400. doi: 10.1038/sj.onc.1206164
Cheng, X., Geng, F., Pan, M., Wu, X., Zhong, Y., Wang, C., et al. (2020). Targeting DGAT1 ameliorates glioblastoma by increasing fat catabolism and oxidative stress. Cell Metab. 32, 229–242.e8. doi: 10.1016/j.cmet.2020.06.002
Chinnaiyan, P., Kensicki, E., Bloom, G., Prabhu, A., Sarcar, B., Kahali, S., et al. (2012). The metabolomic signature of malignant glioma reflects accelerated anabolic metabolism. Cancer Res. 72, 5878–5888. doi: 10.1158/0008-5472.CAN-12-1572-T
Cohen, A., Holmen, S., and Colman, H. (2013). IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 13:345. doi: 10.1007/s11910-013-0345-4
Correia, C. R., Gaifem, J., Oliveira, M. B., Silvestre, R., and Mano, J. F. (2017). Influence of surface modified poly(L-lactic acid) films on the differentiation of human monocytes into macrophages. Biomater. Sci. 5, 551–560. doi: 10.1039/c6bm00920d
Cuddapah, V. A., Robel, S., Watkins, S., and Sontheimer, H. (2014). A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 15, 455–465. doi: 10.1038/nrn3765
Dai, D.-W., Lu, Q., Wang, L.-X., Zhao, W.-Y., Cao, Y.-Q., Li, Y.-N., et al. (2013). Decreased miR-106a inhibits glioma cell glucose uptake and proliferation by targeting SLC2A3 in GBM. BMC Cancer 13:478. doi: 10.1186/1471-2407-13-478
Daneman, R., and Prat, A. (2015). The blood–brain barrier. Cold Spring Harb. Perspect. Biol. 7:a020412. doi: 10.1101/cshperspect.a020412
Davis, M. E. (2016). Glioblastoma: overview of disease and treatment. Clin. J. Oncol. Nurs. 20, S2–S8. doi: 10.1188/16.CJON.S1.2-8
de Gooijer, M. C., Guillén Navarro, M., Bernards, R., Wurdinger, T., and van Tellingen, O. (2018). An experimenter’s guide to glioblastoma invasion pathways. Trends Mol. Med. 24, 763–780. doi: 10.1016/j.molmed.2018.07.003
Dwarakanath, B. S., Singh, D., Banerji, A. K., Sarin, R., Venkataramana, N. K., Jalali, R., et al. (2009). Clinical studies for improving radiotherapy with 2-deoxy-D-glucose: present status and future prospects. J. Cancer Res. Ther. 5(Suppl. 1), S21–S26. doi: 10.4103/0973-1482.55136
Everts, B., Amiel, E., van der Windt, G. J. W., Freitas, T. C., Chott, R., Yarasheski, K. E., et al. (2012). Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 120, 1422–1431. doi: 10.1182/blood-2012-03-419747
Fack, F., Espedal, H., Keunen, O., Golebiewska, A., Obad, N., Harter, P. N., et al. (2015). Bevacizumab treatment induces metabolic adaptation toward anaerobic metabolism in glioblastomas. Acta Neuropathol. 129, 115–131. doi: 10.1007/s00401-014-1352-5
Fatehi Hassanabad, A. (2019). Current perspectives on statins as potential anti-cancer therapeutics: clinical outcomes and underlying molecular mechanisms. Transl. Lung Cancer Res. 8, 692–699. doi: 10.21037/tlcr.2019.09.08
Garcia-Bermudez, J., Baudrier, L., La, K., Zhu, X. G., Fidelin, J., Sviderskiy, V. O., et al. (2018). Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumors. Nat. Cell Biol. 20, 775–781. doi: 10.1038/s41556-018-0118-z
Garnier, D., Renoult, O., Alves-Guerra, M.-C., Paris, F., and Pecqueur, C. (2019). Glioblastoma stem-like cells, metabolic strategy to kill a challenging target. Front. Oncol. 9:118. doi: 10.3389/fonc.2019.00118
Geng, F., Cheng, X., Wu, X., Yoo, J. Y., Cheng, C., Guo, J. Y., et al. (2016). Inhibition of SOAT1 suppresses glioblastoma growth via blocking SREBP-1-mediated lipogenesis. Clin. Cancer Res. 22, 5337–5348. doi: 10.1158/1078-0432.CCR-15-2973
Georgiadou, M., Lilja, J., Jacquemet, G., Guzmán, C., Rafaeva, M., Alibert, C., et al. (2017). AMPK negatively regulates tensin-dependent integrin activity. J. Cell Biol. 216, 1107–1121. doi: 10.1083/jcb.201609066
Ghesquière, B., Wong, B. W., Kuchnio, A., and Carmeliet, P. (2014). Metabolism of stromal and immune cells in health and disease. Nature 511, 167–176. doi: 10.1038/nature13312
Godlewski, J., Nowicki, M. O., Bronisz, A., Nuovo, G., Palatini, J., De Lay, M., et al. (2010). MicroRNA-451 regulates LKB1/AMPK signaling and allows adaptation to metabolic stress in glioma cells. Mol. Cell 37, 620–632. doi: 10.1016/j.molcel.2010.02.018
Goldbrunner, R. H., Bernstein, J. J., and Tonn, J. C. (1998). ECM-mediated glioma cell invasion. Microsc. Res. Tech. 43, 250–257. doi: 10.1002/(SICI)1097-0029(19981101)43:3<250::AID-JEMT7<3.0.CO;2-C
Gong, L., Xiao, Y., Xia, F., Wu, P., Zhao, T., Xie, S., et al. (2019). The mevalonate coordinates energy input and cell proliferation. Cell Death Dis. 10:327. doi: 10.1038/s41419-019-1544-y
Guo, D., Bell, E. H., and Chakravarti, A. (2013). Lipid metabolism emerges as a promising target for malignant glioma therapy. CNS Oncol. 2, 289–299. doi: 10.2217/cns.13.20
Gupta, K., Vuckovic, I., Zhang, S., Xiong, Y., Carlson, B. L., Jacobs, J., et al. (2020). Radiation induced metabolic alterations associate with tumor aggressiveness and poor outcome in glioblastoma. Front. Oncol. 10:535. doi: 10.3389/fonc.2020.00535
Hagemann, C., Anacker, J., Ernestus, R.-I., and Vince, G. H. (2012). A complete compilation of matrix metalloproteinase expression in human malignant gliomas. World J. Clin. Oncol. 3, 67–79. doi: 10.5306/wjco.v3.i5.67
Hale, J. S., Otvos, B., Sinyuk, M., Alvarado, A. G., Hitomi, M., Stoltz, K., et al. (2014). Cancer stem cell-specific scavenger receptor CD36 drives glioblastoma progression. Stem Cells 32, 1746–1758. doi: 10.1002/stem.1716
Hamm, R., Zeino, M., Frewert, S., and Efferth, T. (2014). Up-regulation of cholesterol associated genes as novel resistance mechanism in glioblastoma cells in response to archazolid B. Toxicol. Appl. Pharmacol. 281, 78–86. doi: 10.1016/j.taap.2014.08.033
Han, T., Kang, D., Ji, D., Wang, X., Zhan, W., Fu, M., et al. (2013). How does cancer cell metabolism affect tumor migration and invasion? Cell Adh. Migr. 7, 395–403. doi: 10.4161/cam.26345
Hassanein, M., Hoeksema, M. D., Shiota, M., Qian, J., Harris, B. K., Chen, H., et al. (2013). SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin. Cancer Res. 19, 560–570. doi: 10.1158/1078-0432.CCR-12-2334
Hatoum, A., Mohammed, R., and Zakieh, O. (2019). The unique invasiveness of glioblastoma and possible drug targets on extracellular matrix. Cancer Manag. Res. 11, 1843–1855. doi: 10.2147/CMAR.S186142
Hegazy, A. M., Yamada, D., Kobayashi, M., Kohno, S., Ueno, M., Ali, M. A. E., et al. (2016). Therapeutic strategy for targeting aggressive malignant gliomas by disrupting their energy balance. J. Biol. Chem. 291, 21496–21509. doi: 10.1074/jbc.M116.734756
Hernandez-Anzaldo, S., Brglez, V., Hemmeryckx, B., Leung, D., Filep, J. G., Vance, J. E., et al. (2016). Novel role for matrix metalloproteinase 9 in modulation of cholesterol metabolism. J. Am. Heart Assoc. 5:4228. doi: 10.1161/JAHA.116.004228
Huang, J., Yu, J., Tu, L., Huang, N., Li, H., and Luo, Y. (2019). Isocitrate dehydrogenase mutations in glioma: from basic discovery to therapeutics development. Front. Oncol. 9:506. doi: 10.3389/fonc.2019.00506
Hussain, S. F., Yang, D., Suki, D., Aldape, K., Grimm, E., and Heimberger, A. B. (2006). The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 8, 261–279. doi: 10.1215/15228517-2006-008
Jahangiri, A., Nguyen, A., Chandra, A., Sidorov, M. K., Yagnik, G., Rick, J., et al. (2017). Cross-activating c-Met/β1 integrin complex drives metastasis and invasive resistance in cancer. Proc. Natl. Acad. Sci. U.S.A. 114, E8685–E8694. doi: 10.1073/pnas.1701821114
Jameson, M. (2014). Pilot Study Evaluating Progression-free Survival in Patients Using a Ketogenic Diet While Receiving Chemoradiation for Glioblastoma Multiforme. Available online at: http://apps.who.int/trialsearch/Trial2.aspx?TrialID=ACTRN12614001056684
Jewell, J. L., Kim, Y. C., Russell, R. C., Yu, F.-X., Park, H. W., Plouffe, S. W., et al. (2015). Differential regulation of mTORC1 by leucine and glutamine. Science 347, 194–198. doi: 10.1126/science.1259472
Jiang, P., Du, W., and Wu, M. (2014). Regulation of the pentose phosphate pathway in cancer. Protein Cell 5, 592–602. doi: 10.1007/s13238-014-0082-8
Jiang, W., Finniss, S., Cazacu, S., Xiang, C., Brodie, Z., Mikkelsen, T., et al. (2016). Repurposing phenformin for the targeting of glioma stem cells and the treatment of glioblastoma. Oncotarget 7, 56456–56470. doi: 10.18632/oncotarget.10919
Jin, L., and Zhou, Y. (2019). Crucial role of the pentose phosphate pathway in malignant tumors. Oncol. Lett. 17, 4213–4221. doi: 10.3892/ol.2019.10112
Ju, J. A., Godet, I., Ye, I. C., Byun, J., Jayatilaka, H., Lee, S. J., et al. (2017). Hypoxia selectively enhances integrin receptor expression to promote metastasis. Mol. Cancer Res. 15, 723–734. doi: 10.1158/1541-7786.MCR-16-0338
Kalyanaraman, B. (2017). Teaching the basics of cancer metabolism: developing antitumor strategies by exploiting the differences between normal and cancer cell metabolism. Redox Biol. 12, 833–842. doi: 10.1016/j.redox.2017.04.018
Kasznicki, J., Sliwinska, A., and Drzewoski, J. (2014). Metformin in cancer prevention and therapy. Ann. Transl. Med. 2:57. doi: 10.3978/j.issn.2305-5839.2014.06.01
Kathagen-Buhmann, A., Schulte, A., Weller, J., Holz, M., Herold-Mende, C., Glass, R., et al. (2016). Glycolysis and the pentose phosphate pathway are differentially associated with the dichotomous regulation of glioblastoma cell migration versus proliferation. Neuro Oncol. 18, 1219–1229. doi: 10.1093/neuonc/now024
Kesarwani, P., Kant, S., Prabhu, A., and Chinnaiyan, P. (2017). The interplay between metabolic remodeling and immune regulation in glioblastoma. Neuro Oncol. 19, 1308–1315. doi: 10.1093/neuonc/nox079
Kesarwani, P., Prabhu, A., Kant, S., Kumar, P., Graham, S. F., Buelow, K. L., et al. (2018). Tryptophan metabolism contributes to radiation-induced immune checkpoint reactivation in glioblastoma. Clin. Cancer Res. 24, 3632–3643. doi: 10.1158/1078-0432.CCR-18-0041
Klein, T., and Bischoff, R. (2011). Physiology and pathophysiology of matrix metalloproteases. Amino Acids 41, 271–290. doi: 10.1007/s00726-010-0689-x
Klein, P., Tyrlikova, I., Zuccoli, G., Tyrlik, A., and Maroon, J. C. (2020). Treatment of glioblastoma multiforme with “classic” 4:1 ketogenic diet total meal replacement. Cancer Metabol. 8:24. doi: 10.1186/s40170-020-00230-9
Kowalik, M. A., Columbano, A., and Perra, A. (2017). Emerging role of the pentose phosphate pathway in hepatocellular carcinoma. Front. Oncol. 7:87. doi: 10.3389/fonc.2017.00087
Kuang, R., Jahangiri, A., Mascharak, S., Nguyen, A., Chandra, A., Flanigan, P. M., et al. (2017). GLUT3 upregulation promotes metabolic reprogramming associated with antiangiogenic therapy resistance. JCI Insight 2:e88815. doi: 10.1172/jci.insight.88815
Labak, C. M., Wang, P. Y., Arora, R., Guda, M. R., Asuthkar, S., Tsung, A. J., et al. (2016). Glucose transport: meeting the metabolic demands of cancer, and applications in glioblastoma treatment. Am. J. Cancer Res. 6, 1599–1608.
Lakka, S. S., Bhattacharya, A., Mohanam, S., Boyd, D., and Rao, J. S. (2001). Regulation of the uPA gene in various grades of human glioma cells. Int. J. Oncol. 18, 71–79.
Lakka, S. S., Gondi, C. S., and Rao, J. S. (2005). Proteases and glioma angiogenesis. Brain Pathol. 15, 327–341. doi: 10.1111/j.1750-3639.2005.tb00118.x
Lathia, J. D., Mack, S. C., Mulkearns-Hubert, E. E., Valentim, C. L. L., and Rich, J. N. (2015). Cancer stem cells in glioblastoma. Genes Dev. 29, 1203–1217. doi: 10.1101/gad.261982.115
Lau, L. W., Cua, R., Keough, M. B., Haylock-Jacobs, S., and Yong, V. W. (2013). Pathophysiology of the brain extracellular matrix: a new target for remyelination. Nat. Rev. Neurosci. 14, 722–729. doi: 10.1038/nrn3550
Lee, J. V., Berry, C. T., Kim, K., Sen, P., Kim, T., Carrer, A., et al. (2018). Acetyl-CoA promotes glioblastoma cell adhesion and migration through Ca2+-NFAT signaling. Genes Dev. 32, 497–511. doi: 10.1101/gad.311027.117
Li, K., Ouyang, L., He, M., Luo, M., Cai, W., Tu, Y., et al. (2017). IDH1 R132H mutation regulates glioma chemosensitivity through Nrf2 pathway. Oncotarget 8, 28865–28879. doi: 10.18632/oncotarget.15868
Liang, Y., Han, H., Liu, L., Duan, Y., Yang, X., Ma, C., et al. (2018). CD36 plays a critical role in proliferation, migration and tamoxifen-inhibited growth of ER-positive breast cancer cells. Oncogenesis 7, 1–14. doi: 10.1038/s41389-018-0107-x
Lieu, E. L., Nguyen, T., Rhyne, S., and Kim, J. (2020). Amino acids in cancer. Exp. Mol. Med. 52, 15–30. doi: 10.1038/s12276-020-0375-3
Lin, E. Y., Li, J.-F., Gnatovskiy, L., Deng, Y., Zhu, L., Grzesik, D. A., et al. (2006). Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 66, 11238–11246. doi: 10.1158/0008-5472.CAN-06-1278
Liu, B., Bai, W., Ou, G., and Zhang, J. (2019). Cdh1-mediated metabolic switch from pentose phosphate pathway to glycolysis contributes to sevoflurane-induced neuronal apoptosis in developing brain. ACS Chem. Neurosci. 10, 2332–2344. doi: 10.1021/acschemneuro.8b00644
Loreck, D. J., Galarraga, J., Van der Feen, J., Phang, J. M., Smith, B. H., and Cummins, C. J. (1987). Regulation of the pentose phosphate pathway in human astrocytes and gliomas. Metab. Brain Dis. 2, 31–46. doi: 10.1007/BF00999507
Lu, C.-L., Qin, L., Liu, H.-C., Candas, D., Fan, M., and Li, J. J. (2015). Tumor cells switch to mitochondrial oxidative phosphorylation under radiation via mTOR-mediated hexokinase II inhibition–a Warburg-reversing effect. PLoS One 10:e0121046. doi: 10.1371/journal.pone.0121046
Lussier, D. M., Woolf, E. C., Johnson, J. L., Brooks, K. S., Blattman, J. N., and Scheck, A. C. (2016). Enhanced immunity in a mouse model of malignant glioma is mediated by a therapeutic ketogenic diet. BMC Cancer 16:2337. doi: 10.1186/s12885-016-2337-7
Maiti, R. (2014). Metronomic chemotherapy. J. Pharmacol. Pharmacother. 5, 186–192. doi: 10.4103/0976-500X.136098
Malric, L., Monferran, S., Gilhodes, J., Boyrie, S., Dahan, P., Skuli, N., et al. (2017). Interest of integrins targeting in glioblastoma according to tumor heterogeneity and cancer stem cell paradigm: an update. Oncotarget 8, 86947–86968. doi: 10.18632/oncotarget.20372
Malta, T. M., de Souza, C. F., Sabedot, T. S., Silva, T. C., Mosella, M. S., Kalkanis, S. N., et al. (2018). Glioma CpG island methylator phenotype (G-CIMP): biological and clinical implications. Neuro Oncol. 20, 608–620. doi: 10.1093/neuonc/nox183
Marie, S. K. N., and Shinjo, S. M. O. (2011). Metabolism and brain cancer. Clinics (Sao Paulo) 66, 33–43. doi: 10.1590/S1807-59322011001300005
Marin-Valencia, I., Yang, C., Mashimo, T., Cho, S., Baek, H., Yang, X.-L., et al. (2012). Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse, human glioblastomas in the mouse brain in vivo. Cell Metab. 15, 827–837. doi: 10.1016/j.cmet.2012.05.001
Martin-McGill, K. J., Marson, A. G., Tudur Smith, C., and Jenkinson, M. D. (2017). Ketogenic diets as an adjuvant therapy in glioblastoma (the KEATING trial): study protocol for a randomised pilot study. Pilot Feasibility Stud. 3:209. doi: 10.1186/s40814-017-0209-9
Martin-McGill, K. J., Marson, A. G., Tudur Smith, C., and Jenkinson, M. D. (2018). The modified ketogenic diet in adults with glioblastoma: an evaluation of feasibility and deliverability within the National Health Service. Nutr. Cancer 70, 643–649. doi: 10.1080/01635581.2018.1460677
Maus, A., and Peters, G. J. (2017). Glutamate and α-ketoglutarate: key players in glioma metabolism. Amino Acids 49, 21–32. doi: 10.1007/s00726-016-2342-9
Mergenthaler, P., Lindauer, U., Dienel, G. A., and Meisel, A. (2013). Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 36, 587–597. doi: 10.1016/j.tins.2013.07.001
Mestre-Farrera, A., Bruch-Oms, M., Peña, R., Rodríguez-Morató, J., Alba-Castellón, L., Comerma, L., et al. (2021). Glutamine-directed migration of cancer-activated fibroblasts facilitates epithelial tumor invasion. Cancer Res. 81, 438–451. doi: 10.1158/0008-5472.CAN-20-0622
Miller, T. E., Liau, B. B., Wallace, L. C., Morton, A. R., Xie, Q., Dixit, D., et al. (2017). Transcription elongation factors represent in vivo cancer dependencies in glioblastoma. Nature 547, 355–359. doi: 10.1038/nature23000
Mukherjee, J., Phillips, J. J., Zheng, S., Wiencke, J., Ronen, S. M., and Pieper, R. O. (2013). Pyruvate kinase M2 expression, but not pyruvate kinase activity, is up-regulated in a grade-specific manner in human glioma. PLoS One 8:e57610. doi: 10.1371/journal.pone.0057610
Munn, D. H., Sharma, M. D., Hou, D., Baban, B., Lee, J. R., Antonia, S. J., et al. (2004). Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Invest. 114, 280–290. doi: 10.1172/JCI21583
Nakano, I. (2014). Therapeutic potential of targeting glucose metabolism in glioma stem cells. Expert Opin. Ther. Targets 18, 1233–1236. doi: 10.1517/14728222.2014.944899
Natarajan, S. K., and Venneti, S. (2019). Glutamine metabolism in brain tumors. Cancers (Basel) 11:628. doi: 10.3390/cancers11111628
Nebeling, L. C., Miraldi, F., Shurin, S. B., and Lerner, E. (1995). Effects of a ketogenic diet on tumor metabolism and nutritional status in pediatric oncology patients: two case reports. J. Am. College Nutr. 14, 202–208.
Obara-Michlewska, M., and Szeliga, M. (2020). Targeting glutamine addiction in gliomas. Cancers (Basel) 12:310. doi: 10.3390/cancers12020310
Olar, A., and Aldape, K. D. (2014). Using the molecular classification of glioblastoma to inform personalized treatment. J. Pathol. 232, 165–177. doi: 10.1002/path.4282
Oliva, C. R., Nozell, S. E., Diers, A., McClugage, S. G., Sarkaria, J. N., Markert, J. M., et al. (2010). Acquisition of temozolomide chemoresistance in gliomas leads to remodeling of mitochondrial electron transport chain ∗. J. Biol. Chem. 285, 39759–39767. doi: 10.1074/jbc.M110.147504
Özcan, E., and Çakır, T. (2016). Reconstructed metabolic network models predict flux-level metabolic reprogramming in glioblastoma. Front. Neurosci. 10:156. doi: 10.3389/fnins.2016.00156
Palanichamy, K., and Chakravarti, A. (2017). Diagnostic and prognostic significance of methionine uptake and methionine positron emission tomography imaging in gliomas. Front. Oncol. 7:257. doi: 10.3389/fonc.2017.00257
Palanichamy, K., Thirumoorthy, K., Kanji, S., Gordon, N., Singh, R., Jacob, J. R., et al. (2016). Methionine and kynurenine activate oncogenic kinases in glioblastoma, and methionine deprivation compromises proliferation. Clin. Cancer Res. 22, 3513–3523. doi: 10.1158/1078-0432.CCR-15-2308
Panosyan, E. H., Lin, H. J., Koster, J., and Lasky, J. L. (2017). In search of druggable targets for GBM amino acid metabolism. BMC Cancer 17:3148. doi: 10.1186/s12885-017-3148-1
Paolillo, M., Serra, M., and Schinelli, S. (2016). Integrins in glioblastoma: still an attractive target? Pharmacol. Res. 113, 55–61. doi: 10.1016/j.phrs.2016.08.004
Patel, D., Ahmad, F., Kambach, D. M., Sun, Q., Halim, A. S., Kramp, T., et al. (2019). LXRβ controls glioblastoma cell growth, lipid balance, and immune modulation independently of ABCA1. Sci. Rep. 9:15458. doi: 10.1038/s41598-019-51865-8
Pavlyk, I., Rzhepetskyy, Y., Jagielski, A. K., Drozak, J., Wasik, A., Pereverzieva, G., et al. (2015). Arginine deprivation affects glioblastoma cell adhesion, invasiveness and actin cytoskeleton organization by impairment of β-actin arginylation. Amino Acids 47, 199–212. doi: 10.1007/s00726-014-1857-1
Paw, I., Carpenter, R. C., Watabe, K., Debinski, W., and Lo, H.-W. (2015). Mechanisms regulating glioma invasion. Cancer Lett. 362, 1–7. doi: 10.1016/j.canlet.2015.03.015
Pearce, E. L., and Pearce, E. J. (2013). Metabolic pathways in immune cell activation and quiescence. Immunity 38, 633–643. doi: 10.1016/j.immuni.2013.04.005
Phan, L. M., Yeung, S.-C. J., and Lee, M.-H. (2014). Cancer metabolic reprogramming: importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 11, 1–19. doi: 10.7497/j.issn.2095-3941.2014.01.001
Pirmoradi, L., Seyfizadeh, N., Ghavami, S., Zeki, A. A., and Shojaei, S. (2019). Targeting cholesterol metabolism in glioblastoma: a new therapeutic approach in cancer therapy. J. Invest. Med. 67, 715–719. doi: 10.1136/jim-2018-000962
Pollard, J. W. (2004). Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 4, 71–78. doi: 10.1038/nrc1256
Pombo Antunes, A. R., Scheyltjens, I., Duerinck, J., Neyns, B., Movahedi, K., and Van Ginderachter, J. A. (2020). Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. eLife 9:52176. doi: 10.7554/eLife.52176
Prager, B. C., Bhargava, S., Mahadev, V., Hubert, C. G., and Rich, J. N. (2020). Glioblastoma stem cells: driving resilience through chaos. Trends Cancer 6, 223–235. doi: 10.1016/j.trecan.2020.01.009
Quinones, A., and Le, A. (2018). The multifaceted metabolism of glioblastoma. Adv. Exp. Med. Biol. 1063, 59–72. doi: 10.1007/978-3-319-77736-8_4
Rao, J. S. (2003). Molecular mechanisms of glioma invasiveness: the role of proteases. Nat. Rev. Cancer 3, 489–501. doi: 10.1038/nrc1121
Reeds, P. J., Burrin, D. G., Davis, T. A., and Stoll, B. (1998). Amino acid metabolism and the energetics of growth. Arch. Tierernahr. 51, 187–197. doi: 10.1080/17450399809381918
Rieger, J., Bähr, O., Maurer, G. D., Hattingen, E., Franz, K., Brucker, D., et al. (2014). ERGO: a pilot study of ketogenic diet in recurrent glioblastoma. Int. J. Oncol. 44, 1843–1852. doi: 10.3892/ijo.2014.2382
Santos, J. G., Da Cruz, W. M. S., Schönthal, A. H., Salazar, M. D., Fontes, C. A. P., Quirico-Santos, T., et al. (2018). Efficacy of a ketogenic diet with concomitant intranasal perillyl alcohol as a novel strategy for the therapy of recurrent glioblastoma. Oncol. Lett. 15, 1263–1270. doi: 10.3892/ol.2017.7362
Schlaepfer, I. R., Nambiar, D. K., Ramteke, A., Kumar, R., Dhar, D., Agarwal, C., et al. (2015). Hypoxia induces triglycerides accumulation in prostate cancer cells and extracellular vesicles supporting growth and invasiveness following reoxygenation. Oncotarget 6, 22836–22856.
Schmidt, C., Sciacovelli, M., and Frezza, C. (2019). Fumarate hydratase in cancer: a multifaceted tumour suppressor. Semin. Cell Dev. Biol. 98, 15–25. doi: 10.1016/j.semcdb.2019.05.002
Schousboe, A., Scafidi, S., Bak, L. K., Waagepetersen, H. S., and McKenna, M. C. (2014). Glutamate metabolism in the brain focusing on astrocytes. Adv. Neurobiol. 11, 13–30. doi: 10.1007/978-3-319-08894-5_2
Schwartz, K. A., Noel, M., Nikolai, M., and Chang, H. T. (2018). Investigating the ketogenic diet as treatment for primary aggressive brain cancer: challenges and lessons learned. Front. Nutr. 5:11. doi: 10.3389/fnut.2018.00011
Seliger, C., and Hau, P. (2018). Drug repurposing of metabolic agents in malignant glioma. Int. J. Mol. Sci. 19:2768. doi: 10.3390/ijms19092768
Semonche, A., Shah, A. H., Ivan, M. E., and Komotar, R. J. (2019). Towards a microRNA-based Gene therapy for glioblastoma. Neurosurgery 85, E210–E211. doi: 10.1093/neuros/nyz166
Sferrazzo, G., Di Rosa, M., Barone, E., Li Volti, G., Musso, N., Tibullo, D., et al. (2020). Heme oxygenase-1 in central nervous system malignancies. J. Clin. Med. 9:1562. doi: 10.3390/jcm9051562
Shah, S. S., Rodriguez, G. A., Musick, A., Walters, W. M., de Cordoba, N., Barbarite, E., et al. (2019). Targeting glioblastoma stem cells with 2-deoxy-D-glucose (2-DG) potentiates radiation-induced unfolded protein response (UPR). Cancers (Basel) 11:159. doi: 10.3390/cancers11020159
Shakya, S., Gromovsky, A. D., Hale, J. S., Knudsen, A. M., Prager, B., Wallace, L. C., et al. (2021). Altered lipid metabolism marks glioblastoma stem and non-stem cells in separate tumor niches. Cancer Biol. 9:101. doi: 10.1186/s40478-021-01205-7
Shen, H., Decollogne, S., Dilda, P. J., Hau, E., Chung, S. A., Luk, P. P., et al. (2015). Dual-targeting of aberrant glucose metabolism in glioblastoma. J. Exp. Clin. Cancer Res. 34:14. doi: 10.1186/s13046-015-0130-0
Shi, L., He, C., Li, Z., Wang, Z., and Zhang, Q. (2017). FBP1 modulates cell metabolism of breast cancer cells by inhibiting the expression of HIF-1α. Neoplasma 64, 535–542. doi: 10.4149/neo_2017_407
Shi, Y., Lim, S. K., Liang, Q., Iyer, S. V., Wang, H.-Y., Wang, Z., et al. (2019). Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 567, 341–346. doi: 10.1038/s41586-019-0993-x
Shibuya, K., Okada, M., Suzuki, S., Seino, M., Seino, S., Takeda, H., et al. (2014). Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget 6, 651–661. doi: 10.18632/oncotarget.2892
Silva, L. S., Poschet, G., Nonnenmacher, Y., Becker, H. M., Sapcariu, S., Gaupel, A.-C., et al. (2017). Branched-chain ketoacids secreted by glioblastoma cells via MCT1 modulate macrophage phenotype. EMBO Rep. 18, 2172–2185. doi: 10.15252/embr.201744154
Singer, K., Gottfried, E., Kreutz, M., and Mackensen, A. (2011). Suppression of T-cell responses by tumor metabolites. Cancer Immunol. Immunother. 60, 425–431. doi: 10.1007/s00262-010-0967-1
Smith, R. J. (1990). Glutamine metabolism and its physiologic importance. JPEN J. Parenter. Enteral Nutr. 14, 40S–44S. doi: 10.1177/014860719001400402
Snaebjornsson, M. T., Janaki-Raman, S., and Schulze, A. (2020). Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab. 31, 62–76. doi: 10.1016/j.cmet.2019.11.010
Sousa, B., Pereira, J., and Paredes, J. (2019). The crosstalk between cell adhesion and cancer metabolism. Int. J. Mol. Sci. 20:1933. doi: 10.3390/ijms20081933
Strickland, M., and Stoll, E. A. (2017). Metabolic reprogramming in glioma. Front. Cell Dev. Biol. 5:43. doi: 10.3389/fcell.2017.00043
Strowd, R. E., Cervenka, M. C., Henry, B. J., Kossoff, E. H., Hartman, A. L., and Blakeley, J. O. (2015). Glycemic modulation in neuro-oncology: experience and future directions using a modified Atkins diet for high-grade brain tumors. Neuro-Oncol. Pract. 2, 127–136. doi: 10.1093/nop/npv010
Stupp, R., Lukas, R. V., and Hegi, M. E. (2019). Improving survival in molecularly selected glioblastoma. Lancet 393, 615–617. doi: 10.1016/S0140-6736(18)33211-2
Taïb, B., Aboussalah, A. M., Moniruzzaman, M., Chen, S., Haughey, N. J., Kim, S. F., et al. (2019). Lipid accumulation and oxidation in glioblastoma multiforme. Sci. Rep. 9:19593. doi: 10.1038/s41598-019-55985-z
Tardito, S., Oudin, A., Ahmed, S. U., Fack, F., Keunen, O., Zheng, L., et al. (2015). Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 17, 1556–1568. doi: 10.1038/ncb3272
Thorens, B. (2012). Sensing of glucose in the brain. Handb. Exp. Pharmacol. 12, 277–294. doi: 10.1007/978-3-642-24716-3_12
Tran, A. A., and Prasad, V. (2020). Drug repurposing for cancer treatments: a well-intentioned, but misguided strategy. Lancet Oncol. 21, 1134–1136. doi: 10.1016/S1470-2045(20)30424-1
Ulasov, I., Yi, R., Guo, D., Sarvaiya, P., and Cobbs, C. (2014). The emerging role of MMP14 in brain tumorigenesis and future therapeutics. Biochim. Biophys. Acta 1846, 113–120. doi: 10.1016/j.bbcan.2014.03.002
Valtorta, S., Salvatore, D., Rainone, P., Belloli, S., Bertoli, G., and Moresco, R. M. (2020). Molecular and cellular complexity of glioma. Focus on tumour microenvironment and the use of molecular and imaging biomarkers to overcome treatment resistance. Int. J. Mol. Sci. 21:5631. doi: 10.3390/ijms21165631
van Deijk, A.-L. F., Camargo, N., Timmerman, J., Heistek, T., Brouwers, J. F., Mogavero, F., et al. (2017). Astrocyte lipid metabolism is critical for synapse development and function in vivo. Glia 65, 670–682. doi: 10.1002/glia.23120
van der Louw, E. J. T. M., Olieman, J. F., van den Bemt, P. M. L. A., Bromberg, J. E. C., Oomen-de Hoop, E., Neuteboom, R. F., et al. (2019). Ketogenic diet treatment as adjuvant to standard treatment of glioblastoma multiforme: a feasibility and safety study. Ther. Adv. Med. Oncol. 11:1758835919853958. doi: 10.1177/1758835919853958
Villa, G. R., Hulce, J. J., Zanca, C., Bi, J., Ikegami, S., Cahill, G. L., et al. (2016). An LXR-cholesterol axis creates a metabolic co-dependency for brain cancers. Cancer Cell 30, 683–693. doi: 10.1016/j.ccell.2016.09.008
Vollmann-Zwerenz, A., Leidgens, V., Feliciello, G., Klein, C. A., and Hau, P. (2020). Tumor cell invasion in glioblastoma. Int. J. Mol. Sci. 21:1932. doi: 10.3390/ijms21061932
Wainwright, D. A., Chang, A. L., Dey, M., Balyasnikova, I. V., Kim, C. K., Tobias, A., et al. (2014). Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res. 20, 5290–5301. doi: 10.1158/1078-0432.CCR-14-0514
Wang, G., Fu, X.-L., Wang, J.-J., Guan, R., Sun, Y., and Tony To, S.-S. (2019). Inhibition of glycolytic metabolism in glioblastoma cells by Pt3glc combinated with PI3K inhibitor via SIRT3-mediated mitochondrial and PI3K/Akt-MAPK pathway. J. Cell Physiol. 234, 5888–5903. doi: 10.1002/jcp.26474
Wei, J., Wu, A., Kong, L.-Y., Wang, Y., Fuller, G., Fokt, I., et al. (2011). Hypoxia potentiates glioma-mediated immunosuppression. PLoS One 6:e16195. doi: 10.1371/journal.pone.0016195
Wen, P. Y., and Kesari, S. (2008). Malignant gliomas in adults. N. Engl. J. Med. 359, 492–507. doi: 10.1056/NEJMra0708126
Wibom, C., Surowiec, I., Mörén, L., Bergström, P., Johansson, M., Antti, H., et al. (2010). Metabolomic patterns in glioblastoma and changes during radiotherapy: a clinical microdialysis study. J. Proteome Res. 9, 2909–2919. doi: 10.1021/pr901088r
Wolf, A., Agnihotri, S., and Guha, A. (2010). Targeting metabolic remodeling in glioblastoma multiforme. Oncotarget 1, 552–562. doi: 10.18632/oncotarget.190
Wolf, K. J., Chen, J., Coombes, J., Aghi, M. K., and Kumar, S. (2019). Dissecting and rebuilding the glioblastoma microenvironment with engineered materials. Nat. Rev. Mater. 4, 651–668. doi: 10.1038/s41578-019-0135-y
Woolf, E. C., Syed, N., and Scheck, A. C. (2016). Tumor metabolism, the ketogenic diet and β-hydroxybutyrate: novel approaches to adjuvant brain tumor therapy. Front. Mol. Neurosci. 9:122. doi: 10.3389/fnmol.2016.00122
Wu, F., Chai, R.-C., Wang, Z., Liu, Y.-Q., Zhao, Z., Li, G.-Z., et al. (2019). Molecular classification of IDH-mutant glioblastomas based on gene expression profiles. Carcinogenesis 40, 853–860. doi: 10.1093/carcin/bgz032
Wu, G. (2009). Amino acids: metabolism, functions, and nutrition. Amino Acids 37, 1–17. doi: 10.1007/s00726-009-0269-0
Würth, R., Pattarozzi, A., Gatti, M., Bajetto, A., Corsaro, A., Parodi, A., et al. (2013). Metformin selectively affects human glioblastoma tumor-initiating cell viability. Cell Cycle 12, 145–156. doi: 10.4161/cc.23050
Xiao, M., Yang, H., Xu, W., Ma, S., Lin, H., Zhu, H., et al. (2012). Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 26, 1326–1338. doi: 10.1101/gad.191056.112
Xu, S., Shao, Q.-Q., Sun, J.-T., Yang, N., Xie, Q., Wang, D.-H., et al. (2013). Synergy between the ectoenzymes CD39 and CD73 contributes to adenosinergic immunosuppression in human malignant gliomas. Neuro Oncol. 15, 1160–1172. doi: 10.1093/neuonc/not067
Xu, Y.-Y., Gao, P., Sun, Y., and Duan, Y.-R. (2015). Development of targeted therapies in treatment of glioblastoma. Cancer Biol. Medr. 12, 223–237. doi: 10.7497/j.issn.2095-3941.2015.0020
Yan, A., Joachims, M. L., Thompson, L. F., Miller, A. D., Canoll, P. D., and Bynoe, M. S. (2019). CD73 promotes glioblastoma pathogenesis and enhances its chemoresistance via A2B adenosine receptor signaling. J. Neurosci. 39, 4387–4402. doi: 10.1523/JNEUROSCI.1118-18.2019
Yasumoto, Y., Miyazaki, H., Vaidyan, L. K., Kagawa, Y., Ebrahimi, M., Yamamoto, Y., et al. (2016). Inhibition of fatty acid synthase decreases expression of stemness markers in glioma stem cells. PLoS One 11:e0147717. doi: 10.1371/journal.pone.0147717
Yuan, T., Liao, W., Feng, N.-H., Lou, Y.-L., Niu, X., Zhang, A.-J., et al. (2013). Human induced pluripotent stem cell-derived neural stem cells survive, migrate, differentiate, and improve neurologic function in a rat model of middle cerebral artery occlusion. Stem Cell Res. Ther. 4:73. doi: 10.1186/scrt224
Zhai, L., Lauing, K. L., Chang, A. L., Dey, M., Qian, J., Cheng, Y., et al. (2015). The role of IDO in brain tumor immunotherapy. J. Neurooncol. 123, 395–403. doi: 10.1007/s11060-014-1687-8
Zhang, H., Ma, Y., Wang, H., Xu, L., and Yu, Y. (2019). MMP-2 expression and correlation with pathology and MRI of glioma. Oncol. Lett. 17, 1826–1832. doi: 10.3892/ol.2018.9806
Zhao, G., Wang, B., Liu, Y., Zhang, J.-G., Deng, S.-C., Qin, Q., et al. (2013). miRNA-141, downregulated in pancreatic cancer, inhibits cell proliferation and invasion by directly targeting MAP4K4. Mol. Cancer Ther. 12, 2569–2580. doi: 10.1158/1535-7163.MCT-13-0296
Zhao, W., Kridel, S., Thorburn, A., Kooshki, M., Little, J., Hebbar, S., et al. (2006). Fatty acid synthase: a novel target for antiglioma therapy. Br. J. Cancer 95, 869–878. doi: 10.1038/sj.bjc.6603350
Zhou, H.-M., Nichols, A., Meda, P., and Vassalli, J.-D. (2000). Urokinase-type plasminogen activator and its receptor synergize to promote pathogenic proteolysis. EMBO J. 19, 4817–4826. doi: 10.1093/emboj/19.17.4817
Zhou, W., and Wahl, D. R. (2019). Metabolic abnormalities in glioblastoma and metabolic strategies to overcome treatment resistance. Cancers (Basel) 11:1231. doi: 10.3390/cancers11091231
Zhu, X. Y., Wang, L., Luan, S. H., Zhang, H. S., Huang, W. T., and Wang, N. H. (2014). The PGI-KLF4 pathway regulates self-renewal of glioma stem cells residing in the mesenchymal niches in human gliomas. Neoplasma 61, 401–410. doi: 10.4149/neo_2014_049
Keywords: glioblastoma, invasion, brain tumor, metabolism, microenvironment
Citation: Garcia JH, Jain S and Aghi MK (2021) Metabolic Drivers of Invasion in Glioblastoma. Front. Cell Dev. Biol. 9:683276. doi: 10.3389/fcell.2021.683276
Received: 20 March 2021; Accepted: 19 May 2021;
Published: 01 July 2021.
Edited by:
Tanja Nicole Hartmann, University of Freiburg Medical Center, GermanyReviewed by:
Georg Karpel-Massler, Ulm University Medical Center, GermanyThomas Iskratsch, Queen Mary University of London, United Kingdom
Copyright © 2021 Garcia, Jain and Aghi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manish K. Aghi, bWFuaXNoLmFnaGlAdWNzZi5lZHU=