 Runze Qiu
Runze Qiu Yue Zhong2†
Yue Zhong2† Qingquan Li
Qingquan Li Yingbin Li
Yingbin Li Hongwei Fan
Hongwei Fan- 1Department of Clinical Pharmacology Lab, Nanjing First Hospital, Nanjing Medical University, Nanjing, China
- 2Center of Drug Discovery, State Key Laboratory of Natural Medicines, China Pharmaceutical University, Nanjing, China
- 3Department of Neurosurgery, The Second Affiliated Hospital of Nanjing Medical University, Nanjing, China
During metabolic reprogramming, glioma cells and their initiating cells efficiently utilized carbohydrates, lipids and amino acids in the hypoxic lesions, which not only ensured sufficient energy for rapid growth and improved the migration to normal brain tissues, but also altered the role of immune cells in tumor microenvironment. Glioma cells secreted interferential metabolites or depriving nutrients to injure the tumor recognition, phagocytosis and lysis of glioma-associated microglia/macrophages (GAMs), cytotoxic T lymphocytes, natural killer cells and dendritic cells, promoted the expansion and infiltration of immunosuppressive regulatory T cells and myeloid-derived suppressor cells, and conferred immune silencing phenotypes on GAMs and dendritic cells. The overexpressed metabolic enzymes also increased the secretion of chemokines to attract neutrophils, regulatory T cells, GAMs, and dendritic cells, while weakening the recruitment of cytotoxic T lymphocytes and natural killer cells, which activated anti-inflammatory and tolerant mechanisms and hindered anti-tumor responses. Therefore, brain-targeted metabolic therapy may improve glioma immunity. This review will clarify the metabolic properties of glioma cells and their interactions with tumor microenvironment immunity, and discuss the application strategies of metabolic therapy in glioma immune silence and escape.
Introduction
Glioma is the most common primary intracranial cancer with a 5-year survival rate of less than 10% (Wang J. et al., 2019), occurring in glial cells such as astrocytes, oligodendrocytes and microglia. Glioblastoma multiforme (GBM) arose from astrocytes is the most frequent glioma with high malignancy and drug resistance, which was classified as grade IV in the WHO grade 2016, with a 5-year relative survival rate of only 5% because of rapid relapse after treatment (Gusyatiner and Hegi, 2018). According to gene transcription characteristics, GBM can be further classified into three subtypes: proneural [mutations on isocitrate dehydrogenase (IDH)-1 or tumor suppressor p53, and PDGFRA amplification], mesenchymal (mutation/deficiency of tumor suppressor NF1), and classical [EGFR amplification and CDKN2A (Ink4a/ARF) homozygous deletion] (Wang Q. et al., 2017). NF1 mutation-mediated proneural-mesenchymal transition is the key mechanism of relapse, causing resistance to treatment (Behnan et al., 2019).
Operative resection, although improving overall survival and prognosis in patients with low- and intermediate-grade gliomas (LGGs and IGGs) (Hervey-Jumper and Berger, 2016), shows limited effect on high-grade gliomas (HGGs) including anaplastic gliomas (WHO grade III) and GBM. Ironically, due to the changeable biological properties and the location of gliomas, non-specific interventions including radiation and brain-permeable cytotoxic drugs benefited patients even more than targeted therapies (Chen R. et al., 2017; Touat et al., 2017). Nevertheless, the tumor microenvironment (TME) makes gliomas resistant to chemotherapeutic drugs, and bring about inflammation to further reduce the prognosis of patients (Wu and Dai, 2017; Yang and Lin, 2017). In order to satisfactorily treat gliomas, one needs to be familiar with brain TME, which determine the evolution of tumors (Hirata and Sahai, 2017).
Since peripheral immune cells cannot enter the blood-brain barrier (BBB) and release inflammatory factors into brain under physiological conditions, brain tissues are protected from inflammation (Engelhardt et al., 2017; Figure 1A). Glial cells play an important role in the integrity and damage repair of BBB (Lou et al., 2016), so glioma cells originating from glial cells can impair BBB and allow peripheral immune cells to enter the brain (Figure 1B), forming a unique TME together with intracranial situ cells, tumor-associated vasculature, perivascular niche and lymphatic vessels (Quail and Joyce, 2017). Immune cells are major members of the glioma TME (Magana-Maldonado et al., 2016; Gieryng et al., 2017), which gradually lost tumor clearance duties or became associates when exposed to tumors. To support tumor growth, glioma cells and glioma stem-like cells (GSCs, also known as brain tumor initiating cells) interacted with adaptive immune cells and recruited immunotolerant innate immune cells to inhibit or evade anti-tumor responses (Broekman et al., 2018).
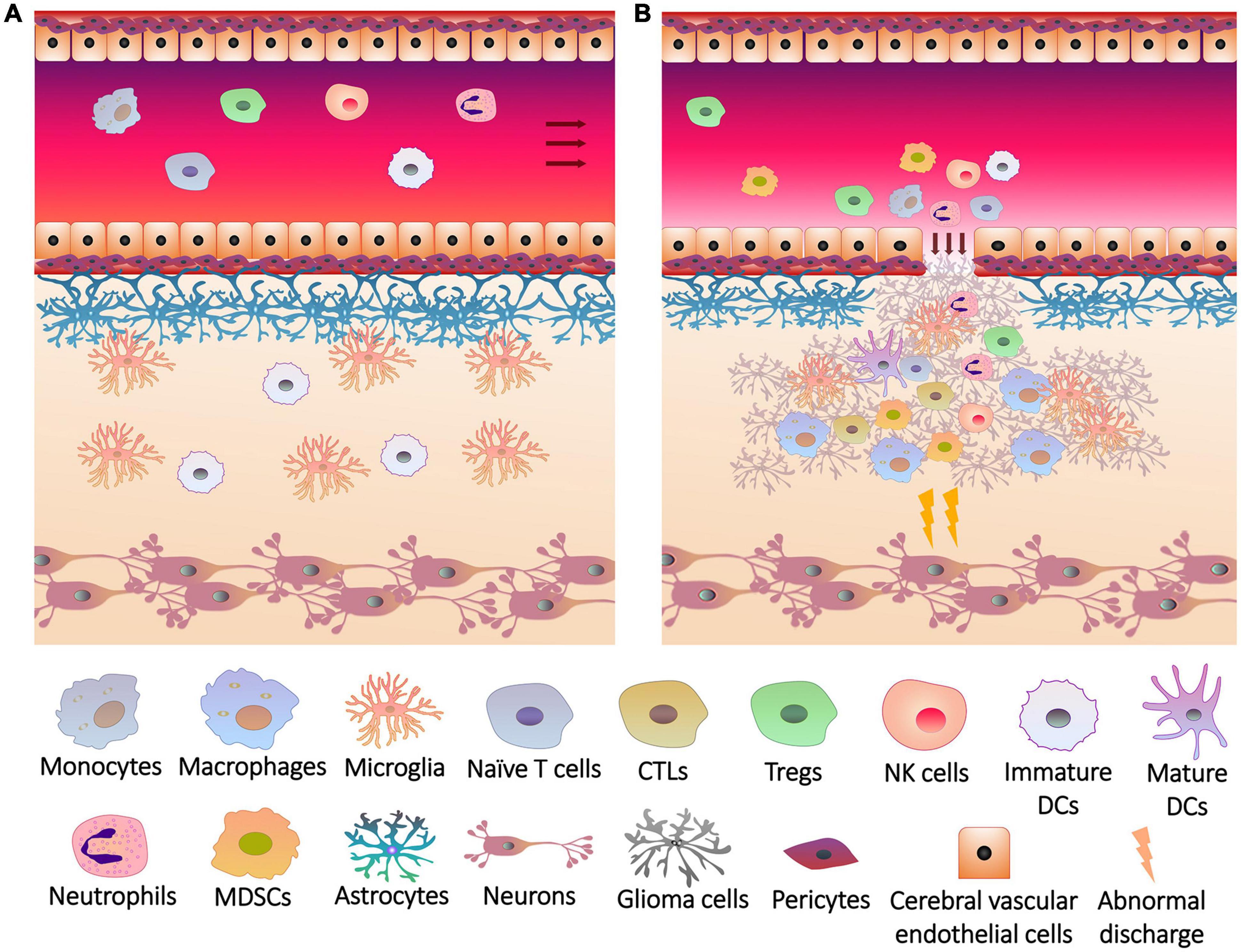
Figure 1. The breakage of blood brain barrier and the infiltration of immune cells into glioma. (A) Under physiological conditions, the blood brain barrier consists of a firm multilayer barrier. Cerebral vascular endothelial cells form a tight junction structure, which is closely connected with pericytes, and is supported by basement membrane underneath. Astrocytes wrap the basement membrane through the end foot, and microglia maintain the integrity of the barrier. The immune cells originally present in the brain are mainly microglia and dendritic cells (DCs). Peripheral immune cells cannot penetrate the blood brain barrier, and brain tissues do not release recruitment signals to the cerebral blood vessels. (B) Rapid growth of glioma cells not only overexcites neurons to induce seizures, but also injures blood brain barrier. Glioma cells release chemokines and other cytokines through the cracks of blood brain barrier to induce the differentiation, expansion and recruitment of peripheral immune cells, including monocytes (in blood)/macrophages (differentiated from monocytes in brain tissue), cytotoxic T lymphocytes (CTLs), regulatory T cells (Tregs), natural killer (NK) cells, neutrophils, DCs and myeloid-derived suppressor cells (MDSCs), which infiltrate the lesions with immune cells in situ.
Although the BBB was damaged, glioma TME reconstructed the blood-brain tumor barrier (BBTB), whose structure is still dense and makes it difficult for antibodies to enter the lesion (van Tellingen et al., 2015). This may be the reason why immunotherapy such as PD-1/PD-L1 monoclonal antibody was less effective in glioma (Jackson et al., 2019). In contrast, metabolic therapies, such as ketogenic diet, significantly benefited patients (Winter et al., 2017). Metabolism is a medium of the communications between glioma and immune cells in TME (Thomas and Yu, 2017). Glioma cells interfere with immune cells through heterogeneous metabolism to mediate tumor growth, invasion, drug resistance and recurrence, which will be reviewed below.
Metabolic Properties of Glioma Cells
Compared with normal tissues, tumors have specific anabolic and catabolic needs due to their rapid and uncontrolled growth. After metabolic reprogramming, tumors tend to gain energy through glycolysis rather than oxidative phosphorylation (OXPHOS) even under aerobic conditions (DeBerardinis and Chandel, 2016), which is called Warburg effect. Recent studies have found that glioma cells that initially grew in an ischemic environment relied on aerobic pentose phosphate pathway (PPP) instead of glycolysis after being exposed to adequate oxygen (Kathagen-Buhmann et al., 2016). When glucose was depleted, glioma cells re-converted the metabolic pattern to OXPHOS via the accumulation of lactate, which is the by-product of glycolysis (Duan et al., 2018). In addition to differentiated glioma cells, GSCs can also switch metabolism between glycolysis and OXPHOS catering to changing circumstances (Shibao et al., 2018), suggesting that the metabolism of glioma cells is environmentally plastic.
Mutations or changed levels of metabolic enzymes and accumulation of metabolites in TME were associated with the malignant progression and epigenetic modifications (Agnihotri and Zadeh, 2016; Masui et al., 2019). Isocitrate dehydrogenase (IDH), a key rate-limiting enzyme in tricarboxylic acid (TCA) cycle regulating carbohydrate, lipid and amino acid metabolism, was widely mutated in proneural GBM cells. Mutations in gliomas often occur in IDH1 and IDH2, both of which caused the conversion of α-ketoglutarate (α-KG) to 2-hydroxyglutarate (2-HG) in a NADPH-dependent manner (Waitkus et al., 2016). The overall survival of patients with proneural subtypes is longer than other subtypes, but the survival became the shortest after excluding IDH mutations, suggesting the positive significance of IDH mutations for prognosis (Behnan et al., 2019). However, IDH mutations may lead to the conversion of LGG to secondary HGG and the emergence of hypermutation phenotypes (Han et al., 2020). As another feature of proneural subtypes, mutations in p53 led to the loss of glycolysis inhibition and promoted tumor cells to adapt to hypoxic environments (Maurer et al., 2019). The expansion of PDGFRA also promoted glycolysis of proneural subtypes (Ran et al., 2013). In the classical GBM subtype, overexpression of EGFR activated PFK1 through PI3K-AKT signaling, leading to the upregulation of GLUT1 and the enhancement of glucose uptake (Lee et al., 2018). Although there is no direct evidence that the inactivation of tumor suppressor NF1 is associated with enhanced glycolysis, the glucose uptake and glycolysis of mesenchymal GBM cells are more active than other subtypes, which explains the increase in the malignancy of GBM caused by proneural-mesenchymal transition. The accumulation of ROS caused by hypoxic lesions can induce this transition, and PI3K-AKT was also stimulated under hypoxia, resulting in active glucose uptake and glycolysis (Talasila et al., 2017; Liu et al., 2020).
Dietary or pharmacological interventions on metabolism, such as ketogenic diet (Poff et al., 2019), dimethylbiguanide, statins and NSAIDs (Gerthofer et al., 2018; Seliger and Hau, 2018) inhibited the growth and invasion of HGGs and the malignant transformation of LGGs, and induced programmed tumor death. In view of the role of cell metabolism in the progression of gliomas, means of metabolomics based on high-throughput analysis has been developed currently (Pandey et al., 2017), whose application requires the familiarity with the metabolic characteristics of glioma cells and GSCs.
Carbohydrate Metabolism
The metabolic trend of glioma cells switches between glycolysis and PPP according to the concentration of oxygen. Under hypoxia, glioma cells overexpressed glycolytic enzymes to maintain energy supply and promote migration, while up-regulating PPP enzymes for rapid proliferation and division under oxygen-rich condition (Kathagen-Buhmann et al., 2016; Kathagen-Buhmann et al., 2018). Glycolysis was highly activated in HGG cells with invasiveness and resistance to conventional treatment (Corbin et al., 2017), but weaker glycolysis was detected in LGG cells with IDH1 mutations, which restricted their energy and made them less aggressive (Fack et al., 2017). Intracellular glucose was catalyzed to glucose-6-phosphate (G6P) by hexokinase (HK), followed by the transformation to 2 glyceraldehyde 3-phosphate (GA3P) via glucose-6-phosphate isomerase, 6-phosphofructokinase (PFK)-1, aldolase and triose phosphate isomerase sequentially, with the cost of 2 ATP. Then GA3P was converted to pyruvate catalyzed by glyceraldehyde 3-phosphate dehydrogenase, phosphaglycerate kinase (PGK), phosphoglycerate mutase 1, enolase and pyruvate kinase M (PKM), during which 4 ATP were generated. The product pyruvate was aerobically converted to acetyl-CoA to enter TCA cycle via pyruvate dehydrogenase (PDH), or became lactate by lactic dehydrogenase (LDH)-A without oxygen. The level of lactate was positively related to the speed of glycolysis, which was significantly higher in HGG cells than neuroblastoma cells and neurons (Kim J. et al., 2015). Apart from energy supply, abundant glycolytic enzymes in malignant glioma cells and GSCs promoted the shift of carbon to ribose-5-phosphate (R5P) for the synthesis of nucleotide (Agnihotri and Zadeh, 2016), supporting tumor growth. To ensure enough raw materials for glycolysis, glioma cells and GSCs took up glucose efficiently via elevated glucose transporter (GLUT) (Libby et al., 2018), of which the level of GLUT3 is characteristic (Xu et al., 2015; Zheng et al., 2016), relating to the resistance to antiangiogenic drugs (Kuang et al., 2017). Glucose is not the only carbohydrate source for glioma cells. They also ingested fructose through GLUT5 and utilized by ketohexokinase for limited growth (Gao et al., 2018; Su et al., 2018).
Carbohydrate uptake and glycolysis of glioma cells were driven by the PI3K–AKT pathway activated by receptor tyrosine kinases including EGFR and c-Met, which up-regulated PFK1 and GLUT1/3 via the activation of PFK2 and glycoprotein synthase kinase (GSK)-3β (Kuang et al., 2017; Lee J.H. et al., 2017; Lee et al., 2018). The mTOR–c-Myc signaling also facilitated glycolysis by glutamine–fructose-6-phosphate aminotransferase 1 (Liu B. et al., 2019). In contrast, the AMP-activated protein kinase (AMPK) inhibited glycolysis and glucose uptake by inhibiting mTORC1 and attenuating transcription coactivator yes-associated protein-induced GLUT3 expression (Wang et al., 2015). As a downstream effector of mTORC2 and a regulator of the PI3K–AKT signaling (Oh et al., 2017; Wang G. et al., 2017), hypoxia-inducible factor (HIF)-1α induced the production of GLUT1/3 and glycolytic enzymes such as HK2 and PDH kinase 1 to drive glucose uptake and glycolysis, and reduced reactive oxygen species (ROS) to resist oxidative stress (Yang et al., 2014; Gabriely et al., 2017). Augmented levels of microRNAs that activate PI3K–AKT–mTOR and mTORC2–c-Myc axis and repress AMPK signaling have been observed in malignant glioma cells (Alfardus et al., 2017), reflecting the transcriptional activation of glycolysis and glucose transport.
As an important part of anabolism, PPP was activated by receptor tyrosine kinases–mTOR pathway by phosphorylated 6-phosphogluconate dehydrogenase (Liu R. et al., 2019), using G6P to produce substrates needed for glioma growth. The consumption of each G6P via 6-phosphogluconate dehydrogenase and 6-phosphate gluconate dehydrogenase produced 1 CO2, 3 H+ and 2 NADPH, maintaining tumor growth by adjusting pH and producing NAPDH for the synthesis of GSH (reduced glutathione) and fatty acids, and the intermediate product R5P was converted into fructose-6-phosphate and GA3P for glycolysis or purine nucleotide synthesis (Payen et al., 2016). Over-activated PPP increased oxygen consumption and made glioma cells more sensitive to hypoxia-induced death (Thiepold et al., 2017). Therefore, irreversible activating mTORC1 to forcibly drive PPP, while obstructing the glycolysis may control gliomas by inducing hypoxic damage.
Lipid Metabolism
As building materials and energy sources, lipids are essential for glioma cells. Exogenous lipids were mainly obtained from intracranial glial cells in the form of lipoproteins through intercellular exchange, but rarely from the periphery (An and Weiss, 2016). The low-density lipoprotein (LDL) receptor-mediated cholesterol uptake supported the survival of glioma cells, which was counteracted by ATP-binding cassette sub-family A member (ABCA)-1-dependent cholesterol efflux promoted by liver X receptor agonists (Villa et al., 2016). Even without cholesterol intake, glioma cells and GSCs can synthesize cholesterol de novo. Cholesterol was synthesized from acetyl-CoA through sterol regulatory element-binding protein (SREBP)-2 and mobilized from endoplasmic reticulum via sterol O-acyltransferase (SOAT) and stored in lipid droplets in the form of cholesterol ester (Geng et al., 2016). Lipid droplets then activated SREBP-1, which was overexpressed in malignant glioma cells and can initiate angiogenesis and the synthesis of lipids on cell membrane and organelle via fatty acid synthase (FASN) (Zhou et al., 2016). Increased levels of polyunsaturated fatty acid synthetase ELOVL2 (elongation of very long chain fatty acids protein) and cholesterol synthase 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) were also found in GSCs, involved in the elongation of fatty acids, synthesis of membrane lipids and facilitation of EGFR signaling to support cell growth (Wang X. et al., 2017; Gimple et al., 2019), and the impediment of fatty acid activator fatty acyl-CoA synthetase VL3 decreased the expression of stem-like phenotype CD133 and self-renewal functional molecules aldehyde dehydrogenase, musashi-1 and SOX2 on GSCs (Sun et al., 2014). The brain fatty acid binding protein (B-FABP, FABP7) participating in the utilization of unsaturated fatty acids also acted as a risk factor to drive the migration and infiltration of glioma cells and the growth of GSCs (Elsherbiny et al., 2013; Morihiro et al., 2013) dependent on the ratio of arachidonic acid (AA) to docosahexaenoic acid (DHA) (Elsherbiny et al., 2018). Prostaglandin E2 (PGE2) is another unsaturated fatty acid and known as an inducer of inflammation and pain, which was catalyzed from AA by cyclooxygenase (COX)-2 and prostaglandin E synthase (PGES) overexpressed in glioma cells, especially mesenchymal cells (Behnan et al., 2019). After binding to their receptors (EPs) in glioma tissues, PGE2 promoted tumor growth, invasion and immune escape (Jiang et al., 2017), and induced the angiogenesis with 20-hydroxyeicosatetraenoic acid (20-HETE), a transformation product of AA mediated by cytochrome P450 4A (CYP4A) (Feng et al., 2017; Wang C. et al., 2019). Inhibition of EPs or application of NSAIDs hindered the growth of gliomas (Seliger and Hau, 2018; Qiu et al., 2019). The synthesis and utilization of fatty acids and cholesterol participated in the malignant progression of gliomas, which were controllable under AMPK blockade (Guo et al., 2009; Kim et al., 2018).
Similar to carbohydrate, lipids were also catabolized by glioma cells to obtain energy. Ketone bodies are intermediate products of fatty acid oxidation, which were transported to brain tissues and converted to acetyl-CoA for vital activities. Considering that brain tumors cannot use ketone bodies and rely on glucose, the ketogenic diet was developed to limit the energy supply of gliomas rather than normal tissues (Poff et al., 2019). However, recent studies have proved that glioma cells can oxidize ketone bodies via up-regulated monocarboxylate transporter (MCT) during ketogenic diet (De Feyter et al., 2016), suggesting the ability of glioma cells to gain energy from lipids. In addition to fast-cycling glioma cells dependent on aerobic glycolysis, a subpopulation of slow-cycling cells with lipid transport and oxidation as the main metabolic mode has been confirmed, which can obtain energy in the absence of glucose (Hoang-Minh et al., 2018). Under oxygen-rich conditions, fatty acid oxidation was an essential energy pathway for glioma cells expressing high levels of fatty acid oxidases such as carnitine palmitoyltransferase 1 to grow independently of glycolysis (Lin et al., 2017; Wu et al., 2019). Nevertheless, the energy-producing efficiency of fatty acid oxidation is far less than glycolysis. Conversion of glycolysis to fatty acid oxidation by activating PPARα eventually led to the depletion of ATP in glioma cells (Wilk et al., 2015).
Distinguishing from conventional lipid metabolism, lipid peroxidation is an over-oxidation of ROS and lipids in the cell membrane and cytoplasm, producing cytotoxic peroxides including malonaldehyde (MDA) and 4-hydroxy-2-nonenal (HNE). Although there is a potential correlation between lipid peroxidation and the grade of gliomas clinically (Atukeren et al., 2017), lipid peroxidation in differentiated glioma cells induced ferroptosis, a type of programmed death. To resist peroxidative damage, glioma cells initiate degradation of peroxidatively modified proteins through proteasome system (Nakayama et al., 2016), and abate ferroptosis by GSH, phospholipid hydroperoxidase glutathione peroxidase 4 (GPX4) and glutamate (Glu)/cystine (Cys) antiporter system Xc–. Depleting GSH and Cys or inhibiting system Xc– and GPX4 impeded the survival of glioma cells and increased their sensitivity to radiation-induced lipid peroxidation (Wang et al., 2018; Ye et al., 2020).
Controlling lipid metabolism resisted the invasion of gliomas, such as the application of phytol, retinol, and quercetin acting on FASN and SREBP1/2 (Facchini et al., 2018; Damiano et al., 2019), and the inhibition of acetyl-CoA carboxylase 1 and HMGCR by oleic acid and hydroxytyrosol (Priore et al., 2017). PPARα activator fenofibrate also inhibited glioma growth by inducing the dependence of tumor cells on fatty acid oxidation instead of glycolysis (Wilk et al., 2015). On this basis, using fatty acid oxidation inhibitors such as etomoxir may limit the leftover energy-producing pathways of glioma cells (Lin et al., 2017; Petovari et al., 2018), which is a potential treatment strategy.
Amino Acid and One-Carbon (C1) Metabolism
As synthesis materials or decomposition products of proteins, amino acids supported and regulated the growth of tumor cells and tumor stem-like cells (Mayers et al., 2016; Jones et al., 2018). The heterogeneous amino acid metabolism of glioma cells is formed during environmental adaptation. In order to eliminate ROS accumulation caused by vigorous glucose metabolism, the level of xCT, the light chain subunit of system Xc– in glioma cells was up-regulated to promote the intake of Cys, providing raw material for the synthesis of GSH. This cytoprotective effect relied on glucose, whose deprivation rapidly depleted NADPH during ingestion of Cys, inducing cell death of GBM cells (Goji et al., 2017). Glu, another raw material of GSH, and its metabolite L-glutamine (Gln) are fuels for glioma growth, both of which can be autonomously synthesized by glioma cells or taken up from metabolites of astrocytes (Tardito et al., 2015). The bioenergy of conversion from Glu to Gln through Gln synthetase were provided by lactate produced during glycolysis of glioma cells and normal astrocytes. The glutaminase mediated the transformation from Gln to Glu, releasing amide nitrogen for the biosynthesis of purines and pyrimidines (Venneti and Thompson, 2017), which was accelerated by the generation of Gln via GSH (Tardito et al., 2015). The excessive secretion of Glu from glioma cells can trigger glioma-related seizures by binding to receptors on neurons around the tumor (Huberfeld and Vecht, 2016). Glu was also released from synaptic neurons as a neurotransmitter, initiating the cascade of AKT and MAPK signaling through the Glu receptor on the surface of glioma cells to promote invasion. Moreover, in a glucose-deficient condition, Glu were metabolized by Glu dehydrogenase (GLUD1)-1 into the intermediate product of TCA cycle, α-KG, which activated inhibitor of nuclear factor kappa-B kinase subunit β and nuclear factor κB (NF-κB) to promote glucose uptake by up-regulating GLUT1 (Wang X. et al., 2019). mTOR2 was activated by high levels of Gln (Liu B. et al., 2019) to regulate Glu/Gln metabolism, promoting Glu secretion, Cys uptake, GSH synthesis and Gln catabolism to obtain energy and transmit growth factor signaling for glioma cells by activating c-Myc (Gu et al., 2017).
As another critical part of amino acid metabolism, serine (Ser)/glycine (Gly) metabolism governs the synthesis of nucleotides, proteins and lipids, and is the hub of glycolysis and folate metabolism (Maddocks et al., 2017). Ser was synthesized from the glycolytic intermediate 3-phosphoglycerate via phosphoglycerate dehydrogenase (PHGDH), and was converted into Gly by mitochondrial serine hydroxymethyltransferase (SHMT2) (Kim D. et al., 2015; Venneti and Thompson, 2017). When Ser was sufficient, PKM2 was stimulated to promote glycolysis of glioma cells, while SHMT2 was activated to counteract augmented TCA cycle activity and save oxygen. Although Gly accumulation caused by SHMT2 was detrimental to cell growth, glioma cells expressed high levels of Gly decarboxylase (GLDC) to decompose Gly into innocuous metabolites, inhibition of which led to the loaded cytotoxic aminoacetone and methylglyoxal. Conversely, when Ser deficiency was sensed, glioma cells stopped cell cycle by activating cyclin-dependent kinase inhibitor p21 through p53 and promoted the synthesis of GSH to maintain survival (Venneti and Thompson, 2017).
Ser is the main source of C1 units during the conversion to Gly and the decomposition of Gly. Other amino acids including Glu, Gln, tryptophan (Trp), and methionine (Met) are also the source of C1 units. Glioma cells expressing high level of IDH3α up-regulated SHMT2 and facilitated the activation of cytosolic SHMT (SHMT1) to promote the release of C1 units (May et al., 2019). miR-940, which obstructs the folate cycle and C1 metabolism by inhibiting methylenetetrahydrofolate dehydrogenase, was also down-regulated in glioma cells (Xu et al., 2019). The abundant C1 units participate in the biosynthesis of nucleotide and produce CO2 and NADPH, enabling glioma cells to survive, proliferate and invade under hypoxic conditions (Wypych and Baranska, 2020). Furthermore, C1 units can be thoroughly utilized by GSCs with rich purine synthases (Wang Q. et al., 2017), promoting the onset and rapid recurrence of GBM.
For malignant invasion, glioma cells overexpressed amino acid metabolic enzymes to resist hypoxia and glucose deficiency. Based on the distinct metabolic characteristics of tumors and normal brain tissues, intervention of amino acid metabolism was a selective means to improve gliomas (Panosyan et al., 2017). Moreover, considering efficient Gln uptake in glioma tissues, PET technology has been developed for imaging, which overcame the limitations of conventional nuclide 18F-FDG in the context of normal brain tissues with similar strong capacity of glucose uptake (Venneti et al., 2015).
The metabolic reprogramming allowed glioma cells to proliferate regardless of the ischemic lesion (Table 1), and endowed them with strong migration capabilities for a better growth condition (Kathagen-Buhmann et al., 2018), enabling the rapid invasion into healthy brain tissues. Concurrently, to cope with changes in nutritional sources, the metabolic plasticity of glioma cells resulted in the resistance to anti-metabolic therapies, including diet and drugs (De Feyter et al., 2016; Shibao et al., 2018). It may be a forward therapeutic strategy to block the adaptively up-regulated metabolic enzymes and activating factors in glioma cells appropriately while limiting the intake of energy substrates.
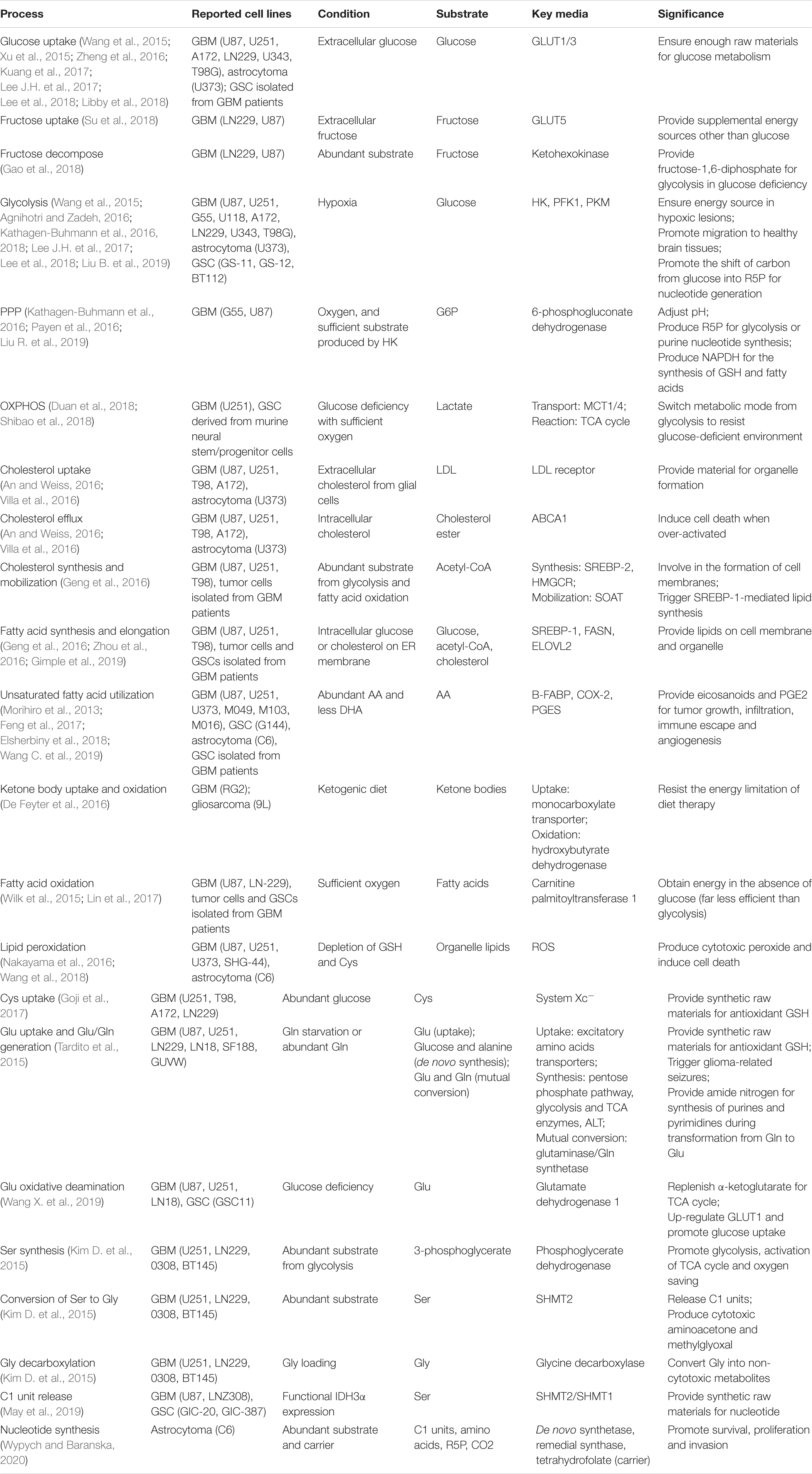
Table 1. Metabolism processes of glioma cells and GSCs.
Glioma Cell Metabolism Links to Immune Cells in Microenvironment
Metabolic plasticity not only promoted the energy supply and the synthesis of substrates required for growth and heredity of glioma cells, but also induced immune evasion (Ganapathy-Kanniappan, 2017). Immune cells accumulating and infiltrating in the glioma tissues include glioma-associated microglia/macrophages (GAMs), T lymphocytes, natural killer (NK) cells, neutrophils, dendritic cells (DCs) and myeloid-derived suppressor cells (MDSCs) (Magana-Maldonado et al., 2016; Gieryng et al., 2017; Figures 2A,B), supporting tumor growth instead of surveillance and annihilation and limiting the prognosis (Zhang et al., 2017; Boussiotis and Charest, 2018; Figure 2C). Metabolic remodeling increased the level of metabolites from glioma cells to induce immune tolerance in the TME (Kesarwani et al., 2017), and drove the production of immunosuppressive factors such as arginase (ARG)-1, IL-10, and TGF-β (Guo et al., 2018; Figure 3A). Moreover, the hypoxia caused by uncontrolled proliferation of metabolic reprogrammed glioma cells reduced the viability of tumor killer cells, further facilitating the survival of glioma cells (Colwell et al., 2017). The interactions between glioma cell metabolism and immune cells are a novel perspective for understanding the immune escape and refractoriness of gliomas.
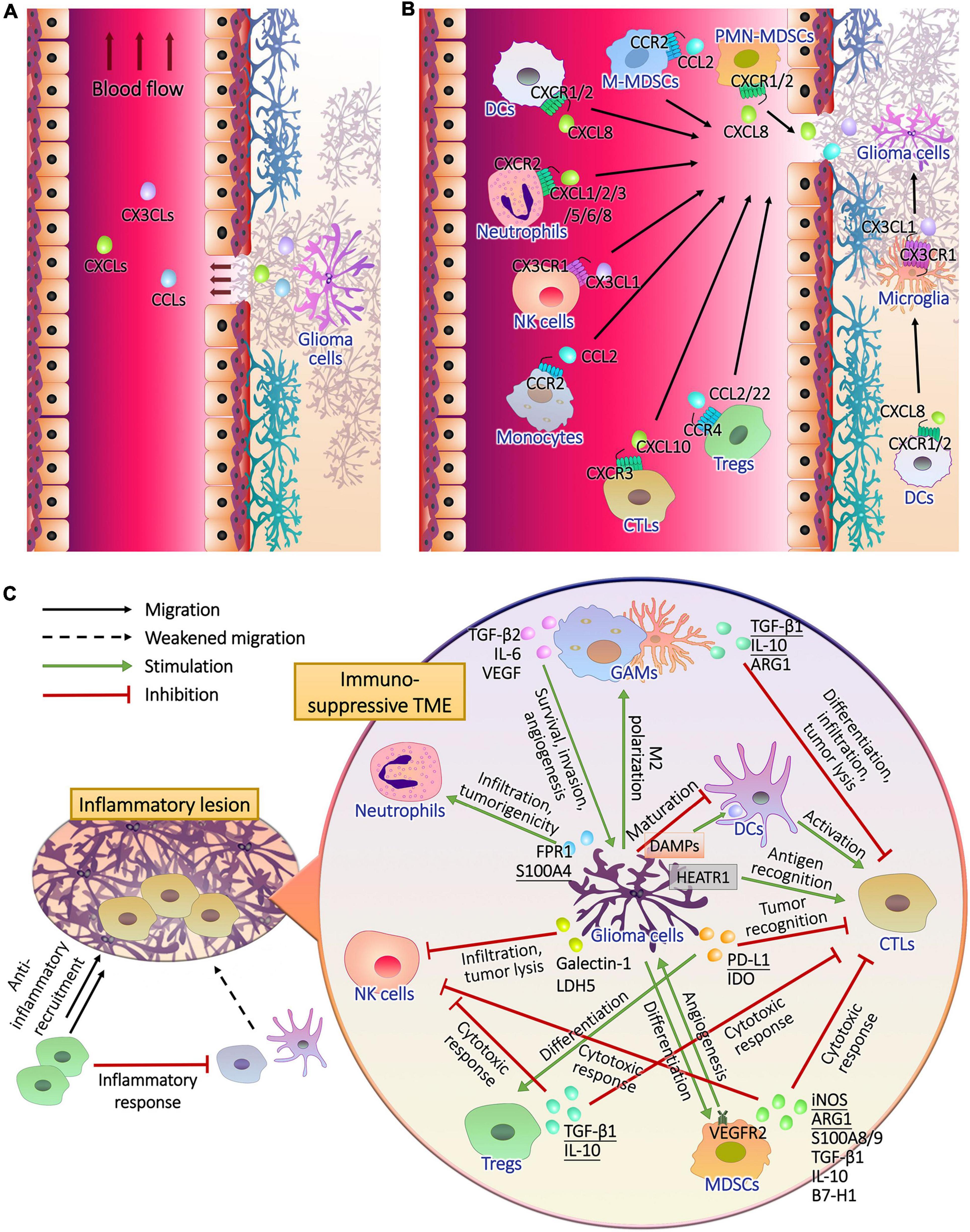
Figure 2. The recruitment of immune cells and the formation of immunosuppressive glioma microenvironment. (A) Glioma cells release chemokines to lesion tissues, part of which entered peripheral blood through the pathological blood brain barrier. (B) After the chemokine receptors of peripheral and intracranial immune cells captured their ligands, they drive the cells to migrate upstream where chemokines are released. (C) During the infiltration of inflammatory T cells, anti-inflammatory cells, mainly regulatory T cells (Tregs), are recruited to inhibit the antigen presentation of dendritic cells (DCs) and T cell activation, resulting in impaired immune response. In addition to the activation of anti-inflammatory mechanisms, glioma cells perform complex intercellular interactions with immune cells in tumor microenvironment. Cytotoxic T lymphocytes (CTLs) should recognize tumor antigen HEAT repeat-containing protein 1 (HEATR1) and kill glioma cells with natural killer (NK) cells, and CTLs were activated by mature DCs during the presentation of damage associated molecular patterns (DAMPs) released by glioma cells. Glioma cells inhibited the maturation of DCs. Furthermore, glioma cells secreted cytokines to induce the generation and recruitment of tumorigenic Tregs, myeloid-derived suppressor cells (MDSCs), neutrophils and M2 polarized microglia and macrophages, and inhibit the infiltration and tumor lysis of CTLs and NK cells.
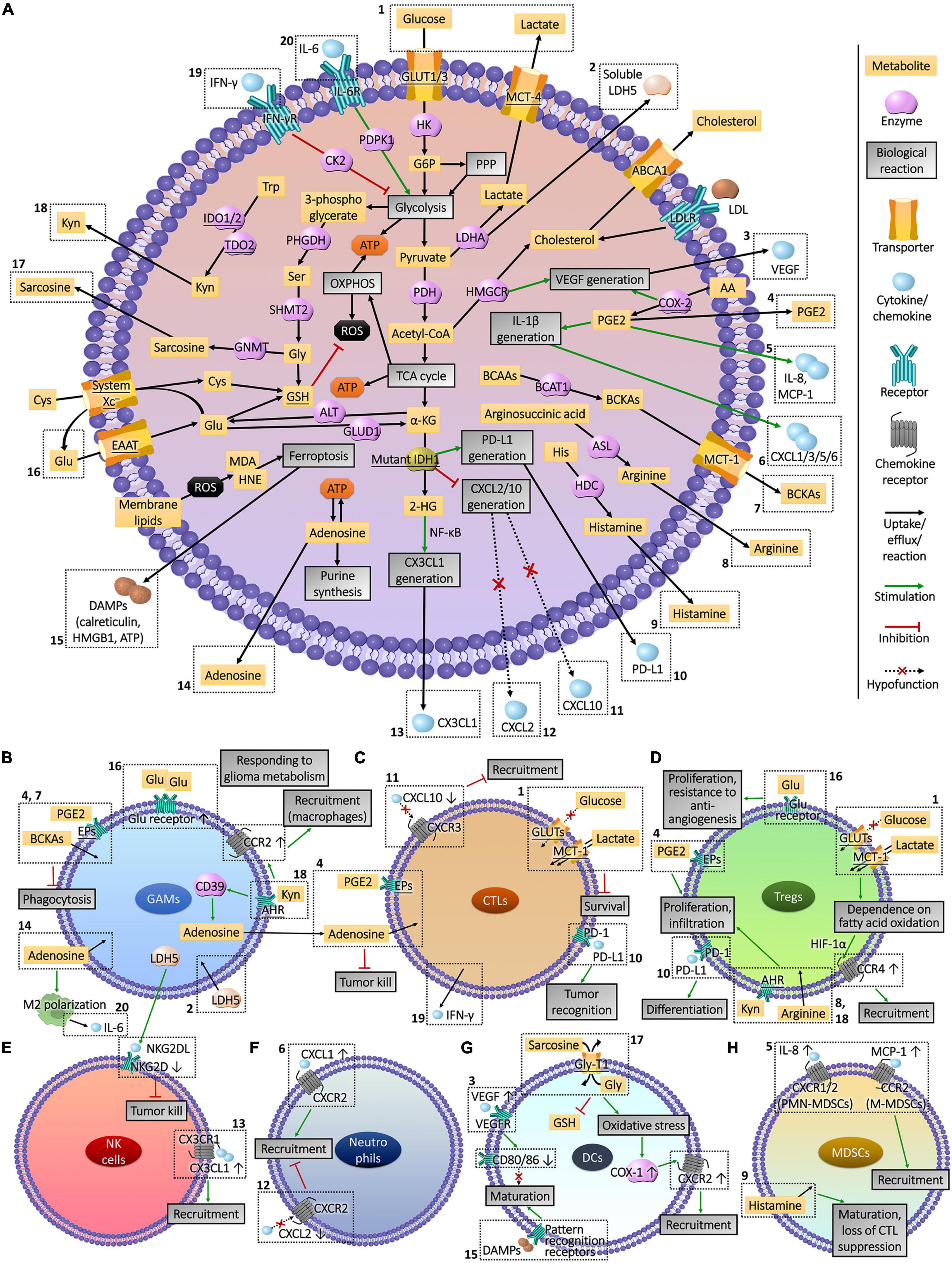
Figure 3. Interactions between glioma cell metabolism and immunomicroenvironment. (A) Metabolic reprogramming enables glioma cells to express high levels of substrate transporters and metabolic enzymes to obtain sufficient energy in the harsh conditions of lesion, resulting in the deprivation of nutrient substrates and accumulation of immune-interfering metabolites in the extracellular fluid. Abnormally expressed or mutated metabolic enzymes also affected immune cells by regulating the production of chemokines and other cytokines. The actions of these molecules that enter the tumor microenvironment on immune cells and the influences of immune cells on glioma cells metabolism are presented in part (B–H). The Arabic numerals in the figure link the metabolism of glioma cells (A) with immune cells (B–H).
GAMs
GAMs account for 30–50% of glioma-infiltrating immune cells, which is the highest proportion in tumor tissues (Hambardzumyan et al., 2016). Microglia were thought to be macrophages settled in the central nervous system (Sankowski et al., 2019), but their origins and phenotypes are different. Microglia originated from the neuroepithelial yolk sac progenitor cells (Gomez Perdiguero et al., 2015) with high level of CX3CR-1, but low level of CD45 and no CCR-2 expression (Hutter et al., 2019). In contrast, macrophages in glioma TME with CX3CR1 expression were differentiated from CX3CR1lo peripheral monocytes entered the cranial cavity (Quail and Joyce, 2017), which highly express CD45 and CCR2 (Hambardzumyan et al., 2016; Chen Z. et al., 2017). Monocyte chemotactic protein 1 (MCP-1, CCL2) secreted by glioma cells mediated the recruitment of CCR2+ monocytes and macrophages (Chen Z. et al., 2017; Vakilian et al., 2017), and CX3CL1 induced the infiltration of CX3CR+ microglia (Hambardzumyan et al., 2016). Tumor recognition and phagocytosis functions of these innate immune cells were declined in glioma milieu (Poon et al., 2017), and cytokines such as TGF-β1 and IL-10 they secreted contributed to the formation of immunosuppressive TME (Roesch et al., 2018).
There is an interaction between GAMs and metabolites from glioma cells. Compared to homologous cells not exposed to the gliomas, up-regulated Glu receptors and GS and decreased xCT were detected in GAMs in response to Glu secreted by tumor cells (Choi et al., 2015). Meanwhile indoleamine 2,3-dioxygenase (IDO)-1/2 and tryptophan 2,3-dioxygenase (TDO)-2 were highly expressed in glioma cells and were proportional to tumor grade (Guastella et al., 2018), which catalyzed the decomposition of Trp into kynurenine (Kyn), a ligand of the aryl hydrocarbon receptor (AHR). The released Kyn induced the expression of CCR2 by activating AHR, and advanced the recruitment of macrophages to tumor sites by enhancing the response to MCP-1 secreted by glioma cells (Takenaka et al., 2019). In addition to the impact on recruitment, metabolism of glioma cells was involved in the regression of innate immune abilities. Branched-chain ketoacids (BCKAs) metabolized from branched-chain amino acids (BCAAs) and unsaturated fatty acid PGE2 released by glioma cells were taken up by GAMs, accompanied by decreased phagocytosis (Ghosh et al., 2010; Silva et al., 2017). On the other hand, Kyn secreted by glioma cells activated AHR in GAMs to inhibit the cytotoxicity of T lymphocytes by up-regulating the production of ectonucleotidase CD39 and adenosine (Takenaka et al., 2019). The polarization of GAMs to immunosuppressive M2 type is a representative tumorigenic process positively bound up with the grading and rapid recurrence of gliomas (Wang Q. et al., 2017; Sorensen et al., 2018), which was widely found in mesenchymal GBM cells and was related to NF1 loss (Wang Q. et al., 2017). M2 polarization was also induced by metabolites of glioma cells such as adenosine (Komohara et al., 2008; Kesarwani et al., 2019), along with the secretion of CSF-1 (Pyonteck et al., 2013), the expression of CCR5 (Laudati et al., 2017), and DNA damage repair (Meng et al., 2019) of glioma cells. However, GAMs polarized into immune-promoting M1 type were not facilely affected by metabolites (Guan et al., 2017). In addition to immunosuppressive functions, GAMs promoted the migration, angiogenesis, and invasion of gliomas via TGF-β2, IL-6, and VEGF (Roesch et al., 2018), which were affected by glioma cell metabolism as well. The secretion of VEGF and TGF-β from GAMs were reduced by blocking CYP4A to inhibit the synthesis of unsaturated fatty acid 20-HETE (Wang C. et al., 2017). The metabolism of glioma cells was also regulated by GAMs. IL-6 secreted by M2 macrophages promoted glycolysis of GBM cells by phosphorylating PGK1 via 3-phosphoinositide-dependent protein kinase 1 (PDPK1) (Zhang et al., 2018), and quinolinic acid secreted by microglia was taken up by glioma cells for the synthesis of NAD+ to resist oxidative stress (Sahm et al., 2013).
Glioma-associated microglia/macrophages are body guards of gliomas from the host. The metabolism of glioma cells is closely related to the recruitment, infiltration, and polarization of GAMs (Figure 3B). Utilizing the communications between glioma cells and GAMs can create the possibility to kill gliomas at close range, such as proper interventions in metabolism.
T Lymphocytes
CD8+ cytotoxic T lymphocytes (CTLs) and CD4+ CD25+ FOXP3+ regulatory T cells (Tregs) are active lineages of glioma-infiltrating T lymphocytes. CTLs are the main contributors that infiltrate and kill tumor cells. In addition to the secretion of tumor-damaging cytokines such as IFN-γ, CTLs can recognize antigens such as HEAT repeat-containing protein 1 (HEATR1) expressed by glioma cells via human leukocyte antigen (HLA)-A2 [or major histocompatibility complex class 1 in animals] and trigger cell lysis (Wu et al., 2014; Shao et al., 2017). Due to less chance of the contact between ER and mitochondria, GSCs expressed low levels of sialylated glycans on cell surface and were more sensitive to CTLs than differentiated glioma cells (Bassoy et al., 2017). As part of TME, the cytotoxicity of CTLs cannot function properly. In response to the IFN-γ secreted by CTLs, glioma cells released PD-L1 through exosomes to interact with T cells for immune escape (Qian et al., 2018). IDH1/2 mutations reduced the release of CXCL10 that attract the accumulation of CTLs (Kohanbash et al., 2017), and increased the production of PD-L1 (Berghoff et al., 2017), hinting the involvement of glioma cell metabolism in the dysfunction of CTLs.
Glioma cells with heterogeneous carbohydrate and amino acid uptake capacity occupied the supply of glucose and amino acids in hypoxic niduses, leading to the exhaustion of CTLs (Mirzaei et al., 2017; Rashidi et al., 2020), and the accumulation of lactate during glycolysis destroyed the intracellular and extracellular concentration gradients, which hindered lactate efflux and reduced the viability of CTLs (Shao et al., 2017). As an immune checkpoint (Kesarwani et al., 2018), the Trp metabolism of glioma cells also limited the infiltration of CTLs through IDO1 (Zhai et al., 2018). In contrast, the histidine (His) metabolism promoted the activation of CTLs, and the decline of His decarboxylase (HDC) activated the inhibition of CTL infiltration by MDSCs (Ahn et al., 2015). The elevated levels of other metabolic enzymes including argininosuccinate lyase (ASL), ARG2, and COX-2 in glioma cells led to augmented synthesis of Trp, arginine and PGE2, which induced the repression of CTLs as well (Eberstal et al., 2014; Authier et al., 2015; Kesarwani et al., 2019). CTLs also affect the metabolism of glioma cells through IFN-γ, which activated casein kinase (CK)-2 of tumor cells to hold up glycolysis, impeding the growth of gliomas (Ghildiyal and Sen, 2017).
Treg is a T cell subtype responsible for anti-inflammatory and immune tolerance, secreting high levels of inhibitory cytokines including TGF-β and IL-10 (Qiu et al., 2020), which allowed glioma cells to escape the cytotoxic damage of CTLs in TME (See et al., 2015). In malignant gliomas, Tregs were associated with the tumor recurrence, resistance to targeted drugs and decrease of survival period (Sayour et al., 2015; Du Four et al., 2016), whose depletion improved the condition of glioma mice by inducing spontaneous rejection of the tumor. Glioma cell metabolism also affects Tregs. Augmented production of Trp and arginine, and elevated expression of metabolic enzymes IDO1, ASL and ARG2 in glioma cells were accompanied by increased infiltration of Tregs (Kesarwani et al., 2019). Mutations in the metabolic enzyme IDH1/2 promoted the secretion of PD-L1 (Berghoff et al., 2017), which induce the differentiation of Tregs (DiDomenico et al., 2018). Differentiated Tregs recruited into tumor region were mediated by chemokines MCP-1 and CCL22 secreted by glioma cells (Jacobs et al., 2010; Chang et al., 2016). Due to the efficient glucose uptake and glycolysis, glucose was deprived in the hypoxic lesions by glioma cells, resulting in Tregs relying on fatty acids for mitochondrial metabolism and migrating to glioma tissues in response to CCL22 in a HIF-1α-dependent manner (Miska et al., 2019). Accumulated lactate further drove the infiltration of Tregs, and the depletion of lactate from glioma cells decreased tumor-infiltrating Tregs (Chirasani et al., 2013). PGE2 synthesis in glioma cells with up-regulated COX-2 were also related to the dilation and infiltration of Tregs (Rolle et al., 2012; Authier et al., 2015). In addition, glioma cells can activate Tregs by enhancing extracellular transport of Glu to survive anti-angiogenic therapy (Long et al., 2020).
CTLs and Tregs are T cell lineages with opposite effects in glioma TME, which were both influenced by the metabolic state of cancer cells (Figures 3C,D). The intervention of glioma cell metabolism can promote the infiltration of CTLs and restore their anti-tumor immunity, meanwhile hinder the recruitment of Treg, relieving the immune tolerance in TME.
NK Cells
Ly6c+ NK cells in the glioma TME are a group of innate tumor killer cells derived from the bone marrow, which are part of glioma-infiltrating lymphocytes. Due to different metabolic properties, the grade of gliomas was inversely proportional to the infiltration and anti-tumor functions of NK cells. Attributing to the recruitment inhibitor galectin-1 secreted by HGG cells (Baker et al., 2016), low levels of infiltrating NK cells were observed (Domingues et al., 2016; Zhu et al., 2019), which possessed faint cytotoxicity and expressed high level of Tim-3 to prevent the tumor-killing helper T cells type 1, shortening the survival time of patients (Pereira et al., 2018). Conversely, mutations of metabolic enzyme IDH1 in LGG cells produced 2-HG and activated NF-κB to promote the secretion of CX3CL1, which attracted CX3CR1-expressing NK cells to infiltrate, making patients with a good prognosis (Ren et al., 2019). The decreased immune functions of NK cells were also related to the metabolism of glioma cells. Soluble lactate metabolic enzyme LDH5 secreted by glioma cells induced tumor infiltrating myeloid cells and circulating monocytes to release the ligand of NK group 2 member D (NKG2D), a surface receptor on NK cells in a long term, leading to the down-regulation of NKG2D and malfunction of tumor lysis (Crane et al., 2014; Figure 3E). According to the grade of gliomas, a suitable metabolic intervention scheme should be adjusted to effectively exert the tumor infiltration and cytotoxicity functions of NK cells.
Neutrophils
High proportion of neutrophils expressing CD11b and Ly6G in glioma tissues increased the tumor malignancy (Spiegel et al., 2016), becoming a powerful indicator of poor prognosis (Massara et al., 2017; Zhang et al., 2017). The S100A4 expressed in GSCs, which is a novel biomarker promoting the transcription of genes involving glycolysis and gluconeogenesis (Chow et al., 2017), induced tumorigenicity of neutrophils involving the promotion of tumor growth, metastasis, and resistance to anti-angiogenic drugs (Liang et al., 2014). Besides, metabolism of glioma cells interfered the recruitment and infiltration of neutrophils (Figure 3F). Neutrophils were recruited to the periphery of the glioma inflammatory region via chemokines including CXCL1/2/3/5/6 and IL-8 (CXCL8), which were secreted from glioma cells due to the expression of IL-1β (Lee S.Y. et al., 2017; Mostofa et al., 2017). Subsequently, neutrophils infiltrated the core area of tumor tissues through formylpeptide receptor 1 (FPR1) secreted by GBM cells to promote tumor growth, invasion and angiogenesis (Liu et al., 2012). The expression of COX-2 in glioma cells promoted the anabolic metabolism of PGE2, up-regulating the expression of IL-1β and CXCL1 (Jiang and Dingledine, 2013) to recruit neutrophils (Mostofa et al., 2017). The IDH1 mutation in glioma cells weakened the CXCL2-mediated recruitment of neutrophils (Amankulor et al., 2017), which may be one of the reasons for the low malignancy of LGGs. Despite the pro-glioma effects, utilizing the performance of directional migration and infiltration to the tumor area through BBB, the localized drug delivery and imaging tracer based on neutrophil carrier raised the treatment and staging diagnosis of glioma to a new height (Osuka and Van Meir, 2017; Xue et al., 2017; Wu et al., 2018), and can inspect the efficacy of metabolic interventions.
Dendritic Cells
As resident antigen presenting cells (APCs) in central nervous system, DCs possess the ability to activate glioma-killing CTLs (Malo et al., 2018a). The decrease of tumor-infiltrating DCs led to reduced survival of GBM mice (Mathios et al., 2016). DC vaccine-based immunotherapy has been widely investigated in the treatment of gliomas (Mitchell et al., 2015; Weller et al., 2017). However, the anti-tumor functions of DCs were covered by the glioma-induced immunosuppression, which limited the effectiveness of DC therapy (Garg et al., 2016; Li et al., 2018). Glioma cells recruited DCs through the CXC chemokine family (Dastmalchi et al., 2019). Under the exposure to glioma cells expressing VEGF, the maturation of DCs were suppressed and antigen presentation and T-cell activation capabilities were diminished (Sheng et al., 2020). What’s more, DCs in the glioma TME exhibited tumor tolerable properties, expressing more IDO to induce Tregs to infiltrate glioma tissues for immune escape (Wainwright et al., 2014; He et al., 2015). The decrease of immunosuppressive glioma-infiltrated DCs via immune checkpoint inhibitors improved T cell responses and survival of GBM mice (Hung et al., 2018).
The recruitment of DCs was affected by amino acid metabolism of glioma cells (Figure 3G). Glycine-N-methyl transferase mediates the conversion of Gly to sarcosine, which released from glioma cells and competed with DCs for Gly uptake through glycine transporter type-1 (Gly-T1) (Dastmalchi et al., 2019). Gly depletion led to decreased GSH and oxidative stress of DCs, leading to the up-regulation of COX-1 to promote CXCR2 expression and responding to IL-8 from glioma region. The immune tolerance and malignant transformation of DCs were also related to glioma cell metabolism. By inhibiting glycolysis or LDHA, the proliferation, migration and infiltration into glioma tissues of malignantly transformed DCs were repressed (Sheng et al., 2020), and these DCs secreted high levels of IL-12 to induce anti-tumor behavior of T cells (Chirasani et al., 2013). In addition, DCs stayed in immature state and co-stimulatory molecules CD80 and CD86 were down-regulated owing to elevated secretion of VEGF from glioma cells (Malo et al., 2018b) expressing elevated metabolic enzymes (He et al., 2015), involving HK2, PHGDH (Wolf et al., 2011; Liu et al., 2013), HMGCR (Slawinska-Brych et al., 2014), COX-2 (Feng et al., 2017), nitric oxide (NO) metabolic regulation enzyme dimethylarginine dimethylaminohydrolase (Boult et al., 2011) and mutated IDH1 (Wang et al., 2014).
The loss of immunogenicity of tumor cells is a cause of the failure of APCs. Inducing the release of damage associated molecular patterns (DAMPs) from glioma cells restored their immunogenicity, and triggered the activation of CTLs by DCs (Dastmalchi et al., 2019). Lipid peroxidation and ferroptosis induced by photodynamic therapy induced dying glioma cells to release DAMPs such as calreticulin, high mobility group protein B1 (HMGB1) and ATP that can be swallowed by DCs, stimulating DC maturation and activation (Turubanova et al., 2019). It can be inferred that the restoration of antigen recognition and presentation abilities of glioma-infiltrating DCs via the domination of glioma cell metabolism is critical to their anti-glioma functions, which could authentically exert the efficacy of DC vaccine on gliomas.
Myeloid-Derived Suppressor Cells
Myeloid-derived suppressor cells are a lineage of immature bone marrow-derived cells (BMDCs) activated under pathological conditions, including CD11b+ Ly6Chi Ly6G– immature monocytes (M-MDSCs) and CD11b+ Ly6Clo Ly6G+ immature polymorphonuclear cells (PMN-MDSCs, also known as G-MDSCs, immature granulocytes) (Marvel and Gabrilovich, 2015). Regardless of their origins, the monocyte marker HLA-DR is hardly expressed in M-MDSCs, while PMN-MDSCs express lectin-type oxidized LDL receptor 1, which is hardly detected in neutrophils (Gabrilovich, 2017). MDSCs account for more than 40% of glioma-infiltrating immune cells (Kamran et al., 2017) and expressed IL-4Rα, inducible nitric oxide synthase (iNOS) and ARG1 to inhibit the responses of glioma-killer T lymphocytes and NK cells (Kohanbash et al., 2013; Gielen et al., 2016), causing patients poor prognosis (Alban et al., 2018). Among MDSCs, PMN-MDSCs induced CD4+ glioma-infiltrating T lymphocytes to express PD-1, exhibiting more prominent T cell suppression (Dubinski et al., 2016). Furthermore, the expression of VEGFR2 in MDSCs promoted the malignant progression of gliomas by inducing angiogenesis (Huang et al., 2017).
The generation, infiltration, and acquisition of immunosuppressive capacity of MDSCs were regulated by the metabolism of unsaturated fatty acids and amino acids of glioma cells (Figure 3H). When exposed to the exosomes released by glioma cells, the expansion of BMDCs with MDSCs phenotype was induced in healthy BMDCs (Gielen et al., 2016; Guo et al., 2019), secreting IL-10, TGF-β, Fas-ligand and B7-H1 to inhibit the activation of T lymphocytes (Chae et al., 2015). The development of ARG1 phenotype in MDSCs was also induced by GSCs in a CXCR2-dependent manner, via the secretion of macrophage migration inhibitory factor (Otvos et al., 2016). Then, M-MDSCs and PMN-MDSCs migrated to TME in response to MCP-1 and IL-8 released from glioma cells separately, which can recruit monocytes and neutrophils expressing same chemokine receptors, CCR2 or CXCR1/2 (Chang et al., 2016; Ding et al., 2019). Anabolism of PGE2 in glioma cells was involved in the generation and recruitment of M-MDSCs and PMN-MDSCs. Depending on the level of COX-2 (Fujita et al., 2011; Kosaka et al., 2014), PGE2 promoted the secretion of IL-8 from glioma cells through autocrine (Venza et al., 2011, 2012), advanced the migratory response of M-MDSCs to MCP-1, and promoted the expression of ARG1 in PMN-MDSCs. However, histamine secreted from glioma cells mediated the maturation of MDSCs and the loss of immunosuppressive functions. The exhaustion of histamine via HDC knockout resulted in augmented infiltration of MDSCs into glioma tissues with suppressed CTLs (Ahn et al., 2015). Studying the relationship between the generation and recruitment of MDSCs and glioma cell metabolism is expected to improve the immune microenvironment of the lesions.
Metabolic Immunotherapy Strategies for Glioma
Metabolic heterogeneity of tumor cells and their initiating cells promoted the rapid invasion and recurrence of gliomas (Kathagen-Buhmann et al., 2016; Shibao et al., 2018). In the hypoxic area of intracranial lesions, these malignant cells made thorough use of glucose, lipoproteins and amino acids to ingest energy, synthesize hereditary substance, and produce antioxidants to resist oxidative stress. Metabolic reprogramming also brought enhanced migratory ability for glioma cells to invade healthy brain tissues for more energy supply. In order to ensure the smoothness of these biological processes, glioma cells took advantage of their metabolic properties for immune tolerance and escape mainly through the following ways.
(1) Their powerful nutrient uptake depleted the energy supply of tumor killer cells, and enhanced the synthesis of immune checkpoints including PD-L1 and IDO, impairing the anti-tumor responses.
(2) The metabolites secreted into TME promoted the infiltration and expansion of inflammatory neutrophils and suppressive Tregs and MDSCs to activate
anti-inflammatory mechanisms and immunotolerant responses.
(3) The metabolites led to malignant transformation of tumor-infiltrating APCs and phagocytes and exerted immunosuppressive functions, indirectly resisting tumor killer cells.
(4) After metabolic remodeling, the ability to recruit immune-tolerant cells was enhanced, but anti-tumor cells were less recruited. On the contrary, peroxidation metabolism enhanced immunogenicity and attract the presentation of tumor antigens (Turubanova et al., 2019).
Energy production and antioxidant responses during metabolism of glioma cells can also be induced by GAMs (Sahm et al., 2013; Zhang et al., 2018), but inhibited by CTLs in TME (Ghildiyal and Sen, 2017), reflecting the bidirectional conversation between glioma cell metabolism and immune microenvironment.
Several strategies for improving glioma TME through metabolic therapy are recommended (Table 2). Glycolysis inhibitors, or drugs targeting GLUT3 and MCT4 can restore the supply of extracellular glucose and reduce the lactate stress, improving the growth of CTLs, and hindering the tendency of Tregs to fatty acid oxidation to inhibit the infiltration of Tregs. Inhibiting inflammation and the release of PGE2 by anti-COX drugs such as NSAIDs can hinder inflammatory infiltration and activate anti-inflammatory mechanisms, restore the phagocytosis of GAMs and tumor lysis of CTLs, and repress the recruitment of Tregs, neutrophils and MDSCs. Inhibiting key enzymes in Trp–Kyn pathway and adenosine synthesis to prevent the loaded Kyn from activating AHR of GAMs and Tregs, and reduce the concentration of adenosine to enhance the cytotoxicity of CTLs is also a good choice. Compounds designed to act on amino acid transporters, including MCT1 and system Xc– can inhibit the release of BCKAs and the uptake of GSH synthetic materials, so as to restore GAMs phagocytosis, and induce the release of glioma-derived DAMPs recognized by DCs during ferroptosis. In addition, local injection of sarcosine and histamine can promote the migration of DCs to glioma region and promote the maturation of MDSCs. For glioma cells with IDH mutations, targeting mutant IDH can restore the release of CXCL10 and inhibit the production of PD-L1, promoting recruitment and tumor recognition of CTLs and inhibiting Tregs. It can be combined with imaging methods such as PET to analyze the metabolic characteristics of the lesions (Venneti et al., 2015) and formulate an individualized metabolic treatment plan.

Table 2. The impact of metabolic remodeling of glioma cells on immune cells.
Discussion
The microenvironment where glioma cells regulate immune cells through metabolic reprogramming is in the brain. Therefore, a challenge for glioma metabolic immunotherapy is the BBTB. In order to selectively act drugs on the lesions and reduce the impact on peripheral tissues, a brain drug delivery system needs to be established. Osmotic BBB disruption based on intra-arterial infusion of hypertonic mannitol solution, intravenous bradykinin analogs that relax tight junctions, drugs coupling to a mediator targeting and shuttling insulin or transferrin receptors, nanoparticle drug delivery system, and direct delivery of drugs to the brain parenchyma or excision cavity are potential methods for metabolic immunotherapeutic drugs to cross BBTB (van Tellingen et al., 2015; Li et al., 2020). Targeting metabolic functional proteins selectively and highly expressed in glioma cells, such as GLUT3 (Xu et al., 2015; Zheng et al., 2016), or using targeted metabolic drug delivery systems based on neutrophils (Osuka and Van Meir, 2017; Xue et al., 2017) or other immune cells accumulating in glioma tissues is also a good solution.
To conclude, glioma cells show different metabolic pattern and immune microenvironment from peripheral tumors (Table 3). Interfering with cell metabolism could not only hinder the growth of glioma cells, but also improve the immune response in the focal area to systematically resist tumor progression, which is expected to become a new direction for clinical treatment of gliomas.

Table 3. Differentials in cell metabolism and immune TME of gliomas compared with peripheral tumors.
Author Contributions
RQ and YZ contributed equally to this work and worked for the first draft of text writing and illustrations. QL participated in the literature search and revised the introduction part of the manuscript. YL and HF provided professional guidance on disease details and treatment status, and edited the manuscript. All of the authors approved the publication of this manuscript.
Funding
This work was supported by the Science and Technology Development Fund of Nanjing Medical University (Grant No. NMUB2019153) and Nanjing Health Youth Talent Fund (Grant No. QRX11032).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
2-HG, 2-hydroxyglutarate; AA, arachidonic acid; ABCA, ATP-binding cassette sub-family A member; BCAA, branched-chain amino acid; BCKA, branched-chain ketoacid; B-FABP, brain fatty acid binding protein; EAAT, excitatory amino acids transporters; ELOVL2, elongation of very long chain fatty acids protein; FASN, fatty acid synthase; FPR, formylpeptide receptor 1; HK, hexokinase; HMGB, high mobility group protein B; MCT, monocarboxylate transporter; PFK, 6-phosphofructokinase; PGES, prostaglandin E synthase; PKM, pyruvate kinase M; R5P, ribose-5-phosphate; SHMT, serine hydroxymethyltransferase; SOAT, sterol O-acyltransferase; SREBP, sterol regulatory element-binding protein; α-KG, α-ketoglutarate.
References
Agnihotri, S., and Zadeh, G. (2016). Metabolic reprogramming in glioblastoma: the influence of cancer metabolism on epigenetics and unanswered questions. Neuro Oncol. 18, 160–172. doi: 10.1093/neuonc/nov125
Ahn, B., Kohanbash, G., Ohkuri, T., Kosaka, A., Chen, X., Ikeura, M., et al. (2015). Histamine deficiency promotes accumulation of immunosuppressive immature myeloid cells and growth of murine gliomas. Oncoimmunology 4:e1047581. doi: 10.1080/2162402X.2015.1047581
Alban, T. J., Alvarado, A. G., Sorensen, M. D., Bayik, D., Volovetz, J., Serbinowski, E., et al. (2018). Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight 3:e122264. doi: 10.1172/jci.insight.122264
Aldana, B. I. (2019). Microglia-specific metabolic changes in neurodegeneration. J. Mol. Biol. 431, 1830–1842. doi: 10.1016/j.jmb.2019.03.006
Alfardus, H., McIntyre, A., and Smith, S. (2017). MicroRNA regulation of glycolytic metabolism in glioblastoma. Biomed. Res. Int. 2017:9157370. doi: 10.1155/2017/9157370
Amankulor, N. M., Kim, Y., Arora, S., Kargl, J., Szulzewsky, F., Hanke, M., et al. (2017). Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 31, 774–786. doi: 10.1101/gad.294991.116
An, Z., and Weiss, W. A. (2016). Cholesterol: an achilles’. Heel for Glioblastoma? Cancer Cell 30, 653–654. doi: 10.1016/j.ccell.2016.10.011
Atukeren, P., Oner, S., Baran, O., Kemerdere, R., Eren, B., Cakatay, U., et al. (2017). Oxidant and anti-oxidant status in common brain tumors: correlation to TP53 and human biliverdin reductase. Clin. Neurol. Neurosurg. 158, 72–76. doi: 10.1016/j.clineuro.2017.05.003
Authier, A., Farrand, K. J., Broadley, K. W., Ancelet, L. R., Hunn, M. K., Stone, S., et al. (2015). Enhanced immunosuppression by therapy-exposed glioblastoma multiforme tumor cells. Int. J. Cancer 136, 2566–2578. doi: 10.1002/ijc.29309
Baker, G. J., Chockley, P., Zamler, D., Castro, M. G., and Lowenstein, P. R. (2016). Natural killer cells require monocytic Gr-1(+)/CD11b(+) myeloid cells to eradicate orthotopically engrafted glioma cells. Oncoimmunology 5:e1163461. doi: 10.1080/2162402X.2016.1163461
Bassoy, E. Y., Kasahara, A., Chiusolo, V., Jacquemin, G., Boydell, E., Zamorano, S., et al. (2017). ER-mitochondria contacts control surface glycan expression and sensitivity to killer lymphocytes in glioma stem-like cells. EMBO J. 36, 1493–1512. doi: 10.15252/embj.201695429
Behnan, J., Finocchiaro, G., and Hanna, G. (2019). The landscape of the mesenchymal signature in brain tumours. Brain 142, 847–866. doi: 10.1093/brain/awz044
Berghoff, A. S., Kiesel, B., Widhalm, G., Wilhelm, D., Rajky, O., Kurscheid, S., et al. (2017). Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro Oncol. 19, 1460–1468. doi: 10.1093/neuonc/nox054
Boult, J. K., Walker-Samuel, S., Jamin, Y., Leiper, J. M., Whitley, G. S., and Robinson, S. P. (2011). Active site mutant dimethylarginine dimethylaminohydrolase 1 expression confers an intermediate tumour phenotype in C6 gliomas. J. Pathol. 225, 344–352. doi: 10.1002/path.2904
Boussiotis, V. A., and Charest, A. (2018). Immunotherapies for malignant glioma. Oncogene 37, 1121–1141. doi: 10.1038/s41388-017-0024-z
Broekman, M. L., Maas, S. L. N., Abels, E. R., Mempel, T. R., Krichevsky, A. M., and Breakefield, X. O. (2018). Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 14, 482–495. doi: 10.1038/s41582-018-0025-8
Chae, M., Peterson, T. E., Balgeman, A., Chen, S., Zhang, L., Renner, D. N., et al. (2015). Increasing glioma-associated monocytes leads to increased intratumoral and systemic myeloid-derived suppressor cells in a murine model. Neuro Oncol. 17, 978–991. doi: 10.1093/neuonc/nou343
Chang, A. L., Miska, J., Wainwright, D. A., Dey, M., Rivetta, C. V., Yu, D., et al. (2016). CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 76, 5671–5682. doi: 10.1158/0008-5472.CAN-16-0144
Chen, R., Smith-Cohn, M., Cohen, A. L., and Colman, H. (2017). Glioma subclassifications and their clinical significance. Neurotherapeutics 14, 284–297. doi: 10.1007/s13311-017-0519-x
Chen, Z., Feng, X., Herting, C. J., Garcia, V. A., Nie, K., Pong, W. W., et al. (2017). Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res. 77, 2266–2278. doi: 10.1158/0008-5472.CAN-16-2310
Chirasani, S. R., Leukel, P., Gottfried, E., Hochrein, J., Stadler, K., Neumann, B., et al. (2013). Diclofenac inhibits lactate formation and efficiently counteracts local immune suppression in a murine glioma model. Int. J. Cancer 132, 843–853. doi: 10.1002/ijc.27712
Choi, J., Stradmann-Bellinghausen, B., Yakubov, E., Savaskan, N. E., and Regnier-Vigouroux, A. (2015). Glioblastoma cells induce differential glutamatergic gene expressions in human tumor-associated microglia/macrophages and monocyte-derived macrophages. Cancer Biol. Ther. 16, 1205–1213. doi: 10.1080/15384047.2015.1056406
Chow, K. H., Park, H. J., George, J., Yamamoto, K., Gallup, A. D., Graber, J. H., et al. (2017). S100A4 is a biomarker and regulator of glioma stem cells that is critical for mesenchymal transition in glioblastoma. Cancer Res. 77, 5360–5373. doi: 10.1158/0008-5472.CAN-17-1294
Colwell, N., Larion, M., Giles, A. J., Seldomridge, A. N., Sizdahkhani, S., Gilbert, M. R., et al. (2017). Hypoxia in the glioblastoma microenvironment: shaping the phenotype of cancer stem-like cells. Neuro Oncol. 19, 887–896. doi: 10.1093/neuonc/now258
Corbin, Z., Spielman, D., and Recht, L. (2017). A metabolic therapy for malignant glioma requires a clinical measure. Curr. Oncol. Rep. 19:84. doi: 10.1007/s11912-017-0637-y
Crane, C. A., Austgen, K., Haberthur, K., Hofmann, C., Moyes, K. W., Avanesyan, L., et al. (2014). Immune evasion mediated by tumor-derived lactate dehydrogenase induction of NKG2D ligands on myeloid cells in glioblastoma patients. Proc. Natl. Acad. Sci. U.S.A. 111, 12823–12828. doi: 10.1073/pnas.1413933111
Damiano, F., Giannotti, L., Gnoni, G. V., Siculella, L., and Gnoni, A. (2019). Quercetin inhibition of SREBPs and ChREBP expression results in reduced cholesterol and fatty acid synthesis in C6 glioma cells. Int. J. Biochem. Cell Biol. 117:105618. doi: 10.1016/j.biocel.2019.105618
Dastmalchi, F., Karachi, A., Yang, C., Azari, H., Sayour, E. J., Dechkovskaia, A., et al. (2019). Sarcosine promotes trafficking of dendritic cells and improves efficacy of anti-tumor dendritic cell vaccines via CXC chemokine family signaling. J. Immunother. Cancer 7:321. doi: 10.1186/s40425-019-0809-4
De Feyter, H. M., Behar, K. L., Rao, J. U., Madden-Hennessey, K., Ip, K. L., Hyder, F., et al. (2016). A ketogenic diet increases transport and oxidation of ketone bodies in RG2 and 9L gliomas without affecting tumor growth. Neuro Oncol. 18, 1079–1087. doi: 10.1093/neuonc/now088
DeBerardinis, R. J., and Chandel, N. S. (2016). Fundamentals of cancer metabolism. Sci. Adv. 2:e1600200. doi: 10.1126/sciadv.1600200
DiDomenico, J., Lamano, J. B., Oyon, D., Li, Y., Veliceasa, D., Kaur, G., et al. (2018). The immune checkpoint protein PD-L1 induces and maintains regulatory T cells in glioblastoma. Oncoimmunology 7:e1448329. doi: 10.1080/2162402X.2018.1448329
Ding, A. S., Routkevitch, D., Jackson, C., and Lim, M. (2019). Targeting myeloid cells in combination treatments for glioma and other tumors. Front. Immunol. 10:1715. doi: 10.3389/fimmu.2019.01715
Domingues, P., Gonzalez-Tablas, M., Otero, A., Pascual, D., Miranda, D., Ruiz, L., et al. (2016). Tumor infiltrating immune cells in gliomas and meningiomas. Brain Behav. Immun. 53, 1–15. doi: 10.1016/j.bbi.2015.07.019
Du Four, S., Maenhout, S. K., Benteyn, D., De Keersmaecker, B., Duerinck, J., Thielemans, K., et al. (2016). Disease progression in recurrent glioblastoma patients treated with the VEGFR inhibitor axitinib is associated with increased regulatory T cell numbers and T cell exhaustion. Cancer Immunol. Immunother. 65, 727–740. doi: 10.1007/s00262-016-1836-3
Duan, K., Liu, Z. J., Hu, S. Q., Huo, H. Y., Xu, Z. R., Ruan, J. F., et al. (2018). Lactic acid induces lactate transport and glycolysis/OXPHOS interconversion in glioblastoma. Biochem. Biophys. Res. Commun. 503, 888–894. doi: 10.1016/j.bbrc.2018.06.092
Dubinski, D., Wolfer, J., Hasselblatt, M., Schneider-Hohendorf, T., Bogdahn, U., Stummer, W., et al. (2016). CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. Neuro Oncol. 18, 807–818. doi: 10.1093/neuonc/nov280
Eberstal, S., Fritzell, S., Sanden, E., Visse, E., Darabi, A., and Siesjo, P. (2014). Immunizations with unmodified tumor cells and simultaneous COX-2 inhibition eradicate malignant rat brain tumors and induce a long-lasting CD8(+) T cell memory. J. Neuroimmunol. 274, 161–167. doi: 10.1016/j.jneuroim.2014.06.019
Elsherbiny, M. E., Chen, H., Emara, M., and Godbout, R. (2018). omega-3 and omega-6 fatty acids modulate conventional and atypical protein kinase C activities in a brain fatty acid binding protein dependent manner in glioblastoma multiforme. Nutrients 10:454. doi: 10.3390/nu10040454
Elsherbiny, M. E., Emara, M., and Godbout, R. (2013). Interaction of brain fatty acid-binding protein with the polyunsaturated fatty acid environment as a potential determinant of poor prognosis in malignant glioma. Prog. Lipid Res. 52, 562–570. doi: 10.1016/j.plipres.2013.08.004
Engelhardt, B., Vajkoczy, P., and Weller, R. O. (2017). The movers and shapers in immune privilege of the CNS. Nat. Immunol. 18, 123–131. doi: 10.1038/ni.3666
Facchini, G., Ignarro, R. S., Rodrigues-Silva, E., Vieira, A. S., Lopes-Cendes, I., Castilho, R. F., et al. (2018). Toxic effects of phytol and retinol on human glioblastoma cells are associated with modulation of cholesterol and fatty acid biosynthetic pathways. J. Neurooncol. 136, 435–443. doi: 10.1007/s11060-017-2672-9
Fack, F., Tardito, S., Hochart, G., Oudin, A., Zheng, L., Fritah, S., et al. (2017). Altered metabolic landscape in IDH-mutant gliomas affects phospholipid, energy, and oxidative stress pathways. EMBO Mol. Med. 9, 1681–1695. doi: 10.15252/emmm.201707729
Feng, X., Yu, Y., He, S., Cheng, J., Gong, Y., Zhang, Z., et al. (2017). Dying glioma cells establish a proangiogenic microenvironment through a caspase 3 dependent mechanism. Cancer Lett. 385, 12–20. doi: 10.1016/j.canlet.2016.10.042
Fujii, T., Khawaja, M. R., DiNardo, C. D., Atkins, J. T., and Janku, F. (2016). Targeting isocitrate dehydrogenase (IDH) in cancer. Discov. Med. 21, 373–380.
Fujita, M., Kohanbash, G., Fellows-Mayle, W., Hamilton, R. L., Komohara, Y., Decker, S. A., et al. (2011). COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. 71, 2664–2674. doi: 10.1158/0008-5472.CAN-10-3055
Gabriely, G., Wheeler, M. A., Takenaka, M. C., and Quintana, F. J. (2017). Role of AHR and HIF-1alpha in glioblastoma metabolism. Trends Endocrinol. Metab. 28, 428–436. doi: 10.1016/j.tem.2017.02.009
Gabrilovich, D. I. (2017). Myeloid-derived suppressor cells. Cancer Immunol. Res. 5, 3–8. doi: 10.1158/2326-6066.CIR-16-0297
Ganapathy-Kanniappan, S. (2017). Linking tumor glycolysis and immune evasion in cancer: emerging concepts and therapeutic opportunities. Biochim. Biophys. Acta Rev. Cancer 1868, 212–220. doi: 10.1016/j.bbcan.2017.04.002
Gao, W., Li, N., Li, Z., Xu, J., and Su, C. (2018). Ketohexokinase is involved in fructose utilization and promotes tumor progression in glioma. Biochem. Biophys. Res. Commun. 503, 1298–1306. doi: 10.1016/j.bbrc.2018.07.040
Garg, A. D., Vandenberk, L., Koks, C., Verschuere, T., Boon, L., Van Gool, S. W., et al. (2016). Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci. Transl. Med. 8:328ra327. doi: 10.1126/scitranslmed.aae0105
Geng, F., Cheng, X., Wu, X., Yoo, J. Y., Cheng, C., Guo, J. Y., et al. (2016). Inhibition of SOAT1 suppresses glioblastoma growth via blocking SREBP-1-Mediated lipogenesis. Clin. Cancer Res. 22, 5337–5348. doi: 10.1158/1078-0432.CCR-15-2973
Gerthofer, V., Kreutz, M., Renner, K., Jachnik, B., Dettmer, K., Oefner, P., et al. (2018). Combined modulation of tumor metabolism by metformin and diclofenac in glioma. Int. J. Mol. Sci. 19:2586. doi: 10.3390/ijms19092586
Ghildiyal, R., and Sen, E. (2017). CK2 induced RIG-I drives metabolic adaptations in IFNgamma-treated glioma cells. Cytokine 89, 219–228. doi: 10.1016/j.cyto.2015.10.009
Ghosh, A., Bhattacharya, M., Sarkar, P., Acharya, S., and Chaudhuri, S. (2010). T11 target structure exerts effector function by activating immune cells in CNS against glioma where cytokine modulation provide favorable microenvironment. Indian J. Exp. Biol. 48, 879–888.
Gielen, P. R., Schulte, B. M., Kers-Rebel, E. D., Verrijp, K., Bossman, S. A., Ter Laan, M., et al. (2016). Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro Oncol. 18, 1253–1264. doi: 10.1093/neuonc/now034
Gieryng, A., Pszczolkowska, D., Walentynowicz, K. A., Rajan, W. D., and Kaminska, B. (2017). Immune microenvironment of gliomas. Lab. Invest. 97, 498–518. doi: 10.1038/labinvest.2017.19
Gimple, R. C., Kidwell, R. L., Kim, L. J. Y., Sun, T., Gromovsky, A. D., Wu, Q., et al. (2019). Glioma stem cell-specific superenhancer promotes polyunsaturated fatty-acid synthesis to support EGFR signaling. Cancer Discov. 9, 1248–1267. doi: 10.1158/2159-8290.CD-19-0061
Goji, T., Takahara, K., Negishi, M., and Katoh, H. (2017). Cystine uptake through the cystine/glutamate antiporter xCT triggers glioblastoma cell death under glucose deprivation. J. Biol. Chem. 292, 19721–19732. doi: 10.1074/jbc.M117.814392
Gomez Perdiguero, E., Klapproth, K., Schulz, C., Busch, K., Azzoni, E., Crozet, L., et al. (2015). Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518, 547–551. doi: 10.1038/nature13989
Gu, Y., Albuquerque, C. P., Braas, D., Zhang, W., Villa, G. R., Bi, J., et al. (2017). mTORC2 regulates amino acid metabolism in cancer by phosphorylation of the cystine-glutamate antiporter xCT. Mol. Cell 67, 128.e7–138.e7. doi: 10.1016/j.molcel.2017.05.030
Guan, J., Zhang, Z., Hu, X., Yang, Y., Chai, Z., Liu, X., et al. (2017). Cholera toxin subunit B enabled multifunctional glioma-targeted drug delivery. Adv. Healthc Mater. 6:1700709. doi: 10.1002/adhm.201700709
Guastella, A. R., Michelhaugh, S. K., Klinger, N. V., Fadel, H. A., Kiousis, S., Ali-Fehmi, R., et al. (2018). Investigation of the aryl hydrocarbon receptor and the intrinsic tumoral component of the kynurenine pathway of tryptophan metabolism in primary brain tumors. J. Neurooncol. 139, 239–249. doi: 10.1007/s11060-018-2869-6
Guo, D., Hildebrandt, I. J., Prins, R. M., Soto, H., Mazzotta, M. M., Dang, J., et al. (2009). The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc. Natl. Acad. Sci. U.S.A. 106, 12932–12937. doi: 10.1073/pnas.0906606106
Guo, X., Qiu, W., Liu, Q., Qian, M., Wang, S., Zhang, Z., et al. (2018). Immunosuppressive effects of hypoxia-induced glioma exosomes through myeloid-derived suppressor cells via the miR-10a/Rora and miR-21/Pten Pathways. Oncogene 37, 4239–4259. doi: 10.1038/s41388-018-0261-9
Guo, X., Qiu, W., Wang, J., Liu, Q., Qian, M., Wang, S., et al. (2019). Glioma exosomes mediate the expansion and function of myeloid-derived suppressor cells through microRNA-29a/Hbp1 and microRNA-92a/Prkar1a pathways. Int. J. Cancer 144, 3111–3126. doi: 10.1002/ijc.32052
Gusyatiner, O., and Hegi, M. E. (2018). Glioma epigenetics: from subclassification to novel treatment options. Semin. Cancer Biol. 51, 50–58. doi: 10.1016/j.semcancer.2017.11.010
Hambardzumyan, D., Gutmann, D. H., and Kettenmann, H. (2016). The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 19, 20–27. doi: 10.1038/nn.4185
Han, S., Liu, Y., Cai, S. J., Qian, M., Ding, J., Larion, M., et al. (2020). IDH mutation in glioma: molecular mechanisms and potential therapeutic targets. Br. J. Cancer 122, 1580–1589. doi: 10.1038/s41416-020-0814-x
He, X. Z., Wang, Q. F., Han, S., Wang, H. Q., Ye, Y. Y., Zhu, Z. Y., et al. (2015). Cryo-ablation improves anti-tumor immunity through recovering tumor educated dendritic cells in tumor-draining lymph nodes. Drug Des. Dev. Ther. 9, 1449–1458. doi: 10.2147/DDDT.S76592
Hervey-Jumper, S. L., and Berger, M. S. (2016). Maximizing safe resection of low- and high-grade glioma. J. Neurooncol. 130, 269–282. doi: 10.1007/s11060-016-2110-4
Hirata, E., and Sahai, E. (2017). Tumor microenvironment and differential responses to therapy. Cold Spring Harb. Perspect. Med. 7:a026781. doi: 10.1101/cshperspect.a026781
Hoang-Minh, L. B., Siebzehnrubl, F. A., Yang, C., Suzuki-Hatano, S., Dajac, K., Loche, T., et al. (2018). Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. 37:e98772. doi: 10.15252/embj.201798772
Huang, Y., Rajappa, P., Hu, W., Hoffman, C., Cisse, B., Kim, J. H., et al. (2017). A proangiogenic signaling axis in myeloid cells promotes malignant progression of glioma. J. Clin. Invest. 127, 1826–1838. doi: 10.1172/JCI86443
Huberfeld, G., and Vecht, C. J. (2016). Seizures and gliomas–towards a single therapeutic approach. Nat. Rev. Neurol. 12, 204–216. doi: 10.1038/nrneurol.2016.26
Hung, A. L., Maxwell, R., Theodros, D., Belcaid, Z., Mathios, D., Luksik, A. S., et al. (2018). TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology 7:e1466769. doi: 10.1080/2162402X.2018.1466769
Hutter, G., Theruvath, J., Graef, C. M., Zhang, M., Schoen, M. K., Manz, E. M., et al. (2019). Microglia are effector cells of CD47-SIRPalpha antiphagocytic axis disruption against glioblastoma. Proc. Natl. Acad. Sci. U.S.A. 116, 997–1006. doi: 10.1073/pnas.1721434116
Jackson, C. M., Choi, J., and Lim, M. (2019). Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat. Immunol. 20, 1100–1109. doi: 10.1038/s41590-019-0433-y
Jacobs, J. F., Idema, A. J., Bol, K. F., Grotenhuis, J. A., de Vries, I. J., Wesseling, P., et al. (2010). Prognostic significance and mechanism of Treg infiltration in human brain tumors. J. Neuroimmunol. 225, 195–199. doi: 10.1016/j.jneuroim.2010.05.020
Jais, A., Solas, M., Backes, H., Chaurasia, B., Kleinridders, A., Theurich, S., et al. (2016). Myeloid-cell-derived VEGF maintains brain glucose uptake and limits cognitive impairment in obesity. Cell 165, 882–895. doi: 10.1016/j.cell.2016.03.033
Jiang, J., and Dingledine, R. (2013). Role of prostaglandin receptor EP2 in the regulations of cancer cell proliferation, invasion, and inflammation. J. Pharmacol. Exp. Ther. 344, 360–367. doi: 10.1124/jpet.112.200444
Jiang, J., Qiu, J., Li, Q., and Shi, Z. (2017). Prostaglandin E2 signaling: alternative target for glioblastoma? Trends Cancer 3, 75–78. doi: 10.1016/j.trecan.2016.12.002
Jones, C. L., Stevens, B. M., D’Alessandro, A., Reisz, J. A., Culp-Hill, R., Nemkov, T., et al. (2018). Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell 34, 724.e4–740.e4. doi: 10.1016/j.ccell.2018.10.005
Kamran, N., Kadiyala, P., Saxena, M., Candolfi, M., Li, Y., Moreno-Ayala, M. A., et al. (2017). Immunosuppressive myeloid cells’ blockade in the glioma microenvironment enhances the efficacy of immune-stimulatory gene therapy. Mol. Ther. 25, 232–248. doi: 10.1016/j.ymthe.2016.10.003
Kathagen-Buhmann, A., Maire, C. L., Weller, J., Schulte, A., Matschke, J., Holz, M., et al. (2018). The secreted glycolytic enzyme GPI/AMF stimulates glioblastoma cell migration and invasion in an autocrine fashion but can have anti-proliferative effects. Neuro Oncol. 20, 1594–1605. doi: 10.1093/neuonc/noy117
Kathagen-Buhmann, A., Schulte, A., Weller, J., Holz, M., Herold-Mende, C., Glass, R., et al. (2016). Glycolysis and the pentose phosphate pathway are differentially associated with the dichotomous regulation of glioblastoma cell migration versus proliferation. Neuro Oncol. 18, 1219–1229. doi: 10.1093/neuonc/now024
Kesarwani, P., Kant, S., Prabhu, A., and Chinnaiyan, P. (2017). The interplay between metabolic remodeling and immune regulation in glioblastoma. Neuro Oncol. 19, 1308–1315. doi: 10.1093/neuonc/nox079
Kesarwani, P., Prabhu, A., Kant, S., and Chinnaiyan, P. (2019). Metabolic remodeling contributes towards an immune-suppressive phenotype in glioblastoma. Cancer Immunol. Immunother. 68, 1107–1120. doi: 10.1007/s00262-019-02347-3
Kesarwani, P., Prabhu, A., Kant, S., Kumar, P., Graham, S. F., Buelow, K. L., et al. (2018). Tryptophan metabolism contributes to radiation-induced immune checkpoint reactivation in glioblastoma. Clin. Cancer Res. 24, 3632–3643. doi: 10.1158/1078-0432.CCR-18-0041
Kim, D., Fiske, B. P., Birsoy, K., Freinkman, E., Kami, K., Possemato, R. L., et al. (2015). SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 520, 363–367. doi: 10.1038/nature14363
Kim, J., Han, J., Jang, Y., Kim, S. J., Lee, M. J., Ryu, M. J., et al. (2015). High-capacity glycolytic and mitochondrial oxidative metabolisms mediate the growth ability of glioblastoma. Int. J. Oncol. 47, 1009–1016. doi: 10.3892/ijo.2015.3101
Kim, S., Jing, K., Shin, S., Jeong, S., Han, S. H., Oh, H., et al. (2018). omega3-polyunsaturated fatty acids induce cell death through apoptosis and autophagy in glioblastoma cells: in vitro and in vivo. Oncol. Rep. 39, 239–246. doi: 10.3892/or.2017.6101
Kohanbash, G., Carrera, D. A., Shrivastav, S., Ahn, B. J., Jahan, N., Mazor, T., et al. (2017). Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J. Clin. Invest. 127, 1425–1437. doi: 10.1172/JCI90644
Kohanbash, G., McKaveney, K., Sakaki, M., Ueda, R., Mintz, A. H., Amankulor, N., et al. (2013). GM-CSF promotes the immunosuppressive activity of glioma-infiltrating myeloid cells through interleukin-4 receptor-alpha. Cancer Res. 73, 6413–6423. doi: 10.1158/0008-5472.CAN-12-4124
Komohara, Y., Ohnishi, K., Kuratsu, J., and Takeya, M. (2008). Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 216, 15–24. doi: 10.1002/path.2370
Kosaka, A., Ohkuri, T., and Okada, H. (2014). Combination of an agonistic anti-CD40 monoclonal antibody and the COX-2 inhibitor celecoxib induces anti-glioma effects by promotion of type-1 immunity in myeloid cells and T-cells. Cancer Immunol. Immunother. 63, 847–857. doi: 10.1007/s00262-014-1561-8
Kuang, R., Jahangiri, A., Mascharak, S., Nguyen, A., Chandra, A., Flanigan, P. M., et al. (2017). GLUT3 upregulation promotes metabolic reprogramming associated with antiangiogenic therapy resistance. JCI Insight 2:e88815. doi: 10.1172/jci.insight.88815
Laudati, E., Curro, D., Navarra, P., and Lisi, L. (2017). Blockade of CCR5 receptor prevents M2 microglia phenotype in a microglia-glioma paradigm. Neurochem. Int. 108, 100–108. doi: 10.1016/j.neuint.2017.03.002
Lee, J. H., Liu, R., Li, J., Wang, Y., Tan, L., Li, X. J., et al. (2018). EGFR-phosphorylated platelet isoform of phosphofructokinase 1 promotes PI3K activation. Mol. Cell 70, 197.e7–210.e7. doi: 10.1016/j.molcel.2018.03.018
Lee, J. H., Liu, R., Li, J., Zhang, C., Wang, Y., Cai, Q., et al. (2017). Stabilization of phosphofructokinase 1 platelet isoform by AKT promotes tumorigenesis. Nat. Commun. 8:949. doi: 10.1038/s41467-017-00906-9
Lee, S. Y., Kim, J. K., Jeon, H. Y., Ham, S. W., and Kim, H. (2017). CD133 regulates IL-1beta signaling and neutrophil recruitment in glioblastoma. Mol. Cells 40, 515–522. doi: 10.14348/molcells.2017.0089
Li, J., Zhao, J., Tan, T., Liu, M., Zeng, Z., Zeng, Y., et al. (2020). Nanoparticle drug delivery system for glioma and its efficacy improvement strategies: a comprehensive review. Int. J. Nanomed. 15, 2563–2582. doi: 10.2147/IJN.S243223
Li, T. F., Li, K., Zhang, Q., Wang, C., Yue, Y., Chen, Z., et al. (2018). Dendritic cell-mediated delivery of doxorubicin-polyglycerol-nanodiamond composites elicits enhanced anti-cancer immune response in glioblastoma. Biomaterials 181, 35–52. doi: 10.1016/j.biomaterials.2018.07.035
Liang, J., Piao, Y., Holmes, L., Fuller, G. N., Henry, V., Tiao, N., et al. (2014). Neutrophils promote the malignant glioma phenotype through S100A4. Clin. Cancer Res. 20, 187–198. doi: 10.1158/1078-0432.CCR-13-1279
Libby, C. J., Zhang, S., Benavides, G. A., Scott, S. E., Li, Y., Redmann, M., et al. (2018). Identification of compounds that decrease glioblastoma growth and glucose uptake in vitro. ACS Chem. Biol. 13, 2048–2057. doi: 10.1021/acschembio.8b00251
Lin, H., Patel, S., Affleck, V. S., Wilson, I., Turnbull, D. M., Joshi, A. R., et al. (2017). Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro Oncol. 19, 43–54. doi: 10.1093/neuonc/now128
Liu, B., Huang, Z. B., Chen, X., See, Y. X., Chen, Z. K., and Yao, H. K. (2019). Mammalian target of rapamycin 2 (MTOR2) and C-MYC Modulate Glucosamine-6-phosphate synthesis in glioblastoma (GBM) cells through glutamine: Fructose-6-phosphate aminotransferase 1 (GFAT1). Cell Mol. Neurobiol. 39, 415–434. doi: 10.1007/s10571-019-00659-7
Liu, J., Gao, L., Zhan, N., Xu, P., Yang, J. A., Yuan, F. E., et al. (2020). Hypoxia induced ferritin light chain (FTL) promoted epithelia mesenchymal transition and chemoresistance of glioma. J. Exp. Clin. Cancer Res. 39:137. doi: 10.1186/s13046-020-01641-8
Liu, J., Guo, S., Li, Q., Yang, L., Xia, Z., Zhang, L., et al. (2013). Phosphoglycerate dehydrogenase induces glioma cells proliferation and invasion by stabilizing forkhead box M1. J. Neurooncol. 111, 245–255. doi: 10.1007/s11060-012-1018-x
Liu, M., Zhao, J., Chen, K., Bian, X., Wang, C., Shi, Y., et al. (2012). G protein-coupled receptor FPR1 as a pharmacologic target in inflammation and human glioblastoma. Int. Immunopharmacol. 14, 283–288. doi: 10.1016/j.intimp.2012.07.015
Liu, R., Li, W., Tao, B., Wang, X., Yang, Z., Zhang, Y., et al. (2019). Tyrosine phosphorylation activates 6-phosphogluconate dehydrogenase and promotes tumor growth and radiation resistance. Nat. Commun. 10:991. doi: 10.1038/s41467-019-08921-8
Long, Y., Tao, H., Karachi, A., Grippin, A. J., Jin, L., Chang, Y. E., et al. (2020). Dysregulation of glutamate transport enhances treg function that promotes VEGF blockade resistance in glioblastoma. Cancer Res. 80, 499–509. doi: 10.1158/0008-5472.CAN-19-1577
Lou, N., Takano, T., Pei, Y., Xavier, A. L., Goldman, S. A., and Nedergaard, M. (2016). Purinergic receptor P2RY12-dependent microglial closure of the injured blood-brain barrier. Proc. Natl. Acad. Sci. U.S.A. 113, 1074–1079. doi: 10.1073/pnas.1520398113
Maddocks, O. D. K., Athineos, D., Cheung, E. C., Lee, P., Zhang, T., van den Broek, N. J. F., et al. (2017). Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544, 372–376. doi: 10.1038/nature22056
Magana-Maldonado, R., Chavez-Cortez, E. G., Olascoaga-Arellano, N. K., Lopez-Mejia, M., Maldonado-Leal, F. M., Sotelo, J., et al. (2016). Immunological evasion in glioblastoma. Biomed. Res. Int. 2016:7487313. doi: 10.1155/2016/7487313
Malo, C. S., Huggins, M. A., Goddery, E. N., Tolcher, H. M. A., Renner, D. N., Jin, F., et al. (2018a). Non-equivalent antigen presenting capabilities of dendritic cells and macrophages in generating brain-infiltrating CD8 (+) T cell responses. Nat. Commun. 9:633. doi: 10.1038/s41467-018-03037-x
Malo, C. S., Khadka, R. H., Ayasoufi, K., Jin, F., AbouChehade, J. E., Hansen, M. J., et al. (2018b). Immunomodulation mediated by anti-angiogenic therapy improves CD8 T Cell immunity against experimental glioma. Front. Oncol. 8:320. doi: 10.3389/fonc.2018.00320
Marvel, D., and Gabrilovich, D. I. (2015). Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J. Clin. Invest. 125, 3356–3364. doi: 10.1172/JCI80005
Massara, M., Persico, P., Bonavita, O., Mollica Poeta, V., Locati, M., Simonelli, M., et al. (2017). Neutrophils in gliomas. Front. Immunol. 8:1349. doi: 10.3389/fimmu.2017.01349
Masui, K., Onizuka, H., Cavenee, W. K., Mischel, P. S., and Shibata, N. (2019). Metabolic reprogramming in the pathogenesis of glioma: update. Neuropathology 39, 3–13. doi: 10.1111/neup.12535
Mathios, D., Kim, J. E., Mangraviti, A., Phallen, J., Park, C. K., Jackson, C. M., et al. (2016). Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci. Transl. Med. 8:370ra180. doi: 10.1126/scitranslmed.aag2942
Maurer, G. D., Heller, S., Wanka, C., Rieger, J., and Steinbach, J. P. (2019). Knockdown of the TP53-Induced glycolysis and apoptosis regulator (TIGAR) Sensitizes glioma cells to hypoxia, irradiation and temozolomide. Int. J. Mol. Sci. 20:1061. doi: 10.3390/ijms20051061
May, J. L., Kouri, F. M., Hurley, L. A., Liu, J., Tommasini-Ghelfi, S., Ji, Y., et al. (2019). IDH3alpha regulates one-carbon metabolism in glioblastoma. Sci. Adv. 5:eaat0456. doi: 10.1126/sciadv.aat0456
Mayers, J. R., Torrence, M. E., Danai, L. V., Papagiannakopoulos, T., Davidson, S. M., Bauer, M. R., et al. (2016). Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 353, 1161–1165. doi: 10.1126/science.aaf5171
Meng, X., Duan, C., Pang, H., Chen, Q., Han, B., Zha, C., et al. (2019). DNA damage repair alterations modulate M2 polarization of microglia to remodel the tumor microenvironment via the p53-mediated MDK expression in glioma. EBioMedicine 41, 185–199. doi: 10.1016/j.ebiom.2019.01.067
Mirzaei, R., Sarkar, S., and Yong, V. W. (2017). T cell exhaustion in glioblastoma: intricacies of immune checkpoints. Trends Immunol. 38, 104–115. doi: 10.1016/j.it.2016.11.005
Miska, J., Lee-Chang, C., Rashidi, A., Muroski, M. E., Chang, A. L., Lopez-Rosas, A., et al. (2019). HIF-1alpha is a metabolic switch between glycolytic-driven migration and oxidative phosphorylation-driven immunosuppression of tregs in glioblastoma. Cell Rep. 27, 226.e4–237.e4. doi: 10.1016/j.celrep.2019.03.029
Mitchell, D. A., Batich, K. A., Gunn, M. D., Huang, M. N., Sanchez-Perez, L., Nair, S. K., et al. (2015). Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 519, 366–369. doi: 10.1038/nature14320
Morihiro, Y., Yasumoto, Y., Vaidyan, L. K., Sadahiro, H., Uchida, T., Inamura, A., et al. (2013). Fatty acid binding protein 7 as a marker of glioma stem cells. Pathol. Int. 63, 546–553. doi: 10.1111/pin.12109
Mostofa, A. G., Punganuru, S. R., Madala, H. R., Al-Obaide, M., and Srivenugopal, K. S. (2017). The process and regulatory components of inflammation in brain oncogenesis. Biomolecules 7:34. doi: 10.3390/biom7020034
Nakayama, N., Yamaguchi, S., Sasaki, Y., and Chikuma, T. (2016). Hydrogen peroxide-induced oxidative stress activates proteasomal trypsin-like activity in Human U373 glioma cells. J. Mol. Neurosci. 58, 297–305. doi: 10.1007/s12031-015-0680-9
Oh, E. T., Kim, C. W., Kim, H. G., Lee, J. S., and Park, H. J. (2017). Brusatol-mediated inhibition of c-Myc Increases HIF-1alpha degradation and causes cell death in colorectal cancer under hypoxia. Theranostics 7, 3415–3431. doi: 10.7150/thno.20861
Osuka, S., and Van Meir, E. G. (2017). Cancer therapy: neutrophils traffic in cancer nanodrugs. Nat. Nanotechnol. 12, 616–618. doi: 10.1038/nnano.2017.82
Otvos, B., Silver, D. J., Mulkearns-Hubert, E. E., Alvarado, A. G., Turaga, S. M., Sorensen, M. D., et al. (2016). Cancer stem cell-secreted macrophage migration inhibitory factor stimulates myeloid derived suppressor cell function and facilitates glioblastoma immune evasion. Stem Cells 34, 2026–2039. doi: 10.1002/stem.2393
Pandey, R., Caflisch, L., Lodi, A., Brenner, A. J., and Tiziani, S. (2017). Metabolomic signature of brain cancer. Mol. Carcinog. 56, 2355–2371. doi: 10.1002/mc.22694
Panosyan, E. H., Lin, H. J., Koster, J., and Lasky, J. L. III (2017). In search of druggable targets for GBM amino acid metabolism. BMC Cancer 17:162. doi: 10.1186/s12885-017-3148-1
Payen, V. L., Porporato, P. E., Baselet, B., and Sonveaux, P. (2016). Metabolic changes associated with tumor metastasis, part 1: tumor pH, glycolysis and the pentose phosphate pathway. Cell Mol. Life Sci. 73, 1333–1348. doi: 10.1007/s00018-015-2098-5
Pereira, M. B., Barros, L. R. C., Bracco, P. A., Vigo, A., Boroni, M., Bonamino, M. H., et al. (2018). Transcriptional characterization of immunological infiltrates and their relation with glioblastoma patients overall survival. Oncoimmunology 7:e1431083. doi: 10.1080/2162402X.2018.1431083
Petovari, G., Hujber, Z., Krencz, I., Danko, T., Nagy, N., Toth, F., et al. (2018). Targeting cellular metabolism using rapamycin and/or doxycycline enhances anti-tumour effects in human glioma cells. Cancer Cell Int. 18:211. doi: 10.1186/s12935-018-0710-0
Poff, A., Koutnik, A. P., Egan, K. M., Sahebjam, S., D’Agostino, D., and Kumar, N. B. (2019). Targeting the Warburg effect for cancer treatment: ketogenic diets for management of glioma. Semin. Cancer Biol. 56, 135–148. doi: 10.1016/j.semcancer.2017.12.011
Poon, C. C., Sarkar, S., Yong, V. W., and Kelly, J. J. P. (2017). Glioblastoma-associated microglia and macrophages: targets for therapies to improve prognosis. Brain 140, 1548–1560. doi: 10.1093/brain/aww355
Priore, P., Gnoni, A., Natali, F., Testini, M., Gnoni, G. V., Siculella, L., et al. (2017). Oleic acid and hydroxytyrosol inhibit cholesterol and fatty acid synthesis in C6 Glioma cells. Oxid. Med. Cell Longev. 2017:9076052. doi: 10.1155/2017/9076052
Pyonteck, S. M., Akkari, L., Schuhmacher, A. J., Bowman, R. L., Sevenich, L., Quail, D. F., et al. (2013). CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 19, 1264–1272. doi: 10.1038/nm.3337
Qian, J., Wang, C., Wang, B., Yang, J., Wang, Y., Luo, F., et al. (2018). The IFN-gamma/PD-L1 axis between T cells and tumor microenvironment: hints for glioma anti-PD-1/PD-L1 therapy. J. Neuroinflammation 15:290. doi: 10.1186/s12974-018-1330-2
Qiu, J., Li, Q., Bell, K. A., Yao, X., Du, Y., Zhang, E., et al. (2019). Small-molecule inhibition of prostaglandin E receptor 2 impairs cyclooxygenase-associated malignant glioma growth. Br. J. Pharmacol. 176, 1680–1699. doi: 10.1111/bph.14622
Qiu, R., Zhou, L., Ma, Y., Zhou, L., Liang, T., Shi, L., et al. (2020). Regulatory T Cell plasticity and stability and autoimmune diseases. Clin. Rev. Allergy Immunol. 58, 52–70. doi: 10.1007/s12016-018-8721-0
Quail, D. F., and Joyce, J. A. (2017). The microenvironmental landscape of brain tumors. Cancer Cell 31, 326–341. doi: 10.1016/j.ccell.2017.02.009
Ran, C., Liu, H., Hitoshi, Y., and Israel, M. A. (2013). Proliferation-independent control of tumor glycolysis by PDGFR-mediated AKT activation. Cancer Res. 73, 1831–1843. doi: 10.1158/0008-5472.CAN-12-2460
Rashidi, A., Miska, J., Lee-Chang, C., Kanojia, D., Panek, W. K., Lopez-Rosas, A., et al. (2020). GCN2 is essential for CD8(+) T cell survival and function in murine models of malignant glioma. Cancer Immunol. Immunother. 69, 81–94. doi: 10.1007/s00262-019-02441-6
Ren, F., Zhao, Q., Huang, L., Zheng, Y., Li, L., He, Q., et al. (2019). The R132H mutation in IDH1 promotes the recruitment of NK cells through CX3CL1/CX3CR1 chemotaxis and is correlated with a better prognosis in gliomas. Immunol. Cell Biol. 97, 457–469. doi: 10.1111/imcb.12225
Roesch, S., Rapp, C., Dettling, S., and Herold-Mende, C. (2018). When immune cells turn bad-tumor-associated microglia/macrophages in glioma. Int. J. Mol. Sci. 19:436. doi: 10.3390/ijms19020436
Rolle, C. E., Sengupta, S., and Lesniak, M. S. (2012). Mechanisms of immune evasion by gliomas. Adv. Exp. Med. Biol. 746, 53–76. doi: 10.1007/978-1-4614-3146-6_5
Sahm, F., Oezen, I., Opitz, C. A., Radlwimmer, B., von Deimling, A., Ahrendt, T., et al. (2013). The endogenous tryptophan metabolite and NAD+ precursor quinolinic acid confers resistance of gliomas to oxidative stress. Cancer Res. 73, 3225–3234. doi: 10.1158/0008-5472.CAN-12-3831
Sankowski, R., Bottcher, C., Masuda, T., Geirsdottir, L., Sagar, Sindram, E., et al. (2019). Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat. Neurosci. 22, 2098–2110. doi: 10.1038/s41593-019-0532-y
Sayour, E. J., McLendon, P., McLendon, R., De Leon, G., Reynolds, R., Kresak, J., et al. (2015). Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunol. Immunother. 64, 419–427. doi: 10.1007/s00262-014-1651-7
See, A. P., Parker, J. J., and Waziri, A. (2015). The role of regulatory T cells and microglia in glioblastoma-associated immunosuppression. J. Neurooncol. 123, 405–412. doi: 10.1007/s11060-015-1849-3
Seliger, C., and Hau, P. (2018). Drug repurposing of metabolic agents in malignant glioma. Int. J. Mol. Sci. 19:2768. doi: 10.3390/ijms19092768
Shao, S., Risch, E., Burner, D., Lu, L., Minev, B., and Ma, W. (2017). IFNgamma enhances cytotoxic efficiency of the cytotoxic T lymphocytes against human glioma cells. Int. Immunopharmacol. 47, 159–165. doi: 10.1016/j.intimp.2017.04.003
Sheng, Y., Jiang, Q., Dong, X., Liu, J., Liu, L., Wang, H., et al. (2020). 3-Bromopyruvate inhibits the malignant phenotype of malignantly transformed macrophages and dendritic cells induced by glioma stem cells in the glioma microenvironment via miR-449a/MCT1. Biomed. Pharmacother. 121:109610. doi: 10.1016/j.biopha.2019.109610
Shibao, S., Minami, N., Koike, N., Fukui, N., Yoshida, K., Saya, H., et al. (2018). Metabolic heterogeneity and plasticity of glioma stem cells in a mouse glioblastoma model. Neuro Oncol. 20, 343–354. doi: 10.1093/neuonc/nox170
Silva, L. S., Poschet, G., Nonnenmacher, Y., Becker, H. M., Sapcariu, S., Gaupel, A. C., et al. (2017). Branched-chain ketoacids secreted by glioblastoma cells via MCT1 modulate macrophage phenotype. EMBO Rep. 18, 2172–2185. doi: 10.15252/embr.201744154
Slawinska-Brych, A., Zdzisinska, B., and Kandefer-Szerszen, M. (2014). Fluvastatin inhibits growth and alters the malignant phenotype of the C6 glioma cell line. Pharmacol. Rep. 66, 121–129. doi: 10.1016/j.pharep.2014.01.002
Sorensen, M. D., Dahlrot, R. H., Boldt, H. B., Hansen, S., and Kristensen, B. W. (2018). Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol. Appl. Neurobiol. 44, 185–206. doi: 10.1111/nan.12428
Spiegel, A., Brooks, M. W., Houshyar, S., Reinhardt, F., Ardolino, M., Fessler, E., et al. (2016). Neutrophils suppress intraluminal NK cell-mediated tumor cell clearance and enhance extravasation of disseminated carcinoma cells. Cancer Discov. 6, 630–649. doi: 10.1158/2159-8290.CD-15-1157
Strickland, M., and Stoll, E. A. (2017). Metabolic reprogramming in glioma. Front. Cell Dev. Biol. 5:43. doi: 10.3389/fcell.2017.00043
Su, C., Li, H., and Gao, W. (2018). GLUT5 increases fructose utilization and promotes tumor progression in glioma. Biochem. Biophys. Res. Commun. 500, 462–469. doi: 10.1016/j.bbrc.2018.04.103
Sun, P., Xia, S., Lal, B., Shi, X., Yang, K. S., Watkins, P. A., et al. (2014). Lipid metabolism enzyme ACSVL3 supports glioblastoma stem cell maintenance and tumorigenicity. BMC Cancer 14:401. doi: 10.1186/1471-2407-14-401
Takenaka, M. C., Gabriely, G., Rothhammer, V., Mascanfroni, I. D., Wheeler, M. A., Chao, C. C., et al. (2019). Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 22, 729–740. doi: 10.1038/s41593-019-0370-y
Talasila, K. M., Røsland, G. V., Hagland, H. R., Eskilsson, E., Flønes, I. H., Fritah, S., et al. (2017). The angiogenic switch leads to a metabolic shift in human glioblastoma. Neuro Oncol. 19, 383–393. doi: 10.1093/neuonc/now175
Tardito, S., Oudin, A., Ahmed, S. U., Fack, F., Keunen, O., Zheng, L., et al. (2015). Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 17, 1556–1568. doi: 10.1038/ncb3272
Thiepold, A. L., Lorenz, N. I., Foltyn, M., Engel, A. L., Dive, I., Urban, H., et al. (2017). Mammalian target of rapamycin complex 1 activation sensitizes human glioma cells to hypoxia-induced cell death. Brain 140, 2623–2638. doi: 10.1093/brain/awx196
Thomas, T. M., and Yu, J. S. (2017). Metabolic regulation of glioma stem-like cells in the tumor micro-environment. Cancer Lett. 408, 174–181. doi: 10.1016/j.canlet.2017.07.014
Touat, M., Idbaih, A., Sanson, M., and Ligon, K. L. (2017). Glioblastoma targeted therapy: updated approaches from recent biological insights. Ann. Oncol. 28, 1457–1472. doi: 10.1093/annonc/mdx106
Turubanova, V. D., Balalaeva, I. V., Mishchenko, T. A., Catanzaro, E., Alzeibak, R., Peskova, N. N., et al. (2019). Immunogenic cell death induced by a new photodynamic therapy based on photosens and photodithazine. J. Immunother. Cancer 7:350. doi: 10.1186/s40425-019-0826-3
Vakilian, A., Khorramdelazad, H., Heidari, P., Sheikh Rezaei, Z., and Hassanshahi, G. (2017). CCL2/CCR2 signaling pathway in glioblastoma multiforme. Neurochem. Int. 103, 1–7. doi: 10.1016/j.neuint.2016.12.013
van Tellingen, O., Yetkin-Arik, B., de Gooijer, M. C., Wesseling, P., Wurdinger, T., and de Vries, H. E. (2015). Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updates 19, 1–12. doi: 10.1016/j.drup.2015.02.002
Venneti, S., Dunphy, M. P., Zhang, H., Pitter, K. L., Zanzonico, P., Campos, C., et al. (2015). Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci. Transl. Med. 7:274ra217. doi: 10.1126/scitranslmed.aaa1009
Venneti, S., and Thompson, C. B. (2017). Metabolic reprogramming in brain tumors. Annu. Rev. Pathol. 12, 515–545. doi: 10.1146/annurev-pathol-012615-044329
Venza, I., Visalli, M., Fortunato, C., Ruggeri, M., Ratone, S., Caffo, M., et al. (2012). PGE2 induces interleukin-8 derepression in human astrocytoma through coordinated DNA demethylation and histone hyperacetylation. Epigenetics 7, 1315–1330. doi: 10.4161/epi.22446
Venza, M., Visalli, M., Alafaci, C., Caffo, M., Caruso, G., Salpietro, F. M., et al. (2011). Interleukin-8 overexpression in astrocytomas is induced by prostaglandin E2 and is associated with the transcription factors CCAAT/enhancer-binding protein-beta and CCAAT/enhancer-binding homologous protein. Neurosurgery 69, 713–721. doi: 10.1227/NEU.0b013e31821954c6 discussion 721,
Villa, G. R., Hulce, J. J., Zanca, C., Bi, J., Ikegami, S., Cahill, G. L., et al. (2016). An LXR-Cholesterol axis creates a metabolic Co-Dependency for Brain Cancers. Cancer Cell 30, 683–693. doi: 10.1016/j.ccell.2016.09.008
Wainwright, D. A., Chang, A. L., Dey, M., Balyasnikova, I. V., Kim, C. K., Tobias, A., et al. (2014). Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin. Cancer Res. 20, 5290–5301. doi: 10.1158/1078-0432.CCR-14-0514
Waitkus, M. S., Diplas, B. H., and Yan, H. (2016). Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol. 18, 16–26. doi: 10.1093/neuonc/nov136
Wang, C., Chen, Y., Wang, Y., Liu, X., Liu, Y., Li, Y., et al. (2019). Inhibition of COX-2, mPGES-1 and CYP4A by isoliquiritigenin blocks the angiogenic Akt signaling in glioma through ceRNA effect of miR-194-5p and lncRNA NEAT1. J. Exp. Clin. Cancer Res. 38:371. doi: 10.1186/s13046-019-1361-2
Wang, C., Li, Y., Chen, H., Zhang, J., Zhang, J., Qin, T., et al. (2017). Inhibition of CYP4A by a novel flavonoid FLA-16 prolongs survival and normalizes tumor vasculature in glioma. Cancer Lett. 402, 131–141. doi: 10.1016/j.canlet.2017.05.030
Wang, G., Sai, K., Gong, F., Yang, Q., Chen, F., and Lin, J. (2014). Mutation of isocitrate dehydrogenase 1 induces glioma cell proliferation via nuclear factor-kappaB activation in a hypoxia-inducible factor 1-alpha dependent manner. Mol. Med. Rep. 9, 1799–1805. doi: 10.3892/mmr.2014.2052
Wang, G., Wang, J. J., Fu, X. L., Guang, R., and To, S. T. (2017). Advances in the targeting of HIF-1alpha and future therapeutic strategies for glioblastoma multiforme (Review). Oncol. Rep. 37, 657–670. doi: 10.3892/or.2016.5309
Wang, J., Xu, S. L., Duan, J. J., Yi, L., Guo, Y. F., Shi, Y., et al. (2019). Invasion of white matter tracts by glioma stem cells is regulated by a NOTCH1-SOX2 positive-feedback loop. Nat. Neurosci. 22, 91–105. doi: 10.1038/s41593-018-0285-z
Wang, Q., Hu, B., Hu, X., Kim, H., Squatrito, M., Scarpace, L., et al. (2017). Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell 32, 42.e6–56.e6. doi: 10.1016/j.ccell.2017.06.003
Wang, W., Xiao, Z. D., Li, X., Aziz, K. E., Gan, B., Johnson, R. L., et al. (2015). AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 17, 490–499. doi: 10.1038/ncb3113
Wang, X., Huang, Z., Wu, Q., Prager, B. C., Mack, S. C., Yang, K., et al. (2017). MYC-Regulated mevalonate metabolism maintains brain tumor-initiating cells. Cancer Res. 77, 4947–4960. doi: 10.1158/0008-5472.CAN-17-0114
Wang, X., Liu, R., Qu, X., Yu, H., Chu, H., Zhang, Y., et al. (2019). alpha-ketoglutarate-activated NF-kappaB signaling promotes compensatory glucose uptake and brain tumor development. Mol. Cell 76, 148.e7–162.e7. doi: 10.1016/j.molcel.2019.07.007
Wang, Z., Ding, Y., Wang, X., Lu, S., Wang, C., He, C., et al. (2018). Pseudolaric acid B triggers ferroptosis in glioma cells via activation of Nox4 and inhibition of xCT. Cancer Lett. 428, 21–33. doi: 10.1016/j.canlet.2018.04.021
Weller, M., Roth, P., Preusser, M., Wick, W., Reardon, D. A., Platten, M., et al. (2017). Vaccine-based immunotherapeutic approaches to gliomas and beyond. Nat. Rev. Neurol. 13, 363–374. doi: 10.1038/nrneurol.2017.64
Wilk, A., Wyczechowska, D., Zapata, A., Dean, M., Mullinax, J., Marrero, L., et al. (2015). Molecular mechanisms of fenofibrate-induced metabolic catastrophe and glioblastoma cell death. Mol. Cell Biol. 35, 182–198. doi: 10.1128/MCB.00562-14
Winter, S. F., Loebel, F., and Dietrich, J. (2017). Role of ketogenic metabolic therapy in malignant glioma: a systematic review. Crit. Rev. Oncol. Hematol. 112, 41–58. doi: 10.1016/j.critrevonc.2017.02.016
Wolf, A., Agnihotri, S., Micallef, J., Mukherjee, J., Sabha, N., Cairns, R., et al. (2011). Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 208, 313–326. doi: 10.1084/jem.20101470
Wu, F., Zhao, Z., Chai, R. C., Liu, Y. Q., Li, G. Z., Jiang, H. Y., et al. (2019). Prognostic power of a lipid metabolism gene panel for diffuse gliomas. J. Cell Mol. Med. 23, 7741–7748. doi: 10.1111/jcmm.14647
Wu, M., Zhang, H., Tie, C., Yan, C., Deng, Z., Wan, Q., et al. (2018). MR imaging tracking of inflammation-activatable engineered neutrophils for targeted therapy of surgically treated glioma. Nat. Commun. 9:4777. doi: 10.1038/s41467-018-07250-6
Wu, T., and Dai, Y. (2017). Tumor microenvironment and therapeutic response. Cancer Lett. 387, 61–68. doi: 10.1016/j.canlet.2016.01.043
Wu, Z. B., Qiu, C., Zhang, A. L., Cai, L., Lin, S. J., Yao, Y., et al. (2014). Glioma-associated antigen HEATR1 induces functional cytotoxic T lymphocytes in patients with glioma. J. Immunol. Res. 2014:131494. doi: 10.1155/2014/131494
Wypych, D., and Baranska, J. (2020). Cross-talk in nucleotide signaling in glioma C6 Cells. Adv. Exp. Med. Biol. 1202, 35–65. doi: 10.1007/978-3-030-30651-9_3
Xu, C. F., Liu, Y., Shen, S., Zhu, Y. H., and Wang, J. (2015). Targeting glucose uptake with siRNA-based nanomedicine for cancer therapy. Biomaterials 51, 1–11. doi: 10.1016/j.biomaterials.2015.01.068
Xu, T., Zhang, K., Shi, J., Huang, B., Wang, X., Qian, K., et al. (2019). MicroRNA-940 inhibits glioma progression by blocking mitochondrial folate metabolism through targeting of MTHFD2. Am. J. Cancer Res. 9, 250–269.
Xue, J., Zhao, Z., Zhang, L., Xue, L., Shen, S., Wen, Y., et al. (2017). Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat. Nanotechnol. 12, 692–700. doi: 10.1038/nnano.2017.54
Yang, F., Zhang, H., Mei, Y., and Wu, M. (2014). Reciprocal regulation of HIF-1alpha and lincRNA-p21 modulates the Warburg effect. Mol. Cell 53, 88–100. doi: 10.1016/j.molcel.2013.11.004
Yang, L., and Lin, P. C. (2017). Mechanisms that drive inflammatory tumor microenvironment, tumor heterogeneity, and metastatic progression. Semin. Cancer Biol. 47, 185–195. doi: 10.1016/j.semcancer.2017.08.001
Ye, L. F., Chaudhary, K. R., Zandkarimi, F., Harken, A. D., Kinslow, C. J., Upadhyayula, P. S., et al. (2020). Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducers. ACS Chem. Biol. 15, 469–484. doi: 10.1021/acschembio.9b00939
Zhai, L., Ladomersky, E., Lenzen, A., Nguyen, B., Patel, R., Lauing, K. L., et al. (2018). IDO1 in cancer: a gemini of immune checkpoints. Cell Mol. Immunol. 15, 447–457. doi: 10.1038/cmi.2017.143
Zhang, C., Cheng, W., Ren, X., Wang, Z., Liu, X., Li, G., et al. (2017). Tumor purity as an underlying key factor in glioma. Clin. Cancer Res. 23, 6279–6291. doi: 10.1158/1078-0432.CCR-16-2598
Zhang, Y., Yu, G., Chu, H., Wang, X., Xiong, L., Cai, G., et al. (2018). Macrophage-associated PGK1 phosphorylation promotes aerobic glycolysis and tumorigenesis. Mol. Cell 71, 201.e7–215.e7. doi: 10.1016/j.molcel.2018.06.023
Zheng, C., Yang, K., Zhang, M., Zou, M., Bai, E., Ma, Q., et al. (2016). Specific protein 1 depletion attenuates glucose uptake and proliferation of human glioma cells by regulating GLUT3 expression. Oncol. Lett. 12, 125–131. doi: 10.3892/ol.2016.4599
Zhou, Y., Jin, G., Mi, R., Zhang, J., Zhang, J., Xu, H., et al. (2016). Inhibition of fatty acid synthase suppresses neovascularization via regulating the expression of VEGF-A in glioma. J. Cancer Res. Clin. Oncol. 142, 2447–2459. doi: 10.1007/s00432-016-2249-6
Keywords: glioma, metabolic reprogramming, tumor microenvironment, immune escape, metabolic therapy
Citation: Qiu R, Zhong Y, Li Q, Li Y and Fan H (2021) Metabolic Remodeling in Glioma Immune Microenvironment: Intercellular Interactions Distinct From Peripheral Tumors. Front. Cell Dev. Biol. 9:693215. doi: 10.3389/fcell.2021.693215
Received: 10 April 2021; Accepted: 19 May 2021;
Published: 11 June 2021.
Edited by:
Hongming Miao, Army Medical University, ChinaReviewed by:
Jianmei Wu Leavenworth, University of Alabama at Birmingham, United StatesHiroaki Shime, Nagoya City University, Japan
Copyright © 2021 Qiu, Zhong, Li, Li and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongwei Fan, ZmFuaG9uZ3dlaTE3OEBuam11LmVkdS5jbg==; Yingbin Li, eWluZ2JpbmxpNjVAc2luYS5jb20=
†These authors have contributed equally to this work and share first authorship