 Yuqing Zhang
Yuqing Zhang De Jin
De Jin Xiaomin Kang2
Xiaomin Kang2- 1Endocrinology Department, Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing, China
- 2Endocrinology Department, Guang’anmen Hospital, Beijing University of Chinese Medicine, Beijing, China
- 3Endocrinology Department, Affiliated Hospital to Changchun University of Chinese Medicine, Changchun University of Chinese Medicine, Changchun, China
Diabetic kidney disease (DKD), as the most common complication of diabetes mellitus (DM), is the major cause of end-stage renal disease (ESRD). Renal interstitial fibrosis is a crucial metabolic change in the late stage of DKD, which is always considered to be complex and irreversible. In this review, we discuss the pathological mechanisms of diabetic renal fibrosis and discussed some signaling pathways that are closely related to it, such as the TGF-β, MAPK, Wnt/β-catenin, PI3K/Akt, JAK/STAT, and Notch pathways. The cross-talks among these pathways were then discussed to elucidate the complicated cascade behind the tubulointerstitial fibrosis. Finally, we summarized the new drugs with potential therapeutic effects on renal fibrosis and listed related clinical trials. The purpose of this review is to elucidate the mechanisms and related pathways of renal fibrosis in DKD and to provide novel therapeutic intervention insights for clinical research to delay the progression of renal fibrosis.
Introduction
Diabetic kidney disease (DKD) is the most common complication of diabetes mellitus (DM), which is characterized by glomerular hyperfiltration, progressive albuminuria, and decreased glomerular filtration rate (GFR), and ultimately leads to end-stage renal disease (ESRD) (Alicic et al., 2017). According to the prevalence survey, about 30–50% of ESRD worldwide is caused by DKD (Tuttle et al., 2014). DKD has become the primary cause of ESRD in middle-aged and elderly people in China (Duan et al., 2020). The pathological changes of DKD include nodular or diffuse glomerulosclerosis, tubular inflammation, atrophy, and interstitial fibrosis (Thomas et al., 2015). Among them, renal interstitial fibrosis is a common metabolic change in the late stage of DKD, which is also a factor that promotes the progression of the disease.
Renal fibrosis is the deposition of fibrotic matrix and the formation of scar in response to severe or persistent injury (Humphreys, 2018). Although it is involved in the wound healing process, continued fibrosis can damage tissue structure and organ function, eventually causing renal failure (Liu, 2011). Chronic injury to the kidney promotes a variety of pathological changes, including epithelial–mesenchymal transition (EMT) (Yang and Liu, 2001; Hertig et al., 2008), endothelial–mesenchymal transition (EndoMT) (Li et al., 2009), and activation of fibroblasts and pericytes. EMT is characterized by the loss of intracellular adhesion, such as E-cadherin, and the acquisition of mesenchymal markers, such as αSMA, fibroblast-specific protein 1 (FSP1), fibronectin, collagen, and vimentin (Zhou and Liu, 2016). EndoMT is a special EMT subset that occurs in endothelial cells, which is similar to EMT (Sun et al., 2016). Fibroblasts and pericytes are always regarded as the major origin of myofibroblasts, whose activation predominantly accelerates the irreversible formation of myofibroblasts (Mack and Yanagita, 2015). These pathological processes transform renal cells into myofibroblasts, which exert their profibrotic function by secreting collagen I, III, and IV, fibronectin, and laminin, leading to extracellular matrix (ECM) accumulation and eventually resulting in tubulointerstitial fibrosis (Figure 1).
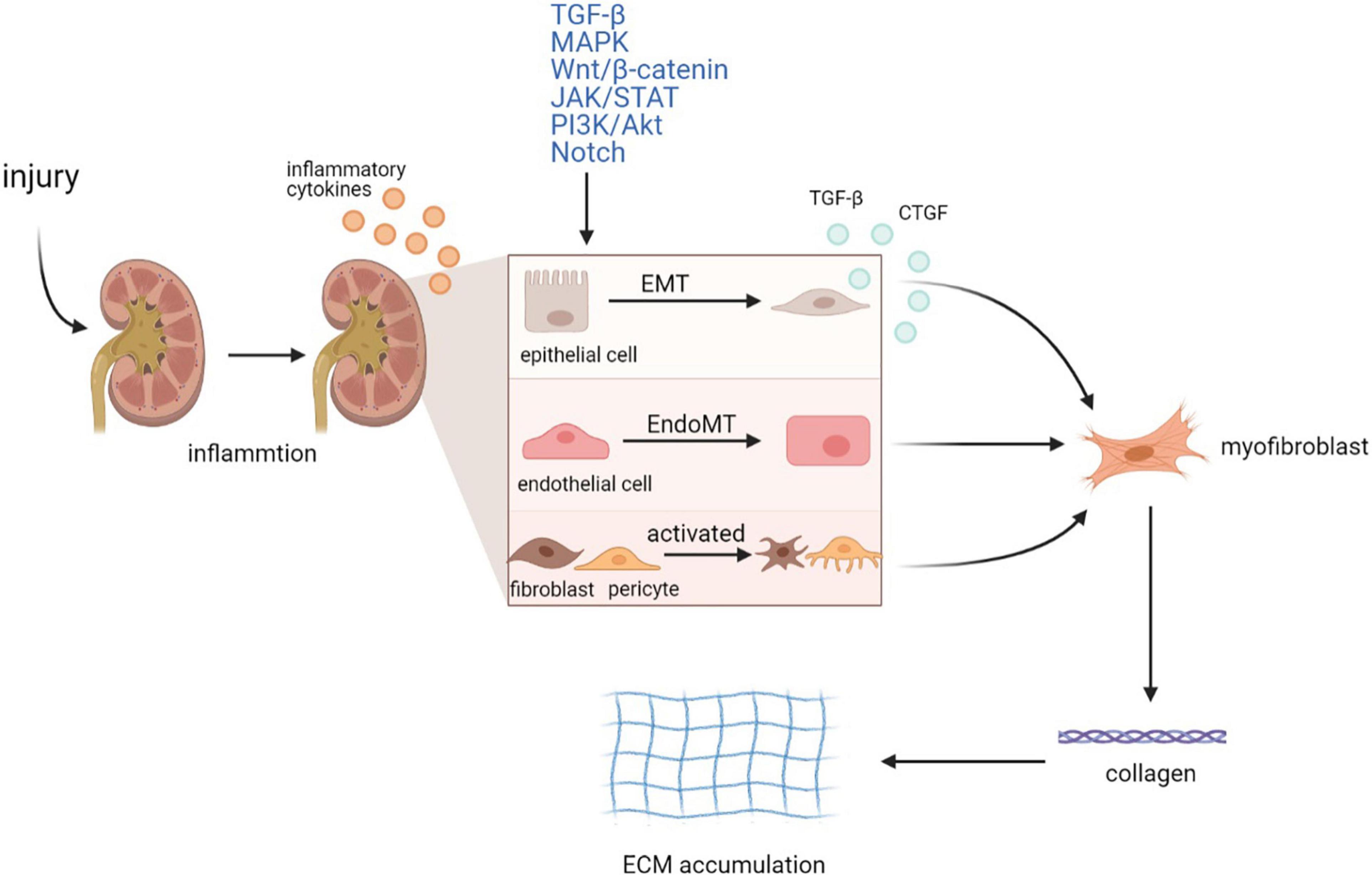
Figure 1. Mechanism of renal fibrosis. Injury to the kidney activates inflammatory response and promotes the secretion of inflammatory cytokines by renal cells. Then, in response to severe or persistent inflammation, renal epithelial cells and endothelial cells undergo phenotypic transitions called EMT and EndoMT, respectively, while fibroblasts and pericytes are activated. Activated intrinsic renal cells also secrete cytokines such as TGF-β and CTGF to further promote the formation of myofibroblasts. Various signaling pathways are involved in these processes, including TGF-β, MAPK, Wnt/β-catenin, PI3K/Akt, JAK/STAT, and Notch pathway. These pathological changes result in the irreversible formation of myofibroblasts, followed by the production of multiple types of collagens and the ECM accumulation, eventually leading to renal tubulointerstitial fibrosis. EMT, epithelial–mesenchymal transition; EndoMT, endothelial–mesenchymal transition.
Renal fibrosis is the major outcome event of nearly all chronic kidney disease and is always considered to be irreversible and remains to be an unsolved clinical conundrum (Francois and Chatziantoniou, 2018). It is well established that many signaling pathways are involved in renal fibrosis through a variety of complex cascades. In this review, we discuss the diagnostic method and the mechanisms of renal fibrosis, especially tubulointerstitial fibrosis, and signaling pathways that are closely related to it, and summarize potential targets as well as new drugs of DKD.
Examination and Quantification Scores of Renal Fibrosis
The native kidney biopsy is the gold standard for the diagnosis of renal interstitial fibrosis, by which the pathological process and the prognosis of patients could be evaluated more accurately (Luciano and Moeckel, 2019). According to the Banff 97 criteria (Racusen et al., 1999), an adequate cortical specimen that contains at least 10 glomeruli and 2 arteries should be obtained by kidney biopsy. Then, staining methods, such as hematoxylin and eosin (HE) stain, periodic acid-Schiff (PAS) stain, and trichrome stain, are performed to improve the identification of the degree of lesion (Farris and Colvin, 2012). Masson trichrome stain is the preferred method for diagnosis of renal interstitial fibrosis in clinical practice because of its simple operation and clear results that can be obtained under a light microscope (Street et al., 2014). Furthermore, with the introduction of immunofluorescence and electron microscopy techniques, the accuracy of qualitative diagnosis and quantitative analysis of kidney biopsy samplings has been improved (Hogan et al., 2016).
The widely used Banff 97 classification proposed by Racusen et al. (1999) divided the lesions into four grades, namely, ci0, ci1, ci2, and ci3, according to the percentage of cortical parenchyma affected. The corresponding percentage of the four levels are 0 to 5, 6 to 25, 26 to 50%, and more than 50%, respectively.
Although kidney biopsy is the only specific diagnostic method for renal fibrosis, non-invasive techniques such as diffusion tensor imaging (DTI) (Nassar et al., 2021) and molecular imaging of ECM (Bülow and Boor, 2019) are valuable in the assessment of renal interstitial fibrosis.
Signaling Pathways Involved in Renal Fibrosis
TGF-β Signaling Pathway
It is well accepted that the TGF-β signaling pathway plays a crucial role in fibrogenesis, especially in renal fibrosis of diabetic kidney disease. TGF-β, a key mediator of fibrosis, exerts its profibrotic effect through the activation of downstream signaling, leading to EMT, EndoMT, and myofibroblast activation, which instigates a loss of adhesion proteins and connexins under high-glucose conditions (Hills et al., 2012). Therefore, ECM accumulates on the cell surface or between cells, which is the key cause of renal fibrosis.
The three isoforms of TGF-β, TGF-β1, TGF-β2, and TGF-β3, all have fibrogenic effects on renal cells and stimulate the production of ECM proteins in renal fibroblasts, renal tubular epithelial cells, and glomerular mesangial cells (Ito et al., 2001). Among them, TGF-β1 is considered to play a major role in fibrogenesis and mediate part of the functions of TGF-β2 and TGF-β3 (Yu et al., 2003). These reports indicate a better therapeutic effect in attenuating renal fibrosis by blocking all three isoforms together. TGF-β1 binds receptor II (T beta RII) to exert its biological functions, and the disruption of T beta RII in renal fibroblasts can reduce the accumulation of ECM and inhibit fibrosis through TGF-β1-induced Smad signaling pathway (Kasuga et al., 2001; Meng et al., 2012a). CaSR, a kind of G protein-coupled receptor, could combine with TβRII to form a CaSR–TβRII complex, which is then translocated from the cell membrane to the cytoplasm. This process leads to the reduction of TβRII on the cell membrane, thereby reducing its binding to its ligand TGFβ1, inhibiting the activation of the TGF-β/Smads signaling pathway and the expression of downstream genes, and finally alleviates the ECM accumulation mediated by TGF-β1 or high glucose (Li et al., 2020a).
The TGF-β signaling pathway could be activated by high-glucose condition (Qi et al., 2007), which significantly increases the expression of TGF-β1 mRNA and induces the synthetic phenotype of mesangial cells (Liu, 2011). The injury of mesangial cells and podocytes caused by diabetic nephropathy can activate the signal transduction cascade of the TGF-β/Smad signaling pathway and stimulate the expression of TGF-1, TSP-1, and TGF-IIR in GEC, thus activating the Smad signaling pathway and leading to increased production of ECM (Kim et al., 2003). Animal studies have also demonstrated that stimulation with TGF-β1 increased the kidney expression of fibrotic genes such as collagen I, collagen IV, and fibronectin in mesangial cells and tubular epithelial cells in UUO mice (Wang et al., 2020a).
On the other hand, treatment with pyrrole-imidazole (PI) polyamides, a transcription inhibitor of TGF-β1, decreased the growth of mesangial cells, which demonstrated the role of TGF-β1 in renal fibrosis (Horikoshi et al., 2020). Treatment with anti-TGF-β antibody (αT), an antibody neutralizing the activity of all three isoforms of TGF-β, reduced the deposition of ECM and alleviated renal interstitial fibrosis (Fukasawa et al., 2004). Furthermore, the application of αT not only prevented early changes in renal histopathology and attenuated the accumulation of ECM with a short-term treatment (Sharma et al., 1996), but also reduced the expression of α1 (IV) collagen and fibronectin in later stages of diabetic nephropathy with chronic administration (Ziyadeh et al., 2000). However, a randomized, double-blind clinical study showed that using a humanized neutralizing monoclonal antibody (TGF-β1mAb) to neutralize active TGF-β1 did not slow the progression of diabetic nephropathy, and the trial was terminated early for lack of efficacy. Although TGF blockers have made some progress in animal experiments, their clinical application is still a conundrum (Voelker et al., 2017).
The Factors That Stimulate the TGF-β Signaling Pathway
In the occurrence and development of diabetic nephropathy, many factors promote the progression of renal fibrosis by stimulating the TGF-β production, such as hyperglycemia, advanced glycation end products (AGEs), reactive oxygen species (ROS), and renin–angiotensin II–aldosterone system (RAAS).
Numerous studies have indicated that hyperglycemia is a key regulatory factor that mediates the TGF-β secretion in both in vitro and in vivo studies (Zhu et al., 2005). High glucose stimulated the expression of TGF-β1 mRNA and increased total TGF-β1 protein production in murine mesangial cells (MMCs) (Hoffman et al., 1998) and human proximal tubular cells (HPTC) (Phillips et al., 1995) in culture. Moreover, blood glucose fluctuation (BGF) plays a significant role in renal TGF-β1 gene expression; even the level of the blood glucose is in the normal range (Qi et al., 2007). Another experiment on animals demonstrated that BGF treatment markedly upregulates TGF-β1 expression, increasing the synthesis of type I collagen and inhibiting collagen degradation. Furthermore, BGF appeared to have a more harmful effect in fibrogenesis of renal cells in diabetic mice than in mice with consistent hyperglycemia (Cheng et al., 2013).
Except for the effects brought by blood glucose, AGEs also upregulated the levels of TGF-β mRNAs partly by the increasing production of ROS and subsequently induced mesangial cell hypertrophy and fibrosis (Yamagishi et al., 2003). On the contrary, the AGE cross-link breaker could reduce EMT in diabetic rats by decreasing tubular AGE and TGF-β expression (Oldfield et al., 2001). Both in vivo and in vitro studies demonstrated that AGEs induced the deposition of matrix protein via induction of TGF-β and Smad2/3 activation in mesangial cells, which was suppressed by the inhibitor of AGE receptor (Lee et al., 2018).
In addition, the results of the previous study suggested that the activated local RAAS contributed to high glucose-induced EMT by activating AT receptors and promoting angiotensin II production. Angiotensin II and activated AT receptors could stimulate TGF-β synthesis in the kidney and upregulated TGF-β receptors. Research also found that the AT receptor antagonist losartan partially inhibited the increases in TGF-β in rat kidney proximal tubular epithelial cell line NRK-52E (Zhou et al., 2010a), which indicated that RAAS played a pivotal role in the TGF-β signaling pathway. Currently, there is no direct way to inhibit the TGF-β system without side effects. Therefore, angiotensin-converting enzyme (ACE) inhibitors and angiotensin type 1 (AT1) receptor blockers are widely used in diabetic nephropathy at present, which partly interfere with TGF-β expression mediated by ANG II (Wolf, 2006). Although Ang II blockade alone reduces renal fibrosis, simultaneous blockade of Ang II and TGF-β takes a better effect in ameliorating renal fibrosis, suggesting that drug combinations will be a future therapeutic direction and novel measures that block the TGF signaling pathway still need further development (Yu et al., 2004).
Downstream Targets of TGF-β Signaling
TGF-β signaling exerts its biological functions through both canonical (Smad-dependent) and non-canonical (Smad-independent) pathways (Derynck and Zhang, 2003). In the Smad-dependent pathway, the active TGF-β binds to its receptor type II and type I serine/threonine kinase receptors and thus phosphorylates Smad2 and Smad3, and oligomeric complexes are formed with Smad4, which translocates into the nucleus, regulating the transcription of target genes. Smad7, one of the inhibitory Smads (I-Smads), exerts its autoinhibitory function by inhibiting the signals from the serine/threonine kinase receptors (Miyazono et al., 2000).
In this process, Smad3 plays a major role in the development of renal fibrosis (Ju et al., 2006). It has provided evidence that the TGF-β/Smad3 signaling pathway is highly activated in renal fibrogenesis. Smad3 exhibited an obvious deposition of collagen, contributing to the development of renal fibrosis in db/db mice, whereas this process could be inhibited by Smad3 knockout (Xu et al., 2020). Further research suggested that Smad3 increased TGFβ1-induced connective tissue growth factor (CTGF) expression and decreased E-cadherin expression. It also cooperated with Smad2 to increase the expression of alpha-SMA (Phanish et al., 2006). HIPK2, a regulator of the TGF-β1/Smad3 pathway, inhibited Smad3 phosphorylation, leading to the mitigation of renal fibrosis (Liu et al., 2017), which indicated that Smad3 is a potential therapeutic target for T2DN. Although Smad2 and Smad3 are both generally considered to be crucial downstream mediators of TGF-β1, the pathological function of Smad2 and Smad3 may be different in terms of collagen matrix deposition and interstitial fibrosis. It has been demonstrated that the deletion of Smad2 had an effect in enhancing TGF-β/Smad3 signaling, which promoted collagen production and fibrosis. On the contrary, the overexpression of Smad2 attenuated this process, indicating a protective mechanism of Smad2 in TGF-mediated fibrosis (Meng et al., 2010). It corresponds to the previous results that Smad2 knockout promoted EMT progression (Ju et al., 2006; Runyan et al., 2009). Smad4, the same as Smad3, promotes fibrogenesis, which was proven by the observation of reduced collagen I expression and inhibited renal fibrosis in the Smad4 knock-out mouse model (Meng et al., 2012b). Although the downstream Smad signaling primarily promotes TGF-β-induced renal fibrosis, Smad7, an inhibitory Smad, negatively regulates fibrotic cytokines expression. The anti-fibrosis mechanism of Smad7 has been demonstrated to be a blockade of Smad2/3 activation via regulating the expression of microRNAs (Lan, 2008; Chung et al., 2013). In general, Smads play a crucial role in TGF-β-mediated renal fibrosis as a downstream signaling pathway (Figure 2), which provides novel and potential therapeutic targets for clinical treatment and drug discovery of renal fibrosis.
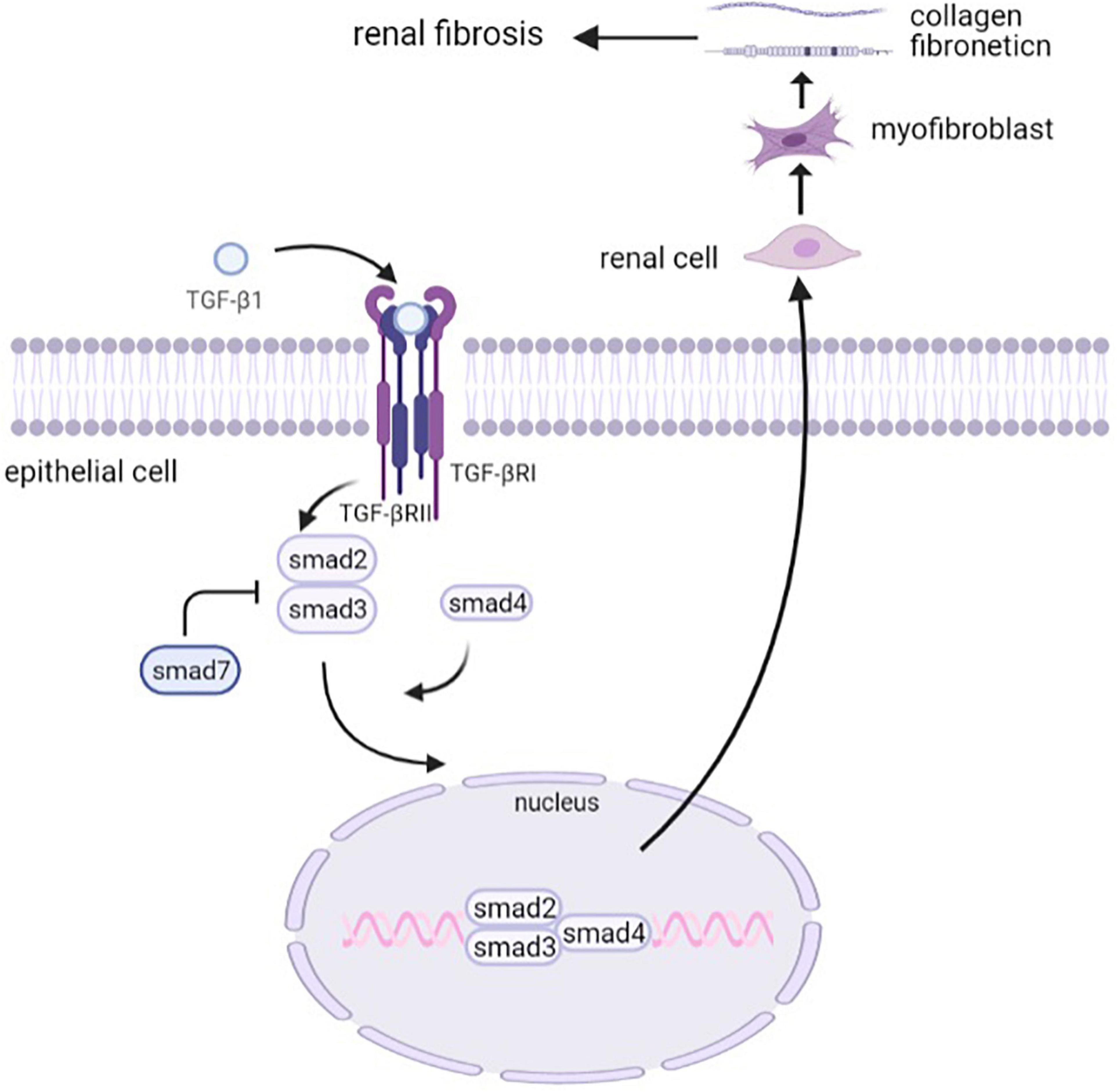
Figure 2. The canonical TGF-β signaling pathway. Activated TGF-β binds to its type II receptor and type I serine/threonine kinase receptors and thus phosphorylates Smad2 and Smad3. Smad7 is one of the inhibitory Smads that inhibit the signals from the serine/threonine kinase receptors. Smad2/3 then forms an oligomeric complex with Smad4, which translocates into the nucleus to regulate the transcription of target genes. The activation of the TGF-β signaling pathway drives renal cells into myofibroblasts, which secrete collagens and fibronectins to promote renal fibrosis.
In addition to the Smad-dependent pathway, multiple non-Smad downstream signaling pathways are involved in TGF-β-mediated renal tubular fibrosis, such as the mitogen-activated protein kinase (MAPK) pathway, Wnt/β-catenin pathway, extracellular signal-regulated kinase (ERK)1/2, c-Jun N-terminal kinase (JNK), and phosphatidylinositol 3-kinase (PI3K) (Sutariya et al., 2016). These pathways cross-talk with TGF-β signaling to exert complex biological functions and together promote the progression of renal interstitial fibrosis.
MAPK Pathway
Mitogen-activated protein kinases consist of a group of serine/threonine protein kinases, controlling various aspects of cellular functions, and can be activated in high-glucose conditions (Haneda et al., 1997). The main subgroups of MAPK are P38MAPK, ERK, and JNK, which together mediate signal transduction in fibrosis (Davis, 1994).
It is reported that the overexpression of EphA1, a modulator of fibrosis, decreased the phosphorylation of ERK1/2 and JNK in the kidney and finally alleviated renal fibrosis of diabetic nephropathy mice (Li et al., 2017c). Zhang et al. (2015) also reported that ERK1/2 MAPK signaling was involved in diabetic nephropathy to mediate fibrogenesis by regulating mesangial cell proliferation and ECM accumulation, which suggested the crucial biological function of the ERK MAPK pathway in tubulointerstitial fibrosis. Another research found that blockade of p38 MAPK exerted a beneficial effect via inhibiting the production of phosphorylated p38 MAPK-positive cells in the early stage of fibrosis in the treated kidney (Wada et al., 2006). Moreover, the single blockade of p38 MAPK was also demonstrated to be effective to alleviate renal fibrosis in the established fibrotic UUO model (Nishida et al., 2008). At the same time, administration of JNK inhibitor, which blocked all JNK isoforms, obviously delayed fibrosis progression via inhibiting accumulation of collagen IV and α-SMA + myofibroblast in an animal model (Ma et al., 2007).
Extracellular signal-regulated kinase, p38 MAPK, and JNK pathways play different but complementary roles in ECM deposition and fibrogenesis. P38 MAPK is more important in regulating Matrix metalloproteinase 1 (MMP1), while the ERK MAPK pathway mainly moderates the production of type I collagen (COL1) in high-glucose conditions (Okano et al., 2010). Moreover, P38 MAPK and JNK, but not ERK signaling, induced thrombospondin-1 (TSP-1) production, which is known to be an activator of the latent TGF-β1 complex (Naito et al., 2004). JNK markedly contributes to TGF-β1-induced CTGF mRNA expression, whereas p38 MAPK and ERK pathways take little effect (Utsugi et al., 2003).
In total, although targeting different aspects of the fibrotic process, the three subgroups of MAPK all contribute to pathological changes of renal fibrosis. Therefore, targeting the MAPK pathway as a novel and effective therapy could be a potential research direction in the future for renal fibrosis of diabetic kidney disease.
Wnt/β-Catenin Signaling Pathway
It is well established that the Wnt signaling pathway plays multiple roles in the injury and repair of renal cells through the mediation of inflammation, fibrosis, angiogenesis, and insulin secretion (Zhou et al., 2010b; Xiao et al., 2013). The canonical pathway of Wnt is β-catenin (Harris and Peifer, 2005), through which Wnt signaling induces the expression of cyclin and glucokinase, contributing to cell proliferation and glucose sensing (Schinner et al., 2009).
Wnt signaling was activated by high glucose both in STZ-induced diabetic rats and in db/db mouse models with an upregulation of β-catenin and WNT protein level, which could be attenuated by lowering the blood glucose levels with the administration of insulin. It was found in an in vitro study that hyperglycemia and oxidative stress were both responsible for the activation of the Wnt pathway in renal tubular epithelial cells. The blockade of the Wnt pathway with the treatment of LDL-receptor-related protein inhibitor was demonstrated to ameliorate renal fibrosis (Zhou et al., 2012). Kameswaran et al. also observed a reduction in fibronectin and α-SMA with the treatment of inhibitor of Wnt/β-catenin signaling transduction (Surendran et al., 2005).
Further studies illuminated the detailed mechanism of Wnt/β-catenin in promoting renal fibrosis. When the signaling is “on,” the Wnt binds to the frizzled receptor, which works together with the low-density lipoprotein receptor-related proteins (LRP)5 and LRP6 co-receptors to inhibit the ubiquitination of destruction complex (Piersma et al., 2015). Therefore, the degradation activity of β-catenin by β-TrCP is limited, causing the accumulation of β-catenin (Bilic et al., 2007). Finally, β-catenin translocates from the cytoplasm to the nucleus to regulate the transcription of target genes (Kawakami et al., 2013), such as Snail and Twist (Liu, 2010), thereby promoting the EMT process. For example, Snail was involved in EMT by downregulating the expression of adhesion protein E-cadherin, which was further demonstrated by an increasing level of E-cadherin expression with the administration of Snail inhibitor (Batlle et al., 2000; Cano et al., 2000). The twist is also associated with EMT and subsequent renal fibrosis by inducing a mesenchymal phenotype in UUO mice, whereas the knockdown of Twist by short interfering RNA (siRNA) markedly attenuated EMT and fibrosis (Kida et al., 2007; Sun et al., 2009).
PI3K/Akt Signaling Pathway
Phosphoinositide 3-kinase (PI3K) is an intracellular phosphatidylinositol kinase, also having serine/threonine (Ser/Thr) kinase activity (Fruman et al., 2017). Akt is a protein kinase, also called protein kinase B (PKB), because of its high homology with protein kinase A (PKA) and protein kinase C (PKC). PI3K is firstly activated by tyrosine kinase to transform phosphatidylinositol 4,5-biphosphate (PIP2) into phosphatidylinositol 3,4,5-triphosphate (PIP3) (Huang et al., 2018), which promotes the accumulation of Akt at the plasma membrane. Then, Thr308 of Akt is phosphorylated with the assistance of 3-phosphoinositide-dependent protein kinase 1 (PDK1). Finally, activated Akt can exert its biological function in multiple physiological processes, including cellular proliferation, differentiation, apoptosis, migration, and metabolism (Xu et al., 2015; Zeng et al., 2019).
It has been observed that PI3K/Akt signaling is associated with ECM accumulation and EMT. Therefore, it has a potential involvement in renal interstitial fibrosis (Xie et al., 2015; Shin et al., 2019). Chen et al. (2016) found that the activation of PI3K/Akt participated in renal tubulointerstitial fibrosis induced by HG condition. Furthermore, Li et al. (2017a) found that knocking down the negative regulator SHIP in human renal tubular epithelial cells (HK2 cells) led to the activation of the PI3K/Akt pathway and subsequently upregulated the expression of TGF-β1, α-SMA, and collagen type 3. Additionally, Zhu et al. also found that high-glucose condition increased the levels of phospho-Akt (Ser 473), phospho-Akt (Thr 308), CTGF, and α-SMA in both STZ-induced diabetic mice and in vitro human renal tubular epithelial cells. However, the upregulation of Ser 473 and Thr 308 expression was inhibited by LY294002, an inhibitor of the PI3K/Akt signal transduction pathway, followed by reduced CTGF, fibronectin, and collagen production (Zhu et al., 2016).
JAK/STAT Signaling Pathway
The Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway is involved in the regulation of cell proliferation, differentiation, apoptosis, and other important biological processes by activating many cytokines, growth factors, and hormones (Bhattacharjee et al., 2016). It has been established that the JAK/STAT signaling pathway is associated with renal fibrosis in both human and mouse diabetic kidney disease (Brosius, 2008). The signaling cascade of the JAK/STAT signaling pathway includes JAK activation, tyrosine phosphorylation, and STAT recruiting (Morris et al., 2018). Activation of the JAK/STAT pathway and related proteins contributed to the proliferation of renal fibroblasts and accelerated the development of renal interstitial fibrosis in rat NRK-49F cells. However, this process could be blocked by the JAK inhibitor AG490 (Sun et al., 2019). Similarly, in DKD patients, enhanced expression of JAK/STAT mRNA was observed, with the pathological changes of fibrosis. There is a possible link between JAK2, a pivotal upstream regulator of the JAK pathway, and tubulointerstitial fibrosis, which was suggested by the temporal association of upregulated JAK2 level and evolution of human DKD (Berthier et al., 2009). Moreover, a phase 2 clinical trial showed that treatment with Baricitinib, an oral selective inhibitor of JAK1 and JAK2, resulted in amelioration of albuminuria, indicating a renal protective effect of JAK signaling blockade (Tuttle et al., 2018).
A further study observed that high glucose exposure directly induced the tyrosine phosphorylation of JAK2, STAT1, STAT3, and STAT5. At the same time, the activation of JAK2 promoted the expression of downstream targets STAT1 and STAT3, but not STAT5 (Wang et al., 2002), showing the signal transduction of JAK/STAT in HG conditions. Another study showed that treatment with probiotic Lactobacillus markedly inhibited the phosphorylation of JAK2 and STAT1 in the renal cortex in STZ-induced diabetic mice and subsequently decreased the expression of α-SMA and fibronectin protein (Lu et al., 2010). In addition, the administration of selective STAT3 inhibitor reduced the profibrotic gene expression of collagen IV, TGF-β1, VEGF, and ACE in tubular epithelial cells in mouse kidneys (Zheng et al., 2019). Zhou et al. (2014) also found that the expression of JAK2, STAT3, and STAT5 markedly increased in STZ-induced mouse kidneys, whereas it decreased after treatment of suppressor of cytokine signaling (SOCS) 2, along with the attenuation of renal fibrosis formation. These results provide evidence that blockade of JAK/STAT signaling reduces collagen accumulation and profibrotic cytokine expression, resulting in the inhibition of HG-induced fibrotic response in STZ-induced mice.
Moreover, the JAK/STAT pathway has a possible involvement in AGE-induced cell proliferation and collagen production in NRK-49F cells (Huang et al., 1999; Lee et al., 2005), which account for its profibrotic function from another aspect. Huang et al. (2001) found that AGE-induced collagen expression was suppressed by either JAK2 inhibitor or STAT1 and STAT3 decoy oligodeoxynucleotides (ODNs) in NRK-49F cells and thus concluded that JAK2-STAT1/STAT3 was responsible for the induction of collagen and promoted renal fibrogenesis.
Notch Signaling Pathway
Notch signaling consists of four transmembrane receptors (Notch1–Notch4), two Jagged family ligands (JAG1 and JAG2), and three delta-like ligands (DLL1, DLL3, and DLL4) in the mammalian system (Hu et al., 2015). It can be reactivated in pathological conditions and involved in a variety of processes, including cellular proliferation, apoptosis, and EMT (Kopan and Ilagan, 2009). Upon the combination of the Notch signaling pathway ligand and the receptor, Notch transforms to the activated form Notch intracellular domain (NICD), which enters the nucleus to regulate the expression of downstream targets and trigger ECM and EMT, and ultimately results in renal fibrogenesis in diabetic kidney disease (Bonegio and Susztak, 2012).
The activation of the Notch signaling pathway was observed both in tubular interstitial fibrosis (TIF) patients and in TIF mouse models. Moreover, the Notch pathway was proved to be both necessary and sufficient for the occurrence and development of TIF. Tian et al. (2018) demonstrated reno-protective functions of Gliquidone, which took effect by blocking the Notch/Snail signaling pathway to delay renal fibrosis. Additionally, with the administration of γ-secretase inhibitor, a pharmacological inhibitor of Notch activation, the cascade reaction of the Notch signaling pathway was blocked (Nishad et al., 2019). The blockade of Notch resulted in the amelioration of renal fibrosis in the folic acid-induced (FA-induced) TIF mouse model, as reflected by the decreased level of fibronectin, collagen, and vimentin (Bielesz et al., 2010). Jing et al. (2020) also found that administration of DAPT, an inhibitor of the Notch pathway, reversed HG-induced protein expression of Jagged1, PGC-1α, and Drpl in renal tubular epithelial cells. It indicated that Notch signaling pathway might accelerate renal fibrosis by regulating oxidative damage and mitochondrial dysfunction.
Among the downstream genes of the Notch signaling pathway, the Snail plays the most important role in inducing fibrosis. It acts as a bridge for inducing EMT in tubular epithelial cells and activating the Notch signaling pathway (Grande et al., 2015). The expression of the Snail promoter is stimulated by the Notch pathway in a dose-dependent manner (Sahlgren et al., 2008), and its overproduction directly leads to the decreased expression of E-cadherin and increased production of alpha-SMA, MMP-2, and MMP-9 (Saad et al., 2010). Yang et al. (2017) found that suppressing the Notch/Snail axis activation with the treatment of berberine (BBR) increased E-cadherin as well as decreased α-SMA level, protecting renal tubular cells against EMT and renal fibrosis both in vivo and in vitro.
Cross-Talks Among Signaling Pathways
In the occurrence and development of renal fibrosis, these pathways above not only play an independent promoting role but also have complex interactions with each other. The complicated cross-talks among them further illuminate the mechanism of renal tubulointerstitial fibrosis in diabetic kidney disease. Cross-talks involving the TGF-β signaling pathway have been highlighted because the TGF-β pathway plays a predominant role in the fibrogenic process and interacts with nearly all fibrosis-related pathways.
Cross-Talk Between MAPK and TGF-β Pathways
In terms of the cross-talk between TGF-β and MAPK, Beek et al. (Chin et al., 2001) found that treatment with exogenous TGF-β1 in mesangial cells markedly induced phosphorylation of p38 MAPK and ERK1/ERK2. The activation of the former one could be blocked by an inhibitor of the TGF-β type I receptor. Blocking of the latter one markedly inhibited the phosphorylation of ERK1/2 induced by high glucose and at the same time attenuated the level of TGF-β1 expression and resulted in alleviation of EndMT (Yu et al., 2017). Utsugi et al. (2003) also found that levels of phosphorylated threonine and tyrosine of JNK and p38MAPK increased by adding TGF-β1 in human lung fibroblasts. Therefore, these findings demonstrated that TGF-β1 has stimulating effects on all the main subgroups of MAPK. Further study found that the ERK/p38 MAPK pathway activity played a critical role in maximal induction of Smad activity by TGF-β1 (Li et al., 2004). The blockade of ERK inhibits R-Smad-dependent transcriptional activation, leading to a reduction in TGF-β-induced transcriptional activity (Hayashida et al., 2003). Also, there is a positive feedback loop between JNK and TGF-β/Smad (Grynberg et al., 2017). The activation of the TGF-β/Smad pathway stimulates JNK in the epithelial cell, while JNK directly induces the phosphorylation of the linker of Smad3, thereby promoting the transcriptional activity of Smad (van der Velden et al., 2011). In addition, extracellular MAPK activation induced by factors such as TNF-α, angiotensin II, and IL-1 can stimulate latent TGF-β in mesangial cells via TSP-1 (Naito et al., 2004) or activator protein 1 (AP-1) (Lee et al., 2006; Rui et al., 2012). In summary, there is a positive cross-talk between MAPK and TGF-β/Smad signaling pathways, which causes synergistic enhancement of TGF-dependent responses.
Cross-Talk Between Wnt and TGF-β Pathways
TGF-β/Smad and Wnt/β-catenin pathways were found to promote renal fibrosis concertedly or independently. The TGF-β pathway could activate the canonical Wnt pathway mainly by downregulating the level of Wnt antagonist Dickkopf-1 (DKK1) (Akhmetshina et al., 2012). The decrease of DKK1 upregulated Wnt1-dependent β-catenin expression (Wang et al., 2011). In addition to the Wnt1-dependent pathway, TGF-β repressed WT1, a negative regulator of β-catenin, to promote the secretion of β-catenin and its target genes, such as MMP-9, Snail1, PAI-1, and Fsp1 (Chang et al., 2008; Kim et al., 2010). TGF-β also promotes β-catenin dephosphorylation and activation by inactivating glycogen synthase kinase-3β (GSK-3β) (Zeng et al., 2005; Dai et al., 2011). Furthermore, the cross-talk between these pathways can be partly indicated by the binding of β-catenin with Smad3 in renal tubular epithelial cells, which results in the progression of EMT, suggesting the profibrotic effects that β-catenin brings for TGF-β1 (Tian et al., 2013). In addition, Wnt signaling pathway components, especially Wnt11, are regulated by a Smad3-dependent mechanism, thus promoting the activation of mesenchymal marker genes, such as Snail1, Zeb1, Pai1, and SMA. It finally leads to pathological changes, including fibroblast proliferation and ECM deposition (Zhang et al., 2012).
The fact that pharmacological blockade of either TGF-β or Wnt would be sufficient to inhibit the other one further demonstrated the synergistic effect of TGF-β and Wnt/β-catenin. Stellor et al. found that deleting TβRII in proximal tubule epithelial cells inhibited the expression of TGF-β and, at the same time, blocked the β-catenin signaling (Nlandu-Khodo et al., 2017). In return, Hoi et al. (2020) identified that the WNT/β-catenin inhibitor, ICG-001, as well as its derivatives blocked TGF-β-induced phosphorylation of Smad2/3, thus ameliorating renal fibrosis triggered both by TGF-β and by Wnt pathways.
Cross-Talk Between PI3K/Akt and TGF-β Pathways
Accumulating evidence has revealed that the PI3K/Akt pathway has a close relationship with TGF-β signaling in promoting ECM deposition and renal fibrosis. The PI3K/Akt pathway was found to be activated in renal tubular cells exposed to high-glucose condition, followed by increased expression of TGF-β1 and production of ECM (Hao et al., 2011). Inhibition of the PI3K/Akt pathway by the chemical inhibitor LY294002 or a small hairpin RNA vector downregulated TGF-β1 production and indirectly alleviated the accumulation of ECM and collagen (Wu et al., 2007). Chen et al. also found that with the treatment of carboxyl-terminal modulator protein (CTMP), an endogenous inhibitor of Akt (Chae et al., 2005), the expression of TGF-β and α-SMA was markedly reduced in cultured renal tubular cells (Chen et al., 2016). On the other hand, PI3K/Akt serves as a pivotal downstream regulator of TGF-β1, whose activation markedly promotes TGF-β1-induced EMT and collagen production (Bakin et al., 2000; Runyan et al., 2004). Kattla et al. (2008) also found that administration of TGF-β1 led to activation of PI3K and Akt, which could be demonstrated by increased phosphorylation levels of Ser473 and GSK-3β. In terms of the mechanism by which TGF-β activates PI3K/Akt, researchers have different opinions. Some researchers hold the view that TGF-β directly activates PI3K and subsequently stimulates Akt production (Higaki and Shimokado, 1999), while others consider TGF-β-induced activation of PI3K as p38MAPK dependent (Horowitz et al., 2004).
Cross-Talk Between JAK/STAT and TGF-β Pathways
In addition to the well-known Smad family, TGF-β also activates JAK1, STAT1, STAT3, and STAT5 to regulate the occurrence of fibrosis (Dong et al., 2016). In return, STAT3 can enhance fibrosis partly by stimulating TGF-β expression (Ogata et al., 2006). Researchers observed that the TGF-β-induced phosphorylation of STAT3 was dependent on the binding of JAK to TβRI in the early stage and was mediated by Smad3 and TβRI kinase activity in the late phase (Itoh et al., 2018; Wiegertjes et al., 2019). It was verified that blockade of JAK2 by either JAK2 antisense or specific inhibitor AG-490 inhibited TGF-β protein expression and fibronectin production in glomerular mesangial cells. Also, depletion of STAT1 by corresponding antisense, but not STAT3, reduced HG-induced TGF-β expression (Wang et al., 2002), suggesting that JAK2-STAT1 signaling has a great influence on the TGF-β pathway.
Cross-Talk Between Notch and TGF-β Pathways
Studies have shown that the Notch pathway is mediated by the TGF-β pathway in renal fibrosis, and these two signaling pathways play a mutual promoting role both in vivo and in vitro. On the one hand, the TGF-β pathway was reported to upregulate the ligand of Notch, such as jagged1 (Liu et al., 2013) and Hey1 (Zavadil et al., 2004) in kidney cells. Moreover, Wang et al. (2017) found that the TGF-β inhibitor reduced the expression of Notch and its target genes, including Notch1, Hes1, and Hes5, in the rats with tissue fibrosis, demonstrating a stimulating function of the TGF-β pathway in Notch signal transduction. On the other hand, a previous study reported that treatment with (−)-epigallocatechin gallate (EGCG) inhibited the Notch pathway in kidney cells, which is mainly through the inhibition of TGFβRII and Smad3 (Zhu et al., 2020). Xiao et al. (2014) also found that the administration of the Notch inhibitor dibenzazepine (DBZ) inhibited the activation of the Notch pathway, as well as the expression of TGF-β and the phosphorylation of Smad2 and Smad3, which markedly ameliorated the severity of renal fibrosis. It was also observed that the inhibitory effect of sh-Notch1 or GSI on the Notch pathway increased the expression and activity of MMP-2 and MMP-9, reduced the level of TGF-β1, and inhibited the expression of type IV collagen and laminin in mouse podocytes (Yao et al., 2015).
Furthermore, studies observed a direct link between TGF-β and Notch signaling pathways in renal fibrosis. NICD, an active form of Notch that is closely related to the extent of tubulointerstitial fibrosis (Murea et al., 2010), was found to interact directly with Smad3, which was enhanced by TGF-β administration (Klüppel and Wrana, 2005). In addition, Blokzijl et al. (2003) discovered that Hes-1, a confirmed Notch target gene, also acts as a direct downstream gene of the TGF-β pathway. The activation of Hes-1 by either the TGF-β or Notch pathway contributes to the fibrogenesis in kidney cells (Zavadil et al., 2004). In summary, these all indicate that Notch signaling and TGF-β signaling have a functional synergism in regulating renal fibrosis.
Cross-Talk Between Notch and Wnt Pathways
It has been reported that the Wnt pathway has a positive cross-talk with the Notch pathway (Bertrand et al., 2012; Perkins et al., 2016). Estrach et al. (2006) concluded that JAG1, a known ligand of the Notch pathway, also acts as a β-catenin target gene that is stimulated by the activation of Wnt/β-catenin signaling. To further verify it, Chen et al. (2010) inhibited the Wnt signaling and observed a reduction in JAG1 expression. Mödder et al. (2011) also found that Wnt10b could promote the activation of Wnt and Notch signaling, indicating that Wnt/β-catenin signaling performed as an upstream mediator of the Notch pathway.
In conclusion, there is a close connection between the TGF-β pathway and other pro-fibrosis pathways, and the mechanism of fibrosis progression with TGF-β as the core has been established (Figure 3).
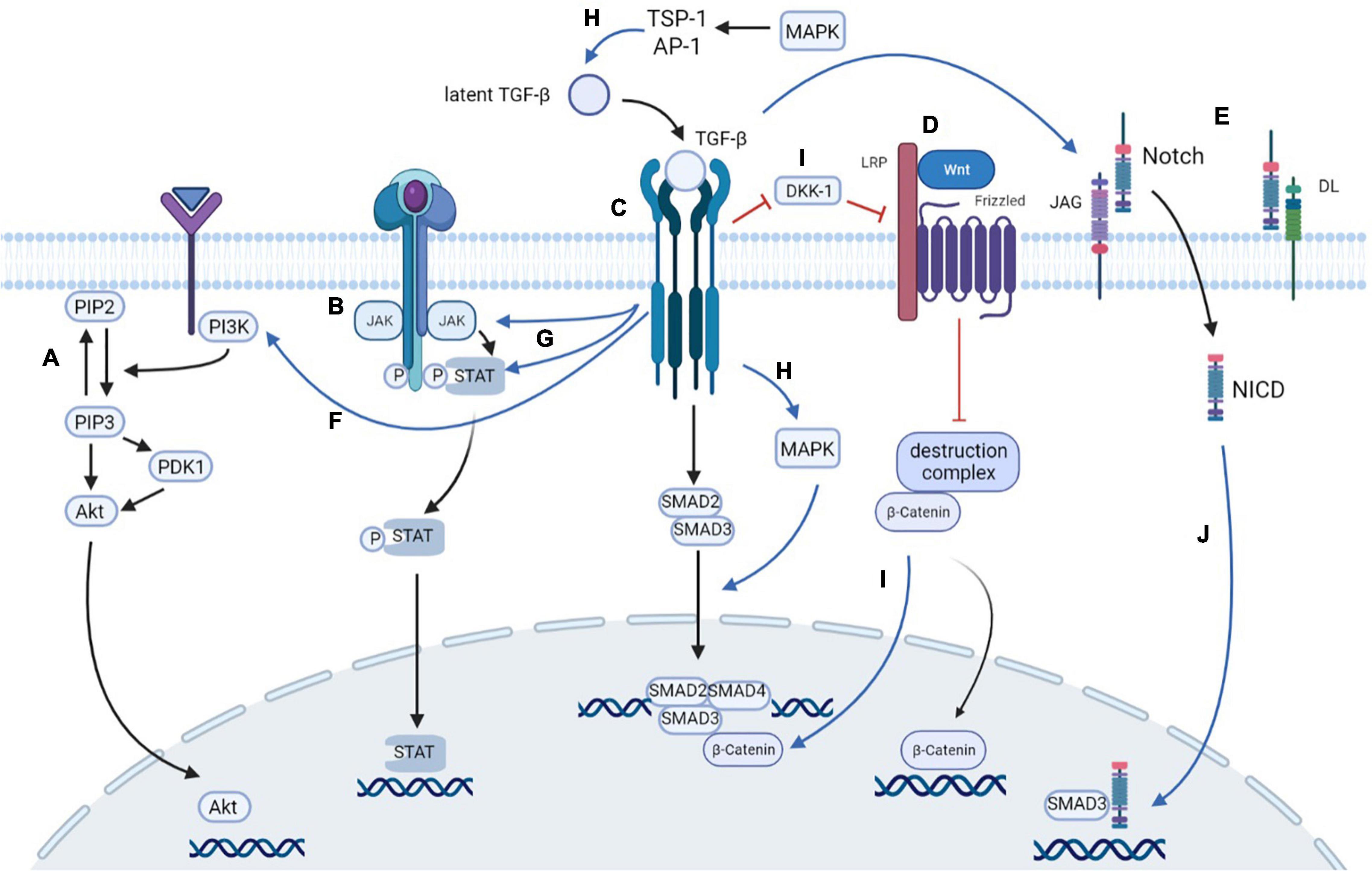
Figure 3. Cross-talks among signaling pathways. (A) PI3K/Akt signaling pathway: PI3K is activated to transform PIP2 into PIP3, which promotes the accumulation of Akt with the assistance of PDK1 at the plasma membrane. Activated Akt then translocates into nucleus to exert its biological function. (B) JAK/STAT signaling pathway: The signaling cascade of JAK/STAT signaling pathway including JAK activation, tyrosine phosphorylation, and STAT recruiting. Then, the STAT is transported to nucleus to regulate gene transcription. (C) TGF-β/Smad signaling pathway: Activated TGF-β binds to its receptors and thus phosphorylates Smad2 and Smad3. Smad2/3 then forms an oligomeric complex with Smad4, which translocates into the nucleus to regulate the transcription of target genes. (D) Wnt/β-catenin signaling pathway: The Wnt binds to the frizzled receptor, which works together with the LRP5 and LRP6 co-receptors to inhibit the ubiquitination of destruction complex. Therefore, the degradation activity of β-catenin is limited, causing the accumulation of β-catenin, which translocates from the cytoplasm to the nucleus to regulate the transcription. (E) Notch signaling pathway: Upon the combination of the receptor Notch, and the ligand JAG and DL, Notch transforms to the activated form NICD, which enters the nucleus to regulate the expression of downstream targets. (F) TGF-β directly activates PI3K and subsequently stimulates Akt production. (G) TGF-β can activate both JAK and STAT, and the activation of JAK subsequently stimulates the phosphorylation of STAT3. (H) The activation of the TGF-β/Smad pathway stimulates MAPK, while MAPK directly induces the phosphorylation of the linker of Smad3, thereby promoting the transcriptional activity of Smad. Extracellular MAPK activation can stimulate latent TGF-β in mesangial cells via TSP-1 or AP-1. (I) TGF-β pathway activates Wnt pathway by downregulating the level of Wnt antagonist DKK1. β-catenin can bind with Smad3, promoting the transcription of Smads. (J) TGF-β pathway upregulates the ligand of Notch, such as JAG, to transform Notch to NICD. NICD can interact directly with Smad3, which is enhanced by TGF-β administration. PI3K, phosphoinositide 3-kinase; PIP2, phosphatidylinositol 4,5-biphosphate; PIP3, phosphatidylinositol 3,4,5-triphosphate; PDK1, 3-phosphoinositide-dependent protein kinase 1; JAK, Janus kinase; STAT, signal transducer and activator of transcription; LRP, low-density lipoprotein receptor-related proteins; NICD, notch intracellular domain; TSP-1, thrombospondin-1; AP-1, activator protein 1; DKK-1, Dickkopf-1.
New Therapies for Renal Fibrosis
New Drugs Designed for Renal Fibrosis
Roxadustat, a new drug designed for renal anemia (Chen et al., 2019b), has been shown to inhibit hypoxia-inducible factor (HIF) and alleviate the progression of renal fibrosis (Packer, 2020). Li et al. (2020b) have implemented animal research to figure out that Roxadustat may retard the progression of fibrosis by regulating Akt/GSK-3β-dependent Nrf2 activation. Li et al. designed a randomized, multicenter, and active-controlled study (NCT02652806) of the efficacy of Roxadustat in anemia treatment in CKD patients (Chen et al., 2019a), but there is still a lack of clinical evidence to support this antifibrotic effect of Roxadustat.
Paricalcitol (PC) is a vitamin D analog that retains similar vitamin D bioactivity but has fewer side effects (Sprague et al., 2003; Fryer et al., 2007). Tan et al. (2009) found that Paricalcitol inhibited the expression of fibronectin, collagen, vimentin, as well as Snail1 in UUO mice, showing a remarkable amelioration in fibrogenesis. Nolan et al. (2015) observed that PC affected the progression of renal fibrosis via targeting the TGF-β1/Smad2 pathway, Notch/Jagged1 pathway, and PI3K pathway in cultured human renal epithelial cells. In comparison, Chung et al. (2017) figured out that PC might also alleviate renal fibrosis through the MAPK signaling pathway. In addition, PC shows a synergistic effect on the blocking of RAAS by ACEI (Martínez-Arias et al., 2021), which possibly participates in the mitigation of EMT and renal fibrosis. A randomized clinical trial (PALIFE NCT01820078) has been designed to explore the effect of Paricalcitol on fibrosis on chronic renal diseases, but the trial has been terminated.
It has been well established that overactivation of the mineralocorticoid receptor (MR) results in renal fibrosis and is closely linked to the progression of renal injury in DKD (Shrestha et al., 2019; Patel et al., 2020). Therefore, new drugs targeting MR are considered as potential therapies that slow the progression of renal fibrosis. Finerenone is a novel, non-steroidal, selective MR antagonist (MRA) with fewer negative effects on serum potassium and renal function in comparison with steroidal MRAs (Rico-Mesa et al., 2020). This medication is in phase 3 clinical trials and shows promising results in CKD progression, composite kidney outcome, and cardiovascular events (Agarwal et al., 2020). The ARTS-DN trial (NCT01874431) conducted by Kabris et al. proved that DKD patients with the administration of Finerenone for 90 days had lower urine albumin creatinine ratio (UACR) in a dose-dependent way compared to placebo (Bakris et al., 2015). The FIGARO-DKD (NCT02545049) trial designed by Agarwal et al. is a randomized, double-blind, placebo-controlled, parallel-group, and multicenter phase 3 study. It assessed the efficacy and safety of Finerenone on the reduction of all-cause mortality and the onset of kidney failure and its influence on eGFR and UACR in DKD patients (Ruilope et al., 2019). The study has been completed, but the results have not been published yet. Another randomized, double-blind phase 3 study (FIDELIO-DKD NCT02540993) has similar inclusion criteria and intervention with FIGARO-DKD. The results indicated that the experimental group had lower composite kidney outcomes (kidney failure, a sustained ≥ 40% decrease in eGFR from baseline, or renal death) (Filippatos et al., 2021) and reduced risks of CKD progression and cardiovascular events compared to placebo (Bakris et al., 2020).
Pirfenidone is an antifibrotic drug widely used in the clinical treatment of idiopathic pulmonary fibrosis (IPF) (Nathan et al., 2019). However, recent studies have figured out that Pirfenidone also exerts its anti-fibrotic effect in renal fibrosis. Takakura et al. (2012) observed that Pirfenidone ameliorated renal interstitial fibrosis partly through decreasing the expression of TGF-β-induced collagen, fibronectin, CTGF, and plasminogen activator inhibitor type 1 (PAI-1) in rat proximal tubular epithelial cells. In addition to the TGF-β pathway, Li et al. (2017c) found that Pirfenidone could attenuate EMT and renal fibrosis by antagonizing the MAPK pathway both in vivo and in vitro. Therefore, Pirfenidone can inhibit related signaling pathways and reduce the expression of fibrotic markers to slow the process of renal fibrosis (Chen et al., 2013). A randomized phase 3 study has been designed to evaluate the efficacy of Pirfenidone on GFR and albuminuria in DKD patients1. This study will assess the improvement of GFR and the changes of 24-h urine microalbuminuria and TGF-β concentrations to determine the therapeutic effects Pirfenidone takes in the progression of DKD and renal fibrosis. It is still on the recruiting stage and has no result yet. Another phase 2 clinical study also in recruitment is to assess the change from baseline in renal fibrosis by diffusion-weighted magnetic resonance imaging (DW-MRI) and urinary markers in CKD patients (2 NCT04258397). Moreover, the trial NCT00063583 has already provided evidence that subjects assigned to the Pirfenidone group (1200 mg/day) had higher mean eGFR than placebo after 6 months of treatment (Sharma et al., 2011; Table 1).
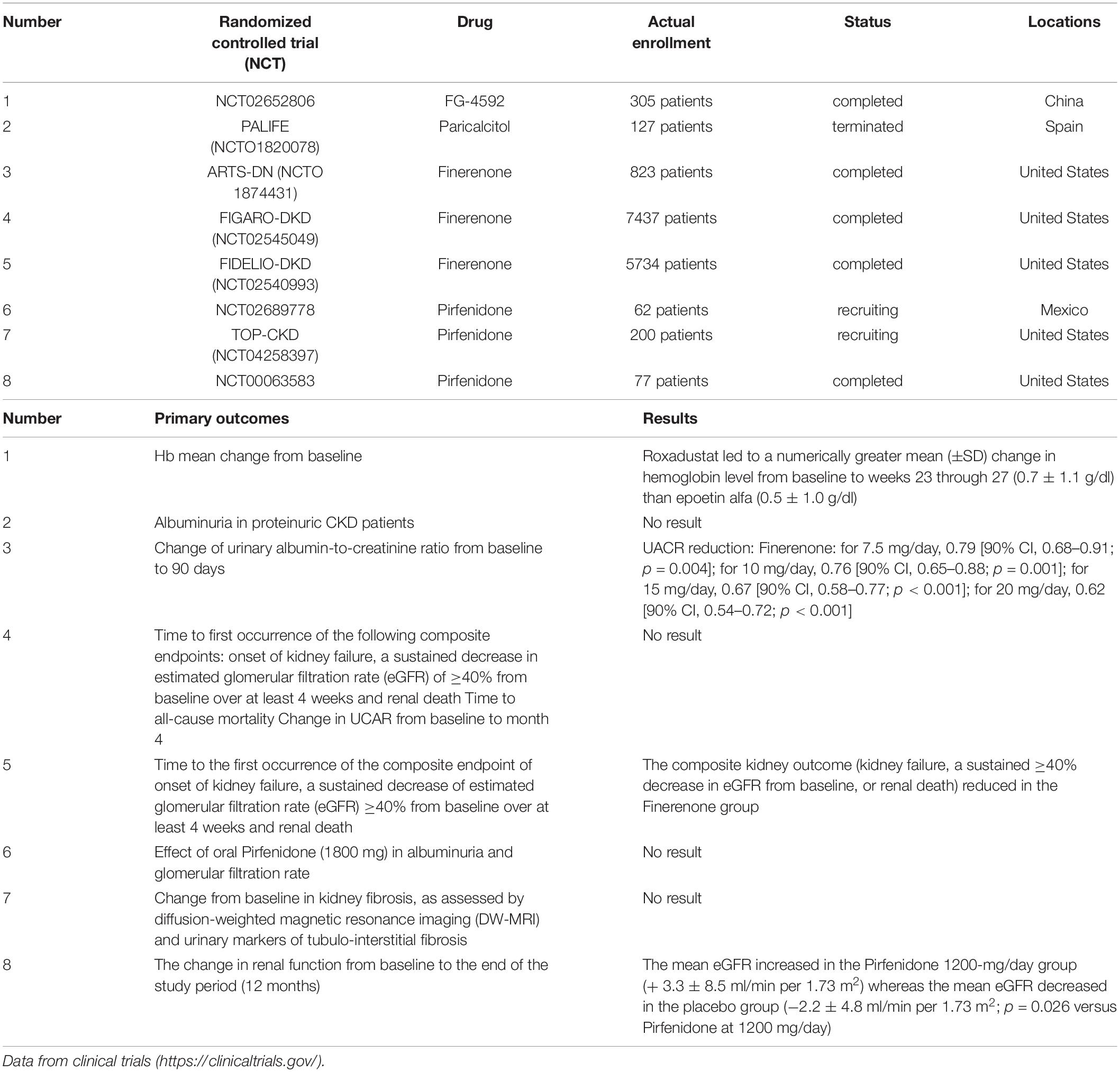
Table 1. New drugs that target renal fibrosis.
Moreover, these drugs have shown anti-fibrosis benefits in other organs besides the kidney. Roxadustat was found to take effect in the pulmonary fibrosis mice model via the inhibition of the TGF-β/Smad signaling pathway (Huang et al., 2020). Similarly, Paricalcitol showed relevant beneficial effects in the alleviation of liver fibrosis (Reiter et al., 2019), peritoneal fibrosis (Ko et al., 2019), and myocardial fibrosis (Panizo et al., 2013) by blocking the TGF-β pathway. Pirfenidone is a broad-spectrum anti-fibrosis drug that has been approved for the treatment of IPF (Rogliani et al., 2016). It inhibits the myofibroblast differentiation and collagen production in the lung by blocking the overexpressed pathways, including the TGF-β signaling pathway (Li et al., 2018), PI3K-Akt pathway (Kurita et al., 2017), and Wnt/β-catenin pathway (Ballester et al., 2019). It is also involved in liver fibrosis by inhibition of the TGF-β pathway and downregulation of Smads (García et al., 2002).
In addition to their anti-fibrosis effects, it is also important to understand the adverse effects of these drugs. According to the previous clinical trials, Roxadustat was generally well tolerated with few treatment-emergent adverse events (TEAEs). Common TEAEs were diarrhea, vomiting, contusion (Akizawa et al., 2020a), back pain, and nasopharyngitis (Akizawa et al., 2020b). It was also reported that patients taking Roxadustat had relatively higher risks of upper respiratory infection, hyperkalemia (Chen et al., 2019a), and metabolic acidosis (Chen et al., 2019b). With the administration of Paricalcitol, patients are more likely to develop hypercalcemia, hyperphosphatemia, and excessive suppression of parathyroid hormone, which could return to normal with the reduction of drug dose (Oblak et al., 2018). The most reported adverse events of Finerenone were diarrhea, muscle spasms, blood creatine phosphokinase increased, dizziness (Bakris et al., 2015), nasopharyngitis, constipation, and bronchitis (Katayama et al., 2017). In terms of adverse effects of Pirfenidone, gastrointestinal disorder (King et al., 2014), skin-related disease, and fatigue (Maher et al., 2020) were most reported. However, the overall percentage of patients with these adverse events was only slightly higher than in the placebo group, indicating an acceptable level of drug safety.
RNA Interference (RNAi) Therapy in Renal Fibrosis
RNA interference technique is an emerging therapeutic by which messenger RNA (mRNA) could be degraded selectively and the expression of specific proteins was inhibited (Saw and Song, 2020). Hence, RNAi is involved in renal fibrosis by inhibiting the overexpression of related genes, most commonly TGF-β1 (Takabatake et al., 2005) along with its downstream genes, such as CTGF (Ren et al., 2015) and proliferator-activated receptor coactivator-1 alpha (PGC-1α) (Wang et al., 2018) and phosphorylation of Drp1 at serine 616 (p-Drp1S616) (Wang et al., 2020b). Researchers found that small interfering RNA (siRNA)-mediated gene silencing effectively suppressed the expression of corresponding mRNA and protein and thus significantly inhibited the secretion of fibrogenic factors and delay the progression of renal fibrosis (Khaja et al., 2016; Isaka, 2018). The RNAi strategy offers a potential therapy for renal fibrosis, but the research is still in its initial stage and requires more attention.
These studies suggest that some progress has been made in the treatment of organ fibrosis, especially renal fibrosis. The process of renal fibrosis is divided into three stages, namely, inflammatory reaction, fibrosis formation, and scar formation (Liu, 2011). Blocking the transformation of renal intrinsic cells into myofibroblasts and degrading fibrous tissue by targeting the fibrotic pathway can block or even reverse the progression of the disease in the first two phases (Sun et al., 2016). However, once renal fibrous tissue develops into scar tissue, it can never be reversed (Nastase et al., 2018). Therefore, we emphasized that renal fibrosis may be blocked and renal function may be improved with effective anti-fibrosis therapy in the early stage of fibrosis.
Kidney biopsy is difficult to perform in clinical trials due to its complexity, high cost, and considerable pain to patients. Therefore, there is a lack of solid evidence to support the anti-fibrosis effects of these drugs. Future studies with repeated biopsies should be conducted to demonstrate whether tubulointerstitial fibrosis can be reversed with novel therapies and to reveal the specific mechanisms of them.
Conclusion
Renal fibrosis is a pivotal pathological change in DKD and markedly increases the mortality rate of late-stage DKD patients. In this paper, we discussed various signaling pathways involved in renal fibrosis, including the TGF-β, MAPK, Wnt/β-catenin, PI3K/Akt, JAK/STAT, and Notch pathways. These pathways all play a significant role in the accumulation of ECM, the expression of collagen and fibronectin, and the secretion of other related proteins. Moreover, an increasing number of novel therapies are under clinical study, and many efforts have been made to delay or even try to reverse the progression of renal fibrosis. Despite this progress, the exact effects of new drugs on renal tubulointerstitial fibrosis are still not clear and need to be further studied employing renal biopsies. With the development of modern medical technology, renal fibrosis is expected to become a reversible process, and the prognosis of DKD is expected to be improved.
Author Contributions
YZ and DJ contributed to the conception and design of the study. YZ wrote the first draft of the manuscript. DJ, XK, RZ, and YS wrote the sections of the manuscript. FL and XT guided the writing of this article. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was funded by the 2015 Traditional Chinese Medicine Scientific Research (No. 201507001-11).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the China Academy of Chinese Medical Sciences for supporting the study. We also appreciate the involvement of all authors in this study.
Footnotes
References
Agarwal, R., Anker, S. D., Bakris, G., Filippatos, G., Pitt, B., Rossing, P., et al. (2020). Investigating new treatment opportunities for patients with chronic kidney disease in type 2 diabetes: the role of finerenone. Nephrol. Dial. Transplant doi: 10.1093/ndt/gfaa294 [Epub ahead of print].
Akhmetshina, A., Palumbo, K., Dees, C., Bergmann, C., Venalis, P., Zerr, P., et al. (2012). Activation of canonical wnt signalling is required for TGF-β-Mediated FIbrosis. Nat. Commun. 3:735. doi: 10.1038/ncomms1734
Akizawa, T., Iwasaki, M., Yamaguchi, Y., Majikawa, Y., and Reusch, M. (2020a). Phase 3, randomized, double-blind, active-comparator (Darbepoetin Alfa) study of oral roxadustat in CKD patients with anemia on hemodialysis in Japan. J. Am. Soc. Nephrol. 31, 1628–1639. doi: 10.1681/ASN.2019060623
Akizawa, T., Otsuka, T., Reusch, M., and Ueno, M. (2020b). Intermittent oral dosing of roxadustat in peritoneal dialysis chronic kidney disease patients with anemia: a randomized, Phase 3, multicenter, open-label study. Ther. Apher. Dial. 24, 115–125. doi: 10.1111/1744-9987.12888
Alicic, R. Z., Rooney, M. T., and Tuttle, K. R. (2017). Diabetic kidney disease: challenges, progress, and possibilities. Clin. J. Am. Soc. Nephrol. CJASN 12:2032.
Bakin, A. V., Tomlinson, A. K., Bhowmick, N. A., Moses, H. L., and Arteaga, C. L. (2000). Phosphatidylinositol 3-Kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 275, 36803–36810. doi: 10.1074/jbc.M005912200
Bakris, G. L., Agarwal, R., Anker, S. D., Pitt, B., Ruilope, L. M., Rossing, P., et al. (2020). Effect of finerenone on chronic kidney disease outcomes in Type 2 diabetes. N. Engl. J. Med. 383, 2219–2229. doi: 10.1056/NEJMoa2025845
Bakris, G. L., Agarwal, R., Chan, J. C., Cooper, M. E., Gansevoort, R. T., Haller, H., et al. (2015). Effect of finerenone on albuminuria in patients with diabetic nephropathy: a randomized clinical trial. JAMA 314, 884–894. doi: 10.1001/jama.2015.10081
Ballester, B., Milara, J., and Cortijo, J. (2019). Idiopathic pulmonary fibrosis and lung cancer: mechanisms and molecular targets. Int. J. Mol. Sci. 20:593. doi: 10.3390/ijms20030593
Batlle, E., Sancho, E., Francí, C., Domínguez, D., Monfar, M., Baulida, J., et al. (2000). The transcription factor snail is a repressor of E-Cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2, 84–89. doi: 10.1038/35000034
Berthier, C. C., Zhang, H., Schin, M., Henger, A., Nelson, R. G., Yee, B., et al. (2009). Enhanced expression of janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes Metab. Res. Rev. 58, 469–477. doi: 10.2337/db08-1328
Bertrand, F. E., Angus, C. W., Partis, W. J., and Sigounas, G. (2012). Developmental pathways in colon Cancer. Cell Cycle 11, 4344–4351. doi: 10.4161/cc.22134
Bhattacharjee, N., Barma, S., Konwar, N., Dewanjee, S., and Manna, P. (2016). Mechanistic insight of diabetic nephropathy and its pharmacotherapeutic targets: an update. Eur. J. Pharmacol. 791, 8–24. doi: 10.1016/j.ejphar.2016.08.022
Bielesz, B., Sirin, Y., Si, H., Niranjan, T., Gruenwald, A., Ahn, S., et al. (2010). Epithelial notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J. Clin. Invest. 120, 4040–4054. doi: 10.1172/JCI43025
Bilic, J., Huang, Y.-L., Davidson, G., Zimmermann, T., Cruciat, C.-M., Bienz, M., et al. (2007). Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 316, 1619–1622. doi: 10.1126/science.1137065
Blokzijl, A., Dahlqvist, C., Reissmann, E., Falk, A., Moliner, A., Lendahl, U., et al. (2003). Cross-talk between the Notch and TGF-beta signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J. Cell Biol. 163, 723–728.
Bonegio, R., and Susztak, K. (2012). Notch signaling in diabetic nephropathy. Exp. Cell Res. 318, 986–992. doi: 10.1016/j.yexcr.2012.02.036
Brosius, F. C. (2008). New insights into the mechanisms of fibrosis and sclerosis in diabetic nephropathy. Rev. Endocrine Metab. Disord. 9, 245–254. doi: 10.1007/s11154-008-9100-9106
Bülow, R. D., and Boor, P. (2019). Extracellular matrix in kidney fibrosis: more than just a scaffold. J. Histochem. Cytochem. 67, 643–661. doi: 10.1369/0022155419849388
Cano, A., Pérez-Moreno, M. A., Rodrigo, I., Locascio, A., Blanco, M. J., del Barrio, M. G., et al. (2000). The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-Cadherin expression. Nat. Cell Biol. 2, 76–83. doi: 10.1038/35000025
Chae, K.-S., Martin-Caraballo, M., Anderson, M., and Dryer, S. E. (2005). Akt activation is necessary for growth factor-induced trafficking of functional K(Ca) channels in developing parasympathetic neurons. J. Neurophysiol. 93, 1174–1182. doi: 10.1152/jn.00796.2004
Chang, H., Gao, F., Guillou, F., Taketo, M. M., Huff, V., and Behringer, R. R. (2008). Wt1 negatively regulates beta-catenin signaling during testis development. Development 135, 1875–1885. doi: 10.1242/dev.018572
Chen, J.-F., Ni, H.-F., Pan, M.-M., Liu, H., Xu, M., Zhang, M.-H., et al. (2013). Pirfenidone inhibits macrophage infiltration in 5/6 nephrectomized rats. Am. J. Physiol. Renal Physiol. 304, F676–F685. doi: 10.1152/ajprenal.00507.2012
Chen, N., Hao, C., Liu, B.-C., Lin, H., Wang, C., Xing, C., et al. (2019a). Roxadustat treatment for anemia in patients undergoing long-term dialysis. N. Engl. J. Med. 381, 1011–1022. doi: 10.1056/NEJMoa1901713
Chen, N., Hao, C., Peng, X., Lin, H., Yin, A., Hao, L., et al. (2019b). Roxadustat for anemia in patients with kidney disease not receiving dialysis. N. Engl. J. Med. 381, 1001–1010. doi: 10.1056/NEJMoa1813599
Chen, N., Hao, J., Li, L., Li, F., Liu, S., and Duan, H. (2016). Carboxy-Terminal modulator protein attenuated extracellular matrix deposit by inhibiting phospho-Akt, TGF-B 1 and α-SMA in kidneys of diabetic mice. Biochem. Biophys. Res. Commun. 474, 753–760. doi: 10.1016/j.bbrc.2016.05.032
Chen, X., Stoeck, A., Lee, S. J., Shih, I.-M., Wang, M. M., and Wang, T.-L. (2010). Jagged1 expression regulated by Notch3 and Wnt/β-Catenin signaling pathways in ovarian Cancer. Oncotarget 1, 210–218. doi: 10.18632/oncotarget.127
Cheng, X., Gao, W., Dang, Y., Liu, X., Li, Y., Peng, X., et al. (2013). Both ERK/MAPK and TGF-Beta/Smad signaling pathways play a role in the kidney fibrosis of diabetic mice accelerated by blood glucose fluctuation. J. Diab. Res. 2013:463740. doi: 10.1155/2013/463740
Chin, B. Y., Mohsenin, A., Li, S. X., Choi, A. M. K., and Choi, M. E. (2001). Stimulation of Pro-A 1(I) collagen by TGF-B 1 in mesangial cells: role of the P38 MAPK pathway. Am. J. Physiol.-Renal Physiol. 280, F495–F504.
Chung, A. C. K., Dong, Y., Yang, W., Zhong, X., Li, R., and Lan, H. Y. (2013). Smad7 suppresses renal fibrosis via altering expression of TGF-Beta/Smad3-Regulated MicroRNAs. Mol. Ther. 21, 388–398. doi: 10.1038/mt.2012.251
Chung, S., Kim, S., Kim, M., Koh, E. S., Shin, S. J., Park, C. W., et al. (2017). Treatment combining aliskiren with paricalcitol is effective against progressive renal tubulointerstitial fibrosis via dual blockade of intrarenal renin. PLoS One 12:e0181757. doi: 10.1371/journal.pone.0181757
Dai, C., Wen, X., He, W., and Liu, Y. (2011). Inhibition of proinflammatory RANTES expression by TGF-Beta1 is mediated by glycogen synthase Kinase-3beta-Dependent beta-catenin signaling. J. Biol. Chem. 286, 7052–7059. doi: 10.1074/jbc.M110.174821
Davis, R. J. (1994). MAPKs: new JNK expands the group. Trends Biochem. Sci. 19, 470–473. doi: 10.1016/0968-0004(94)90132-90135
Derynck, R., and Zhang, Y. E. (2003). Smad-Dependent and smad-independent pathways in TGF-Beta family signalling. Nature 425, 577–584. doi: 10.1038/nature02006
Dong, Z., Tai, W., Lei, W., Wang, Y., Li, Z., and Zhang, T. (2016). IL-27 inhibits the TGF-B 1-Induced epithelial-mesenchymal transition in alveolar epithelial cells. BMC Cell Biol. 17:7. doi: 10.1186/s12860-016-0084-x
Duan, J.-Y., Duan, G.-C., Wang, C.-J., Liu, D.-W., Qiao, Y.-J., Pan, S.-K., et al. (2020). Prevalence and risk factors of chronic kidney disease and diabetic kidney disease in a central chinese urban population: a cross-sectional survey. BMC Nephrol. 21:115. doi: 10.1186/s12882-020-01761-1765
Estrach, S., Ambler, C. A., Lo Celso, C., Hozumi, K., and Watt, F. M. (2006). Jagged 1 is a beta-catenin target gene required for ectopic hair follicle formation in adult epidermis. Development 133, 4427–4438. doi: 10.1242/dev.02644
Farris, A. B., and Colvin, R. B. (2012). Renal interstitial fibrosis: mechanisms and evaluation. Curr. Opin. Nephrol. Hypertens 21, 289–300. doi: 10.1097/MNH.0b013e3283521cfa
Filippatos, G., Anker, S. D., Agarwal, R., Pitt, B., Ruilope, L. M., Rossing, P., et al. (2021). Finerenone and cardiovascular outcomes in patients with chronic kidney disease and Type 2 diabetes. Circulation 143, 540–552.
Francois, H., and Chatziantoniou, C. (2018). Renal fibrosis: recent translational aspects. Matrix Biol. 68–69, 318–332. doi: 10.1016/j.matbio.2017.12.013
Fruman, D. A., Chiu, H., Hopkins, B. D., Bagrodia, S., Cantley, L. C., and Abraham, R. T. (2017). The PI3K pathway in human disease. Cell 170, 605–635. doi: 10.1016/j.cell.2017.07.029
Fryer, R. M., Rakestraw, P. A., Nakane, M., Dixon, D., Banfor, P. N., Koch, K. A., et al. (2007). Differential inhibition of renin MRNA expression by paricalcitol and calcitriol in C57/BL6 mice. Nephron Physiol. 106, 76–81. doi: 10.1159/000104875
Fukasawa, H., Yamamoto, T., Suzuki, H., Togawa, A., Ohashi, N., Fujigaki, Y., et al. (2004). Treatment with Anti-TGF-β antibody ameliorates chronic progressive nephritis by inhibiting Smad/TGF-β signaling. Kidney Int. 65, 63–74. doi: 10.1111/j.1523-1755.2004.00393.x
García, L., Hernández, I., Sandoval, A., Salazar, A., Garcia, J., Vera, J., et al. (2002). Pirfenidone effectively reverses experimental liver fibrosis. J. Hepatol. 37, 797–805. doi: 10.1016/s0168-8278(02)00272-276
Grande, M. T., Sánchez-Laorden, B., López-Blau, C., De Frutos, C. A., Boutet, A., Arévalo, M., et al. (2015). Snail1-Induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 21, 989–997. doi: 10.1038/nm.3901
Grynberg, K., Ma, F. Y., and Nikolic-Paterson, D. J. (2017). The JNK signaling pathway in renal fibrosis. Front. Physiol. 8:829. doi: 10.3389/fphys.2017.00829
Haneda, M., Araki, S., Togawa, M., Sugimoto, T., Isono, M., and Kikkawa, R. (1997). Mitogen-activated protein kinase cascade is activated in glomeruli of diabetic rats and glomerular mesangial cells cultured under high glucose conditions. Diab. Metab. Res. Rev. 46, 847–853. doi: 10.2337/diab.46.5.847
Hao, J., Liu, S., Zhao, S., Liu, Q., Lv, X., Chen, H., et al. (2011). PI3K/Akt pathway mediates high glucose-induced lipogenesis and extracellular matrix accumulation in HKC cells through regulation of SREBP-1 and TGF-B 1. Histochem. Cell Biol. 135, 173–181. doi: 10.1007/s00418-011-0777-773
Harris, T. J. C., and Peifer, M. (2005). Decisions, decisions: beta-catenin chooses between adhesion and transcription. Trends Cell Biol. 15, 234–237. doi: 10.1016/j.tcb.2005.03.002
Hayashida, T., deCaestecker, M., and Schnaper, H. W. (2003). Cross-Talk between ERK MAP kinase and smad-signaling pathways enhances TGF-Beta dependent responses in human mesangial cells. FASEB J. 17, 1576–1578. doi: 10.1096/fj.03-0037fje
Hertig, A., Anglicheau, D., Verine, J., Pallet, N., Touzot, M., Ancel, P.-Y., et al. (2008). Early epithelial phenotypic changes predict graft fibrosis. J. Am. Soc. Nephrol. 19, 1584–1591. doi: 10.1681/ASN.2007101160
Higaki, M., and Shimokado, K. (1999). Phosphatidylinositol 3-Kinase is required for growth factor-induced amino acid uptake by vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 19, 2127–2132. doi: 10.1161/01.atv.19.9.2127
Hills, C. E., Siamantouras, E., Smith, S. W., Cockwell, P., Liu, K.-K., and Squires, P. E. (2012). TGFβ modulates cell-to-cell communication in early epithelial-to-mesenchymal transition. Diabetologia 55, 812–824. doi: 10.1007/s00125-011-2409-2409
Hoffman, B. B., Sharma, K., Zhu, Y., and Ziyadeh, F. N. (1998). Transcriptional activation of transforming growth factor-β1 in mesangial cell culture by high glucose concentration. Kidney Int. 54, 1107–1116. doi: 10.1046/j.1523-1755.1998.00119.x
Hogan, J. J., Mocanu, M., and Berns, J. S. (2016). The native kidney biopsy: update and evidence for best practice. Clin. J. Am. Soc. Nephrol. 11, 354–362. doi: 10.2215/CJN.05750515
Hoi, S., Tsuchiya, H., Itaba, N., Suzuki, K., Oka, H., Morimoto, M., et al. (2020). WNT/β-Catenin Signal Inhibitor IC-2-Derived small-molecule compounds suppress TGF-B 1-Induced fibrogenic response of renal epithelial cells by inhibiting SMAD2/3 signalling. Clin. Exp. Pharmacol. Physiol. 47, 940–946. doi: 10.1111/1440-1681.13270
Horikoshi, S., Fukuda, N., Tsunemi, A., Okamura, M., Otsuki, M., Endo, M., et al. (2020). Contribution of TGF-B 1 and effects of gene silencer pyrrole-imidazole polyamides targeting TGF-B 1 in diabetic nephropathy. Molecules (Basel, Switzerland) 25:950. doi: 10.3390/molecules25040950
Horowitz, J. C., Lee, D. Y., Waghray, M., Keshamouni, V. G., Thomas, P. E., Zhang, H., et al. (2004). Activation of the pro-survival phosphatidylinositol 3-Kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by P38 MAPK-Dependent induction of an autocrine growth factor. J. Biol. Chem. 279, 1359–1367. doi: 10.1074/jbc.M306248200
Hu, C., Sun, L., Xiao, L., Han, Y., Fu, X., Xiong, X., et al. (2015). Insights into the mechanisms involved in the expression and regulation of extracellular matrix proteins in diabetic nephropathy. Curr. Med. Chem. 22, 2858–2870. doi: 10.2174/0929867322666150625095407
Huang, H., Wang, X., Zhang, X., Wang, H., and Jiang, W. (2020). Roxadustat attenuates experimental pulmonary fibrosis in vitro and in vivo. Toxicol. Lett. 331, 112–121. doi: 10.1016/j.toxlet.2020.06.009
Huang, J. S., Guh, J. Y., Chen, H. C., Hung, W. C., Lai, Y. H., and Chuang, L. Y. (2001). Role of receptor for advanced glycation End-Product (RAGE) and the JAK/STAT-Signaling pathway in AGE-Induced collagen production in NRK-49F cells. J. Cell. Biochem. 81, 102–113.
Huang, J. S., Guh, J. Y., Hung, W. C., Yang, M. L., Lai, Y. H., Chen, H. C., et al. (1999). Role of the Janus Kinase (JAK)/Signal transducters and activators of transcription (STAT) cascade in advanced glycation end-product-induced cellular mitogenesis in NRK-49F cells. Biochem. J. 342(Pt 1), 231–238.
Huang, X., Liu, G., Guo, J., and Su, Z. (2018). The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 14, 1483–1496. doi: 10.7150/ijbs.27173
Isaka, Y. (2018). Targeting TGF-Beta signaling in kidney fibrosis. Int. J. Mol. Sci. 19:2532. doi: 10.3390/ijms19092532
Ito, Y., Goldschmeding, R., Bende, R., Claessen, N., Chand, M., Kleij, L., et al. (2001). Kinetics of connective tissue growth factor expression during experimental proliferative glomerulonephritis. J. Am. Soc. Nephrol. 12, 472–484.
Itoh, Y., Saitoh, M., and Miyazawa, K. (2018). Smad3-STAT3 crosstalk in pathophysiological contexts. Acta Biochim. Biophys. Sin (Shanghai) 50, 82–90. doi: 10.1093/abbs/gmx118
Jing, Z., Hu, L., Su, Y., Ying, G., Ma, C., and Wei, J. (2020). Potential signaling pathway through which notch regulates oxidative damage and apoptosis in renal tubular epithelial cells induced by high glucose. J. Recept. Signal Transduct. Res. doi: 10.1080/10799893.2020.1810706 [Epub ahead of print].
Ju, W., Ogawa, A., Heyer, J., Nierhof, D., Yu, L., Kucherlapati, R., et al. (2006). Deletion of Smad2 in mouse liver reveals novel functions in hepatocyte growth and differentiation. Mol. Cell. Biol. 26, 654–667.
Kasuga, H., Ito, Y., Sakamoto, S., Kawachi, H., Shimizu, F., Yuzawa, Y., et al. (2001). Effects of Anti-TGF-Beta Type II receptor antibody on experimental glomerulonephritis. Kidney Int. 60, 1745–1755. doi: 10.1046/j.1523-1755.2001.00990.x
Katayama, S., Yamada, D., Nakayama, M., Yamada, T., Myoishi, M., Kato, M., et al. (2017). A randomized controlled study of finerenone versus placebo in Japanese patients with type 2 diabetes mellitus and diabetic nephropathy. J. Diab. Compl. 31, 758–765. doi: 10.1016/j.jdiacomp.2016.11.021
Kattla, J. J., Carew, R. M., Heljic, M., Godson, C., and Brazil, D. P. (2008). Protein Kinase B/Akt activity is involved in renal TGF-Beta1-Driven epithelial-mesenchymal transition in vitro and in vivo. Am. J. Physiol. Renal Physiol. 295, F215–F225. doi: 10.1152/ajprenal.00548.2007
Kawakami, T., Ren, S., and Duffield, J. S. (2013). Wnt signalling in kidney diseases: dual roles in renal injury and repair. J. Pathol. 229, 221–231. doi: 10.1002/path.4121
Khaja, F., Jayawardena, D., Kuzmis, A., and Önyüksel, H. (2016). Targeted sterically stabilized phospholipid SiRNA nanomedicine for hepatic and renal fibrosis. Nanomaterials (Basel, Switzerland) 6:8. doi: 10.3390/nano6010008
Kida, Y., Asahina, K., Teraoka, H., Gitelman, I., and Sato, T. (2007). Twist relates to tubular epithelial-mesenchymal transition and interstitial fibrogenesis in the obstructed kidney. J. Histochem. Cytochem. 55, 661–673. doi: 10.1369/jhc.6A7157.2007
Kim, J. H., Kim, B. K., Moon, K. C., Hong, H. K., and Lee, H. S. (2003). Activation of the TGF-Beta/Smad signaling pathway in focal segmental glomerulosclerosis. Kidney Int. 64, 1715–1721. doi: 10.1046/j.1523-1755.2003.00288.x
Kim, M. S., Yoon, S. K., Bollig, F., Kitagaki, J., Hur, W., Whye, N. J., et al. (2010). A Novel Wilms Tumor 1 (WT1) target gene negatively regulates the WNT signaling pathway. J. Biol. Chem. 285, 14585–14593. doi: 10.1074/jbc.M109.094334
King, T. E., Bradford, W. Z., Castro-Bernardini, S., Fagan, E. A., Glaspole, I., Glassberg, M. K., et al. (2014). A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 370, 2083–2092. doi: 10.1056/NEJMoa1402582
Klüppel, M., and Wrana, J. L. (2005). Turning it up a notch: cross-talk between TGF beta and notch signaling. Bioessays 27, 115–118. doi: 10.1002/bies.20187
Ko, J., Kang, H.-J., Kim, D.-A., Ryu, E.-S., Yu, M., Lee, H., et al. (2019). Paricalcitol Attenuates TGF-B 1-Induced phenotype transition of human peritoneal mesothelial cells (HPMCs) via modulation of oxidative stress and NLRP3 inflammasome. FASEB J. 33, 3035–3050. doi: 10.1096/fj.201800292RR
Kopan, R., and Ilagan, M. X. G. (2009). The canonical notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233. doi: 10.1016/j.cell.2009.03.045
Kurita, Y., Araya, J., Minagawa, S., Hara, H., Ichikawa, A., Saito, N., et al. (2017). Pirfenidone inhibits myofibroblast differentiation and lung fibrosis development during insufficient mitophagy. Respir. Res. 18:114. doi: 10.1186/s12931-017-0600-603
Lan, H. Y. (2008). Smad7 as a therapeutic agent for chronic kidney diseases. Front. Bioscience-Landmark 13:4984–4992. doi: 10.2741/3057
Lee, C.-I., Guh, J.-Y., Chen, H.-C., Hung, W.-C., Yang, Y.-L., and Chuang, L.-Y. (2005). Advanced glycation end-product-induced mitogenesis and collagen production are dependent on angiotensin II and connective tissue growth factor in NRK-49F cells. J. Cell. Biochem. 95, 281–292. doi: 10.1002/jcb.20380
Lee, E.-J., Kang, M.-K., Kim, D. Y., Kim, Y.-H., Oh, H., and Kang, Y.-H. (2018). Chrysin inhibits advanced glycation end products-induced kidney fibrosis in renal mesangial cells and diabetic kidneys. Nutrients 10:882. doi: 10.3390/nu10070882
Lee, K.-Y., Ito, K., Hayashi, R., Jazrawi, E. P. I., Barnes, P. J., and Adcock, I. M. (2006). NF-KappaB and activator protein 1 response elements and the role of histone modifications in IL-1beta-Induced TGF-Beta1 gene transcription. J. Immunol. 176, 603–615. doi: 10.4049/jimmunol.176.1.603
Li, F., Li, L., Hao, J., Liu, S., and Duan, H. (2017a). Src Homology 2 Domain-Containing Inositol 5′-Phosphatase ameliorates high glucose-induced extracellular matrix deposition via the phosphatidylinositol 3-Kinase/Protein kinase B pathway in renal tubular epithelial cells. J. Cell. Biochem. 118, 2271–2284. doi: 10.1002/jcb.25881
Li, J., Qu, X., and Bertram, J. F. (2009). Endothelial-Myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am. J. Pathol. 175, 1380–1388. doi: 10.2353/ajpath.2009.090096
Li, J., Wu, B., Hu, H., Fang, X., Liu, Z., and Wu, S. (2020a). GdCl3 attenuates the glomerular sclerosis of streptozotocin (STZ) induced diabetic rats via inhibiting TGF-β/Smads signal pathway. J. Pharmacol. Sci. 142, 41–49. doi: 10.1016/j.jphs.2019.06.008
Li, J. H., Huang, X. R., Zhu, H.-J., Oldfield, M., Cooper, M., Truong, L. D., et al. (2004). Advanced glycation end products activate smad signaling via TGF-Beta-Dependent and independent mechanisms: implications for diabetic renal and vascular disease. FASEB J. 18, 176–178. doi: 10.1096/fj.02-1117fje
Li, X., Zou, Y., Xing, J., Fu, Y.-Y., Wang, K.-Y., Wan, P.-Z., et al. (2020b). Pretreatment with roxadustat (FG-4592) attenuates folic acid-induced kidney injury through antiferroptosis via Akt/GSK-3β/Nrf2 Pathway. Oxid. Med. Cell Longev. 2020:6286984. doi: 10.1155/2020/6286984
Li, Y., Li, H., Liu, S., Pan, P., Su, X., Tan, H., et al. (2018). Pirfenidone ameliorates lipopolysaccharide-induced pulmonary inflammation and fibrosis by blocking NLRP3 inflammasome activation. Mol. Immunol. 99, 134–144. doi: 10.1016/j.molimm.2018.05.003
Li, Y., Yan, H., Wang, F., Huang, S., Zhang, Y., Wang, Z., et al. (2017b). Activation of EphA1-Epha receptor axis attenuates diabetic nephropathy in mice. Biochem. Biophys. Res. Commun. 486, 693–699. doi: 10.1016/j.bbrc.2017.03.100
Li, Z., Liu, X., Wang, B., Nie, Y., Wen, J., Wang, Q., et al. (2017c). Pirfenidone suppresses MAPK signalling pathway to reverse epithelial-mesenchymal transition and renal fibrosis. Nephrology (Carlton) 22, 589–597. doi: 10.1111/nep.12831
Liu, L., Gao, C., Chen, G., Li, X., Li, J., Wan, Q., et al. (2013). Notch signaling molecules activate TGF- β in rat mesangial cells under high glucose conditions. J. Diab. Res. 2013:979702. doi: 10.1155/2013/979702
Liu, R., Das, B., Xiao, W., Li, Z., Li, H., Lee, K., et al. (2017). A novel inhibitor of homeodomain interacting protein kinase 2 mitigates kidney fibrosis through inhibition of the TGF- β 1/Smad3 pathway. J. Am. Soc. Nephrol. 28, 2133–2143.
Liu, Y. (2010). New insights into epithelial-mesenchymal transition in kidney fibrosis. J. Am. Soc. Nephrol. 21, 212–222. doi: 10.1681/ASN.2008121226
Liu, Y. (2011). Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 7, 684–696. doi: 10.1038/nrneph.2011.149
Lu, Y.-C., Yin, L.-T., Chang, W.-T., and Huang, J.-S. (2010). Effect of lactobacillus reuteri GMNL-263 treatment on renal fibrosis in diabetic rats. J. Biosci. Bioeng. 110, 709–715. doi: 10.1016/j.jbiosc.2010.07.006
Luciano, R. L., and Moeckel, G. W. (2019). Update on the native kidney biopsy: core curriculum 2019. Am. J. Kidney Dis. 73, 404–415. doi: 10.1053/j.ajkd.2018.10.011
Ma, F. Y., Flanc, R. S., Tesch, G. H., Han, Y., Atkins, R. C., Bennett, B. L., et al. (2007). A pathogenic role for c-jun amino-terminal kinase signaling in renal fibrosis and tubular cell apoptosis. J. Am. Soc. Nephrol. 18, 472–484. doi: 10.1681/ASN.2006060604
Mack, M., and Yanagita, M. (2015). Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 87, 297–307. doi: 10.1038/ki.2014.287
Maher, T. M., Corte, T. J., Fischer, A., Kreuter, M., Lederer, D. J., Molina-Molina, M., et al. (2020). Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double-blind, randomised, placebo-controlled, Phase 2 trial. Lancet Respir. Med. 8, 147–157. doi: 10.1016/S2213-2600(19)30341-30348
Martínez-Arias, L., Panizo, S., Alonso-Montes, C., Martín-Vírgala, J., Martín-Carro, B., and Fernández-Villabrille, S. (2021). Effects of calcitriol and paricalcitol on renal fibrosis in CKD. Nephrol. Dial. Transplant 36, 793–803. doi: 10.1093/ndt/gfaa373
Meng, X. M., Huang, X. R., Chung, A. C. K., Qin, W., Shao, X., Igarashi, P., et al. (2010). Smad2 protects against TGF-Beta/Smad3-Mediated renal fibrosis. J. Am. Soc. Nephrol. 21, 1477–1487. doi: 10.1681/ASN.2009121244
Meng, X.-M., Huang, X. R., Xiao, J., Chen, H., Zhong, X., Chung, A. C. K., et al. (2012a). Diverse roles of TGF-β receptor II in renal fibrosis and inflammation in vivo and in vitro. J. Pathol. 227, 175–188. doi: 10.1002/path.3976
Meng, X.-M., Huang, X. R., Xiao, J., Chung, A. C. K., Qin, W., Chen, H., et al. (2012b). Disruption of Smad4 Impairs TGF-Beta/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int. 81, 266–279. doi: 10.1038/ki.2011.327
Miyazono, K., ten Dijke, P., and Heldin, C. H. (2000). TGF-Beta signaling by smad proteins. Adv. Immunol. 75, 115–157.
Mödder, U. I., Oursler, M. J., Khosla, S., and Monroe, D. G. (2011). Wnt10b activates the Wnt, Notch and NFκB pathways in U2OS osteosarcoma cells. J. Cell. Biochem. 112:1392. doi: 10.1002/jcb.23048
Morris, R., Kershaw, N. J., and Babon, J. J. (2018). The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 27, 1984–2009. doi: 10.1002/pro.3519
Murea, M., Park, J.-K., Sharma, S., Kato, H., Gruenwald, A., Niranjan, T., et al. (2010). Expression of notch pathway proteins correlates with albuminuria. glomerulosclerosis, and renal function. Kidney Int. 78, 514–522. doi: 10.1038/ki.2010.172
Naito, T., Masaki, T., Nikolic-Paterson, D. J., Tanji, C., Yorioka, N., and Kohno, N. (2004). Angiotensin II induces thrombospondin-1 production in human mesangial cells via P38 MAPK and JNK: a mechanism for activation of latent TGF-Beta1. Am. J. Physiol. Renal Physiol. 286, F278–F287. doi: 10.1152/ajprenal.00139.2003
Nassar, M. K., Khedr, D., Abu-Elfadl, H. G., Abdulgalil, E. A., Abdalbary, M., Moustafa, F. E.-H., et al. (2021). Diffusion tensor imaging in early prediction of renal fibrosis in patients with renal disease: functional and histopathological correlations. Int. J. Clin. Pract. 75:e13918. doi: 10.1111/ijcp.13918
Nastase, M. V., Zeng-Brouwers, J., Wygrecka, M., and Schaefer, L. (2018). Targeting renal fibrosis: mechanisms and drug delivery systems. Adv. Drug Deliv. Rev. 129, 295–307. doi: 10.1016/j.addr.2017.12.019
Nathan, S. D., Costabel, U., Albera, C., Behr, J., Wuyts, W. A., Kirchgaessler, K.-U., et al. (2019). Pirfenidone in patients with idiopathic pulmonary fibrosis and more advanced lung function impairment. Respir. Med. 153, 44–51. doi: 10.1016/j.rmed.2019.04.016
Nishad, R., Mukhi, D., Tahaseen, S. V., Mungamuri, S. K., and Pasupulati, A. K. (2019). Growth hormone induces notch1 signaling in podocytes and contributes to proteinuria in diabetic nephropathy. J. Biol. Chem. 294, 16109–16122. doi: 10.1074/jbc.RA119.008966
Nishida, M., Okumura, Y., Sato, H., and Hamaoka, K. (2008). Delayed inhibition of P38 mitogen-activated protein kinase ameliorates renal fibrosis in obstructive nephropathy. Nephrol. Dial. Transplant 23, 2520–2524. doi: 10.1093/ndt/gfn309
Nlandu-Khodo, S., Neelisetty, S., Phillips, M., Manolopoulou, M., Bhave, G., May, L., et al. (2017). Blocking TGF-β and β-Catenin epithelial crosstalk exacerbates CKD. J. Am. Soc. Nephrol. 28, 3490–3503. doi: 10.1681/ASN.2016121351
Nolan, K. A., Brennan, E. P., Scholz, C. C., Cullen, C., Ryan, A., Taylor, C. T., et al. (2015). Paricalcitol protects against TGF-B 1-Induced fibrotic responses in hypoxia and stabilises Hif-α in renal epithelia. Exp. Cell Res. 330, 371–381. doi: 10.1016/j.yexcr.2014.07.034
Oblak, M., Mlinšek, G., Kandus, A., Buturović-Ponikvar, J., and Arnol, M. (2018). Paricalcitol versus placebo for reduction of proteinuria in kidney transplant recipients: a double-blind, randomized controlled trial. Transpl. Int. 31, 1391–1404. doi: 10.1111/tri.13323
Ogata, H., Chinen, T., Yoshida, T., Kinjyo, I., Takaesu, G., Shiraishi, H., et al. (2006). Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-Mediated TGF- β 1 production. Oncogene 25, 2520–2530. doi: 10.1038/sj.onc.1209281
Okano, K., Tsuruta, Y., Yamashita, T., Takano, M., Echida, Y., and Nitta, K. (2010). Suppression of renal fibrosis by Galectin-1 in high glucose-treated renal epithelial cells. Exp. Cell Res. 316, 3282–3291. doi: 10.1016/j.yexcr.2010.08.015
Oldfield, M. D., Bach, L. A., Forbes, J. M., Nikolic-Paterson, D., McRobert, A., Thallas, V., et al. (2001). Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J. Clin. Invest. 108, 1853–1863. doi: 10.1172/JCI11951
Packer, M. (2020). Mutual antagonism of hypoxia-inducible factor isoforms in cardiac, vascular, and renal disorders. JACC Basic Transl. Sci. 5, 961–968. doi: 10.1016/j.jacbts.2020.05.006
Panizo, S., Barrio-Vázquez, S., Naves-Díaz, M., Carrillo-López, N., Rodríguez, I., Fernández-Vázquez, A., et al. (2013). Vitamin D receptor activation, left ventricular hypertrophy and myocardial fibrosis. Nephrol. Dial. Transpl. 28, 2735–2744. doi: 10.1093/ndt/gft268
Patel, V., Joharapurkar, A., and Jain, M. (2020). Role of mineralocorticoid receptor antagonists in kidney diseases. Drug Dev. Res. 82, 341–363. doi: 10.1002/ddr.21760
Perkins, T. N., Dentener, M. A., Stassen, F. R., Rohde, G. G., Mossman, B. T., Wouters, E. F. M., et al. (2016). Alteration of canonical and non-canonical WNT-Signaling by crystalline silica in human lung epithelial cells. Toxicol. Appl. Pharmacol. 301, 61–70. doi: 10.1016/j.taap.2016.04.003
Phanish, M. K., Wahab, N. A., Colville-Nash, P., Hendry, B. M., and Dockrell, M. E. C. (2006). The differential role of Smad2 and Smad3 in the regulation of pro-fibrotic TGF Beta 1 responses in human proximal-tubule epithelial cells. Biochem. J. 393, 601–607. doi: 10.1042/bj20051106
Phillips, A. O., Steadman, R., Topley, N., and Williams, J. D. (1995). Elevated D-Glucose concentrations modulate TGF-Beta 1 synthesis by human cultured renal proximal tubular cells. the permissive role of platelet-derived growth factor. Am. J. Pathol. 147, 362–374.
Piersma, B., Bank, R. A., and Boersema, M. (2015). Signaling in fibrosis: TGF-β, WNT, and YAP/TAZ Converge. Front. Med. (Lausanne) 2:59. doi: 10.3389/fmed.2015.00059
Qi, W., Chen, X., Zhang, Y., Holian, J., Mreich, E., Gilbert, R. E., et al. (2007). High glucose induces macrophage inflammatory Protein-3α in renal proximal tubule cells via a transforming growth Factor-B 1 dependent mechanism. Nephrol. Dial. Transplant. 22, 3147–3153. doi: 10.1093/ndt/gfm365
Racusen, L. C., Solez, K., Colvin, R. B., Bonsib, S. M., Castro, M. C., Cavallo, T., et al. (1999). The Banff 97 working classification of renal allograft pathology. Kidney Int. 55, 713–723. doi: 10.1046/j.1523-1755.1999.00299.x
Reiter, F. P., Ye, L., Bösch, F., Wimmer, R., Artmann, R., Ziesch, A., et al. (2019). Antifibrotic effects of hypocalcemic Vitamin D analogs in murine and human hepatic stellate cells and in the CCl4 mouse model. Lab. Invest. 99, 1906–1917. doi: 10.1038/s41374-019-0310-311
Ren, Y., Du, C., Yan, L., Wei, J., Wu, H., Shi, Y., et al. (2015). CTGF SiRNA ameliorates tubular cell apoptosis and tubulointerstitial fibrosis in obstructed mouse kidneys in a Sirt1-Independent manner. Drug Des. Dev. Ther. 9, 4155–4171. doi: 10.2147/DDDT.S86748
Rico-Mesa, J. S., White, A., Ahmadian-Tehrani, A., and Anderson, A. S. (2020). Mineralocorticoid receptor antagonists: a comprehensive review of finerenone. Curr. Cardiol. Rep. 22:140. doi: 10.1007/s11886-020-01399-1397
Rogliani, P., Calzetta, L., Cavalli, F., Matera, M. G., and Cazzola, M. (2016). Pirfenidone, Nintedanib and N-Acetylcysteine for the treatment of idiopathic pulmonary fibrosis: a systematic review and meta-analysis. Pulm Pharmacol. Ther. 40, 95–103. doi: 10.1016/j.pupt.2016.07.009
Rui, H., Wang, Y., Cheng, H., and Chen, Y. (2012). JNK-Dependent AP-1 activation is required for aristolochic acid-induced TGF-B 1 synthesis in human renal proximal epithelial cells. Am. J. Physiol. Renal Physiol. 302, F1569–F1575. doi: 10.1152/ajprenal.00560.2011
Ruilope, L. M., Agarwal, R., Anker, S. D., Bakris, G. L., Filippatos, G., Nowack, C., et al. (2019). Design and baseline characteristics of the finerenone in reducing cardiovascular mortality and morbidity in diabetic kidney disease trial. Am. J. Nephrol. 50, 345–356. doi: 10.1159/000503712
Runyan, C. E., Hayashida, T., Hubchak, S., Curley, J. F., and Schnaper, H. W. (2009). Role of SARA (SMAD Anchor for Receptor Activation) in maintenance of epithelial cell phenotype. J. Biol. Chem. 284, 25181–25189. doi: 10.1074/jbc.M109.032847
Runyan, C. E., Schnaper, H. W., and Poncelet, A.-C. (2004). The phosphatidylinositol 3-Kinase/Akt pathway enhances smad3-stimulated mesangial cell collagen i expression in response to transforming growth Factor-Beta1. J. Biol. Chem. 279, 2632–2639. doi: 10.1074/jbc.M310412200
Saad, S., Stanners, S. R., Yong, R., Tang, O., and Pollock, C. A. (2010). Notch mediated epithelial to mesenchymal transformation is associated with increased expression of the snail transcription factor. Int. J. Biochem. Cell Biol. 42, 1115–1122. doi: 10.1016/j.biocel.2010.03.016
Sahlgren, C., Gustafsson, M. V., Jin, S., Poellinger, L., and Lendahl, U. (2008). Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. U S A. 105, 6392–6397. doi: 10.1073/pnas.0802047105
Saw, P. E., and Song, E.-W. (2020). SiRNA therapeutics: a clinical reality. Sci. China Life Sci. 63, 485–500. doi: 10.1007/s11427-018-9438-y
Schinner, S., Willenberg, H. S., Schott, M., and Scherbaum, W. A. (2009). Pathophysiological aspects of Wnt-Signaling in endocrine disease. Eur. J. Endocrinol. 160, 731–737. doi: 10.1530/EJE-08-0831
Sharma, K., Ix, J. H., Mathew, A. V., Cho, M., Pflueger, A., Dunn, S. R., et al. (2011). Pirfenidone for diabetic nephropathy. J. Am. Soc. Nephrol. 22, 1144–1151. doi: 10.1681/asn.2010101049
Sharma, K., Jin, Y., Guo, J., and Ziyadeh, F. N. (1996). Neutralization of TGF-β by Anti-TGF-β antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-Induced diabetic mice. Diab. Metab. Res. Rev 45, 522–530. doi: 10.2337/diab.45.4.522
Shin, J. H., Kim, K. M., Jeong, J. U., Shin, J. M., Kang, J. H., Bang, K., et al. (2019). Nrf2-Heme Oxygenase-1 attenuates high-glucose-induced epithelial-to-mesenchymal transition of renal tubule cells by inhibiting ROS-Mediated PI3K/Akt/GSK-3β signaling. J. Diab. Res. 2019:2510105. doi: 10.1155/2019/2510105
Shrestha, A., Che, R.-C., and Zhang, A.-H. (2019). Role of aldosterone in renal fibrosis. Adv. Exp. Med. Biol. 1165, 325–346. doi: 10.1007/978-981-13-8871-2_15
Sprague, S. M., Llach, F., Amdahl, M., Taccetta, C., and Batlle, D. (2003). Paricalcitol versus calcitriol in the treatment of secondary hyperparathyroidism. Kidney Int. 63, 1483–1490. doi: 10.1046/j.1523-1755.2003.00878.x
Street, J. M., Souza, A. C., Alvarez-Prats, A., Horino, T., Hu, X., Yuen, P. S., et al. (2014). Automated quantification of renal fibrosis with sirius red and polarization contrast microscopy. Physiol. Rep. 2:e12088. doi: 10.14814/phy2.12088
Sun, M.-Y., Wang, S.-J., Li, X.-Q., Shen, Y.-L., Lu, J.-R., Tian, X.-H., et al. (2019). CXCL6 promotes renal interstitial fibrosis in diabetic nephropathy by activating JAK/STAT3 signaling pathway. Front. Pharmacol. 10:224. doi: 10.3389/fphar.2019.00224
Sun, S., Ning, X., Zhang, Y., Lu, Y., Nie, Y., Han, S., et al. (2009). Hypoxia-Inducible Factor-1alpha induces twist expression in tubular epithelial cells subjected to hypoxia, leading to epithelial-to-mesenchymal transition. Kidney Int. 75, 1278–1287. doi: 10.1038/ki.2009.62
Sun, Y. B. Y., Qu, X., Caruana, G., and Li, J. (2016). The origin of renal Fibroblasts/Myofibroblasts and the signals that trigger fibrosis. Differentiation 92, 102–107. doi: 10.1016/j.diff.2016.05.008
Surendran, K., Schiavi, S., and Hruska, K. A. (2005). Wnt-Dependent β-Catenin signaling is activated after unilateral ureteral obstruction, and recombinant secreted frizzled-related protein 4 alters the progression of renal fibrosis. JASN 16, 2373–2384. doi: 10.1681/ASN.2004110949
Sutariya, B., Jhonsa, D., and Saraf, M. N. (2016). TGF-Beta: the connecting link between nephropathy and fibrosis. Immunopharmacol. Immunotoxicol. 38, 39–49. doi: 10.3109/08923973.2015.1127382
Takabatake, Y., Isaka, Y., Mizui, M., Kawachi, H., Shimizu, F., Ito, T., et al. (2005). Exploring RNA interference as a therapeutic strategy for renal disease. Gene Ther. 12, 965–973. doi: 10.1038/sj.gt.3302480
Takakura, K., Tahara, A., Sanagi, M., Itoh, H., and Tomura, Y. (2012). Antifibrotic effects of pirfenidone in rat proximal tubular epithelial cells. Ren. Fail 34, 1309–1316. doi: 10.3109/0886022X.2012.718955
Tan, X., He, W., and Liu, Y. (2009). Combination therapy with paricalcitol and trandolapril reduces renal fibrosis in obstructive nephropathy. Kidney Int. 76, 1248–1257. doi: 10.1038/ki.2009.346
Thomas, M. C., Brownlee, M., Susztak, K., Sharma, K., Jandeleit-Dahm, K. A. M., Zoungas, S., et al. (2015). Diabetic kidney disease. Nat. Rev. Dis. Primers 1:15018. doi: 10.1038/nrdp.2015.18
Tian, H., Yang, J., Xie, Z., and Liu, J. (2018). Gliquidone alleviates diabetic nephropathy by inhibiting notch/snail signaling pathway. Cell Physiol. Biochem. 51, 2085–2097. doi: 10.1159/000495827
Tian, X., Zhang, J., Tan, T. K., Lyons, J. G., Zhao, H., Niu, B., et al. (2013). Association of β-Catenin with P-Smad3 but Not LEF-1 dissociates in vitro profibrotic from anti-inflammatory effects of TGF-B 1. J. Cell Sci. 126(Pt 1), 67–76. doi: 10.1242/jcs.103036
Tuttle, K. R., Bakris, G. L., Bilous, R. W., Chiang, J. L., de Boer, I. H., Goldstein-Fuchs, J., et al. (2014). Diabetic kidney disease: a report from an ADA consensus conference. Am. J. Kidney Dis. 64, 510–533. doi: 10.1053/j.ajkd.2014.08.001
Tuttle, K. R., Brosius, F. C., Adler, S. G., Kretzler, M., Mehta, R. L., Tumlin, J. A., et al. (2018). JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: results from a Phase 2 randomized controlled clinical trial. Nephrol. Dial. Transplant. 33, 1950–1959. doi: 10.1093/ndt/gfx377
Utsugi, M., Dobashi, K., Ishizuka, T., Masubuchi, K., Shimizu, Y., Nakazawa, T., et al. (2003). C-Jun-NH2-Terminal kinase mediates expression of connective tissue growth factor induced by transforming growth factor-beta1 in human lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 28, 754–761. doi: 10.1165/rcmb.4892
van der Velden, J. L. J., Alcorn, J. F., Guala, A. S., Badura, E. C. H. L., and Janssen-Heininger, Y. M. W. (2011). C-Jun N-Terminal Kinase 1 promotes transforming Growth Factor-B 1-Induced Epithelial-to-Mesenchymal transition via control of linker phosphorylation and transcriptional activity of Smad3. Am. J. Respir. Cell Mol. Biol. 44, 571–581. doi: 10.1165/rcmb.2009-0282OC
Voelker, J., Berg, P. H., Sheetz, M., Duffin, K., Shen, T., Moser, B., et al. (2017). Anti-TGF-B 1 antibody therapy in patients with diabetic nephropathy. J. Am. Soc. Nephrol. JASN 28, 953–962. doi: 10.1681/ASN.2015111230
Wada, T., Azuma, H., Furuichi, K., Sakai, N., Kitagawa, K., Iwata, Y., et al. (2006). Reduction in chronic allograft nephropathy by inhibition of p38 mitogen-activated protein kinase. Am. J. Nephrol. 26, 319–325. doi: 10.1159/000094365
Wang, B., Ding, X., Ding, C., Tesch, G., Zheng, J., Tian, P., et al. (2020b). WNT1-Inducible-Signaling pathway protein 1 regulates the development of kidney fibrosis through the TGF-B 1 pathway. FASEB J. 34, 14507–14520. doi: 10.1096/fj.202000953R
Wang, D., Dai, C., Li, Y., and Liu, Y. (2011). Canonical Wnt/β-Catenin signaling mediates transforming growth Factor-B 1-Driven podocyte injury and proteinuria. Kidney Int. 80, 1159–1169. doi: 10.1038/ki.2011.255
Wang, J.-L., Chen, C.-W., Tsai, M.-R., Liu, S.-F., Hung, T.-J., Yu-Ju-Hung, et al. (2018). Antifibrotic Role of PGC-1α-SiRNA against TGF-B 1-Induced renal interstitial fibrosis. Exp. Cell Res. 370, 160–167. doi: 10.1016/j.yexcr.2018.06.016
Wang, X., Shaw, S., Amiri, F., Eaton, D. C., and Marrero, M. B. (2002). Inhibition of the Jak/STAT signaling pathway prevents the high glucose-induced increase in Tgf-Beta and fibronectin synthesis in mesangial cells. Diab. Metab. Res. Rev. 51, 3505–3509. doi: 10.2337/diabetes.51.12.3505
Wang, Y., Lu, M., Xiong, L., Fan, J., Zhou, Y., Li, H., et al. (2020a). Drp1-Mediated mitochondrial fission promotes renal fibroblast activation and fibrogenesis. Cell Death Dis. 11:29. doi: 10.1038/s41419-019-2218-2215
Wang, Y., Shen, R.-W., Han, B., Li, Z., Xiong, L., Zhang, F.-Y., et al. (2017). Notch signaling mediated by TGF-β/Smad pathway in concanavalin a-induced liver fibrosis in rats. World J. Gastroenterol. 23, 2330–2336. doi: 10.3748/wjg.v23.i13.2330
Wiegertjes, R., van Caam, A., van Beuningen, H., Koenders, M., van Lent, P., van der Kraan, P., et al. (2019). TGF-β Dampens IL-6 Signaling in articular chondrocytes by decreasing IL-6 receptor expression. Osteoarthritis Cartilage 27, 1197–1207. doi: 10.1016/j.joca.2019.04.014
Wolf, G. (2006). Renal Injury due to renin-angiotensin-aldosterone system activation of the transforming growth factor-beta pathway. Kidney Int. 70, 1914–1919. doi: 10.1038/sj.ki.5001846
Wu, D., Peng, F., Zhang, B., Ingram, A. J., Gao, B., and Krepinsky, J. C. (2007). Collagen I induction by high glucose levels is mediated by epidermal growth factor receptor and phosphoinositide 3-kinase/Akt signalling in mesangial cells. Diabetologia 50, 2008–2018.
Xiao, L., Wang, M., Yang, S., Liu, F., and Sun, L. (2013). A glimpse of the pathogenetic mechanisms of Wnt/β-Catenin signaling in diabetic nephropathy. Biomed. Res. Int. 2013:987064. doi: 10.1155/2013/987064
Xiao, Z., Zhang, J., Peng, X., Dong, Y., Jia, L., Li, H., et al. (2014). The Notch γ-Secretase inhibitor ameliorates kidney fibrosis via inhibition of TGF-β/Smad2/3 signaling pathway activation. Int. J. Biochem. Cell Biol. 55, 65–71. doi: 10.1016/j.biocel.2014.08.009
Xie, X., Xia, W., Fei, X., Xu, Q., Yang, X., Qiu, D., et al. (2015). Relaxin inhibits high glucose-induced matrix accumulation in human mesangial cells by interfering with TGF-B 1 production and mesangial cells phenotypic transition. Biol. Pharmaceut. Bull. 38, 1464–1469. doi: 10.1248/bpb.b15-00127
Xu, B.-H., Sheng, J., You, Y.-K., Huang, X.-R., Ma, R. C. W., Wang, Q., et al. (2020). Deletion of Smad3 prevents renal fibrosis and inflammation in Type 2 diabetic nephropathy. Metab. Clin. Exp. 103:154013. doi: 10.1016/j.metabol.2019.154013
Xu, W., Yang, Z., and Lu, N. (2015). A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh. Migr. 9, 317–324. doi: 10.1080/19336918.2015.1016686
Yamagishi, S.-I., Inagaki, Y., Okamoto, T., Amano, S., Koga, K., and Takeuchi, M. (2003). Advanced glycation end products inhibit de novo protein synthesis and induce TGF-Beta overexpression in proximal tubular cells. Kidney Int. 63, 464–473. doi: 10.1046/j.1523-1755.2003.00752.x
Yang, G., Zhao, Z., Zhang, X., Wu, A., Huang, Y., Miao, Y., et al. (2017). Effect of berberine on the renal tubular epithelial-to-mesenchymal transition by inhibition of the notch/snail pathway in diabetic nephropathy model KKAy mice. Drug Des. Dev. Ther. 11, 1065–1079. doi: 10.2147/DDDT.S124971
Yang, J. W., and Liu, Y. H. (2001). Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am. J. Pathol. 159, 1465–1475.
Yao, M., Wang, X., Wang, X., Zhang, T., Chi, Y., and Gao, F. (2015). The notch pathway mediates the angiotensin II-Induced synthesis of extracellular matrix components in podocytes. Int. J. Mol. Med. 36, 294–300. doi: 10.3892/ijmm.2015.2193
Yu, C.-H., Suriguga, Gong, M., Liu, W.-J., Cui, N.-X., Wang, Y., et al. (2017). High glucose induced endothelial to mesenchymal transition in human umbilical vein endothelial cell. Exp. Mol. Pathol. 102, 377–383. doi: 10.1016/j.yexmp.2017.03.007
Yu, L., Border, W. A., Anderson, I., McCourt, M., Huang, Y., and Noble, N. A. (2004). Combining TGF-beta inhibition and angiotensin II blockade results in enhanced antifibrotic effect. Kidney Int. 66, 1774–1784.
Yu, L., Border, W. A., Huang, Y. F., and Noble, N. A. (2003). TGF-Beta isoforms in renal fibrogenesis. Kidney Int. 64, 844–856. doi: 10.1046/j.1523-1755.2003.00162.x
Zavadil, J., Cermak, L., Soto-Nieves, N., and Böttinger, E. P. (2004). Integration of TGF-Beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 23, 1155–1165. doi: 10.1038/sj.emboj.7600069
Zeng, L.-F., Xiao, Y., and Sun, L. (2019). A glimpse of the mechanisms related to renal fibrosis in diabetic nephropathy. Adv. Exp. Med. Biol. 1165, 49–79. doi: 10.1007/978-981-13-8871-2_4
Zeng, X., Tamai, K., Doble, B., Li, S., Huang, H., Habas, R., et al. (2005). A dual-kinase mechanism for Wnt Co-Receptor phosphorylation and activation. Nature 438, 873–877. doi: 10.1038/nature04185
Zhang, J., Zhong, H.-B., Lin, Y., Yao, W., and Huang, J.-Y. (2015). KLF15 suppresses cell proliferation and extracellular matrix expression in mesangial cells under high glucose. Int. J. Clin. Exp. Med. 8, 20330–20336.
Zhang, P., Cai, Y., Soofi, A., and Dressler, G. R. (2012). Activation of Wnt11 by transforming growth Factor-β drives mesenchymal gene expression through non-canonical wnt protein signaling in renal epithelial cells. J. Biol. Chem. 287, 21290–21302. doi: 10.1074/jbc.M112.357202
Zheng, C., Huang, L., Luo, W., Yu, W., Hu, X., Guan, X., et al. (2019). Inhibition of STAT3 in tubular epithelial cells prevents kidney fibrosis and nephropathy in STZ-induced diabetic mice. Cell Death Dis. 10:848. doi: 10.1038/s41419-019-2085-2080
Zhou, D., and Liu, Y. (2016). Understanding the mechanisms of kidney fibrosis. Nat. Rev. Nephrol. 12:68. doi: 10.1038/nrneph.2015.215
Zhou, L., Xue, H., Yuan, P., Ni, J., Yu, C., Huang, Y., et al. (2010a). Angiotensin AT1 receptor activation mediates high glucose-induced epithelial-mesenchymal transition in renal proximal tubular cells. Clin. Exp. Pharmacol. Physiol. 37, e152–e157. doi: 10.1111/j.1440-1681.2010.05421.x
Zhou, T., He, X., Cheng, R., Zhang, B., Zhang, R. R., Chen, Y., et al. (2012). Implication of dysregulation of the canonical wingless-Type MMTV integration site (WNT) pathway in diabetic nephropathy. Diabetologia 55, 255–266. doi: 10.1007/s00125-011-2314-2312
Zhou, T., Hu, Y., Chen, Y., Zhou, K. K., Zhang, B., Gao, G., et al. (2010b). The pathogenic role of the canonical wnt pathway in age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 51, 4371–4379. doi: 10.1167/iovs.09-4278
Zhou, Y., Lv, C., Wu, C., Chen, F., Shao, Y., and Wang, Q. (2014). Suppressor of Cytokine Signaling (SOCS) 2 attenuates renal lesions in rats with diabetic nephropathy. Acta Histochem. 116, 981–988. doi: 10.1016/j.acthis.2014.04.002
Zhu, L., Zhao, S., Liu, S., Liu, Q., Li, F., and Hao, J. (2016). PTEN regulates renal extracellular matrix deposit via increased CTGF in diabetes mellitus. J. Cell. Biochem. 117, 1187–1198. doi: 10.1002/jcb.25402
Zhu, Q.-Q., Yang, X.-Y., Zhang, X.-J., Yu, C.-J., Pang, Q.-Q., Huang, Y.-W., et al. (2020). EGCG targeting notch to attenuate renal fibrosis via inhibition of TGFβ/Smad3 signaling pathway activation in streptozotocin-induced diabetic mice. Food Funct. 11, 9686–9695. doi: 10.1039/d0fo01542c
Zhu, Y., Casado, M., Vaulont, S., and Sharma, K. (2005). Role of upstream stimulatory factors in regulation of renal transforming growth Factor-B 1. Diab. Metab. Res. Rev. 54, 1976–1984. doi: 10.2337/diabetes.54.7.1976
Ziyadeh, F. N., Hoffman, B. B., Han, D. C., Cruz, M. C., Hong, S. W., Isono, M., et al. (2000). Long-Term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth Factor-β antibody in Db/Db diabetic mice. PNAS 97, 8015–8020. doi: 10.1073/pnas.120055097
Keywords: signaling pathway, renal fibrosis, diabetic kidney disease, TGF-β, cross-talk
Citation: Zhang Y, Jin D, Kang X, Zhou R, Sun Y, Lian F and Tong X (2021) Signaling Pathways Involved in Diabetic Renal Fibrosis. Front. Cell Dev. Biol. 9:696542. doi: 10.3389/fcell.2021.696542
Received: 16 April 2021; Accepted: 08 June 2021;
Published: 12 July 2021.
Edited by:
Albena Todorova Dinkova-Kostova, University of Dundee, United KingdomReviewed by:
Alessio Lepore, University of Leeds, United KingdomMuhammad Nawaz, University of Gothenburg, Sweden
Copyright © 2021 Zhang, Jin, Kang, Zhou, Sun, Lian and Tong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fengmei Lian, Njk0Mzk3NjQ0QHFxLmNvbQ==; Xiaolin Tong, dG9uZ3hpYW9saW42NkBzaW5hLmNvbQ==
†These authors have contributed equally to this work and share first authorship