Abstract
The interaction between the serine protease urokinase-type plasminogen activator (uPA) and its glycolipid-anchored receptor (uPAR) focalizes plasminogen activation to cell surfaces, thereby regulating extravascular fibrinolysis, cell adhesion, and migration. uPAR belongs to the Ly6/uPAR (LU) gene superfamily and the high-affinity binding site for uPA is assembled by a dynamic association of its three consecutive LU domains. In most human solid cancers, uPAR is expressed at the invasive areas of the tumor-stromal microenvironment. High levels of uPAR in resected tumors or shed to the plasma of cancer patients are robustly associated with poor prognosis and increased risk of relapse and metastasis. Over the years, a plethora of different strategies to inhibit uPA and uPAR function have been designed and investigated in vitro and in vivo in mouse models, but so far none have been implemented in the clinics. In recent years, uPAR-targeting with the intent of cytotoxic eradication of uPAR-expressing cells have nonetheless gained increasing momentum. Another avenue that is currently being explored is non-invasive imaging with specific uPAR-targeted reporter-molecules containing positron emitting radionuclides or near-infrared (NIR) florescence probes with the overarching aim of being able to: (i) localize disease dissemination using positron emission tomography (PET) and (ii) assist fluorescence guided surgery using optical imaging. In this review, we will discuss these advancements with special emphasis on applications using a small 9-mer peptide antagonist that targets uPAR with high affinity.
Introduction
The first direct evidence of a high-affinity cellular binding site for the urokinase-type plasminogen activator uPA1 (i.e., uPAR) was reported more than 35 years ago (Stoppelli et al., 1985; Vassalli et al., 1985). That discovery represented the final culmination of year’s research to define the “lytic agent that allowed Rous sarcoma virus transformed cells to liquefy the stroma binding cells together” (Fischer, 1946; Dano et al., 1985). We now understand that the “lytic system responsible for this liquefaction” is in fact uPA-mediated plasminogen activation and that the active protease degrading this stroma (insoluble fibrin) is plasmin in a process called fibrinolysis. The seminal discovery of a cellular binding site for uPA uncovered an important hallmark of this system—it provided a mechanism by which cells can focalize and control uPA-mediated plasminogen activation on their cell membrane. The ability to orchestrate the activity of such a powerful proteolytic system on cell surfaces immediately called upon a role in cell migration and extracellular matrix remodeling (Estreicher et al., 1990). The obvious medical implications thereof prompted an intensive research in the structure-function relationships of this system and targeting uPAR in the context of cancer cell invasion and metastasis became a prime objective. The magnitude of that research is illustrated by the fact that a PubMed search on “uPAR and Cancer” yields more than 1,700 entries.
In this review, we will discuss structure-function relationships in the interactions between uPAR and its two principle biological ligands—the serine protease uPA and the provisional matrix protein vitronectin. Although a plethora of potential ligands for uPAR have been proposed over the years and collectively dubbed the “uPAR interactome” (Eden et al., 2011), we will refrain from discussing the molecular properties of these interactions in detail since no solid structural data are available in the form of co-crystal structures. Furthermore, the functional implications of several of these putative uPAR-interactors are either circumstantial or at best indirect (Ferraris et al., 2014). For more information on non-canonical uPAR ligands, their possible role(s) in cell migration and signaling, and their targeting, the reader is referred to the following comprehensive reviews (Blasi and Sidenius, 2010; Smith and Marshall, 2010; Gonias and Hu, 2015; Li Santi et al., 2021; Yuan et al., 2021). We will instead focus on (i) structure-function relationships in uPAR and (ii) recent developments in targeted imaging of uPAR expression using radionuclide probes for positron emission tomography (PET) scanning or near-infrared (NIR) fluorescent probes for optical imaging to assist precision guided cancer surgery.
Structure of uPAR
In humans, uPAR is encoded by PLAUR on chromosome 19q13 and translation of its 7 exons yields a 335 residue long precursor polypeptide. The mature uPAR protein is, however, truncated to 283 residues by posttranslational removal of both N- and C-terminal signal sequences needed for endoplasmic reticulum translocation and glycosyl-phosphatidylinositol (GPI) membrane anchoring, respectively (Ploug et al., 1991). Other modifications include N-linked glycosylation of Asn52, Asn162, Asn172, and Asn200 (Ploug et al., 1998b; Gardsvoll et al., 2004) and oxidation of 28 cysteine residues to form 14 disulfide bonds.
Member of the LU Domain Protein Superfamily
Sequence alignments, limited proteolysis and disulfide bond assignment (Behrendt et al., 1991; Ploug et al., 1993) provided the first evidence that uPAR is a modular protein with three homologous domains related to Ly-6 antigens and snake venom α-neurotoxins (Ploug and Ellis, 1994; Figure 1A). Finally, the intron-exon organization of PLAUR reveals that each domain is encoded by separate exon-sets flanked by symmetrical phase-1 introns, which replicates the general construction of genes encoding prototypical single LU domain proteins (Casey et al., 1994; Leth et al., 2019a). Of note, human uPAR deviates from the ancestral LU domain consensus motif inasmuch it contains three consecutive LU domains and that its N-terminal domain lacks one of the five plesiotypic disulfide bond (Figure 1A)—a feature shared among all known mammalian orthologues of uPAR. This is indeed remarkable, as that disulfide bond connecting cysteine 7 and 8 is essential for the correct folding and stability of single LU domain proteins such as SLURP-1 (Adeyo et al., 2015), GPIHBP1 (Beigneux et al., 2015; Kristensen et al., 2021), CD59 (Petranka et al., 1996), and κ-bungarotoxin (Grant et al., 1998). Akin to uPAR, other multidomain members of the LU gene superfamily (e.g., Haldisin, C4.4A, TEX101) also lack this particular disulfide bond, but notably only in their N-terminal LU domain (Kjaergaard et al., 2008; Gårdsvoll et al., 2013; Jiang et al., 2020; Masutani et al., 2020). The evolutionary deletion of the 7–8 disulfide bond in uPAR DI has functional consequences as its reintroduction into recombinant human uPAR impairs both uPA-binding and the dynamic association between uPAR domain DI and uPAR domains DIIDIII in the unoccupied receptor (Mertens et al., 2012; Leth et al., 2019b).
FIGURE 1
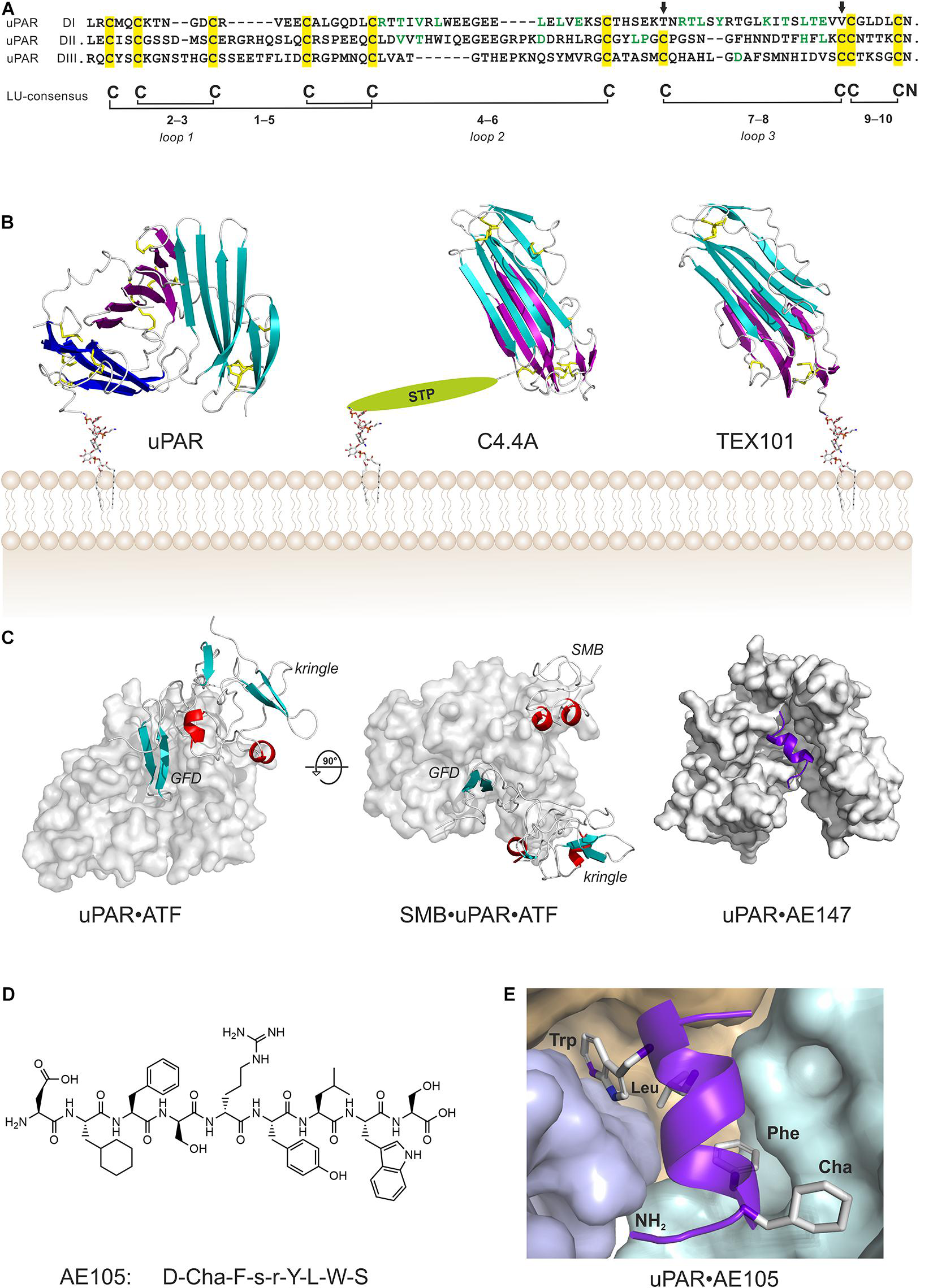
Structure of uPAR in complex with various ligands. (A) Sequence alignment of the three LU domains in human uPAR (inter-domain linker regions are omitted for clarity). Cysteine residues are highlighted in yellow and the conserved disulfide bonding are shown. The arrows mark the position of the missing consensus 7–8 LU-disulfide bond in uPAR DI. This pleisiotypic disulfide bond is also absent from the N-terminal LU domain in all other multidomain members of the Ly6/uPAR gene superfamily, but only in the N-terminal domain (Kjaergaard et al., 2008). Residues facing the hydrophobic ligand-binding cavity are shown in green. (B) The atomic structures of multi-LU-domain members of the Ly6/uPAR gene superfamily: uPAR with three consecutive LU domains (Xu et al., 2012; Zhao et al., 2015) and C4.4A (Jiang et al., 2020) and TEX101 (Masutani et al., 2020) each with two LU domains. The X-ray structures are shown in a cartoon representation with the β-sheets colored cyan (DI; N-terminal domain), magenta (DII), and blue (DIII) while the disulfide bonds are shown as yellow sticks. The C-terminal of the last LU-domain in uPAR and TEX101 is joined directly with a GPI-anchor moiety, while C4.4A is tethered to the GPI-anchor via a Ser/Thr/Pro–rich linker domain (STP) carrying several O-linked glycans (Hansen et al., 2004). (C) Shown are co-crystal structures of uPAR (gray surface representation) in complex with its natural ligand ATF (Huai et al., 2006), and with ATF and SMB (Huai et al., 2008), and in complex with a 13-mer peptide antagonist AE147 (Llinas et al., 2005). (D) The chemical structure of the 9-mer peptide AE105 that antagonizes uPA-binding to uPAR with an IC50 of 7 nM (Ploug et al., 2001). Cha is (L)-cyclohexylalanine, s is (D)-serine, and r is (D)-arginine. (E) The binding pose of AE105 within uPAR’s central ligand-binding cavity. AE105 adopts a short α-helix upon binding (Jorgensen et al., 2004); the binding cavity is assembled by DI (cyan), DII (wheat), and DIII (blue); the hydrophobic side-chains are buried deeply within this cavity (Llinas et al., 2005).
Evolution
The plasminogen system developed at the root of vertebrate evolution as an important mediator of fibrin surveillance securing vascular patency in species with a circulatory system. A plasminogen molecule cognate to that found in mammalians arose 550 million years ago (mya) with cyclostomes (i.e., lampreys), while its prime activators, tPA and uPA, appeared 450 mya with jawed-vertebrates (i.e., cartilaginous fishes). The most primitive orthologue known to resemble mammalian uPAR appeared 370 mya at the branch between lungfish and tetrapods; two lungfish genes were found to encode an uPAR-like protein with three consecutive LU domains, each having all 10 consensus cysteine residues (Chana-Muñoz et al., 2019). The unique loss of the 7–8 disulfide bond in the N-terminal LU domain occurred later with the radiation of tetrapods (amphibians, snakes, lizards, turtles, crocodilians, and mammals). Intriguingly, the elimination of that particular disulfide bond co-evolved with the acquisition of an uPA sequence compatible with receptor binding, as deducted from studies on mammals (Lin et al., 2010; Bager et al., 2012; Leth et al., 2019b). Within the avian lineage, the gene encoding uPAR was lost by chromosomal rearrangements, while uPA with a receptor-binding competent growth-factor like domain (GFD) was conserved (Aimes et al., 2003). The inability of a subgroup of avian species to focus uPA-mediated plasminogen activation on their cell surfaces was further exacerbated by the subsequent loss of a gene encoding a prominent plasminogen binding membrane protein (Plg-RKT) in the galliform lineage (Galliformes), which includes chicken (Sharma et al., 2020).
Glycolipid Membrane Anchoring
In common with many LU domain proteins, uPAR is tethered to the outer leaflet of the lipid bilayer of the cell membrane via a GPI-anchor. This GPI-anchor is added en bloc to Gly283 in a transamidase reaction during the posttranslational removal of the C-terminal signal sequence (Ploug et al., 1991; Kinoshita, 2020). This particular mode of membrane tethering provides uPAR with several distinct features such as (i) a prevalent clustering within membrane microdomains or membrane rafts (Varma and Mayor, 1998; Caiolfa et al., 2007; Suzuki et al., 2012), (ii) a mechanism for the specific shedding of uPAR via cleavage of the GPI-anchor by GDE3, a glycerophosphodiester phosphodiesterase (van Veen et al., 2017), and (iii) a deficiency of uPAR on bone-marrow derived blood cells from patients with the hematologic disorder paroxysmal nocturnal hemoglobinuria (Ploug et al., 1992b; Hill et al., 2017). One important corollary of the GPI-anchoring is that uPAR cannot per se transmit any signal across the cell membrane and therefore have to rely on indirect signaling pathways, which often complicates the interpretation of causality in molecular terms. The widely accepted model that uPAR-mediated signaling is driven by a promiscuous binding to various integrins (Wei et al., 1996; Smith and Marshall, 2010) has even been challenged by Sidenius and coworkers (Ferraris et al., 2014). They found that the mere binding of uPAR to the matrix protein vitronectin was necessary and sufficient to trigger ligand-independent β1 and β3 integrin signaling. A direct molecular engagement between uPAR and integrins was therefore not required per se and the signaling events in that model were relayed by alterations in membrane tension rather than by direct molecular interactions between uPAR and integrins.
Three-Dimensional Protein Structure
The first atomic structures of uPAR were solved by X-ray crystallography with co-crystals of either a 13-mer antagonist peptide of uPA binding (Llinas et al., 2005) or a receptor binding fragment (ATF) of the natural protease ligand uPA (Barinka et al., 2006; Huai et al., 2006; Lin et al., 2010). In these structures, all LU domains of uPAR assemble via a pseudo-3-fold symmetry to form a large hydrophobic ligand binding cavity, which is delimited by the concave faces of the central β-sheets of the individual LU domains (Figures 1B,C). Of note, this assembly is very different to that found in both C4.4A and TEX101, where the two LU domains assemble with a pseudo-2-fold symmetry by an extensive hydrophobic packing of the concave faces of their central β-sheets (Figure 1B). Construction of uPAR’s multi-domain topology with a flexible assembly of its individual LU domains has important functional consequences—it endows uPAR with cooperativity in uPA- and vitronectin-binding (Madsen et al., 2007; Gardsvoll et al., 2011b; Gardsvoll et al., 2011a; Mertens et al., 2012). This flexibility became first evident by the different uPAR conformations that were trapped in the complexes with the antagonist peptide AE147 and ATF (Figure 1C). Later, biophysical studies on uPAR in solution showed that the N-terminal LU domain (DI) has a high propensity for being detached from uPAR domains DII and DIII resulting in an “open” uPAR conformation, which is driven into a closed and compact conformation by uPA binding (Mertens et al., 2012). Crystalizing uPAR in a closed conformation with an empty ligand binding cavity required that the multi-domain topology was stabilized by either a non-natural disulphide bond between DI and DIII as in uPARH47C–N259C (Gardsvoll et al., 2011b; Xu et al., 2012) or by a monoclonal antibody that bound to the flexible linker region between DI and DII (Zhao et al., 2015). Attempts to determine the structure of “native” uPAR without any stabilizing agents resulted in an electron density map from which a compact structure of uPAR DIIDIII could be determined, but no electron densities corresponding to DI were traceable despite DI was present in the crystals (Liu et al., 2019).
From an evolutionary perspective, the flexible association between uPAR DI and DIIDIII is notable since this association is more prevalent when the pleisotypic 7–8 disulfide bond is absent from DI (Leth et al., 2019b). As discussed previously, the “loss” of this particular disulfide bond in uPAR occurred simultaneously with the acquisition of a receptor binding sequence in uPA. The dynamic detachment of DI provides furthermore a molecular explanation for the uPA- and uPAR-dependent cell adhesion to vitronectin matrices (Wei et al., 1994; Gardsvoll and Ploug, 2007; Madsen et al., 2007). Induction of the closed uPAR conformation by uPA-binding thus increases the affinity between uPAR and vitronectin by assembling a composite binding interface comprising elements of DI, DII, and the linker region connecting these LU domains (Gardsvoll and Ploug, 2007; Huai et al., 2008).
Targeting such a large and dynamic binding interface with small-molecule inhibitors obviously represents a major challenge. Building on structural information on the uPA•uPAR interaction, Meroueh et al. nevertheless succeeded in developing a small compound (IPR-3011) that inhibited AE147 binding to uPAR with an inhibition constant Ki = 2.4 ± 0.3 μM (Xu et al., 2017). Interestingly, that compound inhibited uPA-binding to the open conformation of uPARwt with a Ki = 60 ± 5 μM, while it inhibited uPA-binding to the closed conformation of uPARH47C–N259C with a 10-fold improved efficacy Ki = 6.6 ± 0.4 μM (Xu et al., 2021).
The uPA•uPAR Interaction
The uPA•uPAR interaction represents a very high affinity binding with a KD of 19 pM and the complex is long-lived (koff = 2 × 10–4 s–1) as assessed by surface plasmon resonance with purified components (Leth et al., 2019b). This tight binding is replicated on cells with a KD of 55 pM using 125I-labeled uPA and isolated monocytes (Nykjaer et al., 1990) and is well aligned with a plasma concentration of 20 pM uPA in healthy donors2 (Zeitler and Schuster, 1999). The β-hairpin of the growth factor-like domain (GFD) in uPA represents the key uPAR-binding region, with the hot-spot residues (Tyr24, Phe25, Ile28, and Trp30) being buried deeply within the ligand binding cavity (Figure 1C; Huai et al., 2006; Lin et al., 2010). In contrast, more than 15 residues distributed along the surface of uPAR’s ligand binding cavity contribute to the binding affinity for uPA, but none acts as prominent hot spot residues (Gardsvoll et al., 2006). Comparing the atomic structures of unoccupied and uPA-bound uPAR revealed additional flexibility in the multidomain organization. Loop 2 in uPAR DII (residues 130–140) undergoes profound structural shifts to partly cover the entrance of the ligand binding cavity by directly interacting with and covering the β-hairpin of the bound GFD (Barinka et al., 2006; Xu et al., 2012; Zhao et al., 2015). This region, which was dubbed the ligand loading/unloading loop in uPAR, further adds to the complexity of the conformational changes that occurs in the assembly of the LU domains upon uPA binding.
The Vitronectin uPAR Interaction
The first evidence that uPAR facilitates cell adhesion to vitronectin-coated surfaces was reported in 1994 (Waltz and Chapman, 1994; Wei et al., 1994). These early studies reported that uPA-binding stimulated the uPAR-mediated cell adhesion to vitronectin. We now know that vitronectin primarily binds uPAR via its small somatomedin B (SMB) domain and that this interaction is of relatively weak affinity with a KD of 2 μM (Gardsvoll and Ploug, 2007). In agreement with the potentiation of cell adhesion by uPA-binding, the affinity of the uPAR•SMB interaction increases approximately 3-fold by either uPA-binding or by introducing a non-natural disulfide bond in uPARH47C–N259C. Both mechanisms drive uPAR into the closed conformation, lead to robust lamellipodia formations (Gardsvoll et al., 2011a; Gardsvoll et al., 2011b), and increase cell migration on vitronetin-coated matrices (Madsen et al., 2007). Although a 3-fold increase in the affinity of uPAR•uPA for SMB may at first sight appear incremental, its biological impact is likely driven by pronounced avidity effects originating from the binding between uPA–uPAR clusters in lipid rafts and vitronectin molecules embedded in the provisional matrix. Again, inherent flexibility in the assembly of the LU domains in uPAR is required for the allosteric regulation of vitronectin-binding by uPA.
The functional and structural epitope on uPAR for vitronectin-binding is assembled by residues located in uPAR DI (Trp32, Arg58, Ile63), DII (Gln114, Arg116) and the flexible linker region between DI and DII (Arg91, Tyr92), and is well separated from the central uPA-binding cavity (Figure 1C; Gardsvoll and Ploug, 2007; Madsen et al., 2007; Huai et al., 2008). This binding interface is dominated by the burial of Arg91 within a small cavity in SMB where it forms a strong ionic interaction with Asp22 and is flanked by Phe13 and Tyr28. This binding pose resembles the one found between SMB and Arg101 in PAI-1 (Zhou et al., 2003) and the one utilized by the neutralizing monoclonal antibody 8B12 where Asp99 forms ionic interactions with Arg89 and Arg91 in uPAR (Zhao et al., 2015). One puzzling observation is that uPAR-binding of vitronectin completely buries a linear sequence in the linker region between uPAR DI and uPAR DII (–Ser88–Arg89–Ser90–Arg91–Tyr92–), which has been implicated in uPAR-mediated chemotaxis, directional cell migration, and angiogenesis via the formyl-peptide receptor type 1 (Resnati et al., 2002; Bifulco et al., 2010; Minopoli et al., 2019).
uPAR Biology
Baseline expression levels of uPAR are generally low in most homeostatic tissues and its scattered expression is primarily confined to bone-marrow derived white blood cells, pulmonary alveoli, glomeruli, and a few quiescent endothelial cells (Solberg et al., 2001). In the gastrointestinal tract, uPAR is likewise absent from most epithelial compartments, except from the antrum and the transitional cells of the squamo-columnar junction in mice (Alpízar-Alpízar et al., 2020). In contrast, uPAR expression is upregulated in many tissues undergoing active remodeling such as (i) the leading edge keratinocytes during re-epithelialization in wound healing (Romer et al., 1994), (ii) the invasive extravillous trophoblasts during early embryo implantation (Multhaupt et al., 1994; Plaisier et al., 2008), and (iii) the regressing glandular tissue during mammary gland involution (Solberg et al., 2001).
Despite an elevated uPAR expression during tissue remodeling, congenital uPAR deficiency is clearly not detrimental to development, survival, or reproduction as Plaur–/– mice are fertile with no overt early-onset phenotypes (Bugge et al., 1995b, 1996a). The mild phenotypes of uPAR deficiency is very different from those associated with plasminogen deficiency. Humans with type I plasminogen deficiency/severe hypoplasminogenemia (Schott et al., 1998) and Plg–/– mice (Bugge et al., 1995a; Romer et al., 1996; Lund et al., 2000) both display multiple adverse clinical manifestations due to a progressive extravascular fibrin deposition. In long-term studies, mouse strains that were unable to focus plasminogen activation on their cell surfaces due to wholesale gene ablations (Plaur––/–– or Plau––/––) or gene replacement with an uPAR binding–incompetent uPA-variant (PlauGFDhu/GFDhu)3 all developed chronic hepatic inflammation associated with an impaired fibrin surveillance (Connolly et al., 2010). Cooperating this role in extravascular fibrinolysis, uPA and uPAR expressing endothelial cells and macrophages were found to line the surfaces of fibrinoid deposits in the placenta (Pierleoni et al., 2003). In humans, genetic studies find no association between single missense variants in PLAUR and a robust risk for disease predisposition, except for a few publications where PLAUR variants located outside the protein coding regions were correlated to vascular complications in patients with systemic sclerosis (Manetti et al., 2011) and to a decline in pulmonary function in asthma patients (Barton et al., 2009). Collectively, these studies indicate (i) that in healthy individuals uPAR is not critical to the function of vital tissues and (ii) that uPAR assists in long-term fibrin surveillance alleviating chronic inflammation provoked by fibrin deposition. The pathogenesis associated with impaired plasminogen activation is primarily driven by an excessive fibrin deposition since the severe pleiotropic phenotypes of Plg–/– mice were more or less absent in double-deficient Plg–/– Fib–/– mice (Bugge et al., 1996b).
Role in Normal Physiology
Cell Surface Associated Plasminogen Activation
In vitro studies with purified proteins and cultured cells provided an outline of the biochemical pathway orchestrating cell surface associated plasminogen activation (Figure 2A). The hallmark of this pathway is the separate docking of two zymogens on the cell surface by: (i) a specific and high-affinity interaction between uPAR and pro-uPA (KD ∼ 20 pM); and (ii) a low-affinity binding of plasminogen to an array of broadly distributed membrane proteins with C-terminal lysine residues (KD ∼ 1 μM), including Plg-RKT (Miles et al., 2014). Of note, Plg-RKT colocalizes with uPAR on cell surfaces (Andronicos et al., 2010). The concomitant binding of pro-uPA and of plasminogen to cell surfaces provides a template for enhanced plasminogen activation due to an increased efficiency of the reciprocal activation of the two zymogens i.e., pro-uPA activation by plasmin and plasminogen activation by receptor-bound uPA thus forming a positive feedback loop. This arrangement lowers the Km for plasminogen activation from 25 μM in solution to 0.7 μM for cell-bound reactants, which is well aligned with a plasma concentration of 2 μM plasminogen (Ellis et al., 1991). Of note, cell-bound plasmin is refractory to inhibition by α2-antiplasmin. This differential sensitivity to α2-antiplasmin mediated inhibition provides a further drive toward a focal confinement of plasminogen activation on cell surfaces.
FIGURE 2
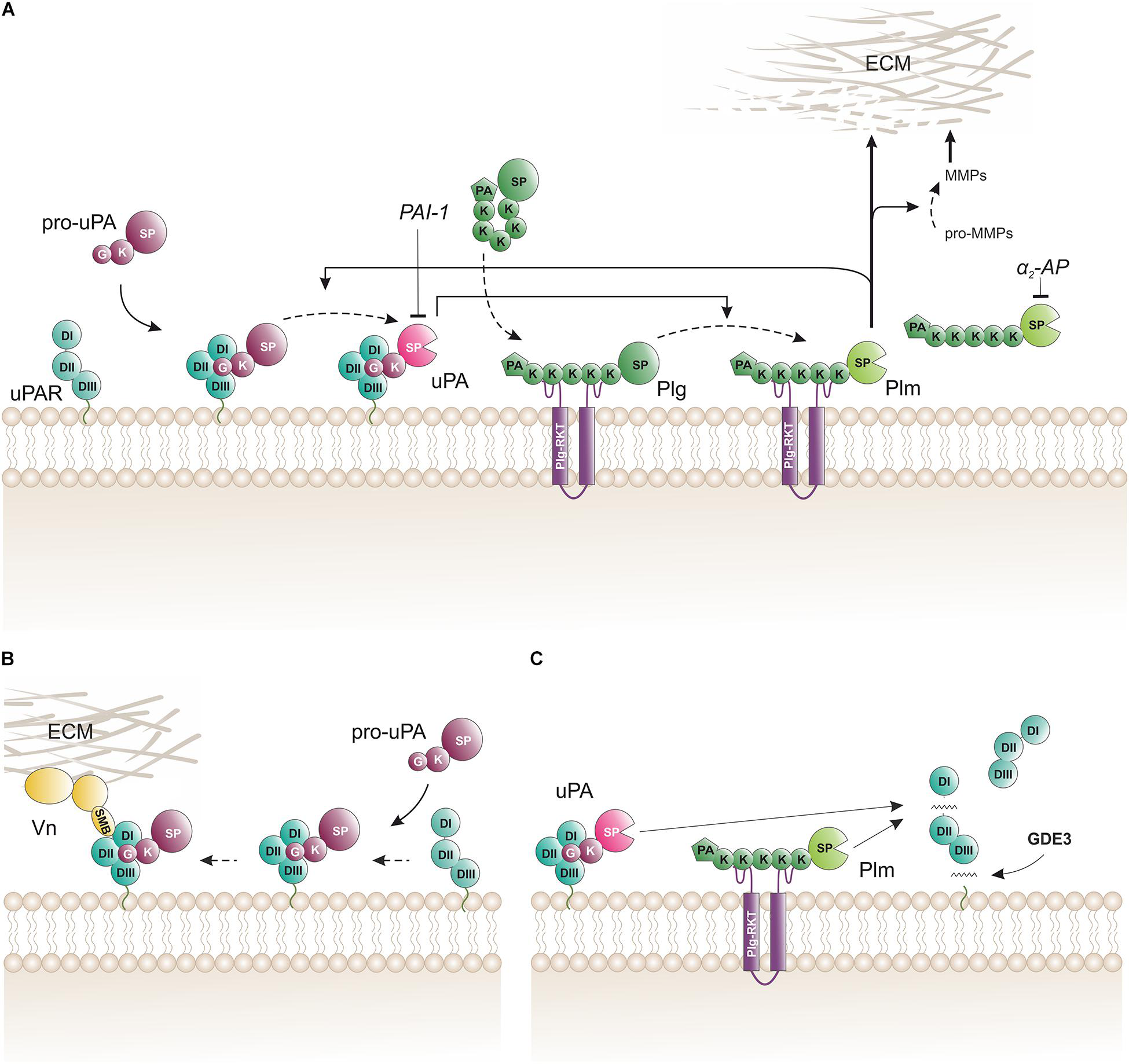
Biochemical pathways for proteolytic function(s) of uPAR on the cell surface. (A) The membrane template for an amplified cell surface associated generation of plasmin activity by reciprocal zymogen activation of pro-uPA and plasminogen (Plg). Fluid-phase propagation of plasmin (Plm) activity is prevented by the inhibition of dissociated Plm by its cognate inhibitor α2-antiplasmin (α2-AP). Regulation of the amplification by this template is also secured by the irreversible inhibition of uPA by a dedicated serpin, the plasminogen activator inhibitor type-1 (PAI-1). (B) Engagement of pro-uPA•uPAR complexes in cell adhesion to vitronectin (Vn) in the provisional matrix. Interactions between the closed conformation of uPAR (induced by pro-uPA binding) and the somatomedin B domain (SMB) of vitronectin tethers the cell to the vitronectin-rich extracellular matrix (ECM) and this presumably causes indirect activation of integrins and relay cell signaling altering cell adhesion and migration (Smith and Marshall, 2010; Ferraris et al., 2014). This uPAR-dependent adhesion is weakened when the cell surface template becomes activated since the proteolytic activity of the generated membrane-bound uPA and Plm will release the SMB domain from vitronectin thus breaking the tether between the cell surface and the matrix (De Lorenzi et al., 2016). (C) Membrane shedding of soluble uPAR variants. The glycerophosphodiester phosphodiesterase GDE3 may shed intact uPAR or uPAR DIIDIII by cleaving their GPI-anchor (van Veen et al., 2017). Proteolytic activity from membrane-bound uPA or Plm efficiently cleaves uPAR in the flexible linker region between DI and DII thus releasing uPAR DI. K, kringle domain; G, growth-factor like domain; SP, serine protease domain; PA, apple domain; SMB, somatomedin B domain; DI, DII, DIII, LU domains in uPAR.
The inherent power of arming cells with such a potent protease system on their cell-surface is clearly demonstrated in vivo by the severe cutaneous phenotypes (phemphigoid lesions) that develop in bitransgenic mice, in which Plaur and Plau transcription in the skin is controlled by the keratin 5 promoter (Zhou et al., 2000). Assembly of a functional cell-surface template with plasminogen and uPAR-bound pro-uPA is required to drive this dermal pathogenesis, since neither of the single transgenic mice nor the bitransgenic mouse crossed into a Plg–/–background develop these lesions (Bolon et al., 2004). The necessity for an intact and functional template to activate plasminogen is also found under non-pathological conditions, since mice with single deficiencies in either Plaur, Plau, or Plg are resistant to an engineered anthrax toxin that requires proteolytic activation by surface bound uPA (Liu et al., 2003). That dependency is further underscored by the lack of toxicity in the PlauGFDhu/GFDhu mouse strain, which produces a catalytic proficient pro-uPA that fails to bind uPAR (Connolly et al., 2010). It is therefore beyond any reasonable doubts that cell-surface plasminogen activation driven by the assembly of pro-uPA•uPAR complexes is operational in vivo. The mild overt phenotypes of Plaur–/– mice and their late-onset is most likely a consequence of functional redundancy, since mice with combined deficiencies for uPAR/tPA or for uPA/tPA (the two prime plasminogen activators) exhibit exacerbated hepatic fibrin deposition compared to Plaur–/– mice (Bugge et al., 1996a).
The membrane-bound template for pro-uPA and plasminogen activation is also implicated in the proteolytic activation of an oncogenic transmembrane receptor denoted CUB domain containing protein 1 (CDCP1) (Kryza et al., 2021). A proteomic search in three different cancer cell lines with a protease-reactive warhead build on CDCP1 sequences, identified uPA and plasmin as the lead candidates for cleaving CDCP1 at Arg368 or Lys369. Proteolytic processing of CDCP1 at these sites leads to potentiation of its pro-metastatic effect. Subsequent mechanistic studies demonstrated that uPA needed to be tethered on the cell surface via uPAR binding to promote the cleavage of CDCP1 (Kryza et al., 2021).
uPAR in Vitronectin-Dependent Cell Adhesion
While the impact of uPAR and uPA•uPAR complexes on cell adhesion and migration is well documented in vitro by a plethora of different cell culture experiments (Madsen et al., 2007; Petzinger et al., 2007; Salasznyk et al., 2007; Smith and Marshall, 2010; De Lorenzi et al., 2016), their impact (if any) under normal physiologic conditions in vivo is less clear. Like Plaur–/– mice, Vtn–/– mice have no overt phenotypes that would support a vital role for vitronectin in cell adhesion and migration during normal development (Zheng et al., 1995). Nonetheless, one study found that Vtn–/– mice had slightly slower dermal wound healing, reduced dermal microvessel density, and focal sites with delayed hemorrhage (Jang et al., 2000). The origin of this mild phenotype was presumably endorsed by an unbalanced activity of tPA and uPA in the wound field allegedly due to a faster latency transition of their shared inhibitor PAI-1 in the absence of vitronectin. An intriguing rendezvous between uPAR-dependent cell adhesion and cell-surface associated plasminogen activation that is mediated by uPA•uPAR complexes provides a possible regulatory mechanism to control the adhesion between uPAR-positive cells and vitronectin-rich provisional matrices. This interplay involves three sequential steps: (i) focal adhesion initiated by the binding between pro-uPA–uPAR complexes on the cell surface and SMB in vitronectin that is deposited as part of the provisional extracellular matrix (Figure 2B; Madsen et al., 2007), (ii) manifest cell adhesion and migration after relay of signals via indirect coupling to integrins, receptor tyrosin kinases, or G-protein-coupled receptors (Smith and Marshall, 2010), and finally (iii) attenuation of cell adhesion and migration by activation of pro-uPA to uPA (De Lorenzi et al., 2016). Of note, zymogen activation within the pro-uPA•uPAR•plasminogen template thus provides a negative feedback loop on cell adhesion by promoting the cleavage of either uPAR in the linker region between DI and DII (Figure 2C) or at the R↓GD-motif in the linker to the SMB domain (Hoyer-Hansen et al., 1992, 1997; De Lorenzi et al., 2016). Both cleavages dismantle the ternary uPA•uPAR•vitronectin complex and are mediated by either uPAR-bound uPA or by cell surface associated plasmin.
uPAR in Pathophysiology
In general, pathologies associated with chronic inflammation, such as rheumatoid arthritis, cancer, and Crohn’s disease, display elevated uPAR expression levels at their lesion sites primarily due to infiltrating immune cells. While evidence for a direct causal effect of uPAR-expression on disease progression often remains uncertain (Almholt et al., 2015), elevated uPAR levels in resected lesions or shed into the circulation are strong and robust surrogate biomarkers of disease severity. Most observational studies find an inverse correlation between elevated plasma levels of soluble uPAR and patient performance and survival (Lund et al., 2011; Madunic, 2018).
Soluble uPAR as a Surrogate Biomarker for Cancer Progression
Building on the seminal discoveries that plasma levels of uPA (Duffy et al., 1990) and soluble uPAR (Stephens et al., 1999) are robust biomarkers for adverse disease progression, accumulating observational studies continue to underscore that high uPA and/or high uPAR levels predict poor prognosis for patients with solid tumors (Grunnet et al., 2014; Dohn et al., 2015; Brungs et al., 2017; Loosen et al., 2018, 2019; Lu et al., 2018; Madunic, 2018). The source(s) of shed soluble uPAR is not always known, but the activated tumor-stromal microenvironment is a possible culprit from which uPAR may be released by proteases and/or hydrolases cleaving the GPI-anchor e.g., GDE3 (Figure 2C). The linker region between uPAR DI and DII is highly susceptible to proteolytic cleavage by e.g., uPA or plasmin, which releases uPAR DI from the cell surface (Hoyer-Hansen et al., 1992). Time-resolved fluorescence immunoassays with high sensitivity and specificity were therefore developed and validated for quantification of intact uPAR and its cleavage products in plasma (Piironen et al., 2004; Thurison et al., 2010). The baseline expression levels of uPAR, uPAR DIIDIII, and uPAR DI in healthy subjects are low; 36.5, 13.9, and 18.6 pM, respectively (Thurison et al., 2015). Subsequent studies showed that these soluble uPAR fragments were independent prognostic biomarkers in pre- and post-operative plasma samples from patients with colorectal cancer (Rolff et al., 2019). For more detailed information on uPAR expression and cancer dissemination the reader is referred to the following reviews (Andreasen et al., 2000; Romer et al., 2004; Allgayer, 2010; Kriegbaum et al., 2011; Madunic, 2018; Li Santi et al., 2021).
Paroxysmal Nocturnal Hemoglobinuria
The etiology of a rare hematological disorder termed paroxysmal nocturnal hemoglobinuria (PNH) is the clonal expansion of hematopoietic stem cells carrying somatic loss-of-function mutations in PIGA, which encodes an enzyme pivotal for the biosynthesis of GPI-anchors (Hill et al., 2017; Kinoshita, 2020). The overarching hallmark of the PNH syndrome is that affected blood cells fail to express GPI-anchored proteins on their cell membranes and instead secrete a soluble truncated variant lacking the C-terminal signal sequence for GPI-anchoring. Accordingly, monocytes and neutrophils affected by PNH are deficient in membrane-tethered uPAR (Ploug et al., 1992b) and they secrete a soluble uPAR (Ploug et al., 1992a) leading to sustained elevated plasma levels of soluble uPAR with an average plasma concentration of 120 pM (Ronne et al., 1995; Sloand et al., 2008). Due to the pleiotropic effects of PIGA deficiency, the molecular causality underlying the clinic manifestations of PNH are often complex and incompletely delineated. Notwithstanding this uncertainty, treatment with a neutralizing anti-C5 antibody (eculizumab) alleviates all of the major adverse clinical complications in PNH i.e., intravascular hemolysis, venous thrombosis, renal dysfunction, and pulmonary hypertension (Hill et al., 2017). It is therefore likely that the principal driver of these pathogenic complications in PNH is intravascular hemolysis due to CD59 deficiency and that eculizumab treatment compensates for the impaired complement regulation. Secondary to episodes of intravascular hemolysis, excessive renal resorption of hemoglobin dimers accompanied by heme cytotoxicity may thus drive the development of acute and chronic kidney disease (Kokoris et al., 2018).
uPAR in Kidney Disease
Soluble uPAR levels in patients with idiopathic focal and segmental glomerulosclerosis (FSGS) has attracted a lot of attention after Reiser et al., proposed that soluble uPAR was a prime candidate for the elusive serum permeability factor responsible for recurrent FSGS after kidney transplantations (Wei et al., 2008, 2011). Among other findings, they based their proposition on (i) that the elevated plasma levels of soluble uPAR in primary and recurrent FSGS were independent of the estimated glomerular filtration rates (eGFR) and (ii) that glomerular uPAR deposits activated podocyte αvβ3 and thereby drove foot process effacement leading to manifest proteinuric glomerular disease. Although this hypothesis initially gained some support, it nevertheless had some inherent weaknesses and it remains highly controversial (Konigshausen and Sellin, 2016; Kronbichler et al., 2016; Saleem, 2018). First, it was questioned whether the increased levels of soluble uPAR in FSGS patients actually were unrelated to decreased eGFR (Meijers et al., 2014; Musetti et al., 2015). Second, the finding that elevated levels of circulating soluble uPAR should be a pathogenic factor per se causing proteinuria and onset of FSGS in mouse model systems could not be reproduced by several independent laboratories (Cathelin et al., 2014; Spinale et al., 2015; Harel et al., 2020). One independent group (Alfano et al., 2015) did, however, report that a single iv injection of 20 μg full-length soluble mouse uPAR induced proteinuria after 24 h, but these studies were conducted in Plaur–/– mice and therefore have little bearings on clinical FSGS—in common with the original studies by Reiser et al. Third, it is difficult to reconcile a causative pathogenic role of soluble uPAR in humans with the fact that patients with PNH have life-long and marked elevations of their plasma uPAR levels, but the renal dysfunction found in PNH patients can be treated with eculizumab showing that it is secondary to complement-mediated hemolysis (Kokoris et al., 2018).
Adding to the complexity in the tale of the pathogenic effects of soluble uPAR in FSGS, Reiser et al., reported that an alternative splice variant of mouse uPAR was the more potent nephrotoxin compared to the conventional soluble uPAR with intact LU-domains (Wei et al., 2011, 2019). Building on our extensive knowledge from protein structure-function analyses on uPAR, this proposal is very surprising. The alleged protein product from this alternative splice variant would thus comprise the N-terminal LU domain, the linker region and only half of the second LU domain and contain an uneven number of cysteine residues. Generally, the folding of LU domain proteins are very sensitive to mutations of their consensus cysteine residues leading to misfolded and aggregated proteins (Leth et al., 2019a). Notwithstanding these concerns, Reiser et al., reported a low resolution (17 Å) structure of this particular uPAR splice variant based on single particle reconstructions from electron microscopy images (Wei et al., 2019). Whether this structure actually represents the elusive uPAR splice variant in question remains nonetheless unclear to us, since their protein preparations also contained at least one dominating protein contamination of 50–80 kDa (as judged by their SDS-PAGE under reducing conditions). Furthermore, their native PAGE reveals a lot of protein aggregation as would be predicted for this construct from our general knowledge of LU domain protein stability (Leth et al., 2019a). As a consequence, we still consider the existence of such a properly folded truncated uPAR splice variant for highly speculative both in vitro and in vivo.
uPAR in Rheumatoid Arthritis
Chronic rheumatoid arthritis (RA) is a progressive inflammatory disease that ultimately leads to irreversible joint destruction by an invading pannus causing cartilage degradation and bone erosion. Studies on biopsies from arthritic lesions in RA patients by in situ zymography revealed pronounced elevations in uPA activity in the hyperproliferative synovial lining (Busso et al., 1997). Accordingly, these lesions express markedly elevated levels of both uPA and uPAR by infiltrating neutrophils, macrophages, and fibroblast-like cells in the inflamed RA synovium (Almholt et al., 2018) and increased plasma levels of soluble uPAR correlate to disease activity (Slot et al., 1999; Enocsson et al., 2021).
Several mouse models have been developed as experimental surrogates of rheumatoid arthritis in humans; please consult Buckley et al. (2019) for a comprehensive review. One widely used mouse model that mimics non-septic systemic polyarthritis in humans is the autoimmune collagen type-II induced arthritis model (CIA). Elegant genetic dissections, provided strong evidence for the causal role of uPA-mediated plasminogen activation in driving inflammatory CIA. First, Plg–/– mice were shown to be resilient to CIA and to a cocktail of mouse monoclonal anti-collagen type-II auto-antibodies, but daily administration of purified human plasminogen restored their sensitivity to these treatments (Li et al., 2005). Second, Plau–/– and Plaur–/– mice showed none or only very mild symptoms of arthritis after being challenged with collagen type-II (Thornton et al., 2017). In all three genotypes, the adaptive immune response was intact and mounted a similar response toward collagen type-II as did littermate wild-type control mice. With reciprocal bone marrow transplantations, the impact of uPAR to disease incidence and severity was traced to cells originating from the bone-marrow compartment with inflammatory monocyte/macrophages being the likely culprits (Thornton et al., 2017).
Prompted by the beneficial impact of Plau–/– and Plaur–/– on the incident and severity of CIA, Almholt et al. (2018) used neutralizing murine monoclonal antibodies toward mouse uPA (mU1) or mouse uPAR (mR1) to treat progression of arthritis in the CIA model. Aligned with the genetic data, inhibition of uPA catalytic activity with mU1 markedly alleviated the pathology and the adverse disease progression of CIA. Unexpectedly, treatment with mR1 showed no beneficial effect, which is unexpected given the beneficial effects observed in either (i) Plaur–/– mice (Thornton et al., 2017) or (ii) in a gene therapy approach competing the uPA•uPAR interaction with a hybrid molecule between human serum albumin (HSA) and the receptor binding amino-terminal fragment (ATF) of uPA (Apparailly et al., 2002). Although speculative, it is possible that the inhibitory properties of mR1 is inadequate for such long-term treatment studies. The inhibitory mechanism of mR1 is unique in the sense that it traps a partially open conformation of unoccupied uPAR by binding to DI, but in the presence of elevated concentrations of uPA a ternary complex will form and this will drive uPAR into the closed conformation and thereby expel the bound mR1 (Gardsvoll et al., 2011a). The reverse scenario, where mR1 displaces uPAR-bound uPA, is not an option, since mR1 does not recognize the closed conformation of uPAR in the uPA•uPAR complex (Pass et al., 2007).
In vivo Targeting of uPAR in Non-Invasive Imaging Modalities
The pronounced expression of uPAR at the invasive tumor-stroma microenvironment of most solid human cancers along with the correlation of high uPAR levels with poor patient prognosis, renders uPAR an attractive target for treatment modalities in aggressive cancers (Romer et al., 2004). Initially, such intervention programs primarily focused on function inhibition to dampen surface associated plasminogen activation (Crowley et al., 1993; Schmiedeberg et al., 2002; Lin et al., 2020; Yuan et al., 2021), but with the limited expression of uPAR in vital tissues, the focus gradually shifted toward targeted interventions based on cytotoxic eradication of uPAR expressing cells. Such uPAR-targeted treatment modalities include (i) recruiting the immune response to eliminate uPAR expressing cells using CAR-T cells (Amor et al., 2020) or priming the adaptive immune response with uPAR-targeted haptens (Rullo et al., 2016), (ii) proteolytic activation of prodrugs by uPAR-bound uPA (Liu et al., 2003; Gerspach et al., 2006; Schafer et al., 2011), (iii) uPAR-mediated internalization of cytotoxin-conjugated uPA-derivatives (Waldron et al., 2012; Zuppone et al., 2020) or antibodies (Harel et al., 2019), and (iv) targeted radiotherapy (Knör et al., 2008; Persson et al., 2012c; LeBeau et al., 2013). Notwithstanding the low uPAR expression in most vital tissues, the baseline expression in the glomeruli of normal kidneys may, however, pose a concern for such cytotoxic treatment modalities.
Targeting uPAR With Peptide Antagonists
Although a wide array of intervention strategies based on uPAR-targeting have been designed and tested in preclinical animal studies, we will in the next sections primarily focus on studies using one particular 9-mer antagonist peptide denoted AE105 (Figures 1D,E) that binds human uPAR with high affinity. A 15-mer precursor peptide of AE105 was originally selected in an unbiased phage-display library and it bound uPAR with a KD of 10 nM (Goodson et al., 1994). Using a functional scanning strategy, this 15-mer peptide was subsequently truncated to a decamer without substantial loss of affinity (Ploug, 1998; Ploug et al., 1998a). That truncated variant was subsequently used as template for the generation of focused combinatorial chemical bead-libraries with a view to affinity maturation and improvement in the biological stability of the peptide (Ploug et al., 2001). Panning these libraries with purified uPAR identified the 9-mer lead peptide (AE105), which bound uPAR with a KD of 7 nM and exhibited a remarkable serum stability due its composition of both (L)-, (D)-, and non-natural amino acids (Figure 1D; Ploug et al., 2001). Derivatives of this 9-mer peptide was instrumental for solving the first crystal structure of human uPAR (Figure 1E; Llinas et al., 2005) and proved their diligence as the highly efficient uPAR-targeting core of several imaging probes used for non-invasive imaging of uPAR expression in patients with solid cancers (Ploug, 2013; Persson et al., 2015). The versatile applications of AE105 as uPAR-targeting moiety is illustrated in Table 1.
TABLE 1
| Compound | Reporter | Application | Model system | References |
|
|
||||
| Non-invasive radionuclide imaging of uPAR expression | ||||
| [Cu2+]-DOTA–AE105 [Cu2+]-DOTA–AE105 [Cu2+]-CB-TE2A–AE105 [Cu2+]-CB-TE2A-PA–AE105 [Cu2+]-CHS1–AE105 [Cu2+]-DOTA–AE105 | 64Cu (PET) 64Cu (PET) 64Cu (PET) 64Cu (PET) 64Cu & Cy5.5 64Cu (PET) | Preclinical Preclinical Preclinical Preclinical Preclinical Phase-1 trial | U87MG, MDA-MB-435 H727, U87MG, HT29 U87MG U87MG U87MG breast & prostate | Li et al., 2008 Persson et al., 2012a Persson et al., 2013a Persson et al., 2013a Sun et al., 2015 Persson et al., 2015 |
| [Ga3+]-DOTA–AE105 [Ga3+]-NODAGA–AE105 [Ga3+]-NOTA–AE105 [Ga3+]-NOTA–AE105 [Ga3+]-NOTA–AE105 | 68Ga (PET) 68Ga (PET) 68Ga (PET) 68Ga (PET) 68Ga (PET) | Preclinical Preclinical Orthotopic Phase-1 trial Phase-2 trial | U87MG U87MG glioblastoma PDX breast, prostate, bladder prostate | Persson et al., 2012b Persson et al., 2012b; Vats et al., 2018Persson et al., 2016Skovgaard et al., 2017; Fosbol et al., 2021a, b |
| [AlF]-NOTA–AE105 | 18F (PET) | Preclinical | PC-3 | Persson et al., 2013b |
| [Tc2+]-DPA–AE105 | 99mTc (SPECT) | Preclinical | PC-3 | Kasten et al., 2016 |
| [In3+]-DOTA–(AE105)2 | 111In (SPECT) | Preclinical | MDA-MB-231 | Liu et al., 2009 |
|
|
||||
| Fluorescence-guided optical imaging of uPAR expression | ||||
|
|
||||
| ICG-linker1–AE105 ICG-linker1–AE105 ICG-linker1–AE105 CH1055-linker2–AE105 IRDye800CW-linker3–AE105 | ICG ICG ICG CH1055 IRDye800CW | Preclinical Orthotopic Orthotopic Orthotopic Orthotopic | U87MG OSC-19-luc2 BxPC3-luc2 U87MG U87MG | Juhl et al., 2016 Christensen et al., 2017 Juhl et al., 2019 Kurbegovic et al., 2018 Kurbegovic et al., 2021 |
|
|
||||
| Targeted radiotherapy toward uPAR expressing cells | ||||
|
|
||||
| [Lu2+]-DOTA–AE105 [Bi3+]-DOTA–(linker4-AE105)2 | 177Lu 213Bi | Preclinical Preclinical | HT29, BxPC3-luc2 OV-MZ-6 | Persson et al., 2012c, 2014Knör et al., 2008 |
|
|
||||
| uPAR-targeting of nanoparticles for drug delivery | ||||
|
|
||||
| Dansyl-linker5–AE105 AE105 Cu2_x-RB@DMSN–AE105 AuNRs@SiO2–AE105 GNS@Ir@P–AE105 | INOP INOP DMSN AuNRs@SiO2 Photoacustic | Cell culture Cell culture Preclinical Preclinical Preclinical | HEK293–uPAR cells PC-3 cells OSC-19 HeLa MDA-MD-231 | Hansen et al., 2013 Ahmed et al., 2017 Zuo et al., 2020 Hu et al., 2019 Yu et al., 2020 |
|
|
||||
| Miscellaneous applications of the uPAR-targeting peptide | ||||
|
|
||||
| Sepharose–(AE105)2 FAM–AE147 (AE105)2 AE147 | Sepharose Fluorescein None Hg modification | Purification FP-assay ELISA Structure | Cell culture media Small molecule inhbitors Human plasma samples uPAR•AE147 crystals | Jacobsen et al., 2007 Mani et al., 2013 Piironen et al., 2004 Llinas et al., 2005 |
Versatility in the applications of the uPAR targeting peptide AE105.
AuNRs@SiO2, mesoporous silica-coated Au nanorods; CB-TE2A, 4,11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane; CB-TE2A-PA, 4-carboxymethyl-11-(1,3-dicarboxypropyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane; DMSN, dendritic mesoporous silica nanoparticles; DOTA, 1,4,7,10-tetraazadodecane-N,N’,N”,N”’-tetraacetic acid; DPA:2,2’, dipicolylamine; ICG, indocyanine green; INOP, ion oxide nanoparticle; Linker1, EE; Linker2, EEEE; Linker3, EE(OEG)2; Linker4, KGSGG; Linker5, KGSGSGG; NODAGA, 2-(4,7-bis(carboxymethyl)-1,4,7-triazonan-1-yl)pentanedioic acid; NOTA, 1,4,7-triazacyclononane-1,4,7-triacetic acid; PDX, patient derived xenograft.
Positron Emission Tomography
Position emission tomography (PET) combined with computed tomography (CT) constitute the central imaging platform in clinical oncology (Duclos et al., 2021). The overarching virtue of PET/CT is that it is (i) non-invasive, (ii) provides an overview of the global expression of a given molecular target or function shortly after injection of the radionuclide tracer, (iii) has no issues with sampling bias, and (iv) is highly versatile in the sense that the collection of molecular targets that can be visualized depends only on the availability of a suitable PET probe. Although 18F-fluorodeoxyglucose (18F-FDG) is the unopposed toiler of PET imaging in nuclear medicine, new peptide-based PET probes are continuously being developed thus widening the range of molecular targets that can be visualized (Askari Rizvi and Zhang, 2021). As alluded to in the previous sections, the increased uPAR-expression in the tumor-stromal microenvironment of invading cancer lesions makes it an attractive target for molecular imaging in the clinical assessment of tumor invasion and metastatic dissemination. As listed in Table 1, a plethora of PET-probes, targeting uPAR via the AE105 moiety, have been designed and explored in preclinical animal models bearing various human cancers (subcutaneous, orthotopic, and patient derived xenografts). These studies include xenografts from different origins (e.g., brain, prostate, lung, and breast) and use PET tracers combining various macrocyclic chelators (DOTA, NOTA, CB-TE2, CB-TE2A-PA) with different radionuclides (64Cu, 68Ga, Al19F) - as shown in Table 1 (Price and Orvig, 2014). All studies showed specific uptake in tumor lesions and the uptake levels correlated to uPAR levels as determined by ELISA or immunohistochemistry in resected subcutaneous tumor xenografts (Persson et al., 2012a, 2013a). It should be emphasized that the endogenous expression of mouse uPAR will not be detected in these imaging studies as the AE105 targeting moiety is strictly specific for human uPAR (Ploug et al., 2001). Building on these encouraging preclinical mouse studies, the first phase-1 clinical trials were conducted in human cancer patients with 64Cu-DOTA–AE105 (Persson et al., 2015) and 68Ga-NOTA–AE105 (Skovgaard et al., 2017). Both studies showed no adverse pharmacological effects and internal radiation burdens were equivalent to that of standard 19F-FDG PET scans. Importantly, specific tracer uptake was noted in primary lesions as well as in several lymph node metastases and subsequent immunohistochemistry on resected tumor tissue confirmed that all these lesions were indeed uPAR positive. Circumstantial observations from these phase-1 clinical trials demonstrate the power of such global and unbiased uPAR–PET imaging (Figure 3). First, one patient enrolled with an indication of breast cancer revealed an unexpected additional tracer uptake in the brain, which was later diagnosed as a primary meningioma (Persson et al., 2015). Second, another breast cancer patient had a robust tracer uptake in two axillary lymph nodes despite previous preoperative staging by ultrasound, fine-needle biopsy, and CT failed to detect any malignant lymph node involvement (Skovgaard et al., 2017). A small prospective phase-2 clinical trial on 68Ga-NOTA–AE105 uptake in the primary prostate lesions of 27 cancer patients found a positive correlation between standard uptake values (SUVmax) and Gleason score based on histopathological grading of biopsy specimens (Fosbol et al., 2021a). In a small follow-up study, baseline SUVmax of 68Ga-NOTA–AE105 in bone metastasis index lesions of 14 prostate cancer patients were measured before two cycles of 223RaCl2 therapy. Despite the low number of patients enrolled in this study, baseline SUVmax correlated to overall survival (Fosbol et al., 2021b). Notwithstanding these promising results, large scale clinical trials are needed to precisely define the clinical utility of uPAR-PET scans in cancer patient management.
FIGURE 3
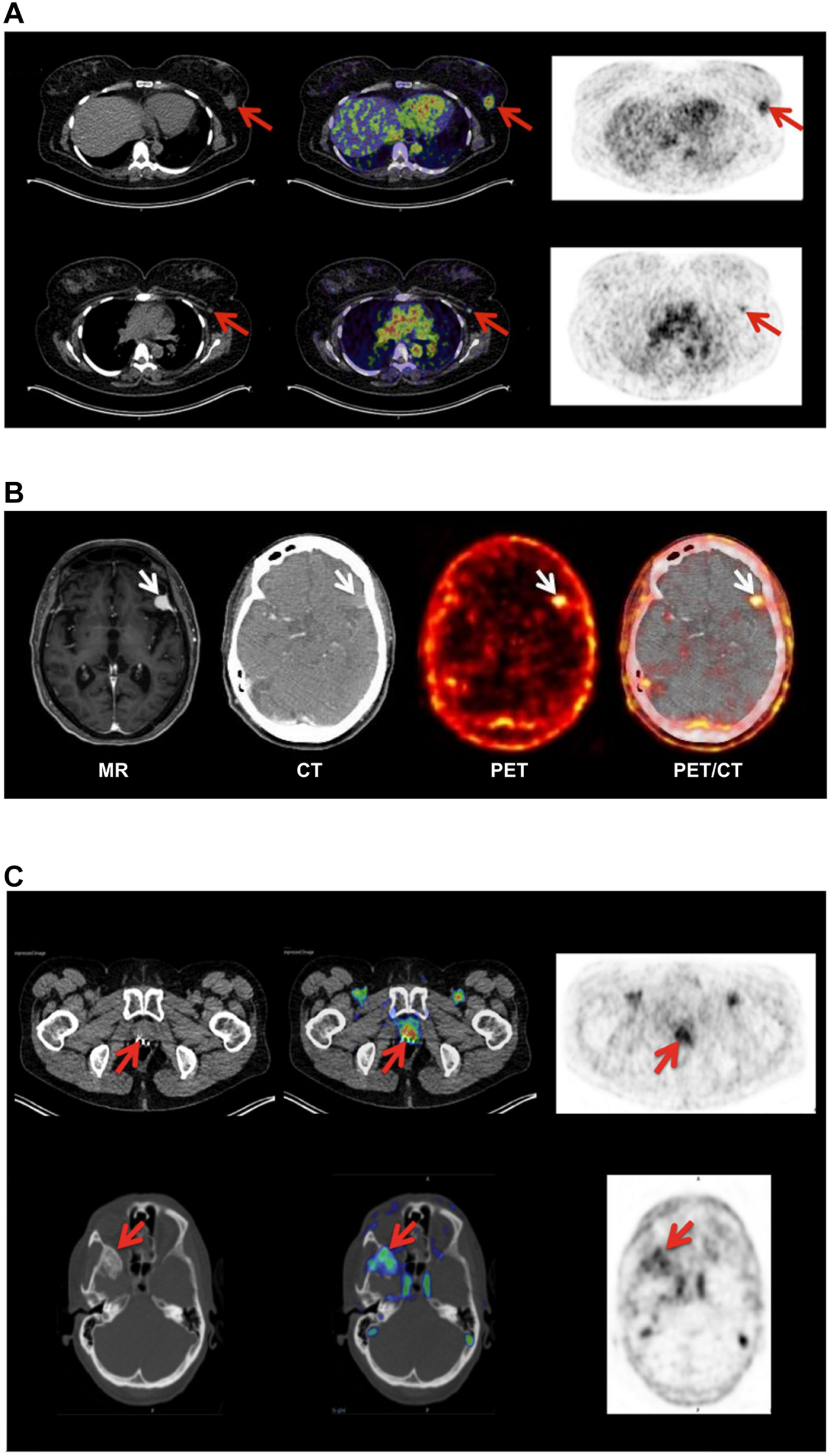
Non-invasive PET imaging of uPAR expression with AE105 as targeting principle. (A) Non-invasive PET/CT imaging of a breast cancer patient 10–60 min post iv injection of 68Ga-NOTA–AE105. Shown to the left are the CT scans, to the right PET scans, and in the middle the merged images. The red arrows highlight (i) the uPAR-positive primary breast cancer lesion (upper row) and (ii) an uPAR-positive axillary lymph node lesion (lower row) that escaped detection by preoperative ultrasound scanning and fine-needle biopsy. (B) Non-invasive PET/CT image of the transversal cross-section of the brain of another breast cancer patient with confirmed primary breast cancer lesion 60 min post iv injection of 64Cu-DOTA–AE105. High tracer uptake was unexpectedly noted in a brain lesion that was later confirmed by magnetic resonance scanning (MR) and diagnosed as a meningioma (white arrow). (C) PET/CT imaging of a prostate cancer patient using 68Ga-NOTA–AE105 to visualize uPAR expression. Red arrows highlight tracer accumulation in the primary prostate lesion (upper row) and in a metastasis lodged in the sphenoid bone (lower row). Images were reproduced from Persson et al. (2015) and Skovgaard et al. (2017) under the CC-BY license.
Due to the short half-lives of traditional positron emitting radionuclides [68Ga (67.7 min), 18F (109.7 min), and 64Cu (12.7 h)], the majority of PET probes rely on small molecules or peptides as their targeting moiety due a fast pharmacokinetic profile with rapid contrast development. The clinical implementation of immunotherapy created, however, an unmet need for immuno-PET imaging to stratify and select patients eligible for antibody-based therapy (Liberini et al., 2021). To create a better match with the slow pharmacokinetics of antibodies, most immuno-PET studies use 89Zr as positron emitting nuclide with a half-life of 3.3 days. So far this technology has not been applied to uPAR-PET imaging, but was used to image its high-affinity protease ligand uPA in subcutaneous tumors using an 89Zr-labeled mouse monoclonal antibody (ATN291) that is specific for human uPA (Yang et al., 2016). It is unclear whether this imaging represents uPA in complex with uPAR on the cells surface of the transplanted human cancer cells. Two different laboratories conducted low resolution imaging with single photon emission computed tomography (SPECT) to visualize uPAR expression in xenografts using the 111In-labeled monoclonal anti-huPAR antibodies ATN658 (Boonstra et al., 2015, 2017) and 2G10 (LeBeau et al., 2013, 2014). Both approaches provided clear primary tumor delineations 72 h post tracer iv injection and 111In-labeled 2G10 detected occult bone metastasis in mice with triple negative human breast cancer xenografts (LeBeau et al., 2013).
Fluorescence-Guided Intraoperative Imaging
The Holy Grail in cancer surgery is the radical removal of all malignant tissue creating tumor free resection margins while preserving as much of the healthy tissue as possible—ultimately leading to improvements in progression-free survival. While PET/CT and MRI imaging platforms are important for staging and localization of disseminated disease, they cannot easily guide surgical procedures in real-time. In the last decade, design of targeted near-infrared (NIR) probes for fluorescence-guided cancer surgery enabled the rapid development of emerging imaging tools that could potentially assist surgical navigation in real-time (Debie and Hernot, 2019). Although still in its infancy, targeted imaging in fluorescence-guided cancer surgery is gaining momentum especially with antibodies that are already applied in therapy or have a strong biomarker potential. But proof of concept studies demonstrating the added beneficial effects for cancer patient survival are still lacking (Hernot et al., 2019).
As alluded to in the previous sections, baseline expression of uPAR is low and scattered in normal healthy tissues while it is robustly upregulated in most active cancer lesions, particularly in the microenvironment of the invading tumor-stromal interface. This expression pattern makes uPAR an ideal target candidate in fluorescence guided intraoperative imaging and several strategies have accordingly been explored in preclinical mouse models with AE105 as the targeting core (Table 1). Using NIR fluorophores as reporter groups increases the depth of the surgical field that can be visualized due to a higher tissue penetration and a decreased auto-fluorescence of wavelengths in the NIR-I (650–900 nm) and NIR-II (1,000–1,700 nm) window. The first generation of uPAR-specific optical probes exploited the clinically approved fluorophore indocyanine green (ICG) as reporter group (Juhl et al., 2016). Although the conjugation of ICG to AE105 reduced the affinity for uPAR by 20-fold, initial optical imaging studies on subcutaneous xenotransplants of the U87MG glioblastoma cell line (Allen et al., 2016) led to a specific probe uptake reaching maximal tumor-to-background ratio (TBR) after 6–24 h post iv administration (Juhl et al., 2016). The ICG–AE105 conjugate also showed promise in the real-time guidance of preclinical surgery of xenotransplanted orthotropic mouse models of head-and-neck and pancreatic cancers (Christensen et al., 2017; Juhl et al., 2019). Building on these promising preclinical studies, a phase 1–2 clinical trial was launched in 2020 exploring the safety and use of ICG–AE105 in patients with glioblastoma (clinicaltrialsregister.eu, EudraCT 2020-003089-38). In second generation AE105-conjugates, ICG was replaced by fluorophores with more optimal spectral properties i.e., CH1055 (NIR-II) and IRDye800CW (Kurbegovic et al., 2018, 2021). Especially IRDye800CW–AE105 was optimized biochemically and analyzed extensively in a surrogate glioblastoma mouse model with U89MG cells as orthotropic transplants (Figures 4A,B) (Kurbegovic et al., 2021). Testing different linker sequences, the IRDye800CW-linker3–AE105 conjugate was selected (Table 1), which binds uPAR with only 3-fold lower affinity as compared to unconjugated AE105. Dependent on the amount of probe administered, maximal TBRs of 6.6 to 7.0 were reached within 1–3 h post iv injection. The fast kinetics in generating optimal contrast is remarkable and is most likely a consequence of the relatively small size and hydrophilicity of this probe as compared to ICG-AE105 (more hydrophobic) and the larger antibody based probes, where optimal contrast for the latter typically requires a much longer probe washout (3–5 days).
FIGURE 4
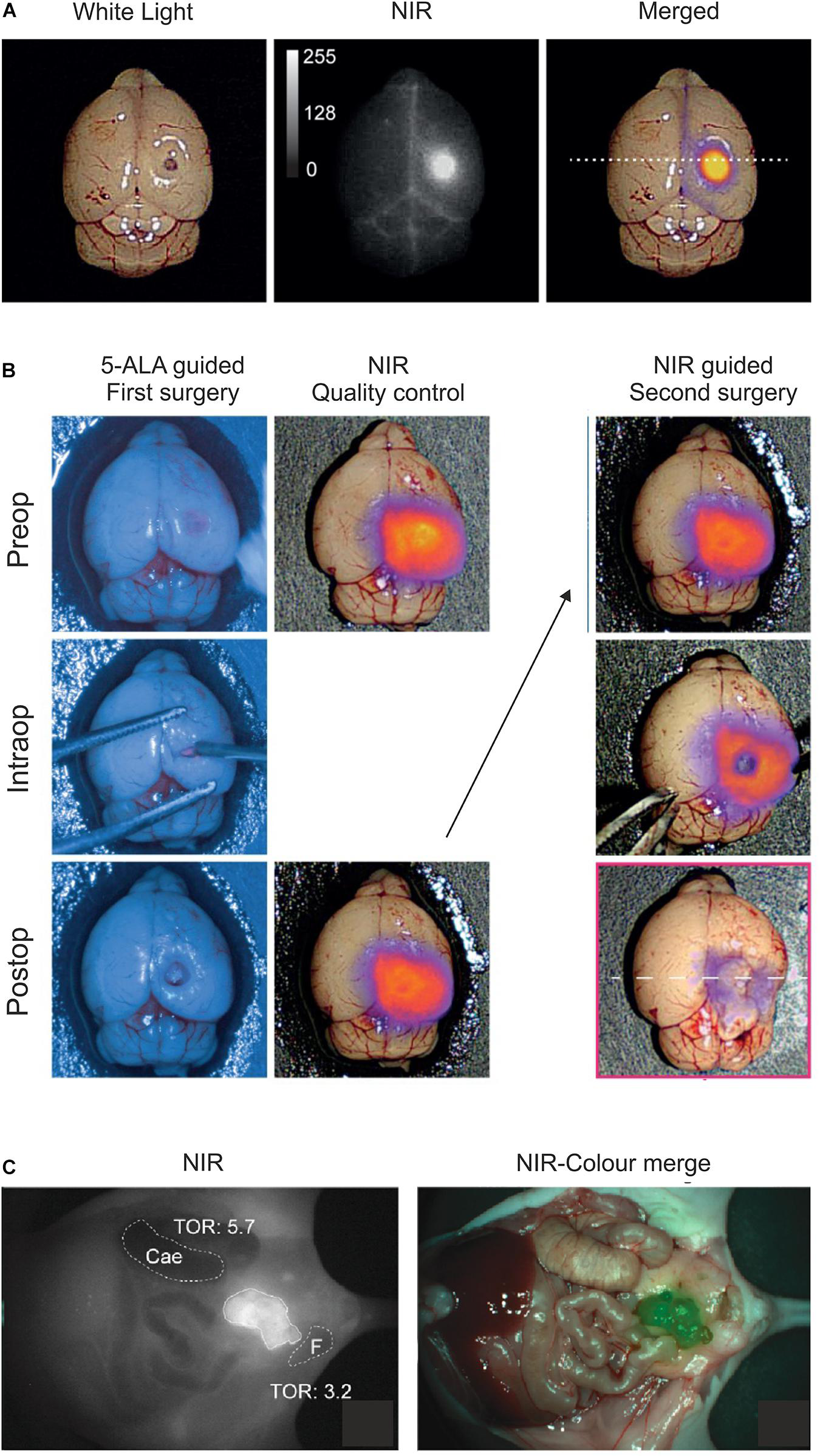
Optical imaging of uPAR expression with AE105 as targeting principle in mouse models. (A) Intact resected mouse brain with an orthotropic xenotransplant of U87MG cells imaged 1.5 h after iv injection of 6 nmol IRDye800CW-linker3–AE105. Left panel shows image with white light; middle panel shows NIR image; and right panel shows the merged image using pseudo-colors to illustrate relative tracer uptake—low to high uptake: blue→red→yellow. (B) Fluorescence guided intraoperative imaging following the surgical resection of an orthotropic U87MG transplant. Left panel shows the initial surgical brain resection guided by the real-time fluorescence signal from protoporphyrin IX after 5-aminolevulinic acid administration (5-ALA, Gliolan®); middle panel shows the corresponding NIR control images from IRDye800CW-linker3–AE105 fluorescence of the surgical bed before and after resection guided by 5-ALA; and the right panel shows the subsequent surgical resection guided by NIR fluorescence and visualized by a modified EleVisionTMIR system (Medtronic, MN, United States). (C) NIR and merged images recorded 3 days post iv injection of 1 nmol IRDye800CW–ATN658 (a humanized monoclonal anti-uPAR antibody) in a orthotropic mouse model of urothelial cell carcinoma. (A,B) were reproduced and modified from Kurbegovic et al. (2021) and (C) from Baart et al. (2021) according to the CC-BY license.
Besides peptide-based targeting of uPAR—the topic of this review—several macromolecular uPAR-targeting moieties have been developed for optical imaging and therapeutic targeting including (i) monoclonal anti-uPAR antibodies (Figure 4C; LeBeau et al., 2014; Boonstra et al., 2015; Baart et al., 2021) and (ii) various nanoparticles carrying receptor binding fragments of uPA (ATF) or peptide derivatives (AE105) (Hansen et al., 2007; Yang et al., 2013; Gao et al., 2017; Zuo et al., 2020). Due to the inherently long “washout times” (days rather than hours) needed for these probes to reach maximal TBR, they are probably less suited in the daily clinical workflow as imaging modalities, but they are optimally suited for targeted delivery of cytotoxic payloads.
Targeting uPAR With Therapeutic Intention
In the last two decades, a variety of different targeted intervention strategies have been developed to eradicate uPAR-expressing cells. Specificity of the cytotoxic insult were devised by targeting uPAR directly or by exploiting the proteolytic activity of uPAR-bound uPA (Liu et al., 2003; Rustamzadeh et al., 2007; Schafer et al., 2011; Morodomi et al., 2012; LeBeau et al., 2013; Jing et al., 2017; Harel et al., 2019; Amor et al., 2020; Zuppone et al., 2020). Several of these treatment modalities have shown promising results in preclinical mouse models bearing human cancer cell xenografts. One small study showed beneficial effects on canine oral mucosal melanomas inducing stable disease in all five dogs treated with uPA- and MMP2-activated anthrax toxin (Nishiya et al., 2020). But none of these modalities have so far entered clinical trials in humans. We predict that the non-invasive uPAR–PET imaging platforms discussed in this review will greatly facilitate the future clinical translation of a given uPAR-targeted therapy regimen—assisting in both initial patient selection based of target availability as well as response marker monitoring treatment efficacy.
Conclusion
Research performed in the last two decades have provided a detailed outline for the structure-function relationships in components responsible for cell surface associated plasminogen activation. This knowledge has been instrumental for designing a plethora of intervention strategies to target these components or their function with special emphasis on treatment modalities in oncology. Opportunities to target uPAR in inflammatory diseases beyond cancer should, however, also be considered (Baart et al., 2020).
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Author contributions
JL and MP wrote the manuscript. Both authors contributed to the article and approved the submitted version.
Funding
Research in the authors’ laboratories that was instrumental for writing this review was supported by grants from The John and Birthe Meyer Foundation and the Innovation Fund Denmark.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
1.^ATF, amino-terminal fragment of uPA; DI-DII-DIII, the first, second and third LU domain in uPAR; GFD, growth factor -like domain; GPI, glycosyl-phosphatidylinositol; ICG, indocyanine green; LU, Ly6/uPAR; NIR, near-infrared; PET, positron emission tomography; PNH, paroxysmal nocturnal hemoglobinuria; SMB, somatomedin B; tPA, tissue-type plasminogen activator; uPA, urokinase-type plasminogen activator; uPAR, uPA receptor.
2.^A baseline concentration of 20 pM prouPA—slightly below or equal to the KD for its interaction with uPAR—makes this interaction optimally suited to sense and respond to variations in prouPA concentrations during e.g., chronic inflammation.
3.^ Plau is the mouse gene encoding uPA where the key residues for binding to mouse uPAR (Tyr, Arg, Arg, and Arg) were replaced by the corresponding residues from human uPA (Asn, Asn, His, and Trp), which leads to an 400-fold decrease in the affinity for mouse uPAR (Connolly et al., 2010; Lin et al., 2010) thus essentially uncoupling that reaction.
References
1
Adeyo O. Oberer M. Ploug M. Fong L. G. Young S. G. Beigneux A. P. (2015). Heterogeneity in the properties of mutant secreted lymphocyte antigen 6/urokinase receptor-related protein 1 (SLURP1) in Mal de Meleda.Br. J. Dermatol.1731066–1069. 10.1111/bjd.13868
2
Ahmed M. S. U. Salam A. B. Yates C. Willian K. Jaynes J. Turner T. et al (2017). Double-receptor-targeting multifunctional iron oxide nanoparticles drug delivery system for the treatment and imaging of prostate cancer.Int. J. Nanomed.126973–6984. 10.2147/ijn.s139011
3
Aimes R. T. Regazzoni K. Quigley J. P. (2003). Human/chicken urokinase chimeras demonstrate sequences outside the serine protease domain that dictate autoactivation.Thromb. Haemost.89382–392. 10.1055/s-0037-1613456
4
Alfano M. Cinque P. Giusti G. Proietti S. Nebuloni M. Danse S. et al (2015). Full-length soluble urokinase plasminogen activator receptor down-modulates nephrin expression in podocytes.Sci. Rep.5:13647.
5
Allen M. Bjerke M. Edlund H. Nelander S. Westermark B. (2016). Origin of the U87MG glioma cell line: good news and bad news.Sci. Transl. Med.8:354re353. 10.1126/scitranslmed.aaf6853
6
Allgayer H. (2010). Translational research on u-PAR.Eur. J. Cancer461241–1251.
7
Almholt K. Hebsgaard J. B. Nansen A. Andersson C. Rønø B. et al (2018). Antibody-mediated neutralization of uPA proteolytic function reduces disease progression in mouse arthritis models.J. Immunol.200957–965. 10.4049/jimmunol.1701317
8
Almholt K. Lærum O. D. Nielsen B. S. Lund I. K. Lund L. R. Rømer J. et al (2015). Spontaneous lung and lymph node metastasis in transgenic breast cancer is independent of the urokinase receptor uPAR.Clin. Exp. Metastasis32543–554. 10.1007/s10585-015-9726-1
9
Alpízar-Alpízar W. Skindersoe M. E. Rasmussen L. Kriegbaum M. C. Christensen I. J. Lund I. K. et al (2020). Helicobacter pylori colonization drives urokinase receptor (uPAR) expression in murine gastric epithelium during early pathogenesis.Microorganisms8:1019. 10.3390/microorganisms8071019
10
Amor C. Feucht J. Leibold J. Ho Y. J. Zhu C. Alonso-Curbelo D. et al (2020). Senolytic CAR T cells reverse senescence-associated pathologies.Nature583127–132. 10.1038/s41586-020-2403-9
11
Andreasen P. A. Egelund R. Petersen H. H. (2000). The plasminogen activation system in tumor growth, invasion, and metastasis.Cell Mol. Life Sci.5725–40. 10.1007/s000180050497
12
Andronicos N. M. Chen E. I. Baik N. Bai H. Parmer C. M. Kiosses W. B. et al (2010). Proteomics-based discovery of a novel, structurally unique, and developmentally regulated plasminogen receptor, Plg-RKT, a major regulator of cell surface plasminogen activation.Blood1151319–1330. 10.1182/blood-2008-11-188938
13
Apparailly F. Bouquet C. Millet V. Noel D. Jacquet C. Opolon P. et al (2002). Adenovirus-mediated gene transfer of urokinase plasminogen inhibitor inhibits angiogenesis in experimental arthritis.Gene Ther.9192–200. 10.1038/sj.gt.3301628
14
Askari Rizvi S. F. Zhang H. (2021). Emerging trends of receptor-mediated tumor targeting peptides: a review with perspective from molecular imaging modalities.Eur. J. Med. Chem.221:113538. 10.1016/j.ejmech.2021.113538
15
Baart V. M. Houvast R. D. de Geus-Oei L. F. Quax P. H. A. Kuppen P. J. K. Vahrmeijer A. L. et al (2020). Molecular imaging of the urokinase plasminogen activator receptor: opportunities beyond cancer.EJNMMI Res.10:87. 10.1186/s13550-020-00673-7
16
Baart V. M. van der Horst G. Deken M. M. Bhairosingh S. S. Schomann T. Sier V. Q. et al (2021). A multimodal molecular imaging approach targeting urokinase plasminogen activator receptor for the diagnosis, resection and surveillance of urothelial cell carcinoma.Eur. J. Cancer14611–20. 10.1016/j.ejca.2021.01.001
17
Bager R. Kristensen T. K. Jensen J. K. Szczur A. Christensen A. Andersen L. M. et al (2012). Urokinase-type plasminogen activator-like proteases in teleosts lack genuine receptor-binding epidermal growth factor-like domains.J. Biol. Chem.28727526–27536. 10.1074/jbc.m112.369207
18
Barinka C. Parry G. Callahan J. Shaw D. E. Kuo A. Bdeir K. et al (2006). Structural basis of interaction between urokinase-type plasminogen activator and its receptor.J. Mol. Biol.363482–495.
19
Barton S. J. Koppelman G. H. Vonk J. M. Browning C. A. Nolte I. M. Stewart C. E. et al (2009). PLAUR polymorphisms are associated with asthma, PLAUR levels, and lung function decline.J. Allergy Clin. Immunol.1231391–1400. 10.1016/j.jaci.2009.03.014
20
Behrendt N. Ploug M. Patthy L. Houen G. Blasi F. Danø K. (1991). The ligand-binding domain of the cell surface receptor for urokinase-type plasminogen activator.J. Biol. Chem.2667842–7847. 10.1016/s0021-9258(20)89526-x
21
Beigneux A. P. Fong L. G. Bensadoun A. Davies B. S. Oberer M. Gårdsvoll H. et al (2015). GPIHBP1 missense mutations often cause multimerization of GPIHBP1 and thereby prevent lipoprotein lipase binding.Circ. Res.116624–632. 10.1161/circresaha.116.305085
22
Bifulco K. Longanesi-Cattani I. Gala M. Di Carluccio G. Masucci M. T. Pavone M. et al (2010). The soluble form of urokinase receptor promotes angiogenesis through its Ser88-Arg-Ser-Arg-Tyr92 chemotactic sequence.J. Thromb. Haemost.82789–2799. 10.1111/j.1538-7836.2010.04075.x
23
Blasi F. Sidenius N. (2010). The urokinase receptor: focused cell surface proteolysis, cell adhesion and signaling.FEBS Lett.5841923–1930. 10.1016/j.febslet.2009.12.039
24
Bolon I. Zhou H. M. Charron Y. Wohlwend A. Vassalli J. D. (2004). Plasminogen mediates the pathological effects of urokinase-type plasminogen activator overexpression.Am. J. Pathol.1642299–2304. 10.1016/s0002-9440(10)63786-8
25
Boonstra M. C. van Driel P. B. van Willigen D. M. Stammes M. A. Prevoo H. A. Tummers Q. R. J. G. et al (2015). uPAR-targeted multimodal tracer for pre- and intraoperative imaging in cancer surgery.Oncotarget614260–14273. 10.18632/oncotarget.3680
26
Boonstra M. C. Van Driel P. Keereweer S. Prevoo H. Stammes M. A. Baart V. M. et al (2017). Preclinical uPAR-targeted multimodal imaging of locoregional oral cancer.Oral Oncol.661–8. 10.1016/j.oraloncology.2016.12.026
27
Brungs D. Chen J. Aghmesheh M. Vine K. L. Becker T. M. Carolan M. G. et al (2017). The urokinase plasminogen activation system in gastroesophageal cancer: a systematic review and meta-analysis.Oncotarget823099–23109. 10.18632/oncotarget.15485
28
Buckley B. J. Ali U. Kelso M. J. Ranson M. (2019). The urokinase plasminogen activation system in rheumatoid arthritis: pathophysiological roles and prospective therapeutic targets.Curr. Drug Targets20970–981. 10.2174/1389450120666181204164140
29
Bugge T. H. Flick M. J. Danton M. J. Daugherty C. C. Rømer J. Danø K. et al (1996a). Urokinase-type plasminogen activator is effective in fibrin clearance in the absence of its receptor or tissue-type plasminogen activator.Proc. Natl. Acad. Sci. U.S.A.935899–5904.
30
Bugge T. H. Flick M. J. Daugherty C. C. Degen J. L. (1995a). Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction.Genes Dev.9794–807. 10.1101/gad.9.7.794
31
Bugge T. H. Kombrinck K. W. Flick M. J. Daugherty C. C. Danton M. J. Degen J. L. (1996b). Loss of fibrinogen rescues mice from the pleiotropic effects of plasminogen deficiency.Cell87709–719. 10.1016/s0092-8674(00)81390-2
32
Bugge T. H. Suh T. T. Flick M. J. Daugherty C. C. Rømer J. Solberg H. et al (1995b). The receptor for urokinase-type plasminogen activator is not essential for mouse development or fertility.J. Biol. Chem.27016886–16894.
33
Busso N. Peclat V. So A. Sappino A. P. (1997). Plasminogen activation in synovial tissues: differences between normal, osteoarthritis, and rheumatoid arthritis joints.Ann. Rheum. Dis.56550–557. 10.1136/ard.56.9.550
34
Caiolfa V. R. Zamai M. Malengo G. Andolfo A. Madsen C. D. Sutin J. et al (2007). Monomer dimer dynamics and distribution of GPI-anchored uPAR are determined by cell surface protein assemblies.J. Cell Biol.1791067–1082. 10.1083/jcb.200702151
35
Casey J. R. Petranka J. G. Kottra J. Fleenor D. E. Rosse W. F. (1994). The structure of the urokinase-type plasminogen activator receptor gene.Blood841151–1156.
36
Cathelin D. Placier S. Ploug M. Verpont M. C. Vandermeersch S. Luque Y. et al (2014). Administration of recombinant soluble urokinase receptor per se is not sufficient to induce podocyte alterations and proteinuria in mice.J. Am. Soc. Nephrol.251662–1668. 10.1681/asn.2013040425
37
Chana-Muñoz A. Jendroszek A. Sønnichsen M. Wang T. Ploug M. Jensen J. K. et al (2019). Origin and diversification of the plasminogen activation system among chordates.BMC Evol. Biol.19:27. 10.1186/s12862-019-1353-z
38
Christensen A. Juhl K. Persson M. Charabi B. W. Mortensen J. Kiss K. et al (2017). uPAR-targeted optical near-infrared (NIR) fluorescence imaging and PET for image-guided surgery in head and neck cancer: proof-of-concept in orthotopic xenograft model.Oncotarget815407–15419. 10.18632/oncotarget.14282
39
Connolly B. M. Choi E. Y. Gårdsvoll H. Bey A. L. Currie B. M. Chavakis T. et al (2010). Selective abrogation of the uPA-uPAR interaction in vivo reveals a novel role in suppression of fibrin-associated inflammation.Blood1161593–1603. 10.1182/blood-2010-03-276642
40
Crowley C. W. Cohen R. L. Lucas B. K. Liu G. Shuman M. A. Levinson A. D. (1993). Prevention of metastasis by inhibition of the urokinase receptor.Proc. Natl. Acad. Sci. U.S.A.905021–5025. 10.1073/pnas.90.11.5021
41
Dano K. Andreasen P. A. Grøndahl-Hansen J. Kristensen P. Nielsen L. S. Skriver L. (1985). Plasminogen activators, tissue degradation, and cancer.Adv. Cancer Res.44139–266. 10.1016/s0065-230x(08)60028-7
42
De Lorenzi V. Sarra Ferraris G. M. Madsen J. B. Lupia M. Andreasen P. A. Sidenius N. (2016). Urokinase links plasminogen activation and cell adhesion by cleavage of the RGD motif in vitronectin.EMBO Rep.17982–998. 10.15252/embr.201541681
43
Debie P. Hernot S. (2019). Emerging fluorescent molecular tracers to guide intra-operative surgical decision-making.Front. Pharmacol.10:510. 10.3389/fphar.2019.00510
44
Dohn L. H. Illemann M. Høyer-Hansen G. Christensen I. J. Hostmark J. Litlekalsoy J. et al (2015). Urokinase-type plasminogen activator receptor (uPAR) expression is associated with t-stage and survival in urothelial carcinoma of the bladder.Urol. Oncol.165e15–e24. 10.1016/j.urolonc.2014.12.001
45
Duclos V. Iep A. Gomez L. Goldfarb L. Besson F. L. (2021). PET molecular imaging: a holistic review of current practice and emerging perspectives for diagnosis, therapeutic evaluation and prognosis in clinical oncology.Int. J. Mol. Sci.224159–4185. 10.3390/ijms22084159
46
Duffy M. J. Reilly D. O’Sullivan C. O’Higgins N. Fennelly J. J. Andreasen P. (1990). Urokinase-plasminogen activator, a new and independent prognostic marker in breast cancer.Cancer Res.506827–6829.
47
Eden G. Archinti M. Furlan F. Murphy R. Degryse B. (2011). The urokinase receptor interactome.Curr. Pharm. Des.171874–1889. 10.2174/138161211796718215
48
Ellis V. Behrendt N. Danø K. (1991). Plasminogen activation by receptor-bound urokinase. a kinetic study with both cell-associated and isolated receptor.J. Biol. Chem.26612752–12758. 10.1016/s0021-9258(18)98963-5
49
Enocsson H. Lukic T. Ziegelasch M. Kastbom A. (2021). Serum levels of the soluble urokinase plasminogen activator receptor (suPAR) correlates with disease activity in early rheumatoid arthritis and reflects joint damage over time.Transl. Res.232142–149. 10.1016/j.trsl.2021.02.007
50
Estreicher A. Muhlhauser J. Carpentier J. L. Orci L. Vassalli J. D. (1990). The receptor for urokinase type plasminogen activator polarizes expression of the protease to the leading edge of migrating monocytes and promotes degradation of enzyme inhibitor complexes.J. Cell Biol.111783–792. 10.1083/jcb.111.2.783
51
Ferraris G. M. Schulte C. Buttiglione V. De Lorenzi V. Piontini A. Piontini A. et al (2014). The interaction between uPAR and vitronectin triggers ligand-independent adhesion signalling by integrins.EMBO J.332458–2472. 10.15252/embj.201387611
52
Fischer A. (1946). Mechanism of the proteolytic activity of malignant tissue cells.Nature157:442. 10.1038/157442c0
53
Fosbol M. O. Kurbegovic S. Johannesen H. H. Røder M. A. Hansen A. E. Mortensen J. et al (2021a). Urokinase-type plasminogen activator receptor (uPAR) PET/MRI of prostate cancer for noninvasive evaluation of aggressiveness: comparison with gleason score in a prospective phase 2 clinical trial.J. Nucl. Med.62354–359. 10.2967/jnumed.120.248120
54
Fosbol M. O. Mortensen J. Petersen P. M. Loft A. Madsen J. Kjær A. (2021b). uPAR PET/CT for prognostication and response assessment in patients with metastatic castration-resistant prostate cancer undergoing radium-223 therapy: a prospective phase II study.Diagnostics11:1087. 10.3390/diagnostics11061087
55
Gao N. Bozeman E. N. Qian W. Wang L. Chen H. Lipowska M. et al (2017). Tumor penetrating theranostic nanoparticles for enhancement of targeted and image-guided drug delivery into peritoneal tumors following intraperitoneal delivery.Theranostics71689–1704. 10.7150/thno.18125
56
Gardsvoll H. Ploug M. (2007). Mapping of the vitronectin-binding site on the urokinase receptor: involvement of a coherent receptor interface consisting of residues from both domain I and the flanking interdomain linker region.J. Biol. Chem.28213561–13572. 10.1074/jbc.m610184200
57
Gardsvoll H. Gilquin B. Le, Du M. H. Ménèz A. Jørgensen T. J. et al (2006). Characterization of the functional epitope on the urokinase receptor. complete alanine scanning mutagenesis supplemented by chemical cross-linking.J. Biol. Chem.28119260–19272. 10.1074/jbc.m513583200
58
Gardsvoll H. Jacobsen B. Kriegbaum M. C. Behrendt N. Engelholm L. Østergård S. et al (2011a). Conformational regulation of urokinase receptor function: impact of receptor occupancy and epitope-mapped monoclonal antibodies on lamellipodia induction.J. Biol. Chem.28633544–33556. 10.1074/jbc.m111.220087
59
Gardsvoll H. Kjaergaard M. Jacobsen B. Kriegbaum M. C. Huang M. Ploug M. (2011b). Mimicry of the regulatory role of urokinase in lamellipodia formation by introduction of a non-native interdomain disulfide bond in its receptor.J. Biol. Chem.28643515–43526. 10.1074/jbc.m111.300020
60
Gårdsvoll H. Kriegbaum M. C. Hertz E. P. Alpízar-Alpízar W. Ploug M. (2013). The urokinase receptor homolog haldisin is a novel differentiation marker of stratum granulosum in squamous epithelia.J. Histochem. Cytochem.61802–813. 10.1369/0022155413501879
61
Gardsvoll H. Werner F. Søndergård L. Danø K. Ploug M. (2004). Characterization of low-glycosylated forms of soluble human urokinase receptor expressed in Drosophila Shneider 2 cells after deletion of glycosylation-sites.Protein Expr. Purif.34284–295. 10.1016/j.pep.2003.12.002
62
Gerspach J. Nemeth J. Munkel S. Wajant H. Pfizenmaier K. (2006). Target-selective activation of a TNF prodrug by urokinase-type plasminogen activator (uPA) mediated proteolytic processing at the cell surface.Cancer Immunol. Immunother.551590–1600. 10.1007/s00262-006-0162-6
63
Gonias S. L. Hu J. (2015). Urokinase receptor and resistance to targeted anticancer agents.Front. Pharmacol.6:154. 10.3389/fphar.2015.00154
64
Goodson R. J. Doyle M. V. Kaufman S. E. Rosenberg S. (1994). High-affinity urokinase receptor antagonists identified with bacteriophage peptide display.Proc. Natl. Acad. Sci. U.S.A.917129–7133. 10.1073/pnas.91.15.7129
65
Grant G. A. Luetje C. W. Summers R. Xu X. L. (1998). Differential roles for disulfide bonds in the structural integrity and biological activity of kappa-bungarotoxin, a neuronal nicotinic acetylcholine receptor antagonist.Biochemistry3712166–12171. 10.1021/bi981227y
66
Grunnet M. Christensen I. J. Lassen U. Jensen L. H. Lydolph M. Lund I. K. et al (2014). Prognostic significance of circulating intact and cleaved forms of urokinase plasminogen activator receptor in inoperable chemotherapy treated cholangiocarcinoma patients.Clin. Biochem.47599–604. 10.1016/j.clinbiochem.2014.01.030
67
Hansen L. V. Gårdsvoll H. Nielsen B. S. Lund L. R. Danø K. Jensen O. N. et al (2004). Structural analysis and tissue localization of human C4.4A: a protein homologue of the urokinase receptor.Biochem. J.380(Pt 3), 845–857. 10.1042/bj20031478
68
Hansen L. V. Skov B. G. Ploug M. Pappot H. (2007). Tumour cell expression of C4.4A, a structural homologue of the urokinase receptor, correlates with poor prognosis in non-small cell lung cancer.Lung Cancer58260–266. 10.1016/j.lungcan.2007.06.025
69
Hansen L. Larsen E. K. Nielsen E. H. Iversen F. Liu Z. Thomsen K. et al (2013). Targeting of peptide conjugated magnetic nanoparticles to urokinase plasminogen activator receptor (uPAR) expressing cells.Nanoscale58192–8201. 10.1039/c3nr32922d
70
Harel E. T. Drake P. M. Barfield R. M. Lui I. Farr-Jones S. Veer L. V. et al (2019). Antibody-drug conjugates targeting the urokinase receptor (uPAR) as a possible treatment of aggressive breast cancer.Antibodies8:54. 10.3390/antib8040054
71
Harel E. Shoji J. Abraham V. Miller L. Laszik Z. G. King A. et al (2020). Further evidence that the soluble urokinase plasminogen activator receptor does not directly injure mice or human podocytes.Transplantation10454–60. 10.1097/tp.0000000000002930
72
Hernot S. van Manen L. Debie P. Mieog J. S. D. Vahrmeijer A. L. (2019). Latest developments in molecular tracers for fluorescence image-guided cancer surgery.Lancet Oncol.20e354–e367. 10.1016/S1470-2045(19)30317-1
73
Hill A. DeZern A. E. Kinoshita T. Brodsky R. A. (2017). Paroxysmal nocturnal haemoglobinuria.Nat. Rev. Dis. Primers3:17028. 10.1038/nrdp.2017.28
74
Hoyer-Hansen G. Ploug M. Behrendt N. Rønne E. Danø K. (1997). Cell-surface acceleration of urokinase-catalyzed receptor cleavage.Eur. J. Biochem.24321–26. 10.1111/j.1432-1033.1997.0021a.x
75
Hoyer-Hansen G. Rønne E. Solberg H. Behrendt N. Ploug M. Lund L. R. et al (1992). Urokinase plasminogen activator cleaves its cell surface receptor releasing the ligand-binding domain.J. Biol. Chem.26718224–18229. 10.1016/s0021-9258(19)37176-5
76
Hu X. Mandika C. He L. You Y. Chang Y. Wang J. et al (2019). Construction of urokinase-type plasminogen activator receptor-targeted heterostructures for efficient photothermal chemotherapy against cervical cancer to achieve simultaneous anticancer and antiangiogenesis.ACS Appl. Mater. Interfaces1139688–39705. 10.1021/acsami.9b15751
77
Huai Q. Mazar A. P. Kuo A. Parry G. C. Shaw D. E. Callahan J. et al (2006). Structure of human urokinase plasminogen activator in complex with its receptor.Science311656–659. 10.1126/science.1121143
78
Huai Q. Zhou A. Lin L. Mazar A. P. Parry G. C. Callanhan J. et al (2008). Crystal structures of two human vitronectin, urokinase and urokinase receptor complexes.Nat. Struct. Mol. Biol.15422–423. 10.1038/nsmb.1404
79
Jacobsen B. Gårdsvoll H. Juhl Funch G. Østergaard S. Barkholt V. Ploug M. (2007). One-step affinity purification of recombinant urokinase-type plasminogen activator receptor using a synthetic peptide developed by combinatorial chemistry.Protein Expr. Purif.52286–296. 10.1016/j.pep.2006.08.011
80
Jang Y. C. Tsou R. Gibran N. S. Isik F. F. (2000). Vitronectin deficiency is associated with increased wound fibrinolysis and decreased microvascular angiogenesis in mice.Surgery127696–704. 10.1067/msy.2000.105858
81
Jiang Y. Lin L. Chen S. Jiang L. Kriegbaum M. C. Gårdsvoll H. et al (2020). Crystal structures of human C4.4A reveal the unique association of Ly6/uPAR/α-neurotoxin domain.Int. J. Biol. Sci.16981–993. 10.7150/ijbs.39919
82
Jing Y. Chavez V. Ban Y. Acquavella N. El-Ashry D. Pronin A. et al (2017). Molecular effects of stromal-selective targeting by uPAR-retargeted oncolytic virus in breast cancer.Mol. Cancer Res.151410–1420. 10.1158/1541-7786.mcr-17-0016
83
Jorgensen T. J. Gårdsvoll H. Danø K. Roepstorff P. Ploug M. (2004). Dynamics of urokinase receptor interaction with peptide antagonists studied by amide hydrogen exchange and mass spectrometry.Biochemistry4315044–15057. 10.1021/bi048706j
84
Juhl K. Christensen A. Persson M. Ploug M. Kjaer A. (2016). Peptide-based optical uPAR imaging for surgery: in vivo testing of ICG-Glu-Glu-AE105.PLoS One11:e0147428. 10.1371/journal.pone.0147428
85
Juhl K. Christensen A. Rubek N. Karnov K. K. S. von Buchwald C. Kjær A. (2019). Improved surgical resection of metastatic pancreatic cancer using uPAR targeted in vivo fluorescent guidance: comparison with traditional white light surgery.Oncotarget106308–6316. 10.18632/oncotarget.27220
86
Kasten B. B. Ma X. Cheng K. Bu L. Slocumb W. S. Hayes T. R. et al (2016). Isothiocyanate-functionalized bifunctional chelates and fac-[MI(CO)3]+ (M = Re, 99mTc) complexes for targeting uPAR in prostate cancer.Bioconjug. Chem.27130–142. 10.1021/acs.bioconjchem.5b00531
87
Kinoshita T. (2020). Biosynthesis and biology of mammalian GPI-anchored proteins.Open Biol.10:190290. 10.1098/rsob.190290
88
Kjaergaard M. Hansen L. V. Jacobsen B. Gårdsvoll H. Ploug M. (2008). Structure and ligand interactions of the urokinase receptor (uPAR).Front. Biosci.135441–5461. 10.2741/3092
89
Knör S. Sato S. Huber T. Morgenstern A. Bruchertseifer F. Schmitt M. et al (2008). Development and evaluation of peptidic ligands targeting tumour-associated urokinase plasminogen activator receptor (uPAR) for use in alpha-emitter therapy for disseminated ovarian cancer.Eur. J. Nucl. Med. Mol. Imaging3553–64. 10.1007/s00259-007-0582-3
90
Kokoris S. I. Gavriilaki E. Miari A. Travlou A. Kyriakou E. Anagnostopoulos A. et al (2018). Renal involvement in paroxysmal nocturnal hemoglobinuria: an update on clinical features, pathophysiology and treatment.Hematology23558–566. 10.1080/10245332.2018.1444563
91
Konigshausen E. Sellin L. (2016). Circulating permeability factors in primary focal segmental glomerulosclerosis: a review of proposed candidates.Biomed. Res. Int.2016:3765608. 10.1155/2016/3765608
92
Kriegbaum M. C. Persson M. Haldager L. Alpízar-Alpízar W. Jacobsen B. Gårdsvoll H. et al (2011). Rational targeting of the urokinase receptor (uPAR): development of antagonists and non-invasive imaging probes.Curr. Drug Targets121711–1728. 10.2174/138945011797635812
93
Kristensen K. K. Leth-Espensen K. Z. Kumari A. Grønnemose A. L. Winther A. M. L. Young S. G. et al (2021). GPIHBP1 and ANGPTL4 utilize protein disorder to orchestrate order in plasma triglyceride metabolism and regulate compartmentalization of LPL activity.Front. Cell Dev. Biol.9:702508. 10.3389/fcell.2021.702508
94
Kronbichler A. Saleem M. A. Meijers B. Shin J. I. (2016). Soluble urokinase receptors in focal segmental glomerulosclerosis: a review on the scientific point of view.J. Immunol. Res.2016:2068691. 10.1155/2016/2068691
95
Kryza T. Khan T. Lovell S. Harrington B. S. Yin J. Porazinski S. et al (2021). Substrate-biased activity-based probes identify proteases that cleave receptor CDCP1.Nat. Chem. Biol.17776–783. 10.1038/s41589-021-00783-w
96
Kurbegovic S. Juhl K. Chen H. Qu C. Ding B. Leth J. M. et al (2018). Molecular targeted NIR-II probe for image-guided brain tumor surgery.Bioconjug. Chem.293833–3840. 10.1021/acs.bioconjchem.8b00669
97
Kurbegovic S. Juhl K. Sørensen K. K. Leth J. Willemoe G. L. Christensen A. et al (2021). IRDye800CW labeled uPAR-targeting peptide for fluorescence-guided glioblastoma surgery: preclinical studies in orthotopic xenografts.Theranostics117159–7174. 10.7150/thno.49787
98
LeBeau A. M. Duriseti S. Murphy S. T. Pepin F. Hann B. Gray J. W. et al (2013). Targeting uPAR with antagonistic recombinant human antibodies in aggressive breast cancer.Cancer Res.732070–2081. 10.1158/0008-5472.can-12-3526
99
LeBeau A. M. Sevillano N. King M. L. Duriseti S. Murphy S. T. Craik C. S. et al (2014). Imaging the urokinase plasminongen activator receptor in preclinical breast cancer models of acquired drug resistance.Theranostics4267–279. 10.7150/thno.7323
100
Leth J. M. Leth-Espensen K. Z. Kristensen K. K. Kumari A. Lund Winther A. M. Young S. G. et al (2019a). Evolution and medical significance of LU domain-containing proteins.Int. J. Mol. Sci.20:2760. 10.3390/ijms20112760
101
Leth J. M. Mertens H. D. T. Leth-Espensen K. Z. Jørgensen T. J. D. Ploug M. (2019b). Did evolution create a flexible ligand-binding cavity in the urokinase receptor through deletion of a plesiotypic disulfide bond?J. Biol. Chem.2947403–7418. 10.1074/jbc.ra119.007847
102
Li Santi A. Napolitano F. Montuori N. Ragno P. (2021). The urokinase receptor: a multifunctional receptor in cancer cell biology. therapeutic implications.Int. J. Mol. Sci.22:4111. 10.3390/ijms22084111
103
Li J. Ny A. Leonardsson G. Nandakumar K. S. Holmdahl R. Ny T. (2005). The plasminogen activator/plasmin system is essential for development of the joint inflammatory phase of collagen type II-induced arthritis.Am. J. Pathol.166783–792. 10.1016/s0002-9440(10)62299-7
104
Li Z. B. Niu G. Wang H. He L. Yang L. Ploug M. et al (2008). Imaging of urokinase-type plasminogen activator receptor expression using a 64Cu-labeled linear peptide antagonist by microPET.Clin. Cancer Res.144758–4766. 10.1158/1078-0432.ccr-07-4434
105
Liberini V. Laudicella R. Capozza M. Huellner M. W. Burger I. A. Baldari S. et al (2021). The future of cancer diagnosis, treatment and surveillance: a systemic review on immunotherapy and immuno-PET radiotracers.Molecules26:2201. 10.3390/molecules26082201
106
Lin C. M. Arancillo M. Whisenant J. Burgess K. (2020). Unconventional secondary structure mimics: ladder-rungs.Angew. Chem. Int. Ed. Engl.599398–9402. 10.1002/anie.202002639
107
Lin L. Gårdsvoll H. Huai Q. Huang M. Ploug M. (2010). Structure-based engineering of species selectivity in the interaction between urokinase and its receptor: implication for preclinical cancer therapy.J. Biol. Chem.28510982–10992. 10.1074/jbc.m109.093492
108
Liu D. Overbey D. Watkinson L. Giblin M. F. (2009). Synthesis and characterization of an 111In-labeled peptide for the in vivo localization of human cancers expressing the urokinase-type plasminogen activator receptor (uPAR).Bioconjug. Chem.20888–894. 10.1021/bc800433y
109
Liu M. Lin L. Høyer-Hansen G. Ploug M. Li H. Jiang L. et al (2019). Crystal structure of the unoccupied murine urokinase-type plasminogen activator receptor (uPAR) reveals a tightly packed DII-DIII unit.FEBS Lett.5931236–1247. 10.1002/1873-3468.13397
110
Liu S. Aaronson H. Mitola D. J. Leppla S. H. Bugge T. H. (2003). Potent antitumor activity of a urokinase-activated engineered anthrax toxin.Proc. Natl. Acad. Sci. U.S.A.100657–662. 10.1073/pnas.0236849100
111
Llinas P. Hélène, Le, Du M. Gårdsvoll H. Danø K. et al (2005). Crystal structure of the human urokinase plasminogen activator receptor bound to an antagonist peptide.EMBO J.241655–1663. 10.1038/sj.emboj.7600635
112
Loosen S. H. Tacke F. Binnebosel M. Leyh C. Heitkamp F. et al (2018). Serum levels of soluble urokinase plasminogen activator receptor (suPAR) predict outcome after resection of colorectal liver metastases.Oncotarget927027–27038. 10.18632/oncotarget.25471
113
Loosen S. H. Tacke F. Puthe N. Binneboesel M. Wiltberger G. Alizai P. H. et al (2019). High baseline soluble urokinase plasminogen activator receptor (suPAR) serum levels indicate adverse outcome after resection of pancreatic adenocarcinoma.Carcinogenesis40947–955. 10.1093/carcin/bgz033
114
Lu J. J. Guo H. Gao B. Zhang Y. Lin Q. L. Shi J. et al (2018). Prognostic value of urokinase plasminogen activator system in non-small cell lung cancer: a systematic review and meta-analysis.Mol. Clin. Oncol.8127–132.
115
Lund I. K. Illemann M. Thurison T. Christensen I. J. Høyer-Hansen G. (2011). uPAR as anti-cancer target: evaluation of biomarker potential, histological localization, and antibody-based therapy.Curr. Drug Targets121744–1760. 10.2174/138945011797635902
116
Lund L. R. Bjørn S. F. Sternlicht M. D. Nielsen B. S. Solberg H. Usher P. A. et al (2000). Lactational competence and involution of the mouse mammary gland require plasminogen.Development1274481–4492. 10.1242/dev.127.20.4481
117
Madsen C. D. Ferraris G. M. Andolfo A. Cunningham O. Sidenius N. (2007). uPAR-induced cell adhesion and migration: vitronectin provides the key.J. Cell Biol.177927–939. 10.1083/jcb.200612058
118
Madunic J. (2018). The urokinase plasminogen activator system in human cancers: an overview of its prognostic and predictive role.Thromb. Haemost.1182020–2036. 10.1055/s-0038-1675399
119
Manetti M. Allanore Y. Revillod L. Fatini C. Guiducci S. Cuomo G. et al (2011). A genetic variation located in the promoter region of the uPAR (CD87) gene is associated with the vascular complications of systemic sclerosis.Arthritis Rheum.63247–256. 10.1002/art.30101
120
Mani T. Liu D. Zhou D. Li L. Knabe W. E. Wang F. et al (2013). Probing binding and cellular activity of pyrrolidinone and piperidinone small molecules targeting the urokinase receptor.ChemMedChem81963–1977. 10.1002/cmdc.201300340
121
Masutani M. Sakurai S. Shimizu T. Ohto U. (2020). Crystal structure of TEX101, a glycoprotein essential for male fertility, reveals the presence of tandemly arranged Ly6/uPAR domains.FEBS Lett.5943020–3031. 10.1002/1873-3468.13875
122
Meijers B. Maas R. J. Sprangers B. Claes K. Poesen R. Bammens B. et al (2014). The soluble urokinase receptor is not a clinical marker for focal segmental glomerulosclerosis.Kidney Int.85636–640. 10.1038/ki.2013.505
123
Mertens H. D. Kjaergaard M. Mysling S. Gårdsvoll H. Jørgensen T. J. Svegun D. I. et al (2012). A flexible multidomain structure drives the function of the urokinase-type plasminogen activator receptor (uPAR).J. Biol. Chem.28734304–34315. 10.1074/jbc.m112.398404
124
Miles L. A. Lighvani S. Baik N. Parmer C. M. Khaldoyanidi S. Mueller B. M. et al (2014). New insights into the role of Plg-RKT in macrophage recruitment.Int. Rev. Cell Mol. Biol.309259–302. 10.1016/b978-0-12-800255-1.00005-3
125
Minopoli M. Polo A. Ragone C. Ingangi V. Ciliberto G. Pessi A. et al (2019). Structure-function relationship of an urokinase receptor-derived peptide which inhibits the formyl peptide receptor type 1 activity.Sci. Rep.9:12169. 10.1038/s41598-019-47900-3
126
Morodomi Y. Yano T. Kinoh H. Harada Y. Saito S. Kyuragi R. et al (2012). Bioknife, a uPA activity-dependent oncolytic Sendai virus, eliminates pleural spread of malignant mesothelioma via simultaneous stimulation of uPA expression.Mol. Ther.20769–777. 10.1038/mt.2011.305
127
Multhaupt H. A. Mazar A. Cines D. B. Warhol M. J. McCrae K. R. (1994). Expression of urokinase receptors by human trophoblast. A histochemical and ultrastructural analysis.Lab. Invest.71392–400.
128
Musetti C. Quaglia M. Cena T. Chiocchetti A. Monti S. Clemente N. et al (2015). Circulating suPAR levels are affected by glomerular filtration rate and proteinuria in primary and secondary glomerulonephritis.J. Nephrol.28299–305. 10.1007/s40620-014-0137-1
129
Nishiya A. T. Nagamine M. K. Fonseca I. Miraldo A. C. Scattone N. V. Guerra J. L. et al (2020). Inhibitory effects of a reengineered anthrax toxin on canine oral mucosal melanomas.Toxins12:157. 10.3390/toxins12030157
130
Nykjaer A. Petersen C. M. Christensen E. I. Davidsen O. Gliemann J. (1990). Urokinase receptors in human monocytes.Biochim. Biophys. Acta1052399–407. 10.1016/0167-4889(90)90149-8
131
Pass J. Jögi A. Lund I. K. Rønø B. Rasch M. G. Gårdsvoll H. et al (2007). Murine monoclonal antibodies against murine uPA receptor produced in gene-deficient mice: inhibitory effects on receptor-mediated uPA activity in vitro and in vivo.Thromb. Haemost.971013–1022. 10.1160/th06-11-0644
132
Persson M. Hosseini M. Madsen J. Jørgensen T. J. Jensen K. J. Kjær A. et al (2013a). Improved PET imaging of uPAR expression using new 64Cu-labeled cross-bridged peptide ligands: comparative in vitro and in vivo studies.Theranostics3618–632. 10.7150/thno.6810
133
Persson M. Juhl K. Rasmussen P. Brandt-Larsen M. Madsen J. Ploug M. et al (2014). uPAR targeted radionuclide therapy with 177Lu-DOTA-AE105 inhibits dissemination of metastatic prostate cancer.Mol. Pharm.112796–2806. 10.1021/mp500177c
134
Persson M. Liu H. Madsen J. Cheng Z. Kjær A. (2013b). First 18F-labeled ligand for PET imaging of uPAR: in vivo studies in human prostate cancer xenografts.Nucl. Med. Biol.40618–624. 10.1016/j.nucmedbio.2013.03.001
135
Persson M. Madsen J. Østergård S. Jensen M. M. Jørgensen J. T. Juhl K. et al (2012a). Quantitative PET of human urokinase-type plasminogen activator receptor with 64Cu-DOTA-AE105: implications for visualizing cancer invasion.J. Nucl. Med.53138–145. 10.2967/jnumed.110.083386
136
Persson M. Madsen J. Østergard S. Ploug M. Kjær A. (2012b). 68Ga-labeling and in vivo evaluation of a uPAR binding DOTA- and NODAGA-conjugated peptide for PET imaging of invasive cancers.Nucl. Med. Biol.39560–569. 10.1016/j.nucmedbio.2011.10.011
137
Persson M. Nedergaard M. K. Brandt-Larsen M. Skovgaard D. Jørgensen J. T. Michaelsen S. R. et al (2016). Urokinase-type plasminogen activator receptor as a potential PET biomarker in glioblastoma.J. Nucl. Med.57272–278. 10.2967/jnumed.115.161703
138
Persson M. Rasmussen P. Madsen J. Ploug M. Kjær A. (2012c). New peptide receptor radionuclide therapy of invasive cancer cells: in vivo studies using 177Lu-DOTA-AE105 targeting uPAR in human colorectal cancer xenografts.Nucl. Med. Biol.39962–969. 10.1016/j.nucmedbio.2012.05.007
139
Persson M. Skovgaard D. Brandt-Larsen M. Christensen C. Madsen J. Nielsen C. H. et al (2015). First-in-human uPAR PET: imaging of cancer aggressiveness.Theranostics51303–1316. 10.7150/thno.12956
140
Petranka J. Zhao J. Norris J. Tweedy N. B. Ware R. E. Sims P. J. et al (1996). Structure-function relationships of the complement regulatory protein. CD59.Blood Cells Mol. Dis.22281–296. 10.1006/bcmd.1996.0111
141
Petzinger J. Saltel F. Hersemeyer K. Daniel J. M. Preissner K. T. Wehle-Haller B. et al (2007). Urokinase receptor (CD87) clustering in detergent-insoluble adhesion patches leads to cell adhesion independently of integrins.Cell Commun. Adhes.14137–155. 10.1080/15419060701557487
142
Pierleoni C. Castellucci M. Kaufmann P. Lund L. R. Schnack Nielsen B. (2003). Urokinase receptor is up-regulated in endothelial cells and macrophages associated with fibrinoid deposits in the human placenta.Placenta24677–685. 10.1016/s0143-4004(03)00082-1
143
Piironen T. Laursen B. Pass J. List K. Gårdsvoll H. Ploug M. et al (2004). Specific immunoassays for detection of intact and cleaved forms of the urokinase receptor.Clin. Chem.502059–2068. 10.1373/clinchem.2004.038232
144
Plaisier M. Koolwijk P. Willems F. Helmerhorst F. M. van Hinsbergh V. W. (2008). Pericellular-acting proteases in human first trimester decidua.Mol. Hum. Reprod.1441–51. 10.1093/molehr/gam085
145
Ploug M. (1998). Identification of specific sites involved in ligand binding by photoaffinity labeling of the receptor for the urokinase-type plasminogen activator. residues located at equivalent positions in uPAR domains I and III participate in the assembly of a composite ligand-binding site.Biochemistry3716494–16505. 10.1021/bi981203r
146
Ploug M. (2013). Structure-driven design of radionuclide tracers for non-invasive imaging of uPAR and targeted radiotherapy. The tale of a synthetic peptide antagonist.Theranostics3467–476. 10.7150/thno.3791
147
Ploug M. Ellis V. (1994). Structure-function relationships in the receptor for urokinase-type plasminogen activator. comparison to other members of the Ly-6 family and snake venom alpha-neurotoxins.FEBS Lett.349163–168. 10.1016/0014-5793(94)00674-1
148
Ploug M. Eriksen J. Plesner T. Hansen N. E. Danø K. (1992a). A soluble form of the glycolipid-anchored receptor for urokinase-type plasminogen activator is secreted from peripheral blood leukocytes from patients with paroxysmal nocturnal hemoglobinuria.Eur. J. Biochem.208397–404. 10.1111/j.1432-1033.1992.tb17200.x
149
Ploug M. Kjalke M. Rønne E. Weidle U. Høyer-Hansen G. Danø K. (1993). Localization of the disulfide bonds in the NH2-terminal domain of the cellular receptor for human urokinase-type plasminogen activator. a domain structure belonging to a novel superfamily of glycolipid-anchored membrane proteins.J. Biol. Chem.26817539–17546. 10.1016/s0021-9258(19)85366-8
150
Ploug M. Østergård S. Gårdsvoll H. Kovalski K. Holst-Hansen C. Holst-Hansen C. et al (2001). Peptide-derived antagonists of the urokinase receptor. affinity maturation by combinatorial chemistry, identification of functional epitopes, and inhibitory effect on cancer cell intravasation.Biochemistry4012157–12168. 10.1021/bi010662g
151
Ploug M. Østergård S. Hansen L. B. Holm A. Danø K. (1998a). Photoaffinity labeling of the human receptor for urokinase-type plasminogen activator using a decapeptide antagonist. evidence for a composite ligand-binding site and a short interdomain separation.Biochemistry373612–3622. 10.1021/bi972787k
152
Ploug M. Plesner T. Rønne E. Ellis V. Høyer-Hansen G. Pedersen W. M. et al (1992b). The receptor for urokinase-type plasminogen activator is deficient on peripheral blood leukocytes in patients with paroxysmal nocturnal hemoglobinuria.Blood791447–1455. 10.1182/blood.v79.6.1447.bloodjournal7961447
153
Ploug M. Rahbek-Nielsen H. Nielsen P. F. Roepstorff P. Danø K. (1998b). Glycosylation profile of a recombinant urokinase-type plasminogen activator receptor expressed in chinese hamster ovary cells.J. Biol. Chem.27313933–13943. 10.1074/jbc.273.22.13933
154
Ploug M. Rønne E. Behrendt N. Jensen A. L. Blasi F. Danø K. (1991). Cellular receptor for urokinase plasminogen activator. Carboxyl-terminal processing and membrane anchoring by glycosyl-phosphatidylinositol.J. Biol. Chem.2661926–1933. 10.1016/s0021-9258(18)52382-6
155
Price E. W. Orvig C. (2014). Matching chelators to radiometals for radiopharmaceuticals.Chem. Soc. Rev.43260–290. 10.1039/c3cs60304k
156
Resnati M. Pallavicini I. Wang J. M. Oppenheim J. Serhan C. N. Romano M. et al (2002). The fibrinolytic receptor for urokinase activates the G protein-coupled chemotactic receptor FPRL1/LXA4R.Proc. Natl. Acad. Sci. U.S.A.991359–1364. 10.1073/pnas.022652999
157
Rolff H. C. Christensen I. J. Svendsen L. B. Wilhelmsen M. Lund I. K. Thurison T. et al (2019). The concentration of the cleaved suPAR forms in pre- and postoperative plasma samples improves the prediction of survival in colorectal cancer: a nationwide multicenter validation and discovery study.J. Surg. Oncol.1201404–1411. 10.1002/jso.25733
158
Romer J. Bugge T. H. Pyke C. Lund L. R. Flick M. J. Degen J. L. et al (1996). Impaired wound healing in mice with a disrupted plasminogen gene.Nat. Med.2287–292. 10.1038/nm0396-287
159
Romer J. Lund L. R. Eriksen J. Pyke C. Kristensen P. Danø K. (1994). The receptor for urokinase-type plasminogen activator is expressed by keratinocytes at the leading edge during re-epithelialization of mouse skin wounds.J. Invest. Dermatol.102519–522. 10.1111/1523-1747.ep12373187
160
Romer J. Nielsen B. S. Ploug M. (2004). The urokinase receptor as a potential target in cancer therapy.Curr. Pharm. Des.102359–2376. 10.2174/1381612043383962
161
Ronne E. Pappot H. Grøndahl-Hansen J. Høyer-Hansen G. Plesner T. Hansen N. E. et al (1995). The receptor for urokinase plasminogen activator is present in plasma from healthy donors and elevated in patients with paroxysmal nocturnal haemoglobinuria.Br. J. Haematol.89576–581. 10.1111/j.1365-2141.1995.tb08366.x
162
Rullo A. F. Fitzgerald K. J. Muthusamy V. Liu M. Yuan C. Huang M. et al (2016). Re-engineering the immune response to metastatic cancer: antibody-recruiting small molecules targeting the urokinase receptor.Angew. Chem. Int. Ed. Engl.553642–3646. 10.1002/anie.201510866
163
Rustamzadeh E. Hall W. A. Todhunter D. A. Vallera V. D. Low W. C. Liu H. et al (2007). Intracranial therapy of glioblastoma with the fusion protein DTAT in immunodeficient mice.Int. J. Cancer120411–419. 10.1002/ijc.22278
164
Salasznyk R. M. Zappala M. Zheng M. Yu L. Wilkins-Port C. McKeown-Longo P. J. (2007). The uPA receptor and the somatomedin B region of vitronectin direct the localization of uPA to focal adhesions in microvessel endothelial cells.Matrix Biol.26359–370. 10.1016/j.matbio.2007.01.009
165
Saleem M. A. (2018). What is the role of soluble urokinase-type plasminogen activator in renal disease?Nephron139334–341. 10.1159/000490118
166
Schafer J. M. Peters D. E. Morley T. Liu S. Molinolo A. A. Leppla S. H. et al (2011). Efficient targeting of head and neck squamous cell carcinoma by systemic administration of a dual uPA and MMP-activated engineered anthrax toxin.PLoS One6:e20532. 10.1371/journal.pone.0020532
167
Schmiedeberg N. Schmitt M. Rölz C. Truffault V. Sukopp M. Bürgle M. et al (2002). Synthesis, solution structure, and biological evaluation of urokinase type plasminogen activator (uPA)-derived receptor binding domain mimetics.J. Med. Chem.454984–4994. 10.1021/jm020254q
168
Schott D. Dempfle C. E. Beck P. Liermann A. Mohr-Pennert A. Goldner M. et al (1998). Therapy with a purified plasminogen concentrate in an infant with ligneous conjunctivitis and homozygous plasminogen deficiency.N. Engl. J. Med.3391679–1686. 10.1056/nejm199812033392305
169
Sharma S. Shinde S. S. Teekas L. Vijay N. (2020). Evidence for the loss of plasminogen receptor kt gene in chicken.Immunogenetics72507–515. 10.1007/s00251-020-01186-2
170
Skovgaard D. Persson M. Brandt-Larsen M. Christensen C. Madsen J. Klausen T. L. et al (2017). Safety, dosimetry, and tumor detection ability of 68Ga-NOTA-AE105: first-in-human study of a novel radioligand for uPAR PET imaging.J. Nucl. Med.58379–386. 10.2967/jnumed.116.178970
171
Sloand E. M. Pfannes L. Scheinberg P. More K. Wu C. O. Horne M. et al (2008). Increased soluble urokinase plasminogen activator receptor (suPAR) is associated with thrombosis and inhibition of plasmin generation in paroxysmal nocturnal hemoglobinuria (PNH) patients.Exp. Hematol.361616–1624. 10.1016/j.exphem.2008.06.016
172
Slot O. Brünner N. Locht H. Oxholm P. Stephens R. W. (1999). Soluble urokinase plasminogen activator receptor in plasma of patients with inflammatory rheumatic disorders: increased concentrations in rheumatoid arthritis.Ann. Rheum. Dis.58488–492. 10.1136/ard.58.8.488
173
Smith H. W. Marshall C. J. (2010). Regulation of cell signalling by uPAR.Nat. Rev. Mol. Cell Biol.1123–36. 10.1038/nrm2821
174
Solberg H. Ploug M. Høyer-Hansen G. Nielsen B. S. Lund L. R. (2001). The murine receptor for urokinase-type plasminogen activator is primarily expressed in tissues actively undergoing remodeling.J. Histochem. Cytochem.49237–246. 10.1177/002215540104900211
175
Spinale J. M. Mariani L. H. Kapoor S. Zhang J. Weyant R. Song P. X. et al (2015). A reassessment of soluble urokinase-type plasminogen activator receptor in glomerular disease.Kidney Int.87564–574. 10.1038/ki.2014.346
176
Stephens R. W. Nielsen H. J. Christensen I. J. Thorlacius-Ussing O. Sørensen S. Danø K. et al (1999). Plasma urokinase receptor levels in patients with colorectal cancer: relationship to prognosis.J. Natl. Cancer Inst.91869–874. 10.1093/jnci/91.10.869
177
Stoppelli M. P. Corti A. Soffientini A. Cassani G. Blasi F. Assoian R. K. (1985). Differentiation-enhanced binding of the amino-terminal fragment of human urokinase plasminogen activator to a specific receptor on u937 monocytes.Proc. Natl. Acad. Sci. U.S.A.824939–4943. 10.1073/pnas.82.15.4939
178
Sun Y. Ma X. Cheng K. Wu B. Duan J. Chen H. et al (2015). Strained cyclooctyne as a molecular platform for construction of multimodal imaging probes.Angew. Chem. Int. Ed. Engl.545981–5984. 10.1002/anie.201500941
179
Suzuki K. G. Kasai R. S. Hirosawa K. M. Nemoto Y. L. Ishibashi M. Miwa Y. et al (2012). Transient GPI-anchored protein homodimers are units for raft organization and function.Nat. Chem. Biol.8774–783. 10.1038/nchembio.1028
180
Thornton S. Raghu H. Cruz C. Frederick M. D. Palumbo J. S. Mullins E. S. et al (2017). Urokinase plasminogen activator and receptor promote collagen-induced arthritis through expression in hematopoietic cells.Blood Adv.1545–556. 10.1182/bloodadvances.2016004002
181
Thurison T. Christensen I. J. Lund I. K. Nielsen H. J. Høyer-Hansen G. (2015). Circulating intact and cleaved forms of the urokinase-type plasminogen activator receptor: biological variation, reference intervals and clinical useful cut-points.Clin. Chim. Acta43984–90. 10.1016/j.cca.2014.10.004
182
Thurison T. Lomholt A. F. Rasch M. G. Lund I. K. Nielsen H. J. Christensen I. J. et al (2010). A new assay for measurement of the liberated domain i of the urokinase receptor in plasma improves the prediction of survival in colorectal cancer.Clin. Chem.561636–1640. 10.1373/clinchem.2010.144410
183
van Veen M. Matas-Rico E. van de Wetering K. Leyton-Puig D. Kedziora K. M. De Lorenzi V. et al (2017). Negative regulation of urokinase receptor activity by a GPI-specific phospholipase C in breast cancer cells.Elife6:e23649. 10.7554/eLife.23649
184
Varma R. Mayor S. (1998). GPI-anchored proteins are organized in submicron domains at the cell surface.Nature394798–801. 10.1038/29563
185
Vassalli J. D. Baccino D. Belin D. (1985). A cellular binding site for the Mr 55,000 form of the human plasminogen activator, urokinase.J. Cell Biol.10086–92. 10.1083/jcb.100.1.86
186
Vats K. Sharma R. Sarma H. D. Satpati D. Dash A. (2018). 68Ga-labeled HEBED-CC variant of uPAR targeting peptide AE105 compared with 68Ga-NODAGA-AE105.Anticancer Agents Med. Chem.181289–1294. 10.2174/1871520618666180316152618
187
Waldron N. N. Oh S. Vallera D. A. (2012). Bispecific targeting of EGFR and uPAR in a mouse model of head and neck squamous cell carcinoma.Oral Oncol.481202–1207. 10.1016/j.oraloncology.2012.06.002
188
Waltz D. A. Chapman H. A. (1994). Reversible cellular adhesion to vitronectin linked to urokinase receptor occupancy.J. Biol. Chem.26914746–14750. 10.1016/s0021-9258(17)36688-7
189
Wei C. El Hindi S. Li J. Fornoni A. Goes N. Sageshima J. et al (2011). Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis.Nat. Med.17952–960.
190
Wei C. Li J. Adair B. D. Zhu K. Cai J. Merchant M. et al (2019). uPAR isoform 2 forms a dimer and induces severe kidney disease in mice.J. Clin. Invest.1291946–1959. 10.1172/jci124793
191
Wei C. Moller C. C. Altintas M. M. Li J. Schwarz K. Zacchigna S. et al (2008). Modification of kidney barrier function by the urokinase receptor.Nat. Med.1455–63. 10.1038/nm1696
192
Wei Y. Lukashev M. Simon D. I. Bodary S. C. Rosenberg S. Doyle M. V. et al (1996). Regulation of integrin function by the urokinase receptor.Science2731551–1555. 10.1126/science.273.5281.1551
193
Wei Y. Waltz D. A. Rao N. Drummond R. J. Rosenberg S. Chapman H. A. (1994). Identification of the urokinase receptor as an adhesion receptor for vitronectin.J. Biol. Chem.26932380–32388. 10.1016/s0021-9258(18)31646-6
194
Xu D. Bum-Erdene K. Leth J. M. Ghozayel M. K. Ploug M. Meroueh S. O. (2021). Small-molecule inhibition of the uPAR uPA interaction by conformational selection.ChemMedChem16377–387. 10.1002/cmdc.202000558
195
Xu D. Bum-Erdene K. Si Y. Zhou D. Ghozayel M. K. Meroueh S. O. (2017). Mimicking intermolecular interactions of tight protein-protein complexes for small-molecule antagonists.ChemMedChem121794–1809. 10.1002/cmdc.201700572
196
Xu X. Gårdsvoll H. Yuan C. Lin L. Ploug M. Huang M. (2012). Crystal structure of the urokinase receptor in a ligand-free form.J. Mol. Biol.416629–641. 10.1016/j.jmb.2011.12.058
197
Yang D. Severin G. W. Dougherty C. A. Lombardi R. Chen D. van Dort M. E. et al (2016). Antibody-based PET of uPA/uPAR signaling with broad applicability for cancer imaging.Oncotarget773912–73924. 10.18632/oncotarget.12528
198
Yang L. Sajja H. K. Cao Z. Qian W. Bender L. Marcus A. I. et al (2013). uPAR-targeted optical imaging contrasts as theranostic agents for tumor margin detection.Theranostics4106–118. 10.7150/thno.7409
199
Yu S. Huang G. Yuan R. Chen T. (2020). A uPAR targeted nanoplatform with an NIR laser-responsive drug release property for tri-modal imaging and synergistic photothermal-chemotherapy of triple-negative breast cancer.Biomater. Sci.8720–738. 10.1039/c9bm01495k
200
Yuan C. Guo Z. Yu S. Jiang L. Huang M. (2021). Development of inhibitors for uPAR: blocking the interaction of uPAR with its partners.Drug Discov. Today261076–1085. 10.1016/j.drudis.2021.01.016
201
Zeitler P. Schuster V. (1999). Plasma values for u-PA in children.Eur. J. Pediatr.158(Suppl. 3), S205–S208. 10.1007/pl00014360
202
Zhao B. Gandhi S. Yuan C. Luo Z. Li R. Gårdsvoll H. et al (2015). Stabilizing a flexible interdomain hinge region harboring the SMB binding site drives uPAR into its closed conformation.J. Mol. Biol.4271389–1403. 10.1016/j.jmb.2015.01.022
203
Zheng X. Saunders T. L. Camper S. A. Samuelson L. C. Ginsburg D. (1995). Vitronectin is not essential for normal mammalian development and fertility.Proc. Natl. Acad. Sci. U.S.A.9212426–12430. 10.1073/pnas.92.26.12426
204
Zhou A. Huntington J. A. Pannu N. S. Carrell R. W. Read R. J. (2003). How vitronectin binds PAI-1 to modulate fibrinolysis and cell migration.Nat. Struct. Biol.10541–544. 10.1038/nsb943
205
Zhou H. M. Nichols A. Meda P. Vassalli J. D. (2000). Urokinase-type plasminogen activator and its receptor synergize to promote pathogenic proteolysis.EMBO J.194817–4826. 10.1093/emboj/19.17.4817
206
Zuo J. Huo M. Wang L. Li J. Chen Y. Xiong P. (2020). Photonic hyperthermal and sonodynamic nanotherapy targeting oral squamous cell carcinoma.J. Mater. Chem. B.1072411–2502.
207
Zuppone S. Assalini C. Minici C. Bertagnoli S. Branduardi P. Degano M. et al (2020). The anti-tumoral potential of the saporin-based uPAR-targeting chimera ATF-SAP.Sci. Rep.10:2521. 10.1038/s41598-020-59313-8
Summary
Keywords
PET imaging, fluorescence guided surgery, LU domain, uPAR, optical imaging
Citation
Leth JM and Ploug M (2021) Targeting the Urokinase-Type Plasminogen Activator Receptor (uPAR) in Human Diseases With a View to Non-invasive Imaging and Therapeutic Intervention. Front. Cell Dev. Biol. 9:732015. doi: 10.3389/fcell.2021.732015
Received
28 June 2021
Accepted
26 July 2021
Published
20 August 2021
Volume
9 - 2021
Edited by
Anil K. Bamezai, Villanova University, United States
Reviewed by
Cynthia L. Bristow, Alpha-1 Biologics, United States; Marcelo Coutinho De Miranda, Albert Einstein College of Medicine, United States
Updates
Copyright
© 2021 Leth and Ploug.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Ploug, m-ploug@finsenlab.dk
This article was submitted to Signaling, a section of the journal Frontiers in Cell and Developmental Biology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.