 Hao Zhang†
Hao Zhang† Ruisi Xu
Ruisi Xu- Endoscopy Center, China-Japan Union Hospital of Jilin University, Changchun, China
Gastric cancer is a common malignant tumor of the digestive system with no specific symptoms. Due to the limited knowledge of pathogenesis, patients are usually diagnosed in advanced stage and do not have effective treatment methods. Proteome has unique tissue and time specificity and can reflect the influence of external factors that has become a potential biomarker for early diagnosis. Therefore, discovering gastric cancer-related proteins could greatly help researchers design drugs and develop an early diagnosis kit. However, identifying gastric cancer-related proteins by biological experiments is time- and money-consuming. With the high speed increase of data, it has become a hot issue to mine the knowledge of proteomics data on a large scale through computational methods. Based on the hypothesis that the stronger the association between the two proteins, the more likely they are to be associated with the same disease, in this paper, we constructed both disease similarity network and protein interaction network. Then, Graph Convolutional Networks (GCN) was applied to extract topological features of these networks. Finally, Xgboost was used to identify the relationship between proteins and gastric cancer. Results of 10-cross validation experiments show high area under the curve (AUC) (0.85) and area under the precision recall (AUPR) curve (0.76) of our method, which proves the effectiveness of our method.
Introduction
Gastric cancer is a worldwide disease with high incidence rate and mortality rate, especially in East Asia (Villanueva, 2011). According to the data from GLOBOCAN in 2018, there were 1,033,701 new cases of and 782,685 deaths from gastric cancer in the world (Bray et al., 2018). At present, the early diagnosis of gastric cancer is limited; patients are usually diagnosed in advanced stage. Therefore, early diagnosis is the key to improve the prognosis of patients, which is also the goal pursued by many researchers (Jin et al., 2015). Biomarkers refer to the substances that can reflect the physiological, biochemical, immune, genetic, and other molecular changes in the organism (Szász et al., 2016; Liang et al., 2019; Cheng et al., 2021). The levels of biomarkers in patients’ samples (such as blood, plasma, saliva, and urine) can reflect the health or disease status of patients, as well as the response to anticancer treatment. Due to the strong heterogeneity of gastric cancer, the use of proteomics technology to find new specific biomarkers will greatly improve the sensitivity and accuracy of the diagnosis of patients (Gullo et al., 2018).
Although many researchers tend to reveal diseases pathogenic mechanism by genomics (Peng and Zhao, 2020; Zhao et al., 2020b; Zhou et al., 2020), changes in protein quality in diseases reflect the progression of the disease and are also the product of genes (Zhao et al., 2020c). Unlike those studies that research diseases through gene expression (Zhao et al., 2021b), protein quantification is more accurate and has the potential to become a biomarker. Researchers have used various protein separation techniques, such as two-dimensional gel electrophoresis (2-DE) (Gygi et al., 2000), two-dimensional fluorescence difference gel electrophoresis (2D-DIGE) (Tannu and Hemby, 2006), isobaric tags for relative and absolute quantitation (iTRAQ), hydrophilic interaction liquid chromatography (HILIC) screening of potential target proteins of new gastric cancer biomarkers, and then Western blotting and enzyme-linked immunosorbent assay or immunohistochemistry (IHC) methods are further validated, and biomarkers that play a key role in the occurrence of malignant tumors can be discovered.
Ryu et al. (2003) used the tumor proteomics technology of antibody microarrays to identify inflammatory protein markers of gastric cancer. They found that 14 proteins have different expressions between normal gastric mucosa and tumor gastric mucosa. The proteome can be regarded as the functional cell equivalent of the genome. Proteomics is useful in discovering biomarkers and improving the diagnostic efficiency of early gastric cancer and has obvious advantages. At present, the prognosis and treatment methods of gastric cancer are guided by genome. Surgical resection is still the most common strategy of gastric cancer, but due to the high risk of disease progression in stage II or III patients, it becomes important to increase adjuvant therapy. The strong heterogeneity of gastric cancer makes the therapeutic effect heterogeneous. Therefore, although the TNM system can help the prognosis of gastric cancer, many researchers tend to discover biomarkers to predict treatment outcomes more accurately (Pang et al., 2018). For example, Balluff et al. (2011) used matrix-assisted laser desorption/ionization (MALDI) imaging technology to analyze tissue samples and found that cysteine-rich intestinal protein 1 (CRIP1) and human neutrophil peptide-1 (HNP-1) were prognostic factors for gastric cancer. Human epidermal growth factor receptor 2 (HER2) is an important biomarker in gastric tumors, which can be specifically targeted for treatment with trastuzumab monoclonal antibody (mAb). For patients with advanced gastric cancer or gastroesophageal junction cancer, trastuzumab combined with chemotherapy can improve the survival rate of patients (Park et al., 2018).
There are still few proteins known to be related to gastric cancer. With the explosive growth of various types of omics data (Mo et al., 2020; Zhao et al., 2020a,2021a), computational methods are widely used to identify disease-related biomolecules. Mining disease-related molecules based on the protein interaction networks has become a universal method. Sang et al. (2011) discovered genes and pathways of ciliopathy disease based on protein network. Seyfried et al. (2017) constructed protein network to identify protein-specific co-expression in Alzheimer’s disease. With the development of Graph Convolutional Networks (GCN), an increasing number of researchers tend to use this method to process the complex topological features of the biological network. Its core point of view is to make the entire graph converge through the dissemination of node information and then make predictions on the basis of it. It has been widely used in prediction of biomolecular interaction (Tianyi et al., 2020). Therefore, we proposed a GCN-based method in this paper, named “GXGCP” (Gcn-Xgboost for Gastric Cancer-related Proteins identification) to identify gastric cancer-related proteins.
Materials and Methods
There are four steps to implement GXGCP. Step 1 is to construct disease similarity network and protein interaction network. Step 2 is using GCN to extract topological features of disease similarity network and protein interaction network, respectively. Step 3 is to reduce the dimension of protein and gastric cancer features by principal component analysis (PCA). Step 4 is to identify gastric cancer-related proteins based on the features of protein and gastric cancer by Xgboost. The workflow of GXGCP is shown in Figure 1.
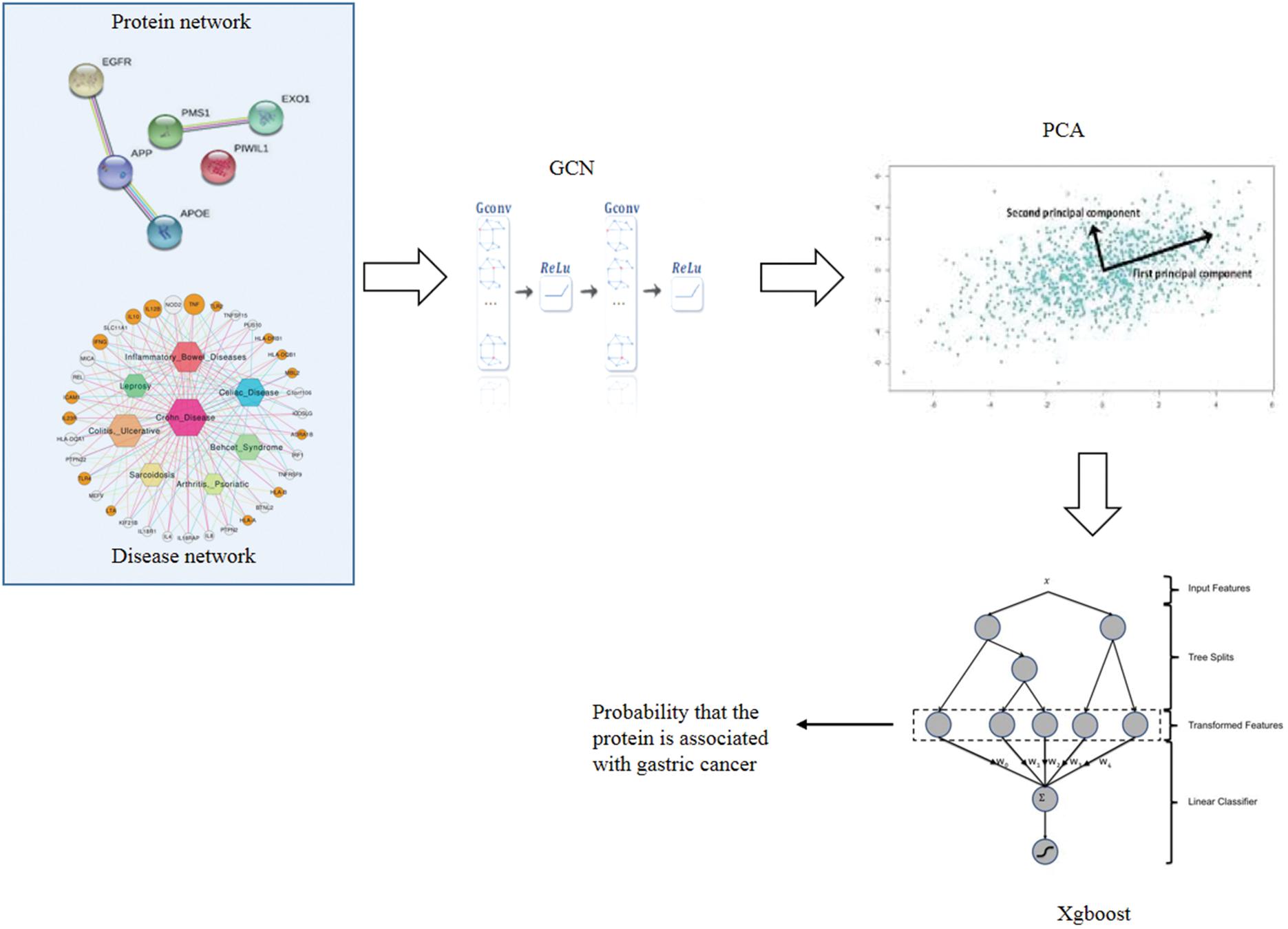
Figure 1. Workflow of GXGCP (Gcn-Xgboost for Gastric Cancer-related Proteins identification).
Construction of Network
We used SemFunsim (Cheng et al., 2014) to obtain diseases that are similar to gastric cancer. This method considers both disease semantic association and gene association. The detailed calculation process will not be repeated in this paper. A total of 327 diseases were found to be similar to gastric cancer. Based on the similarity, we constructed disease network, in which the edges are similarity and nodes are diseases. Therefore, the network has weight.
We downloaded protein interaction information from Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) (Mering et al., 2003). Based on the interaction, we constructed a protein network. If a protein can interact with the other one, there would be an edge to connect each other. Since the intensity of interaction between different proteins is different, this network also has weight.
Extracting Topological Features by Graph Convolutional Networks
To fully extract topological features of protein and disease network, GCN was applied (Han et al., 2019). The aim to implement GCN is to convert network topology into a vector output:
where H(0) is node’s feature in the network.
First, Laplace transform should be done on the network:
where D is the degree matrix of the network, and A is the adjacency matrix.
Then, normalization should be implemented on the Laplacian matrix:
Lsym is defined as:
With the Laplacian matrix, we can perform spectral convolution on the network. We need to find a suitable convolution kernel so that f() can reduce the loss of classification after the convolution transformation of the convolution kernel. The core of the machine learning task on the graph is to find a convolution kernel that can reduce the loss, regard h(λ1),…h(λn) as the parameters of the model, and apply the gradient descent method to update these parameters.
The final formula of GCN would be:
where σ() is the activation function, and W(l) is the parameter to be trained.
Reduction Dimension by Principal Component Analysis
Since the dimension of metabolites and gastric cancer features are large, we used PCA to reduce the dimension. There are four steps to apply PCA (Tipping and Bishop, 1999; Cheng et al., 2019). The first step is feature centralization. That is, the data of each dimension are subtracted from the mean value of that dimension, and the mean value of each dimension becomes 0 after the transformation. The second step is to calculate covariance matrix. The third step is to calculate the eigenvalues and eigenvectors of the covariance matrix. The last step is to select the feature vector corresponding to the large feature value to obtain a new data set.
Classification of Gastric Cancer-Related Proteins by Xgboost
Xgboost is a sparse perception algorithm that can be used for parallel tree learning (Chen and Guestrin, 2016). Since the features of gastric cancer and proteins are sparse, Xgboost is very suitable for the classification.
Xgboost is a tree ensemble model. It sums the results of K (the number of trees) as the final predicted value.
Assuming that a given sample set has n samples and m features, then
where xi represents the i-th sample, yi represents the i-th category label, and the space F of the regression tree (CART tree) is:
where q represents the structure of each tree, it maps the sample to the corresponding leaf node; T is the number of leaf nodes of the corresponding tree; f(x) corresponds to the structure q of the tree and the leaf node weight w. Therefore, the predicted value of Xgboost is the sum of the values of the leaf nodes corresponding to each tree.
Our goal is to learn these k trees, so we minimize the following objective function with regular terms:
where
Experiment Results
We implemented 10-cross validation experiments to test the performance of GXGCP. We divided our data into 10 groups. We used nine of 10 groups’ data to train the model and the data of the remaining one to test the model. After repeating this process 10 times, each group has been tested once. To show the accuracy of our model, we compared GXGCP with several other methods such as RWXGCP, GXGCP without PCA, GSVMCP, GANNCP, and GCNNCP. RWXGCP replaces the GCN part of GXGCP with random walk (RW). GSVMCP replaces the Xgboost part of GXGCP with support vector machine (SVM). GANNCP replaces the Xgboost part of GXGCP with artificial neural network (ANN). GCNNCP replaces the Xgboost part of GXGCP with convolutional neural network (CNN).
The area under the curve (AUC) and area under the precision recall (AUPR) curve of GXGCP are shown in Figure 2. The comparison results are listed in Table 1.
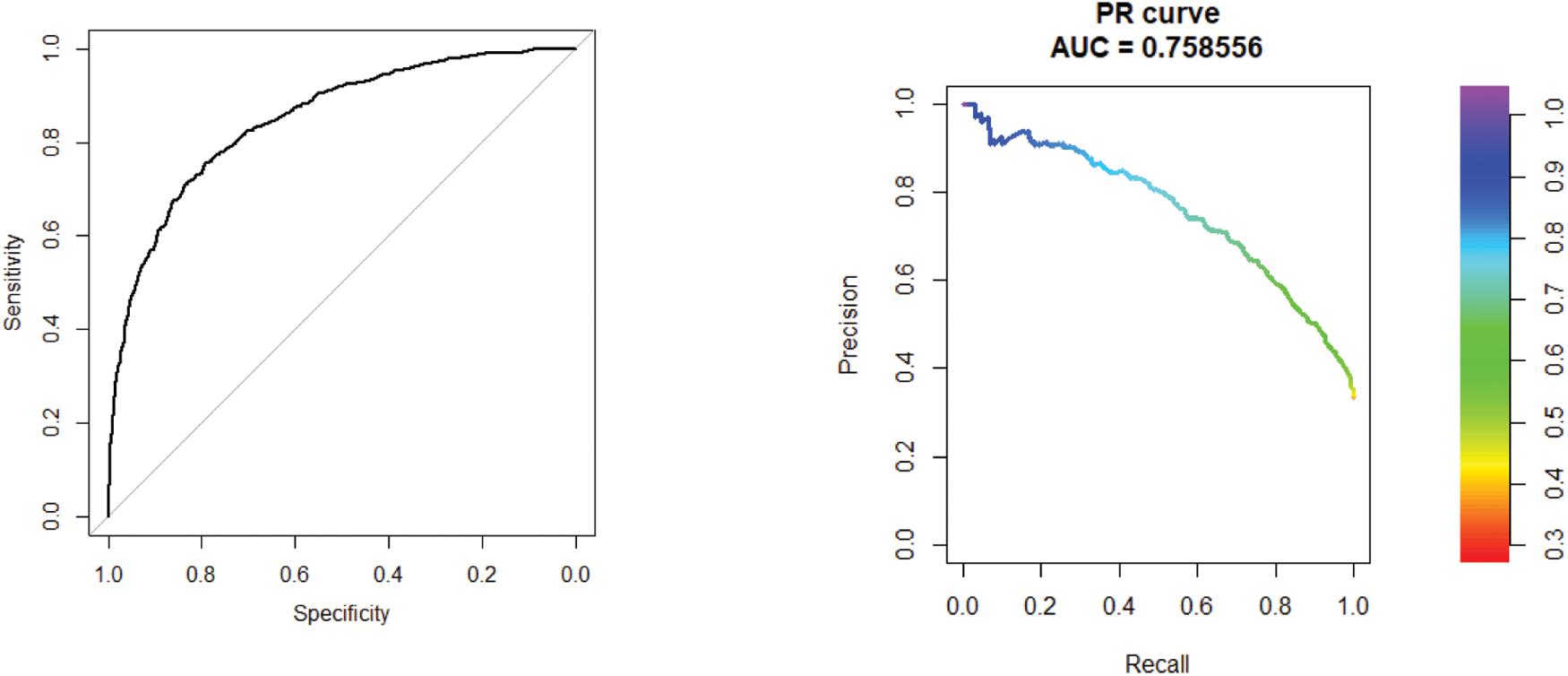
Figure 2. Receiver operating characteristic (ROC) curve of 10-cross validation experiments. The AUC and AUPR curve of GXGCP are 0.85 and 0.76, respectively. The comparison results are listed in Table 1.
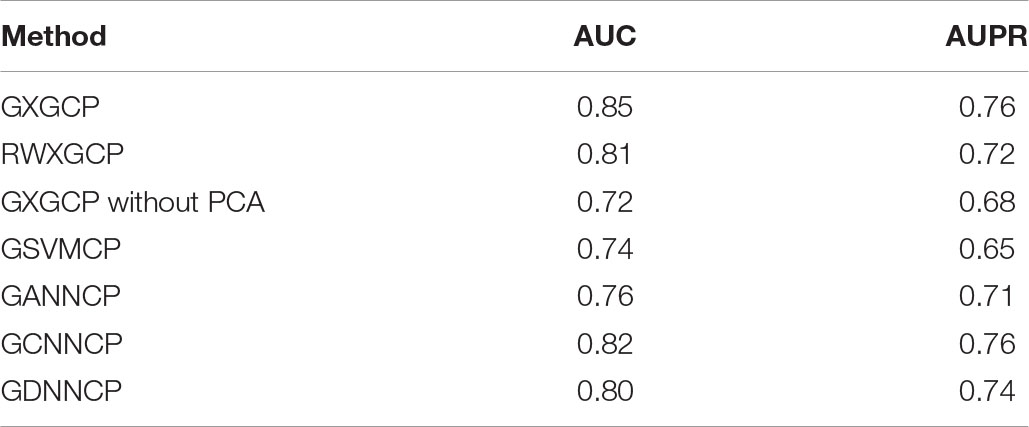
Table 1. Comparison results.
As shown in Table 1, GXGCP performed best among these five methods. These results show that GCN is more suitable for encoding network than RW, and Xgboost is more suitable for building model by sparse data than SVM and ANN.
Conclusion
Protein is the main executor of life activities. To decrypt the genome, you must first systematically understand the proteome. Identifying gastric cancer-related proteins can greatly help develop screening or testing tools for tumor detection, early diagnosis or differential diagnosis, prognostic analysis, efficacy evaluation, etc. Due to the high cost of biological experiments, we proposed GXGCP that fuses GCN, Xgboost, and PCA to identify gastric cancer-related proteins. To verify the accuracy of our method, we did 10-cross validation experiments. The results show that the AUC of GXGCP reached 0.85 and AUPR reached 0.76. To show the superiority of GXGCP, we compared it with several other methods, and GXGCP performed best. Overall, we propose a novel, efficient, and accurate method for large-scale identification of gastric cancer-related proteins, which would greatly benefit the study of the pathogenic mechanism and clinical research of gastric cancer.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics Statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author Contributions
HZ and RX wrote this manuscript and did experiments. MD and YZ provided important ideas. All authors read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Balluff, B., Rauser, S., Meding, S., Elsner, M., Schöne, C., Feuchtinger, A., et al. (2011). MALDI imaging identifies prognostic seven-protein signature of novel tissue markers in intestinal-type gastric cancer. Am. J. Pathol. 179, 2720–2729. doi: 10.1016/j.ajpath.2011.08.032
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. doi: 10.3322/caac.21492
Chen, T., and Guestrin, C. (2016). “Xgboost: a scalable tree boosting system,” in Proceedings of the 22nd ACM Sigkdd International Conference on Knowledge Discovery and Data Mining, (New York, NY: Association for Computing Machinery), 785–794.
Cheng, L., Han, X., Zhu, Z., Qi, C., Wang, P., and Zhang, X. (2021). Functional alterations caused by mutations reflect evolutionary trends of SARS-CoV-2. Brief. Bioinform. 22, 1442–1450. doi: 10.1093/bib/bbab042
Cheng, L., Li, J., Ju, P., Peng, J., and Wang, Y. (2014). SemFunSim: a new method for measuring disease similarity by integrating semantic and gene functional association. PLoS One 9:e99415. doi: 10.1371/journal.pone.0099415
Cheng, L., Zhao, H., Wang, P., Zhou, W., Luo, M., Li, T., et al. (2019). Computational methods for identifying similar diseases. Mol. Ther. Nucleic Acids 18, 590–604. doi: 10.1016/j.omtn.2019.09.019
Gullo, I., Carneiro, F., Oliveira, C., and Almeida, G. M. (2018). Heterogeneity in gastric cancer: from pure morphology to molecular classifications. Pathobiology 85, 50–63. doi: 10.1159/000473881
Gygi, S. P., Corthals, G. L., Zhang, Y., Rochon, Y., and Aebersold, R. (2000). Evaluation of two-dimensional gel electrophoresis-based proteome analysis technology. Proc. Natl. Acad. Sci. U.S.A. 97, 9390–9395. doi: 10.1073/pnas.160270797
Han, P., Yang, P., Zhao, P., Shang, S., Liu, Y., Zhou, J., et al. (2019). “GCN-MF: disease-gene association identification by graph convolutional networks and matrix factorization,” in Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, (New York, NY: Association for Computing Machinery), 705–713.
Jin, Z., Jiang, W., and Wang, L. (2015). Biomarkers for gastric cancer: Progression in early diagnosis and prognosis. Oncol. Lett. 9, 1502–1508. doi: 10.3892/ol.2015.2959
Liang, C., Changlu, Q., He, Z., Tongze, F., and Xue, Z. (2019). gutMDisorder: a comprehensive database for dysbiosis of the gut microbiota in disorders and interventions. Nucleic Acids Res. 48:7603. doi: 10.1093/nar/gkz843
Mering, C. V., Huynen, M., Jaeggi, D., Schmidt, S., Bork, P., and Snel, B. (2003). STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 31, 258–261. doi: 10.1093/nar/gkg034
Mo, F., Luo, Y., Fan, D. A., Zeng, H., Zhao, Y. N., Luo, M., et al. (2020). Integrated analysis of mRNA-seq and miRNA-seq to identify c-MYC, YAP1 and miR-3960 as major players in the anticancer effects of caffeic acid phenethyl ester in human small cell lung cancer cell line. Curr. Gene Ther. 20, 15–24. doi: 10.2174/1566523220666200523165159
Pang, L., Wang, J., Fan, Y., Xu, R., Bai, Y., and Bai, L. (2018). Correlations of TNM staging and lymph node metastasis of gastric cancer with MRI features and VEGF expression. Cancer Biomark. 23, 53–59. doi: 10.3233/cbm-181287
Park, J. S., Lee, N., Beom, S. H., Kim, H. S., Lee, C.-K., Rha, S. Y., et al. (2018). The prognostic value of volume-based parameters using 18 F-FDG PET/CT in gastric cancer according to HER2 status. Gastric Cancer 21, 213–224. doi: 10.1007/s10120-017-0739-0
Peng, J., and Zhao, T. (2020). Reduction in TOM1 expression exacerbates Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 117, 3915–3916. doi: 10.1073/pnas.1917589117
Ryu, J. W., Kim, H. J., Lee, Y. S., Myong, N. H., Hwang, C. H., Lee, G. S., et al. (2003). The proteomics approach to find biomarkers in gastric cancer. J. Korean Med. Sci. 18, 505–509. doi: 10.3346/jkms.2003.18.4.505
Sang, L., Miller, J. J., Corbit, K. C., Giles, R. H., Brauer, M. J., Otto, E. A., et al. (2011). Mapping the NPHP-JBTS-MKS protein network reveals ciliopathy disease genes and pathways. Cell 145, 513–528. doi: 10.1016/j.cell.2011.04.019
Seyfried, N. T., Dammer, E. B., Swarup, V., Nandakumar, D., Duong, D. M., Yin, L., et al. (2017). A multi-network approach identifies protein-specific co-expression in asymptomatic and symptomatic Alzheimer’s disease. Cell Syst. 4, 60–72.e4. doi: 10.1016/j.cels.2016.11.006
Szász, A. M., Lánczky, A., Nagy, Á, Förster, S., Hark, K., Green, J. E., et al. (2016). Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget 7, 49322–49333. doi: 10.18632/oncotarget.10337
Tannu, N. S., and Hemby, S. E. (2006). Two-dimensional fluorescence difference gel electrophoresis for comparative proteomics profiling. Nat. Protoc. 1, 1732–1742. doi: 10.1038/nprot.2006.256
Tianyi, Z., Yang, H., Valsdottir, L. R., Tianyi, Z., and Jiajie, P. (2020). Identifying drug–target interactions based on graph convolutional network and deep neural network. Brief. Bioinform. 22, 2141–2150. doi: 10.1093/bib/bbaa044
Tipping, M. E., and Bishop, C. M. (1999). Mixtures of probabilistic principal component analyzers. Neural. Comput. 11, 443–482. doi: 10.1162/089976699300016728
Villanueva, M. T. (2011). Combination therapy: update on gastric cancer in East Asia. Nat. Rev. Clin. Oncol. 8:690. doi: 10.1038/nrclinonc.2011.171
Zhao, T., Hu, Y., and Cheng, L. (2020a). Deep-DRM: a computational method for identifying disease-related metabolites based on graph deep learning approaches. Brief. Bioinform. 22:bbaa212. doi: 10.1093/bib/bbaa212
Zhao, T., Hu, Y., Peng, J., and Cheng, L. (2020b). DeepLGP: a novel deep learning method for prioritizing lncRNA target genes. Bioinformatics 36, 4466–4472. doi: 10.1093/bioinformatics/btaa428
Zhao, T., Hu, Y., Zang, T., and Wang, Y. (2020c). Identifying protein biomarkers in blood for Alzheimer’s disease. Front. Cell Dev. Biol. 8:472. doi: 10.3389/fcell.2020.00472
Zhao, T., Liu, J., Zeng, X., Wang, W., Li, S., Zang, T., et al. (2021a). Prediction and collection of protein–metabolite interactions. Brief. Bioinform. bbab014. doi: 10.1093/bib/bbab014
Zhao, T., Lyu, S., Lu, G., Juan, L., Zeng, X., Wei, Z., et al. (2021b). SC2disease: a manually curated database of single-cell transcriptome for human diseases. Nucleic Acids Res. 49, D1413–D1419. doi: 10.1093/nar/gkaa838
Keywords: gastric cancer, protein, proteomics data, graph convolutional network, Xgboost
Citation: Zhang H, Xu R, Ding M and Zhang Y (2021) Prediction of Gastric Cancer-Related Proteins Based on Graph Fusion Method. Front. Cell Dev. Biol. 9:739715. doi: 10.3389/fcell.2021.739715
Received: 11 July 2021; Accepted: 02 August 2021;
Published: 01 November 2021.
Edited by:
Lei Deng, Central South University, ChinaReviewed by:
Ningyi Zhang, Harbin Institute of Technology, ChinaHong Ju, Heilongjiang Vocational College of Biology Science and Technology, China
Copyright © 2021 Zhang, Xu, Ding and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Meng Ding, MzA5NDc4NTA2QHFxLmNvbQ==; Ying Zhang, NTE1MDcwNzg5QHFxLmNvbQ==
†These authors have contributed equally to this work