 Fulin Wang1,2,3,4,5†
Fulin Wang1,2,3,4,5† Qiuhuan Yuan1,2,3,4,5†Fengying Chen6*Jiaojiao Pang1,2,3,4,5
Qiuhuan Yuan1,2,3,4,5†Fengying Chen6*Jiaojiao Pang1,2,3,4,5 Chang Pan1,2,3,4,5
Chang Pan1,2,3,4,5 Feng Xu1,2,3,4,5*
Feng Xu1,2,3,4,5* Yuguo Chen1,2,3,4,5*
Yuguo Chen1,2,3,4,5*- 1Department of Emergency Medicine, Qilu Hospital, Shandong University, Jinan, China
- 2Chest Pain Center, Qilu Hospital, Shandong University, Jinan, China
- 3Shandong Provincial Clinical Research Center for Emergency and Critical Care Medicine, Institute of Emergency and Critical Care Medicine of Shandong University, Qilu Hospital, Shandong University, Jinan, China
- 4Key Laboratory of Emergency and Critical Care Medicine of Shandong Province, Key Laboratory of Cardiopulmonary-Cerebral Resuscitation Research of Shandong Province, Shandong Provincial Engineering Laboratory for Emergency and Critical Care Medicine, Qilu Hospital, Shandong University, Jinan, China
- 5The Key Laboratory of Cardiovascular Remodeling and Function Research, Chinese Ministry of Education, Chinese Ministry of Health and Chinese Academy of Medical Sciences, The State and Shandong Province Joint Key Laboratory of Translational Cardiovascular Medicine, Qilu Hospital, Shandong University, Jinan, China
- 6Emergency Department, The Affiliated Hospital of Inner Mongolia Medical University, Hohhot, China
Nitrosative stress, as an important oxygen metabolism disorder, has been shown to be closely associated with cardiovascular diseases, such as myocardial ischemia/reperfusion injury, aortic aneurysm, heart failure, hypertension, and atherosclerosis. Nitrosative stress refers to the joint biochemical reactions of nitric oxide (NO) and superoxide (O2–) when an oxygen metabolism disorder occurs in the body. The peroxynitrite anion (ONOO–) produced during this process can nitrate several biomolecules, such as proteins, lipids, and DNA, to generate 3-nitrotyrosine (3-NT), which further induces cell death. Among these, protein tyrosine nitration and polyunsaturated fatty acid nitration are the most studied types to date. Accordingly, an in-depth study of the relationship between nitrosative stress and cell death has important practical significance for revealing the pathogenesis and strategies for prevention and treatment of various diseases, particularly cardiovascular diseases. Here, we review the latest research progress on the mechanisms of nitrosative stress-mediated cell death, primarily involving several regulated cell death processes, including apoptosis, autophagy, ferroptosis, pyroptosis, NETosis, and parthanatos, highlighting nitrosative stress as a unique mechanism in cardiovascular diseases.
Introduction
Cell death is a manifestation of the irreversible cessation and end of life, which occurs widely during development and pathological processes (Fricker et al., 2018; Green, 2019). Recent studies have shown that nitrosative stress plays an important role in the pathophysiological processes of cell death. Under physiological conditions, low-level nitrosative stress can be detected in almost all cells in vivo, including cardiomyocytes, endothelial cells, fibroblasts, mesenchymal stem cells, and vascular smooth muscle cells (Brunner and Wolkart, 2003; Borbely et al., 2005; Steinberg, 2013). However, little is known about nitrosative stress in pathological conditions and the degree to which nitrosative stress changes the structure and function of cells. Furthermore, the type of cell death caused by stress is also less known. Additionally, what we still have to prove is that nitrosative stress and not other stresses related to oxygen metabolism disorders affects cell death. This review focuses on the biochemistry of nitrosative stress, as well as several new forms of cell death, and highlights its role in cell death, which provides important clues for studying the mechanism of cell death-related diseases (Table 1).

Table 1. Cell death caused by nitrosative stress.
Nitrosative Stress
There are various oxygen metabolism disorders in human body, including hypoxia (West, 2017), oxidative stress (Sohal and Allen, 1990), nitrosative stress (Lim, 2019), endoplasmic reticulum stress (ERS) (Marciniak and Ron, 2006), mitochondria dysfunction (Rowlands, 2016), and carbonyl stress (Suzuki et al., 2001; Miyata and de Strihou, 2010), which are listed in Table 2. Nitrosative stress is closely associated with oxidative stress. Reactive oxygen species (ROS), such as the superoxide anion (O), singlet oxygen (1O2), hydroxyl radical (OH), hydrogen peroxide (H2O2), peroxynitrite anion (ONOO–), and nitric oxide (NO), which are involved in oxidative stress, overlap with the formation and scavenging pathways of reactive nitrogen species (RNS) and regulate each other reciprocally (Espey et al., 2000; Maes et al., 2011). The ONOO– produced can nitrate several biomolecules, including proteins, lipids, and DNA, to generate 3-nitrotyrosine (3-NT). The most important characteristic of nitrosative stress is tyrosine nitration. Protein nitration modification is a post-translational modification of proteins caused by their interaction with RNS/ROS (Ischiropoulos, 2003; Velsor et al., 2003). In recent years, various studies have shown that the formation of 3-NT is a specific biomarker of nitrosative stress. It can be used to monitor the intracellular production and localization of ONOO– and the severity of cell death (Ahsan, 2013; Zhang and Wei, 2013).

Table 2. Comparison of different types of abnormal oxygen metabolism.
A Brief History and the Biochemistry of Nitrosative Stress
Ohshima et al. (1990) reported the detection of 3-NT and 3-nitrophenylacetic acid in human urine and proposed that 3-NT could be used as a marker for endogenous protein nitration. In 1992, the Beckman group confirmed that ONOO– can promote the nitrification of proteins such as superoxide dismutase (SOD) and CuZnSOD in vitro and proposed that endogenous nitrifiers can cause protein nitrification in the body (Ischiropoulos et al., 1992). Thereafter, Beckman proposed the use of the antibody method to identify nitrifying proteins and confirmed their existence in human atherosclerotic lesions and acute respiratory distress syndrome lung tissues using the immunohistochemical method (Ye et al., 1996). Subsequently, increasing studies have focused on protein tyrosine nitration from biochemical and biomedical standpoints. As extensive studies have been carried out on protein nitration in nitrosative stress injury, we primarily reviewed the biological characteristics of protein nitration. Multiple pathways can lead to protein nitration, among which ONOO– and NO2–/H2O2/heme peroxidase are considered the most important pathways (Herold, 2004). The process by which NO and O2– produce ONOO– is irreversible and does not require enzyme catalysis. The protein nitration pathways induced by ONOO– can be broadly divided into three categories: direct redox reactions, reactions with carbon dioxide (CO2), and homolysis after protonation (Alvarez and Radi, 2003; Figure 1A). Protein nitration via the NO2–/H2O2/heme peroxidase system also involves the formation of oxygen radicals (Bian et al., 2003; Figure 1B).
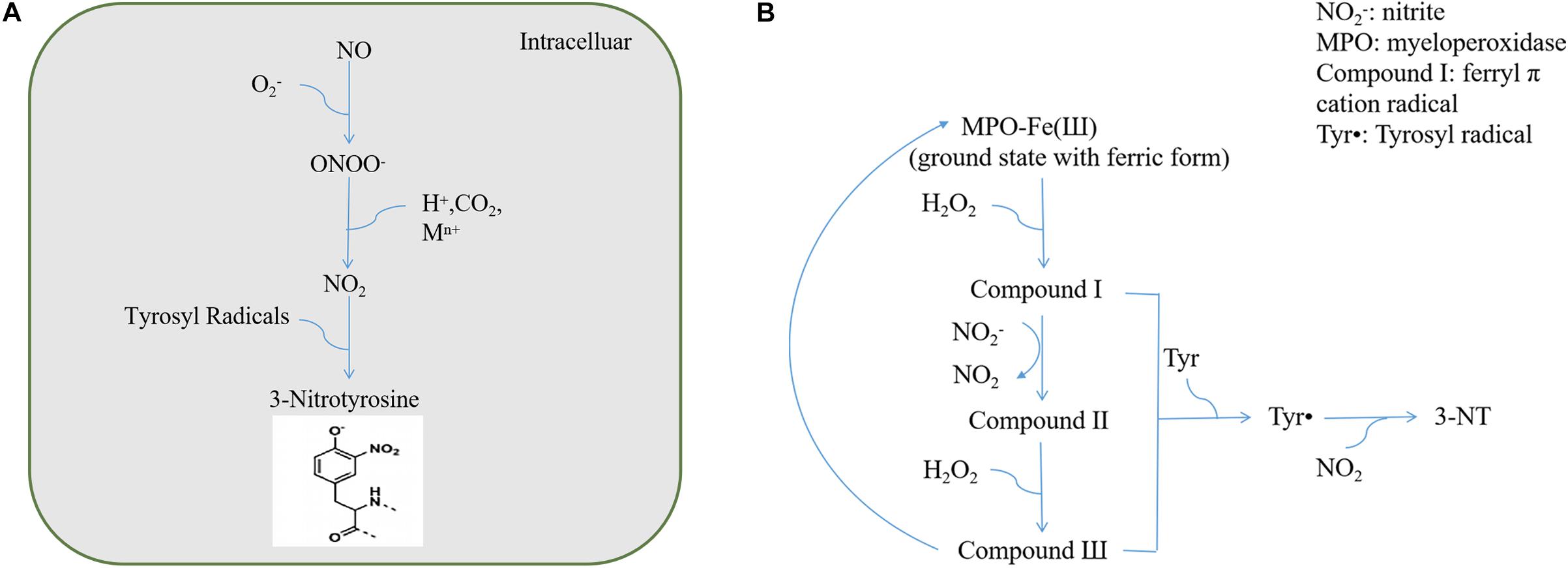
Figure 1. (A) ONOO– reaction pathways. (B) NO2–/H2O2/heme peroxidase system.
The Meaning of Nitrosative Stress
Nitrosative stress can promote protein tyrosine nitration, resulting in lipid peroxidation, DNA strand breaks, cell membrane damage, the inactivation of functional enzymes, and activation of cascade signal responses of cell death. Based on these basic biological functions, nitrosative stress affects diseases through various processes such as signal transduction in cells, mitochondrial energy metabolism, messenger ribonucleic acid transcription, protein post-translational modification, and ion channel function (Nobrega et al., 2019; Schiattarella et al., 2019; Paulus, 2020).
Cell Death
Cell death is defined by the phenomenon of the irreversible cessation of life, which is one of the leading causes of treatment failure and death in multiple diseases. The process of cell death comprises many steps, and the mechanism is complicated and has become the focus of life science and medical research (Chen et al., 2020). With the deepening of research, new progress has been made on the formation and mechanisms of cell death. To date, there are at least a dozen types of cell death, including apoptosis, necroptosis, pyroptosis, oncosis, ferroptosis, entotic cell death, NETotic cell death, parthanatos, phagocytosis, lysosome-dependent cell death, autophagy-dependent cell death, alkaliptosis, and oxeiptosis (Fricker et al., 2018; Tang et al., 2019; Bedoui et al., 2020).
Mechanisms of the Cell Death Caused by Nitrosative Stress
Apoptosis
Nitrosative stress-mediated apoptosis is an important apoptotic pathway newly discovered following intrinsic and extrinsic apoptosis pathways associated with the pathogenesis of various diseases (Andreka et al., 2004). An increasing number of studies have shown that nitrosative stress-mediated apoptosis is involved in the pathogenesis of many diseases. To some extent, 3-NT can be used as a marker of apoptosis (Jumper et al., 2002; Zhang and Wei, 2013).
Mitoptosis
Studies have shown that nitrosative stress affects enzyme activity, including that of complex I (NADH dehydrogenase), complex III (cytochrome c reductase), complex IV (cytochrome c oxidase), and complex V (ATP synthase), in the mitochondrial respiratory chain and increases cytochrome c release from mitochondria into the cytoplasm, indicating mitochondrial dysfunction (Arnaiz et al., 1999; Mastrocola et al., 2005). Furthermore, severe nitrosative stress leads to excessive consumption of ATP, resulting in an irreversible decline in mitochondrial membrane potential (ΔΨm), which eventually leads to mitochondrial apoptosis. This indicates that nitrosative stress not only can directly cause mitochondrial damage but also can act synergistically with oxidative stress (Tao et al., 2014; Ma et al., 2018a). At present, mitochondrial permeability transition pore (mPTP) opening is considered a common pathway for endogenous apoptosis after cell injury. The generation of RNS and the opening of the mPTP are a vicious cycle of mutual promotion. Abnormal mPTP opening due to external or pathological brings about the production of large numbers of ROS, RNS, and malondialdehyde (MDA) in cells, which may lead to the conformational changes in structural proteins on the mPTP, mPTP opening, and an increase in mitochondrial permeability, which in turn leads to apoptosis (Salimi et al., 2019).
Endoplasmic Reticulum Stress
Nitrosative stress can synergistically induce cell apoptosis under oxidative and ERS. C/EBP-homologous protein (CHOP) and caspase-12 are key proteins involved in ERS-mediated apoptosis. Thus, nitrosative stress induces ERS-dependent apoptosis through ERS-mediated JNK activation, CHOP transcriptional activation, and caspase-12 activation (Zhao et al., 2013; Tao et al., 2014; Lv et al., 2020).
Caspases/Bcl-2 Family Proteins
Studies have shown that the activity of caspase-3 in nitrosative stress induced by H2O2 increases and positively correlates with the apoptosis ratio. As a direct substrate of caspase-3, Bcl-2 plays a vital role in apoptosis. Nitrosative stress causes a reduction in the expression levels of STAT3, which regulates the transcription of the Bcl-2 family of anti-apoptotic genes (Rosati et al., 2019). Additionally, nitrosative stress leads to the activation of inducible nitric oxide synthase (iNOS)/Bax/caspase-3-mediated apoptosis in the L132 cell line (Anand et al., 2014). Another study showed that α-ZAL, a natural phytoestrogen, reduces Bax expression and the expression and activity of caspase-9. These effects may be related to the inhibition of nitrosative stress (Liu et al., 2013).
Other Pathways in Apoptosis Signaling
Peroxisome proliferator-activated receptor gamma (PPARγ), a transcriptional regulator of energy balance, can regulate nitrosative stress and inflammation in endothelial cells. A recent study showed that salusin-β, a bioactive peptide composed of 20 amino acid residues, regulates PPARγ to attenuate nitrosative stress, thereby reducing apoptosis (Sun et al., 2017). Another protein kinase, NF-κB, stimulates IL-1β transferase protease and TNF-α gene expression and induces apoptosis. Nitrosative stress causes abnormal activation of the NF-κB signaling pathway, and its transcription activity increases, thereby triggering apoptosis signals, resulting in apoptosis (Cheng et al., 2014). Several studies found that 3-NT can bind to the C-terminus of α-tubulin through tubulin tyrosine ligase to produce tyrosine tubulin, which is insufficient to maintain microtubule stability, causing cell degeneration, eventually leading to apoptosis (Bisig et al., 2002; Peluffo et al., 2004; Blanchard-Fillion et al., 2006). Another study showed that 3-NT can be used as an indirect inhibitor of tubulin-specific carboxypeptidase, thereby blocking the production of glutamate microtubules and further causing apoptosis (Chang et al., 2002). In recent years, researchers utilize this mechanism to develop related antitumor drugs, which induce tumor cell apoptosis by mediating the nitration of tubulin in tumor cells (Xue et al., 2020).
Autophagy
Autophagy can be divided into macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) according to the mode of transportation of degraded substances to lysosomes (Filomeni et al., 2015; Doherty and Baehrecke, 2018). In macroautophagy, intracellular autophagy-associated proteins (Atg) form autophagosomes by wrapping damaged proteins or organelles in the cytoplasm, and autophagosomes fuse with lysosomes to form autolysosomes. Autophagy lysosomes degrade the encapsulated substances, and some small molecular substances can be reused by cells. Microautophagy is a method by which lysosomes actively and directly engulf cytoplasmic components, which has rarely been explored in mammals. In CMA, some molecular chaperones, such as hsp70, can help unfolded proteins translocate into lysosomes. In recent years, with advances in research, some autophagy forms with a specific selectivity for substrate degradation, including mitochondrial autophagy (mitophagy), peroxisome autophagy (pexophagy), aggregate autophagy (aggrephagy), endoplasmic reticulum autophagy (reticulophagy), and ribosomal autophagy (ribophagy), have emerged (Kiffin et al., 2006; Coliva et al., 2019). Various studies have shown that nitrosative stress is involved in the occurrence and development of autophagy and that it can induce autophagy through multiple signal transduction pathways.
Macroautophagy
ROS/RNS can induce autophagy by inhibiting the protein kinase B (PKB)-Akt pathway and the Akt-mTOR pathway (Ma et al., 2018b). RNS is related to the post-translational modification of target proteins in cells, including protein thiol oxidation or S-nitrosylation related to the autophagy pathway. The accumulation of ROS/RNS generates carbonyls, whose accumulation is positively correlated with the development of autophagy (Di Meo et al., 2016; Feng et al., 2017). The autophagy-related proteins Atg3, 7, and 10 use cysteine residues to catalyze ubiquitin transfer at their catalytic sites (Ornatowski et al., 2020). The sulfhydryl groups on cysteine residues are particularly vulnerable to the modification of ROS/RNS oxidation; therefore, these proteins may be sensitive to redox signals (Filomeni et al., 2015; Varga et al., 2015). Notch 1 signaling is a key regulator of autophagy and reduces nitrosative stress. For example, genistein (Gen), a natural biologically active flavonoid found in soy, attenuates burn-induced myocardial autophagy by activating cardiac Notch 1 signaling and reducing nitrosative stress (Fang et al., 2019). In addition, nitrosative stress potentiates autophagy-lysosome signaling, which involves Beclin1, LC3, LAMP2, and cathepsin B (Han et al., 2011).
Mitophagy
Nitrosative stress can decrease ΔΨm and cause mitochondrial dysfunction. When ΔΨm decreases, PTEN-induced putative kinase 1 (PINK1) accumulates in the outer membrane and forms a large complex on the outer membrane surface to recruit Parkin to the damaged mitochondria, thereby causing mitochondrial autophagy (Redmann et al., 2016; Song et al., 2018). In a cerebral ischemia/reperfusion model, ONOO– induced the tyrosine nitration of dynamin-related protein 1 (Drp1) peptide and recruitment of Drp1 to damage mitochondria and subsequently induce PINK1/Parkin-mediated autophagy activation, thereby promoting brain ischemia/reperfusion injury (Feng et al., 2018a,b).
Pexophagy
Peroxisomes are critical metabolic organelles found in nearly all eukaryotic cells and are involved in ROS/RNS metabolism. Many peroxisomes cause severe nitrosative stress during metabolism (Jo et al., 2015; Sandalio and Romero-Puertas, 2015; Olmedilla and Sandalio, 2019). A recent study showed that certain miRNAs are associated with age-related degenerative changes. MiR-142 targets endothelial PAS domain protein 1 (Epas1), which is a known pexophagy regulatory protein. MiR-142 induces RNS/ROS accumulation by inducing pexophagy (Houri et al., 2020). The accumulation of ROS/RNS inhibits mTORC1 activity in the peroxisome, leading to the active translocation of the transcription factor EB (TFEB) into the nucleus, ultimately promoting autophagic flux and specifically inducing pexophagy through ubiquitin designation (Ganguli et al., 2019; Germain and Kim, 2020).
Ferroptosis
Ferroptosis was initially identified in small molecule erastin-induced cell death, which blocks cystine import, leading to glutathione depletion and inactivation of glutathione peroxidase 4 (GPX4) (Ursini et al., 1982; Yang et al., 2014). Ferroptosis is characterized by the accumulation of lethal lipid peroxidation, which is iron-dependent (Dixon et al., 2012; Stockwell et al., 2017; Lei et al., 2020). As the major component of lipid peroxidation, nitrosative stress was found to be related to the occurrence of ferroptosis (Uribe et al., 2018; Wu et al., 2020). Ferroptosis has been reported to be the prominent form of hepatocyte damage in Concanavalin A (ConA)-induced acute immune hepatitis, which is accompanied by RNS accumulation (Deng et al., 2020; Zeng et al., 2020). More importantly, the administration of an iNOS inhibitor or ONOO– scavenger diminishes RNS levels and reduces hepatocyte ferroptosis, suggesting that RNS, downstream of Caveolin-1 (Cav-1), is an important mediator that drives the hepatocellular ferroptosis induced by ConA. In addition, nitrosative stress is regulated by indoleamine 2,3-dioxygenase 1 (IDO1) during ferroptosis. IDO1 is an important cellular heme enzyme induced by pro-inflammatory mediators in response to inflammation and contributes to restraining the T and NK cells. Upregulation of IDO1 and nitrosative stress in ConA-induced hepatic damage are suppressed by ferroptosis abolishment. DO1 deficiency leads to ferroptosis inhibition by increasing the expression of solute carrier family 7 member 11 and RNS production, and the IDO1 inhibitor reduces iNOS and 3-NT expression during ferroptosis suppression (Zeng et al., 2020). Although few studies have paid close attention to the role of nitrosative stress in ferroptosis, the nature of ferroptosis caused by lethal lipid peroxidation suggests that nitrosative stress might significantly contribute to ferroptosis in various pathological conditions.
Pyroptosis
Nitrosative stress induces the activation of the nod-like receptor protein-3 inflammasome, which can cause pyroptosis through caspase-1- and caspase-11-mediated cytoplasmic protein gasdermin D (Mari and Colell, 2021). When acute and chronic inflammation occurs, the production rate of ONOO– exceeds the ability of the endogenous ONOO– defense system to scavenge it (Dedon and Tannenbaum, 2004; Galloway et al., 2014). Excessive ONOO– leads to an increase in inflammatory cytokine levels and induces tyrosine nitration, ultimately exacerbating the initial damage. A previous report suggested that TNF-α can activate various transcription factors, including NF-κB, activator protein 1, and interferon regulator factor 3, which induce the expression of target genes that encode various inflammatory factors (Pei et al., 2015). Furthermore, TNF-α induces nitrosative stress by upregulating the intercellular expression of adhesion molecule-1. Adiponectin (APN), an adipocyte-derived cytokine, attenuates TNF-α-induced inflammatory response through Cav-1-mediated ceramidase recruitment and activation in an AdipoR1-dependent manner (Wang et al., 2014). In addition, the upregulation of NF-E2-related factor-2, which is mainly secreted by macrophages, reduces nitrosative stress by blocking the NF-κB signal transduction (Li et al., 2019; Nadeem et al., 2020). The inflammatory response mediates the activation of phosphoinositide 3-kinase and the AKT (PI3K/AKT) and mitogen-activated protein kinase (MAPK) signaling pathways, which may also participate in the suppression of nitrosative stress (Ma et al., 2018b; Sarkozy et al., 2018).
NETosis
Some studies have shown that excessive intracellular RNS production in neutrophils is key to NETosis occurrence (Manda-Handzlik and Demkow, 2015; Lenin et al., 2018). Nitrosative stress mediates neutrophil activation, and activated neutrophils release neutrophil extracellular traps (NETs), a reticular structure that exerts a protective effect by surrounding and degrading pro-inflammatory cytokines (Boeltz et al., 2019). Recent research has shown that the formation of NETs induced by RNS depends on the activities of phosphoinositide 3-kinase and myeloperoxidase, as well as the selective degradation of histones H2A and H2B by neutrophil elastase (Chakraborty et al., 2011; Manda-Handzlik et al., 2020).
Parthanatos
A recent report showed that the overactivation of poly adenosine diphosphate-ribose polymerase-1 (PARP-1) leads to cell death. It is a new form of programmed cell death, termed “parthanatos,” which differs from apoptosis and other forms of cell death (Lee et al., 2013, 2015; Tang et al., 2019). At present, research on parthanatos is in its infancy, and the molecular mechanism of its signaling pathway is still unclear. As a unique cell death pathway, parthanatos is characterized by the overactivation of PARP-1, accumulation of cytoplasmic PAR polymers, mitochondrial depolarization, and apoptosis-inducing factor (AIF) nuclear translocation. When AIF enters the nucleus, it interacts with DNA through an as-of-yet unidentified PAAN, leading to large-scale DNA fragmentation and chromatin condensation (Fatokun et al., 2014). As DNA bases are sensitive to nitrosative stress, they are often modified by ONOO–. One study showed that DNA nitration is upstream of PARP-1 activation. Spermidine is a naturally occurring polyamine that is widely involved in DNA replication, transcription, and translation. The use of exogenous spermidine effectively inhibits the activation of PARP-1 and DNA nitrosative stress (Kim, 2017). Under nitrosative stress, ONOO– not only induces DNA single-strand breaks but also activates PARP (Buelow et al., 2009), which consumes a large amount of NAD+, resulting in energy depletion and necrotic cell death. One study showed that DNA nitration is upstream of PARP-1 activation (Barany et al., 2017; Horvath et al., 2018). The initial signaling of PARP-1 in mitochondria is mediated by the downstream activation of JNK-1, a stress-activated protein kinase (SAPK). When nitrosative stress occurs, JNK-1 is activated and participates in the regulation of parthanatos by regulating the intracellular ROS/RNS levels (Zheng et al., 2017).
Nitrosative Stress and Cardiovascular Diseases
Cardiovascular diseases have become the most important causes of death worldwide. Therefore, clarifying its pathophysiological mechanisms is crucial for formulating prevention and treatment strategies. From a cardiovascular aspect, nitrosative stress is associated with the pathophysiological processes. Nitrosative stress can lead to key protein tyrosine nitration related to contractility, metabolism, and antioxidant defense mechanisms of the myocardium and skeletal muscle (Breitkreuz and Hamdani, 2015; Mozos and Luca, 2017). Furthermore, it induces myocardial hypertrophy, fibrosis, or cell death by activating inflammatory responses and stress signals (such as apoptosis and autophagy) (Frati et al., 2017). Myocardial ischemia/reperfusion injury and heart failure are the two main pathophysiological alterations associated with nitrosative stress. Recent studies demonstrated that melatonin receptor 2 (MT2) protects cardiomyocytes from nitrosative stress injury through the MT2/Notch1/Hes1/RORIα signaling pathway in a mouse myocardial ischemia/reperfusion model. Moreover, TNF-α-converting enzyme (TACE) protects against MRI via Notch1-mediated suppression of nitrosative stress. These results demonstrate that the upregulation of Notch1 may alleviate nitrosative stress-mediated myocardial ischemia/reperfusion injury (Pei et al., 2015; Yu et al., 2018; Zhang et al., 2020). In addition, ONOO– can interfere with the cellular calcium transport system, such as the oxidation of sulfhydryl groups of Na+/Ca2+ exchangers, resulting in Na+/Ca2+ exchanger protein dysfunction (Kitao et al., 2010). In heart failure, nitrosative stress causes sarcoplasmic reticulum Ca2+-ATPase tyrosine nitration (Schoneich et al., 1999; Lokuta et al., 2005), further aggravating the dysfunction of left ventricular and vascular systolic functions (Kennedy et al., 2015; Schiattarella et al., 2019). Heart failure induces excessive production of iNOS by macrophages and cardiomyocytes, leading to the loss of myocardial contractility and decreased reactivity of β-adrenergic receptors (Kingery et al., 2017). Nitrosative stress adversely affects the regulation of muscle fiber energy. Myofibril creatine kinase plays an essential role in regulating cardiomyocyte contraction in patients with heart failure and is more susceptible to nitration modification. Creatine kinase activity can be inhibited by nitrosative stress, resulting in the decline of myocardial contractile function and promotion of the occurrence and development of heart failure (Mihm et al., 2001a,b).
Conclusion
Nitrosative stress falls under oxidative stress in the traditional category. However, in recent years, a growing body of knowledge has revealed that nitrosative stress, as an independent special biochemical phenomenon in the process of cell death, has unique pathophysiological characteristics that differ from those of oxidative stress in a general sense. This is because of the high responsiveness and short half-life of RNS. However, current research methods have not been able to accurately identify the RNS that induces nitrosative stress. In addition, there is a lack of practical methodologies for the positioning and quantification of nitrosative stress. Although a few antibodies can detect 3-NT, protein tyrosine nitration does not fully reflect the level of nitrosative stress. Therefore, future breakthroughs in biological methodology for detecting nitrosative stress will promote the development of research in this field and deepen our understanding of nitrosative stress biology. During nitrosative stress, the specific target protein tyrosine nitration in different diseases differs. Further studies on the relationship between specific protein tyrosine nitration and diseases will help reveal the pathogenesis of related diseases and identify new targets for drugs. The mechanisms underlying cell death due to nitrosative stress include target protein tyrosine nitration, mitochondrial dysfunction, and cell membrane destruction, which eventually lead to cell death. Revealing the mechanism of cell death induced by nitrosative stress is of great significance for the development of drugs targeting nitrosative stress. In recent years, significant progress has been made in determining the role of nitrosative stress injury in cell death; however, some key problems, such as the concentration threshold of ONOO– damage to various important cellular organs and the corresponding pathophysiological effects, have not been addressed. In summary, research on the mechanism of cell death induced by nitrosative stress helps us to further reveal the pathogenesis of cell injury, explore new sub-therapeutic targets, and provide new ways to prevent and treat diseases related to cell death. Cardiovascular disease is the first enemy of human health. Understanding nitrosative stress will help bridge the gap in the relationship between cell death and cardiovascular disease, thereby providing vital information for improving human health.
Author Contributions
YC, FX, and FC designed the framework. FW and QY wrote the manuscript. JP and CP revised the manuscript. All authors read and approved the final version of the manuscript for publication.
Funding
This work was supported by the National Natural Science Foundation of China (81873950, 82072144, 81900435, 81772036, and 81873953), the State Key Program of the National Natural Science Foundation of China (82030059), the National Key R&D Program of China (2020YFC1512700, 2020YFC1512705, and 2020YFC1512703), National S&T Fundamental Resources Investigation Project (2018FY100600 and 2018FY100602), the Taishan Young Scholar Program of Shandong Province (tsqn20161065 and tsqn201812129), the Taishan Pandeng Scholar Program of Shandong Province (tspd20181220), and the Interdisciplinary Young Researcher Groups Program of Shandong University (2020QNQT004).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahsan, H. (2013). 3-Nitrotyrosine: a biomarker of nitrogen free radical species modified proteins in systemic autoimmunogenic conditions. Hum. Immunol. 74, 1392–1399. doi: 10.1016/j.humimm.2013.06.009
Alvarez, B., and Radi, R. (2003). Peroxynitrite reactivity with amino acids and proteins. Amino Acids 25, 295–311. doi: 10.1007/s00726-003-0018-8
Anand, T., Pandareesh, M. D., Bhat, P. V., and Venkataramana, M. (2014). Anti-apoptotic mechanism of bacoside rich extract against reactive nitrogen species induced activation of iNOS/Bax/caspase 3 mediated apoptosis in L132 cell line. Cytotechnology 66, 823–838. doi: 10.1007/s10616-013-9634-7
Andreka, P., Tran, T., Webster, K. A., and Bishopric, N. H. (2004). Nitric oxide and promotion of cardiac myocyte apoptosis. Mol. Cell. Biochem. 263, 35–53. doi: 10.1023/b:mcbi.0000041847.63338.b8
Arnaiz, S. L., Coronel, M. F., and Boveris, A. (1999). Nitric oxide, superoxide, and hydrogen peroxide production in brain mitochondria after haloperidol treatment. Nitric Oxide 3, 235–243. doi: 10.1006/niox.1999.0229
Barany, T., Simon, A., Szabo, G., Benkõ, R., Mezei, Z., Molnár, L., et al. (2017). Oxidative stress-related parthanatos of circulating mononuclear leukocytes in heart failure. Oxid. Med. Cell. Longev. 2017:1249614.
Bedoui, S., Herold, M. J., and Strasser, A. (2020). Emerging connectivity of programmed cell death pathways and its physiological implications. Nat. Rev. Mol. Cell Biol. 21, 678–695. doi: 10.1038/s41580-020-0270-8
Bian, K., Gao, Z., Weisbrodt, N., and Murad, F. (2003). The nature of heme/iron-induced protein tyrosine nitration. Proc. Natl. Acad. Sci. U.S.A. 100, 5712–5717. doi: 10.1073/pnas.0931291100
Bisig, C. G., Purro, S. A., Contin, M. A., Barra, H. S., and Arce, C. A. (2002). Incorporation of 3-nitrotyrosine into the C-terminus of alpha-tubulin is reversible and not detrimental to dividing cells. Eur. J. Biochem. 269, 5037–5045. doi: 10.1046/j.1432-1033.2002.03220.x
Blanchard-Fillion, B., Prou, D., Polydoro, M., Spielberg, D., Tsika, E., Wang, Z., et al. (2006). Metabolism of 3-nitrotyrosine induces apoptotic death in dopaminergic cells. J. Neurosci. 26, 6124–6130. doi: 10.1523/jneurosci.1038-06.2006
Boeltz, S., Amini, P., Anders, H. J., Andrade, F., Bilyy, R., Chatfield, S., et al. (2019). To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 26, 395–408.
Borbely, A., Toth, A., Edes, I., Virág, L., Papp, J. G., Varró, A., et al. (2005). Peroxynitrite-induced alpha-actinin nitration and contractile alterations in isolated human myocardial cells. Cardiovasc. Res. 67, 225–233.
Breitkreuz, M., and Hamdani, N. (2015). A change of heart: oxidative stress in governing muscle function? Biophys. Rev. 7, 321–341. doi: 10.1007/s12551-015-0175-5
Brunner, F., and Wolkart, G. (2003). Peroxynitrite-induced cardiac depression: role of myofilament desensitization and cGMP pathway. Cardiovasc. Res. 60, 355–364. doi: 10.1016/j.cardiores.2003.08.001
Buelow, B., Uzunparmak, B., Paddock, M., and Scharenberg, A. M. (2009). Structure/function analysis of PARP-1 in oxidative and nitrosative stress-induced monomeric ADPR formation. PLoS One 4:e6339. doi: 10.1371/journal.pone.0006339
Chakraborty, A., Chowdhury, S., and Bhattacharyya, M. (2011). Effect of metformin on oxidative stress, nitrosative stress and inflammatory biomarkers in type 2 diabetes patients. Diabetes Res. Clin. Pract. 93, 56–62. doi: 10.1016/j.diabres.2010.11.030
Chang, W., Webster, D. R., Salam, A. A., Gruber, D., Prasad, A., Eiserich, J. P., et al. (2002). Alteration of the C-terminal amino acid of tubulin specifically inhibits myogenic differentiation. J. Biol. Chem. 277, 30690–30698. doi: 10.1074/jbc.m204930200
Chen, Y., Hua, Y., Li, X., Arslan, I. M., Zhang, W., and Meng, G. (2020). Distinct types of cell death and the implication in diabetic cardiomyopathy. Front. Pharmacol. 11:42. doi: 10.3389/fphar.2020.00042
Cheng, C. Y., Lin, J. G., Tang, N. Y., Kao, S. T., and Hsieh, C. L. (2014). Electroacupuncture-like stimulation at the Baihui (GV20) and Dazhui (GV14) acupoints protects rats against subacute-phase cerebral ischemia-reperfusion injuries by reducing S100B-mediated neurotoxicity. PLoS One 9:e91426. doi: 10.1371/journal.pone.0091426
Coliva, G., Duarte, S., Perez-Sala, D., and Fedorova, M. (2019). Impact of inhibition of the autophagy-lysosomal pathway on biomolecules carbonylation and proteome regulation in rat cardiac cells. Redox Biol. 23:101123. doi: 10.1016/j.redox.2019.101123
Dedon, P. C., and Tannenbaum, S. R. (2004). Reactive nitrogen species in the chemical biology of inflammation. Arch. Biochem. Biophys. 423, 12–22. doi: 10.1016/j.abb.2003.12.017
Deng, G., Li, Y., Ma, S., Gao, Z., Zeng, T., Chen, L., et al. (2020). Caveolin-1 dictates ferroptosis in the execution of acute immune-mediated hepatic damage by attenuating nitrogen stress. Free Radic. Biol. Med. 148, 151–161.
Di Meo, S., Reed, T. T., Venditti, P., and Victor, V. M. (2016). Role of ROS and RNS sources in physiological and pathological conditions. Oxid. Med. Cell. Longev. 2016:1245049.
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. doi: 10.1016/j.cell.2012.03.042
Espey, M. G., Miranda, K. M., Feelisch, M., Fukuto, J., Grisham, M. B., Vitek, M. P., et al. (2000). Mechanisms of cell death governed by the balance between nitrosative and oxidative stress. Ann. N. Y. Acad. Sci. 899, 209–221.
Fang, Z., Wu, G., Zhang, D., Wang, K., Deng, X., Liu, M., et al. (2019). Genistein protects against burn-induced myocardial injury via Notch1 mediated suppression of oxidative/nitrative stress. Shock 54, 277–279
Fatokun, A. A., Dawson, V. L., and Dawson, T. M. (2014). Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 171, 2000–2016. doi: 10.1111/bph.12416
Feng, J., Chen, X., and Shen, J. (2017). Reactive nitrogen species as therapeutic targets for autophagy: implication for ischemic stroke. Expert Opin. Ther. Targets 21, 305–317. doi: 10.1080/14728222.2017.1281250
Feng, J., Chen, X., Guan, B., Li, C., Qiu, J., and Shen, J. (2018a). Inhibition of peroxynitrite-induced mitophagy activation attenuates cerebral ischemia-reperfusion injury. Mol. Neurobiol. 55, 6369–6386. doi: 10.1007/s12035-017-0859-x
Feng, J., Chen, X., Lu, S., Li, W., Yang, D., Su, W., et al. (2018b). Naringin attenuates cerebral ischemia-reperfusion injury through inhibiting peroxynitrite-mediated mitophagy activation. Mol. Neurobiol. 55, 9029–9042. doi: 10.1007/s12035-018-1027-7
Filomeni, G., De Zio, D., and Cecconi, F. (2015). Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 22, 377–388. doi: 10.1038/cdd.2014.150
Frati, G., Schirone, L., Chimenti, I., Yee, D., Biondi-Zoccai, G., Volpe, M., et al. (2017). An overview of the inflammatory signalling mechanisms in the myocardium underlying the development of diabetic cardiomyopathy. Cardiovasc. Res. 113, 378–388. doi: 10.1093/cvr/cvx011
Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M., and Brown, G. C. (2018). Neuronal cell death. Physiol. Rev. 98, 813–880.
Galloway, C. A., Lee, H., Brookes, P. S., and Yoon, Y. (2014). Decreasing mitochondrial fission alleviates hepatic steatosis in a murine model of nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 307, G632–G641.
Ganguli, G., Mukherjee, U., and Sonawane, A. (2019). Peroxisomes and oxidative stress: their implications in the modulation of cellular immunity during mycobacterial infection. Front. Microbiol. 10:1121. doi: 10.3389/fmicb.2019.01121
Germain, K., and Kim, P. K. (2020). Pexophagy: a model for selective autophagy. Int. J. Mol. Sci. 21:578. doi: 10.3390/ijms21020578
Green, D. R. (2019). The coming decade of cell death research: five riddles. Cell 177, 1094–1107. doi: 10.1016/j.cell.2019.04.024
Han, F., Chen, Y. X., Lu, Y. M., Huang, J. Y., Zhang, G. S., Tao, R. R., et al. (2011). Regulation of the ischemia-induced autophagy-lysosome processes by nitrosative stress in endothelial cells. J. Pineal Res. 51, 124–135. doi: 10.1111/j.1600-079x.2011.00869.x
Herold, S. (2004). Nitrotyrosine, dityrosine, and nitrotryptophan formation from metmyoglobin, hydrogen peroxide, and nitrite. Free Radic. Biol. Med. 36, 565–579. doi: 10.1016/j.freeradbiomed.2003.10.014
Horvath, E. M., Magenheim, R., Beres, N. J., Benkõ, R., Pék, T., Tabák, ÁG., et al. (2018). Oxidative-nitrative stress and poly (ADP-ribose) polymerase activation 3 years after pregnancy. Oxid. Med. Cell. Longev. 2018:1743253.
Houri, K., Mori, T., Onodera, Y., Tsujimoto, T., Takehara, T., Nakao, S., et al. (2020). miR-142 induces accumulation of reactive oxygen species (ROS) by inhibiting pexophagy in aged bone marrow mesenchymal stem cells. Sci. Rep. 10:3735.
Ischiropoulos, H. (2003). Biological selectivity and functional aspects of protein tyrosine nitration. Biochem. Biophys. Res. Commun. 305, 776–783. doi: 10.1016/s0006-291x(03)00814-3
Ischiropoulos, H., Zhu, L., Chen, J., Tsai, M., Martin, J. C., Smith, C. D., et al. (1992). Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch. Biochem. Biophys. 298, 431–437. doi: 10.1016/0003-9861(92)90431-u
Jo, D. S., Bae, D. J., Park, S. J., Seo, H. M., Kim, H. B., Oh, J. S., et al. (2015). Pexophagy is induced by increasing peroxisomal reactive oxygen species in 1’10-phenanthroline-treated cells. Biochem. Biophys. Res. Commun. 467, 354–360. doi: 10.1016/j.bbrc.2015.09.153
Jumper, J., Kalns, J., Batey, K., Lane, R., and Reed, J. (2002). The 3-nitrotyrosine marker of apoptosis shows time-dependent and tissue-specific variation in a porcine retinal laser injury model. J. Investig. Ophthalmol. Vis. Sci. 43:4528.
Kennedy, D. J., Shrestha, K., Sheehey, B., Li, X. S., Guggilam, A., Wu, Y., et al. (2015). Elevated plasma marinobufagenin, an endogenous cardiotonic steroid, is associated with right ventricular dysfunction and nitrative stress in heart failure. Circ. Heart Fail. 8, 1068–1076. doi: 10.1161/circheartfailure.114.001976
Kiffin, R., Bandyopadhyay, U., and Cuervo, A. M. (2006). Oxidative stress and autophagy. Antioxid. Redox Signal. 8, 152–162.
Kim, J. (2017). Spermidine is protective against kidney ischemia and reperfusion injury through inhibiting DNA nitration and PARP1 activation. Anat. Cell Biol. 50, 200–206. doi: 10.5115/acb.2017.50.3.200
Kingery, J. R., Hamid, T., Lewis, R. K., Ismahil, M. A., Bansal, S. S., Rokosh, G., et al. (2017). Leukocyte iNOS is required for inflammation and pathological remodeling in ischemic heart failure. Basic Res. Cardiol. 112:19. doi: 10.1016/b978-0-12-800039-7.00002-5
Kitao, T., Takuma, K., Kawasaki, T., Inoue, Y., Ikehara, A., Nashida, T., et al. (2010). The Na+/Ca2+ exchanger-mediated Ca2+ influx triggers nitric oxide-induced cytotoxicity in cultured astrocytes. Neurochem. Int. 57, 58–66. doi: 10.1016/j.neuint.2010.04.016
Lee, Y., Karuppagounder, S. S., Shin, J. H., Lee, Y. I., Ko, H. S., Swing, D., et al. (2015). Corrigendum: parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat. Neurosci. 18:1861. doi: 10.1038/nn1215-1861a
Lee, Y., Karuppagounder, S. S., Shin, J. H., Lee, Y. I., Ko, H. S., Swing, D., et al. (2013). Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat. Neurosci. 16, 1392–1400. doi: 10.1038/nn.3500
Lei, J., Chen, Z., Song, S., Sheng, C., Song, S., and Zhu, J. (2020). Insight into the role of ferroptosis in non-neoplastic neurological diseases. Front. Cell. Neurosci. 14:231. doi: 10.3389/fncel.2020.00231
Lenin, R., Thomas, S. M., and Gangaraju, R. (2018). Endothelial activation and oxidative stress in neurovascular defects of the retina. Curr. Pharm. Des. 24, 4742–4754. doi: 10.2174/1381612825666190115122622
Li, Y., Deng, S. L., Lian, Z. X., and Yu, K. (2019). Roles of Toll-like receptors in nitroxidative stress in mammals. Cells 8:576. doi: 10.3390/cells8060576
Lim, G. B. (2019). New mouse model reveals nitrosative stress as a novel driver of HFpEF. Nat. Rev. Cardiol. 16:383. doi: 10.1038/s41569-019-0203-4
Liu, T., Hou, D. D., Zhao, Q., Liu, W., Zhen, P. P., Xu, J. P., et al. (2013). Phytoestrogen alpha-zearalanol attenuates homocysteine-induced apoptosis in human umbilical vein endothelial cells. Biomed. Res. Int. 2013:813450.
Lokuta, A. J., Maertz, N. A., Meethal, S. V., Potter, K. T., Kamp, T. J., Valdivia, H. H., et al. (2005). Increased nitration of sarcoplasmic reticulum Ca2+-ATPase in human heart failure. Circulation 111, 988–995. doi: 10.1161/01.cir.0000156461.81529.d7
Lv, S., Wu, N., Wang, Q., and Yang, L. H. (2020). Endogenous hydrogen sulfide alleviates methotrexate-induced cognitive impairment by attenuating endoplasmic reticulum stress-induced apoptosis via CHOP and caspase-12. Fundam. Clin. Pharmacol. 34, 559–570. doi: 10.1111/fcp.12543
Ma, L. L., Li, Y., Yin, P. P., Kong, F. J., Guo, J. J., Shi, H. T., et al. (2018a). Hypertrophied myocardium is vulnerable to ischemia/reperfusion injury and refractory to rapamycin-induced protection due to increased oxidative/nitrative stress. Clin. Sci. 132, 93–110. doi: 10.1042/cs20171471
Ma, L. L., Ma, X., Kong, F. J., Guo, J. J., Shi, H. T., Zhu, J. B., et al. (2018b). Mammalian target of rapamycin inhibition attenuates myocardial ischaemia-reperfusion injury in hypertrophic heart. J. Cell. Mol. Med. 22, 1708–1719. doi: 10.1111/jcmm.13451
Maes, M., Galecki, P., Chang, Y. S., and Berk, M. (2011). A review on the oxidative and nitrosative stress (O&NS) pathways in major depression and their possible contribution to the (neuro)degenerative processes in that illness. Prog. Neuropsychopharmacol. Biol. Psychiatry 35, 676–692.
Manda-Handzlik, A., and Demkow, U. (2015). Neutrophils: the role of oxidative and nitrosative stress in health and disease. Adv. Exp. Med. Biol. 857, 51–60. doi: 10.1007/5584_2015_117
Manda-Handzlik, A., Bystrzycka, W., Cieloch, A., Glodkowska-Mrowka, E., Jankowska-Steifer, E., Heropolitanska-Pliszka, E., et al. (2020). Nitric oxide and peroxynitrite trigger and enhance release of neutrophil extracellular traps. Cell. Mol. Life Sci. 77, 3059–3075. doi: 10.1007/s00018-019-03331-x
Marciniak, S. J., and Ron, D. (2006). Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 86, 1133–1149. doi: 10.1152/physrev.00015.2006
Mari, M., and Colell, A. (2021). Mitochondrial oxidative and nitrosative stress as a therapeutic target in diseases. Antioxidants 10:314 doi: 10.3390/antiox10020314
Mastrocola, R., Restivo, F., Vercellinatto, I., Danni, O., Brignardello, E., Aragno, M., et al. (2005). Oxidative and nitrosative stress in brain mitochondria of diabetic rats. J. Endocrinol. 187, 37–44. doi: 10.1677/joe.1.06269
Mihm, M. J., Coyle, C. M., Schanbacher, B. L., Weinstein, D. M., and Bauer, J. A. (2001a). Peroxynitrite induced nitration and inactivation of myofibrillar creatine kinase in experimental heart failure. Cardiovasc. Res. 49, 798–807. doi: 10.1016/s0008-6363(00)00307-2
Mihm, M. J., Yu, F., Carnes, C. A., Reiser, P. J., McCarthy, P. M., Van Wagoner, D. R., et al. (2001b). Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation 104, 174–180. doi: 10.1161/01.cir.104.2.174
Miyata, T., and de Strihou, C. (2010). Diabetic nephropathy: a disorder of oxygen metabolism? Nat. Rev. Nephrol. 6, 83–95. doi: 10.1038/nrneph.2009.211
Mozos, I., and Luca, C. T. (2017). Crosstalk between oxidative and nitrosative stress and arterial stiffness. Curr. Vasc. Pharmacol. 15, 446–456.
Nadeem, A., Ahmad, S. F., Al-Ayadhi, L. Y., Attia, S. M., Al-Harbi, N. O., Alzahrani, K. S., et al. (2020). Differential regulation of Nrf2 is linked to elevated inflammation and nitrative stress in monocytes of children with autism. Psychoneuroendocrinology 113:104554. doi: 10.1016/j.psyneuen.2019.104554
Nobrega, N., Araujo, N. F., Reis, D., Facine, L. M., Miranda, C. A. S., Mota, G. C., et al. (2019). Hydrogen peroxide and nitric oxide induce anticontractile effect of perivascular adipose tissue via renin angiotensin system activation. Nitric Oxide 84, 50–59.
Ohshima, H., Friesen, M., Brouet, I., and Bartsch, H. (1990). Nitrotyrosine as a new marker for endogenous nitrosation and nitration of proteins. Food Chem. Toxicol. 28, 647–652. doi: 10.1016/0278-6915(90)90173-k
Olmedilla, A., and Sandalio, L. M. (2019). Selective autophagy of peroxisomes in plants: from housekeeping to development and stress responses. Front. Plant Sci. 10:1021. doi: 10.3389/fpls.2019.01021
Ornatowski, W., Lu, Q., Yegambaram, M., Garcia, A. E., Zemskov, E. A., Maltepe, E., et al. (2020). Complex interplay between autophagy and oxidative stress in the development of pulmonary disease. Redox Biol. 36:101679. doi: 10.1016/j.redox.2020.101679
Paulus, W. J. (2020). Unfolding discoveries in heart failure. N. Engl. J. Med. 382:679–682. doi: 10.1056/nejmcibr1913825
Pei, H., Song, X., Peng, C., Tan, Y., Li, Y., Li, X., et al. (2015). TNF-alpha inhibitor protects against myocardial ischemia/reperfusion injury via Notch1-mediated suppression of oxidative/nitrative stress. Free Radic. Biol. Med. 82, 114–121. doi: 10.1016/j.freeradbiomed.2015.02.002
Peluffo, H., Shacka, J. J., Ricart, K., Bisig, C. G., Martìnez-Palma, L., Pritsch, O., et al. (2004). Induction of motor neuron apoptosis by free 3-nitro-L-tyrosine. J. Neurochem. 89, 602–612. doi: 10.1046/j.1471-4159.2004.02363.x
Redmann, M., Darley-Usmar, V., and Zhang, J. (2016). The Role of autophagy, mitophagy and lysosomal functions in modulating bioenergetics and survival in the context of redox and proteotoxic damage: implications for neurodegenerative diseases. Aging Dis. 7, 150–162. doi: 10.14336/ad.2015.0820
Rosati, R., Shahab, M., Neumann, W. L., and Jamesdaniel, S. (2019). Inhibition of protein nitration prevents cisplatin-induced inactivation of STAT3 and promotes anti-apoptotic signaling in organ of Corti cells. Exp. Cell Res. 381, 105–111. doi: 10.1016/j.yexcr.2019.05.008
Rowlands, D. J. (2016). Mitochondria dysfunction: a novel therapeutic target in pathological lung remodeling or bystander? Pharmacol. Ther. 166, 96–105. doi: 10.1016/j.pharmthera.2016.06.019
Salimi, A., Neshat, M. R., Naserzadeh, P., and Pourahmad, J. (2019). Mitochondrial permeability transition pore sealing agents and antioxidants protect oxidative stress and mitochondrial dysfunction induced by naproxen, diclofenac and celecoxib. Drug Res. 69, 598–605. doi: 10.1055/a-0866-9356
Sandalio, L. M., and Romero-Puertas, M. C. (2015). Peroxisomes sense and respond to environmental cues by regulating ROS and RNS signalling networks. Ann. Bot. 116, 475–485. doi: 10.1093/aob/mcv074
Sarkozy, M., Kovacs, Z. Z. A., Kovacs, M. G., Gaspar, R., Szucs, G., and Dux, L. (2018). Mechanisms and modulation of oxidative/nitrative stress in type 4 cardio-renal syndrome and renal sarcopenia. Front. Physiol. 9:1648. doi: 10.3389/fphys.2018.01648
Schiattarella, G. G., Altamirano, F., Tong, D., French, K. M., Villalobos, E., Kim, S. Y., et al. (2019). Nitrosative stress drives heart failure with preserved ejection fraction. Nature 568, 351–356.
Schoneich, C., Viner, R. I., Ferrington, D. A., and Bigelow, D. J. (1999). Age-related chemical modification of the skeletal muscle sarcoplasmic reticulum Ca-ATPase of the rat. Mech. Ageing Dev. 107, 221–231. doi: 10.1016/s0047-6374(98)00158-4
Sohal, R. S., and Allen, R. G. (1990). Oxidative stress as a causal factor in differentiation and aging: a unifying hypothesis. Exp. Gerontol. 25, 499–522. doi: 10.1016/0531-5565(90)90017-v
Song, L., Huang, Y., Hou, X., Yang, Y., Kala, S., Qiu, Z., et al. (2018). PINK1/Parkin-mediated mitophagy promotes resistance to sonodynamic therapy. Cell. Physiol. Biochem. 49, 1825–1839. doi: 10.1159/000493629
Steinberg, S. F. (2013). Oxidative stress and sarcomeric proteins. Circ. Res. 112, 393–405. doi: 10.1161/circresaha.111.300496
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi: 10.1016/j.cell.2017.09.021
Sun, H. J., Chen, D., Wang, P. Y., Wan, M. Y., Zhang, C. X., Zhang, Z. X., et al. (2017). Salusin-beta is involved in diabetes mellitus-induced endothelial dysfunction via degradation of peroxisome proliferator-activated receptor gamma. Oxid. Med. Cell. Longev. 2017:6905217.
Tang, D., Kang, R., Berghe, T. V., Vandenabeele, P., and Kroemer, G. (2019). The molecular machinery of regulated cell death. Cell Res. 29, 347–364. doi: 10.1038/s41422-019-0164-5
Tao, X., Wan, X., Xu, Y., Xu, L., Qi, Y., Yin, L., et al. (2014). Dioscin attenuates hepatic ischemia-reperfusion injury in rats through inhibition of oxidative-nitrative stress, inflammation and apoptosis. Transplantation 98, 604–611. doi: 10.1097/tp.0000000000000262
Uribe, P., Cabrillana, M. E., Fornes, M. W., Treulen, F., Boguen, R., Isachenko, V., et al. (2018). Nitrosative stress in human spermatozoa causes cell death characterized by induction of mitochondrial permeability transition-driven necrosis. Asian J. Androl. 20, 600–607. doi: 10.4103/aja.aja_29_18
Ursini, F., Maiorino, M., Valente, M., Ferri, L., and Gregolin, C. (1982). Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim. Biophys. Acta 710, 197–211. doi: 10.1016/0005-2760(82)90150-3
Varga, Z. V., Giricz, Z., Liaudet, L., Hasko, G., Ferdinandy, P., and Pacher, P. (2015). Interplay of oxidative, nitrosative/nitrative stress, inflammation, cell death and autophagy in diabetic cardiomyopathy. Biochim. Biophys. Acta 1852, 232–242. doi: 10.1016/j.bbadis.2014.06.030
Velsor, L. W., Ballinger, C. A., Patel, J., and Postlethwait, E. M. (2003). Influence of epithelial lining fluid lipids on NO(2)-induced membrane oxidation and nitration. Free Radic. Biol. Med. 34, 720–733. doi: 10.1016/s0891-5849(02)01370-9
Wang, Y., Wang, X., Lau, W. B., Yuan, Y., Booth, D., Li, J. J., et al. (2014). Adiponectin inhibits tumor necrosis factor-alpha-induced vascular inflammatory response via caveolin-mediated ceramidase recruitment and activation. Circ. Res. 114, 792–805. doi: 10.1161/circresaha.114.302439
Wu, Y., Zhang, S., Gong, X., Tam, S., Xiao, D., Liu, S., et al. (2020). The epigenetic regulators and metabolic changes in ferroptosis-associated cancer progression. Mol. Cancer 19:39.
Xue, L., Zeng, Y., Fang, C., Cheng, W., and Li, Y. (2020). Effect of TTLL12 on tubulin tyrosine nitration as a novel target for screening anticancer drugs in vitro. Oncol. Lett. 20:340.
Yang, W. S., SriRamaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. doi: 10.1016/j.cell.2013.12.010
Ye, Y. Z., Strong, M., Huang, Z. Q., and Beckman, J. S. (1996). Antibodies that recognize nitrotyrosine. Methods Enzymol. 269, 201–209. doi: 10.1016/s0076-6879(96)69022-3
Yu, L., Li, Z., Dong, X., Xue, X., Liu, Y., Xu, S., et al. (2018). Polydatin protects diabetic heart against ischemia-reperfusion injury via Notch1/Hes1-mediated activation of Pten/Akt signaling. Oxid. Med. Cell. Longev. 2018:2750695.
Zeng, T., Deng, G., Zhong, W., Gao, Z., Ma, S., Mo, C., et al. (2020). Indoleamine 2, 3-dioxygenase 1enhanceshepatocytes ferroptosis in acute immune hepatitis associated with excess nitrative stress. Free Radic. Biol. Med. 152, 668–679. doi: 10.1016/j.freeradbiomed.2020.01.009
Zhang, C., Yang, J. B., Quan, W., Feng, Y. D., Feng, J. Y., Cheng, L. S., et al. (2020). Activation of paraventricular melatonin receptor 2 mediates melatonin-conferred cardioprotection against myocardial ischemia/reperfusion injury. J. Cardiovasc. Pharmacol. 76, 197–206. doi: 10.1097/fjc.0000000000000851
Zhang, Y. L., and Wei, J. R. (2013). 3-nitrotyrosine, a biomarker for cardiomyocyte apoptosis induced by diabetic cardiomyopathy in a rat model. Mol. Med. Rep. 8, 989–994. doi: 10.3892/mmr.2013.1644
Zhao, S., Xiong, Z., Mao, X., Meng, D., Lei, Q., Li, Y., et al. (2013). Atmospheric pressure room temperature plasma jets facilitate oxidative and nitrative stress and lead to endoplasmic reticulum stress dependent apoptosis in HepG2 cells. PLoS One 8:e73665. doi: 10.1371/journal.pone.0073665
Keywords: nitrosative stress, peroxynitrite, reactive nitrogen species, cell death, nitric oxide, cardiovascular diseases
Citation: Wang F, Yuan Q, Chen F, Pang J, Pan C, Xu F and Chen Y (2021) Fundamental Mechanisms of the Cell Death Caused by Nitrosative Stress. Front. Cell Dev. Biol. 9:742483. doi: 10.3389/fcell.2021.742483
Received: 16 July 2021; Accepted: 19 August 2021;
Published: 20 September 2021.
Edited by:
Zhi Qi, Nankai University, ChinaReviewed by:
Heng Ma, Fourth Military Medical University, ChinaJinwei Tian, The Second Affiliated Hospital of Harbin Medical University, China
Copyright © 2021 Wang, Yuan, Chen, Pang, Pan, Xu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Xu, eHVmZW5nc2R1QDEyNi5jb20=; Yuguo Chen, Y2hlbjkxOTA4NUBzZHUuZWR1LmNu; Fengying Chen, ZnljaGVuNjI3QHNvaHUuY29t
†These authors have contributed equally to this work