 Lin Lin1
Lin Lin1 Chao Li
Chao Li Yun-lun Li
Yun-lun Li- 1Chinese Medicine Innovation Research Institute, Shandong University of Traditional Chinese Medicine, Jinan, China
- 2The First Clinical Medical College, Shandong University of Traditional Chinese Medicine, Jinan, China
- 3College of Traditional Chinese Medicine, Shandong University of Traditional Chinese Medicine, Jinan, China
- 4Department of Cardiovascular, Affiliated Hospital of Shandong University of Traditional Chinese Medicine, Jinan, China
Atherosclerosis is a chronic inflammatory disorder characterized by the gradual buildup of plaques within the vessel wall of middle-sized and large arteries. The occurrence and development of atherosclerosis and the rupture of plaques are related to the injury of vascular cells, including endothelial cells, smooth muscle cells, and macrophages. Autophagy is a subcellular process that plays an important role in the degradation of proteins and damaged organelles, and the autophagy disorder of vascular cells is closely related to atherosclerosis. Pyroptosis is a proinflammatory form of regulated cell death, while ferroptosis is a form of regulated nonapoptotic cell death involving overwhelming iron-dependent lipid peroxidation. Both of them exhibit distinct features from apoptosis, necrosis, and autophagy in morphology, biochemistry, and genetics. However, a growing body of evidence suggests that pyroptosis and ferroptosis interact with autophagy and participate in the development of cancers, degenerative brain diseases and cardiovascular diseases. This review updated the current understanding of autophagy, pyroptosis, and ferroptosis, finding potential links and their effects on atherogenesis and plaque stability, thus providing ways to develop new pharmacological strategies to address atherosclerosis and stabilize vulnerable, ruptured plaques.
1 Introduction
Atherosclerosis, caused by the accumulation of low-density lipoprotein (LDL) in the subendothelial matrix, is a progressive disease characterized by endothelial damage, inflammatory cell infiltration, cell proliferation, and fat deposition (Lusis, 2000). Proinflammatory and anti-inflammatory mechanisms, along with inadequate cellular inflammation resolution, are the initial factors that promote and accelerate the process of atherosclerotic plaque formation (Back et al., 2019). The composition and vulnerability of a plaque play a principally decisive role in plaque stability, thrombosis, and thrombus-mediated acute coronary events. The functional status, survival and death of vascular cells, including endothelial cells (ECs), vascular smooth muscle cells (VSMCs), and macrophages, can affect the formation and stability of plaques, thus affecting the progression of atherosclerosis.
The endothelial lining of lesion-prone areas of the arterial vasculature is highly susceptible to risk factors associated with atherosclerosis. Oxidized LDL (ox-LDL) is recognized by pattern-recognition receptors in ECs and induces endothelial dysfunction by triggering a cascade of oxidative stress and inflammatory responses (Marchio et al., 2019). Moreover, hyperlipidemia stimulates EC activation before monocyte recruitment (Yin et al., 2015) and activated ECs promote the infiltration of inflammatory cells into atherosclerotic lesions by increasing the release of adhesion molecules, resulting in plaque aggregation and eventually atherosclerosis (Libby et al., 2011). The subendothelial accumulation of foam cells is a major hallmark of early atherosclerotic lesions. It has been recently found that more than 50% of foam cells in plaques are contributed by VSMCs (Allahverdian et al., 2014; Wang Y. et al., 2019), which is the major constituent of the fibrous cap (Pan and Reilly, 2019). Therefore, the death of VSMCs and VSMCs-derived foam cells decreases lesion cellularity, weakens the fibrous cap of the plaque, and increases plaque instability (Clarke et al., 2010). Advanced plaques contain many macrophages with a proinflammatory phenotype, which secrete stromal degradation enzymes, growth factors, cytokines, and intracellular lipids into the extracellular space, leading to plaque instability, plaque rupture, and thrombotic events. Moreover, in advanced atherosclerotic plaques, up to 50% of dead cells are macrophages; the mechanisms of macrophage death involve the decay of lesion cellularity and the promotion of inflammation. Macrophage death is also a significant feature of advanced plaques in atherosclerosis and serves as a catalyst for the formation of necrotic core and plaque instability (Lutgens et al., 1999; Martinet et al., 2019). Atherosclerosis is still the leading cause of death worldwide because of the acute occlusion caused by the formation of a thrombus or blood clot. Hence, the different processes and mechanisms involved in plaque stabilization need to be further explored to find new therapies for atherosclerosis.
Regulated cell death (RCD) is a highly regulated cellular response that controls cell fate in multicellular organisms after they are subjected to various cellular pressures and/or external stimuli. The most common modulated form of cell death is called apoptosis (Kerr et al., 1972); other forms of RCD have been gradually discovered, such as autophagy (Ashford and Porter, 1962), pyroptosis (Fink and Cookson, 2005), and ferroptosis (Dixon et al., 2012). Different types of RCD display distinct features, meanwhile sharing many similar characteristics with considerable overlap and crosstalk (Table 1). The interactions between these cell death programs play a role in controlling the ultimate outcome during cell death (Lockshin and Zakeri, 2004). Under normal circumstances, various forms of RCD remove damaged or infected cells from the affected tissues so that the surrounding healthy cells can better perform their proper functions. Nevertheless, the loss of control over single or mixed types of RCD can lead to human diseases, such as cancer, neurodegeneration, autoimmune diseases, infectious diseases, and cardiovascular diseases. Multiple types of RCD have been identified in the pathological process of atherosclerosis, but the intricate overlapping effects and interactions have rarely been systematically summarized. Based on this, the present review provided a current overview of the known signal cascade and interaction of the common patterns of RCD, namely autophagy, pyroptosis, and ferroptosis, and the latest understanding of their functional role and significance in atherosclerosis.
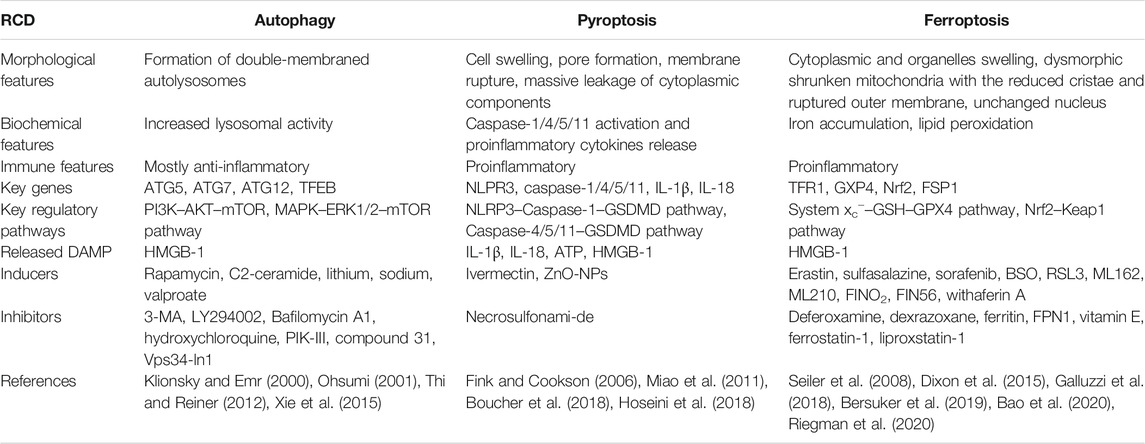
TABLE 1. The main features of autophagy, pyroptosis, and ferroptosis.
2 The Roles of Autophagy in Atherosclerosis
2.1 Overview of Autophagy
Autophagy is an evolutionarily conservative homeostatic mechanism consisting of three general subtypes: macroautophagy, microautophagy and chaperone-mediated autophagy (CMA) (Klionsky et al., 2016). Macroautophagy (hereafter referred to as autophagy) is a lysosome-dependent intracellular degradation system by which cytoplasmic organelles, macromolecules, proteins, and invading pathogens are degraded in the lysosome, with the ability to produce new building blocks and energy for cellular renovation and homeostasis (Mizushima and Komatsu, 2011; Parzych and Klionsky, 2014). Morphologically, autophagy begins in the pre-autophagosomal structure in the cytoplasm and gradually develops into a phagophore, followed by an autophagosome, a double-membraned vacuole containing damaged organelles and denatured macromolecules. Subsequently, the outer membrane of autophagosome fuses with the lysosomal membrane to form autolysosome, and the inner membrane and encapsulated substances of autophagosome enter the lysosomal cavity and are degraded by activated lysosomal hydrolases (Klionsky and Emr, 2000).
Autophagy is a continuous and dynamic process that tightly regulated by autophagy-related genes (ATG) (Kang et al., 2011; Xie et al., 2015). Mammalian target of rapamycin (mTOR) is negatively involved in autophagy regulation. When the cell in the absence of growth factors and amino acids or is stimulated by rapamycin, unc-51 like autophagy-activating kinase 1 (ULK1) is separated from mTOR and undergoes rapid dephosphorylation; activated ULK1 promotes ATG13 phosphorylation and autophagy (Jung et al., 2009; Kim et al., 2011). The class III phosphatidylinositol 3-kinase (Class III PI3K) complex, which includes phosphatidylinositol 3-kinase, catalytic subunit type 3 (PIK3C3)/VPS34, Beclin-1, and ATG14, promotes the nucleation of phagophores by generating the phosphatidylinositol-3-phosphate and recruiting other factors involved in the process of autophagosome formation (Thi and Reiner, 2012; McKnight and Zhenyu, 2013; Bernard and Klionsky, 2014). Subsequently, the phagophore expands by membrane addition though two ubiquitin-like modification processes, among which ATG12-conjugation and microtubule-associated protein 1A/1B-light chain 3 (LC3)-modification play the most crucial roles (Ohsumi, 2001; Tanida et al., 2004). ATG12 is activated by ATG7, and then combines with ATG5 and ATG16L to form a ATG12-ATG5-ATG16L complex at phagophores. In the process of LC3-modification, proLC3 is first processed into LC3-I with the help of ATG4, activated by ATG7, transported to ATG3, and then processed into LC3-II, which is the membrane-bound form localized on preautophagosomes and autophagosomes. After the extension, the outer membrane of autophagosome fuses with lysosomes to form autolysosome and degrade encapsulated substances.
Autophagy plays a dual role, either preventing or promoting cell death, depending on the environment (Fitzwalter and Thorburn, 2015). On the one hand, autophagy-related stress tolerance protects cells against various kinds of cellular death stimuli. Free amino acids and fatty acids produced by the autophagy degradation of unused protein aggregates or damaged organelles can be used for protein synthesis and energy production to adapt to environmental stress (Kroemer et al., 2010). On the other hand, excessive or uncontrolled autophagy can result in the degradation of prosurvival proteins and promote cell death, termed “autophagy-dependent cell death” (Denton and Kumar, 2019). Autophagy regulates inflammatory responses and cell death pathways, thereby influencing the pathogenesis of diseases, including atherosclerosis.
2.2 Effects of Autophagy on Atherosclerosis
Autophagy is closely associated with atherosclerosis, which has been confirmed by the expression profile of ARGs (Chen Y. et al., 2021). Multiple autophagy triggers are present in atherosclerotic plaques, such as reactive oxygen species (ROS), LDL, inflammatory mediators, and tumor necrosis factor-α (TNF-α) (Shao et al., 2016; Zhu et al., 2019). Mild cellular stress, including oxidative stress, ox-LDL, and endoplasmic reticulum (ER) stress, stimulates mild adaptive autophagy of vascular cells (Menghini et al., 2014), which promotes cell survival by degrading damaged organelles and proteins, thereby protecting vascular tissues from oxidative stress or inflammation. It has been confirmed that autophagy can diminish the atherosclerotic plaque area and effectively preserve the stable plaque phenotype, including reduced lipid deposition and proinflammatory macrophages; increased anti-inflammatory macrophages, collagen content, and VSMCs; and lessened cell death within the plaques (Li et al., 2013). Nevertheless, similarly, dysfunction of autophagy has also been found to occur in the progression of atherosclerosis, regardless of the presence or absence of these autophagy stimulators in atherosclerotic plaques. The expression of autophagy markers ATG13 and LC3 in aortic intimal ECs with severe atherosclerosis was found to be significantly higher than the expression in those without atherosclerosis (Chen et al., 2017). Severe oxidative stress or inflammation stimulates excessive autophagy, leading to autophagy-dependent cell death, decreased collagen synthesis, thinning of the fibrous cap, plaque instability, lesions thrombosis, restenosis, and acute coronary events (Cai et al., 2018; Ge et al., 2021). Thus, the resistance to excessive autophagy in an early stage is critical in preventing arteriosclerosis, further leading to serious cardiovascular complications. Except excessive autophagy, the loss of autophagy is more prevalent and may be an important determinant of atherosclerosis. In addition, autophagy may also become insufficient, especially in advanced lesions with a large amount of oxidative stress. In this case, autophagy is functional, but is subsequently unable to cope with excess stress in the plaques, leading to cell apoptosis (Mollace et al., 2015). Therefore, the cytoprotective autophagic pathway may be transformed into a maladaptive pathway depending on the developmental stage of the plaques (Figure 1).
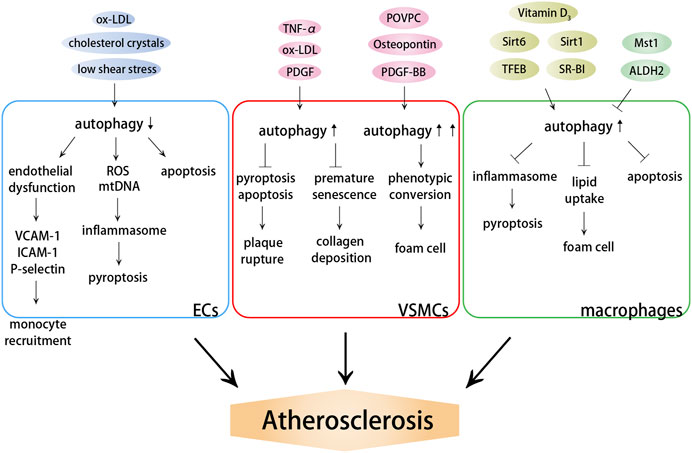
FIGURE 1. Effects of autophagy on atherosclerosis. Autophagy dysfunction induces endothelial dysfunction and inflammation, promoting monocyte recruitment and cell death. Autophagy dysfunction aggravates plaque instability and promotes the formation of foam cells by influencing VSMC death and phenotypic conversion. Autophagy dysfunction induces lipid accumulation, macrophage foam cell formation and cell death.
2.2.1 Autophagy Dysfunction Induces Endothelial Dysfunction and Inflammation
Endothelial dysfunction, leukocyte adhesion, and foam macrophage formation are the main pathogeneses of atherosclerosis. Endothelial dysfunction, in particular, is thought to be the first step in atherosclerosis (Gimbrone and Garcia-Cardena, 2016). The known mechanisms related to atherosclerosis and endothelial dysfunction include impaired eNOS ability to produce NO, increased oxidative stress, inflammation and impaired autophagy. EC autophagy deficiency improves the levels of vascular cell adhesion molecule-1 (VCAM-1), intercellular adhesion molecule-1 (ICAM-1), von Willebrand factor, and P-selectin, thus promoting the infiltration of macrophages and foam cells, as well as increasing the risk of arterial thrombosis (Wu Q. et al., 2019; Perrotta et al., 2020). Furthermore, Vion et al. reported that specific deficiency in endothelial autophagy only stimulated the development of atherosclerotic lesions in atheroresistant areas, but not in atheroprone areas, in which the endothelial autophagic flux was already blocked (Vion et al., 2017). Thus, increasing basal autophagy and preventing the age-dependent decline in the autophagic flux are effective strategies for restoring endothelial health and inhibiting atherogenesis.
Current evidence supports the central role of EC inflammation in atherosclerosis. Oxidative stress is considered as a key signal in the progression of inflammation, and autophagy is a protective mechanism that protects plaque cells and ECs from certain stimulation on the arterial wall, especially oxidative damage (Cho and Choi, 2019). During oxidant stress, damaged mitochondrial DNA (mtDNA) that escaped autophagy resulted in a potent inflammatory response in atherosclerosis (Ding et al., 2013a). Increased autophagy significantly suppressed chronic vascular inflammation, restrained the formation of plaques, decreased plaque area, and attenuated atherogenesis (Pankratz et al., 2018; Meng Q. et al., 2021), whereas the inhibition or impairment of autophagy exacerbated the inflammation response (Guo F. X. et al., 2019). These studies implied that autophagy was a protective mechanism against endothelial inflammation and a potential target for treating atherosclerosis and related cardiovascular diseases.
Given the protective effect of autophagy on atherosclerotic resistance, the mechanisms of EC autophagy regulation in atherosclerosis have been gradually revealed. Arterial wall shear stress is considered to be one of the important factors in regulating EC autophagy. High shear stress triggers protective autophagy to limit atherosclerotic plaque formation by preventing endothelial apoptosis, senescence, and inflammation, whereas low shear stress, a potential proatherogenic factor, suppresses autophagy by activating the mTOR pathway (Vion et al., 2017; Yuan et al., 2020). The defect in endothelial autophagy may be the missing link between low shear stress and atherosclerosis. Moreover, caveolin-1 (CAV1) plays a critical role in atherogenesis by controlling NO production, vascular inflammation, and extracellular matrix remodeling (Pavlides et al., 2014; Ramirez et al., 2019). A recent study further found that CAV1 could interact with the ATG5–ATG12 complex and regulate the autophagosome formation by influencing the cellular localization of autophagosome components in lipid rafts. The increased autophagy activation and the autophagic flux of ECs caused by CAV1 deficiency mediated atheroprotection by attenuating EC activation in response to proatherogenic cytokines, inhibiting inflammation and macrophage recruitment (Zhang X. et al., 2020). Sun et al. reported that the deficiency of proprotein convertase subtilisin/kexin type 9 (PCSK9), which bound to the LDL reporter (LDLR) and thereby promoted its intracellular degradation, could affect atherogenesis by modulating the levels and properties of apolipoprotein B (ApoB)-containing lipoproteins dependent on increased autophagy signaling pathway and autophagic flux, rather on the effect of PCSK9 on cell surface LDLRs (Sun H. et al., 2018). This result revealed a critical mechanism by which PCSK9 inhibited atherosclerosis, in which autophagy was a major participant. Moreover, microRNAs (miRNAs), which have been identified as the critical regulators of gene expression in many organisms and biological processes, were studied extensively. MiR-214-3p regulates ox-LDL-initiated EC autophagy by directly targeting 3′-UTR of ATG5, which may play an appropriate role in the pathogenesis of atherosclerosis (Wang et al., 2018). Santovito et al. reported a noncanonical inhibition of caspase-3 by miR-126-5p, which was transported into the nucleus and bound to caspase-3, preventing caspase dimerization and downregulating activity to limit apoptosis. The aforementioned antiapoptotic effect conferred endothelial protection by autophagy in atherosclerosis, and the nuclear import of miR-126-5p was blocked by the ablation of ATG5 (Santovito et al., 2020), reflecting the potential effect of ECs autophagy in cell death resistance and atheroprotection.
2.2.2 Autophagy Dysfunction Aggravates Plaque Instability by Influencing Vascular Smooth Muscle Cell Death and Phenotypic Conversion
Defective autophagy caused by VSMC-specific HuR knockout has recently been reported to trigger off plaque formation and plaque instability (Liu S. et al., 2021). Actually, the death of VSMCs and VSMC-derived foam cells is adverse to maintaining plaque stability, while autophagy promotes cell survival. Autophagy and mitophagy induced by oxidized lipids in the plaques protect VSMCs against apoptosis (Martinet et al., 2008; Swiader et al., 2016). In addition, several cytokines and growth factors, such as TNF-α and platelet-derived growth factor (PDGF), also promote autophagy to resist cell death (Jia et al., 2006; Salabei et al., 2013). Compared with healthy human VSMCs, VSMCs isolated from carotid plaque specimens showed increased autophagy (Yu et al., 2017). However, a fivefold decrease in LC3β expression was observed when comparing carotid plaques in symptomatic and asymptomatic patients, suggesting that decreased autophagy was associated with the clinical stage of atherosclerosis (Swaminathan et al., 2014). VSMC-specific ATG7 knockout in ApoE−/− mice significantly increased the susceptibility of VSMCs to apoptotic cell death, leading to plaque growth, medial disruption and aneurysm formation, which provided the evidence that autophagy deficiency of VSMCs aggravated atherosclerosis via inducing VSMC death (Osonoi et al., 2018). ATG7 deletion also accelerated the development of premature senescence, which was characterized by increased total collagen deposition, nuclear hypertrophy, and CDKN2A-RB-mediated G1 proliferative arrest (Grootaert et al., 2015). Furthermore, VSMC autophagy in ApoE−/− mice stimulated cholesterol efflux and inhibited lipid accumulation and necrotic core formation (Michiels et al., 2016), reflecting a regulatory effect of VSMC autophagy on lipid metabolism.
Phenotypic conversion promotes the migration and proliferation of VSMCs and the formation of VSMC-derived foam cells (Rong et al., 2003). Autophagy may play a critical role in the phenotypic conversion of VSMCs, as diverse stimuli promoting phenotypic conversions, such as ROS, oxidized lipids, and metabolic stress, are also associated with autophagy (Tai et al., 2016). Some autophagy-inducing stimuli, including PDGF-BB, POVPC, and osteopontin, promoted the loss of contractile phenotype and motivated VSMC proliferation and migration, whereas inhibiting autophagy could promote the maintenance of the contractile phenotype and prevent hyperproliferation (Chaulet et al., 2001; Li et al., 2012; Salabei and Hill, 2013). Furthermore, Wang et al. reported that nicotine-mediated autophagy in VSMCs triggered off phenotypic transition and accelerated atherosclerosis via the nAChRs/ROS/NF-κB signaling pathway (Wang Z. et al., 2019). Moreover, nicotine promoted the synthesis and secretion of cathepsin S that stimulated VSMC migration by activating autophagy (Ni et al., 2020). These results reflected that nicotine-modulated autophagy disorder in VSMCs induced and aggravated atherosclerosis, revealing the potential role of smoking in VSMCs and vascular diseases through the autophagic-lysosomal machinery. In brief, autophagy served as a safeguarding mechanism against VCMCs senescence and death; however, excessive activation of autophagy might aggravate atherosclerosis by triggering a synthetic phenotype of VSMCs.
2.2.3 Autophagy Dysfunction Induces Lipid Accumulation and Macrophage Foam Cell Formation
In macrophages, ox-LDL and one of its primary oxysterols, 7-ketonecholesterol, can directly or indirectly stimulate autophagy through ER stress, promoting the survival of macrophages by promoting the clearance of damaged proteins and organelles. Sergin et al. first described the functional status of the autophagy-lysosome system in murine and human atherosclerotic plaques through two key autophagy markers LC3 and SQSTM1/p62 (Sergin et al., 2017). They found that early atherosclerotic lesions had high levels of LC3 co-localized with SQSTM1, indicating the recruitment of autophagy as a stress response to burgeoning plaques. However, advanced lesions were characterized by reduced LC3 and dissociation from SQSTM1, suggesting an extensive dysfunction during the autophagic process (Sergin et al., 2017).
Autophagy of macrophages plays an important role in the pathogenesis of atherosclerosis by inhibiting oxidative stress, inflammation, and the formation of foam cells. For example, macrophage-specific overexpression of TFEB, a master transcriptional regulator of autophagy-lysosome biogenesis, induces a broad atheroprotection by reversing the autophagy dysfunction, including reduction of overall plaque burden and characteristics of plaque complexity, while the effect cannot be achieved in the absence of ATG5 or SQSTM1 (Sergin et al., 2017). Moreover, TFEB also modulates the inflammatory status and enhances the antioxidative capacity by increasing the transcription of genes encoding antioxidant factors (Lu et al., 2017). Arsenic trioxide promoted autophagy and autophagosome-lysosome fusion by triggering the nuclear translocation of TFEB, thus preventing atherosclerosis (Fang et al., 2021). MiR-761 regulates autophagy via the mTOR–ULK1 pathway and subsequently represses the production of inflammatory cytokines, as well as the formation of foam cells (Wang C. et al., 2020), reflecting the critical role of macrophage autophagy in inflammatory resistance and foam cell formation. The activation of autophagy in monocytes also prevents plaque vulnerability and subsequent plaque rupture (Cheng et al., 2015).
The formation of macrophage-derived foam cells is the initial stage of atherosclerosis. Autophagy activation of macrophages caused by sirtuin 6 (Sirt6) overexpression can restrain apoptosis, reduce macrophage foam cell formation, and stabilize atherosclerosis plaques (He et al., 2017; Wang T. et al., 2020). The upregulation of Sirt1 expression or enhanced interaction between Sirt1 and FOXO1 also promotes autophagy and subsequently modulates macrophage polarization, thus attenuating foam cell formation (Luo et al., 2020; Hui et al., 2021). Vitamin D3 can recover ox-LDL-impaired autophagy and attenuate the lipid accumulation in macrophages, and scavenger receptor class B type I (SR-BI) can also promote autophagy by regulating Tfeb expression and recruiting VPS34-Beclin-1 complex, which all inhibit the conversion of macrophages into foam cells (Kumar et al., 2021; Tao et al., 2021).
The differences in the polarization of macrophages lead to differences in the number and distribution of polarized macrophages in plaques. Classically activated (M1) and alternatively activated (M2) macrophages connect to produce atherosclerotic plaques, and M2 macrophages can suppress foam cell transformation (Moore et al., 2013). Sun et al. found that rapamycin stimulated M1 macrophages and inhibited early atherosclerosis by inducing autophagy, and FTY720 could transform foam cells into M2 macrophages through the autophagy pathway, alleviating advanced atherosclerosis (Sun R. Z. et al., 2018). Therefore, autophagy played a role in alleviating atherosclerosis by selectively removing macrophages or changing the polarization state within plaques.
The accumulation of lipid-rich macrophages is considered as a marker of plaque instability, and intact autophagic machinery is essential in limiting lipid uptake by macrophages. Macrophage-specific ATG7-deficient mice exhibited increased susceptibility to atherosclerosis by increasing macrophage LDL uptake and foam cell formation. Compared with ApoE−/− mice, the suppressed autophagic flux caused by mammalian Ste20-like kinase 1 (Mst1) overexpression increased plaque area, lipid core, and macrophage accumulation (Wang et al., 2016). Furthermore, acetaldehyde dehydrogenase 2 (ALDH2) has been reported to interact with adenosine monophosphate-activated protein kinase (AMPK) and then disturb lysosomal function and autophagy, thus repressing cholesterol hydrolysis in lysosomes and promoting lipid deposition and foam cell formation. LDLR can block the effect of ALDH2 through interaction with AMPK, whereas ALDH2 rs671 mutant attenuates this interaction and further promotes the release of ALDH2 to the nucleus where ALDH2 inhibits the transcription of a lysosomal proton pump protein ATP6V0E2 (Zhong et al., 2019). This result provides a possible molecular mechanism to explain why East Asians carrying the single nucleotide polymorphism ALDH2 rs671 have an increased risk of cardiovascular disease (Han et al., 2013). To sum up, the data revealed the function of macrophage autophagy in atherosclerosis and supported several practical methods of enhancing autophagy and degradative capacity of macrophages as a therapy for atherosclerotic vascular disease.
2.2.4 Effect of Lipophagy and Mitophagy in Atherosclerosis
In atherosclerosis, a majority of lipids stored in the foam cells in the form of lipid droplets consist mainly of free cholesterol and cholesterol esters. Therefore, promoting lipid droplet degradation and cholesterol efflux is an effective strategy to restrain foam cell formation and atherosclerosis process. Lipophagy prevents cellular lipid accumulation by autophagic degradation of lipid droplets via lysosomal acid lipases, thus playing a protective role in atherosclerosis (Ouimet et al., 2017). Sirt6 overexpression in foam cells accelerates cholesterol efflux via the autophagic pathway, which can be largely reversed by miR-33 (He et al., 2017). Ouimet et al. found that miR-33 restrained lipid droplet catabolism and cholesterol mobilization by repressing lipophagy. Importantly, the effect of miR-33 in regulating lipophagy lies the upstream of its effect on the ATP-binding cassette transporter A1-dependent cholesterol efflux, as miR-33 inhibitors cannot enhance the cholesterol efflux in autophagy-deficient macrophages, highlighting the importance of lipophagy in cellular lipid metabolism (Ouimet et al., 2017). Moreover, with aging and lipid accumulation, lipophagy activity decreases, leading to the excessive accumulation of lipid droplets, which further impairs lipophagy activity. Thus, improving the decreased activity of lipophagy may play a critical role in the blockage of foam cells formation and atherosclerosis development.
Aging within the aorta during normolipidemia results in mitochondrial dysfunction and an increase in the IL-6 level, accompanied by elevated mitophagy. Enhanced mitophagy pharmacologically during hyperlipidemia increased aortic mitochondrial function and inhibited atherosclerosis by removing damaged or dysfunctional mitochondria, implying that novel therapies to improve vascular mitochondrial bioenergy prior to hyperlipidemia may attenuate age-related atherosclerosis (Tyrrell et al., 2020). Furthermore, mitophagy in macrophages induced by melatonin scavenged mitochondrial ROS and subsequently repressed prolonged NLRP3 inflammasome activation, thus markedly attenuating atherosclerotic plaque area and vulnerability (Ma et al., 2018). These findings confirmed that mitophagy avails to atherosclerosis resistance. However, ox-LDL-stimulated nuclear receptor subfamily 4 group A member 1 (NR4A1) overexpression caused Parkin-mediated mitophagy through the post-transcriptional modification of Ca2+/calmodulin-dependent protein kinase II, while excessive mitophagy significantly consumed mitochondrial mass, resulting in energy shortage and mitochondrial dysfunction and thus aggravating endothelial apoptosis and atherosclerosis (Li P. et al., 2018).
Taken together, the dysregulation of autophagy is present in atherosclerosis. Basic autophagy protects plaque cells from oxidative stress by degrading damaged intracellular substances, whereas overstimulated or deficient autophagic flux may lead to the death of vascular cells and plaque instability in a variety of ways, including promoting inflammation and lipid accumulation. Skyrocketing researches provide insight into the regulation of autophagic homeostasis as a critical way for therapeutic intervention for atherosclerosis. However, notably, in contrast to the proatherogenic role, a counter-regulatory response of autophagy disruption occurs in dendritic cells or T cells, which maintains immune homeostasis and limits atherogenesis (Amersfoort et al., 2018; Clement et al., 2019). Therefore, different types of cell-specific autophagy regulation have different effects on the development of atherosclerosis.
3 The Roles of Pyroptosis in Atherosclerosis
3.1 Overview of Pyroptosis
Pyroptosis is a new caspase-dependent proinflammatory form of regulatory cell death, characterized by cell swelling, pore formation, and membrane rupture, resulting in the massive leakage of cytoplasmic components (Hoseini et al., 2018). Different from the immunologically silent cell death presented by apoptosis, pyroptosis is a response to pathogen-associated molecular patterns (PAMPs) derived from invading pathogens or damage-associated molecular patterns (DAMPs) induced by endogenous stress that leads to RCD and inflammatory response following the release of cytokines (Tavakoli Dargani et al., 2018). Caspases are cysteine proteases that initiate or execute cellular programs causing inflammation or cell death (Schroder and Tschopp, 2010). Pro-caspase-1 is stored in secretory lysosomes, where it awaits exocrine-induced stimulation; otherwise, these molecules may undergo lysosomal degradation (Andrei et al., 1999).
Caspase-dependent pyroptosis requires the activation of the typical inflammatory response (Xue et al., 2019). As proinflammatory caspases, the catalytic activity of caspase-1/4/5/11 is strictly regulated by signal-dependent autoactivation within multiprotein complexes called inflammasomes (Martinon et al., 2002). Inflammasomes are molecular platforms activated in response to cellular infection, toxic insults impinging on the cell, or intracellular stress releasing interleukin (IL)-1 family members, triggering the maturation of proinflammatory cytokines and inducing cell death in the form of pyroptosis (Schroder and Tschopp, 2010; Del Re et al., 2019). An important mechanism of inflammation is the induction of inflammasomes, which requires two signals: the priming signal occurs in response to receptor activation, including interferon (IFN) signaling and Toll-like receptors (TLR) ligands, and induces the transcriptional upregulation of inflammasome components via nuclear factor κB (NF-κB); then, the activating signal provided by an inflammasome activator in the form of a PAMP (e.g., Gram-positive and -negative bacteria, bacterial toxins, DNA and RNA viruses, and fungi) or DAMP (e.g., ATP, uric acid crystals, silica crystals, cholesterol crystals, saturated fatty acids, extracellular histones, lysophosphatidylcholine, mitochondrial ROS and DNA, and aluminum hydroxide) induces the assembly of the inflammasome mediated by kinase NEK7, apoptosis-associated speck-like protein (ASC), and caspase-1 (Bauernfeind et al., 2009; Mathur et al., 2018; Tang et al., 2019). Typical inflammasomes, such as NLRP1b, NLRP3, NAIP-NLRC4, AIM2, and Pyrin, have been found to activate caspase-1, ultimately inducing pyroptosis (Schroder and Tschopp, 2010).
Once activated in response to many stimuli, caspase-1 cleaves the N-terminal and C-terminal domains of the executive protein of pyroptosis, named gasdermin D (GSDMD). These peptides contain the N-terminal active domain of GSDMD, which oligomerizes on the cytoplasmic membrane, generating pores that act as the mediators of pyroptosis and as direct conduits for the transport of IL-1β and IL-18 (Shi J. et al., 2015; Boucher et al., 2018), ultimately causing pyroptosis and inflammatory response (Liu X. et al., 2016). Moreover, caspase-1 results in the cleavage of pro-IL-1β and pro-IL-18 and the release of active IL-1β and IL-18 (Miao et al., 2011), which recruit inflammatory cells and further amplify the inflammatory response. The plasma membrane pores allow the flow of ions, leading to the usual equilibrium of ion gradients between the intracellular and extracellular environments; the water then enters the cell, causing swelling and dissolution (Fink and Cookson, 2006). In the noncanonical caspase-4/5/11-dependent pathway of pyroptosis, the inflammatory agents, such as bacterial lipopolysaccharides (LPS), activate caspase-4/5/11, cleave GSDMD, and release GSDMD-p30 (Kayagaki et al., 2011; Kayagaki et al., 2015), which binds to the plasma membrane and the mitochondrial membrane to kill cells by forming the pyroptotic pores (Aglietti et al., 2016). Besides GSDMD, other members of the gasdermin family, such as GSDMA, GSDMB, GSDMC, GSDME/DFNA5, and GSDMA3, have similar functions in the cytotoxicity of cell membrane disruption (Ding et al., 2016). In the absence or blocking of canonical or noncanonical pyroptosis pathway (caspase1/4/5/11-GSDMD), pyroptosis can be engaged by mechanisms such as caspase-8-GSDMD (Orning et al., 2018) and caspase-3-GSDME (Wang et al., 2017), but their contribution to pyroptosis has not been determined in vivo.
Although originally discovered in bacterial immunity, pyroptosis has gradually become a recognized form of RCD in biological scenarios. Pyroptosis serves as an effective antimicrobial defense for the host during infection (Aachoui et al., 2013). However, the mutations in NOD-like receptor (NLR) proteins or the persistence of sterile inflammatory stimuli can lead to excessive pyroptosis, which is harmful to the host and can increase the levels of inflammatory mediators IL-1β, IL-18, and alarmin high mobility group box-1 (HMGB-1), leading to disease if left unchecked (Bergsbaken et al., 2009).
3.2 Effects of Pyroptotic Cell Death and Inflammation on Atherosclerosis
Inflammation is a primary response of innate immunity and is considered to be the initiator and driver of atherosclerosis. Pyroptosis, an inflammatory form of cell death, has been shown to be involved in atherosclerosis (An et al., 2019). Important functional molecules that result in pyroptosis, including NLRP3, AIM2, and caspase-1, are abundantly expressed and activated in atherosclerosis and are associated with the occurrence, accumulation, and destabilization of atherosclerotic plaques (Ozaki et al., 2015; Hoseini et al., 2018; Pan et al., 2018). The expression of NLRP1 and NLRC4 genes is upregulated in patients with atherosclerosis, and their receptors may result in the systemic alteration in inflammasome activation (Borborema et al., 2020). Caspase-11-GSDMD-mediated pyroptosis and the subsequent proinflammatory response in the pathogenesis of atherosclerosis have also been proved (Jiang M. et al., 2021). Considering that inflammasome and caspase-1 are important factors that induce pyroptosis, both the absence of inflammasome and caspase-1 deficiency alleviate atherosclerosis (Zheng et al., 2014). Caspase-1 deficiency reduces endothelial cell activation and infiltration of monocytes into intima (Gage et al., 2012; Yin et al., 2015) and attenuates ox-LDL-induced VSMC pyroptosis and IL-1β processing (Li et al., 2020), which all suppress the development of atherosclerosis. The pyroptosis of ECs, VSMCs, and monocytes/macrophages is involved in the repair and injury of the intima, but plays different roles in the progression of atherosclerosis (Figure 2).
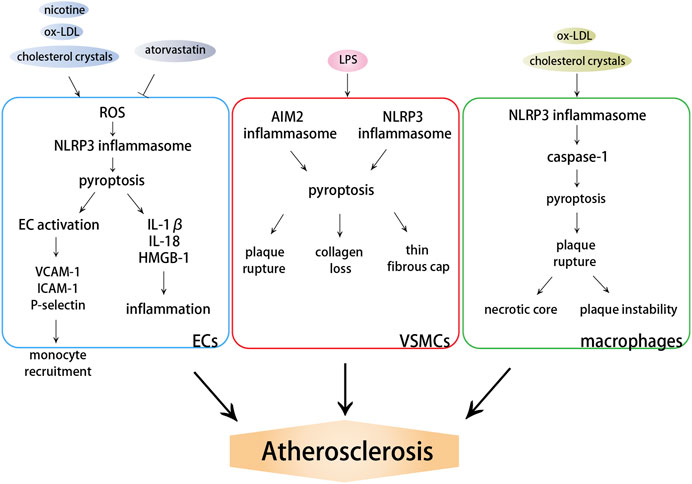
FIGURE 2. Effects of pyroptosis on atherosclerosis. DAMPs induce the activation of NLRP3 or AIM2 inflammasome, which further activate caspase-dependent pyroptosis. ECs pyroptosis triggers monocyte recruitment into the intima in early atherogenesis. VSMC pyroptosis weakens fibrous caps and contributes to pathological instability in atherosclerosis. Macrophage pyroptosis promotes the formation of necrotic core and aggravates plaque instability in advanced atherosclerotic lesions.
3.2.1 Endothelial Cells Pyroptosis Triggers Monocyte Recruitment Into the Intima in Early Atherogenesis
Caspase-1-dependent pyroptotic cell death of ECs and resulting increased inflammatory response are involved in atherosclerosis, especially in early atherosclerotic vascular injury. In the context of dyslipidemia and inflammation, the caspase-1–inflammasome pathway in ECs can sense the elevated lipids or DAMPs and other inflammatory mediators and activate ECs (Yin et al., 2015), which is the foremost step in the progress of atherosclerosis. The significantly increased levels of VCAM-1 and ICAM-1 caused by EC activation in ApoE−/− mice fed a high-fat diet were significantly reduced after caspase-1 knockout, indicating that hyperlipidemic stimulation triggered endothelial activation and upregulated adhesion molecule expression and cytokine secretion by activating caspase-1 and promoting EC pyroptosis (Yin et al., 2015). The upregulated expression of these adhesion molecules, which might be caused by pyroptosis, triggered monocyte adhesion. Based on this, EC pyroptosis promoted the recruitment of monocytes into the intima, exposure to hyperlipidemia, uptake of lipids, differentiation into macrophages, and lipid plaque formation, thus contributing to the formation and progression of atherosclerosis (Lopez-Pastrana et al., 2015; Yin et al., 2015). However, if caspase-1 activation and pyroptosis of ECs were attenuated, monocytes were not recruited into the intima and macrophages were removed from the atherosclerotic plaques (Potteaux et al., 2011; Lopez-Pastrana et al., 2015). In addition, the inhibition of caspase-1 in dyslipidemic and inflammatory environments prevented EC pyroptosis and improved EC survival and angiogenesis mediated by vascular endothelial growth factor receptor 2, thereby preserving vascular intima integrity (Lopez-Pastrana et al., 2015). This inhibited the migration and deposition of lipids, monocytes, and SMCs into the intima and further attenuated the progression of atherosclerosis. A recent study found that the protective effects against atherosclerosis of atorvastatin, as a commonly used lipid-lowering drug in clinical practice, did not depend on cholesterol-lowering capacity, but on the ability to restrain the pyroptosis of ECs through the lncRNA NEXN-AS1/NEXN pathway (Wu L. M. et al., 2020). These results proved that caspase-1-dependent pyroptosis induced by lipid metabolism disorder played a critical role in EC activation, inflammation, and early atherosclerosis.
Except for hyperlipidemia, ECs are affected by many other factors. Smoking has long been considered a risk factor for atherosclerosis. Recent studies revealed that the pyroptosis of ECs and macrophages might be a cellular mechanism for the pro-atherosclerotic properties of nicotine with the stimulation of ROS and activation of the NLRP3 inflammasome as the upstream mediators (Wu et al., 2018; Mao et al., 2021). Moreover, trimethylamine N-oxide (TMAO), an endothelial toxic factor, promoted EC pyroptosis through the increased ROS level induced by mitochondrial respiratory complex II subunit succinate dehydrogenase B (SDHB) oxidation, thereby promoting the development of atherosclerosis (Wu P. et al., 2020). The upregulation of SDHB expression and activity could also be induced by low shear stress-mediated TET2 deletion and reduced recruitment of histone deacetylase 2 (Chen J. et al., 2021). A low shear force also could to promote EC pyroptosis through the miR-181b-5p/STAT-3 axis, causing damage to the vascular wall and tissue remodeling and hence leading to the onset and development of atherosclerosis (Xu et al., 2021). In conclusion, ECs play a central role in mediating inflammation. Hyperlipidemia and other proatherogenic risk factors can promote the activation of the inflammasome and pyroptosis of ECs, increase the secretion of adhesion factors, and significantly promote monocyte recruitment and atherosclerosis progression.
3.2.2 Vascular Smooth Muscle Cell Pyroptosis Weakens Fibrous Caps and Contributes to Pathological Instability in Atherosclerosis
The death of VSMCs in atherosclerotic plaques can lead to significant thinning of fibrous caps, loss of collagen and extracellular matrix, and severe vascular inflammation, aggravating plaque instability. Except apoptosis, the pyroptosis of VSMCs is also tied up with the pathogenesis of atherosclerosis. The active pyroptotic indicators, including caspase-1 and IL-1β, overlapping with SMC marker α smooth muscle actin (α-SMA), were examined in human and mice atherosclerotic plaques, especially near the necrosis core, on the plaque surface, and in the intraplaque hemorrhage area, providing evidence that VSMC pyroptosis participated in atherosclerosis and contributed to plaque instability (Li et al., 2020). Pan et al. also reported the increased expression of GSDMD-N in ApoE−/− mice. AIM2 irritates GSDMD activity and pyroptosis through the ASC-caspase-1 pathway in VSMCs, thereby accelerating the atherosclerotic plaque progression (Pan et al., 2018). LPS derived from porphyromonas gingivalis (Pg-LPS) caused the pyroptosis of VSMCs and instability of atherosclerotic plaques, which played an important role in the occurrence and development of atherosclerosis (Liu G. et al., 2016). However, the blockage of circular RNA serine/threonine protein phosphatase PP1-gamma catalytic subunit (circPPP1CC) relieved the Pg-LPS-induced pyroptosis of VSMCs and atherosclerosis by inhibiting the HMGB-1/TLR9/AIM2 pathway (Liu J. et al., 2021). In advanced plaques, the proliferation of VSMCs promoted fibrous cap formation and contributed to plaque stability (Basatemur et al., 2019). To sum up, as a form of cell death of VSMCs in plaques, pyroptosis weakened the fibrous cap and promoted plaque rupture.
3.2.3 Macrophage Pyroptosis Promotes the Formation of Necrotic Core and Aggravates Plaque Instability in Advanced Atherosclerotic Lesions
Ox-LDL and cholesterol crystals could also trigger NLRP3 inflammasome activation and caspase-1 production in macrophages, leading to caspase-1-mediated pyroptosis (Duewell et al., 2010; Rajamaki et al., 2010). Rajamäki et al. reported that the silencing of NLRP3 receptors, one of the key components of the NLRP3 inflammasome, completely eliminated the cholesterol crystals-induced IL-1β secretion, thus confirming NLRP3 inflammasome as the cholesterol crystal-reactive element in macrophages (Rajamaki et al., 2010). The effect of cholesterol crystals and ox-LDL to activate the NLRP3 inflammasome depends on the destruction of lysosomes and the leakage of cathepsin, all of which promote the cleavage and activation of pro-caspase-1 and pyroptosis (Duewell et al., 2010; Karasawa and Takahashi, 2017; Grebe et al., 2018). Sequential pyroptosis occurs in response to ox-LDL or cholesterol crystals, in which one macrophage cannot digest the crystals and dies via pyroptosis. Then, another macrophage engulfs the same crystals and dies via pyroptosis repeatedly. This circulation reduces lesion cellularity, causes and exacerbates pronounced inflammation by releasing inflammatory mediators from pyroptotic cells, and promotes lesion instability (Chang et al., 2013).
Growing evidence shows that the pyroptosis of macrophages increases the risk of atherosclerotic plaque rupture, hence contributing to the formation of necrotic core and plaque instability in atherosclerotic lesions (Lu et al., 2021). Proinflammatory cytokines and mediators relying on caspase-1 in response to proatherogenic risk factors, such as ox-LDL and cholesterol crystals, trigger off pyroptosis of macrophage foam cells, resulting in the formation of lipid-rich pools, the necrotic core of plaques, in late stages of the lesion (Yan and Hansson, 2007). Additionally, the molecules including NLRP3, ASC, caspase-1, IL-1β, and IL-18 are more expressed in unstable plaques compared with stable plaques (Shi X. et al., 2015). Subsequently, Varghese et al. also demonstrated the close relationship between the upregulation of NLRP3 inflammasome-related components and plaque vulnerability, suggesting the possibility of NLRP3 inflammasome activation in promoting plaque instability (Paramel Varghese et al., 2016). Inhibiting NLRP3 expression and reducing macrophage pyroptosis could stabilize atherosclerotic plaques, which undoubtedly hinted at the potential value of blocking pyroptosis (Lin et al., 2021).
Vulnerable plaques are characterized by the accumulation of dead cells, in which up to 50% cells are macrophages. A large majority of dead cells in human atherosclerotic plaques had a typical cell lysis ultrastructure compared with apoptosis (Naghavi et al., 2003). The ruptured plaque showed strong immunoreactivity to caspase-1, but weak immunoreactivity to caspase-3, the executor caspase of apoptosis (Kolodgie et al., 2000). In plaque macrophages, the suppression of apoptosis by the targeted deletion of caspase-3 or p53, which all induced apoptosis, did not subside inflammation and atherosclerotic lesions (Merched et al., 2003). Conversely, macrophages showed the characteristics of cells undergoing pyroptosis accompanied by the increased caspase-1 activation in response to triglyceride treatment, suggesting that the cells in plaque died mainly via pyroptosis, not apoptosis (Son et al., 2013). Taken together, these data suggested that pyroptosis was involved in macrophage death in advanced atherosclerotic lesions and exacerbated inflammation through proinflammatory cytokines, ultimately leading to plaque rupture and acute coronary syndromes.
3.2.4 Recent Discovery of Inhibiting Pyroptosis and Improving Atherosclerosis
Based on the aforementioned understanding, the focus on novel targets and mechanisms has increased in recent years, so as to discover new approaches to intervene pyroptosis and atherosclerosis. For example, IFN regulatory factor-1, which has been found to be upregulated in macrophages of patients with acute coronary syndrome (Guo M. et al., 2019), facilitated macrophage pyroptosis and plaque rupture in atherosclerosis by restraining circ_0029589 through promoting its m6A modification (Guo et al., 2020). Brain-derived neurotrophic factor, which is found at lower levels in patients with coronary artery disease (CAD) than in those without CAD, inhibited ox-LDL-induced NLRP3 inflammasome formation and pyroptosis of ECs through KLF2/HK1-mediated regulation of glucose metabolism and mitochondrial homeostasis (Jin et al., 2021). Aldehyde dehydrogenase 2 has been reported to suppress ox-LDL-induced NLRP3 inflammasome priming and activation by attenuating oxidative stress (Xu et al., 2020). MicroRNA-302c-3p could directly target NLRP3 and ameliorate aortic inflammation and pyroptosis in ApoE−/− mice (Bai et al., 2021). Further studies are needed on the detailed mechanism of pyroptosis promoting atherosclerosis to find out better targets for atherosclerosis intervention.
4 The Roles of Ferroptosis in Atherosclerosis
4.1 Overview of Ferroptosis
Ferroptosis is an iron-dependent form of RCD resulting from the overwhelming accumulation of lipid hydroperoxides and redox imbalance, characterized by a loss of plasma membrane integrity, cytoplasmic and organelles swelling, dysmorphic shrunken mitochondria with the loss of cristae, as well as condensed and ruptured outer membrane, but the morphology of the nucleus remains unchanged (Dixon et al., 2012; Dixon and Stockwell, 2014; Galluzzi et al., 2018). Ferroptosis occurs through iron-catalyzed lipid peroxidation initiated by nonenzymatic mechanisms such as Fenton reaction, and enzymatic mechanisms including lipoxygenases (LOXs) and EGLN prolyl hydroxylases (PHD), which are enzymes responsible for lipid peroxidation and oxygen homeostasis. The increased uptake of iron by transferrin receptor 1 (TFR1) and reduced iron export by ferroportin-1 (FPN1) trigger off iron overload, in which Fenton reaction-generated ferric iron (Fe3+) increases ROS, activates LOXs or PHD, and results in ferroptosis (Yang and Stockwell, 2008; Gao et al., 2015; Yang et al., 2016). Therefore, the lipid peroxidation and ferroptosis can be prevented by iron chelators such as deferoxamine and dexrazoxane (Dixon et al., 2012; Friedmann Angeli et al., 2014) or by knocking down TFR1 (Gao et al., 2015), whereas decreased expression of ferritin or FPN1 has been testified to be sensitized to ferroptosis (Bao et al., 2020; Chen et al., 2020). In addition to LOXs and PHD, the two other enzymes involved in phospholipid metabolism, especially in the oxidation of specific phosphatidylethanolamine-containing polyunsaturated fatty acids (PUFAs), acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3), also contribute to ferroptosis (Dixon et al., 2015; Doll et al., 2017; Muller et al., 2017; Xiao et al., 2019). In the presence of iron, PUFAs generate lipid hydroperoxides and then produce toxic lipid free radicals, which transfer protons near PUFAs and trigger a new round of lipid oxidation reactions (Yang et al., 2016). Some radical-trapping antioxidants, such as vitamin E, ferrostatin-1, and liproxstatin-1, exhibit oxidation resistance and ferroptosis resistance (Dixon et al., 2012; Hofmans et al., 2016); they directly trap chain-carrying peroxyl radicals rather than repressing LOXs (Zilka et al., 2017).
Except for increasing iron release into the labile iron pool (LIP) mentioned earlier, impeding the activation of glutathione peroxidase 4 (GPX4) is the classical channel that acts on the occurrence of ferroptosis. As the major protective mechanism of biomembranes against lipid peroxidation by catalyzing the conversion of lipid hydroperoxides into lipid alcohols, the function of GPX4 depends on its cofactor glutathione (GSH) and the plasma membrane cystine/glutamate antiporter system xc−, which imports cystine in exchange for intracellular glutamate and generates GSH (Seiler et al., 2008; Yang et al., 2014). GPX4 can be inactivated by the deprivation of GSH through the inhibition of the system xc−, as a result of the restrained function of overloaded extracellular glutamate, erastin, sulfasalazine, sorafenib, p53, and activating transcription factor 3 (ATF3) (Yang et al., 2014; Jiang et al., 2015; Hayano et al., 2016; Wang L. et al., 2020). Buthionine-(S, R)-sulfoximine (BSO) is also able to decrease GSH synthesis (Yang et al., 2014), furthermore, some compounds that directly bind and inactivate GPX4, such as RAS-selective-lethal compound 3 (RSL3), ML162, ML210, FINO2, FIN56, and withaferin A, also have the ability to inhibit GPX4 and subsequently induce lipid peroxidation and ferroptosis (Weiwer et al., 2012; Friedmann Angeli et al., 2014; Yang et al., 2014; Shimada et al., 2016; Gaschler et al., 2018; Hassannia et al., 2018). In contrast, the Nrf2–Keap1 pathway has been confirmed to stimulate the system xc− and enhance resistance to ferroptosis through Nrf2 overexpression or Keap1 downregulation (Fan et al., 2017). Moreover, several other pathways that manage to regulate the peroxide state and prevent ferroptosis are gradually being discovered and studied. A newly discovered ferroptosis-resistance factor, FSP1, has also been shown to confer protection against ferroptosis as an oxidoreductase that catalyzes the regeneration of ubiquinone (also known as coenzyme Q10, CoQ10), which can trap lipid peroxyl radicals that mediate lipid peroxidation (Bersuker et al., 2019; Doll et al., 2019). GTP cyclohydrolase-1 (GCH1) and its metabolic derivatives tetrahydrobiopterin/dihydrobiopterin (BH4/BH2) have also recently been found to protect against ferroptosis because of the effect of BH4 that selectively deters the autoxidation of phospholipids and accelerates CoQ10 (Kraft et al., 2020). These redox pathways act on the progress of ferroptosis, but their roles and in-depth mechanisms in the pathophysiology of ferroptosis need to be further explored.
Ferroptosis induces the opening of plasma membrane pores, which may be caused by the lipid peroxidation-induced conformational changes in lipid domains and plasma membrane regions (Runas et al., 2016; Agmon et al., 2018). The formation of plasma membrane pores contributes to solute exchange with the external environment, resulting in cell swelling, and then ferroptotic cells undergo rupture and death (Riegman et al., 2020). Moreover, the intercellular propagation of the ferroptosis-inducing signal, including erastin and C’ dot nanoparticles but not GPX4 inhibitor, occurs upstream of cell rupture and involves the spread of cell swelling effects through cell populations in an iron- and lipid peroxide-dependent manner (Dolma et al., 2003; Kim et al., 2016; Riegman et al., 2020). Because this wave-like propagation may cause widespread tissue damage (Katikaneni et al., 2020), ferroptosis has been demonstrated to be involved in multiple acute injury and degenerative diseases in various tissues including kidney, liver, heart, and brain (Abdalkader et al., 2018; Fang et al., 2019; Wang M. et al., 2020; Li et al., 2021), and also be a target in the treatment of cancers (Badgley et al., 2020).
4.2 Effects of Ferroptosis on Atherosclerosis
Ferroptosis is a nonapoptotic form of RCD driven by abnormal iron metabolism and lipid peroxidation, which are related to the pathogenesis of atherosclerosis (Vinchi et al., 2020) (Figure 3). Nontransferrin-bound serum iron-induced iron overload triggers off ROS production and stimulates monocyte recruitment, leading to vascular oxidative stress and plaque formation (Vinchi et al., 2020). Lipid peroxidation, intraplaque hemorrhages, and iron deposition, which are hallmarks of advanced plaques (Martinet et al., 2019), are also considered as indirect evidence for the onset of ferroptosis. Therefore, it is reasonable to believe that ferroptosis may participate in the occurrence and development of atherosclerosis. Indeed, a recent study confirmed this hypothesis and revealed that the inhibition of ferroptosis alleviated atherosclerosis by reducing lipid peroxidation and endothelial dysfunction in mouse aortic ECs (Bai T. et al., 2020).
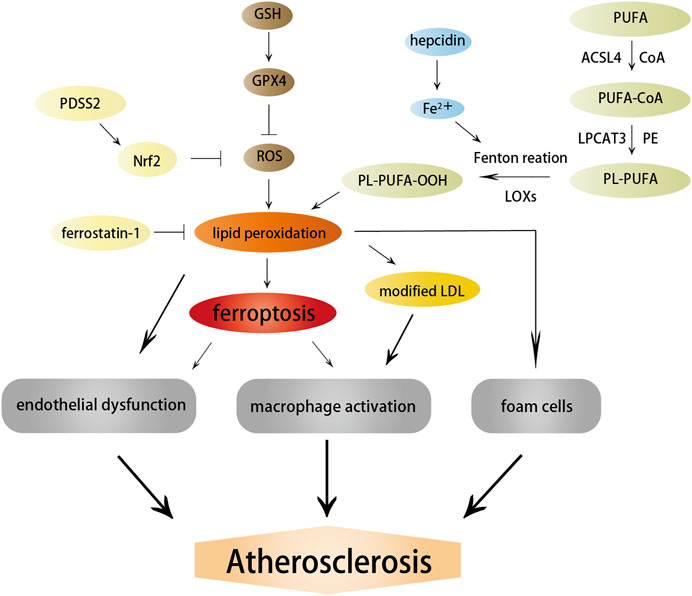
FIGURE 3. Effects of ferroptosis on atherosclerosis. Ferroptosis and lipid peroxidation induce endothelial dysfunction, macrophage activation, and foam cell formation, contributing to the generation of atherosclerosis. ACSL4, LOXs, and hepcidin promote atherosclerosis by inducing lipid peroxidation and iron overload; in contrast, PDSS2 and ferrostatin-1 can inhibit ferroptosis and atherosclerosis by suppressing lipid peroxidation.
Studies on the pathways and regulatory factors related to ferroptosis can also reflect the correlation between ferroptosis and atherosclerosis. GPX4 is responsible for transforming lipid hydroperoxides into nontoxic lipid alcohols, which can effectively abate the damage caused by oxidized lipids to vascular cells. A decreased GPX level has been demonstrated in atherosclerosis. Guo et al. found that the overexpression of GPX4 that could suppress ferroptosis reduced lipid peroxidation and alleviated atherosclerotic lesions in ApoE−/− mice (Guo et al., 2008). Moreover, the positive correlation between the severity of atherosclerosis and ACSL4 also implied that ferroptosis regulated the occurrence and progression of atherosclerosis (Zhou et al., 2021). Increased expression of ferroptosis-associated LOXs promoted atherosclerosis, while inhibiting 12/15-LOX significantly diminished ox-LDL subendothelial deposition and weakened the development of atherosclerosis (Li C. et al., 2018). Yang et al. recently found that prenyldiphosphate synthase subunit 2 (PDSS2), which was a key enzyme for the synthesis of CoQ10, could alleviate ferroptosis and atherosclerosis by activating antioxidant Nrf2 and suppressing ROS release and iron content (Yang et al., 2021). Radical-trapping antioxidant ferrostatin-1 has also been found to ameliorate iron overload, lipid peroxidation, and upregulation of ROS production induced by high glucose and high lipids, thereby effectively reducing ferroptosis and atherosclerosis (Meng Z. et al., 2021).
Ferritin-mediated iron homeostasis is instrumental in the cardiovascular system (Vinokur et al., 2016), and excessively increased ferritin levels can promote early atherosclerosis and its related complications (Sung et al., 2012). A clinical study found that circulating ferritin levels were independently associated with the changes in carotid intima-media thickness, suggesting that it could be used to monitor the risk of atherosclerosis (Prats-Puig et al., 2016). TFR1 promoted iron absorption and ferritin synthesis, and was found to accumulate significantly in the nuclear region of many foam cells, contributing to the development and rupture of atherosclerotic plaques (Li et al., 2008). Hepcidin, another hormone modulator of iron homeostasis, served as a catalyst for ferroptosis by increasing intracellular iron levels (Nemeth et al., 2004) and enhanced inflammatory atherosclerosis by inducing iron deposition (Habib et al., 2015). Therefore, the factors related to iron metabolism are important targets to the resistance of atherosclerosis, but whether they protect against atherosclerosis by affecting ferroptosis needs further exploration.
5 Possible Association Between Autophagy and Pyroptosis in Atherosclerosis
As shown in Figure 4, the level of autophagy has a regulatory effect on pyroptosis. The effect has been verified and applied in treating tumors, infectious diseases, and cardiovascular and cerebrovascular diseases. In recent years, great strides have been made in the mutual regulation of autophagy and pyroptosis in atherosclerosis. Inducing an increase in autophagic activity in vulnerable atherosclerotic plaques restrains NLRP3 inflammasome activation and secretion of inflammatory cytokines, thus ameliorating inflammation and attenuating lipid deposition and pyroptosis (Peng et al., 2018; Cong et al., 2020). In contrast, repressing autophagy aggravates NLRP3 inflammasome activation and pyroptosis (Jiang et al., 2018). As previously mentioned, autophagy defects or autophagy deficiency are normally present in the progression of atherosclerosis. Autophagy deficiency has been shown to lead to atherosclerosis through multiple complementary mechanisms, including overactivation of the inflammasome, accumulation of cytotoxic protein aggregates, and impaired lipid degradation, which are all triggers of pyroptosis.
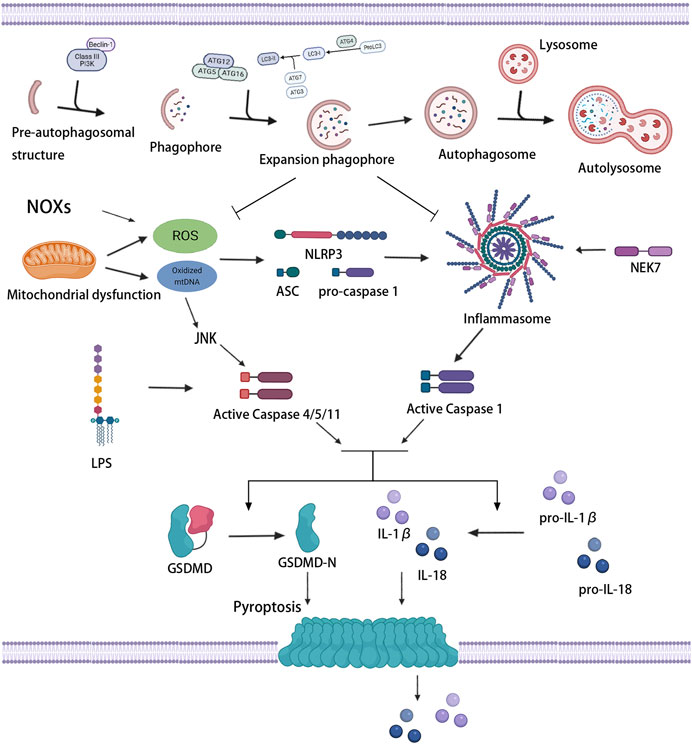
FIGURE 4. Association between autophagy and pyroptosis. Mitochondrial dysfunction and NOXs induce the massive production of ROS and stimulate the activation of inflammasome and caspase-1. LPS and oxidized mtDNA induce caspase-4/5/11 activation. Activated caspase-1/4/5/11 cleaves GSDMD, induces pyroptotic cell death, and promotes the release of IL-1β and IL-18. Mild oxidative stress triggers off autophagy, which inhibits inflammasome signaling and ROS production, thus protecting against pyroptosis.
5.1 Autophagy, Pyroptosis and Oxidative Stress
Various signaling pathways of pyroptosis regulated by autophagy have always been a hot spot for research, especially oxidative stress signaling pathways. Oxidative stress occurs when the production of pro-oxidants or ROS exceeds the capacity of endogenous antioxidants. ROS, which are produced mainly by mitochondria and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), refer to the general term of oxygen-containing free radicals and peroxides that can easily form free radicals related to oxygen metabolism in the organism. Wang et al. found that NOX-mediated accumulation of ROS which stimulated by hypoxia promoted pyroptosis, meanwhile, autophagic activity was lessened in this process, suggesting that severe oxidative stress induced the upregulation of pyroptosis, as well as the inhibition of autophagy (Wang S. et al., 2019).
The increase in the levels of intracellular ROS give rise to pyroptosis through the execution of caspase (Li et al., 2015). The activation of caspase depends on the inflammasome, and oxidative stress is an initial signal that induces inflammasome activation and influences the priming phase of inflammasomes (Bauernfeind et al., 2011; Swanson et al., 2019). The massive accumulation of ROS can trigger off the activation of the NLRP3 inflammasome and then induce caspase-1-dependent pyroptosis (Qiu et al., 2017), which may be related to an ROS-sensitive NLRP3 ligand, thioredoxin-interacting protein (TXNIP/VDUP1) (Zhou et al., 2010). Ox-LDL, which is abundant in atherosclerotic lesions, has been certified to increase the activity of ROS and promote the activation of inflammasome and caspase-1, leading to pyroptosis (Ding et al., 2013b; Lin et al., 2013). Furthermore, increased mitochondria-derived ROS and oxidation of liberated mtDNA from damaged or dysfunctional mitochondria can also drive the assembly and activation of the NLRP3 inflammasome (Zhou et al., 2011; Zhong et al., 2018). Oxidized mtDNA was found to bind to NLRP3 directly, promoting inflammasome function and subsequent caspase-1 activation (Shimada et al., 2012). In contrast, the downregulation of NLRP3 inflammasome activation by ROS scavenger could effectively suppress caspase-1 cleavage, IL-1β production, and pyroptosis (Chen et al., 2016; Long et al., 2020). Antioxidants extracellular superoxide dismutase and catalase also showed the ability to prevent caspase-1 activation and pyroptosis (Xi et al., 2016). In addition, c-Jun N-terminal kinase (JNK) signaling played a role in the noncanonical caspase-4/5/11-dependent pathway of pyroptosis (Abe and Morrell, 2016). ROS activated JNK and transferred it into the nucleus, promoted the expression of caspase-11, and initiated pyroptosis (Lupfer et al., 2014). These results suggested that the activation of inflammasomes associated with oxidative stress induced the initiation of pyroptosis, and the extent of pyroptosis seemed to increase with the increase in inflammasome stimulation.
ROS mediates NLRP3 inflammasome activation, but also upregulates autophagy to inhibit the excessive activation of NLPR3 inflammasome (Jiang L. et al., 2021). For autophagy, ROS is one of the upstream modulator (Szumiel, 2011). Under acute or chronic oxidative stress, autophagy is increased or decreased, respectively (Mitter et al., 2014). Mild oxidative stress triggers some cellular survival and repair mechanisms, including autophagy pathways. Indeed, autophagy increases the bioavailability of NO and restrains oxidative stress and vascular inflammation (LaRocca et al., 2012). Autophagy can also selectively clear dysfunctional mitochondria which leads to massive production of ROS, thus inhibiting ROS accumulation and subsequent inflammasome activation (Li Q. et al., 2018; Lin et al., 2019). In addition, as a mitochondrial outer membrane protein that directly interacted with LC3, NIX was found to promote the degradation and removal of excess mitochondria and ROS by regulating autophagy, thus inhibiting ox-LDL-induced pyroptosis of macrophages in atherosclerosis (Peng et al., 2020). However, tumor necrosis factor receptor-associated factor 3 (TRAF3) has been reported to form a complex with TRAF2 and cIAP1 and mediate ubiquitin and degradation of ULK1, in which mitochondrial ROS production and pyroptosis were promoted (Shen et al., 2020). Therefore, ROS-activated autophagic machinery may represent a negative feedback mechanism that limits ROS-regulated caspase-1 activation through removing ROS-damaged organelles and proteins. Nevertheless, conversely, severe oxidative stress produces a large number of damaged proteins, which may lead to an overload of the autophagosome system, resulting in a reduced autophagic activity. Wang et al. reported that a decreased miR-103 level in oxidatively stressed ECs suppresses the interaction between autophagosomes and lysosomes, restraining the end-stage of autophagy, promoting the accumulation of inflammatory mediators, and thus increasing the occurrence of pyroptosis (Wang Y. et al., 2020).
Accordingly, considering that ROS can promote inflammasome activity but autophagy can suppress ROS production, the suppression of autophagy on inflammasome activity and pyroptosis are related to oxidative stress. ROS blockade with subsequently suppressed inflammasome activity by autophagy is one of the important mechanisms to antagonize pyroptosis. An important feedback network exists between autophagy, NF-κB signaling pathway, and pyroptosis (Gao Y. et al., 2020). Wang et al. reported that enhanced autophagy prevented pyroptotic cell death by eliminating excess ROS and negatively mediating the nuclear translocation of NF-κB P65 and NLRP3 inflammasome activation (Wang X. et al., 2019). Li et al. found that adrenomedullin protected the steroidogenic functions of Leydig cells against pyroptosis by activating autophagy through the ROS-AMPK-mTOR axis (Li et al., 2019). Autophagy activation reversed the expression of pyroptosis-related proteins by downregulating the level of IL-13 and inhibiting the Janus kinase (JAK)-1/signal transducers and activators of transcription (STAT)-1 signaling pathway (Gao C. et al., 2020). Nrf2/ARE, one of the key pathways involved in oxidative stress, can regulate the expression of scavenger receptor-B in the macrophages of ApoE−/− Nrf2+/+ mice, stimulate the rapid uptake of ox-LDL by macrophages, and promote the formation of atherosclerotic plaques (Bozaykut et al., 2014). A recent study further revealed that activation of the Nrf2/ARE pathway might be influenced by autophagy. The blockage of autophagy promoted the pyroptosis of ox-LDL-treated macrophages through the malignant activation between p62 and Nrf2/ARE pathway, which might provide a novel therapeutic target for atherosclerosis (Liu J. et al., 2021). Thereout, autophagy suppresses pyroptosis partly due to its regulation of oxidative stress, while devastating oxidative stress induces pyroptosis by inhibiting autophagy.
5.2 Autophagy, Pyroptosis and Inflammation
Atherosclerosis is a chronic inflammatory disease, and pyroptosis can release the cytoplasmic contents of dead host cells, thus providing an effective signal to initiate an inflammatory cascade, which exacerbates vascular inflammation and atherosclerosis (Man et al., 2017). Some proinflammatory cytokines are expressed in human atherosclerotic lesions, particularly in relation to infiltrating macrophages. Macrophages are considered as the primary sources of IL-1β and IL-18, which are two major substrates of caspase-1 and are mainly responsible for caspase-1-mediated pyroptosis in atherosclerosis, promoting plaque destruction and arterial thrombosis through the release of chemokines, matrix metalloproteinases, and proteases (Dinarello, 2011). Vromman et al. found that IL-1β inhibition promoted the tilt of monocytes toward a less inflammatory state during atherosclerosis and reduced the size of established atherosclerosis (Vromman et al., 2019). Moreover, IL-1β inhibition could recede the expression of VCAM-1 and monocyte chemotactic protein-1 (MCP-1), thus preventing the recruitment of monocytes or macrophages into the intima and significantly reducing atherosclerotic lesions (Kirii et al., 2003). The Canakinumab Anti-inflammatory Thrombosis Outcome Study trial involving 10,061 patients with previous myocardial infarction also revealed that anti-inflammatory therapy targeting the IL-1β innate immune pathway resulted in a significant reduction in the recurrence rate of cardiovascular events, independent of a decrease in lipid levels (Ridker et al., 2017). These results established a pivotal role for IL-1β in driving inflammation during atherogenesis and the evolution of advanced atheroma. Similarly, IL-18 has been found to promote the development of atherosclerosis by promoting lipoprotein oxidation, macrophage-derived foam cell formation, and immune cell activation (Bahrami et al., 2021). Even so, Gomez et al. demonstrated the atheroprotective effects of IL-1β in advanced atherosclerotic lesions. The results highlighted the importance of promoting inflammatory resolution, while the excessive repression of inflammation disrupted the formation and maintenance of SMC- and collagen-rich fibrous caps, leading to atherosclerotic plaque instability (Gomez et al., 2018). Thus, the excessive release of IL-1β and IL-18 attributed to pyroptosis is one of the factors that aggravate atherosclerosis, but meanwhile, the over-inhibition of inflammation also leads to plaque instability.
Pyroptosis and the release of these inflammatory factors requires the activation of inflammasome. Recent evidence has revealed a complex interplay between autophagy and inflammasome. Autophagy may be accompanied by the activation of inflammasomes. Pseudomonas aeruginosa can activate the NLRP3 inflammasome to trigger macrophage autophagy so as to escape intracellular killing (Deng et al., 2016). In addition, as mentioned earlier, mitochondria are involved in inflammasome activation; mitophagy is also stimulated in this process and regulates inflammasome activity by removing damaged mitochondria, thereby providing an important negative feedback mechanism (Green et al., 2011; Nakahira et al., 2011; Zhong et al., 2016). Autophagy moderates inflammation by eliminating active inflammasomes; however, lack of autophagy leads to the opposite effect and thus exacerbates inflammation. Resolvin D2, which is an innate suppressor of inflammation, has been found to promote the degradation of NLRP3 in an autophagy dependent manner; the suppression of autophagy could reverse the inhibition of NLRP3 inflammasome (Cao et al., 2021). LPS-dependent inflammasomes are activated in macrophages whose autophagy is blocked by the genetic ablation of the autophagy regulators ATG16L1 or ATG7, but not in wild-type macrophages, suggesting that autophagy generally suppresses inflammasome activation induced by LPS (Saitoh et al., 2008). The NLRP3 inflammasome can also be activated by oxidative or apoptosis-induced mtDNA, which is released during autophagy repression (Sollberger et al., 2015). Impaired CMA has also been proven to increase NLRP3 inflammasome activation and secretion of IL-1β, promoting vascular inflammation and atherosclerosis (Qiao et al., 2021). Therefore, autophagy can negatively regulate pyroptosis by restraining the activation of the inflammasome and caspase-1/GSDMD pathway (Wang X. et al., 2020). A clinical trial found that excessive inflammation and the pyroptosis of vascular ECs triggered off atherosclerosis in postmenopausal women, which could be ameliorated with estrogen treatment by alleviating EC pyroptosis through estrogen receptor α-mediated activation of autophagy (Meng Q. et al., 2021). For the mechanism underlying autophagy-dependent inflammation inhibition, it has been suggested that autophagy typically restrains inflammasome signaling and IL-1β production by directing ubiquitinated inflammasome complexes and pro-IL-1β to lysosomes for degradation (Katsnelson et al., 2016; Kimura et al., 2017). In other words, the inflammasome itself may serve as a target for autophagic degradation, thereby facilitating the resolution of the inflammatory responses.
In spite of an activation effect on autophagy, the NLRP3 inflammasome has also been found to suppress protective autophagy through the E2/ERβ/AMPK/mTOR pathway or through caspase-1-mediated cleavage of the TLR adaptor (Lai et al., 2018; Wei et al., 2019). Moreover, the triggering of pyroptosis releases a large number of proinflammatory factors, further aggravating the inflammatory response, which in turn aggravates the autophagy defect; the two synergistically exacerbate atherosclerosis. For example, a high level of myeloid cells trigger receptors 1 (TREM-1) and a low level of Sirt6 are associated with increased risk for all-cause mortality and major adverse cardiovascular events. Also, TREM-1 negatively regulates the expression of Sirt6. TREM-1-mediated pyroptosis may underlie the proatherogenic effects of ox-LDL, which may be limited by Sirt6-induced EC autophagy (Zi et al., 2019). In conclusion, autophagy activation could inhibit pyroptosis and excessive inflammation, thereby improving atherosclerosis and reducing the incidence of adverse cardiovascular events, which associated with decreased inflammasome activity.
5.3 Autophagy, Pyroptosis and Lipid Accumulation
Dyslipidemia, which involves pathologically elevated concentrations of cholesterol and other lipids, is a major risk factor for atherosclerotic diseases (Kopin and Lowenstein, 2017). The inflammasome may be a potential target of atherosclerosis induced by abnormal lipid metabolism. The NLRP3 inflammasome is positively correlated with the severity and prognosis of atherosclerosis in patients with acute coronary syndrome (Afrasyab et al., 2016). When the LDLR−/− mice were transplanted with NLRP3-deficient, ASC-deficient, or IL-1α/β-deficient bone marrow and fed a high cholesterol diet, they had significantly reduced early atherosclerosis and inflammasome-dependent IL-18 levels (Duewell et al., 2010), suggesting that the deficiency of the NLRP3 inflammasome significantly protected proatherogenic mice from disease. Given that pyroptosis is an inflammasome-dependent form of cell death, possible lipotoxic mediators including ox-LDL, cholesterol crystals, saturated fatty acids, and palmitic acid were found to induce pyroptosis in ECs (Hu et al., 2018), which involve mitochondrial injury, oxidative stress, activation of JNK and release of DAMPs. LDL entered the arterial wall and was transformed into ox-LDL due to changes in endothelial permeability and extracellular matrix composition located under the endothelium (Tabas et al., 2007). Ox-LDL might induce EC pyroptosis and promote the development of atherosclerosis by regulating the miR-125a-5p/ten-eleven translocation 2 (TET2) pathway, resulting in mitochondrial dysfunction, increased ROS generation, and NF-κB nuclear transposition that triggered the activation of the NLRP3 inflammasome and inflammatory response (Zhaolin et al., 2019). The effect of ox-LDL on the NLRP3 inflammasome also depended on the upregulated expression of mixed lineage kinase domain-like protein (Wu Q. et al., 2020). Cholesterol crystals, a key pathological marker of atherosclerotic plaque vulnerability, which were observed in human atherosclerotic lesions as they progressed from fatty streaks to more advanced lesions (Katz et al., 1976), could induce ROS production, NLRP3 inflammasome activation, and ultimately pyroptosis of ECs (Yang et al., 2020). The major lipid component of the plasma membrane, lysophosphatidylcholine, was also proved to elicit the activation of the inflammasome and pyroptosis though potassium efflux and lysosomal damage in both ECs and monocytes; this process was involved in the maintenance of inflammation in tissues adjacent to blood vessels and the pathological process of atherosclerosis (Correa et al., 2019). These results suggested that dyslipidemia was an important factor to induce pyroptosis, and inflammasome activation might be an important link between lipid metabolism and inflammation in atherosclerotic lesions. Therefore, regulation of lipid metabolism and inhibition of lipid overaccumulation are important ways to inhibit inflammasome activation and pyroptosis in atherosclerosis.
Besides pyroptosis, lipid accumulation may also lead to defects in autophagy. In advanced human carotid atherosclerosis, accompanied by lipid accumulation, the levels of autophagic proteins ATG5 and LC3β were decreased, while the levels of dysfunctional autophagy markers SQSTM1/p62 and ubiquitin were increased (Li et al., 2016). Defects in autophagic machinery might occur during the formation and maturation of autophagosomes or during the fusion of autophagosomes with lysosomes or lysosomal-mediated degradation. It was widely considered that an autophagy defect in atherosclerosis was attributable to defects in the lysosomal-dependent pathway. Abundant cholesterol crystals in atherosclerotic plaques jeopardized the lysosomal membrane, consequently impeding the autophagy process (Duewell et al., 2010). These findings suggested that lysosomal-mediated cytoplasmic degradation might be restrained in advanced atherosclerosis, rather than the initiation of autophagy itself. Therefore, autophagy may still be actuated in response to the autophagy activator in the plaques, such as ROS and oxidative lipids, but the process becomes dysfunctional in the second stage of lysosomal-dependent degradation.
Endothelial autophagy represents an important mechanism to regulate excess, exogenous lipids and limit lipid accumulation in the vascular wall. Oxidized and natural LDL promotes the formation of autophagosomes, while excess natural or modified LDL appears to be engulfed in the autophagic structure, which is intercepted by endothelial-specific ATG7 knockdown, showing an increase in lipid deposition (Torisu et al., 2016). Recently, Kim et al. found that endothelial lipid accumulation in atherosclerosis is caused by autophagy-related lysosomal dysfunction (Kim et al., 2021). Moreover, autophagy deficiency disturbs the degradation of CAV1, which interacts with caveolae associated protein 1 (CAVIN1) and LC3B, and is used to form more caveolae in the cell membrane, contributing to LDL transcytosis across ECs (Bai X. et al., 2020). Therefore, the absence of autophagy of ECs is responsible for the prolonged retention of oxidized and natural LDL and increased atherosclerotic burden. Some researchers reported that ox-LDL in atherosclerosis induced EC autophagy, accompanied by apoptosis and increased monolayer permeability; however, when ox-LDL-mediated autophagy was partly inhibited, apoptosis and monolayer permeability were further increased (Wei et al., 2013; Wang Y. et al., 2019).
A recent study demonstrated that activation of the P2RY12 receptor reduced cholesterol efflux and promoted the formation of VSMC-derived foam cells by blocking autophagy in advanced atherosclerosis, suggesting that autophagy is essential in maintaining lipophagic flux and cholesterol homeostasis in VSMCs (Pi et al., 2021). Similarly, as mentioned earlier, intact autophagic machinery is also essential in limiting lipid uptake by macrophages. C1q/tumor necrosis factor-related protein 13 can diminish ox-LDL uptake, retention, and foam cell formation, meanwhile reducing inflammatory responses and lesion areas, which is attributed to the enhanced autophagy in macrophages and accelerated autophagy-lysosome-dependent degradation of CD36 (Wang C. et al., 2019). Gustafsson and colleagues also found that perilipin-2, whose Pro251 variant had a negative correlation with intima-media thickness at baseline and over 30 months of follow-up, initiated a feed-forward cycle in which liver-X-receptor and autophagy activate each other, thus influencing the susceptibility to atherosclerosis through the increased activation of autophagy and stimulation of cholesterol efflux (Saliba-Gustafsson et al., 2019). Based on this, we have reason to believe that enhancing autophagy can inhibit pyroptosis by regulating lipid accumulation in vascular cells, although there is no clear literature confirming this viewpoint.
6 Possible Association Between Autophagy and Ferroptosis in Atherosclerosis
6.1 Regulation of Autophagy on Ferroptosis
It has shown that the knockout or knockdown of ATG5 and ATG7 prevents erastin-induced ferroptosis by diminishing intracellular iron levels and lipid peroxidation (Hou et al., 2016), suggesting that autophagy may play a critical role in the progress of ferroptosis. As shown in Figure 5, Song et al. found that the autophagy regulator Beclin-1 was phosphorylated by AMPK and translocated to the plasma membrane, which ultimately facilitated ferroptosis through binding and blocking the activity of system xc− (Song et al., 2018). Autophagy also decreases intracellular GSH levels, and thus, autophagy inhibitors such as 3-MA and Baf-A1 can prevent GSH depletion-dependent ferroptosis (Desideri et al., 2012; Sun Y. et al., 2018). Since lysosomes recycle endogenous iron sources like ferritin and mitochondria, it participates in ferroptosis by influencing cellular iron equilibria and ROS generation (Torii et al., 2016). STAT3 positively regulated ferroptosis by mediating cathepsin B (CTSB) expression. Erastin increased lysosomal membrane permeability and subsequent lysosomal cell death, which could be inhibited by the pharmacological blockade of cathepsin activity. The repression of lysosomal activity and the release of acidic hydrolases into the cytoplasm could restrain ferroptosis (Torii et al., 2016; Gao et al., 2018).
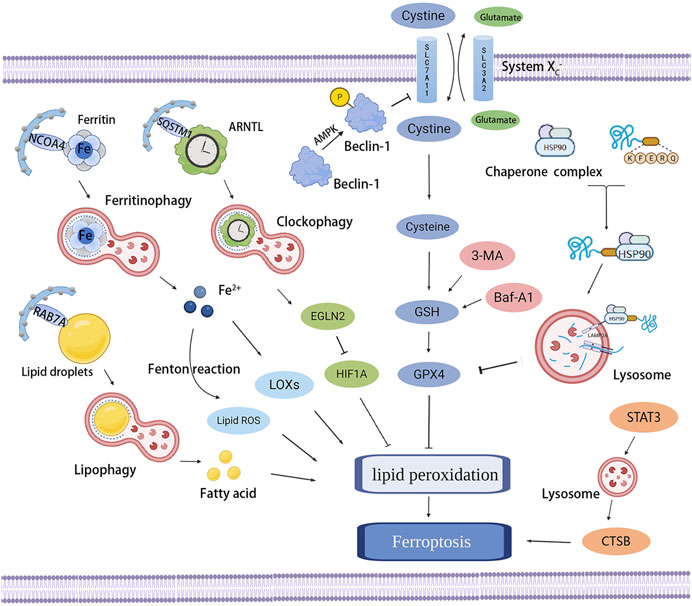
FIGURE 5. Association between autophagy and ferroptosis. Several types of selective autophagy, including ferritinophagy, lipophagy, clockophagy, and CMA, promote lipid peroxidation and ferroptosis by promoting the degradation of ferritin, lipid droplets, ARNTL, and GPX4, respectively. Beclin-1 is phosphorylated by AMPK and then facilitates ferroptosis by binding and blocking the activity of system xc−. Autophagy inhibitors (e.g., 3-MA, Baf-A1) can prevent GSH depletion-dependent ferroptosis. STAT3-mediated CTSB expression and lysosomal cell death promote ferroptosis.
Apart from nonselective autophagy, the excessive activation of selective autophagy that recognizes specific targets and recruits corresponding receptors for subsequent lysosomal degradation mediates ferroptosis. Excess iron can prompt the overproduction of mitochondrial ROS and actuate ferroptosis. Ferritin is the main iron storage protein, which inhibits oxidative stress caused by Fe2+-mediated Fenton reaction and maintains iron homeostasis. Excessive degradation of ferritin can significantly increase intracellular unstable iron levels and enhance sensitivity to ferroptosis (Dufrusine et al., 2019). Ferritin-targeted autophagy, known as ferritinophagy, induces ferroptosis by promoting the release of iron stored in ferritin to the LIP and also lipid peroxidation (Tang et al., 2018). An increasing number of studies reported that activating ferritinophagy promoted ferroptosis by degrading ferritin and inducing TFR1 expression (Du et al., 2019; Sui et al., 2019; Sui et al., 2021), while ferritinophagy deficiency led to cell survival during ferroptosis (Masaldan et al., 2018; Zhang Z. et al., 2020). Ferritinophagy is mediated by nuclear receptor coactivator 4 (NCOA4), which is a cargo receptor for the degradation of ferritin. NCOA4 increases the amount of intracellular iron by binding to FTH1 in autophagosomes and sending the autophagosomes to lysosomes to degrade ferritin (Mancias et al., 2014). Therefore, silencing NCOA4 prevents ferroptosis by abrogating the accumulation of iron and ROS, while the overexpression of NCOA4 increases ferritin degradation and promotes ferroptosis (Gao et al., 2016; Qin et al., 2021). Furthermore, Park et al. confirmed that ferritinophagy was indeed actuated by erastin-induced ROS, providing evidence that the activation of ferritinophagy induced by the ferroptosis inducer was necessary for the initiation of ferroptosis (Park and Chung, 2019).
In addition to ferritinophagy, Yang et al. first revealed that clockophagy, which was a novel mode of selective autophagy, could degrade the core circadian clock protein aryl hydrocarbon receptor nuclear translocator-like protein 1 (ARNTL) depending on the cargo receptor sequestosme 1 (SQSTM1), and favored lipid peroxidation and ferroptosis resulting from type II ferroptosis activators such as RSL3 and FIN56 (Yang et al., 2019). ARNTL could directly repress transcriptional factor egl-9 family hypoxia inducible factor 2 (EGLN2) expression and subsequently increase the level of prosurvival factor hypoxia-inducible factor 1 subunit α (HIF1A), while HIF1A suppressed ferroptosis by regulating lipid metabolism and facilitating the storage of lipids in droplets (Bai et al., 2019). Thus, the clockophagy-mediated degradation of ARNTL induced ferroptosis through the activation of lipid peroxidation. In addition, lipid droplets or increased lipid storage protected against ferroptosis (Kamili et al., 2015). Lipophagy was found to promote ferroptosis by degrading lipid droplets into free fatty acids that then trigger off mitochondrial oxidation, which could be suppressed by the knockout of ATG5 (Bai et al., 2019). In lipophagy, as the key mediator of transporting multivesicular bodies and lysosomes to the lipid droplet surface, RAB7A (a member of the RAS oncogene family) promoted lipid peroxidation and ferroptosis (Schroeder et al., 2015; Bai et al., 2019). Moreover, CMA is a selective autophagy that uses molecular chaperone such as heat shock proteins (HSPs) to stimulate the direct translocation of certain cytoplasmic proteins across the lysosomal membrane by recognizing specific amino acid sequences. It has been shown that autophagy of HSP70 stabilizes lysosomes and inhibits ferroptosis by promoting the transformation of lysosomal redox-active iron into a non-redox-active form (Kurz and Brunk, 2009). HSP member 5 (HSPA5) protected against GPX4 degradation and lipid peroxidation by bounding GPX4 (Zhu et al., 2017). However, Wu et al. found that ferroptosis activators facilitated the degradation of GPX4 protein by increasing the levels of lysosome-associated membrane protein 2a (LAMP2A) and inducing CMA, besides, the inhibition of CMA stabilizes GPX4 and reduces ferroptotic cell death through HSP90 inhibitor (Wu Z. et al., 2019), supporting that HSPs-mediated CMA plays a critical role in ferroptosis. In a nutshell, these findings overturned the previous conclusion that ferroptosis was non-autophagic cell death and confirmed that ferroptosis was an autophagy-mediated form of cell death. The initiation of ferroptosis required autophagy, and ferroptosis was an autophagic process.
6.2 Effect of Ferroptosis on Inflammation
Like pyroptosis, ferroptosis can also promote sterile inflammation and the development of inflammatory diseases by releasing DAMPs (Gong et al., 2020), in which GSH and GPX4 play an important role. As the most abundant antioxidant, GSH can buffer increasing ROS and protect cell from inflammation-associated damage (Mak et al., 2017). Moreover, GPX4 inhibited inflammatory response by suppressing arachidonic acid oxidation and NF-κB pathway activation, meanwhile reducing ROS production (Li C. et al., 2018). In contrast, the disruption of GPX4 could upregulate the expression of 12-lipoxygenase and cyclooxygenase 1, thereby triggering an inflammatory response (Chen et al., 2003). Kang et al. have found that the deficiency of GPX4 in myeloid cells increased the production of caspase-1/11-mediated GSDMD and promoted pyroptosis, which also could promote inflammatory response by releasing plenty of inflammatory factors (Kang et al., 2018).
Ferroptosis-associated peroxidation of LDL induces endothelial dysfunction and macrophages activation; besides, lipid peroxidation increases ROS, decreases NO, and promotes the formation of foam cells, all of these contributing to atherosclerosis (Yu et al., 2021). A recent study found that activation of Sirt1 inhibited excess iron-induced ferroptosis of foam cells through autophagy, providing a novel therapeutic target for atherosclerosis (Su et al., 2021). However, as mentioned earlier, autophagy induces ferroptosis through a variety of pathways, but the role of these regulatory mechanisms of autophagy in ferroptosis of vascular cells in atherosclerosis remains unclear. Furthermore, although we have manifested a correlation between atherosclerosis and ferroptosis, of course, which mainly involved the ferroptosis-related factors including iron overload and redox pathways, whether ferroptosis is active or passive, in other words, whether it is an automatic response to a stimulus that disrupts metabolic homeostasis or it is a stimulus that directly disrupts the homeostasis is still controversial. Therefore, further studies are needed to explore the exact relationship between ferroptosis and pathophysiological conditions of atherosclerosis, and also the interaction between autophagy and ferroptosis in atherosclerosis.
7 Conclusions and Future Perspectives
Oxidative stress plays a key role in the pathogenesis of numerous types of vascular diseases including atherosclerosis. Once exceeding the capacity of the cellular reductase mechanisms, an overwhelming concentration of ROS triggers off direct or indirect functional damage to different molecules, such as proteins, lipids, and nucleic acids, eventually leading to cell death, which unfortunately leads to, rather than ameliorating, atherosclerosis and plaque instability. ROS-induced inflammasome activation and lipid peroxidation are important features of atherosclerosis, therefore, pyroptosis and ferroptosis as nexus linking oxidative stress, inflammation and lipid metabolism are inevitable to play important roles in the pathogenesis of atherosclerosis. Autophagy is a promising pathway for maintaining plaque cell survival and function. Increasing evidence has shown the crosstalk between autophagy, pyroptosis, and ferroptosis, but whether these different modes of RCD can be integrated into a complete regulatory network remains to be explored.
In atherosclerosis, excessive oxidative stress, inflammation, and lipid peroxidation lead to autophagy dysfunction, pyroptosis, and ferroptosis. The regulation of autophagy, pyroptosis, and ferroptosis is thought to be the potential therapeutic option of atherosclerosis. However, the current studies still have significant limitations. Firstly, atherosclerosis is a complex pathological process involving multiple cells, and the results of a single cell type or a single cell process can hardly explain the actual disease. It is a pity that although we have found clues from separate studies, the specific mechanism of mutual regulation of multiple RCDs and the mutual influence of multiple vascular cells on atherosclerosis has not been implemented. Second, although some studies have used animal models, a large number of experiments in vivo are still urgently needed, especially clinical experiments.
In summary, the abnormal states of autophagy, pyroptosis, and ferroptosis in vascular cells including ECs, VSMCs and macrophages associated with oxidative stress, inflammation, and lipid peroxidation are important pathogenesis of atherosclerosis. However, their regulatory mechanisms of atherosclerosis need to be further explored, especially the interaction between multiple RCDs. Therefore, further researches on RCD will develop a broader field for exploring the pathogenesis of atherosclerosis and provide a better clinical treatment for atherosclerosis.
Author Contributions
LL, M-XZ, and LZ took the lead in writing the manuscript. DZ, CL, and Y-LL discussed the contents and edited the manuscript.
Funding
This work received support by the National Natural Science Foundation of China (81974566, 82004276), Jinan independent innovation team—blood circulation and blood vessel reconstruction innovation team (2020GXRC017), Natural Science Foundation of the Shandong Province (ZR2020KH034), China Postdoctoral Science Foundation (2020M672125, 2021T140429) and Shandong Province Postdoctoral Innovation Project (202003027), Medical and Health Science and Technology Development Project of Shandong Province (2019WS553).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
This work is supported by Shandong Provincial Engineering Laboratory of TCM Precision Therapy for Cardiovascular Diseases.
References
Aachoui, Y., Sagulenko, V., Miao, E. A., and Stacey, K. J. (2013). Inflammasome-mediated Pyroptotic and Apoptotic Cell Death, and Defense against Infection. Curr. Opin. Microbiol. 16 (3), 319–326. doi:10.1016/j.mib.2013.04.004
Abdalkader, M., Lampinen, R., Kanninen, K. M., Malm, T. M., and Liddell, J. R. (2018). Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front. Neurosci. 12, 466. doi:10.3389/fnins.2018.00466
Abe, J., and Morrell, C. (2016). Pyroptosis as a Regulated Form of Necrosis: PI+/Annexin V-/High Caspase 1/Low Caspase 9 Activity in Cells = Pyroptosis. Circ. Res. 118 (10), 1457–1460. doi:10.1161/CIRCRESAHA.116.308699
Afrasyab, A., Qu, P., Zhao, Y., Peng, K., Wang, H., Lou, D., et al. (2016). Correlation of NLRP3 with Severity and Prognosis of Coronary Atherosclerosis in Acute Coronary Syndrome Patients. Heart Vessels 31 (8), 1218–1229. doi:10.1007/s00380-015-0723-8
Aglietti, R. A., Estevez, A., Gupta, A., Ramirez, M. G., Liu, P. S., Kayagaki, N., et al. (2016). GsdmD P30 Elicited by Caspase-11 during Pyroptosis Forms Pores in Membranes. Proc. Natl. Acad. Sci. U S A. 113 (28), 7858–7863. doi:10.1073/pnas.1607769113
Agmon, E., Solon, J., Bassereau, P., and Stockwell, B. R. (2018). Modeling the Effects of Lipid Peroxidation during Ferroptosis on Membrane Properties. Sci. Rep. 8 (1), 5155. doi:10.1038/s41598-018-23408-0
Allahverdian, S., Chehroudi, A. C., McManus, B. M., Abraham, T., and Francis, G. A. (2014). Contribution of Intimal Smooth Muscle Cells to Cholesterol Accumulation and Macrophage-like Cells in Human Atherosclerosis. Circulation 129 (15), 1551–1559. doi:10.1161/CIRCULATIONAHA.113.005015
Amersfoort, J., Douna, H., Schaftenaar, F. H., Foks, A. C., Kroner, M. J., van Santbrink, P. J., et al. (2018). Defective Autophagy in T Cells Impairs the Development of Diet-Induced Hepatic Steatosis and Atherosclerosis. Front. Immunol. 9, 2937. doi:10.3389/fimmu.2018.02937
An, N., Gao, Y., Si, Z., Zhang, H., Wang, L., Tian, C., et al. (2019). Regulatory Mechanisms of the NLRP3 Inflammasome, a Novel Immune-Inflammatory Marker in Cardiovascular Diseases. Front. Immunol. 10, 1592. doi:10.3389/fimmu.2019.01592
Andrei, C., Dazzi, C., Lotti, L., Torrisi, M. R., Chimini, G., and Rubartelli, A. (1999). The Secretory Route of the Leaderless Protein Interleukin 1beta Involves Exocytosis of Endolysosome-Related Vesicles. Mol. Biol. Cel 10 (5), 1463–1475. doi:10.1091/mbc.10.5.1463
Ashford, T. P., and Porter, K. R. (1962). Cytoplasmic Components in Hepatic Cell Lysosomes. J. Cel Biol 12, 198–202. doi:10.1083/jcb.12.1.198
Back, M., Yurdagul, A., Tabas, I., Oorni, K., and Kovanen, P. T. (2019). Inflammation and its Resolution in Atherosclerosis: Mediators and Therapeutic Opportunities. Nat. Rev. Cardiol. 16 (7), 389–406. doi:10.1038/s41569-019-0169-2
Badgley, M. A., Kremer, D. M., Maurer, H. C., DelGiorno, K. E., Lee, H. J., Purohit, V., et al. (2020). Cysteine Depletion Induces Pancreatic Tumor Ferroptosis in Mice. Science 368 (6486), 85–89. doi:10.1126/science.aaw9872
Bahrami, A., Sathyapalan, T., and Sahebkar, A. (2021). The Role of Interleukin-18 in the Development and Progression of Atherosclerosis. Curr. Med. Chem. 28 (9), 1757–1774. doi:10.2174/0929867327666200427095830
Bai, B., Yang, Y., Ji, S., Wang, S., Peng, X., Tian, C., et al. (2021). MicroRNA-302c-3p Inhibits Endothelial Cell Pyroptosis via Directly Targeting NOD-, LRR- and Pyrin Domain-Containing Protein 3 in Atherosclerosis. J. Cel Mol Med 25 (9), 4373–4386. doi:10.1111/jcmm.16500
Bai, T., Li, M., Liu, Y., Qiao, Z., and Wang, Z. (2020). Inhibition of Ferroptosis Alleviates Atherosclerosis through Attenuating Lipid Peroxidation and Endothelial Dysfunction in Mouse Aortic Endothelial Cell. Free Radic. Biol. Med. 160, 92–102. doi:10.1016/j.freeradbiomed.2020.07.026
Bai, X., Yang, X., Jia, X., Rong, Y., Chen, L., Zeng, T., et al. (2020). CAV1-CAVIN1-LC3B-mediated Autophagy Regulates High Glucose-Stimulated LDL Transcytosis. Autophagy 16 (6), 1111–1129. doi:10.1080/15548627.2019.1659613
Bai, Y., Meng, L., Han, L., Jia, Y., Zhao, Y., Gao, H., et al. (2019). Lipid Storage and Lipophagy Regulates Ferroptosis. Biochem. Biophys. Res. Commun. 508 (4), 997–1003. doi:10.1016/j.bbrc.2018.12.039
Bao, W. D., Zhou, X. T., Zhou, L. T., Wang, F., Yin, X., Lu, Y., et al. (2020). Targeting miR-124/Ferroportin Signaling Ameliorated Neuronal Cell Death through Inhibiting Apoptosis and Ferroptosis in Aged Intracerebral Hemorrhage Murine Model. Aging Cell 19 (11), e13235. doi:10.1111/acel.13235
Basatemur, G. L., Jorgensen, H. F., Clarke, M. C. H., Bennett, M. R., and Mallat, Z. (2019). Vascular Smooth Muscle Cells in Atherosclerosis. Nat. Rev. Cardiol. 16 (12), 727–744. doi:10.1038/s41569-019-0227-9
Bauernfeind, F., Bartok, E., Rieger, A., Franchi, L., Nunez, G., and Hornung, V. (2011). Cutting Edge: Reactive Oxygen Species Inhibitors Block Priming, but Not Activation, of the NLRP3 Inflammasome. J. Immunol. 187 (2), 613–617. doi:10.4049/jimmunol.1100613
Bauernfeind, F. G., Horvath, G., Stutz, A., Alnemri, E. S., MacDonald, K., Speert, D., et al. (2009). Cutting Edge: NF-kappaB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J. Immunol. 183 (2), 787–791. doi:10.4049/jimmunol.0901363
Bergsbaken, T., Fink, S. L., and Cookson, B. T. (2009). Pyroptosis: Host Cell Death and Inflammation. Nat. Rev. Microbiol. 7 (2), 99–109. doi:10.1038/nrmicro2070
Bernard, A., and Klionsky, D. J. (2014). Defining the Membrane Precursor Supporting the Nucleation of the Phagophore. Autophagy 10 (1), 1–2. doi:10.4161/auto.27242
Bersuker, K., Hendricks, J. M., Li, Z., Magtanong, L., Ford, B., Tang, P. H., et al. (2019). The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nature 575 (7784), 688–692. doi:10.1038/s41586-019-1705-2
Borborema, M. E. A., Crovella, S., Oliveira, D., and de Azevedo Silva, J. (2020). Inflammasome Activation by NLRP1 and NLRC4 in Patients with Coronary Stenosis. Immunobiology 225 (3), 151940. doi:10.1016/j.imbio.2020.151940
Boucher, D., Monteleone, M., Coll, R. C., Chen, K. W., Ross, C. M., Teo, J. L., et al. (2018). Caspase-1 Self-Cleavage Is an Intrinsic Mechanism to Terminate Inflammasome Activity. J. Exp. Med. 215 (3), 827–840. doi:10.1084/jem.20172222
Bozaykut, P., Karademir, B., Yazgan, B., Sozen, E., Siow, R. C., Mann, G. E., et al. (2014). Effects of Vitamin E on Peroxisome Proliferator-Activated Receptor Gamma and Nuclear Factor-Erythroid 2-related Factor 2 in Hypercholesterolemia-Induced Atherosclerosis. Free Radic. Biol. Med. 70, 174–181. doi:10.1016/j.freeradbiomed.2014.02.017
Cai, X., She, M., Xu, M., Chen, H., Li, J., Chen, X., et al. (2018). GLP-1 Treatment Protects Endothelial Cells from Oxidative Stress-Induced Autophagy and Endothelial Dysfunction. Int. J. Biol. Sci. 14 (12), 1696–1708. doi:10.7150/ijbs.27774
Cao, L., Wang, Y., Wang, Y., Lv, F., Liu, L., and Li, Z. (2021). Resolvin D2 Suppresses NLRP3 Inflammasome by Promoting Autophagy in Macrophages. Exp. Ther. Med. 22 (5), 1222. doi:10.3892/etm.2021.10656
Chang, W., Lin, J., Dong, J., and Li, D. (2013). Pyroptosis: an Inflammatory Cell Death Implicates in Atherosclerosis. Med. Hypotheses 81 (3), 484–486. doi:10.1016/j.mehy.2013.06.016
Chaulet, H., Desgranges, C., Renault, M. A., Dupuch, F., Ezan, G., Peiretti, F., et al. (2001). Extracellular Nucleotides Induce Arterial Smooth Muscle Cell Migration via Osteopontin. Circ. Res. 89 (9), 772–778. doi:10.1161/hh2101.098617
Chen, C. J., Huang, H. S., and Chang, W. C. (2003). Depletion of Phospholipid Hydroperoxide Glutathione Peroxidase Up-Regulates Arachidonate Metabolism by 12S-Lipoxygenase and Cyclooxygenase 1 in Human Epidermoid Carcinoma A431 Cells. FASEB J. 17 (12), 1694–1696. doi:10.1096/fj.02-0847fje
Chen, H., Lu, Y., Cao, Z., Ma, Q., Pi, H., Fang, Y., et al. (2016). Cadmium Induces NLRP3 Inflammasome-dependent Pyroptosis in Vascular Endothelial Cells. Toxicol. Lett. 246, 7–16. doi:10.1016/j.toxlet.2016.01.014
Chen, P. H., Wu, J., Ding, C. C., Lin, C. C., Pan, S., Bossa, N., et al. (2020). Kinome Screen of Ferroptosis Reveals a Novel Role of ATM in Regulating Iron Metabolism. Cell Death Differ 27 (3), 1008–1022. doi:10.1038/s41418-019-0393-7
Chen, P. S., Wang, K. C., Chao, T. H., Chung, H. C., Tseng, S. Y., Luo, C. Y., et al. (2017). Recombinant Thrombomodulin Exerts Anti-autophagic Action in Endothelial Cells and Provides Anti-atherosclerosis Effect in Apolipoprotein E Deficient Mice. Sci. Rep. 7 (1), 3284. doi:10.1038/s41598-017-03443-z
Chen, J., Zhang, J., Wu, J., Zhang, S., Liang, Y., Zhou, B., et al. (2021). Low Shear Stress Induced Vascular Endothelial Cell Pyroptosis by TET2/SDHB/ROS Pathway. Free Radic. Biol. Med. 162, 582–591. doi:10.1016/j.freeradbiomed.2020.11.017
Chen, Y., Zeng, A., He, S., He, S., Li, C., Mei, W., et al. (2021). Autophagy-Related Genes in Atherosclerosis. J. Healthc. Eng. 2021, 6402206. doi:10.1155/2021/6402206
Cheng, J., Qiao, L., Xu, X., Zhai, C., Zhao, K., Ji, X., et al. (2015). Lower AMP-Activated Protein Kinase Level Is Associated with the Vulnerability of Coronary Atherosclerotic Plaques by Attenuating the Expression of Monocyte Autophagy. Coron. Artery Dis. 26 (4), 322–327. doi:10.1097/MCA.0000000000000243
Cho, K., and Choi, S. H. (2019). ASK1 Mediates Apoptosis and Autophagy during oxLDL-CD36 Signaling in Senescent Endothelial Cells. Oxid Med. Cel Longev 2019, 2840437. doi:10.1155/2019/2840437
Clarke, M. C., Talib, S., Figg, N. L., and Bennett, M. R. (2010). Vascular Smooth Muscle Cell Apoptosis Induces Interleukin-1-Directed Inflammation: Effects of Hyperlipidemia-Mediated Inhibition of Phagocytosis. Circ. Res. 106 (2), 363–372. doi:10.1161/CIRCRESAHA.109.208389
Clement, M., Raffort, J., Lareyre, F., Tsiantoulas, D., Newland, S., Lu, Y., et al. (2019). Impaired Autophagy in CD11b(+) Dendritic Cells Expands CD4(+) Regulatory T Cells and Limits Atherosclerosis in Mice. Circ. Res. 125 (11), 1019–1034. doi:10.1161/CIRCRESAHA.119.315248
Cong, L., Gao, Z., Zheng, Y., Ye, T., Wang, Z., Wang, P., et al. (2020). Electrical Stimulation Inhibits Val-boroPro-Induced Pyroptosis in THP-1 Macrophages via Sirtuin3 Activation to Promote Autophagy and Inhibit ROS Generation. Aging (Albany NY) 12 (7), 6415–6435. doi:10.18632/aging.103038
Correa, R., Silva, L. F. F., Ribeiro, D. J. S., Almeida, R. D. N., Santos, I. O., Correa, L. H., et al. (2019). Lysophosphatidylcholine Induces NLRP3 Inflammasome-Mediated Foam Cell Formation and Pyroptosis in Human Monocytes and Endothelial Cells. Front. Immunol. 10, 2927. doi:10.3389/fimmu.2019.02927
Del Re, D. P., Amgalan, D., Linkermann, A., Liu, Q., and Kitsis, R. N. (2019). Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 99 (4), 1765–1817. doi:10.1152/physrev.00022.2018
Deng, Q., Wang, Y., Zhang, Y., Li, M., Li, D., Huang, X., et al. (2016). Pseudomonas aeruginosa Triggers Macrophage Autophagy to Escape Intracellular Killing by Activation of the NLRP3 Inflammasome. Infect. Immun. 84 (1), 56–66. doi:10.1128/IAI.00945-15
Denton, D., and Kumar, S. (2019). Autophagy-dependent Cell Death. Cel Death Differ 26 (4), 605–616. doi:10.1038/s41418-018-0252-y
Desideri, E., Filomeni, G., and Ciriolo, M. R. (2012). Glutathione Participates in the Modulation of Starvation-Induced Autophagy in Carcinoma Cells. Autophagy 8 (12), 1769–1781. doi:10.4161/auto.22037
Dinarello, C. A. (2011). A Clinical Perspective of IL-1beta as the Gatekeeper of Inflammation. Eur. J. Immunol. 41 (5), 1203–1217. doi:10.1002/eji.201141550
Ding, J., Wang, K., Liu, W., She, Y., Sun, Q., Shi, J., et al. (2016). Pore-forming Activity and Structural Autoinhibition of the Gasdermin Family. Nature 535 (7610), 111–116. doi:10.1038/nature18590
Ding, Z., Liu, S., Wang, X., Khaidakov, M., Dai, Y., and Mehta, J. L. (2013a). Oxidant Stress in Mitochondrial DNA Damage, Autophagy and Inflammation in Atherosclerosis. Sci. Rep. 3, 1077. doi:10.1038/srep01077
Ding, Z., Wang, X., Schnackenberg, L., Khaidakov, M., Liu, S., Singla, S., et al. (2013b). Regulation of Autophagy and Apoptosis in Response to Ox-LDL in Vascular Smooth Muscle Cells, and the Modulatory Effects of the microRNA Hsa-Let-7 G. Int. J. Cardiol. 168 (2), 1378–1385. doi:10.1016/j.ijcard.2012.12.045
Dixon, S. J., Lemberg, K. M., Lamprecht, M. R., Skouta, R., Zaitsev, E. M., Gleason, C. E., et al. (2012). Ferroptosis: an Iron-dependent Form of Nonapoptotic Cell Death. Cell 149 (5), 1060–1072. doi:10.1016/j.cell.2012.03.042
Dixon, S. J., and Stockwell, B. R. (2014). The Role of Iron and Reactive Oxygen Species in Cell Death. Nat. Chem. Biol. 10 (1), 9–17. doi:10.1038/nchembio.1416
Dixon, S. J., Winter, G. E., Musavi, L. S., Lee, E. D., Snijder, B., Rebsamen, M., et al. (2015). Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem. Biol. 10 (7), 1604–1609. doi:10.1021/acschembio.5b00245
Doll, S., Freitas, F. P., Shah, R., Aldrovandi, M., da Silva, M. C., Ingold, I., et al. (2019). FSP1 Is a Glutathione-independent Ferroptosis Suppressor. Nature 575 (7784), 693–698. doi:10.1038/s41586-019-1707-0
Doll, S., Proneth, B., Tyurina, Y. Y., Panzilius, E., Kobayashi, S., Ingold, I., et al. (2017). ACSL4 Dictates Ferroptosis Sensitivity by Shaping Cellular Lipid Composition. Nat. Chem. Biol. 13 (1), 91–98. doi:10.1038/nchembio.2239
Dolma, S., Lessnick, S. L., Hahn, W. C., and Stockwell, B. R. (2003). Identification of Genotype-Selective Antitumor Agents Using Synthetic Lethal Chemical Screening in Engineered Human Tumor Cells. Cancer Cell 3 (3), 285–296. doi:10.1016/s1535-6108(03)00050-3
Du, J., Wang, T., Li, Y., Zhou, Y., Wang, X., Yu, X., et al. (2019). DHA Inhibits Proliferation and Induces Ferroptosis of Leukemia Cells through Autophagy Dependent Degradation of Ferritin. Free Radic. Biol. Med. 131, 356–369. doi:10.1016/j.freeradbiomed.2018.12.011
Duewell, P., Kono, H., Rayner, K. J., Sirois, C. M., Vladimer, G., Bauernfeind, F. G., et al. (2010). NLRP3 Inflammasomes Are Required for Atherogenesis and Activated by Cholesterol Crystals. Nature 464 (7293), 1357–1361. doi:10.1038/nature08938
Dufrusine, B., Di Francesco, A., Oddi, S., Scipioni, L., Angelucci, C. B., D'Addario, C., et al. (2019). Iron-Dependent Trafficking of 5-Lipoxygenase and Impact on Human Macrophage Activation. Front. Immunol. 10, 1347. doi:10.3389/fimmu.2019.01347
Fan, Z., Wirth, A. K., Chen, D., Wruck, C. J., Rauh, M., Buchfelder, M., et al. (2017). Nrf2-Keap1 Pathway Promotes Cell Proliferation and Diminishes Ferroptosis. Oncogenesis 6 (8), e371. doi:10.1038/oncsis.2017.65
Fang, S., Wan, X., Zou, X., Sun, S., Hao, X., Liang, C., et al. (2021). Arsenic Trioxide Induces Macrophage Autophagy and Atheroprotection by Regulating ROS-dependent TFEB Nuclear Translocation and AKT/mTOR Pathway. Cell Death Dis 12 (1), 88. doi:10.1038/s41419-020-03357-1
Fang, X., Wang, H., Han, D., Xie, E., Yang, X., Wei, J., et al. (2019). Ferroptosis as a Target for protection against Cardiomyopathy. Proc. Natl. Acad. Sci. U S A. 116 (7), 2672–2680. doi:10.1073/pnas.1821022116
Fink, S. L., and Cookson, B. T. (2005). Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description of Dead and Dying Eukaryotic Cells. Infect. Immun. 73 (4), 1907–1916. doi:10.1128/IAI.73.4.1907-1916.2005
Fink, S. L., and Cookson, B. T. (2006). Caspase-1-dependent Pore Formation during Pyroptosis Leads to Osmotic Lysis of Infected Host Macrophages. Cell Microbiol 8 (11), 1812–1825. doi:10.1111/j.1462-5822.2006.00751.x
Fitzwalter, B. E., and Thorburn, A. (2015). Recent Insights into Cell Death and Autophagy. FEBS J. 282 (22), 4279–4288. doi:10.1111/febs.13515
Friedmann Angeli, J. P., Schneider, M., Proneth, B., Tyurina, Y. Y., Tyurin, V. A., Hammond, V. J., et al. (2014). Inactivation of the Ferroptosis Regulator Gpx4 Triggers Acute Renal Failure in Mice. Nat. Cel Biol 16 (12), 1180–1191. doi:10.1038/ncb3064
Gage, J., Hasu, M., Thabet, M., and Whitman, S. C. (2012). Caspase-1 Deficiency Decreases Atherosclerosis in Apolipoprotein E-Null Mice. Can. J. Cardiol. 28 (2), 222–229. doi:10.1016/j.cjca.2011.10.013
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cel Death Differ 25 (3), 486–541. doi:10.1038/s41418-017-0012-4
Gao, H., Bai, Y., Jia, Y., Zhao, Y., Kang, R., Tang, D., et al. (2018). Ferroptosis Is a Lysosomal Cell Death Process. Biochem. Biophys. Res. Commun. 503 (3), 1550–1556. doi:10.1016/j.bbrc.2018.07.078
Gao, M., Monian, P., Pan, Q., Zhang, W., Xiang, J., and Jiang, X. (2016). Ferroptosis Is an Autophagic Cell Death Process. Cell Res 26 (9), 1021–1032. doi:10.1038/cr.2016.95
Gao, M., Monian, P., Quadri, N., Ramasamy, R., and Jiang, X. (2015). Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cel 59 (2), 298–308. doi:10.1016/j.molcel.2015.06.011
Gao, C., Yan, Y., Chen, G., Wang, T., Luo, C., Zhang, M., et al. (2020). Autophagy Activation Represses Pyroptosis through the IL-13 and JAK1/STAT1 Pathways in a Mouse Model of Moderate Traumatic Brain Injury. ACS Chem. Neurosci. 11 (24), 4231–4239. doi:10.1021/acschemneuro.0c00517
Gao, Y., You, X., Liu, Y., Gao, F., Zhang, Y., Yang, J., et al. (2020). Induction of Autophagy Protects Human Dental Pulp Cells from Lipopolysaccharide-Induced Pyroptotic Cell Death. Exp. Ther. Med. 19 (3), 2202–2210. doi:10.3892/etm.2020.8475
Gaschler, M. M., Andia, A. A., Liu, H., Csuka, J. M., Hurlocker, B., Vaiana, C. A., et al. (2018). FINO2 Initiates Ferroptosis through GPX4 Inactivation and Iron Oxidation. Nat. Chem. Biol. 14 (5), 507–515. doi:10.1038/s41589-018-0031-6
Ge, P., Gao, M., Du, J., Yu, J., and Zhang, L. (2021). Downregulation of microRNA-512-3p Enhances the Viability and Suppresses the Apoptosis of Vascular Endothelial Cells, Alleviates Autophagy and Endoplasmic Reticulum Stress as Well as Represses Atherosclerotic Lesions in Atherosclerosis by Adjusting Spliced/unspliced Ratio of X-Box Binding Protein 1 (XBP-1S/XBP-1U). Bioengineered 12 (2), 12469–12481. doi:10.1080/21655979.2021.2006862
Gimbrone, M. A., and Garcia-Cardena, G. (2016). Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 118 (4), 620–636. doi:10.1161/CIRCRESAHA.115.306301
Gomez, D., Baylis, R. A., Durgin, B. G., Newman, A. A. C., Alencar, G. F., Mahan, S., et al. (2018). Interleukin-1beta Has Atheroprotective Effects in Advanced Atherosclerotic Lesions of Mice. Nat. Med. 24 (9), 1418–1429. doi:10.1038/s41591-018-0124-5
Gong, T., Liu, L., Jiang, W., and Zhou, R. (2020). DAMP-sensing Receptors in Sterile Inflammation and Inflammatory Diseases. Nat. Rev. Immunol. 20 (2), 95–112. doi:10.1038/s41577-019-0215-7
Grebe, A., Hoss, F., and Latz, E. (2018). NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 122 (12), 1722–1740. doi:10.1161/CIRCRESAHA.118.311362
Green, D. R., Galluzzi, L., and Kroemer, G. (2011). Mitochondria and the Autophagy-Inflammation-Cell Death axis in Organismal Aging. Science 333 (6046), 1109–1112. doi:10.1126/science.1201940
Grootaert, M. O., da Costa Martins, P. A., Bitsch, N., Pintelon, I., De Meyer, G. R., Martinet, W., et al. (2015). Defective Autophagy in Vascular Smooth Muscle Cells Accelerates Senescence and Promotes Neointima Formation and Atherogenesis. Autophagy 11 (11), 2014–2032. doi:10.1080/15548627.2015.1096485
Guo, M., Yan, R., Ji, Q., Yao, H., Sun, M., Duan, L., et al. (2020). IFN Regulatory Factor-1 Induced Macrophage Pyroptosis by Modulating m6A Modification of Circ_0029589 in Patients with Acute Coronary Syndrome. Int. Immunopharmacol 86, 106800. doi:10.1016/j.intimp.2020.106800
Guo, F. X., Wu, Q., Li, P., Zheng, L., Ye, S., Dai, X. Y., et al. (2019). The Role of the LncRNA-Fa2h-2-MLKL Pathway in Atherosclerosis by Regulation of Autophagy Flux and Inflammation through mTOR-dependent Signaling. Cel Death Differ 26 (9), 1670–1687. doi:10.1038/s41418-018-0235-z
Guo, M., Yan, R., Yao, H., Duan, L., Sun, M., Xue, Z., et al. (2019). IFN Regulatory Factor 1 Mediates Macrophage Pyroptosis Induced by Oxidized Low-Density Lipoprotein in Patients with Acute Coronary Syndrome. Mediators Inflamm. 2019, 2917128. doi:10.1155/2019/2917128
Guo, Z., Ran, Q., Roberts, L. J., Zhou, L., Richardson, A., Sharan, C., et al. (2008). Suppression of Atherogenesis by Overexpression of Glutathione Peroxidase-4 in Apolipoprotein E-Deficient Mice. Free Radic. Biol. Med. 44 (3), 343–352. doi:10.1016/j.freeradbiomed.2007.09.009
Habib, A., Polavarapu, R., Karmali, V., Guo, L., Van Dam, R., Cheng, Q., et al. (2015). Hepcidin-ferroportin axis Controls Toll-like Receptor 4 Dependent Macrophage Inflammatory Responses in Human Atherosclerotic Plaques. Atherosclerosis 241 (2), 692–700. doi:10.1016/j.atherosclerosis.2015.06.025
Han, H., Wang, H., Yin, Z., Jiang, H., Fang, M., and Han, J. (2013). Association of Genetic Polymorphisms in ADH and ALDH2 with Risk of Coronary Artery Disease and Myocardial Infarction: a Meta-Analysis. Gene 526 (2), 134–141. doi:10.1016/j.gene.2013.05.002
Hassannia, B., Wiernicki, B., Ingold, I., Qu, F., Van Herck, S., Tyurina, Y. Y., et al. (2018). Nano-targeted Induction of Dual Ferroptotic Mechanisms Eradicates High-Risk Neuroblastoma. J. Clin. Invest. 128 (8), 3341–3355. doi:10.1172/JCI99032
Hayano, M., Yang, W. S., Corn, C. K., Pagano, N. C., and Stockwell, B. R. (2016). Loss of Cysteinyl-tRNA Synthetase (CARS) Induces the Transsulfuration Pathway and Inhibits Ferroptosis Induced by Cystine Deprivation. Cel Death Differ 23 (2), 270–278. doi:10.1038/cdd.2015.93
He, J., Zhang, G., Pang, Q., Yu, C., Xiong, J., Zhu, J., et al. (2017). SIRT6 Reduces Macrophage Foam Cell Formation by Inducing Autophagy and Cholesterol Efflux under Ox-LDL Condition. FEBS J. 284 (9), 1324–1337. doi:10.1111/febs.14055
Hofmans, S., Vanden Berghe, T., Devisscher, L., Hassannia, B., Lyssens, S., Joossens, J., et al. (2016). Novel Ferroptosis Inhibitors with Improved Potency and ADME Properties. J. Med. Chem. 59 (5), 2041–2053. doi:10.1021/acs.jmedchem.5b01641
Hoseini, Z., Sepahvand, F., Rashidi, B., Sahebkar, A., Masoudifar, A., and Mirzaei, H. (2018). NLRP3 Inflammasome: Its Regulation and Involvement in Atherosclerosis. J. Cel Physiol 233 (3), 2116–2132. doi:10.1002/jcp.25930
Hou, W., Xie, Y., Song, X., Sun, X., Lotze, M. T., Zeh, H. J., et al. (2016). Autophagy Promotes Ferroptosis by Degradation of Ferritin. Autophagy 12 (8), 1425–1428. doi:10.1080/15548627.2016.1187366
Hu, Q., Zhang, T., Yi, L., Zhou, X., and Mi, M. (2018). Dihydromyricetin Inhibits NLRP3 Inflammasome-dependent Pyroptosis by Activating the Nrf2 Signaling Pathway in Vascular Endothelial Cells. Biofactors 44 (2), 123–136. doi:10.1002/biof.1395
Hui, B., Hou, X., Liu, R., Liu, X. H., and Hu, Z. (2021). Gypenoside Inhibits Ox-LDL Uptake and Foam Cell Formation through Enhancing Sirt1-FOXO1 Mediated Autophagy Flux Restoration. Life Sci. 264, 118721. doi:10.1016/j.lfs.2020.118721
Jia, G., Cheng, G., Gangahar, D. M., and Agrawal, D. K. (2006). Insulin-like Growth Factor-1 and TNF-Alpha Regulate Autophagy through C-Jun N-Terminal Kinase and Akt Pathways in Human Atherosclerotic Vascular Smooth Cells. Immunol. Cel Biol 84 (5), 448–454. doi:10.1111/j.1440-1711.2006.01454.x
Jiang, C., Jiang, L., Li, Q., Liu, X., Zhang, T., Dong, L., et al. (2018). Acrolein Induces NLRP3 Inflammasome-Mediated Pyroptosis and Suppresses Migration via ROS-dependent Autophagy in Vascular Endothelial Cells. Toxicology 410, 26–40. doi:10.1016/j.tox.2018.09.002
Jiang, L., Kon, N., Li, T., Wang, S. J., Su, T., Hibshoosh, H., et al. (2015). Ferroptosis as a P53-Mediated Activity during Tumour Suppression. Nature 520 (7545), 57–62. doi:10.1038/nature14344
Jiang, L., Luo, S., Qiu, T., Li, Q., Jiang, C., Sun, X., et al. (2021). Bidirectional Role of Reactive Oxygen Species during Inflammasome Activation in Acrolein-Induced Human EAhy926 Cells Pyroptosis. Toxicol. Mech. Methods 31 (9), 680–689. doi:10.1080/15376516.2021.1953204
Jiang, M., Sun, X., Liu, S., Tang, Y., Shi, Y., Bai, Y., et al. (2021). Caspase-11-Gasdermin D-Mediated Pyroptosis Is Involved in the Pathogenesis of Atherosclerosis. Front. Pharmacol. 12, 657486. doi:10.3389/fphar.2021.657486
Jin, H., Zhu, Y., Wang, X. D., Luo, E. F., Li, Y. P., Wang, B. L., et al. (2021). BDNF Corrects NLRP3 Inflammasome-Induced Pyroptosis and Glucose Metabolism Reprogramming through KLF2/HK1 Pathway in Vascular Endothelial Cells. Cell Signal 78, 109843. doi:10.1016/j.cellsig.2020.109843
Jung, C. H., Jun, C. B., Ro, S. H., Kim, Y. M., Otto, N. M., Cao, J., et al. (2009). ULK-Atg13-FIP200 Complexes Mediate mTOR Signaling to the Autophagy Machinery. Mol. Biol. Cel 20 (7), 1992–2003. doi:10.1091/mbc.E08-12-1249
Kamili, A., Roslan, N., Frost, S., Cantrill, L. C., Wang, D., Della-Franca, A., et al. (2015). TPD52 Expression Increases Neutral Lipid Storage within Cultured Cells. J. Cel Sci 128 (17), 3223–3238. doi:10.1242/jcs.167692
Kang, R., Zeh, H. J., Lotze, M. T., and Tang, D. (2011). The Beclin 1 Network Regulates Autophagy and Apoptosis. Cel Death Differ 18 (4), 571–580. doi:10.1038/cdd.2010.191
Kang, R., Zeng, L., Zhu, S., Xie, Y., Liu, J., Wen, Q., et al. (2018). Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host Microbe 24 (1), 97–108. e104. doi:10.1016/j.chom.2018.05.009
Karasawa, T., and Takahashi, M. (2017). Role of NLRP3 Inflammasomes in Atherosclerosis. J. Atheroscler. Thromb. 24 (5), 443–451. doi:10.5551/jat.RV17001
Katikaneni, A., Jelcic, M., Gerlach, G. F., Ma, Y., Overholtzer, M., and Niethammer, P. (2020). Lipid Peroxidation Regulates Long-Range Wound Detection through 5-lipoxygenase in Zebrafish. Nat. Cel Biol 22 (9), 1049–1055. doi:10.1038/s41556-020-0564-2
Katsnelson, M. A., Lozada-Soto, K. M., Russo, H. M., Miller, B. A., and Dubyak, G. R. (2016). NLRP3 Inflammasome Signaling Is Activated by Low-Level Lysosome Disruption but Inhibited by Extensive Lysosome Disruption: Roles for K+ Efflux and Ca2+ Influx. Am. J. Physiol. Cel Physiol 311 (1), C83–C100. doi:10.1152/ajpcell.00298.2015
Katz, S. S., Shipley, G. G., and Small, D. M. (1976). Physical Chemistry of the Lipids of Human Atherosclerotic Lesions. Demonstration of a Lesion Intermediate between Fatty Streaks and Advanced Plaques. J. Clin. Invest. 58 (1), 200–211. doi:10.1172/JCI108450
Kayagaki, N., Stowe, I. B., Lee, B. L., O'Rourke, K., Anderson, K., Warming, S., et al. (2015). Caspase-11 Cleaves Gasdermin D for Non-canonical Inflammasome Signalling. Nature 526 (7575), 666–671. doi:10.1038/nature15541
Kayagaki, N., Warming, S., Lamkanfi, M., Vande Walle, L., Louie, S., Dong, J., et al. (2011). Non-canonical Inflammasome Activation Targets Caspase-11. Nature 479 (7371), 117–121. doi:10.1038/nature10558
Kerr, J. F., Wyllie, A. H., and Currie, A. R. (1972). Apoptosis: a Basic Biological Phenomenon with Wide-Ranging Implications in Tissue Kinetics. Br. J. Cancer 26 (4), 239–257. doi:10.1038/bjc.1972.33
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR Regulate Autophagy through Direct Phosphorylation of Ulk1. Nat. Cel Biol 13 (2), 132–141. doi:10.1038/ncb2152
Kim, S. E., Zhang, L., Ma, K., Riegman, M., Chen, F., Ingold, I., et al. (2016). Ultrasmall Nanoparticles Induce Ferroptosis in Nutrient-Deprived Cancer Cells and Suppress Tumour Growth. Nat. Nanotechnol 11 (11), 977–985. doi:10.1038/nnano.2016.164
Kim, S., Lee, W., and Cho, K. (2021). P62 Links the Autophagy Pathway and the Ubiquitin-Proteasome System in Endothelial Cells during Atherosclerosis. Int. J. Mol. Sci. 22 (15), 7791. doi:10.3390/ijms22157791
Kimura, T., Jia, J., Kumar, S., Choi, S. W., Gu, Y., Mudd, M., et al. (2017). Dedicated SNAREs and Specialized TRIM Cargo Receptors Mediate Secretory Autophagy. EMBO J. 36 (1), 42–60. doi:10.15252/embj.201695081
Kirii, H., Niwa, T., Yamada, Y., Wada, H., Saito, K., Iwakura, Y., et al. (2003). Lack of Interleukin-1beta Decreases the Severity of Atherosclerosis in ApoE-Deficient Mice. Arterioscler Thromb. Vasc. Biol. 23 (4), 656–660. doi:10.1161/01.ATV.0000064374.15232.C3
Klionsky, D. J., Abdelmohsen, K., Abe, A., Abedin, M. J., Abeliovich, H., Acevedo Arozena, A., et al. (2016). Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (3rd Edition). Autophagy 12 (1), 1–222. doi:10.1080/15548627.2015.1100356
Klionsky, D. J., and Emr, S. D. (2000). Autophagy as a Regulated Pathway of Cellular Degradation. Science 290 (5497), 1717–1721. doi:10.1126/science.290.5497.1717
Kolodgie, F. D., Narula, J., Burke, A. P., Haider, N., Farb, A., Hui-Liang, Y., et al. (2000). Localization of Apoptotic Macrophages at the Site of Plaque Rupture in Sudden Coronary Death. Am. J. Pathol. 157 (4), 1259–1268. doi:10.1016/S0002-9440(10)64641-X
Kopin, L., and Lowenstein, C. (2017). Dyslipidemia. Ann. Intern. Med. 167 (11), ITC81–ITC96. doi:10.7326/AITC201712050
Kraft, V. A. N., Bezjian, C. T., Pfeiffer, S., Ringelstetter, L., Muller, C., Zandkarimi, F., et al. (2020). GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent. Sci. 6 (1), 41–53. doi:10.1021/acscentsci.9b01063
Kroemer, G., Marino, G., and Levine, B. (2010). Autophagy and the Integrated Stress Response. Mol. Cel 40 (2), 280–293. doi:10.1016/j.molcel.2010.09.023
Kumar, S., Nanduri, R., Bhagyaraj, E., Kalra, R., Ahuja, N., Chacko, A. P., et al. (2021). Vitamin D3-VDR-PTPN6 axis Mediated Autophagy Contributes to the Inhibition of Macrophage Foam Cell Formation. Autophagy 17 (9), 2273–2289. doi:10.1080/15548627.2020.1822088
Kurz, T., and Brunk, U. T. (2009). Autophagy of HSP70 and Chelation of Lysosomal Iron in a Non-redox-active Form. Autophagy 5 (1), 93–95. doi:10.4161/auto.5.1.7248
Lai, M., Yao, H., Shah, S. Z. A., Wu, W., Wang, D., Zhao, Y., et al. (2018). The NLRP3-Caspase 1 Inflammasome Negatively Regulates Autophagy via TLR4-TRIF in Prion Peptide-Infected Microglia. Front. Aging Neurosci. 10, 116. doi:10.3389/fnagi.2018.00116
LaRocca, T. J., Henson, G. D., Thorburn, A., Sindler, A. L., Pierce, G. L., and Seals, D. R. (2012). Translational Evidence that Impaired Autophagy Contributes to Arterial Ageing. J. Physiol. 590 (14), 3305–3316. doi:10.1113/jphysiol.2012.229690
Li, C., Chen, J. W., Liu, Z. H., Shen, Y., Ding, F. H., Gu, G., et al. (2018). CTRP5 Promotes Transcytosis and Oxidative Modification of Low-Density Lipoprotein and the Development of Atherosclerosis. Atherosclerosis 278, 197–209. doi:10.1016/j.atherosclerosis.2018.09.037
Li, C., Deng, X., Xie, X., Liu, Y., Friedmann Angeli, J. P., and Lai, L. (2018). Activation of Glutathione Peroxidase 4 as a Novel Anti-inflammatory Strategy. Front. Pharmacol. 9, 1120. doi:10.3389/fphar.2018.01120
Li, P., Bai, Y., Zhao, X., Tian, T., Tang, L., Ru, J., et al. (2018). NR4A1 Contributes to High-Fat Associated Endothelial Dysfunction by Promoting CaMKII-Parkin-Mitophagy Pathways. Cell Stress Chaperones 23 (4), 749–761. doi:10.1007/s12192-018-0886-1
Li, Q., Gao, S., Kang, Z., Zhang, M., Zhao, X., Zhai, Y., et al. (2018). Rapamycin Enhances Mitophagy and Attenuates Apoptosis after Spinal Ischemia-Reperfusion Injury. Front. Neurosci. 12, 865. doi:10.3389/fnins.2018.00865
Li, H., Huang, S., Wang, S., Zhao, J., Su, L., Zhao, B., et al. (2013). Targeting Annexin A7 by a Small Molecule Suppressed the Activity of Phosphatidylcholine-specific Phospholipase C in Vascular Endothelial Cells and Inhibited Atherosclerosis in Apolipoprotein E(-)/(-)mice. Cel Death Dis 4, e806. doi:10.1038/cddis.2013.317
Li, H., Li, J., Li, Y., Singh, P., Cao, L., Xu, L. J., et al. (2012). Sonic Hedgehog Promotes Autophagy of Vascular Smooth Muscle Cells. Am. J. Physiol. Heart Circ. Physiol. 303 (11), H1319–H1331. doi:10.1152/ajpheart.00160.2012
Li, L., Tan, J., Miao, Y., Lei, P., and Zhang, Q. (2015). ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cell Mol Neurobiol 35 (5), 615–621. doi:10.1007/s10571-015-0166-x
Li, M. Y., Zhu, X. L., Zhao, B. X., Shi, L., Wang, W., Hu, W., et al. (2019). Adrenomedullin Alleviates the Pyroptosis of Leydig Cells by Promoting Autophagy via the ROS-AMPK-mTOR axis. Cel Death Dis 10 (7), 489. doi:10.1038/s41419-019-1728-5
Li, S., Zheng, L., Zhang, J., Liu, X., and Wu, Z. (2021). Inhibition of Ferroptosis by Up-Regulating Nrf2 Delayed the Progression of Diabetic Nephropathy. Free Radic. Biol. Med. 162, 435–449. doi:10.1016/j.freeradbiomed.2020.10.323
Li, W., Sultana, N., Siraj, N., Ward, L. J., Pawlik, M., Levy, E., et al. (2016). Autophagy Dysfunction and Regulatory Cystatin C in Macrophage Death of Atherosclerosis. J. Cel Mol Med 20 (9), 1664–1672. doi:10.1111/jcmm.12859
Li, W., Xu, L. H., Forssell, C., Sullivan, J. L., and Yuan, X. M. (2008). Overexpression of Transferrin Receptor and Ferritin Related to Clinical Symptoms and Destabilization of Human Carotid Plaques. Exp. Biol. Med. (Maywood) 233 (7), 818–826. doi:10.3181/0711-RM-320
Li, Y., Niu, X., Xu, H., Li, Q., Meng, L., He, M., et al. (2020). VX-765 Attenuates Atherosclerosis in ApoE Deficient Mice by Modulating VSMCs Pyroptosis. Exp. Cel Res 389 (1), 111847. doi:10.1016/j.yexcr.2020.111847
Libby, P., Ridker, P. M., and Hansson, G. K. (2011). Progress and Challenges in Translating the Biology of Atherosclerosis. Nature 473 (7347), 317–325. doi:10.1038/nature10146
Lin, J., Shou, X., Mao, X., Dong, J., Mohabeer, N., Kushwaha, K. K., et al. (2013). Oxidized Low Density Lipoprotein Induced Caspase-1 Mediated Pyroptotic Cell Death in Macrophages: Implication in Lesion Instability. PLoS One 8 (4), e62148. doi:10.1371/journal.pone.0062148
Lin, Q., Li, S., Jiang, N., Shao, X., Zhang, M., Jin, H., et al. (2019). PINK1-parkin Pathway of Mitophagy Protects against Contrast-Induced Acute Kidney Injury via Decreasing Mitochondrial ROS and NLRP3 Inflammasome Activation. Redox Biol. 26, 101254. doi:10.1016/j.redox.2019.101254
Lin, Y., Liu, M., Chen, E., Jiang, W., Shi, W., and Wang, Z. (2021). Bone Marrow-Derived Mesenchymal Stem Cells Microvesicles Stabilize Atherosclerotic Plaques by Inhibiting NLRP3-Mediated Macrophage Pyroptosis. Cell Biol Int 45 (4), 820–830. doi:10.1002/cbin.11526
Liu, G., Deng, J., Zhang, Q., Song, W., Chen, S., Lou, X., et al. (2016). Porphyromonas Gingivalis Lipopolysaccharide Stimulation of Vascular Smooth Muscle Cells Activates Proliferation and Calcification. J. Periodontol. 87 (7), 828–836. doi:10.1902/jop.2016.150602
Liu, X., Zhang, Z., Ruan, J., Pan, Y., Magupalli, V. G., Wu, H., et al. (2016). Inflammasome-activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature 535 (7610), 153–158. doi:10.1038/nature18629
Liu, J., Wang, C., Li, J., Yu, Y., Liu, Y., Liu, H., et al. (2021). Autophagy Blockage Promotes the Pyroptosis of Ox-LDL-Treated Macrophages by Modulating the p62/Nrf2/ARE axis. J. Physiol. Biochem. 77 (3), 419–429. doi:10.1007/s13105-021-00811-2
Liu, J., Wang, Y., Liao, Y., Zhou, Y., and Zhu, J. (2021). Circular RNA PPP1CC Promotes Porphyromonas Gingivalis-Lipopolysaccharide-Induced Pyroptosis of Vascular Smooth Muscle Cells by Activating the HMGB1/TLR9/AIM2 Pathway. J. Int. Med. Res. 49 (3), 300060521996564. doi:10.1177/0300060521996564
Liu, S., Jiang, X., Cui, X., Wang, J., Liu, S., Li, H., et al. (2021). Smooth Muscle-specific HuR Knockout Induces Defective Autophagy and Atherosclerosis. Cel Death Dis 12 (4), 385. doi:10.1038/s41419-021-03671-2
Lockshin, R. A., and Zakeri, Z. (2004). Apoptosis, Autophagy, and More. Int. J. Biochem. Cel Biol 36 (12), 2405–2419. doi:10.1016/j.biocel.2004.04.011
Long, Y., Liu, X., Tan, X. Z., Jiang, C. X., Chen, S. W., Liang, G. N., et al. (2020). ROS-induced NLRP3 Inflammasome Priming and Activation Mediate PCB 118- Induced Pyroptosis in Endothelial Cells. Ecotoxicol Environ. Saf. 189, 109937. doi:10.1016/j.ecoenv.2019.109937
Lopez-Pastrana, J., Ferrer, L. M., Li, Y. F., Xiong, X., Xi, H., Cueto, R., et al. (2015). Inhibition of Caspase-1 Activation in Endothelial Cells Improves Angiogenesis: A Novel Therapeutic Potential for Ischemia. J. Biol. Chem. 290 (28), 17485–17494. doi:10.1074/jbc.M115.641191
Lu, H., Fan, Y., Qiao, C., Liang, W., Hu, W., Zhu, T., et al. (2017). TFEB Inhibits Endothelial Cell Inflammation and Reduces Atherosclerosis. Sci. Signal. 10 (464), eaah4214. doi:10.1126/scisignal.aah4214
Lu, L. Q., Tian, J., Luo, X. J., and Peng, J. (2021). Targeting the Pathways of Regulated Necrosis: a Potential Strategy for Alleviation of Cardio-Cerebrovascular Injury. Cell Mol Life Sci 78 (1), 63–78. doi:10.1007/s00018-020-03587-8
Luo, Y., Lu, S., Gao, Y., Yang, K., Wu, D., Xu, X., et al. (2020). Araloside C Attenuates Atherosclerosis by Modulating Macrophage Polarization via Sirt1-Mediated Autophagy. Aging (Albany NY) 12 (2), 1704–1724. doi:10.18632/aging.102708
Lupfer, C. R., Anand, P. K., Liu, Z., Stokes, K. L., Vogel, P., Lamkanfi, M., et al. (2014). Reactive Oxygen Species Regulate Caspase-11 Expression and Activation of the Non-canonical NLRP3 Inflammasome during Enteric Pathogen Infection. Plos Pathog. 10 (9), e1004410. doi:10.1371/journal.ppat.1004410
Lutgens, E., de Muinck, E. D., Kitslaar, P. J., Tordoir, J. H., Wellens, H. J., and Daemen, M. J. (1999). Biphasic Pattern of Cell Turnover Characterizes the Progression from Fatty Streaks to Ruptured Human Atherosclerotic Plaques. Cardiovasc. Res. 41 (2), 473–479. doi:10.1016/s0008-6363(98)00311-3
Ma, S., Chen, J., Feng, J., Zhang, R., Fan, M., Han, D., et al. (2018). Melatonin Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome Inhibition. Oxid Med. Cel Longev 2018, 9286458. doi:10.1155/2018/9286458
Mak, T. W., Grusdat, M., Duncan, G. S., Dostert, C., Nonnenmacher, Y., Cox, M., et al. (2017). Glutathione Primes T Cell Metabolism for Inflammation. Immunity 46 (4), 675–689. doi:10.1016/j.immuni.2017.03.019
Man, S. M., Karki, R., and Kanneganti, T. D. (2017). Molecular Mechanisms and Functions of Pyroptosis, Inflammatory Caspases and Inflammasomes in Infectious Diseases. Immunol. Rev. 277 (1), 61–75. doi:10.1111/imr.12534
Mancias, J. D., Wang, X., Gygi, S. P., Harper, J. W., and Kimmelman, A. C. (2014). Quantitative Proteomics Identifies NCOA4 as the Cargo Receptor Mediating Ferritinophagy. Nature 509 (7498), 105–109. doi:10.1038/nature13148
Mao, C., Li, D., Zhou, E., Zhang, J., Wang, C., and Xue, C. (2021). Nicotine Exacerbates Atherosclerosis through a Macrophage-Mediated Endothelial Injury Pathway. Aging (Albany NY) 13 (5), 7627–7643. doi:10.18632/aging.202660
Marchio, P., Guerra-Ojeda, S., Vila, J. M., Aldasoro, M., Victor, V. M., and Mauricio, M. D. (2019). Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid Med. Cel Longev 2019, 8563845. doi:10.1155/2019/8563845
Martinet, W., Coornaert, I., Puylaert, P., and De Meyer, G. R. Y. (2019). Macrophage Death as a Pharmacological Target in Atherosclerosis. Front. Pharmacol. 10, 306. doi:10.3389/fphar.2019.00306
Martinet, W., Schrijvers, D. M., Timmermans, J. P., and Bult, H. (2008). Interactions between Cell Death Induced by Statins and 7-ketocholesterol in Rabbit Aorta Smooth Muscle Cells. Br. J. Pharmacol. 154 (6), 1236–1246. doi:10.1038/bjp.2008.181
Martinon, F., Burns, K., and Tschopp, J. (2002). The Inflammasome: a Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-Beta. Mol. Cel 10 (2), 417–426. doi:10.1016/s1097-2765(02)00599-3
Masaldan, S., Clatworthy, S. A. S., Gamell, C., Meggyesy, P. M., Rigopoulos, A. T., Haupt, S., et al. (2018). Iron Accumulation in Senescent Cells Is Coupled with Impaired Ferritinophagy and Inhibition of Ferroptosis. Redox Biol. 14, 100–115. doi:10.1016/j.redox.2017.08.015
Mathur, A., Hayward, J. A., and Man, S. M. (2018). Molecular Mechanisms of Inflammasome Signaling. J. Leukoc. Biol. 103 (2), 233–257. doi:10.1189/jlb.3MR0617-250R
McKnight, N. C., and Zhenyu, Y. (2013). Beclin 1, an Essential Component and Master Regulator of PI3K-III in Health and Disease. Curr. Pathobiol Rep. 1 (4), 231–238. doi:10.1007/s40139-013-0028-5
Meng, Q., Li, Y., Ji, T., Chao, Y., Li, J., Fu, Y., et al. (2021). Estrogen Prevent Atherosclerosis by Attenuating Endothelial Cell Pyroptosis via Activation of Estrogen Receptor Alpha-Mediated Autophagy. J. Adv. Res. 28, 149–164. doi:10.1016/j.jare.2020.08.010
Meng, Q., Pu, L., Lu, Q., Wang, B., Li, S., Liu, B., et al. (2021). Morin Hydrate Inhibits Atherosclerosis and LPS-Induced Endothelial Cells Inflammatory Responses by Modulating the NFkappaB Signaling-Mediated Autophagy. Int. Immunopharmacol 100, 108096. doi:10.1016/j.intimp.2021.108096
Meng, Z., Liang, H., Zhao, J., Gao, J., Liu, C., Ma, X., et al. (2021). HMOX1 Upregulation Promotes Ferroptosis in Diabetic Atherosclerosis. Life Sci. 284, 119935. doi:10.1016/j.lfs.2021.119935
Menghini, R., Casagrande, V., Marino, A., Marchetti, V., Cardellini, M., Stoehr, R., et al. (2014). MiR-216a: a Link between Endothelial Dysfunction and Autophagy. Cel Death Dis 5, e1029. doi:10.1038/cddis.2013.556
Merched, A. J., Williams, E., and Chan, L. (2003). Macrophage-specific P53 Expression Plays a Crucial Role in Atherosclerosis Development and Plaque Remodeling. Arterioscler Thromb. Vasc. Biol. 23 (9), 1608–1614. doi:10.1161/01.ATV.0000084825.88022.53
Miao, E. A., Rajan, J. V., and Aderem, A. (2011). Caspase-1-induced Pyroptotic Cell Death. Immunol. Rev. 243 (1), 206–214. doi:10.1111/j.1600-065X.2011.01044.x
Michiels, C. F., Kurdi, A., Timmermans, J. P., De Meyer, G. R. Y., and Martinet, W. (2016). Spermidine Reduces Lipid Accumulation and Necrotic Core Formation in Atherosclerotic Plaques via Induction of Autophagy. Atherosclerosis 251, 319–327. doi:10.1016/j.atherosclerosis.2016.07.899
Mitter, S. K., Song, C., Qi, X., Mao, H., Rao, H., Akin, D., et al. (2014). Dysregulated Autophagy in the RPE Is Associated with Increased Susceptibility to Oxidative Stress and AMD. Autophagy 10 (11), 1989–2005. doi:10.4161/auto.36184
Mizushima, N., and Komatsu, M. (2011). Autophagy: Renovation of Cells and Tissues. Cell 147 (4), 728–741. doi:10.1016/j.cell.2011.10.026
Mollace, V., Gliozzi, M., Musolino, V., Carresi, C., Muscoli, S., Mollace, R., et al. (2015). Oxidized LDL Attenuates Protective Autophagy and Induces Apoptotic Cell Death of Endothelial Cells: Role of Oxidative Stress and LOX-1 Receptor Expression. Int. J. Cardiol. 184, 152–158. doi:10.1016/j.ijcard.2015.02.007
Moore, K. J., Sheedy, F. J., and Fisher, E. A. (2013). Macrophages in Atherosclerosis: a Dynamic Balance. Nat. Rev. Immunol. 13 (10), 709–721. doi:10.1038/nri3520
Muller, T., Dewitz, C., Schmitz, J., Schroder, A. S., Brasen, J. H., Stockwell, B. R., et al. (2017). Necroptosis and Ferroptosis Are Alternative Cell Death Pathways that Operate in Acute Kidney Failure. Cel Mol Life Sci 74 (19), 3631–3645. doi:10.1007/s00018-017-2547-4
Naghavi, M., Libby, P., Falk, E., Casscells, S. W., Litovsky, S., Rumberger, J., et al. (2003). From Vulnerable Plaque to Vulnerable Patient: a Call for New Definitions and Risk Assessment Strategies: Part II. Circulation 108 (15), 1772–1778. doi:10.1161/01.CIR.0000087481.55887.C9
Nakahira, K., Haspel, J. A., Rathinam, V. A., Lee, S. J., Dolinay, T., Lam, H. C., et al. (2011). Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the NALP3 Inflammasome. Nat. Immunol. 12 (3), 222–230. doi:10.1038/ni.1980
Nemeth, E., Tuttle, M. S., Powelson, J., Vaughn, M. B., Donovan, A., Ward, D. M., et al. (2004). Hepcidin Regulates Cellular Iron Efflux by Binding to Ferroportin and Inducing its Internalization. Science 306 (5704), 2090–2093. doi:10.1126/science.1104742
Ni, H., Xu, S., Chen, H., and Dai, Q. (2020). Nicotine Modulates CTSS (Cathepsin S) Synthesis and Secretion through Regulating the Autophagy-Lysosomal Machinery in Atherosclerosis. Arterioscler Thromb. Vasc. Biol. 40 (9), 2054–2069. doi:10.1161/ATVBAHA.120.314053
Ohsumi, Y. (2001). Molecular Dissection of Autophagy: Two Ubiquitin-like Systems. Nat. Rev. Mol. Cel Biol 2 (3), 211–216. doi:10.1038/35056522
Orning, P., Weng, D., Starheim, K., Ratner, D., Best, Z., Lee, B., et al. (2018). Pathogen Blockade of TAK1 Triggers Caspase-8-dependent Cleavage of Gasdermin D and Cell Death. Science 362 (6418), 1064–1069. doi:10.1126/science.aau2818
Osonoi, Y., Mita, T., Azuma, K., Nakajima, K., Masuyama, A., Goto, H., et al. (2018). Defective Autophagy in Vascular Smooth Muscle Cells Enhances Cell Death and Atherosclerosis. Autophagy 14 (11), 1991–2006. doi:10.1080/15548627.2018.1501132
Ouimet, M., Ediriweera, H., Afonso, M. S., Ramkhelawon, B., Singaravelu, R., Liao, X., et al. (2017). microRNA-33 Regulates Macrophage Autophagy in Atherosclerosis. Arterioscler Thromb. Vasc. Biol. 37 (6), 1058–1067. doi:10.1161/ATVBAHA.116.308916
Ozaki, E., Campbell, M., and Doyle, S. L. (2015). Targeting the NLRP3 Inflammasome in Chronic Inflammatory Diseases: Current Perspectives. J. Inflamm. Res. 8, 15–27. doi:10.2147/JIR.S51250
Pan, H., and Reilly, M. P. (2019). A Protective Smooth Muscle Cell Transition in Atherosclerosis. Nat. Med. 25 (8), 1194–1195. doi:10.1038/s41591-019-0541-0
Pan, J., Han, L., Guo, J., Wang, X., Liu, D., Tian, J., et al. (2018). AIM2 Accelerates the Atherosclerotic Plaque Progressions in ApoE-/- Mice. Biochem. Biophys. Res. Commun. 498 (3), 487–494. doi:10.1016/j.bbrc.2018.03.005
Pankratz, F., Hohnloser, C., Bemtgen, X., Jaenich, C., Kreuzaler, S., Hoefer, I., et al. (2018). MicroRNA-100 Suppresses Chronic Vascular Inflammation by Stimulation of Endothelial Autophagy. Circ. Res. 122 (3), 417–432. doi:10.1161/CIRCRESAHA.117.311428
Paramel Varghese, G., Folkersen, L., Strawbridge, R. J., Halvorsen, B., Yndestad, A., Ranheim, T., et al. (2016). NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J. Am. Heart Assoc. 5 (5), e003031. doi:10.1161/JAHA.115.003031
Park, E., and Chung, S. W. (2019). ROS-mediated Autophagy Increases Intracellular Iron Levels and Ferroptosis by Ferritin and Transferrin Receptor Regulation. Cel Death Dis 10 (11), 822. doi:10.1038/s41419-019-2064-5
Parzych, K. R., and Klionsky, D. J. (2014). An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 20 (3), 460–473. doi:10.1089/ars.2013.5371
Pavlides, S., Gutierrez-Pajares, J. L., Iturrieta, J., Lisanti, M. P., and Frank, P. G. (2014). Endothelial Caveolin-1 Plays a Major Role in the Development of Atherosclerosis. Cell Tissue Res 356 (1), 147–157. doi:10.1007/s00441-013-1767-7
Peng, S., Xu, L. W., Che, X. Y., Xiao, Q. Q., Pu, J., Shao, Q., et al. (2018). Atorvastatin Inhibits Inflammatory Response, Attenuates Lipid Deposition, and Improves the Stability of Vulnerable Atherosclerotic Plaques by Modulating Autophagy. Front. Pharmacol. 9, 438. doi:10.3389/fphar.2018.00438
Peng, X., Chen, H., Li, Y., Huang, D., Huang, B., and Sun, D. (2020). Effects of NIX-Mediated Mitophagy on Ox-LDL-Induced Macrophage Pyroptosis in Atherosclerosis. Cel Biol Int 44 (7), 1481–1490. doi:10.1002/cbin.11343
Perrotta, P., Van der Veken, B., Van Der Veken, P., Pintelon, I., Roosens, L., Adriaenssens, E., et al. (2020). Partial Inhibition of Glycolysis Reduces Atherogenesis Independent of Intraplaque Neovascularization in Mice. Arterioscler Thromb. Vasc. Biol. 40 (5), 1168–1181. doi:10.1161/ATVBAHA.119.313692
Pi, S., Mao, L., Chen, J., Shi, H., Liu, Y., Guo, X., et al. (2021). The P2RY12 Receptor Promotes VSMC-Derived Foam Cell Formation by Inhibiting Autophagy in Advanced Atherosclerosis. Autophagy 17 (4), 980–1000. doi:10.1080/15548627.2020.1741202
Potteaux, S., Gautier, E. L., Hutchison, S. B., van Rooijen, N., Rader, D. J., Thomas, M. J., et al. (2011). Suppressed Monocyte Recruitment Drives Macrophage Removal from Atherosclerotic Plaques of Apoe-/- Mice during Disease Regression. J. Clin. Invest. 121 (5), 2025–2036. doi:10.1172/JCI43802
Prats-Puig, A., Moreno, M., Carreras-Badosa, G., Bassols, J., Ricart, W., Lopez-Bermejo, A., et al. (2016). Serum Ferritin Relates to Carotid Intima-Media Thickness in Offspring of Fathers with Higher Serum Ferritin Levels. Arterioscler Thromb. Vasc. Biol. 36 (1), 174–180. doi:10.1161/ATVBAHA.115.306396
Qiao, L., Ma, J., Zhang, Z., Sui, W., Zhai, C., Xu, D., et al. (2021). Deficient Chaperone-Mediated Autophagy Promotes Inflammation and Atherosclerosis. Circ. Res. 129 (12), 1141–1157. doi:10.1161/CIRCRESAHA.121.318908
Qin, X., Zhang, J., Wang, B., Xu, G., Yang, X., Zou, Z., et al. (2021). Ferritinophagy Is Involved in the Zinc Oxide Nanoparticles-Induced Ferroptosis of Vascular Endothelial Cells. Autophagy 17 (12), 4266–4285. doi:10.1080/15548627.2021.1911016
Qiu, Z., Lei, S., Zhao, B., Wu, Y., Su, W., Liu, M., et al. (2017). NLRP3 Inflammasome Activation-Mediated Pyroptosis Aggravates Myocardial Ischemia/Reperfusion Injury in Diabetic Rats. Oxid Med. Cel Longev 2017, 9743280. doi:10.1155/2017/9743280
Rajamaki, K., Lappalainen, J., Oorni, K., Valimaki, E., Matikainen, S., Kovanen, P. T., et al. (2010). Cholesterol Crystals Activate the NLRP3 Inflammasome in Human Macrophages: a Novel Link between Cholesterol Metabolism and Inflammation. PLoS One 5 (7), e11765. doi:10.1371/journal.pone.0011765
Ramirez, C. M., Zhang, X., Bandyopadhyay, C., Rotllan, N., Sugiyama, M. G., Aryal, B., et al. (2019). Caveolin-1 Regulates Atherogenesis by Attenuating Low-Density Lipoprotein Transcytosis and Vascular Inflammation Independently of Endothelial Nitric Oxide Synthase Activation. Circulation 140 (3), 225–239. doi:10.1161/CIRCULATIONAHA.118.038571
Ridker, P. M., Everett, B. M., Thuren, T., MacFadyen, J. G., Chang, W. H., Ballantyne, C., et al. (2017). Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 377 (12), 1119–1131. doi:10.1056/NEJMoa1707914
Riegman, M., Sagie, L., Galed, C., Levin, T., Steinberg, N., Dixon, S. J., et al. (2020). Ferroptosis Occurs through an Osmotic Mechanism and Propagates Independently of Cell Rupture. Nat. Cel Biol 22 (9), 1042–1048. doi:10.1038/s41556-020-0565-1
Rong, J. X., Shapiro, M., Trogan, E., and Fisher, E. A. (2003). Transdifferentiation of Mouse Aortic Smooth Muscle Cells to a Macrophage-like State after Cholesterol Loading. Proc. Natl. Acad. Sci. U S A. 100 (23), 13531–13536. doi:10.1073/pnas.1735526100
Runas, K. A., Acharya, S. J., Schmidt, J. J., and Malmstadt, N. (2016). Addition of Cleaved Tail Fragments during Lipid Oxidation Stabilizes Membrane Permeability Behavior. Langmuir 32 (3), 779–786. doi:10.1021/acs.langmuir.5b02980
Saitoh, T., Fujita, N., Jang, M. H., Uematsu, S., Yang, B. G., Satoh, T., et al. (2008). Loss of the Autophagy Protein Atg16L1 Enhances Endotoxin-Induced IL-1beta Production. Nature 456 (7219), 264–268. doi:10.1038/nature07383
Salabei, J. K., Cummins, T. D., Singh, M., Jones, S. P., Bhatnagar, A., and Hill, B. G. (2013). PDGF-mediated Autophagy Regulates Vascular Smooth Muscle Cell Phenotype and Resistance to Oxidative Stress. Biochem. J. 451 (3), 375–388. doi:10.1042/BJ20121344
Salabei, J. K., and Hill, B. G. (2013). Implications of Autophagy for Vascular Smooth Muscle Cell Function and Plasticity. Free Radic. Biol. Med. 65, 693–703. doi:10.1016/j.freeradbiomed.2013.08.003
Saliba-Gustafsson, P., Pedrelli, M., Gertow, K., Werngren, O., Janas, V., Pourteymour, S., et al. (2019). Subclinical Atherosclerosis and its Progression Are Modulated by PLIN2 through a Feed-Forward Loop between LXR and Autophagy. J. Intern. Med. 286 (6), 660–675. doi:10.1111/joim.12951
Santovito, D., Egea, V., Bidzhekov, K., Natarelli, L., Mourao, A., Blanchet, X., et al. (2020). Noncanonical Inhibition of Caspase-3 by a Nuclear microRNA Confers Endothelial protection by Autophagy in Atherosclerosis. Sci. Transl Med. 12 (546), eaaz2294. doi:10.1126/scitranslmed.aaz2294
Schroder, K., and Tschopp, J. (2010). The Inflammasomes. Cell 140 (6), 821–832. doi:10.1016/j.cell.2010.01.040
Schroeder, B., Schulze, R. J., Weller, S. G., Sletten, A. C., Casey, C. A., and McNiven, M. A. (2015). The Small GTPase Rab7 as a central Regulator of Hepatocellular Lipophagy. Hepatology 61 (6), 1896–1907. doi:10.1002/hep.27667
Seiler, A., Schneider, M., Forster, H., Roth, S., Wirth, E. K., Culmsee, C., et al. (2008). Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab 8 (3), 237–248. doi:10.1016/j.cmet.2008.07.005
Sergin, I., Evans, T. D., Zhang, X., Bhattacharya, S., Stokes, C. J., Song, E., et al. (2017). Exploiting Macrophage Autophagy-Lysosomal Biogenesis as a Therapy for Atherosclerosis. Nat. Commun. 8, 15750. doi:10.1038/ncomms15750
Shao, B. Z., Han, B. Z., Zeng, Y. X., Su, D. F., and Liu, C. (2016). The Roles of Macrophage Autophagy in Atherosclerosis. Acta Pharmacol. Sin 37 (2), 150–156. doi:10.1038/aps.2015.87
Shen, Y., Liu, W. W., Zhang, X., Shi, J. G., Jiang, S., Zheng, L., et al. (2020). TRAF3 Promotes ROS Production and Pyroptosis by Targeting ULK1 Ubiquitination in Macrophages. FASEB J. 34 (5), 7144–7159. doi:10.1096/fj.201903073R
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 526 (7575), 660–665. doi:10.1038/nature15514
Shi, X., Xie, W. L., Kong, W. W., Chen, D., and Qu, P. (2015). Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis. J. Stroke Cerebrovasc. Dis. 24 (11), 2455–2466. doi:10.1016/j.jstrokecerebrovasdis.2015.03.024
Shimada, K., Crother, T. R., Karlin, J., Dagvadorj, J., Chiba, N., Chen, S., et al. (2012). Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 36 (3), 401–414. doi:10.1016/j.immuni.2012.01.009
Shimada, K., Skouta, R., Kaplan, A., Yang, W. S., Hayano, M., Dixon, S. J., et al. (2016). Global Survey of Cell Death Mechanisms Reveals Metabolic Regulation of Ferroptosis. Nat. Chem. Biol. 12 (7), 497–503. doi:10.1038/nchembio.2079
Sollberger, G., Strittmatter, G. E., Grossi, S., Garstkiewicz, M., Auf Dem Keller, U., French, L. E., et al. (2015). Caspase-1 Activity Is Required for UVB-Induced Apoptosis of Human Keratinocytes. J. Invest. Dermatol. 135 (5), 1395–1404. doi:10.1038/jid.2014.551
Son, S. J., Rhee, K. J., Lim, J., Kim, T. U., Kim, T. J., and Kim, Y. S. (2013). Triglyceride-induced Macrophage Cell Death Is Triggered by Caspase-1. Biol. Pharm. Bull. 36 (1), 108–113. doi:10.1248/bpb.b12-00571
Song, X., Zhu, S., Chen, P., Hou, W., Wen, Q., Liu, J., et al. (2018). AMPK-mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc(-) Activity. Curr. Biol. 28 (15), 2388–2399 e2385. doi:10.1016/j.cub.2018.05.094
Su, G., Yang, W., Wang, S., Geng, C., and Guan, X. (2021). SIRT1-autophagy axis Inhibits Excess Iron-Induced Ferroptosis of Foam Cells and Subsequently Increases IL-1Beta and IL-18. Biochem. Biophys. Res. Commun. 561, 33–39. doi:10.1016/j.bbrc.2021.05.011
Sui, M., Xu, D., Zhao, W., Lu, H., Chen, R., Duan, Y., et al. (2021). CIRBP Promotes Ferroptosis by Interacting with ELAVL1 and Activating Ferritinophagy during Renal Ischaemia-Reperfusion Injury. J. Cel Mol Med 25 (13), 6203–6216. doi:10.1111/jcmm.16567
Sui, S., Zhang, J., Xu, S., Wang, Q., Wang, P., and Pang, D. (2019). Ferritinophagy Is Required for the Induction of Ferroptosis by the Bromodomain Protein BRD4 Inhibitor (+)-JQ1 in Cancer Cells. Cel Death Dis 10 (5), 331. doi:10.1038/s41419-019-1564-7
Sun, H., Krauss, R. M., Chang, J. T., and Teng, B. B. (2018). PCSK9 Deficiency Reduces Atherosclerosis, Apolipoprotein B Secretion, and Endothelial Dysfunction. J. Lipid Res. 59 (2), 207–223. doi:10.1194/jlr.M078360
Sun, R. Z., Fan, Y., Liang, X., Gong, T. T., Wang, Q., Liu, H., et al. (2018). Rapamycin and FTY720 Alleviate Atherosclerosis by Cross Talk of Macrophage Polarization and Autophagy. Biomed. Res. Int. 2018, 1010248. doi:10.1155/2018/1010248
Sun, Y., Zheng, Y., Wang, C., and Liu, Y. (2018). Glutathione Depletion Induces Ferroptosis, Autophagy, and Premature Cell Senescence in Retinal Pigment Epithelial Cells. Cel Death Dis 9 (7), 753. doi:10.1038/s41419-018-0794-4
Sung, K. C., Kang, S. M., Cho, E. J., Park, J. B., Wild, S. H., and Byrne, C. D. (2012). Ferritin Is Independently Associated with the Presence of Coronary Artery Calcium in 12,033 Men. Arterioscler Thromb. Vasc. Biol. 32 (10), 2525–2530. doi:10.1161/ATVBAHA.112.253088
Swaminathan, B., Goikuria, H., Vega, R., Rodriguez-Antiguedad, A., Lopez Medina, A., Freijo Mdel, M., et al. (2014). Autophagic Marker MAP1LC3B Expression Levels Are Associated with Carotid Atherosclerosis Symptomatology. PLoS One 9 (12), e115176. doi:10.1371/journal.pone.0115176
Swanson, K. V., Deng, M., and Ting, J. P. (2019). The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 19 (8), 477–489. doi:10.1038/s41577-019-0165-0
Swiader, A., Nahapetyan, H., Faccini, J., D'Angelo, R., Mucher, E., Elbaz, M., et al. (2016). Mitophagy Acts as a Safeguard Mechanism against Human Vascular Smooth Muscle Cell Apoptosis Induced by Atherogenic Lipids. Oncotarget 7 (20), 28821–28835. doi:10.18632/oncotarget.8936
Szumiel, I. (2011). Autophagy, Reactive Oxygen Species and the Fate of Mammalian Cells. Free Radic. Res. 45 (3), 253–265. doi:10.3109/10715762.2010.525233
Tabas, I., Williams, K. J., and Boren, J. (2007). Subendothelial Lipoprotein Retention as the Initiating Process in Atherosclerosis: Update and Therapeutic Implications. Circulation 116 (16), 1832–1844. doi:10.1161/CIRCULATIONAHA.106.676890
Tai, S., Hu, X. Q., Peng, D. Q., Zhou, S. H., and Zheng, X. L. (2016). The Roles of Autophagy in Vascular Smooth Muscle Cells. Int. J. Cardiol. 211, 1–6. doi:10.1016/j.ijcard.2016.02.128
Tang, D., Kang, R., Berghe, T. V., Vandenabeele, P., and Kroemer, G. (2019). The Molecular Machinery of Regulated Cell Death. Cel Res 29 (5), 347–364. doi:10.1038/s41422-019-0164-5
Tang, M., Chen, Z., Wu, D., and Chen, L. (2018). Ferritinophagy/ferroptosis: Iron-Related Newcomers in Human Diseases. J. Cel Physiol 233 (12), 9179–9190. doi:10.1002/jcp.26954
Tanida, I., Ueno, T., and Kominami, E. (2004). LC3 Conjugation System in Mammalian Autophagy. Int. J. Biochem. Cel Biol 36 (12), 2503–2518. doi:10.1016/j.biocel.2004.05.009
Tao, H., Yancey, P. G., Blakemore, J. L., Zhang, Y., Ding, L., Jerome, W. G., et al. (2021). Macrophage SR-BI Modulates Autophagy via VPS34 Complex and PPARalpha Transcription of Tfeb in Atherosclerosis. J. Clin. Invest. 131 (7), e94229. doi:10.1172/JCI94229
Tavakoli Dargani, Z., Singla, R., Johnson, T., Kukreja, R., and Singla, D. K. (2018). Exosomes Derived from Embryonic Stem Cells Inhibit Doxorubicin and Inflammation-Induced Pyroptosis in Muscle Cells. Can. J. Physiol. Pharmacol. 96 (3), 304–307. doi:10.1139/cjpp-2017-0340
Thi, E. P., and Reiner, N. E. (2012). Phosphatidylinositol 3-kinases and Their Roles in Phagosome Maturation. J. Leukoc. Biol. 92 (3), 553–566. doi:10.1189/jlb.0212053
Torii, S., Shintoku, R., Kubota, C., Yaegashi, M., Torii, R., Sasaki, M., et al. (2016). An Essential Role for Functional Lysosomes in Ferroptosis of Cancer Cells. Biochem. J. 473 (6), 769–777. doi:10.1042/BJ20150658
Torisu, K., Singh, K. K., Torisu, T., Lovren, F., Liu, J., Pan, Y., et al. (2016). Intact Endothelial Autophagy Is Required to Maintain Vascular Lipid Homeostasis. Aging Cell 15 (1), 187–191. doi:10.1111/acel.12423
Tyrrell, D. J., Blin, M. G., Song, J., Wood, S. C., Zhang, M., Beard, D. A., et al. (2020). Age-Associated Mitochondrial Dysfunction Accelerates Atherogenesis. Circ. Res. 126 (3), 298–314. doi:10.1161/CIRCRESAHA.119.315644
Vinchi, F., Porto, G., Simmelbauer, A., Altamura, S., Passos, S. T., Garbowski, M., et al. (2020). Atherosclerosis Is Aggravated by Iron Overload and Ameliorated by Dietary and Pharmacological Iron Restriction. Eur. Heart J. 41 (28), 2681–2695. doi:10.1093/eurheartj/ehz112
Vinokur, V., Weksler-Zangen, S., Berenshtein, E., Eliashar, R., and Chevion, M. (2016). The Loss of Myocardial Benefit Following Ischemic Preconditioning Is Associated with Dysregulation of Iron Homeostasis in Diet-Induced Diabetes. PLoS One 11 (7), e0159908. doi:10.1371/journal.pone.0159908
Vion, A. C., Kheloufi, M., Hammoutene, A., Poisson, J., Lasselin, J., Devue, C., et al. (2017). Autophagy Is Required for Endothelial Cell Alignment and Atheroprotection under Physiological Blood Flow. Proc. Natl. Acad. Sci. U S A. 114 (41), E8675–E8684. doi:10.1073/pnas.1702223114
Vromman, A., Ruvkun, V., Shvartz, E., Wojtkiewicz, G., Santos Masson, G., Tesmenitsky, Y., et al. (2019). Stage-dependent Differential Effects of Interleukin-1 Isoforms on Experimental Atherosclerosis. Eur. Heart J. 40 (30), 2482–2491. doi:10.1093/eurheartj/ehz008
Wang, J., Wang, W. N., Xu, S. B., Wu, H., Dai, B., Jian, D. D., et al. (2018). MicroRNA-214-3p: A Link between Autophagy and Endothelial Cell Dysfunction in Atherosclerosis. Acta Physiol. (Oxf) 222 (3), e12973. doi:10.1111/apha.12973
Wang, C., Xu, W., Liang, M., Huang, D., and Huang, K. (2019). CTRP13 Inhibits Atherosclerosis via Autophagy-lysosome-dependent Degradation of CD36. FASEB J. 33 (2), 2290–2300. doi:10.1096/fj.201801267RR
Wang, S., Ji, L. Y., Li, L., and Li, J. M. (2019). Oxidative Stress, Autophagy and Pyroptosis in the Neovascularization of Oxygeninduced Retinopathy in Mice. Mol. Med. Rep. 19 (2), 927–934. doi:10.3892/mmr.2018.9759
Wang, X., Jiang, L., Shi, L., Yao, K., Sun, X., Yang, G., et al. (2019). Zearalenone Induces NLRP3-dependent Pyroptosis via Activation of NF-kappaB Modulated by Autophagy in INS-1 Cells. Toxicology 428, 152304. doi:10.1016/j.tox.2019.152304
Wang, Y., Che, J., Zhao, H., Tang, J., and Shi, G. (2019). Paeoniflorin Attenuates Oxidized Low-Density Lipoprotein-Induced Apoptosis and Adhesion Molecule Expression by Autophagy Enhancement in Human Umbilical Vein Endothelial Cells. J. Cel Biochem 120 (6), 9291–9299. doi:10.1002/jcb.28204
Wang, Y., Dubland, J. A., Allahverdian, S., Asonye, E., Sahin, B., Jaw, J. E., et al. (2019). Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler Thromb. Vasc. Biol. 39 (5), 876–887. doi:10.1161/ATVBAHA.119.312434
Wang, Z., Liu, B., Zhu, J., Wang, D., and Wang, Y. (2019). Nicotine-mediated Autophagy of Vascular Smooth Muscle Cell Accelerates Atherosclerosis via nAChRs/ROS/NF-kappaB Signaling Pathway. Atherosclerosis 284, 1–10. doi:10.1016/j.atherosclerosis.2019.02.008
Wang, C., Yang, W., Liang, X., Song, W., Lin, J., Sun, Y., et al. (2020). MicroRNA-761 Modulates Foam Cell Formation and Inflammation through Autophagy in the Progression of Atherosclerosis. Mol. Cel Biochem 474 (1-2), 135–146. doi:10.1007/s11010-020-03839-y
Wang, L., Liu, Y., Du, T., Yang, H., Lei, L., Guo, M., et al. (2020). ATF3 Promotes Erastin-Induced Ferroptosis by Suppressing System Xc−. Cel Death Differ 27 (2), 662–675. doi:10.1038/s41418-019-0380-z
Wang, M., Liu, C. Y., Wang, T., Yu, H. M., Ouyang, S. H., Wu, Y. P., et al. (2020). (+)-Clausenamide Protects against Drug-Induced Liver Injury by Inhibiting Hepatocyte Ferroptosis. Cel Death Dis 11 (9), 781. doi:10.1038/s41419-020-02961-5
Wang, T., Sun, C., Hu, L., Gao, E., Li, C., Wang, H., et al. (2020). Sirt6 Stabilizes Atherosclerosis Plaques by Promoting Macrophage Autophagy and Reducing Contact with Endothelial Cells. Biochem. Cel Biol 98 (2), 120–129. doi:10.1139/bcb-2019-0057
Wang, X., Li, H., Li, W., Xie, J., Wang, F., Peng, X., et al. (2020). The Role of Caspase-1/gsdmd-Mediated Pyroptosis in Taxol-Induced Cell Death and a Taxol-Resistant Phenotype in Nasopharyngeal Carcinoma Regulated by Autophagy. Cell Biol Toxicol 36 (5), 437–457. doi:10.1007/s10565-020-09514-8
Wang, Y., Song, X., Li, Z., Liu, N., Yan, Y., Li, T., et al. (2020). MicroRNA-103 Protects Coronary Artery Endothelial Cells against H2O2-Induced Oxidative Stress via BNIP3-Mediated End-Stage Autophagy and Antipyroptosis Pathways. Oxid Med. Cel Longev 2020, 8351342. doi:10.1155/2020/8351342
Wang, T., Zhang, L., Hu, J., Duan, Y., Zhang, M., Lin, J., et al. (2016). Mst1 Participates in the Atherosclerosis Progression through Macrophage Autophagy Inhibition and Macrophage Apoptosis Enhancement. J. Mol. Cel Cardiol 98, 108–116. doi:10.1016/j.yjmcc.2016.08.002
Wang, Y., Gao, W., Shi, X., Ding, J., Liu, W., He, H., et al. (2017). Chemotherapy Drugs Induce Pyroptosis through Caspase-3 Cleavage of a Gasdermin. Nature 547 (7661), 99–103. doi:10.1038/nature22393
Wei, D. H., Jia, X. Y., Liu, Y. H., Guo, F. X., Tang, Z. H., Li, X. H., et al. (2013). Cathepsin L Stimulates Autophagy and Inhibits Apoptosis of Ox-LDL-Induced Endothelial Cells: Potential Role in Atherosclerosis. Int. J. Mol. Med. 31 (2), 400–406. doi:10.3892/ijmm.2012.1201
Wei, Q., Zhu, R., Zhu, J., Zhao, R., and Li, M. (2019). E2-Induced Activation of the NLRP3 Inflammasome Triggers Pyroptosis and Inhibits Autophagy in HCC Cells. Oncol. Res. 27 (7), 827–834. doi:10.3727/096504018X15462920753012
Weiwer, M., Bittker, J. A., Lewis, T. A., Shimada, K., Yang, W. S., MacPherson, L., et al. (2012). Development of Small-Molecule Probes that Selectively Kill Cells Induced to Express Mutant RAS. Bioorg. Med. Chem. Lett. 22 (4), 1822–1826. doi:10.1016/j.bmcl.2011.09.047
Wu, X., Zhang, H., Qi, W., Zhang, Y., Li, J., Li, Z., et al. (2018). Nicotine Promotes Atherosclerosis via ROS-NLRP3-Mediated Endothelial Cell Pyroptosis. Cel Death Dis 9 (2), 171. doi:10.1038/s41419-017-0257-3
Wu, Q., Hu, Y., Jiang, M., Wang, F., and Gong, G. (2019). Effect of Autophagy Regulated by Sirt1/FoxO1 Pathway on the Release of Factors Promoting Thrombosis from Vascular Endothelial Cells. Int. J. Mol. Sci. 20 (17), 4132. doi:10.3390/ijms20174132
Wu, Z., Geng, Y., Lu, X., Shi, Y., Wu, G., Zhang, M., et al. (2019). Chaperone-mediated Autophagy Is Involved in the Execution of Ferroptosis. Proc. Natl. Acad. Sci. U S A. 116 (8), 2996–3005. doi:10.1073/pnas.1819728116
Wu, L. M., Wu, S. G., Chen, F., Wu, Q., Wu, C. M., Kang, C. M., et al. (2020). Atorvastatin Inhibits Pyroptosis through the lncRNA NEXN-AS1/NEXN Pathway in Human Vascular Endothelial Cells. Atherosclerosis 293, 26–34. doi:10.1016/j.atherosclerosis.2019.11.033
Wu, P., Chen, J., Chen, J., Tao, J., Wu, S., Xu, G., et al. (2020). Trimethylamine N-Oxide Promotes apoE(-/-) Mice Atherosclerosis by Inducing Vascular Endothelial Cell Pyroptosis via the SDHB/ROS Pathway. J. Cel Physiol 235 (10), 6582–6591. doi:10.1002/jcp.29518
Wu, Q., He, X., Wu, L. M., Zhang, R. Y., Li, L. M., Wu, C. M., et al. (2020). MLKL Aggravates Ox-LDL-Induced Cell Pyroptosis via Activation of NLRP3 Inflammasome in Human Umbilical Vein Endothelial Cells. Inflammation 43 (6), 2222–2231. doi:10.1007/s10753-020-01289-8
Xi, H., Zhang, Y., Xu, Y., Yang, W. Y., Jiang, X., Sha, X., et al. (2016). Caspase-1 Inflammasome Activation Mediates Homocysteine-Induced Pyrop-Apoptosis in Endothelial Cells. Circ. Res. 118 (10), 1525–1539. doi:10.1161/CIRCRESAHA.116.308501
Xiao, F. J., Zhang, D., Wu, Y., Jia, Q. H., Zhang, L., Li, Y. X., et al. (2019). miRNA-17-92 Protects Endothelial Cells from Erastin-Induced Ferroptosis through Targeting the A20-ACSL4 axis. Biochem. Biophys. Res. Commun. 515 (3), 448–454. doi:10.1016/j.bbrc.2019.05.147
Xie, Y., Kang, R., Sun, X., Zhong, M., Huang, J., Klionsky, D. J., et al. (2015). Posttranslational Modification of Autophagy-Related Proteins in Macroautophagy. Autophagy 11 (1), 28–45. doi:10.4161/15548627.2014.984267
Xu, X., Yang, Y., Wang, G., Yin, Y., Han, S., Zheng, D., et al. (2021). Low Shear Stress Regulates Vascular Endothelial Cell Pyroptosis through miR-181b-5p/STAT-3 axis. J. Cel Physiol 236 (1), 318–327. doi:10.1002/jcp.29844
Xu, Y., Yuan, Q., Cao, S., Cui, S., Xue, L., Song, X., et al. (2020). Aldehyde Dehydrogenase 2 Inhibited Oxidized LDL-Induced NLRP3 Inflammasome Priming and Activation via Attenuating Oxidative Stress. Biochem. Biophys. Res. Commun. 529 (4), 998–1004. doi:10.1016/j.bbrc.2020.06.075
Xue, Y., Enosi Tuipulotu, D., Tan, W. H., Kay, C., and Man, S. M. (2019). Emerging Activators and Regulators of Inflammasomes and Pyroptosis. Trends Immunol. 40 (11), 1035–1052. doi:10.1016/j.it.2019.09.005
Yan, Z. Q., and Hansson, G. K. (2007). Innate Immunity, Macrophage Activation, and Atherosclerosis. Immunol. Rev. 219, 187–203. doi:10.1111/j.1600-065X.2007.00554.x
Yang, K., Song, H., and Yin, D. (2021). PDSS2 Inhibits the Ferroptosis of Vascular Endothelial Cells in Atherosclerosis by Activating Nrf2. J. Cardiovasc. Pharmacol. 77 (6), 767–776. doi:10.1097/FJC.0000000000001030
Yang, M., Chen, P., Liu, J., Zhu, S., Kroemer, G., Klionsky, D. J., et al. (2019). Clockophagy Is a Novel Selective Autophagy Process Favoring Ferroptosis. Sci. Adv. 5 (7), eaaw2238. doi:10.1126/sciadv.aaw2238
Yang, M., Lv, H., Liu, Q., Zhang, L., Zhang, R., Huang, X., et al. (2020). Colchicine Alleviates Cholesterol Crystal-Induced Endothelial Cell Pyroptosis through Activating AMPK/SIRT1 Pathway. Oxid Med. Cel Longev 2020, 9173530. doi:10.1155/2020/9173530
Yang, W. S., Kim, K. J., Gaschler, M. M., Patel, M., Shchepinov, M. S., and Stockwell, B. R. (2016). Peroxidation of Polyunsaturated Fatty Acids by Lipoxygenases Drives Ferroptosis. Proc. Natl. Acad. Sci. U S A. 113 (34), E4966–E4975. doi:10.1073/pnas.1603244113
Yang, W. S., SriRamaratnam, R., Welsch, M. E., Shimada, K., Skouta, R., Viswanathan, V. S., et al. (2014). Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 156 (1-2), 317–331. doi:10.1016/j.cell.2013.12.010
Yang, W. S., and Stockwell, B. R. (2008). Synthetic Lethal Screening Identifies Compounds Activating Iron-dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 15 (3), 234–245. doi:10.1016/j.chembiol.2008.02.010
Yin, Y., Li, X., Sha, X., Xi, H., Li, Y. F., Shao, Y., et al. (2015). Early Hyperlipidemia Promotes Endothelial Activation via a Caspase-1-Sirtuin 1 Pathway. Arterioscler Thromb. Vasc. Biol. 35 (4), 804–816. doi:10.1161/ATVBAHA.115.305282
Yu, E. P. K., Reinhold, J., Yu, H., Starks, L., Uryga, A. K., Foote, K., et al. (2017). Mitochondrial Respiration Is Reduced in Atherosclerosis, Promoting Necrotic Core Formation and Reducing Relative Fibrous Cap Thickness. Arterioscler Thromb. Vasc. Biol. 37 (12), 2322–2332. doi:10.1161/ATVBAHA.117.310042
Yu, Y., Yan, Y., Niu, F., Wang, Y., Chen, X., Su, G., et al. (2021). Ferroptosis: a Cell Death Connecting Oxidative Stress, Inflammation and Cardiovascular Diseases. Cell Death Discov 7 (1), 193. doi:10.1038/s41420-021-00579-w
Yuan, P., Hu, Q., He, X., Long, Y., Song, X., Wu, F., et al. (2020). Laminar Flow Inhibits the Hippo/YAP Pathway via Autophagy and SIRT1-Mediated Deacetylation against Atherosclerosis. Cel Death Dis 11 (2), 141. doi:10.1038/s41419-020-2343-1
Zhang, X., Ramirez, C. M., Aryal, B., Madrigal-Matute, J., Liu, X., Diaz, A., et al. (2020). Cav-1 (Caveolin-1) Deficiency Increases Autophagy in the Endothelium and Attenuates Vascular Inflammation and Atherosclerosis. Arterioscler Thromb. Vasc. Biol. 40 (6), 1510–1522. doi:10.1161/ATVBAHA.120.314291
Zhang, Z., Guo, M., Li, Y., Shen, M., Kong, D., Shao, J., et al. (2020). RNA-binding Protein ZFP36/TTP Protects against Ferroptosis by Regulating Autophagy Signaling Pathway in Hepatic Stellate Cells. Autophagy 16 (8), 1482–1505. doi:10.1080/15548627.2019.1687985
Zhaolin, Z., Jiaojiao, C., Peng, W., Yami, L., Tingting, Z., Jun, T., et al. (2019). OxLDL Induces Vascular Endothelial Cell Pyroptosis through miR-125a-5p/TET2 Pathway. J. Cel Physiol 234 (5), 7475–7491. doi:10.1002/jcp.27509
Zheng, F., Xing, S., Gong, Z., Mu, W., and Xing, Q. (2014). Silence of NLRP3 Suppresses Atherosclerosis and Stabilizes Plaques in Apolipoprotein E-Deficient Mice. Mediators Inflamm. 2014, 507208. doi:10.1155/2014/507208
Zhong, S., Li, L., Zhang, Y. L., Zhang, L., Lu, J., Guo, S., et al. (2019). Acetaldehyde Dehydrogenase 2 Interactions with LDLR and AMPK Regulate Foam Cell Formation. J. Clin. Invest. 129 (1), 252–267. doi:10.1172/JCI122064
Zhong, Z., Liang, S., Sanchez-Lopez, E., He, F., Shalapour, S., Lin, X. J., et al. (2018). New Mitochondrial DNA Synthesis Enables NLRP3 Inflammasome Activation. Nature 560 (7717), 198–203. doi:10.1038/s41586-018-0372-z
Zhong, Z., Umemura, A., Sanchez-Lopez, E., Liang, S., Shalapour, S., Wong, J., et al. (2016). NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 164 (5), 896–910. doi:10.1016/j.cell.2015.12.057
Zhou, R., Tardivel, A., Thorens, B., Choi, I., and Tschopp, J. (2010). Thioredoxin-interacting Protein Links Oxidative Stress to Inflammasome Activation. Nat. Immunol. 11 (2), 136–140. doi:10.1038/ni.1831
Zhou, R., Yazdi, A. S., Menu, P., and Tschopp, J. (2011). A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 469 (7329), 221–225. doi:10.1038/nature09663
Zhou, Y., Zhou, H., Hua, L., Hou, C., Jia, Q., Chen, J., et al. (2021). Verification of Ferroptosis and Pyroptosis and Identification of PTGS2 as the Hub Gene in Human Coronary Artery Atherosclerosis. Free Radic. Biol. Med. 171, 55–68. doi:10.1016/j.freeradbiomed.2021.05.009
Zhu, L., Wu, G., Yang, X., Jia, X., Li, J., Bai, X., et al. (2019). Low Density Lipoprotein Mimics Insulin Action on Autophagy and Glucose Uptake in Endothelial Cells. Sci. Rep. 9 (1), 3020. doi:10.1038/s41598-019-39559-7
Zhu, S., Zhang, Q., Sun, X., Zeh, H. J., Lotze, M. T., Kang, R., et al. (2017). HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 77 (8), 2064–2077. doi:10.1158/0008-5472.CAN-16-1979
Zi, Y., Yi-An, Y., Bing, J., Yan, L., Jing, T., Chun-Yu, G., et al. (2019). Sirt6-induced Autophagy Restricted TREM-1-Mediated Pyroptosis in Ox-LDL-Treated Endothelial Cells: Relevance to Prognostication of Patients with Acute Myocardial Infarction. Cel Death Discov 5, 88. doi:10.1038/s41420-019-0168-4
Keywords: atherosclerosis, cell death, autophagy, pyroptosis, ferroptosis
Citation: Lin L, Zhang M-X, Zhang L, Zhang D, Li C and Li Y-l (2022) Autophagy, Pyroptosis, and Ferroptosis: New Regulatory Mechanisms for Atherosclerosis. Front. Cell Dev. Biol. 9:809955. doi: 10.3389/fcell.2021.809955
Received: 05 November 2021; Accepted: 27 December 2021;
Published: 13 January 2022.
Edited by:
Zhi Qi, Nankai University, ChinaReviewed by:
Xianwei Wang, Xinxiang Medical University, ChinaAleksandr E. Vendrov, University of Michigan, United States
W. Gray Jerome, Vanderbilt University Medical Center, United States
Tatjana Radosavljević, University of Belgrade, Serbia
Copyright © 2022 Lin, Zhang, Zhang, Zhang, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yun-lun Li, eXVubHVuLmxlZUBob3RtYWlsLmNvbQ==; Dan Zhang, emhhbmdkYW5zZHRjbUBhbGl5dW4uY29t; Chao Li, bGljaGFvNzE3OTVAaG90bWFpbC5jb20=