Abstract
Metals are key elements for the survival and normal development of humans but can also be toxic to cells when mishandled. In fact, even mild disruption of metal homeostasis causes a wide array of disorders. Many of the metals essential to normal physiology are required in mitochondria for enzymatic activities and for the formation of essential cofactors. Copper is required as a cofactor in the terminal electron transport chain complex cytochrome c oxidase, iron is required for the for the formation of iron-sulfur (Fe-S) clusters and heme, manganese is required for the prevention of oxidative stress production, and these are only a few examples of the critical roles that mitochondrial metals play. Even though the targets of these metals are known, we are still identifying transporters, investigating the roles of known transporters, and defining regulators of the transport process. Mitochondria are dynamic organelles whose content, structure and localization within the cell vary in different tissues and organisms. Our knowledge of the impact that alterations in mitochondrial physiology have on metal content and utilization in these organelles is very limited. The rates of fission and fusion, the ultrastructure of the organelle, and rates of mitophagy can all affect metal homeostasis and cofactor assembly. This review will focus of the emerging areas of overlap between metal homeostasis, cofactor assembly and the mitochondrial contact site and cristae organizing system (MICOS) that mediates multiple aspects of mitochondrial physiology. Importantly the MICOS complexes may allow for localization and organization of complexes not only involved in cristae formation and contact between the inner and outer mitochondrial membranes but also acts as hub for metal-related proteins to work in concert in cofactor assembly and homeostasis.
Introduction
Mitochondria are a hub of metabolism and signaling (Cunningham and Rutter, 202020). At the heart of many of these activities are metal cofactors such as heme, iron sulfur clusters and other metalloenzymes (Pierrel et al., 2007a). Therefore, coordinating the flux of metals and controlling cofactor assembly should be closely linked with overall mitochondrial health. While the mitochondrial outer membrane (OM) is porous thus allowing for gated diffusion of metabolites, the inner membrane (IM) is folded into cristae and is impermeable to most small molecules and especially metals, Figure 1. Yet the internal matrix compartment requires metals for processing proteases as well as enzymes of the tricarboxcylic acid cycle. The IM houses many metalloenzymes and contains the highest ratio of proteins to lipids of all eukaryotic membranes (Kühlbrandt, 2015). The assembly and insertion of the cofactors into the complexes of electron transport chain requires assembly proteins localized in the intermembrane space (IMS) and the matrix (Vercellino and Sazanov, 2022). Some cofactors are produced (heme and Fe-S) or stored in the matrix (copper) and they must be redistributed to the IMS for assembly into the target enzymes (Cobine et al., 2021). Therefore, coordinating multiple proteins in multiple compartments with the availability of the metals/cofactors is a task that requires strategies to position the proteins involved at the right time in the right place.
FIGURE 1
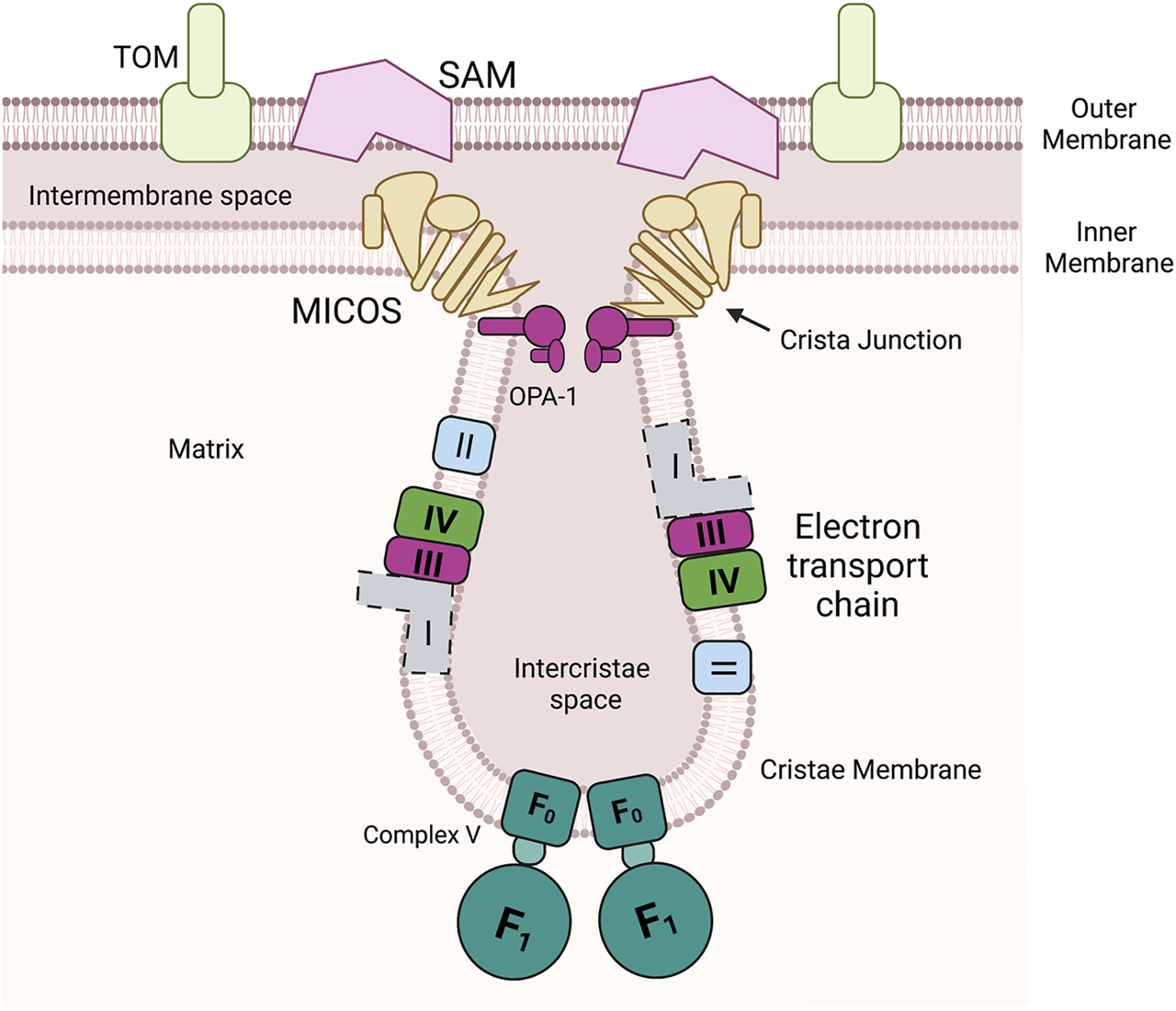
Mitochondria have multiple unique compartments. Outer membrane is a porous membrane that allows exchange of metabolites and small molecules with the cytosol but restricts the entry of proteins to the intermembrane space. The inner membrane is a highly protein loaded membrane that folds into invaginations known as cristae. The cristae house the assembled components of the oxidative phosphorylation (Complex I, II, III, and IV and ATP synthase (F0F1 ATPase, Complex V). In mammals this includes a membrane spanning NADH oxidoreductase (Complex I) (shown with dotted lines due differences between yeast and mammals), in yeast this enzyme is soluble in the matrix. The mitochondrial contact site and cristae organizing system (MICOS) and Opa-1 cooperate to form cristae junctions that encloses the intercristae space. MICOS also provides a structural point of contact between the inner and outer membrane via an interaction with the sorting and assembly machinery (SAM).
The processes involved in mitochondrial ultrastructure, energetics, and metal homeostasis are conserved from yeast to mammals. While the nomenclature and naming of the proteins has been partially unified, examples of differently named homologs still exist. In this review we discuss a mixture of model systems that have been used to further our understanding of mitochondrial physiology. We have noted when evidence is from yeast or mammals in the text and have used all caps for protein names from mammals (including mouse and human) (e.g., SLC25A3) and have used sentence case (e.g., Pic2) for yeast proteins.
Multiple pathways evolved to facilitate the correct localization of the mitochondrial proteome (Dudek et al., 2013). Importantly, all the import pathways require proteins to be unfolded and therefore they must receive the cofactors required for activity in the organelle. Import and insertion is tightly regulated by protein complexes that allow transit of unfolded polypeptides to the correct location without causing disruption of the membrane potential as depolarization of mitochondria is a well described inducer of cell death. Using a combination of cytosolic and mitochondrial proteins, newly translated proteins enter the mitochondrion via translocase of the outer membrane (TOM). Then multiple pathways exist for the delivery of proteins to their final localization (Dudek et al., 2013). Two different TIM complexes, either Tim22-or Tim23-containing complexes, sort and facilitate the insertion of IM and matrix proteins, while the β-barrel sorting and assembly machinery (SAM) is required for OM protein insertion (Dudek et al., 2013). Cysteine-containing IMS localized proteins have a dedicated pathway called the mitochondrial intermembrane space import and assembly (MIA) machinery. Mia40 is the major protein that participates in import via transient disulfide bond formation with the incoming targets and then, in the final stages of the import process, facilitates the formation of disulfide bonds within the IMS protein (Dickson-Murray et al., 2021). In addition, the proteins known as the small TIMs assist in directing OM and IM proteins to the distinct complexes that facilitate insertion. The concerted action of all these complexes allows for the insertion and activity of the ∼1,000 nuclear encoded proteins that function in mitochondria.
Mitochondrial genomes content can vary in different organisms but generally it encodes ribosomal RNAs, tRNAs and select subunits of the OXPHOS machinery. The mRNAs encode the proteins that are core elements of the OXPHOS complexes (3 subunits of complex IV, 3 subunits of complex V, 1 subunit complex III in yeast; 3 subunits of complex IV, 2 subunits of complex V, 1 subunit complex III, 7 subunits of complex I in humans) are transcribed, translated within the matrix by a dedicated machinery. Once transcribed and translated these proteins are inserted into the IM to form distinct complexes and assembly intermediates that form in a modular fashion. OXA1 is essential for the correct insertion and assembly of many of these proteins (Hennon et al., 2015; Thompson et al., 2018). Mutations in human OXA1 result in fatal encephalitis, hypotonia, and developmental delay due to assembly defects in complexes I, IV and V (Thompson et al., 2018). Both human and yeast Oxa1 have been shown to interact with the mitochondrial ribosome. In yeast, mtDNA encoded subunits of complex V are synthesized on the cristae membrane, while complex III and IV components were synthesized both at the inner boundary membrane and cristae membrane as defined by the localization of the specific translational activators (Eaglesfield and Tokatlidis, 2021). Using fluorescent noncanonical amino acid tagging it was shown that in human cells the majority of mtDNA encoded proteins are synthesized at the cristae membrane (Zorkau et al., 2021). The authors of this study note that while the technique cannot follow synthesis in real time it is unlikely that the assembly intermediates could pass through the cristae junctions (Zorkau et al., 2021). Therefore, the cofactor containing subunits of OXPHOS requires the coordination of these translational factors, insertases, the available cofactor at specific localizations to ensure correct assembly.
Mitochondrial Morphology and Organization
Aberrant mitochondrial morphology is associated with cellular dysfunction suggesting that mitochondrial architecture and function are intimately linked. The mitochondrial contact site and cristae organizing system (MICOS) complex consists of numerous proteins essential for formation of mitochondrial structure, Figure 2 (Harner et al., 2011; Eramo et al., 2020). The MICOS complex establishes and maintains the inner membrane architecture, provides contacts between the IM and OM, and facilitates the closure of the cristae junction (Eramo et al., 2020). It is known that membrane potential, lipid and protein composition affect cristae formation and that the maintenance of cristae shape is linked to content of ATP synthase. The conserved dynamin related GTPase, OPA1 catalyzes membrane fusion and is found at crista junctions and regulates both the number of cristae and the release of cristae contents (Ehses et al., 2009).
FIGURE 2
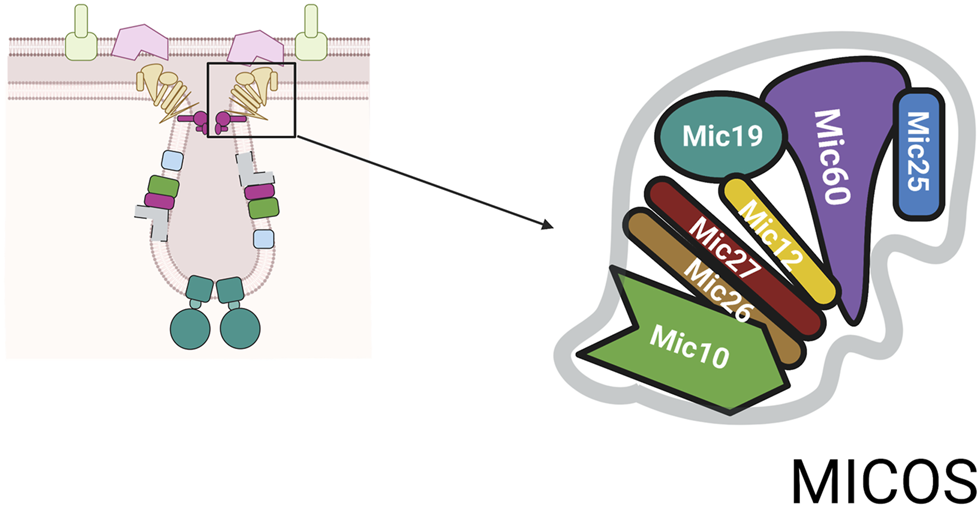
MICOS complex. The MICOS complex consists of multiple proteins named for their molecular weight. The numbers shown are based on mammalian system and are slightly different in yeast system.
Depending on the organism the number of core components identified to date varies (Eramo et al., 2020). The core components of MICOS are: Mic10, Mic12, Mic19, Mic25, Mic26, Mic27, and Mic60, Figure 2 (Pfanner et al., 2014). In addition, to the role in closing the cristae junction, the complex is required for the formation of contact sites between the IM and OM to facilitate communication between these two mitochondrial compartments. MICOS proteins also interact with proteins such as Ugo1, a part of mitochondrial fusion machinery, and porin, the abundant OM channel for small metabolites (Muñoz-Gómez et al., 2015). Through its links with multiple proteins, the MICOS complex functions as an organizer of mitochondrial architecture and integration platform for processes that are centered on mitochondrial membranes.
A recurring theme in mitochondria is that large complexes interact and work together exchanging proteins into and out the complexes to regulate functions critical to cellular physiology. This MICOS network includes interactions of Mic60 with the protein translocases TOM, SAM, and the oxidoreductase Mia40. Mic60 stimulates the TOM-SAM-mediated import of β-barrel proteins into the outer membrane forming a complex with TOM and SAM. In yeast, Tim23 and Mic60 interact to facilitate precursor handover from the TOM complex to the Tim23 complex by bringing the OM and IM into close contact. While TIM22 and MICOS work synergistically in human mitochondria to drive the translocation of mitochondria carrier family (MCF/SLC25) proteins (Callegari et al., 2019). Mic60 also interacts with the MIA machinery to mediate the import of intermembrane space proteins that have containing characteristic cysteine motifs (Rampelt et al., 2017). Since Mic60 transiently binds to both TOM and Mia40, Mic60 helps to position the receptor Mia40 to the intermembrane space side of the TOM complex. This spatial coupling function of Mic60 facilitates the formation of intermolecular disulfide bonds between Mia40 and the preprotein near the TOM complex preventing the exposure of potentially redox-sensitive cysteine residues in the preproteins. Thus, Mia40 can rapidly bind to precursor proteins passing through the TOM channel and promote their efficient import (Schorr and van der Laan, 2018). Mia40 pathway plays critical roles in the import of multiple metalloprotein and metalloprotein related assembly proteins. Therefore, MICOS has a critical role in the translocation of most mitochondrial proteins. This review will focus on the roles MICOS complexes have on metal homeostasis and the roles metalloproteins and metals have on MICOS stability.
Transporters in Mitochondria
Membranes provide the greatest barrier to metal distribution in cells (Vest et al., 2013a). Therefore, understanding metal homoeostasis starts with understanding the transporters that facilitate entry and exit of the metals/cofactors. Transporters have the potential to act as scaffolds for loading metals however limitations in our ability to monitor these interactions, or a lack of knowledge about the transporters in particular compartments has slowed our progress in defining such roles. Mitochondria have five major classes of transporters in the IM that include the mitochondrial carrier family (MCF/SLC25), ATP-binding cassette (ABC) transporters, mitochondrial pyruvate carrier (MPC/SLC54), mitochondrial cation/H+ exchangers (LETM/SLC55), and sideroflexins (SLC56) (Cunningham and Rutter, 202020; Gyimesi and Hediger, 2020). Here we will focus on transporters involved in metals in mitochondria with the MPC and LETM families discussed elsewhere (Gyimesi and Hediger, 2020).
Mitochondrial Carrier Family
Mitochondria contribute to intracellular signaling and regulatory networks in part by coordinating trafficking of metabolites into and out of mitochondria (Cunningham and Rutter, 202020). The mitochondrial carrier family (MCF/Slc25 family) form the largest group of transporters in the IM and are responsible for most of the metabolic flux to, and from, the organelle. In humans there are 53 members that are collectively responsible for the transport of numerous substrates including TCA cycle intermediates, nucleoside di- and triphosphates, amino acids, and metals (Cunningham and Rutter, 202020). Sixteen of the 53 MCFs have no known substrate and the established promiscuity of some of the characterized transporters raises the possibility that even those 37 with known substrates may have additional substrates. Notable members of this family involved in metals and metal cofactor assembly are SLC25A3/Pic2 which is a copper and phosphate transporter (Vest et al., 2013b; Boulet et al., 2018), SLC25A37/MITOFERRIN1/Mrs3 and SLC25A28/MITOFERRIN2/Mrs4 which are involved in mitochondrial iron homeostasis and necessary for iron transport (Shaw et al., 2006; Paradkar et al., 2009; Troadec et al., 2011; Christenson et al., 2018; Seguin et al., 2020), as well as SLC25A38 which transports glycine for heme synthesis (Guernsey et al., 2009).
MCF transporters have a conserved fold consisting of three repeats of approximately 100-amino acids that contain two transmembrane helices connected by a loop with a short α-helix (Ruprecht and Kunji, 2020). This structural insights into the family of transporters is based on the structure determined for ADP/ATP exchanger that was crystalized with inhibitors that stabilized the protein in either an open to the IMS state (c-state) or open to the matrix state (m-state) (Pebay-Peyroula et al., 2003; Ruprecht et al., 2019). The MCF fold is stabilized by salt bridges that form at the closed end of the substrate binding pocket depending on which state the protein adopts. The first,third, and fifth transmembrane helices contains a conserved PX (D/E)XX (R/K) motif that forms salt bridge and hydrogen bonding contacts on the matrix side of the binding pocket. A complementary (Y/F) (D/E) XX (R/K) motif in second,fourth, and sixth helices is found on the IMS side (Ruprecht and Kunji, 2020). The strength of these salt bridge interactions is an important predictor of the requirements and directionality of transport. Two examples exist of single amino-acids changes in MCF that give rise to changes in specificity. In yeast, Rim2 is a bifunctional pyrimidine and iron transporter but E248A mutant specifically disrupts mitochondrial iron transport activity while K299A mutant specifically abrogated pyrimidine nucleotide transport and exchange (Knight et al., 2019). Similarly, a L175A mutation can make the bifunctional copper and phosphate transporter SLC25A3 specific for copper transport (Zhu et al., 2021).
The metal transporting MCF SLC25A3 and SLC25A37 are predicted to be uniporters that import metals into the matrix. Both SLC25A3 and SLC25A37 have been successfully reconstituted into proteoliposomes to demonstrate transport activity (Boulet et al., 2018; Christenson et al., 2018). The yeast homologs, Pic2 and Mrs3 and mammalian SLC25A3 have also been expressed in L. lactis. Expression in L. lactis allows for monitoring import of metals into this organism. Both Pic2 and Mrs3 can transport copper in L. lactis although for Mrs3 to facilitate copper transport iron had to be chelated/limited presumably to prevent competition (Vest et al., 2016). The plant variant of SLC25A37 was also shown to have the ability to transport iron and copper (Bashir et al., 2011a; Bashir et al., 2011b). But our understanding about the interplay between these transporters in vivo is limited.
In addition to a role in transporting metals the MCF family has the potential to form a scaffold for protein-protein interactions to enhance the assembly of the mitochondrial metalloproteome. The MCF proteins have been isolated in many distinct large complexes. The large interactome of these proteins is exemplified by the ADP/ATP exchange proteins (Claypool et al., 2008; Claypool, 2009; Senoo et al., 2020). These proteins play critical role in delivery of the energy currency to the cell and have been isolated in large multiple protein complexes. These ADP/ATP exchanger protein complexes can also include other MCFs including the copper and phosphate carrier (SLC25A3), calcium dependent carriers (SLC25A13, SLC25A24, SLC25A25), the tricarboxylate carrier (SLC25A1), and the carnitine/acylcarnitine carrier (SLC25A20) (Claypool et al., 2008). How these interactions affect and/or regulate the specificity and/or transport activity is unknown due to the difficulties in assessing these biochemical characteristics in vivo or with purified components.
ATP-Binding Cassette-Transporters
There are 4 mitochondrially-localized ABC-transporters (ABCB6, ABCB7, ABCB8 and ABCB10) which are part of a large superfamily of proteins that mediate nucleotide-dependent transport (Cunningham and Rutter, 202020). ABC-transporters have four core domains for function. Two transmembrane domains that provide specificity by forming a binding site, and two nucleotide binding domains to hydrolyze ATP to facilitate transport of the bound ligand. Conservation is found in the nucleotide binding domains with variability in the transmembrane domains to allow for a broad number of substrates (Wilkens, 2015). The four mitochondrial ABC-transporters are involved in moving Fe-S clusters and heme related metabolites into and out of mitochondria. ABCB6 is localized to the OM (and other cellular locations including the cell surface). ABCB6 has been proposed as porphyrin transporters (Krishnamurthy et al., 2006) and has been shown to specify the Langereis blood group (Helias et al., 2012).
ABCB7, ABCB8 and ABCB10 are all localized to the IM and the exact molecule transported by each is still unclear. ABCB7 links to cofactor assembly are because it was identified as a candidate causative gene for X-linked sideroblastic anemia with spinocerebellar ataxia and the yeast homolog ATM1 is regulated by iron and participates in maintaining Fe-S cluster homeostasis (Pondarre et al., 2007; Lill, 2020). Similarly phenotypic data links ABCB8 to iron homeostasis as a mutation of Abcb8 results in cardiomyopathy with increased mitochondrial iron accumulation with decreased activity of cytosolic Fe-S clusters proteins. Finally, the Abcb10 knockout in mouse is embryonic lethal due to anemia. In support of a role of this transporter in iron homeostasis, Abcb10 forms a complex with MITOFERRIN1 (SLC25A38) and ferrochelatase (FECH) that enhances heme synthesis, Figure 3 (Yamamoto et al., 2014; Medlock et al., 2015; Maio et al., 2019). While the substrate for ABCB10 has not be definitively identified, multiple studies suggest different substrates related to heme synthesis and it is clear this protein plays an important role in iron metabolism. The characterization of a larger complex that includes multiple heme synthesis related proteins is a continuation of the theme observed for the MCF proteins, that complexes of transporters and targets can form a potential regulatory mechanism for metal cofactor assembly or metal homeostasis.
FIGURE 3
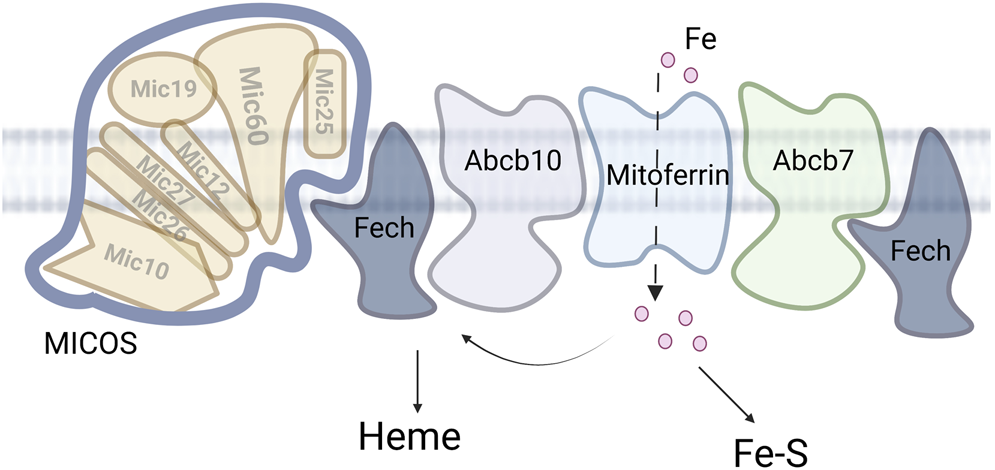
MICOS and mitochondrial iron. Iron enters the mitochondria via the MCF mitoferrin. The iron is then partitioned between heme synthesis, Fe-S assembly and other iron. Accumulation of the other iron is deleterious in multiple disease states.
Other Transporters
Other notable mitochondrial transporters involved with metals are sideroflexins (Slc56). One of the five sideroflexin proteins, sideroflexin-1, has been shown to have a role in serine transport in mitochondria, and both sideroflexin-1 and sideroflexin-3 have been linked to iron homeostasis and Fe-S cluster biogenesis (Tifoun et al., 2021). Sideroflexins are 5 transmembrane domain proteins that contain a conserved mitochondrial tricarboxylate/iron carrier domain (PFAM: 03820). All members share the same topology with the amino-terminal inside and the carboxy-terminal outside. Sideroflexins 1–4 have sites of post-translational modification including acetylation that regulate activity and stability.
Cellular zinc transporters have been reported to localize to different mitochondrial membrane in some cell types. Zinc transporters can be broadly categorized in two groups ZIP (Irt-like) and ZnT (Kambe et al., 2014). ZIP transporters generally act as in zinc import while ZnT class act as zinc exporters. ZIP family members contain 8 transmembrane domains, with the proposed metal-binding residues embedded in transmembrane helices 4 and 5 and a cytoplasmic (inside/IMS in mitochondria) region between transmembrane 3 and transmembrane 4, that is, important for regulation including protein stability the exact roles of this domain are still under investigation (Kambe et al., 2014). It is also important to recognize that the ZIP family homologs have been shown to transport a variety of divalent ions including but not limited to iron, manganese, copper. Therefore, these transporters could have effects on other metals in mitochondria.
Metals in Mitochondria
Copper
Copper is found within each of the mitochondrial compartments. In the IM, cytochrome c oxidase is the multi-subunit complex that catalyzes the final steps of the electron transport chain (ETC). It accounts for about 25% of mitochondrial copper and is the major cuproenzyme present in this organelle (Cobine et al., 2004). In addition, a small percentage of cellular Cu, Zn superoxide dismutase (Sod1) and its copper chaperone, Ccs1, is localized to the IMS. However, these proteins do not account for a significant proportion of the total mitochondrial Cu. The majority (up to 70%) is found in the matrix and is bound to a biochemically characterized but structurally undefined complex known as the copper ligand (CuL) (Cobine et al., 2021). The copper in the matrix is transported by MCF SLC25A3/Pic2, that is, capable of transporting Cu and CuL as a substrate (Vest et al., 2013b; Boulet et al., 2018; Zhu et al., 2021). The data in multiple experimental models shows that the copper in the matrix is redistributed to the IMS for the assembly of cytochrome c oxidase (COX) and superoxide dismutase (Sod1). The matrix distribution model, that is, based on phenotypic data from mutants of the copper importer and biochemical competition assays in yeast under copper stress. These phenotypes are reproduced in mammalian cell culture without the need for copper restriction (Boulet et al., 2018). However, the identity of the transporter responsible for copper export is unknown and therefore the regulation of this pathway is poorly understood. An alternative pathway that feeds copper directly from the cytosol into these enzymes has been suggested based on purification of copper complexes that are thought to be in the IMS, this alternative pathway could operate under conditions where copper is abundant (Lindahl and Moore, 2016; Cobine et al., 2021).
Copper availability in the IMS is critical for the assembly of COX (Cobine et al., 2021). However, copper levels are restricted and therefore chaperone proteins are required to enhance the assembly process. This evidence for the copper delivery pathway is a combination of in vitro observations and genetic suppression experiments. In the IMS, Cox17 presents copper to Sco1 and Cox11 for assembly of the copper sites in COX called CuA and CuB sites, respectively (Horng et al., 2004; Cobine et al., 2021). Cox17 is a soluble protein that adopts a coiled coil-helix-coiled coil-helix fold stabilized by two disulfide bonds. The Cox17-Sco1 interaction is the most thoroughly studied copper transfer reaction that occurs in the IMS. Cox17 donates copper to the exposed CxxxC site on Sco1. Studies have shown that the reactions proceed from the Cu-loaded, partially oxidized conformer of human COX17 to SCO1 via transient interactions (Banci et al., 2007; Banci et al., 2008; Banci et al., 2011). This Cu-delivery pathway is further supported by the observation that SCO1 overexpression can rescue the phenotype of a COX17 mutant cell (Glerum et al., 1996). Sco1 subsequently insert the Cu into Cox2 to form the CuA site in COX. Multiple redox related steps are involved in the COX assembly process. Mammalian COA6 (and yeast Coa6) and SCO2 are two of the proteins with roles in regulating the reduction state of assembly proteins and the target COX2 (Pacheu-Grau et al., 2015; Soma et al., 2019; Swaminathan and Gohil, 2022). Most recently an additional interplay between the Cu assembly process and heme has been identified with a role discovered for COA7, a heme binding protein, that transiently interacts with the copper metallochaperones SCO1 and SCO2 and catalyzes the reduction of disulfide bonds within these proteins (Formosa et al., 2022). To build the CuB site, cells use a different battery of assembly proteins. Cox11 is a membrane bound factor required for the insertion of the Cu in COX1 and is also a recipient of copper from Cox17 (Baker et al., 2017a). Like Sco1 pathway specific accessory factors are required to maintain the redox state of the proteins in the pathway. Cox19 is required for the reduction of two critical copper binding cysteines in Cox11 (Bode et al., 2015). Copper transfer occurs via an interface than, that is, distinct from that used for interaction with Sco1 (Horng et al., 2004; Cobine et al., 2006). The requirement for this multi-step interplay of protein-protein interactions in the IMS to facilitate copper loading reinforces the idea that limited copper availability is a major complication for the correct metalation of cuproenzymes. It is also important to note that mutation in SCO proteins can cause remodeling of cellular copper homeostasis. The mutant cells and mice show copper deficiencies due to inappropriate degradation of the high affinity importer CTR1 or excess activity of the copper exporter ATP7A (Leary et al., 2007; Hlynialuk et al., 2015; Baker et al., 2017a; Baker et al., 2017b). During the cellular copper deficiency mitochondrial copper is maintained suggesting a possible prioritization of the matrix copper under these conditions (Dodani et al., 2011).
To date at least six gene deletions in yeast have been shown to limit matrix copper accumulation based on analysis of the purified mitochondria and mitochondrial targeted copper responsive reporters. The proteins implicated in copper import/maintenance are the MCF proteins: Pic2, Mrs3 and the assembly factors Coa1, Coa4, Coa6 and Shy1 (Pierrel et al., 2007b; Bestwick et al., 2010; Ghosh et al., 2014). Coa6 is a Cx9C containing protein of the IMS, that is, responsible for regulating the redox state of proteins involved in Cox2 assembly machinery (Ghosh et al., 2016; Soma et al., 2019; Swaminathan and Gohil, 2022). Coa1, Coa4 and Shy1 are all linked to the translation and assembly of Cox1 (Pierrel et al., 2007b). The mammalian homologs of Shy1 (SURF1) and Coa1 (COA1/MITRAC15) are part of the mitochondrial translation regulation assembly intermediate of cytochrome c oxidase (MITRAC) complex (Mick et al., 2012). This complex is another example of a large protein complexes that form in mitochondria to facilitate more efficient processing and/or substrate channeling. The assembly of the respiratory chain complexes proceeds in modular fashion and require complex-specific assembly factors to stabilize the intermediates for the complete maturation of complexes. Because maturation of the enzyme complexes is a sequential process during which new components and cofactors are added, the composition of the MITRAC complex changes during the process (Mick et al., 2012). Since MITRAC is facilitating the translation and assembly of COX1 and this is the heme and copper containing subunit it is intriguing to proposed that this complex could be critical for regulating total mitochondrial copper. It should be noted that only Coa1, Shy1 deletion and not other MITRAC homologs in yeast cause the copper deficiency.
Iron
Iron enters mitochondria through multiple mechanisms. The most well-studied example of mitochondrial iron import is via MCF proteins MITOFERRIN1 in mammals and Mrs3 and Mrs4 in yeast (Dietz et al., 2021a). MITOFERRIN and Mrs3 have been shown to modulate the transport of iron in vivo and in vitro while Mrs4 is transcriptionally activated by iron depletion, suggesting a role for homeostasis of iron in mitochondria. Once iron crosses the IM it is used for the synthesis of heme, Fe-S or is bound by other ligands for storage. Genetic experiments suggested that FRAXATIN (or at least the yeast homolog Yfh1) played a role as a chaperone directing iron to produce Fe-S cluster largely based on an iron accumulation phenotype and the activation of Fe-regulated transcription in yeast in YFH1 mutants (Llorens et al., 2019). However additional experimental evidence suggests that this is not a required function of Yfh1. The phenotypes of yfh1∆ can be bypassed completely by a compensatory mutation in the Fe-S cluster scaffold protein IscU (Yoon et al., 2015). This combined with other approaches suggested the primary function of Yfh1 is sulfur delivery to form Fe-S clusters. Once iron reaches the iron-sulfur cluster machinery it is synthesized into 2Fe-2S clusters that are then inserted in mitochondrial targets or exported to the cytosol where the cytosolic iron-sulfur assembly (CIA) machinery matures them to 4Fe-4S clusters and/or inserts them into other targets (Lill, 2020). The other major iron cofactor is heme which is synthesized when iron is inserted into protoporphyrin IX by ferrochelatase (Dailey and Meissner, 2013). Protoporphyrin IX is synthesized by a series of enzymes that are split between the cytosol and the mitochondria with the initial steps of porphyrin synthesis taking place in mitochondria before export of the intermediates for maturation in the cytosol until the final steps occur back in the mitochondrion.
Zinc and Manganese
The mechanisms of zinc and manganese uptake into the mitochondrial matrix are not well understood. Zinc is required in the mitochondria for multiple functions including electron transport chain function (COX), ATP synthesis (Atp32), protection against oxidative stress in the IMS (Sod1), lipid transport (Yme1L), mitochondrial dynamics (Oma1, Yme1L), processing peptidase (MPP), and mitochondrial intermediate peptidase (MIP). A genetic screen to identify a transporter using chemical sensors of available zinc uncovered an unexpected connection between Complex III assembly and mitochondrial zinc. A gene designated MZM1 was identified and characterized to show the gene product was required for both zinc maintenance and complex III assembly (Atkinson et al., 2010; Atkinson et al., 2011). However, this did not result in identification of a zinc transporter in mitochondria. Some studies have suggested that zinc can be imported to mitochondria by the calcium uniporter (MCU) (Gazaryan et al., 2007; Ji et al., 2020). While others have reported dual location of zinc transporters suggesting that zinc entry to the IMS could be facilitated by Zip1 that was shown to localize to the OM (Cho et al., 2019). Further still the zinc transporters Zip7 and ZnT7 have also been shown to have dual localization in mitochondria and ER and expression of Zip7 and ZnT7 contribute to cellular zinc exchange between the organelles in certain cell types (Tuncay et al., 2019). Finally Znt9 (SLC30A9) was identified as a zinc exporter in mitochondria (Deng et al., 2021) and SLC25A25 has been implicated in at least regulating uptake of zinc as deletion can reverse phenotypes induced by SLC30A9 deletion (Ma et al., 2022). Perhaps redundancy is the reason for the failure of the yeast genetic screens to yield a single candidate.
Manganese has a single target in the mitochondria in the form of superoxide dismutase 2 (SOD2). This matrix localized enzyme is responsible for protection against a subset of the reactive oxygen species generated. Deletion of SOD2 in mammalian models is embryonic lethal demonstrating its essential role. However, to date no definitive data exists as the identity of the manganese transporter that provides this metal for the matrix for SOD2 assembly and activity. In yeast deletion of the gene MTM1, which encodes a MCF, caused decreased activity of Sod2 but did not prevent manganese accumulation (Luk et al., 2003). It was subsequently shown that deletion of MTM1 changed iron availability resulting in mismetallation and inactivation of Sod2 by iron (Yang et al., 2006; Naranuntarat et al., 2009). Critically this is an example of the complex requirements for essential cofactor assembly in mitochondria and highlights the need for additional research on regulatory mechanisms that change availability.
Cofactor Assembly
Mitochondria are essential for the synthesis of cofactors required for normal cellular function. It is widely accepted that synthesis of Fe-S clusters in mitochondria is the sole essential function for the organelle in eukaryotes. This is because divergent eukaryotes which have lost other mitochondrial function retain an organelle with Fe-S cluster assembly machinery. In addition to Fe-S clusters, mitochondria also house the machinery for assembly of heme and molybdenum cofactor (MoCo). Heme is required for all aerobic eukaryotes and is involved in oxygen metabolism, sterols synthesis, and amino acid biogenesis amongst other processes. MoCo is involved in multiple cellular functions including sulfur, drug, and nucleotide metabolism.
Heme Assembly
Heme or Fe-protoporphyrin IX is an essential cofactor required for a plethora of cellular processes in eukaryotes. In metazoans, the heme biosynthesis is typically partitioned between the cytosol and mitochondria, with the first and final steps taking place in the mitochondrion. Key enzymes in the pathway that are found in the mitochondrial matrix are aminolevulinic acid synthase, which utilizes glycine and succinyl-CoA to produce aminolevulinic acid, and FECH, which utilizes iron and protoporphyrin IX to make heme (Phillips, 2019). Overall, the pathway has been well studied and all the biosynthetic enzymes structurally characterized. Nevertheless, understanding of the regulation of heme synthesis in different cells and factors that influence this process remains incomplete. Some details of the transcriptional regulation in the context of erythroid development are known, but transcriptional regulation in most other cell types or cellular conditions is much less understood (Dailey and Meissner, 2013). Recent work supports post-translational mechanisms for the regulation of the mitochondrial heme biosynthesis enzymes (Chung et al., 2017). Studies have shown protein-protein interactions regulate heme precursor levels (Medlock et al., 2015) and the incorporation or disruption of cofactors into key enzymes (Shah et al., 2012; Kardon et al., 2020).
Iron-Sulfur Cofactor Assembly
Fe-S clusters are essential cofactors for the ETC and many other biochemical reactions and processes (Lill, 2020). The cofactor assembly systems are required to sequester, chaperone, and regulate delivery of the cofactor. The assembly proteins are not strictly required for building Fe-S cluster, because the clusters will self-assemble in solution, but is thought these proteins are necessary to circumvent the toxicity and indiscriminate reactivity of free iron and sulfide. Defects in the biosynthesis in Fe-S cluster cause dysfunction in mitochondria, neurodegenerative and cardiovascular disease, genomic instability, and the development of aging and cancer.
Mitochondria assemble not only mitochondrial Fe-S clusters but are also involved in the biosynthesis of Fe-S clusters for proteins in the cytosol and in the nucleus (Lill, 2020). These cytosolic and nuclear Fe-S cluster proteins with essential functions for example, ABC protein Rli1, which participates in ribosome assembly and ribosome recycling during termination of polypeptide synthesis (Kispal et al., 2005). Eukaryotic replicative DNA polymerases which also contain a Fe-S cluster in their C-terminal domain (Paul and Lill, 2015). The Fe-S cluster cofactor appears to be indispensable for efficient interaction with their accessory proteins during DNA replication.
Molybdenum Cofactor
Molybdenum is a versatile redox element, that is, used by enzymes to catalyze diverse reactions. While not a focus of this review we will briefly discuss molybdenum cofactor (MoCo) as it is synthesized in mitochondria and has links to iron and copper. MoCo is an ancient cofactor involved in sulfur, drug, and nucleotide metabolism. MoCo synthesis begins in mitochondria with guanosine 5′-triphosphate which is converted via the two proteins to cyclic pyranopterin monophosphate (cPMP). Both mitochondrial enzymes in humans, MOCS1A and MOCS1B, are encoded by a single gene alternatively spliced to yield either protein. For MOCS1A exon one encodes the mitochondrial localization signal, and for MOCS1B localization is designated via exon 10 (Mayr et al., 2020). Interestingly MOCS1A can also be localized to the cytosol in primates via exon 1b, and while it appears to be functional its physiological role in the cytosol is unclear. MOCS1A belongs to the radical SAM enzyme superfamily and has two iron-sulfur clusters (Hänzelmann et al., 2004), specifically 4Fe-4S clusters, thus is connected to iron and iron-sulfur cluster homeostasis. cPMP is then exported across the mitochondria membranes to the cytosol where it is further modified (Mendel, 2013). In plants this is mediated by ATM3 which is the homolog of yeast Atm1 and mammalian ABCB7. Once in the cytosol MPT synthase transfers the two sulfurs to cPMP to create molydopterin (MPT). This intermediate can bind copper, and the copper may act as a protecting group as the cofactor synthesis proceeds (Mendel, 2013). The next step in synthesis is formation of the of adenylated-MPT before the final step of Mo insertion. Therefore, MoCo synthesis has overlap with copper and iron both utilizing cofactors and sharing a transport pathway.
The Connection Between Metals and Organization
Mitochondria have a clear role in multiple aspects of metal homeostasis as described above. But many of the experimental approaches and techniques used have been “static” in nature and did not necessarily consider the dynamics of mitochondria. This includes consideration of total content, structure, and localization within the cell in different tissues and organisms. In a “two-way street” we have limited understanding of the impact that alterations in mitochondrial physiology have on metal content and utilization or how dynamic changes in mitochondria alter cofactor synthesis. Rates of fission and fusion, the ultrastructure of the organelle, and rates of mitophagy all have the possibility to impinge on metal homeostasis and cofactor assembly. An emerging area of overlap is between metal homeostasis/cofactor assembly and MICOS. As described previously the coordination of the proteins in larger complexes is a developing theme and we will discuss two interactions of MICOS with iron and copper proteins that regulate both cofactor assembly and mitochondrial function.
Ferrochelatase-Mitochondrial Contact Site and Cristae Organizing System
The terminal enzyme of the heme biosynthesis pathway, ferrochelatase, has been identified to be part of a multi-protein complex or metabolon in the mitochondrial matrix. This complex has been studied in both mammalian cells (FECH) (Medlock et al., 2015) as well as yeast (Hem15) (Dietz et al., 2021b). In terms of the mammalian cells, most work has focused on the metabolon in developing erythroid cells, as these cells make a large amount of heme. An interesting finding was that mitochondrial heme metabolon interacted with MICOS proteins including MIC60, Figure 3 (Piel et al., 2016; Dietz et al., 2021b). Further work in yeast showed that loss of MICOS negatively impacts Hem15 activity, it affects the size of the Hem15 high-mass complex, and results in accumulation of reactive and potentially toxic porphyrins (which arise from the heme intermediates porphyrinogens) that may cause oxidative damage. Restoring intermembrane connectivity using a heterologously expressed protein capable of tethering the IM and OM “artificially” in MICOS-deficient cells mitigates these cytotoxic effects (Dietz et al., 2021b). The artificial tether clearly shows the importance of mitochondrial ultrastructure to the function of this enzyme.
The localization of ferrochelatase to MICOS also provides a connection to iron and Fe-S cluster trafficking at these sites. Ferrochelatase was shown to interact with SLC25A37/MITOFERRIN1/Mrs3, ABCB10, and ABCB7 (Taketani et al., 2003; Chen et al., 2010; Medlock et al., 2015; Maio et al., 2019)). The connection with MITOFERRIN and ABCB10 is clear as iron is a substrate for the enzyme and thus can be directly trafficked for heme synthesis. The role of FECH and ABCB7 (Banci et al., 2007; Banci et al., 2008) interaction is less clear, except that FECH itself has an 2Fe-2S cluster (Dailey et al., 1994) and thus may regulate or be regulated by Fe-S cluster flux in the matrix. Overall, these data provide new insights into how heme biosynthetic machinery is organized and regulated, linking mitochondrial architecture-organizing factors to heme synthesis for the efficient import of heme precursors and export of heme.
COX17-Mitochondrial Contact Site and Cristae Organizing System
In addition to the role Cox17 plays in COX assembly, it also been linked to a copper dependent interaction with the MICOS complex (Vanisova et al., 2019; Chojnacka et al., 2015). Cox17 interacts with Mic60 to modulate MICOS complex integrity (Chojnacka et al., 2015). This interaction does not involve Sco1 or Cox11. However, the Cox17-MICOS interaction is regulated by copper, Figure 4. Cox17 is therefore a factor involved in maintaining the architecture of mitochondria via the MICOS complex. The regulation of the MICOS complex and therefore IM structure by Cox17 and copper was unexpected. However, it further consolidates a link between the mitochondrial morphology, ETC integrity, and metal homeostasis. It was proposed that Cox17 may be facilitating protein-protein interactions that enhance MICOS stability or that it could be delivering of copper to the MICOS complex (Chojnacka et al., 2015). This would require a yet to be identified copper-binding site in the complex. Cryptic sites of copper binding leading to metalloallosteric regulation have recently been described in multiple well studied processes (lipolysis, proliferation, and autophagy) and therefore a precedent exists to identify these new sites (Ge et al., 2022). Another possible model would be that the presence of excess Cu-Cox17 would indicate that COX is maximally metaled and therefore closing of cristae junctions should proceed to allow for enhanced activity of the OXPHOS in the enclosed intercristae space, Figure 4.
FIGURE 4
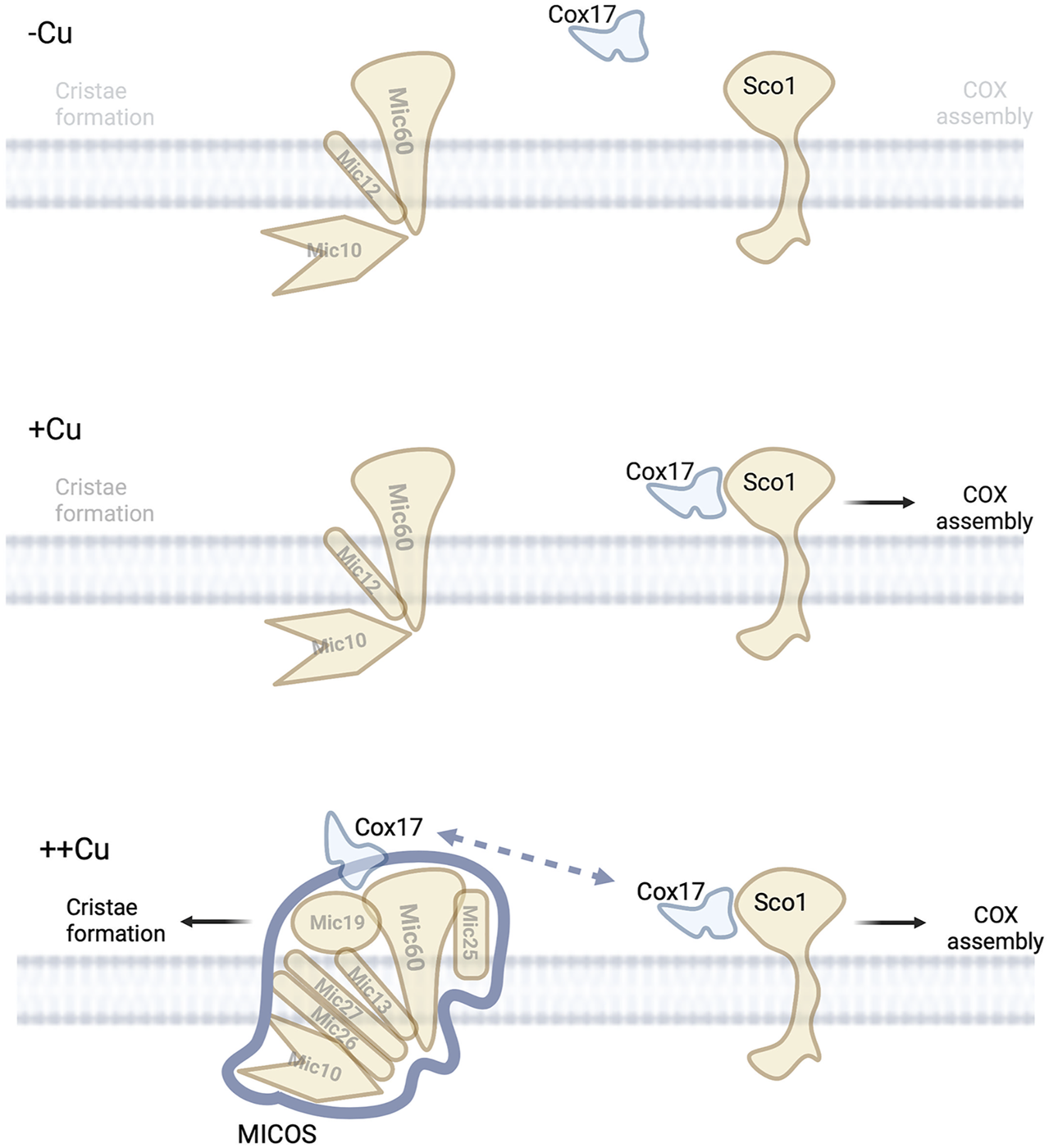
Copper and MICOS complex. Under low copper conditions no copper is available in the IMS to mediate the assembly of COX or allow for efficient assembly of MICOS suggesting low numbers of mitochondria cristae.
SOD1-Mitochondrial Contact Site and Cristae Organizing System
The regulation of metal homeostasis is important to avoid the potential formation of reactive oxygen species (ROS). Superoxide dismutase (Sod1) is involved in the scavenging of ROS in the cytosol and in the IMS of mitochondria. Sod1 requires zinc and copper ions and an intramolecular disulfide bond to catalyzes the conversion of superoxide into hydrogen peroxide and water. The activation, localization, and retention (in IMS) of Sod1 is largely dependent upon the copper chaperone Ccs1 (Sturtz et al., 2001; Gross et al., 2011). Ccs1 is involved in the transfer of copper and disulfide bond formation in the maturation of Sod1. The assembly of Sod1 is also dependent on Mia40. Mia40 is responsible for import and oxidation of Ccs1 to the IMS. Ccs1 is responsible to “fold and trap” Sod1 in the IMS. An additional role for mitochondrial ultrastructure in Sod1 activity was uncovered. Mitochondria that are lacking any components of the MICOS complex can create unusual separations in the IMS. MICOS mutants accumulate a disproportionate amount of oxidized Sod1 (Varabyova et al., 2013). The authors speculate that the reduction of the disulfide bond in Sod1 is prevented by mitochondrial ultrastructure/compartmentalization (Varabyova et al., 2013). The metalation state of Sod1 in the MICOS mutants is unknown and perhaps mitochondrial copper status in MICOS mutations play some role in oxidation states of the Sod1. It is known that decreased mitochondrial copper can change the steady state levels of IMS localized Sod1.
Many mutations in human SOD1 are associated with amyotrophic lateral sclerosis (ALS), a nervous system disease that causes loss of muscle control. This debilitating disease has meant that numerous studies have investigated the role of mitochondrial SOD1 is disease progression. The accumulation of wildtype reduced Sod1 was dependent on the cristae architecture controlled by MICOS and mitochondria with MICOS defects showed an increase in mitochondrial accumulation of ALS-related reduced variants of Sod1 (Varabyova et al., 2013). This accumulation resulted in the accumulation of toxic superoxide and mitochondrial dysfunction (Varabyova et al., 2013). Further suggesting a role for correct mitochondrial ultrastructure in this disorder.
Future Directions and Unanswered Questions
Until now our knowledge of metal homeostasis in mitochondria has been largely based on static measurements and the interpretation of genetic experiments. These have proven to be excellent resources for establishing the components of the pathways and identifying multiple interactors and suppressor that can bypass individual steps. However, recent technological advances in our ability to robustly detect protein complexes and our confidence that these interactions have bona fide physiological consequences have meant that we have learned that maintaining specific complexes in the highly crowded “real estate” of the mitochondrial IM is a recurring theme. The existence of interactions between copper and iron machinery and MICOS, that can serve as “a signpost to the most desirable neighborhood”, have triggered many additional questions about how mitochondrial morphology and metals are intertwined.
An outstanding question is what is the role of copper in the Cox17-MICOS interaction? Does this metal bind directly to stabilize a structure or perhaps enhance assembly or is the role of copper in stabilizing a fold of Cox17. The alpha-fold prediction of Mic60 does present multiple cysteine residues (as potential donors of thiol ligands) within 3–6 Å of each other (Jumper et al., 2021). The roles of these residues in metal binding has not been investigated. There have not been consistent reports of mitochondria morphology changes in cells lacking the major copper transporters such as CTR1, which restricts all available copper stores. However, this has not been systematically measured. In SLC25A3 mutant cells, which have a localized copper deficiency in mitochondria, it has been reported that a less interconnected mitochondrial network and a mitochondrial fusion defect exists, that is, not explained by altered abundance of OPA1 or MITOFUSIN 1/2 or relative amount of different OPA1 forms (Seifert et al., 2016). This defect could be due to decreased mitochondrial copper which leads to changes in downstream targets such as COX17-MICOS complex. Additionally, yeast with lacking MDM38, a membrane-associated mitochondrial ribosome receptor with a role as K+/H+ exchange, have a mitochondrial fragmentation defect that can be suppressed by overexpression of PIC2 and MRS3 (Bauerschmitt et al., 2010; Zotova et al., 2010). The mechanism of that rescue is linked to multiple physiological changes, but it is possible to speculate that PIC2 and MRS3 could be contributing to this rescue via copper transport function of these proteins. Further investigation would be required to pinpoint the mechanisms of defects and rescue of morphology in these mutants.
A major challenge for defining additional roles of the copper transporting MCF SLC25A3/Pic2 is the fact it has been identified in many complexes. In fact, SLC25A3 was included in the Contaminant Repository for Affinity Purification (CRAPome) due it is abundance and “stickiness” (Mellacheruvu et al., 2013). So, determining which complexes are “real” requires directed hypotheses. These hypotheses could include phenotypes such as the mitochondrial morphology under multiple conditions. While these experiments can be labor intensive, armed with the knowledge gained to date, it is now reasonable to invest the time. One observation that could be further investigated is SLC25A3 was found associated with MITRAC but not included as an interactor due to abundance in control samples (Mick et al., 2012). The possibility of SLC25A3 interaction to MITRAC complex would form an attractive link between COA1 and SURF1 (MITRAC members with mitochondrial copper defect) and copper. The mechanism of this is under investigation in yeast where coa1∆ cells have decreased copper availability (Pierrel et al., 2007b). The copper deficit in mitochondria would limit the ability of Cox17 to complete its copper dependent interaction with MICOS raising the possibility of ultrastructure defect in this mutant. Unravelling this connection could further link MICOS to copper.
The interactions between MICOS and SAM and the affects that this could have on IMS content and connectivity to facilitate export of matrix components is understudied in metal homeostasis. The current model for copper and iron is that they are transported into the matrix by MCF proteins localized in the IM (Cobine et al., 2021). While transporters of iron in the OM have been proposed the directionality of the transport has not been completely resolved. For copper the model has always suggested that porins would act as IMS importers. If porin, or other proteins of the OM, were affected then the source of copper for SLC25A3/Pic2 could be disrupted. In addition, the model suggests that the copper chaperones (Cox17, Sco1, Cox11) are bypassed during import but used during export. Therefore, a link to mitochondrial ultrastructure and compartmentalization would help solve part of this bypass conundrum.
The assembly of heme and Fe-S are critical to aerobic eukaryotes and efforts to understand how the soluble metal is inserted into a hydrophobic molecule like protoporphyrin IX have long been discussed. The observation that this is mediated through a megacomplex answers some of the unresolved questions such as how do the substrates channel to the correct location and in part may also explain how a promiscuous enzyme such as ferrochelatase, that has been shown to insert multiple metals into the ring, can maintain specificity in vivo. Linking cofactor assembly to cristae morphology and therefore mitochondrial health is a way to coordinate investments of energy. If mitochondria are unable to produce heme or Fe-S then they can be eliminated as by mitophagy or at least reset by increased fission or fusion rates.
Clearly copper and iron are linked to mitochondrial ultrastructure changes. In addition, the uptake of zinc, manganese, and the export of the MoCo assembly intermediates could all depend on specific aspects of the MICOS machinery due to the fact that all must traverse the IMS during each process. Further understanding of the details of the metal and cofactor trafficking and the interactions with MICOS can provide information as to the regulation of these processes. It is possible that by locating these assembly proteins with a major complex that mediates critical morphology changes in the IM structure, means that metals and the cofactor assembly proteins have positioned themselves in an exclusive neighborhood and therefore are able to monitor, dictate and respond to changes in a dynamic manner to maintain cellular homeostasis and physiology.
Statements
Author contributions
PC, AM, JH, and TB wrote and the manuscript. PC and AM edited the final content. PC and JH constructed the figures.
Funding
This work is supported by the grants from National Institutes of Health [GM120211 to PC and DK111653 to AM]. PC is supported by the Alabama Agricultural Experiment Station.
Acknowledgments
Figures were created using BioRender.com and exported under a paid subscription. Thanks to Xinyu Zhu (Auburn University) for providing feedback on a version of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
AtkinsonA.KhalimonchukO.SmithP.SabicH.EideD.WingeD. R. (2010). Mzm1 Influences a Labile Pool of Mitochondrial Zinc Important for Respiratory Function. J. Biol. Chem.285, 19450–19459. 10.1074/jbc.m110.109793
2
AtkinsonA.SmithP.FoxJ. L.CuiT.-Z.KhalimonchukO.WingeD. R. (2011). The LYR Protein Mzm1 Functions in the Insertion of the Rieske Fe/S Protein in Yeast Mitochondria. Mol. Cell Biol.31, 3988–3996. 10.1128/mcb.05673-11
3
BakerZ. N.CobineP. A.LearyS. C. (2017). The Mitochondrion: a Central Architect of Copper Homeostasis. Metallomics9, 1501–1512. 10.1039/c7mt00221a
4
BakerZ. N.JettK.BouletA.HossainA.CobineP. A.KimB.-E.et al (2017). The Mitochondrial Metallochaperone SCO1 Maintains CTR1 at the Plasma Membrane to Preserve Copper Homeostasis in the Murine Heart. Hum. Mol. Genet.26, 4617–4628. 10.1093/hmg/ddx344
5
BanciL.BertiniI.CefaroC.Ciofi-BaffoniS.GalloA. (2011). Functional Role of Two Interhelical Disulfide Bonds in Human Cox17 Protein from a Structural Perspective. J. Biol. Chem.286, 34382–34390. 10.1074/jbc.m111.246223
6
BanciL.BertiniI.Ciofi-BaffoniS.HadjiloiT.MartinelliM.PalumaaP. (2008). Mitochondrial Copper(I) Transfer from Cox17 to Sco1 Is Coupled to Electron Transfer. Proc. Natl. Acad. Sci. U.S.A.105, 6803–6808. 10.1073/pnas.0800019105
7
BanciL.BertiniI.Ciofi-BaffoniS.LeontariI.MartinelliM.PalumaaP.et al (2007). Human Sco1 Functional Studies and Pathological Implications of the P174L Mutant. Proc. Natl. Acad. Sci. U.S.A.104, 15–20. 10.1073/pnas.0606189103
8
BashirK.IshimaruY.NishizawaN. K. (2011). Identification and Characterization of the Major Mitochondrial Fe Transporter in Rice. Plant Signal. Behav.6, 1591–1593. 10.4161/psb.6.10.17132
9
BashirK.IshimaruY.ShimoH.NagasakaS.FujimotoM.TakanashiH.et al (2011). The Rice Mitochondrial Iron Transporter Is Essential for Plant Growth. Nat. Commun.2, 322. 10.1038/ncomms1326
10
BauerschmittH.MickD. U.DeckersM.VollmerC.FunesS.KehreinK.et al (2010). Ribosome-binding Proteins Mdm38 and Mba1 Display Overlapping Functions for Regulation of Mitochondrial Translation. MBoC21, 1937–1944. 10.1091/mbc.e10-02-0101
11
BestwickM.JeongM.-Y.KhalimonchukO.KimH.WingeD. R. (2010). Analysis of Leigh Syndrome Mutations in the Yeast SURF1 Homolog Reveals a New Member of the Cytochrome Oxidase Assembly Factor Family. Mol. Cell Biol.30, 4480–4491. 10.1128/mcb.00228-10
12
BodeM.WoellhafM. W.BohnertM.LaanM. v. d.SommerF.JungM.et al (2015). Redox-regulated Dynamic Interplay between Cox19 and the Copper-Binding Protein Cox11 in the Intermembrane Space of Mitochondria Facilitates Biogenesis of Cytochrome C Oxidase. MBoC26, 2385–2401. 10.1091/mbc.e14-11-1526
13
BouletA.VestK. E.MaynardM. K.GammonM. G.RussellA. C.MathewsA. T.et al (2018). The Mammalian Phosphate Carrier SLC25A3 Is a Mitochondrial Copper Transporter Required for Cytochrome C Oxidase Biogenesis. J. Biol. Chem.293, 1887–1896. 10.1074/jbc.ra117.000265
14
CallegariS.MüllerT.SchulzC.LenzC.JansD. C.WisselM.et al (2019). A MICOS-TIM22 Association Promotes Carrier Import into Human Mitochondria. J. Mol. Biol.431, 2835–2851. 10.1016/j.jmb.2019.05.015
15
ChenW.DaileyH. A.PawB. H. (2010). Ferrochelatase Forms an Oligomeric Complex with Mitoferrin-1 and Abcb10 for Erythroid Heme Biosynthesis. Blood116, 628–630. 10.1182/blood-2009-12-259614
16
ChoH. M.RyuJ. R.JoY.SeoT. W.ChoiY. N.KimJ. H.et al (2019). Drp1-Zip1 Interaction Regulates Mitochondrial Quality Surveillance System. Mol. Cell73, 364–376. 10.1016/j.molcel.2018.11.009
17
ChojnackaM.GornickaA.OeljeklausS.WarscheidB.ChacinskaA. (2015). Cox17 Protein Is an Auxiliary Factor Involved in the Control of the Mitochondrial Contact Site and Cristae Organizing System. J. Biol. Chem.290, 15304–15312. 10.1074/jbc.m115.645069
18
ChristensonE. T.GallegosA. S.BanerjeeA. (2018). In Vitro reconstitution, Functional Dissection, and Mutational Analysis of Metal Ion Transport by Mitoferrin-1. J. Biol. Chem.293, 3819–3828. 10.1074/jbc.m117.817478
19
ChungJ.WittigJ. G.GhamariA.MaedaM.DaileyT. A.BergoniaH.et al (2017). Erythropoietin Signaling Regulates Heme Biosynthesis. Elife6. 10.7554/eLife.24767
20
ClaypoolS. M. (2009). Cardiolipin, a Critical Determinant of Mitochondrial Carrier Protein Assembly and Function. Biochimica Biophysica Acta (BBA) - Biomembr.1788, 2059–2068. 10.1016/j.bbamem.2009.04.020
21
ClaypoolS. M.OktayY.BoontheungP.LooJ. A.KoehlerC. M. (2008). Cardiolipin Defines the Interactome of the Major ADP/ATP Carrier Protein of the Mitochondrial Inner Membrane. J. Cell Biol.182, 937–950. 10.1083/jcb.200801152
22
CobineP. A.MooreS. A.LearyS. C. (2021). Getting Out what You Put in: Copper in Mitochondria and its Impacts on Human Disease. Biochimica Biophysica Acta (BBA) - Mol. Cell Res.1868, 118867. 10.1016/j.bbamcr.2020.118867
23
CobineP. A.OjedaL. D.RigbyK. M.WingeD. R. (2004). Yeast Contain a Non-proteinaceous Pool of Copper in the Mitochondrial Matrix. J. Biol. Chem.279, 14447–14455. 10.1074/jbc.m312693200
24
CobineP. A.PierrelF.LearyS. C.SasarmanF.HorngY.-C.ShoubridgeE. A.et al (2006). The P174L Mutation in Human Sco1 Severely Compromises Cox17-dependent Metallation but Does Not Impair Copper Binding. J. Biol. Chem.281, 12270–12276. 10.1074/jbc.m600496200
25
CunninghamC. N.RutterJ. (2020). 20,000 Picometers under the OMM: Diving into the Vastness of Mitochondrial Metabolite Transport. EMBO Rep.21, e50071. 10.15252/embr.202050071
26
DaileyH. A.FinneganM. G.JohnsonM. K. (1994). Human Ferrochelatase Is an Iron-Sulfur Protein. Biochemistry33, 403–407. 10.1021/bi00168a003
27
DaileyH. A.MeissnerP. N. (2013). Erythroid Heme Biosynthesis and its Disorders. Cold Spring Harb. Perspect. Med.3, a011676. 10.1101/cshperspect.a011676
28
DengH.QiaoX.XieT.FuW.LiH.ZhaoY.et al (2021). SLC-30A9 Is Required for Zn(2+) Homeostasis, Zn(2+) Mobilization, and Mitochondrial Health. Proc. Natl. Acad. Sci. U. S. A.118. 10.1073/pnas.2023909118
29
Dickson-MurrayE.NedaraK.ModjtahediN.TokatlidisK. (2021). The Mia40/CHCHD4 Oxidative Folding System: Redox Regulation and Signaling in the Mitochondrial Intermembrane Space. Antioxidants (Basel)10. 10.3390/antiox10040592
30
DietzJ. V.FoxJ. L.KhalimonchukO. (2021). Down the Iron Path: Mitochondrial Iron Homeostasis and beyond. Cells10. 10.3390/cells10092198
31
DietzJ. V.WilloughbyM. M.PielR. B.3rdRossT. A.BohovychI.AddisH. G.et al (2021). Mitochondrial Contact Site and Cristae Organizing System (MICOS) Machinery Supports Heme Biosynthesis by Enabling Optimal Performance of Ferrochelatase. Redox Biol.46, 102125. 10.1016/j.redox.2021.102125
32
DodaniS. C.LearyS. C.CobineP. A.WingeD. R.ChangC. J. (2011). A Targetable Fluorescent Sensor Reveals that Copper-Deficient SCO1 and SCO2 Patient Cells Prioritize Mitochondrial Copper Homeostasis. J. Am. Chem. Soc.133, 8606–8616. 10.1021/ja2004158
33
DudekJ.RehlingP.van der LaanM. (2013). Mitochondrial Protein Import: Common Principles and Physiological Networks. Biochimica Biophysica Acta (BBA) - Mol. Cell Res.1833, 274–285. 10.1016/j.bbamcr.2012.05.028
34
EaglesfieldR.TokatlidisK. (2021). Targeting and Insertion of Membrane Proteins in Mitochondria. Front. Cell Dev. Biol.9, 803205. 10.3389/fcell.2021.803205
35
EhsesS.RaschkeI.MancusoG.BernacchiaA.GeimerS.TonderaD.et al (2009). Regulation of OPA1 Processing and Mitochondrial Fusion by M-AAA Protease Isoenzymes and OMA1. J. Cell Biol.187, 1023–1036. 10.1083/jcb.200906084
36
EramoM. J.LisnyakV.FormosaL. E.RyanM. T. (2020). The 'mitochondrial Contact Site and Cristae Organising System' (MICOS) in Health and Human Disease. J. Biochem.167, 243–255. 10.1093/jb/mvz111
37
FormosaL. E.MaghoolS.SharpeA. J.ReljicB.Muellner-WongL.StroudD. A.et al (2022). Mitochondrial COA7 Is a Heme-Binding Protein with Disulfide Reductase Activity, Which Acts in the Early Stages of Complex IV Assembly. Proc. Natl. Acad. Sci. U. S. A.119. 10.1073/pnas.2110357119
38
GazaryanI. G.KrasinskayaI. P.KristalB. S.BrownA. M. (2007). Zinc Irreversibly Damages Major Enzymes of Energy Production and Antioxidant Defense Prior to Mitochondrial Permeability Transition. J. Biol. Chem.282, 24373–24380. 10.1074/jbc.m611376200
39
GeE. J.BushA. I.CasiniA.CobineP. A.CrossJ. R.DeNicolaG. M.et al (2022). Connecting Copper and Cancer: from Transition Metal Signalling to Metalloplasia. Nat. Rev. Cancer22, 102–113. 10.1038/s41568-021-00417-2
40
GhoshA.PrattA. T.SomaS.TheriaultS. G.GriffinA. T.TrivediP. P.et al (2016). Mitochondrial Disease genesCOA6,COX6BandSCO2have Overlapping Roles in COX2 Biogenesis. Hum. Mol. Genet.25, 660–671. 10.1093/hmg/ddv503
41
GhoshA.TrivediP. P.TimbaliaS. A.GriffinA. T.RahnJ. J.ChanS. S. L.et al (2014). Copper Supplementation Restores Cytochrome C Oxidase Assembly Defect in a Mitochondrial Disease Model of COA6 Deficiency. Hum. Mol. Genet.23, 3596–3606. 10.1093/hmg/ddu069
42
GlerumD. M.ShtankoA.TzagoloffA. (1996). SCO1 and SCO2 Act as High Copy Suppressors of a Mitochondrial Copper Recruitment Defect in Saccharomyces cerevisiae. J. Biol. Chem.271, 20531–20535. 10.1074/jbc.271.34.20531
43
GrossD. P.BurgardC. A.ReddehaseS.LeitchJ. M.CulottaV. C.HellK. (2011). Mitochondrial Ccs1 Contains a Structural Disulfide Bond Crucial for the Import of This Unconventional Substrate by the Disulfide Relay System. Mol. Biol. Cell22, 3758–3767. 10.1091/mbc.E11-04-0296
44
GuernseyD. L.JiangH.CampagnaD. R.EvansS. C.FergusonM.KelloggM. D.et al (2009). Mutations in Mitochondrial Carrier Family Gene SLC25A38 Cause Nonsyndromic Autosomal Recessive Congenital Sideroblastic Anemia. Nat. Genet.41, 651–653. 10.1038/ng.359
45
GyimesiG.HedigerM. A. (2020). Sequence Features of Mitochondrial Transporter Protein Families. Biomolecules10. 10.3390/biom10121611
46
HänzelmannP.HernándezH. L.MenzelC.García-SerresR.HuynhB. H.JohnsonM. K.et al (2004). Characterization of MOCS1A, an Oxygen-Sensitive Iron-Sulfur Protein Involved in Human Molybdenum Cofactor Biosynthesis. J. Biol. Chem.279, 34721–34732. 10.1074/jbc.m313398200
47
HarnerM.KörnerC.WaltherD.MokranjacD.KaesmacherJ.WelschU.et al (2011). The Mitochondrial Contact Site Complex, a Determinant of Mitochondrial Architecture. EMBO J.30, 4356–4370. 10.1038/emboj.2011.379
48
HeliasV.SaisonC.BallifB. A.PeyrardT.TakahashiJ.TakahashiH.et al (2012). ABCB6 Is Dispensable for Erythropoiesis and Specifies the New Blood Group System Langereis. Nat. Genet.44, 170–173. 10.1038/ng.1069
49
HennonS. W.SomanR.ZhuL.DalbeyR. E. (2015). YidC/Alb3/Oxa1 Family of Insertases. J. Biol. Chem.290, 14866–14874. 10.1074/jbc.r115.638171
50
HlynialukC. J.LingB.BakerZ. N.CobineP. A.YuL. D.BouletA.et al (2015). The Mitochondrial Metallochaperone SCO1 Is Required to Sustain Expression of the High-Affinity Copper Transporter CTR1 and Preserve Copper Homeostasis. Cell Rep.10, 933–943. 10.1016/j.celrep.2015.01.019
51
HorngY.-C.CobineP. A.MaxfieldA. B.CarrH. S.WingeD. R. (2004). Specific Copper Transfer from the Cox17 Metallochaperone to Both Sco1 and Cox11 in the Assembly of Yeast Cytochrome C Oxidase. J. Biol. Chem.279, 35334–35340. 10.1074/jbc.m404747200
52
JiS. G.MedvedevaY. V.WeissJ. H. (2020). Zn2+ Entry through the Mitochondrial Calcium Uniporter Is a Critical Contributor to Mitochondrial Dysfunction and Neurodegeneration. Exp. Neurol.325, 113161. 10.1016/j.expneurol.2019.113161
53
JumperJ.EvansR.PritzelA.GreenT.FigurnovM.RonnebergerO.et al (2021). Highly Accurate Protein Structure Prediction with AlphaFold. Nature596, 583–589. 10.1038/s41586-021-03819-2
54
KambeT.HashimotoA.FujimotoS. (2014). Current Understanding of ZIP and ZnT Zinc Transporters in Human Health and Diseases. Cell. Mol. Life Sci.71, 3281–3295. 10.1007/s00018-014-1617-0
55
KardonJ. R.MorocoJ. A.EngenJ. R.BakerT. A. (2020). Mitochondrial ClpX Activates an Essential Biosynthetic Enzyme through Partial Unfolding. Elife9. 10.7554/eLife.54387
56
KispalG.SiposK.LangeH.FeketeZ.BedekovicsT.JanákyT.et al (2005). Biogenesis of Cytosolic Ribosomes Requires the Essential Iron-Sulphur Protein Rli1p and Mitochondria. EMBO J.24, 589–598. 10.1038/sj.emboj.7600541
57
KnightS. A. B.YoonH.PandeyA. K.PainJ.PainD.DancisA. (2019). Splitting the Functions of Rim2, a Mitochondrial Iron/pyrimidine Carrier. Mitochondrion47, 256–265. 10.1016/j.mito.2018.12.005
58
KrishnamurthyP. C.DuG.FukudaY.SunD.SampathJ.MercerK. E.et al (2006). Identification of a Mammalian Mitochondrial Porphyrin Transporter. Nature443, 586–589. 10.1038/nature05125
59
KühlbrandtW. (2015). Structure and Function of Mitochondrial Membrane Protein Complexes. BMC Biol.13, 89. 10.1186/s12915-015-0201-x
60
LearyS. C.CobineP. A.KaufmanB. A.GuercinG.-H.MattmanA.PalatyJ.et al (2007). The Human Cytochrome C Oxidase Assembly Factors SCO1 and SCO2 Have Regulatory Roles in the Maintenance of Cellular Copper Homeostasis. Cell Metab.5, 9–20. 10.1016/j.cmet.2006.12.001
61
LillR. (2020). From the Discovery to Molecular Understanding of Cellular Iron-Sulfur Protein Biogenesis. Biol. Chem.401, 855–876. 10.1515/hsz-2020-0117
62
LindahlP. A.MooreM. J. (2016). Labile Low-Molecular-Mass Metal Complexes in Mitochondria: Trials and Tribulations of a Burgeoning Field. Biochemistry55, 4140–4153. 10.1021/acs.biochem.6b00216
63
LlorensJ. V.SorianoS.Calap-QuintanaP.Gonzalez-CaboP.MoltóM. D. (2019). The Role of Iron in Friedreich's Ataxia: Insights from Studies in Human Tissues and Cellular and Animal Models. Front. Neurosci.13, 75. 10.3389/fnins.2019.00075
64
LukE.CarrollM.BakerM.CulottaV. C. (2003). Manganese Activation of Superoxide Dismutase 2 in Saccharomyces cerevisiae Requires MTM1 , a Member of the Mitochondrial Carrier Family. Proc. Natl. Acad. Sci. U.S.A.100, 10353–10357. 10.1073/pnas.1632471100
65
MaT.ZhaoL.ZhangJ.TangR.WangX.LiuN.et al (2022). A Pair of Transporters Controls Mitochondrial Zn2+ Levels to Maintain Mitochondrial Homeostasis. Protein Cell13, 180–202. 10.1007/s13238-021-00881-4
66
MaioN.KimK. S.Holmes-HamptonG.SinghA.RouaultT. A. (2019). Dimeric Ferrochelatase Bridges ABCB7 and ABCB10 Homodimers in an Architecturally Defined Molecular Complex Required for Heme Biosynthesis. Haematologica104, 1756–1767. 10.3324/haematol.2018.214320
67
MayrS. J.RöperJ.SchwarzG. (2020). Alternative Splicing of the Bicistronic Gene Molybdenum Cofactor Synthesis 1 (MOCS1) Uncovers a Novel Mitochondrial Protein Maturation Mechanism. J. Biol. Chem.295, 3029–3039. 10.1074/jbc.ra119.010720
68
MedlockA. E.ShiferawM. T.MarceroJ. R.VashishtA. A.WohlschlegelJ. A.PhillipsJ. D.et al (2015). Identification of the Mitochondrial Heme Metabolism Complex. PLoS One10, e0135896. 10.1371/journal.pone.0135896
69
MellacheruvuD.WrightZ.CouzensA. L.LambertJ.-P.St-DenisN. A.LiT.et al (2013). The CRAPome: a Contaminant Repository for Affinity Purification-Mass Spectrometry Data. Nat. Methods10, 730–736. 10.1038/nmeth.2557
70
MendelR. R. (2013). The Molybdenum Cofactor. J. Biol. Chem.288, 13165–13172. 10.1074/jbc.r113.455311
71
MickD. U.DennerleinS.WieseH.ReinholdR.Pacheu-GrauD.LorenziI.et al (2012). MITRAC Links Mitochondrial Protein Translocation to Respiratory-Chain Assembly and Translational Regulation. Cell151, 1528–1541. 10.1016/j.cell.2012.11.053
72
Muñoz-GómezS. A.SlamovitsC. H.DacksJ. B.WidemanJ. G. (2015). The Evolution of MICOS: Ancestral and Derived Functions and Interactions. Commun. Integr. Biol.8, e1094593. 10.1080/19420889.2015.1094593
73
NaranuntaratA.JensenL. T.PazicniS.Penner-HahnJ. E.CulottaV. C. (2009). The Interaction of Mitochondrial Iron with Manganese Superoxide Dismutase. J. Biol. Chem.284, 22633–22640. 10.1074/jbc.m109.026773
74
Pacheu-GrauD.BarethB.DudekJ.JurisL.VögtleF.-N.WisselM.et al (2015). Cooperation between COA6 and SCO2 in COX2 Maturation during Cytochrome C Oxidase Assembly Links Two Mitochondrial Cardiomyopathies. Cell Metab.21, 823–833. 10.1016/j.cmet.2015.04.012
75
ParadkarP. N.ZumbrennenK. B.PawB. H.WardD. M.KaplanJ. (2009). Regulation of Mitochondrial Iron Import through Differential Turnover of Mitoferrin 1 and Mitoferrin 2. Mol. Cell Biol.29, 1007–1016. 10.1128/mcb.01685-08
76
PaulV. D.LillR. (2015). Biogenesis of Cytosolic and Nuclear Iron-Sulfur Proteins and Their Role in Genome Stability. Biochimica Biophysica Acta (BBA) - Mol. Cell Res.1853, 1528–1539. 10.1016/j.bbamcr.2014.12.018
77
Pebay-PeyroulaE.Dahout-GonzalezC.KahnR.TrézéguetV.LauquinG. J.-M.BrandolinG. (2003). Structure of Mitochondrial ADP/ATP Carrier in Complex with Carboxyatractyloside. Nature426, 39–44. 10.1038/nature02056
78
PfannerN.van der LaanM.AmatiP.CapaldiR. A.CaudyA. A.ChacinskaA.et al (2014). Uniform Nomenclature for the Mitochondrial Contact Site and Cristae Organizing System. J. Cell Biol.204, 1083–1086. 10.1083/jcb.201401006
79
PhillipsJ. D. (2019). Heme Biosynthesis and the Porphyrias. Mol. Genet. Metabolism128, 164–177. 10.1016/j.ymgme.2019.04.008
80
PielR. B.3rdShiferawM. T.VashishtA. A.MarceroJ. R.PraissmanJ. L.PhillipsJ. D.et al (2016). A Novel Role for Progesterone Receptor Membrane Component 1 (PGRMC1): A Partner and Regulator of Ferrochelatase. Biochemistry55, 5204–5217. 10.1021/acs.biochem.6b00756
81
PierrelF.BestwickM. L.CobineP. A.KhalimonchukO.CriccoJ. A.WingeD. R. (2007). Coa1 Links the Mss51 Post-translational Function to Cox1 Cofactor Insertion in Cytochrome C Oxidase Assembly. EMBO J.26, 4335–4346. 10.1038/sj.emboj.7601861
82
PierrelF.CobineP. A.WingeD. R. (2007). Metal Ion Availability in Mitochondria. Biometals20, 675–682. 10.1007/s10534-006-9052-9
83
PondarreC.CampagnaD. R.AntiochosB.SikorskiL.MulhernH.FlemingM. D. (2007). Abcb7, the Gene Responsible for X-Linked Sideroblastic Anemia with Ataxia, Is Essential for Hematopoiesis. Blood109, 3567–3569. 10.1182/blood-2006-04-015768
84
RampeltH.ZerbesR. M.van der LaanM.PfannerN. (2017). Role of the Mitochondrial Contact Site and Cristae Organizing System in Membrane Architecture and Dynamics. Biochimica Biophysica Acta (BBA) - Mol. Cell Res.1864, 737–746. 10.1016/j.bbamcr.2016.05.020
85
RuprechtJ. J.KingM. S.ZöggT.AleksandrovaA. A.PardonE.CrichtonP. G.et al (2019). The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell176, 435–447. 10.1016/j.cell.2018.11.025
86
RuprechtJ. J.KunjiE. R. S. (2020). The SLC25 Mitochondrial Carrier Family: Structure and Mechanism. Trends Biochem. Sci.45, 244–258. 10.1016/j.tibs.2019.11.001
87
SchorrS.van der LaanM. (2018). Integrative Functions of the Mitochondrial Contact Site and Cristae Organizing System. Seminars Cell & Dev. Biol.76, 191–200. 10.1016/j.semcdb.2017.09.021
88
SeguinA.JiaX.EarlA. M.LiL.WallaceJ.QiuA.et al (2020). The Mitochondrial Metal Transporters Mitoferrin1 and Mitoferrin2 Are Required for Liver Regeneration and Cell Proliferation in Mice. J. Biol. Chem.295, 11002–11020. 10.1074/jbc.ra120.013229
89
SeifertE. L.GálA.AcobaM. G.LiQ.Anderson-PullingerL.GolenárT.et al (2016). Natural and Induced Mitochondrial Phosphate Carrier Loss. J. Biol. Chem.291, 26126–26137. 10.1074/jbc.m116.744714
90
SenooN.KandasamyS.OgunbonaO. B.BaileM. G.LuY.ClaypoolS. M. (2020). Cardiolipin, Conformation, and Respiratory Complex-dependent Oligomerization of the Major Mitochondrial ADP/ATP Carrier in Yeast. Sci. Adv.6, eabb0780. 10.1126/sciadv.abb0780
91
ShahD. I.Takahashi-MakiseN.CooneyJ. D.LiL.SchultzI. J.PierceE. L.et al (2012). Mitochondrial Atpif1 Regulates Haem Synthesis in Developing Erythroblasts. Nature491, 608–612. 10.1038/nature11536
92
ShawG. C.CopeJ. J.LiL.CorsonK.HerseyC.AckermannG. E.et al (2006). Mitoferrin Is Essential for Erythroid Iron Assimilation. Nature440, 96–100. 10.1038/nature04512
93
SomaS.MorgadaM. N.NaikM. T.BouletA.RoeslerA. A.DziubaN.et al (2019). COA6 Is Structurally Tuned to Function as a Thiol-Disulfide Oxidoreductase in Copper Delivery to Mitochondrial Cytochrome C Oxidase. Cell Rep.29, 4114–4126. 10.1016/j.celrep.2019.11.054
94
SturtzL. A.DiekertK.JensenL. T.LillR.CulottaV. C. (2001). A Fraction of Yeast Cu,Zn-Superoxide Dismutase and its Metallochaperone, CCS, Localize to the Intermembrane Space of Mitochondria. J. Biol. Chem.276, 38084–38089. 10.1074/jbc.m105296200
95
SwaminathanA. B.GohilV. M. (2022). The Role of COA6 in the Mitochondrial Copper Delivery Pathway to Cytochrome C Oxidase. Biomolecules12. 10.3390/biom12010125
96
TaketaniS.KakimotoK.UetaH.MasakiR.FurukawaT. (2003). Involvement of ABC7 in the Biosynthesis of Heme in Erythroid Cells: Interaction of ABC7 with Ferrochelatase. Blood101, 3274–3280. 10.1182/blood-2002-04-1212
97
ThompsonK.MaiN.OláhováM.ScialóF.FormosaL. E.StroudD. A.et al (2018). OXA1L Mutations Cause Mitochondrial Encephalopathy and a Combined Oxidative Phosphorylation Defect. EMBO Mol. Med.10. 10.15252/emmm.201809060
98
TifounN.De Las HerasJ. M.GuillaumeA.BouleauS.MignotteB.Le FlochN. (2021). Insights into the Roles of the Sideroflexins/SLC56 Family in Iron Homeostasis and Iron-Sulfur Biogenesis. Biomedicines9. 10.3390/biomedicines9020103
99
TroadecM.-B.WarnerD.WallaceJ.ThomasK.SpangrudeG. J.PhillipsJ.et al (2011). Targeted Deletion of the Mouse Mitoferrin1 Gene: from Anemia to Protoporphyria. Blood117, 5494–5502. 10.1182/blood-2010-11-319483
100
TuncayE.BitirimC. V.OlgarY.DurakA.RutterG. A.TuranB. (2019). Zn2+-transporters ZIP7 and ZnT7 Play Important Role in Progression of Cardiac Dysfunction via Affecting Sarco(endo)plasmic Reticulum-Mitochondria Coupling in Hyperglycemic Cardiomyocytes. Mitochondrion44, 41–52. 10.1016/j.mito.2017.12.011
101
VanisovaM.BurskaD.KrizovaJ.DanhelovskaT.DosoudilovaZ.ZemanJ.et al (2019). Stable COX17 Downregulation Leads to Alterations in Mitochondrial Ultrastructure, Decreased Copper Content and Impaired Cytochrome C Oxidase Biogenesis in HEK293 Cells. Folia Biol. (Praha)65, 181–187.
102
VarabyovaA.TopfU.KwiatkowskaP.WrobelL.Kaus-DrobekM.ChacinskaA. (2013). Mia40 and MINOS Act in Parallel with Ccs1 in the Biogenesis of Mitochondrial Sod1. FEBS J.280, 4943–4959. 10.1111/febs.12409
103
VercellinoI.SazanovL. A. (2022). The Assembly, Regulation and Function of the Mitochondrial Respiratory Chain. Nat. Rev. Mol. Cell Biol.23, 141–161. 10.1038/s41580-021-00415-0
104
VestK. E.HashemiH. F.CobineP. A. (2013). The Copper Metallome in Eukaryotic Cells. Met. Ions Life Sci.12, 451–478. 10.1007/978-94-007-5561-1_13
105
VestK. E.LearyS. C.WingeD. R.CobineP. A. (2013). Copper Import into the Mitochondrial Matrix in Saccharomyces cerevisiae Is Mediated by Pic2, a Mitochondrial Carrier Family Protein. J. Biol. Chem.288, 23884–23892. 10.1074/jbc.m113.470674
106
VestK. E.WangJ.GammonM. G.MaynardM. K.WhiteO. L.CobineJ. A.et al (2016). Overlap of Copper and Iron Uptake Systems in Mitochondria in Saccharomyces cerevisiae. Open Biol.6, 150223. 10.1098/rsob.150223
107
WilkensS. (2015). Structure and Mechanism of ABC Transporters. F1000Prime Rep.7, 14. 10.12703/P7-14
108
YamamotoM.ArimuraH.FukushigeT.MinamiK.NishizawaY.TanimotoA.et al (2014). Abcb10 Role in Heme Biosynthesis In Vivo : Abcb10 Knockout in Mice Causes Anemia with Protoporphyrin IX and Iron Accumulation. Mol. Cell Biol.34, 1077–1084. 10.1128/mcb.00865-13
109
YangM.CobineP. A.MolikS.NaranuntaratA.LillR.WingeD. R.et al (2006). The Effects of Mitochondrial Iron Homeostasis on Cofactor Specificity of Superoxide Dismutase 2. EMBO J.25, 1775–1783. 10.1038/sj.emboj.7601064
110
YoonH.KnightS. A. B.PandeyA.PainJ.TurkarslanS.PainD.et al (2015). Turning Saccharomyces cerevisiae into a Frataxin-independent Organism. PLoS Genet.11, e1005135. 10.1371/journal.pgen.1005135
111
ZhuX.BouletA.BuckleyK. M.PhillipsC. B.GammonM. G.OldfatherL. E.et al (2021). Mitochondrial Copper and Phosphate Transporter Specificity Was Defined Early in the Evolution of Eukaryotes. Elife10. 10.7554/elife.64690
112
ZorkauM.AlbusC. A.Berlinguer-PalminiR.Chrzanowska-LightowlersZ. M. A.LightowlersR. N. (2021). High-resolution Imaging Reveals Compartmentalization of Mitochondrial Protein Synthesis in Cultured Human Cells. Proc. Natl. Acad. Sci. U. S. A.118. 10.1073/pnas.2008778118
113
ZotovaL.AleschkoM.SponderG.BaumgartnerR.ReipertS.PrinzM.et al (2010). Novel Components of an Active Mitochondrial K+/H+ Exchange. J. Biol. Chem.285, 14399–14414. 10.1074/jbc.m109.059956
Summary
Keywords
metals, copper, iron, heme, MICOS complex
Citation
Medlock AE, Hixon JC, Bhuiyan T and Cobine PA (2022) Prime Real Estate: Metals, Cofactors and MICOS. Front. Cell Dev. Biol. 10:892325. doi: 10.3389/fcell.2022.892325
Received
08 March 2022
Accepted
02 May 2022
Published
20 May 2022
Volume
10 - 2022
Edited by
Michał Wasilewski, The International Institute of Molecular Mechanisms and Machines Polish Academy of Sciences (IMol), Poland
Reviewed by
David Pacheu-Grau, University of Zaragoza, Spain
Martin Ott, Stockholm University, Sweden
Updates
Copyright
© 2022 Medlock, Hixon, Bhuiyan and Cobine.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul A. Cobine, paul.cobine@auburn.edu
This article was submitted to Cellular Biochemistry, a section of the journal Frontiers in Cell and Developmental Biology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.