 Yasunobu Okada1,2,3,4,5*
Yasunobu Okada1,2,3,4,5* Tomohiro Numata2
Tomohiro Numata2 Ravshan Z. Sabirov6
Ravshan Z. Sabirov6 Makiko Kashio1,3
Makiko Kashio1,3 Peter G. Merzlyak6
Peter G. Merzlyak6 Kaori Sato-Numata2
Kaori Sato-Numata2- 1National Institute for Physiological Sciences (NIPS), Okazaki, Japan
- 2Department of Integrative Physiology, Graduate School of Medicine, AkitaUniversity, Akita, Japan
- 3Department of Physiology, School of Medicine, Aichi Medical Uniersity, Nagakute, Japan
- 4Department of Physiology, Kyoto Prefectural University of Medicine, Kyoto, Japan
- 5Cardiovascular Research Institute, Yokohama City University, Yokohama, Japan
- 6Institute of Biophysics and Biochemistry, National University of Uzbekistan, Tashkent, Uzbekistan
Cell volume regulation (CVR) is a prerequisite for animal cells to survive and fulfill their functions. CVR dysfunction is essentially involved in the induction of cell death. In fact, sustained normotonic cell swelling and shrinkage are associated with necrosis and apoptosis, and thus called the necrotic volume increase (NVI) and the apoptotic volume decrease (AVD), respectively. Since a number of ubiquitously expressed ion channels are involved in the CVR processes, these volume-regulatory ion channels are also implicated in the NVI and AVD events. In Part 1 and Part 2 of this series of review articles, we described the roles of swelling-activated anion channels called VSOR or VRAC and acid-activated anion channels called ASOR or PAC in CVR and cell death processes. Here, Part 3 focuses on therein roles of Ca2+-permeable non-selective TRPM2 and TRPM7 cation channels activated by stress. First, we summarize their phenotypic properties and molecular structure. Second, we describe their roles in CVR. Since cell death induction is tightly coupled to dysfunction of CVR, third, we focus on their participation in the induction of or protection against cell death under oxidative, acidotoxic, excitotoxic, and ischemic conditions. In this regard, we pay attention to the sensitivity of TRPM2 and TRPM7 to a variety of stress as well as to their capability to physicall and functionally interact with other volume-related channels and membrane enzymes. Also, we summarize a large number of reports hitherto published in which TRPM2 and TRPM7 channels are shown to be involved in cell death associated with a variety of diseases or disorders, in some cases as double-edged swords. Lastly, we attempt to describe how TRPM2 and TRPM7 are organized in the ionic mechanisms leading to cell death induction and protection.
Introduction
Animal cells must regulate their cell volume even under physiological conditions with constant extracellular osmolarity. First, it is because the fluctuation of intracellular osmolarity is inevitably induced by cell activity per se which constantly requires osmolyte transport across the cell membrane and cell metabolism (anabolism and catabolism). Second, it is because cell volume changes are coupled to cell migration and cell proliferation connoting mitosis. After cell swelling and shrinkage, animal cells can shortly regulate their volume. The mechanisms of cell volume regulation are called the regulatory volume decrease (RVD) and the regulatory volume increase (RVI) that are attained by water movements driven by KCl efflux and NaCl influx, respectively (Hoffmann et al., 2009; Lang et al., 1998; Okada, 2004). In most types of mammalian cells, such volume-regulatory KCl and NaCl transports are principally accomplished by the activities of ubiquitously expressed anion and cation channels.
When the CVR mechanisms are persistently impaired, cells cannot survive. Sustained cell shrinkage and swelling are major hallmarks of the early, presumably earliest, events of necrotic and apoptotic cell death, and thus called the apoptotic volume decrease (AVD) (Maeno et al., 2000) and the necrotic volume increase (NVI) (Barros et al., 2001; Okada et al., 2001), respectively. Thus, these cell death processes are associated with dysregulation of a variety of ion transport mechanisms especially ion channels (Ritter et al., 2021). In the previous Part 1 article (Okada et al., 2021a) and Part 2 article (Okada et al., 2021b), we summarized the roles of the volume-sensitive outwardly rectifying anion channel (VSOR) (Okada, 1997), also called the volume-regulated anion channel (VRAC) (Nilius et al., 1997a), and those of the acid-sensitive outwardly rectifying anion channel (ASOR) (Wang et al., 2007), also called the proton-activated anion channel (PAC) (Yang et al., 2019), respectively, in CVR and cell death induction. To attain net KCl and NaCl transports, these anion channels should operate in parallel with K+ channels and Na+-permeable cation channels due to the electroneutrality constraint. In fact, altered activities of TRPM2 and TRPM7 cation channels are known to be implicated in CVR function/dysfunction and cell death induction. Here, we review the roles of TRPM2 and TRPM7 channels in the processes of CVR and cell death induction in this Part 3 article.
Phenotypic properties and molecular structures of TRPM2 and TRPM7
Chanzymes TRPM2 and TRPM7 as the members of sensor TRP channels
The Transient Receptor Potential (TRP) ion channel family consists of a large number (28 for human) of members, and is subdivided into 6 subfamilies in mammals: TRPC (“Canonical” or “Classical”), TRPM (“Melastatin”), TRPV (“Vanilloid”), TRPA (“Ankyrin”), TRPML (“MucoLipin”), and TRPP (PKD or “Polycystin”). TRP channels have a tetrameric subunit stoichiometry, with each subunit containing the cytoplasmic N- and C-terminal regions, six transmembrane domains (S1∼S6), and a pore-forming region between S5 and S6. TRP channels are non-selective cation-conductive membrane proteins and play a central role in physiological processes involving ionic signals. TRP channels are polymodal ion channels that have the role of integrating and transmitting a variety of environmental stimuli, including physical stimuli such as mechanical and thermal, and/or chemical stimuli such as pH and plant-derived compounds. These features serve as sensors to monitor the body’s extrinsic and intrinsic abnormalities while providing the basis for maintaining homeostasis that controls adaptive signals. Therefore, these are closely linked to health and disease and are attractive targets for drug discovery.
TRPM, the largest TRP subfamily, contains four melastatin domains, that are TRPM homology regions (MHR1-4), at the N-terminus and functions in a wide variety of cells throughout our body, including homeostasis-related cell proliferation, metabolism, cell death, and cancer (Dhakal and Lee, 2019; Fliniaux et al., 2018; Hantute-Ghesquier et al., 2018; Wong et al., 2019). As a main example, TRPM1: ON bipolar function of the retina (Koike et al., 2010), TRPM2: oxidative stress sensor function of cells and tissues (Naziroğlu, 2007), TRPM3: thermal sensor function (Vriens and Voets, 2019), TRPM4: regulator of cardiac conduction (Wang C. et al., 2018), TRPM5: taste sensor and blood glucose control capability (Vennekens et al., 2018), TRPM6: regulation of magnesium homeostasis (de Baaij et al., 2015), TRPM7: mechano-sensor function in cells (Numata et al., 2017a; Numata et al., 2017b) as well as synaptic and cognitive functions in the nervous system (Abumaria et al., 2019), and TRPM8: cold sensor function (Liu et al., 2020). Among these sensor TRPM channels, TRPM2, TRPM3, TRPM6, TRPM7, and TRPM8 are Ca2+-permeable cation channels activated by stress. In particular, TRPM2 and TRPM7 are unique ion channels possessing both ion channel and enzyme structures/activities and are called chanzymes
Physiological roles of TRPM2 and TRPM7
The activities of TRPM2 and TRPM7 are involved in a variety of physiological functions, as shortly summarized below.
TRPM2
The TRPM2 channel was first cloned by Nagamine et al. (Nagamine et al., 1998) and is a homo-tetrameric, non-voltage-activated, and non-selective cation channel expressed in a variety of cell types including neurons, pancreatic β cells, cardiomyocytes, and immune cells including monocytes/macrophages and neutrophils. The unique characteristic feature of TRPM2 is its temperature sensitivity (Togashi et al., 2006), and the temperature threshold decreases from around 47°C in the absence of oxidative stress to around 35.5°C in the presence of 100 μM H2O2 (Kashio et al., 2012). Thus, TRPM2 contributes to body temperature regulation (Song et al., 2016; Tan and McNaughton, 2016). TRPM2 is also involved in a variety of physiological functions including immunological cell responses, insulin secretion, and oxytocin release (Kashio and Tominaga, 2017; Szollosi, 2021).
TRPM7
TRPM7 is first cloned by three separate groups (Nadler et al., 2001; Runnels et al., 2001; Yamaguchi et al., 2001) and is a constitutively active, homo-tetrameric, non-selective cation channel with protein serine/threonine kinase activity (Nadler et al., 2001; Runnels et al., 2001; Schmitz et al., 2003). The TRPM7 channel is expressed in almost all tissues, including brain, heart, liver, kidney, lung, and spleen (Nadler et al., 2001; Runnels et al., 2001). Accumulating evidence has shown that TRPM7 is essentially involved in a variety of fundamental physiological cell functions, as listed below: cell viability and growth (Nadler et al., 2001; Hanano et al., 2004; Elizondo et al., 2005; Che et al., 2014), cell adhesion (Clark et al., 2006; Su et al., 2006), cytoskeletal regulation (Nadler et al., 2001; Clark et al., 2006), cell migration (Wei et al., 2009; Kuras et al., 2012), cellular and systemic Mg2+ homeostasis (Schmitz et al., 2003; He et al., 2005; Ryazanova et al., 2010; Mittermeier et al., 2019), permeation of trace metal ions into cells (Nadler et al., 2001; Monteilh-Zoller et al., 2003; Schmitz et al., 2003), neurotransmitter release (Krapivinsky et al., 2006; Brauchi et al., 2008), axonal growth (Turlova et al., 2016), and the activation and differentiation of immune cells (see Review: Nadolni and Zierler, 2018).
Biophysical properties of TRPM2 and TRPM7
The monomers of chanzymes TRPM2 and TRPM7 have largely common domain structures, these tetramers both operate as Ca2+-permeable cation channels with different biophysical properties, as summarized below.
TRPM2
TRPM2 has MHR1-4 with the IQ-motif at the large N-terminus, conserved six transmembrane segment (S1-S6) regions, and a pore-forming loop domain between S5 and S6 as well as the C-terminus composed of the TRP helix containing TRP box1 and TRP box2, a coiled-coil domain (CCD), and a unique enzymatic NUDT9 homology (NUDT9-H) domain (Figure 1). The IQ-like motif, located at amino acids (AA) 406–416 of MHR1-4 in human TRPM2 (hTRPM2), is important for Ca-CaM binding (Tong et al., 2006). An additional Ca-CaM-binding motif, W1355-I1368, was found in the NUDT9-H domain of hTRPM2 (Gattkowski et al., 2019). In the pore region, the FGQI motif (AA 979–982) was recently identified as the selectivity filter of hTRPM2 channel (Yu et al., 2021). The CCD is critical for the heteromeric assembly formation of TRPM2 (Mei et al., 2006). In addition, TRPM2 contains a structure of ADP-ribose (ADPR) pyrophosphatase enzyme, although this domain of hTRPM2 is catalytically inactive (Iordanov et al., 2016), and is activated by cytosolic ADPR (cADPR) and reactive oxygen species (ROS) (Hara et al., 2002). Recently, it became clear that ADPR binds not only to the C-terminal NUDT9-H domain (AA 1197–1503 for hTRPM2) but also to MHR1-2 located at amino acids 1–422, and it was revealed that the channel activity is greatly affected by the latter binding (Huang et al., 2018b; Huang et al., 2019).
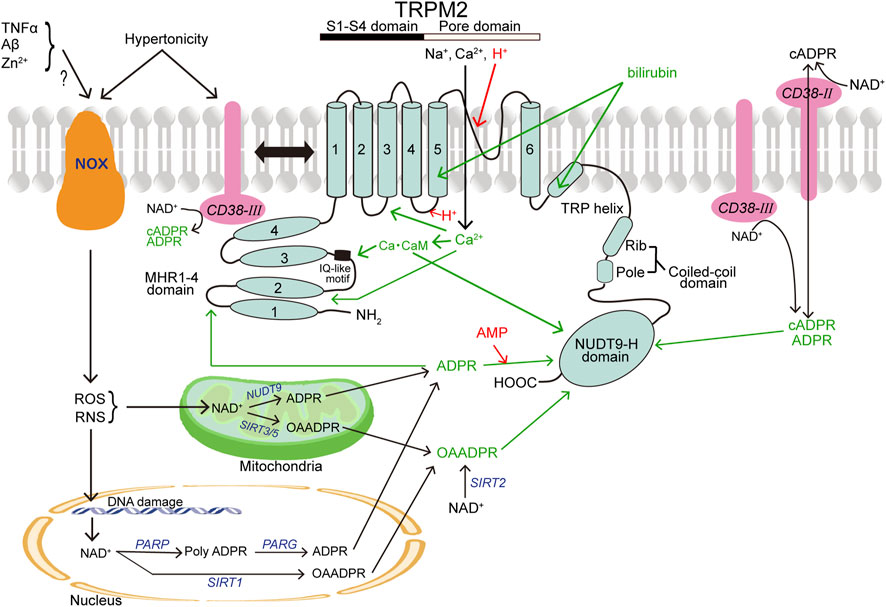
FIGURE 1. Topology model of a monomeric subunit of TRPM2 channel and its modulators. CB38 (painted in pink) and NOX (in brown) represent two other membrane-spanning proteins that are involved in activation of TRPM2 (in light blue). The names of TRPM2 activators and inhibitors are written in green and red, respectively. In light of the involvement in production of TRPM2 activators, a mitochondrion and a nucleus are also depicted. (See the text for details.)
Human TRPM2 exhibits a rapid inactivation time course after activation even in the persistent presence of activators (Csanády and Törocsik, 2009), whereas sea anemone Nematostella vectensis TRPM2 (nvTRPM2) does not show such inactivation kinetics (Zhang et al., 2018). The existence of electroneutral residues of G984 and Y985 in the putative selectivity filter of hTRPM2 is judged to be responsible for the inactivation time course, based on the following observations: First, hTRPM2 currents became non-inactivating, when these residues were replaced with acidic residues (G984D and Y985E) (Tóth and Csanády, 2012). Second, the corresponding residues (D1041 and E1042) for an ancient type of nvTRPM2 are acidic or negatively charged (Iordanov et al., 2019).
The TRPM2 single-channel current exhibits a unitary conductance of about 50–80 pS and a linear current-voltage (I-V) relationship. The whole-cell current also shows a linear I-V relationship. This channel has permeabilities not only to monovalent cations such as Na+, K+ and Cs+ but also to divalent cations such as Ca2+ and Mg2+ with the permeability ratios to a monovalent cation Na+ of 0.67–0.9 and 0.47–0.5, respectively (Kraft et al., 2004; Sano et al., 2001; Xia et al., 2008). Such prominent divalent cation permeabilities are attained by interaction with glutamate, glutamine, and aspartate residues (E960, Q981, D987, and E1022 for hTRPM2) (Belrose and Jackson, 2018; Sumoza-Toledo and Penner, 2011; Turlova et al., 2018; Xia et al., 2008), the residues of which are forming the selectivity filter in the vestibule of the TRPM2 channel pore region (AA 952-1022 for hTRPM2; AA 949-1019 for mouse TRPM2).
TRPM7
TRPM7 has MHR1-4 at the large N-terminus followed by the conserved six transmembrane segment (S1-S6) regions containing the pore-forming loop domain between S5 and S6 and the C-terminus (Figure 2). The C-terminus contains the TRP helix, CCD, and a unique α-kinase domain with an Mg2+‧ATP-binding site (Simon et al., 2013; Yee et al., 2014). TRPM7’s characteristic α-kinase activity and its channel function are shown to be independent of each other by site-directed mutagenesis analyses combined with biochemical and electrophysiological studies (Schmitz et al., 2003; Demeuse et al., 2006). On the other hand, the role of α-kinase activity is, in a manner independent of ion channel activity, involved in phosphorylation of downstream signaling molecules such as annexin A1 (Dorovkov and Ryazanov, 2004), myosin II (Clark et al., 2006; Clark et al., 2008a; Clark et al., 2008b), SMAD2 (Romagnani et al., 2017), and PLCγ2 (Deason-Towne et al., 2012) as well as some store-operated Ca2+ entry (SOCE) components related to STIM or Orai, thereby regulating SOCE (Faouzi et al., 2017).
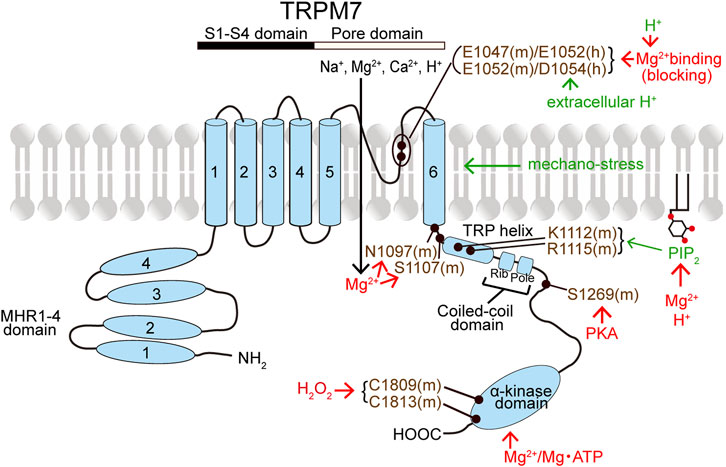
FIGURE 2. Topology model of a monomeric subunit of TRPM7 channel and its modulators. The names of TRPM7 activators and inhibitors are written in green and red, respectively. TRPM7 residues written in violet represent the putative action sites of TRPM7 activators and inhibitors. (See the text for details.)
The single-channel current of TRPM7 has a unitary conductance of around 40 pS at negative voltages (of around −70 mV) in the absence of extracellular Mg2+ (Kozak et al., 2005; Li et al., 2006). The whole-cell currents show a nearly linear I-V relationship with weak inward rectification in the absence of extracellular divalent cations, but a non-linear I-V relationship with strong outward rectification in the presence of extracellular divalent cations (Nadler et al., 2001; Runnels et al., 2001; Aarts et al., 2003; Monteilh-Zoller et al., 2003; Schmitz et al., 2003) and especially in the presence of extracellular Mg2+ due to its open channel blocking action (Kerschbaum et al., 2003). The residues of D1054 and E1052 within the pore-forming region of human TRPM7 provide the binding sites and the selective filter for Mg2+ and Ca2+ (Numata and Okada, 2008a). On the other hand, in the case of mouse TRPM7, E1047 and E1052 were shown to represent the selectivity filter for Mg2+ and Ca2+ (Li et al., 2007). Thus, TRPM7 exhibits permeabilities both to monovalent cations such as Na+ and Cs+ and to divalent metal cations such as Mg2+, Ca2+, Ba2+, Sr2+, Cd2+, and Zn2+. TRPM7 localized in intracellular vesicles mediates Zn2+ release from the vesicles upon ROS-induced TRPM7 activation (Abiria et al., 2017). TRPM7 is characteristic in its permeability to protons (Jiang et al., 2005; Numata and Okada, 2008b). The channel permeability to cations including divalent cations is initiated by attraction to the glutamate and aspartate residues, which are negative charges in the pore vestibule of the TRPM7 channel (Li et al., 2007; Numata and Okada, 2008a; Schlingmann et al., 2007; Yee et al., 2014). The TRPM7 channel exhibits inward proton conductance (Jiang et al., 2005) in a manner sensitive to the presence of extracellular Mg2+ and Ca2+ (Numata and Okada, 2008b). It appears that the proton conductivity of TRPM7 is mediated by the pore per se, because the charge-neutralizing mutation of Asp-1054 (D1054A) of human TRPM7 abolished its proton conductance (Numata and Okada, 2008b).
Activation and inactivation signals for TRPM2 and TRPM7
TRPM2 and TRPM7 channel activities are stimulated or inhibited by numbers of intracellular signaling molecules that are generated or mobilized in response to various environmental stresses, as detailed below.
TRPM2
Adenosine diphosphate ribose (ADPR) is a key gating molecule of the TRPM2 channel (Perraud et al., 2001) with an EC50 of 10–15 μM (Kolisek et al., 2005; Beck et al., 2006) and exerts the action through the binding not only to the C-terminal NUDT9-H domain of TRPM2 (Wehage et al., 2002; Heiner et al., 2003; Perraud et al., 2003; Kraft et al., 2004; Kolisek et al., 2005; Fliegert et al., 2017b) but also to the N-terminal MHR1/2 (Huang et al., 2018b; Huang et al., 2019) (Figure 1). A site-directed mutagenesis study showed that hydrogen bonding of R1433 and Y1349 in the C-terminus is required for hTRPM2 activation induced by the C-terminal binding of ADPR (Fliegert et al., 2017b). For the N-terminal ADPR-binding site, M215, Y295, and R302 were identified as the key residues (Huang et al., 2019). In response to stimulation by extracellular signals, especially oxidative stress (reactive oxygen species: ROS) and nitrosative stress (reactive nitrogen species: RNS), ADPR is generated from NAD+ in the cytosol via a special type of CD38 (see below), mitochondria via mitochondrial NUDT9 (Perraud et al., 2003; Perraud et al., 2005), and nuclei through the sequential actions of nuclear enzyme poly-ADPR polymerase (PARP) and poly-ADPR glycohydrolase (PARG) in response to DNA damage and PARP stimulation (Caiafa et al., 2009; Esposito and Cuzzocrea, 2009; Fauzee et al., 2010) (Figure 1). TRPM2 is also activated by ADPR upon stimulation with other extracellular stimuli including TNFα in mouse cardiac ventricular myocytes (Roberge et al., 2014), amyloid β-peptide (Aβ) in rat striatal cells (Fonfria et al., 2005), and Zn2+ in mouse microglial cells (Mortadza et al., 2017). ADPR-induced TRPM2 activation depends on the presence of phosphatidylinositol 4,5-bisphosphate (PIP2) in the inner leaflet of cell membrane (Tóth and Csanády, 2012; Zhang et al., 2018) and cytosolic Ca2+ (Du et al., 2009a; McHugh et al., 2003; Starkus et al., 2007; Tong et al., 2006). ADPR cannot activate TRPM2 in the absence of Ca2+ (McHugh et al., 2003; Starkus et al., 2007) and of calmodulin (CaM) (Tong et al., 2006). CaM directly interacts with the IQ-like motif (AA406-416) in the N-terminus of TRPM2 (Tong et al., 2006) as well as with the Trp-1355∼Ile-1368 motif in the NUDT9-H domain of hTRPM2 with a Kd of 110 nM (Gattkowski et al., 2019) (Figure 1). Intracellular Ca2+ alone was also reported to activate TRPM2 in a manner independent of ADPR and the ADPR-binding site of the C-terminus of TRPM2 by Du et al. (Du et al., 2009a), although such was not observed by other groups (Csanády and Törocsik, 2009; McHugh et al., 2003; Starkus et al., 2007). In any case, not only ADPR binding but also Ca2+ binding are prerequisite for TRPM2 channel gating (Csanády and Törocsik, 2009). By making site-directed mutagenesis combining with patch-clamp functional assay, it was suggested that the D267-D268 motif in the N-terminus as a Ca2+-binding EF-loop is also critical for hTRPM2 channel activation induced by an unrealistically high concentration (50 mM) of Ca2+ (Luo et al., 2018). In addition, cryo-EM study recently revealed that the Ca2+-binding site is located at the intracellular border of the channel in between S2, S3, and the TRP helix coordinated by E843 and Q846 of S2, N809 of S3, and E1073 of the TRP helix of hTRPM2 (Wang L. et al., 2018). ROS and RNS were shown to stimulate the TRPM2 channel in intact cells (Hara et al., 2002; Wehage et al., 2002) in an indirect manner through promotion of ADPR generation (Fonfria et al., 2004; Perraud et al., 2005; Buelow et al., 2008; Blenn et al., 2011) (Figure 1). However, it is noteworthy that a splice variant of TRPM2 with deletion of the C-terminus can still be activated by H2O2 (though not by ADPR), suggesting that there exists some ADPR-independent activation mechanism. Of note, H2O2 reduces temperature thresholds for TRPM2 activation, thereby inducing TRPM2 activation at body temperature (37°C), in a manner independent of ADPR (Kashio et al., 2012). In connection with this, exposure to H2O2 was shown to induce tyrosine phosphorylation of TRPM2 with activation of TRPM2 channels (Zhang et al., 2007), though the detailed mechanism is not clarified as yet. TRPM2 is also known to be activated by nicotinamide dinucleotide (NAD+) (Sano et al., 2001; Hara et al., 2002; Wehage et al., 2002) and by its metabolite, nicotinic acid adenine dinucleotide phosphate (NAADP) (Beck et al., 2006) as well as by cADPR (Fleig and Penner, 2004; Mei et al., 2006; Perraud et al., 2001; Xia et al., 2008). However, NAD+-induced TRPM2 activation observed previously (Sano et al., 2001; Hara et al., 2002; Naziroğlu and Lückhoff, 2008) is now considered to be due to contamination with ADPR or metabolism of NAD+ (Beck et al., 2006; Grubisha et al., 2006), because after affinity-purification, NAD+ and NAADP were found to be incapable of stimulating TRPM2 even at concentrations considerably higher than their cytosolic concentrations (Tóth et al., 2015). In contrast, cADPR can directly gate the TRPM2 channel at high concentrations (Kolisek et al., 2005) with an EC50 of 60–120 μM (Beck et al., 2006) and therefore seems to exert as a non-physiological low affinity agonist (Yu et al., 2019). O-acetyl-ADP ribose (OAADPR), which is generated in response to ROS/RNS by protein deacetylase sirtuins in the cytosol (via SIRT2), mitochondria (via SIRT1), and nuclei (via SIRT3/5), can also directly activate TRPM2 by binding to the NUDT9-H domain (Grubisha et al., 2006; Tong and Denu, 2010) (Figure 1). TRPM2 activity involves a multifunctional single-pass transmembrane glycoprotein enzyme, CD38, which converts NAD+ and NAAD into cADPR and then hydrolyzes cADPR to ADPR (Kim et al., 1993; Takasawa et al., 1993; Zocchi et al., 1993). Also, it is noted that CD38 is not only a generator of cADPR and ADPR but also a transmembrane transporter of cADPR and ADPR (Franco et al., 1998; Guida et al., 2004), thereby mediating the intracellular actions of cADPR and ADPR generated extracellularly (Figure 1). The most widely known type of CD38 (Type II) is ectoenzyme with the catalytic domain facing outside. In recent years, it was shown that there exists another type of CD38 (Type III) with an opposite orientation of which the catalytic domain is facing the cytosol, thereby catalyzing the synthesis of intracellular cADPR (Liu et al., 2017; Zhao et al., 2012). We showed, for the first time, that TRPM2 physically interacts to CD38, and activity of the ∆C-variant of TRPM2 (TRPM2-∆C) functioning as a hypertonicity-induced cation channel (HICC) is regulated by this direct interaction between TRPM2 and CD38 (Numata et al., 2012). Indeed, the CD38-TRPM2 interaction was shown to play important roles in oxytocin secretion (Higashida et al., 2018), natural killer cell activity (Rah et al., 2015), and chronic inflammation (García-Rodríguez et al., 2018; Balinas et al., 2019). Also, it is noted that hypertonic stimulation induces ROS production in kidney cells (Ikari et al., 2013; Yang et al., 2005) as well as HeLa cells (Numata et al., 2012) and a marked increase in the intracellular cADPR concentration (Numata et al., 2012). Moreover, both cationic currents activated by hypertonicity and by cADPR were inhibited not only by TRPM2-siRNA but also by CD38-siRNA (Numata et al., 2012). Thus, it is concluded that hypertonic stress induces increases in intracellular cADPR, presumably via activation of CD38, thereby activating the TRPM2 channel as HICC (Figure 1). 2′-Deoxy-ADPR, which is an endogenous nucleotide synthesized from nicotinamide mononucleotide (NMN) and deoxy-ATP by consecutive action of an NMN adenylyl transferase and CD38, was identified as an additional agonist of TRPM2 (Fliegert et al., 2017a), although its binding site is not identified as yet. One type of growth factors, vascular endothelial growth factor (VEGF), was shown to activate TRPM2 thereby inducing Ca2+ influx in vascular endothelial cells leading to cadherin phosphorylation (Mittal et al., 2015) and angiogenesis (Negri et al., 2019). Most recently, bilirubin and its derivatives were found to activate TRPM2 channels from the extracellular side by directly interacting with K928 and D1069 existing in the S5 and TRP helix regions of TRPM2, respectively (Figure 1), presumably after getting into an intramembrane deep cavity surrounded by the S3, S5 and TRP helix (Liu et al., 2023).
TRPM2 channel activity is sensitive to extracellular and intracellular acidification. Du et al. (Du et al., 2009b) found that extracellular protons inhibit TRPM2 with an IC50 of pH 5.3 by interacting at H958, D964, and E994 existing in the outer vestibule of the TRPM2 pore, whereas Yang et al. (Yang et al., 2010) found that extracellular proton-induced TRPM2 inhibition (with IC50 of pH 4.7) is mediated by binding to several residues in the outer vestibule of the pore especially K953 and D1002. In contrast, Starkus et al. (Starkus et al., 2010) reported that extracellular acidification inhibits TRPM2 with an IC50 of pH 6.5 after permeating the TRPM2 pore and interacting with an intracellular site. Intracellular protons were observed to completely suppress TRPM2 activity at pH 6 by competitively antagonizing intracellular Ca2+ binding by the latter group (Starkus et al., 2010) and with an IC50 of pH 6.7 by interacting at D933 in the S4-S5 linker region thereby decreasing sensitivity to intracellular Ca2+ and/or intracellular ADPR by the former group (Du et al., 2009b) (Figure 1). AMP, which is a breakdown product of ADPR, is known to specifically antagonize ADPR-induced activation of TRPM2 with an IC50 of 10–70 μM (Kolisek et al., 2005; Beck et al., 2006; Lange et al., 2008) through a competition for the Nudix domain (Figure 1). A most abundant trace metal, Zn2+, inactivates the channel activity of TRPM2 overexpressed in HEK293 cells, in a manner dependent on membrane potentials, by the extracellular application (Yang et al., 2011). Zn2+-induced suppression was found to be full for the inward currents but only partial for the outward currents, suggesting an open-channel blocking action. Positively charged Lys952 and negatively-charged Asp1002 in the outer pore region may provide the blocking site of Zn2+, because charge-neutralizing mutations of these residues (K952A and D1002A) were observed to strongly attenuate the Zn2+-induced suppression (Yang et al., 2011). On the other hand, another trace metal, Cu2+, inhibited, in a manner independent of voltages, hTRPM2 currents with an IC50 of 2.6 μM, when applied extracellularly but not intracellularly (Zeng et al., 2012). However, Cu2+ was found to fail to affect the activity of mouse TRPM2 (mTRPM2), in which the residue corresponding to H995 of hTRPM2 is Q992, though the inhibitory effect of Cu2+ on hTRPM2 was bolstered (Yu et al., 2014). In fact, H995 was found to be critical for Cu2+-induced hTRPM2 inactivation, because charge-neutralizing mutation of His995 (H995A or H995Q) in the pore-forming region abolished the Cu2+-induced suppression (Yu et al., 2014).
TRPM7
The constitutive activity of TRPM7 channel is maintained by PIP2 (Runnels et al., 2002; Kozak et al., 2005; Gwanyanya et al., 2006). Thus, TRPM7 currents are inactivated by PIP2 depletion caused by PLC-coupled GPCR stimulation (Runnels et al., 2002; Langeslag et al., 2007) and by expression of voltage-sensitive phosphatase (VSP) (Xie et al., 2011). PIP2 is known to bind to cationic residues of some TRPs, including TRPV1, TRPM8, and TRPM4 (Rohács et al., 2005; Bousova et al., 2015; Poblete et al., 2015). Similarly, K1112 and R1115 existing in the TRP domain were suggested to be required for PIP2 dependence of mouse TRPM7 (Figure 2), because TRPM7 currents were found to be ablated by the K1112Q/R1115Q double mutation (Xie et al., 2011). In contrast to the inhibitory effect of PIP2 depletion, transient PIP2 hydrolysis was rather found to augment TRPM7 currents (Langeslag et al., 2007).
Intracellular free Mg2+ and Mg‧ATP suppress, in a manner independent of voltages, TRPM7 channel activity (Nadler et al., 2001; Runnels et al., 2001; Hermosura et al., 2002; Kozak and Cahalan, 2003; Schmitz et al., 2003). Depletion of intracellular Mg2+ or Mg‧ATP augments TRPM7 currents (Nadler et al., 2001; Kozak and Cahalan, 2003; Demeuse et al., 2006; Langeslag et al., 2007). Intracellular free Mg2+ completely inhibits TRPM7 currents at millimolar concentrations (Nadler et al., 2001) and suppresses the currents in a dually concentration-dependent manner with two independent sites. Such dual Mg2+-induced inhibitory effects were thus described by two IC50 values of 10–25 and 90–165 μM in Jurkat T lymphocytes (Chokshi et al., 2012b; Chokshi et al., 2012c) or of 5.6–6.5 and 467–558 μM in TRPM7-overexpressing HEK293 cells (Inoue et al., 2021; Inoue et al., 2014). Extracellular Mg2+ also inhibits TRPM7 currents (Nadler et al., 2001) in a manner dependent on voltages with IC50 values of 3.2 μM at −40 mV and 0.11 mM at +80 mV (Numata et al., 2007b), indicating voltage-dependent Mg2+ block of the TRPM7 channel pore. Intracellular Mg‧ATP inhibits TRPM7 activity with an IC50 of 2 mM (Demeuse et al., 2006). Sensitivity to intracellular Mg2+ and Mg‧ATP may be mediated by C-terminal sites (Figure 2), one within and another outside the kinase domain (Schmitz et al., 2003; Demeuse et al., 2006; Yu H. et al., 2013). The kinase activity is not essential for the TRPM7 channel activity, because mutation of two autophosphorylation sites or of a key catalytic site that abolished kinase activity never affected the channel activity (Matsushita et al., 2005). However, the interaction between the kinase domain and channel domain is involved in the modulation of channel activity by altering the sensitivity to Mg2+ and Mg‧ATP (Demeuse et al., 2006; Yu H. et al., 2013). In fact, recently, it was clarified that the channel domain-kinase domain interaction increases TRPM7 currents by decreasing Mg2+-induced inhibition (Inoue et al., 2021). In this study, after cleaving the kinase domain, the channel domain (AA 1-1509) per se was found to be sensitive to intracellular Mg2+ with an IC50 of 3.0 μM, and the interaction of the channel domain with the kinase domain was shown to rather decrease intracellular Mg2+ sensitivity (Inoue et al., 2021). Based on the truncation studies, the CCD of zebrafish TRPM7 (drTRPM7) was suggested to be involved in the channel’s regulation by Mg2+ and Mg‧ATP (Jansen et al., 2016). An involvement of the inter-subunit region between S6 and the TRP domain in the intracellular Mg2+ sensitivity of TRPM7 gating was also suggested, because the S1107E mutant of mouse TRPM7 (mTRPM7) exhibited constitutively active channels in a manner insensitive to intracellular Mg2+ (Hofmann et al., 2014). Indeed, the Mg2+-insensitive S1107E mutant of mTRPM7 was shown to become less sensitive to PIP2 depletion (Zhelay et al., 2018). Recently, N1097 of mTRPM7 was also indicated to form the intracellular Mg2+ regulatory site, because the N1097Q mutation abrogated the inhibition of TRPM7 channel by physiological intracellular Mg2+ concentration (Schmidt et al., 2022). In addition, intracellular Mg2+ was reported to inhibit TRPM7 channel activity by screening the negatively charged PIP2 (Kozak et al., 2005) and thereby disrupting the PIP2-TRPM7 interaction (Zhelay et al., 2018) (Figure 2). Recently, cAMP/PKA was shown to downregulate the TRPM7 activity and expression by phosphorylating TRPM7 at S1269 existing near the CCD region (Tian et al., 2018; Broertjes et al., 2019) (Figure 2). Furthermore, TRPM7 channel activity was demonstrated to be inhibited by ADP-ribosylation factor-like GTPase 15 (ARL15) through forming a macromolecular complex together with TRPM7 and cystathione-β-synthase (CBS)-pair domain divalent metal cation transport mediator (CNNM) (Kollewe et al., 2021; Mahbub et al., 2023).
TRPM7 currents are activated by cytosolic alkalinization and inactivated by intracellular acidification (Kozak et al., 2005). Cytosolic protons inhibit TRPM7 channel with an IC50 of pH 6.3 (Chokshi et al., 2012c) by a charge screening of PIP2, thereby disrupting the PIP2-TRPM7 interaction (Kozak et al., 2005; Zhelay et al., 2018). In contrast, extracellular acidification potentiates TRPM7 presumably by proton-induced unbinding of Ca2+ and Mg2+, thereby removing the blocking effects of Ca2+ and Mg2+ (J. Jiang et al., 2005), at the divalent cation binding sites, the negatively charged E1047 and E1052 for mTRPM7 (Li et al., 2007), and E1052 and D1054 for human TRPM7 (hTRPM7) (Numata and Okada, 2008a) in the pore region (Figure 2). In fact, hTRPM7 activation by extracellular protons was abolished by electro-neutralizing D1054 mutation, but not charge-preserving D1054E mutation (Numata et al., 2019).
TRPM7 was shown to be activated by ROS under anoxic conditions (Aarts et al., 2003). Such an enhancing effect of ROS may be caused by increased expression of TRPM7 mRNA and protein in cells exposed to oxidant agents (Wuensch et al., 2010; Nuñez-Villena et al., 2011). In contrast, recently, increased extracellular H2O2 concentrations were found to inhibit TRPM7 currents in a manner dependent on Mg2+ (with an IC50 of 16 μM) but not on ATP (Inoue et al., 2014). The Mg2+-insensitive S1107E mutant of mTRPM7 is not affected by H2O2 (Inoue et al., 2021). Therefore, it is likely that the inhibitory effect of H2O2 is based on the enhancement of intracellular Mg2+ sensitivity. Supportively, C1809 and C1813 locating in the kinase domain of mTRPM7, which are essential not only for the Mg2+ sensitivity but also for kinase activity (Runnels et al., 2001), were shown to exert as the oxidative stress sensor in the presence of intracellular Mg2+ (H. Inoue et al., 2021).
Mechano-stress is an additional activating factor for TRPM7. Under the whole-cell recordings, osmotic cell swelling and shear stress induced by perfusion of bath solution were found to augment Mg2+-sensitive cation currents in hTRPM7-transfected, but not mock-transfected, HEK293 cells (Numata et al., 2007a). The shear stress-induced augmentation of whole-cell TRPM7 current was not affected by an exocytosis-blocking reagent brefeldin A under the experimental conditions employed in this study, although laminar flow-induced shear stress was reported to cause exocytotic translocation of TRPM7 to the plasma membrane in some cell types (Oancea et al., 2006). It is noteworthy that even under the cell-free inside-out configuration, membrane stretch was found to directly activate single-channel activity of TRPM7 by increasing the open probability (Po) (Numata et al., 2007a). Moreover, similar Mg2+-sensitive single-channel and whole-cell cation currents were also observed upon application of membrane stretch and hypotonic stress, respectively, in human epithelial HeLa cells, in which TRPM7 is endogenously expressed, in a manner sensitive to siRNA-mediated knockdown of TRPM7 (Numata et al., 2007b). In contrast, it was suggested that TRPM7 senses the osmotic gradient rather than membrane stretch in HEK293 cells transfected with hTRPM7, on the basis of observations that whole-cell currents were only slightly increased by cell ballooning induced by intracellular pressure application during observation of TRPM7 activity under the whole-cell configuration (Bessac and Fleig, 2007). In this study, however, there remains a possibility that whole-cell TRPM7 currents were largely pre-activated by cell swelling caused by oncotic pressure due to the cytosolic presence of considerable amount of non-diffusible large organic (colloidal) osmolytes under the experimental conditions where the intracellular pipette solution and the extracellular bath solution had the identical osmolarity. Also, swelling-activated and shrinkage-inhibited Cl− currents might have been, at least in part, contaminated in the whole-cell currents recorded under Cl−-rich conditions. The fact that TRPM7 directly senses mechanical stimulation was, in fact, confirmed in human bone marrow mesenchymal stem cells by measuring suction-induced and hydrostatic pressure-induced membrane currents (Xiao et al., 2015) and in HEK293 cells transfected with mTRPM7 by observing pressure-induced cytosolic Ca2+ increases (R. Zhao et al., 2019). Mechano-sensitivity of TRPM7 was also shown by Ca2+ imaging in mouse mesenchymal stroma cells in response to fluid shear stress (Liu et al., 2015), in rat odontoblasts in response to hypotonic stimulation (Won et al., 2018), and in human MDA-MB-231 adenocarcinoma cells in response to a hydrostatic pressure increase (Zhao et al., 2019). The mechano-sensitivity of TRPM7 might be implicated in hypertension, since molecular TRPM7 expression in vascular smooth muscle cells was found to be decreased in spontaneously hypertensive rats (Touyz et al., 2006).
Pharmacological properties of TRPM2 and TRPM7
For TRPM2 and TRPM7, a large number of potent antagonists have been identified, but only several agonists were found, as listed below. However, most of them are not so specific to TRPM2 or TRPM7. Most of the binding sites for antagonists and agonists of TRPM2 and TRPM7 await future identification.
TRPM2
Since ADPR and cADPR are endogenous activators of TRPM2 channels, it is quite natural that TRPM2 activity is inhibited by ADPR analogs, 8-bromo-ADP-ribose (8-Br-ADPR), at 900 μM (Partida-Sanchez et al., 2007), 8-phenyl-2′-deoxy-ADPR with an IC50 of 3 μM (Moreau et al., 2013), and Compound 7i and 8a at 5–6 μM (Luo et al., 2018) as well as by a cADPR analog, 8-bromo-cyclic ADP-ribose (8-Br-cADPR), at ≥100 μM (Kolisek et al., 2005; Beck et al., 2006) by antagonizing the binding of ADPR and cADPR. Recently, 8-Br-cADPR was shown to exert an inhibitory action by binding not to the C-terminal binding site, the NUDT9-H domain, but to the N-terminal binding site, the U-shaped MHR1/2 domain (Huang et al., 2019). PARP inhibitors, SB750139-13, PJ34, and DPQ, are also effective to suppress TRPM2 activity with IC50 values of 25.1 nM, 2.0 μM, and 15.8 μM, respectively (Fonfria et al., 2004). TRPM2 inhibition was observed to be abolished by exposure to a cell-permeable peptide targeting the Nudix motif of TRPM2, tat-M2NX, at 100 μM (Shimizu et al., 2016). Hydroxyl radical scavengers, dimethylthiourea (DMTU) and N-2-mercaptopropyonyl glycine (MPG), were shown to inhibit TRPM2 activity (Smith et al., 2003; Ishii et al., 2006). However, it must be noted that these chemicals exert inhibitory actions not directly to TRPM2 but indirectly via scavenging hydroxyl radicals. A Janus kinase 2 (JAK2) inhibitor tyrphostin, AG490, was also found to indirectly antagonize, in a manner independent of JAK2, TRPM2 channel activity by scavenging hydroxyl radicals (Shimizu et al., 2014). AG490-related compounds, AG555 and AG556, blocked H2O2-induced activation of TRPM2 channels more strongly than AG490 (Toda et al., 2016). A known phospholipase A2 (PLA2) inhibitor, N-(p-amylcinnamoyl) anthranilic acid (ACA), also blocks TRPM2 currents, in a manner independent of inhibition of PLA2, when applied extracellularly (but not intracellularly), with an IC50 of 1.7 μM (Kraft et al., 2006). However, it must be noted that ACA is not specific for TRPM2 but is also known to inhibit activities of TRPM8 and TRPC6 (Kraft et al., 2006). Recently, one of the derivatives of ACA, called compound A23, was found to be a more effective and selective blocker for TRPM2 with an IC50 of 788 nM (Zhang et al., 2021).
TRPM2 currents are blocked by an antipyretic acid-derivative nonsteroidal anti-inflammatory drug (NSAID), flufenamic acid (FFA), at 50–1000 μM with an IC50 of 70 μM (Hill et al., 2004a). FFA also inhibits HICC/TRPM2-∆C activity (Wehner et al., 2003b) with an IC50 of 117 μM (Numata et al., 2007c). However, it must be noted that FFA affects not only TRPM2 but also other ion channels including some chloride, sodium, potassium and calcium channels (Guinamard et al., 2013). FFA analogs, mefenamic acid (MFA) and niflumic acid (NFA), are also effective to inhibit TRPM2 channels with IC50 values of 76 and ∼120 μM, respectively (Chen et al., 2012). Another fenamate analog, 2-aminoethoxydiphenyl borate (2-APB), suppresses TRPM2 activity with an IC50 of 1.2 μM (Togashi et al., 2008) and HICC activity with an IC50 of 525 μM (Numata et al., 2007c). 2-APB is not specific for TRPM2, because it also inhibits other TRPM members (Togashi et al., 2008) including TRPM7 (Li et al., 2006) and multiple TRPC channels (Xu et al., 2005) but activates several TRPV channels (Hu et al., 2004). In addition, a natural plant-derived polyphenol, curcumin, was more recently found to inhibit TRPM2 channels with an IC50 of around 50 nM (Kheradpezhouh et al., 2016). However, curcumin has been shown to inhibit a variety of ion channels such as K+ channels, Ca2+ channels, CFTR, and VSOR/VRAC Cl− channels (Zhang et al., 2014). Furthermore, TRPM2 activity is sensitive to antifungal agents, clotrimazole and econazole, with IC50 values of 3–30 μM (Hill et al., 2004b) as well as miconazole with IC50 of <3 μM (Togashi et al., 2008). However, these antifungal agents are not specific TRPM2 blockers but are known to block TRPV5 (Nilius et al., 2001), Ca2+-activated IK channels (Jensen et al., 1998), ATP-sensitive K+ channel (Jäger et al., 2004), and L-type Ca2+ channels (Thomas et al., 1999).
In contrast to these TRPM2 inhibitors, an analgesic and antipyretic drug, acetaminophen, was found to activate TRPM2 in rat hepatocytes at high concentrations (10–15 μM) (Kheradpezhouh et al., 2014).
TRPM7
TRPM7 inhibitors are divided into five categories: 1. In vivo metabolites such as sphingosine and spermine. 2. Natural products such as waixenicin A, carvacrol, and ginsenoside-Rd (GS-Rd) as well as quinine. 3. Non-specific channel blockers including NS8593, SKF-9635, and 2-APB. 4. Enzyme antagonists such as nafamostat, CCT128930, 5-ligoxygenase (5-LOX) inhibitors, and TG100-115. 5. Anesthesia-related drugs including lidocaine and midazolam.
Sphingosine is an amino alcohol forming a cell membrane phospholipid and potently inhibits TRPM7 currents with an IC50 of 590 nM (Qin et al., 2013). Sphingosine also blocks TRPM6 (IC50 of 460 nM) but neither TRPM2 nor TRPM4. Its structural analog fingolimod FTY720, which is an immunosuppressant and the first oral drug for treatment of multiple sclerosis, blocks TRPM7 with an IC50 of 720 nM (Qin et al., 2013). These facts may suggest their inhibiting actions are mediated by interacting with membrane phospholipids. Spermine is a tetravalent cationic polyamine and can block, in a voltage-dependent manner, TRPM7 channel activity with an IC50 of 2.3 μM from the extracellular side (Kozak et al., 2002) but not by the intracellular application (Kerschbaum et al., 2003). Single substitution of Ser-1107, which is known to be the site for sensitivity to intracellular Mg2+ (Hofmann et al., 2014), of TRPM7 by Glu (S1107E) was found to reduce the sensitivity not only to Mg2+ but also to spermine (Zhelay et al., 2018). Thus, it is conceivable that spermine-induced inhibition of TRPM7 activity is, in a manner similar to intracellular Mg2+, mediated by electrostatic screening and resultant disruption of interaction between PIP2 and TRPM7 (Zhelay et al., 2018).
Several natural products have been shown to block TRPM7 channels. Among them, the most potent and specific blocker is waixenicin A which is a xenicane diterpenoide derived from marine soft coral (Zierler et al., 2011). Waixenicin A inhibits, in a manner dependent on the intracellular Mg2+ concentration ([Mg2+]i), TRPM7 with IC50 values of 16 nM and 7 μM in the presence and absence of 0.7 mM [Mg2+]i, respectively. Since K1648R mutation of Mg2+-binding site on the kinase domain increased the IC50 value to 2.5 μM in the absence of [Mg2+]i, it is suggested that intracellular Mg2+ facilitates the binding of waixenicin A to TRPM7 (Zierler et al., 2011). Waixenicin A is exceptionally selective to TRPM7 against other TRP channels including TRPM6 (Zierler et al., 2011; Beesetty et al., 2018) and even against zebrafish TRPM7 (Jansen et al., 2016). TRPM7 is inhibited by carvacrol, which is a monoterpene phenolic compound derived from plant volatile oils, with an IC50 of 306 μM (Parnas et al., 2009). GS-Rd isolated from kampo herbal medicine ginseng was shown to inhibit TRPM7 with an IC50 of 170–178 μM (Kim, 2013). A plant alkaloid quinine, which is isolated from the bark of a cinchona, is used to treat malaria and a known blocker for Ca2+-activated K+ channels, can effectively inhibit TRPM7 channels at 30 μM (Chubanov et al., 2012).
A non-selective cation channel blocker NS8593, an aminobenzimidazole derivative, inhibits TRPM7 currents, in a manner sensitive to intracellular Mg2+, with IC50 values of 1.6 and 5.9 μM in the absence and presence of 0.3 mM [Mg2+]i, respectively (Chubanov et al., 2012). Since the mutation of Tyr-1049 on the pore-forming loop (Y1049P) resulted in reduction of IC50 values to 0.47 and 1.9 μM in the absence and presence of 0.3 mM [Mg2+]i, respectively, the TRPM7 pore loop is likely to be involved in the interaction between NS8593 and TRPM7 (Chubanov et al., 2012). NS8593 was shown to be selective at 10 μM to TRPM7 against other TRPs including TRPM2, TRPM3, TRPM5, TRPM8, TRPC6, TRPV1, and TRPA1 (Chubanov et al., 2012). Another non-selective cation channel blocker, 2-APB, inhibits TRPM7 channels with an IC50 of 178 μM but enhances TRPM6 channels with an EC50 of 205 μM (Li et al., 2006). Its inhibiting action is not direct to TRPM7 but indirect through an intracellular acidification (Chokshi et al., 2012a). 2-APB also inhibits the other multiple TRP members especially TRPM2, as mentioned in the above section. A classical non-selective cation channel blocker SKF-96365 exhibits irreversible full inhibition of TRPM7 at 20 μM (Kozak et al., 2002). However, it is noted that SKF-96365 can block voltage-dependent T-type Ca2+ channels as well (Singh et al., 2010).
Some blockers for a variety of enzymes have been shown to block TRPM7. A synthetic broad-spectrum serine protease inhibitor, nafamostat, inhibits TRPM7 currents in a manner dependent on voltages and on extracellular divalent cations (Chen X. et al., 2010). In this study, the IC50 values were found to be 15 μM at −100 mV and 121 μM at +100 mV in Ca2+- and Mg2+-free bathing solution, whereas those values increased to 514 μM at −100 mV and 617 μM at +100 mV in the presence of extracellular 1 mM Ca2+ and Mg2+ in TRPM7-transfected HEK293 cells. Nafamostat-induced inhibition was largely depressed by the charge-neutralizing mutation of Glu-1052 (E1052A) (Chen X. et al., 2010). Thus, it is concluded that Glu-1052 is one of negatively charged residues important for inhibition of TRPM7 channels not only by divalent cations (Numata and Okada, 2008a) but also by nafamostat. Endogenous TRPM7 expressed in mouse hippocampal neurons was inhibited by nafamostat with an IC50 of 27 μM in the presence of a low concentration (0.1 mM) of Ca2+ and Mg2+ in a bath solution, whereas the channels were unexpectedly found to be augmented by pre-application of nafamostat (Chen X. et al., 2010). A potent AKT inhibitor CCT128930 preferentially blocks, in an Mg2+-independent manner, TRPM7 with an IC50 of 0.63–0.86 μM, compared to TRPM6 and TRPM8, presumably by interacting with multiple residues in the selectivity filter (Guan et al., 2021). 5-LOX inhibitors such as NDGA, AA861, and MK886, were also found to inhibit TRPM7 channel activity, in a manner independent of the effects on 5-LOX, at 10–20 μM (Chen H. C. et al., 2010). In addition, TG100-115, which is a PI3Kγ/δ inhibitor, was found to inhibit not only the TRPM7 kinase activity with an IC50 of 1.07 μM but also the TRPM7 channel activity (Song et al., 2017).
Several anesthetic drugs were identified to be effective inhibitors of TRPM7 channels. Local anesthetic lidocaine inhibits TRPM7 currents with an IC50 of 11.1–11.6 mM in a voltage-independent and frequency-dependent manner (Leng et al., 2015). A widely used clinical anesthetic benzodiazepine, midazolam, can suppress TRPM7 currents by treatment for seconds and also inhibit TRPM7 expression by treatment for 48 h (Chen J. et al., 2016).
In addition to these TRPM7 inhibitors belonging to five categories, VER155008, which is an adenosine-derivative inhibitor of heat shock protein 70 (Hsp70), was recently shown to potently suppress TRPM7 channel activity in a manner independent of the kinase activity without affecting TRPM2, TRPM3, TRPM6, and TRPM8 channels (Rössig et al., 2022).
A δ-opioid receptor antagonist, naltriben, was found to be an effective activator of TRPM7 (Hofmann et al., 2014). Naltriben voltage-independently activated TRPM7 channels with an EC50 of 20.7 μM in a manner independent of intracellular Mg2+ and competitive with NS8593, but had no effect on TRPM2, TRPM8, and TRPV1 channels. The site of naltriben action is most likely localized in or near the TRP domain, because the S1107E mutant, which is a constitutively active channel insensitive to intracellular Mg2+, became insensitive to naltriben. A benzimidazole compound, milbefradil, which displays structural homology to benzimidazole NS8593, activated TRPM7-mediated Ca2+ entry with an EC50 of 53 μM and activated TRPM7 currents at 100 μM with a physiological intracellular Mg2+ concentration (0.9 mM) but failed to activate with a higher [Mg2+]i (1.8 mM), whereas milbefradil failed to activate TRPM3, TRPA1, and TRPV1 channels (Schäfer et al., 2016). This study also showed that the S1104E mutation of TRPM7 became insensitive to melbefradil. However, it must be pointed out that milbefradil inhibits voltage-dependent T-type Ca2+ channels (Bezprozvanny and Tsien, 1995; Viana et al., 1997) and volume-sensitive VSOR/VRAC Cl− channels (Nilius et al., 1997b) as well.
Three-dimensional structures of TRPM2 and TRPM7
In the last several years, the three-dimensional structures of TRP channels have become well elucidated owing to the studies with applying cryo-electron microscopy (cryo-EM), as recently reviewed (Cao, 2020; Huffer et al., 2020; Zubcevic, 2020). Here, such cryo-EM structures of TRPM2 and TRPM7 are shortly summarized below.
TRPM2
The three-dimensional structure of TRPM2 channel has been established by cryo-EM (Huang et al., 2020; Szollosi, 2021) using orthologous recombinant proteins from starlet sea anemone N. vectensis (nvTRPM2) (Zhang et al., 2018), zebrafish Danio rerio (drTRPM2) (Huang et al., 2018b; Yin et al., 2019), and human Homo sapience (hTRPM2 or hsTRPM2) (Huang et al., 2019; Wang L. et al., 2018; Yu et al., 2021). Like other six-transmembrane domain cation channels, TRPM2 proteins formed homotetramers with an overall shape reminiscent of a square prism or a bell with a height of up to 16 nm and a large cytosolic part (∼80%) (Figure 3A). The structure of the voltage sensor-like domain (formed by S1-S4) and of the pore domain (formed by S5-S6) resembles that of other voltage-gated cation channels (Figure 3B). However, unlike voltage-gated K+ channels, in TRPM2 (like in other TRPs), the voltage sensor-like domain lacks the conserved array of positively charged arginine and lysine and interacts with the S5-S6 of the adjacent subunit in a domain-swapping manner (Cao, 2020; Xia et al., 2019). Thus, TRPM2 is largely insensitive to voltage. The transmembrane portion of the pore is ∼5 nm long. It begins with an external vestibule with a diameter of ∼1 nm (for nvTRPM2) lined by a double ring of negative charges. The vestibule is followed by a short (∼1 nm) selectivity filter with a constriction of ∼0.52 nm in diameter, which was mostly invariant for all orthologues viewed in detergent micelles (Figure 3C). However, the ligand-free hTRPM2 reconstituted into the lipid nanodiscs had much narrower and ion-impermeable selectivity filter of ∼0.11 nm (Figure 3C), suggesting that it may serve as an upper gate which allowed passage of cysteine-modifying Ag+ only in the open but not in the closed state (Yu et al., 2021). The open TRPM2 pore is large enough to pass hydrated Na+ ions and tetramethylammonium but not N-methyl-D-glucosamine (NMDG). At around 3.5 nm from the entrance is located the gate which was interpreted as a lower gate by Yu et al. (Yu et al., 2021) formed by the S6 helices. Here, the pore is too tight (diameter is less than 0.2 nm for all orthologues in the closed state) to pass even a single water molecule. In the open state of the drTRPM2 bound to Ca2+ and ADP-ribose (ADPR), the lower gate widens up to a diameter of ∼0.9 nm allowing passage of hydrated Na+ and Ca2+ ions. This gate is followed by an internal vestibule, which is also negatively charged but narrower (diameter of 0.47 nm for nvTRPM2) compared to the external entrance. After passing the inner vestibule, ions come to the cytoplasmic cavity that has upper and lower chambers. This part is best described for the nvTRPM2 (Zhang et al., 2018). A bent tunnel with a diameter of over 0.4 nm connects the upper chamber of the cytoplasmic cavity with the Ca2+-binding site. This site is located near the membrane-cytosol interface but outside the central pore axis (Figure 3A). It is formed mostly by the ends of S2 helices and is accessible not only from the cytoplasmic cavity but also from the outside the protein via peripheral tunnels. Thus, the cytosolic parts of the channel form a porous structure with a complex system of cavities. The cytosolic Ca2+ ions first reach the Ca2+-binding site via peripheral tunnels moving nearly parallel to the membrane surface, then come to the upper cytoplasmic cavity, and only then may access the selectivity filter when the gate is open. Although a calmodulin-mediated mechanism was suggested for TRPM2 activation by cytosolic Ca2+ (Du et al., 2009a), mutations of the negatively charged amino acids at the Ca2+-binding site (E893A in nvTRPM2) of the channel protein itself greatly reduced or even abolished Ca2+-induced activation (Zhang et al., 2018), suggesting a key role of Ca2+ coordination within the Ca2+-binding pocket in the channel activation mechanism. ADPR, an obligate co-activator, binds to the U-shaped MHR1/2 and to the C-terminal NUDT9-H domain, which is unique for TRPM2 and is homologous to the mitochondrial ADPR pyrophosphatase NUDT9 (Huang et al., 2019; Huang et al., 2020; Szollosi, 2021) (Figures 3A, B). Based on the previous finding that nvTRPM2 but not hTRPM2 is activated by ADPR even after deletion of the NUDT9-H domain (Kühn et al., 2016) and on the comparison between the ADPR-binding sites of sea anemone, zebrafish, and human orthologues, it is speculated that the ADPR-binding site could be evolutionarily shifted from the MHR1/2 to the NUDT9-H domain of TRPM2 protein (Fliniaux et al., 2018) and that MHR1/2 could bind cADPR, which synergistically enhances the effect of ADPR (Fliegert et al., 2020). The ROS-dependent increase in ADPR production may link TRPM2-mediated Ca2+ influx to the inflammasome activation (Wang et al., 2020). MHR3/4 domains link the agonist-sensing regions (NUDT9-H and MHR1/2) with the pore and thus transduces chemical activation signals to the channel opening (Huang et al., 2018b; Huang et al., 2020). PIP2 binding is also necessary for TRPM2 channel activity and is thought to occur near the Ca2+-binding site (Huang et al., 2020; Szollosi, 2021; Zhang et al., 2018), although its exact site is undetermined. Molecular and structural nature of the temperature sensing by TRPM2 awaits future elucidation, although a role of a dynamic protein-membrane lipid relationship was suggested as the general concept (Zubcevic, 2020).
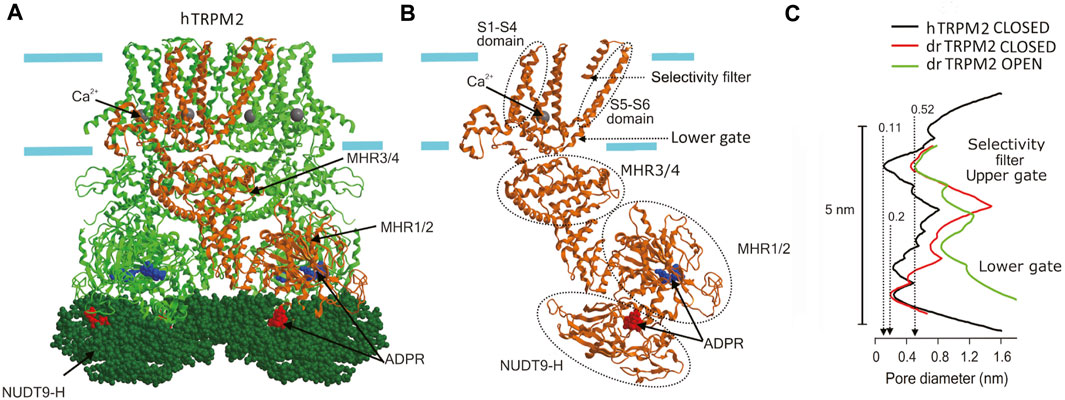
FIGURE 3. Cryo-EM structure of TRPM2. (A) The structure of hTRPM2 viewed parallel to the membrane plane. The drawing is based on the 6PUS.pdb file downloaded from https://www.rcsb.org/structure/6PUS. (B) Single subunit [depicted in (A) in brown] showing the structural elements mentioned in the text. (C) The pore radius along the central axis for drTRPM2 in the closed (apo, red line) and open (ADPR/Ca2+-bound, green line) states (Huang et al., 2018b) and for hTRPM2 reconstituted into the lipid nanodiscs in closed (black line) state (Yu et al., 2021). Approximate positions of the selectivity filter (upper gate) and the lower gate [also shown in (B)] are indicated.
TRPM7
Unlike TRPM2, TRPM7 is an Mg2+-permeable channel, which, in addition, possesses the protein kinase domain which is enzymatically active. The cryo-EM structure of the slightly truncated mTRPM7 protein fused to the maltose binding protein revealed an overall similarity to other TRP channels (Figure 4A) in terms of tetrameric assembly and orientation of helices (Duan et al., 2018; Huang et al., 2020). The tight lower gate at the cytoplasmic end of S6 (N1097) is similar to that of the TRPM2. The conduction pathway did not change upon removal of Mg2+ and other divalent cations (Figure 4C). The Mg2+-binding site is formed by the negative charge of Glu (E1047 for mTRPM7) and backbone carbonyl of Gly (G1046 for mTRPM7), which along with F1045 form the selectivity filter within the pore region (P-loop) of S5-S6 (Figure 2; Figure 4B). This region also contains a disulfide bond, which is stabilized by Mg2+ and important for regulation by glutathione. The structure of the mouse TRPM7 resolved by Duan et al. (Duan et al., 2018) did not contain the C-terminal kinase domain which possesses cysteine residues (C1809 and C1813 for mTRPM7) important for the second low-affinity Mg2+-binding site and for TRPM7 channel inhibition by oxidative stress (Inoue et al., 2021; Inoue et al., 2014) (Figure 2). Very recently, Nadezhdin et al. (Nadezhdin et al., 2023) analyzed cryo-EM structures of constitutively activated mTRPM7 channels with using the gain-of-function N1098Q mutant (Schmidt et al., 2022) and wild type mTRPM7 stimulated with a potent TRPM7 activator, naltriben. This study showed that the open-state of TRPM7 channel is associated with an increase in the pore size at the lower gate (near N1097) to ∼0.23 nm and turning of the tyrosine (Y1085) hydroxyls towards the ion conducting pathway thereby forming the narrowest point of the pore (Figure 4C). This study also identified a binding site for highly potent inhibitors, VER155008 and NS8593, which stabilize the closed conformation of TRPM7 channel. Although the cryo-EM structure of the kinase domain of mouse TRPM7 is still missing, this structure was previously revealed by X-ray crystallography and found to be similar to classical protein kinases such as PKA (Yamaguchi et al., 2001).
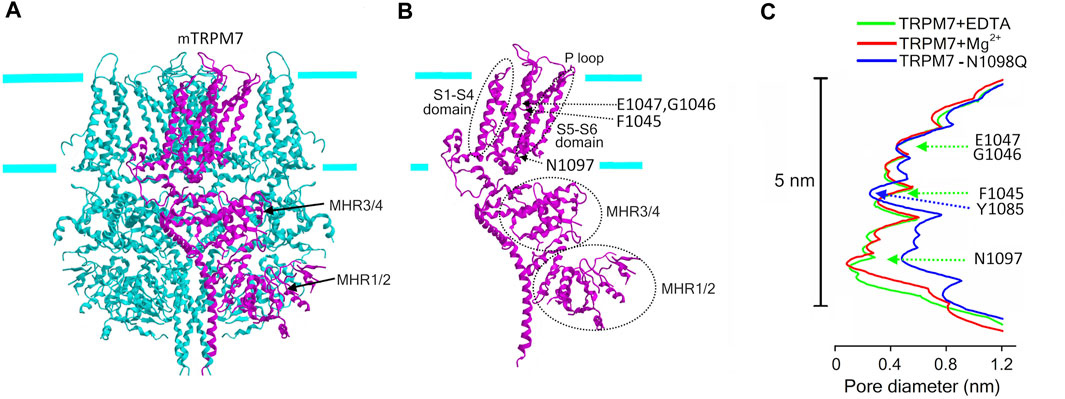
FIGURE 4. Cryo-EM structure of TRPM7. (A) The structure of mTRPM7 viewed parallel to the membrane plane. The C-terminal protein kinase domain is truncated in this structure. The drawing is based on the 5ZX5. pdb file downloaded from https://www.rcsb.org/structure/5ZX5. (B) Single subunit (depicted in (A) in magenta) showing the structural elements mentioned in the text. (C) The pore radius along the central axis for mTRPM7 in the presence of EDTA (green line) and high Mg2+ (red line) and for the constitutively active N1098Q mutant (blue line) (Duan et al., 2018; Nadezhdin et al., 2023). Indicated amino acids mark the selectivity filter (F1045, G1046, E1047) with the Mg2+-binding site formed by G1046 and E1047, and the lower gate (N1097), and the narrowest point of the pore of open-state channel (Y1085).
Roles of TRPM2 and TRPM7 in cell volume regulation and cell death induction/protection
Roles of TRPM2 and TRPM7 in cell volume regulation
Animal cells must cope with fluctuations of osmotic gradient across the cell membrane by two types of cell volume regulation mechanisms: cell volume recovery after osmotic shrinkage called RVI and that after osmotic swelling called RVD (see Reviews: Hoffmann et al., 2009; Okada, 2004; Wehner et al., 2003a). After hypertonic and hypotonic challenges, animal cells attain RVI and RVD mainly by net gain of most abundant extracellular small osmolytes Na+ and Cl− and by net loss of most abundant intracellular small osmolytes K+ and Cl−, respectively, with accompanying fluxes of osmotically obligated water, thereby readjusting the intracellular osmolarity to the extracellular osmolarity. In these volume regulation processes, TRPM2 and TRPM7 play important roles in animal cells.
RVI and TRPM2
RVI is known to be accomplished by Na+-conductive hypertonicity-induced cation channels (HICCs) and/or Na+-permeable electroneutral transporters such as Na+/H+ antiporter (NHE), Na+-K+-Cl− symporter (NKCC) and Na+-Cl− symporter (NCC) (see Reviews: Hoffmann et al., 2009; Okada, 2004). Among them, HICC was shown to be the most effective mechanism of RVI (Wehner et al., 2006; Plettenberg et al., 2008). To attain volume-regulatory NaCl influx, parallel activation of some unidentified type of anion channel, tentatively labelled the hypertonicity-induced anion channel (HIAC) (Okada, 2016), is to be required. Such HIAC-like currents were notably observed in human hepatoma HepG2 cells (Bondarava et al., 2009).
HICC currents were, for the first time, recorded in response to an isotonic challenge in Intestine 407 cells equilibrated under hypotonic conditions, and were suggested to be involved in the post-RVD RVI process (Okada and Hazama, 1989). So far, two groups of HICCs have been reported: the one is amiloride-sensitive and Gd3+-insensitive, and another is amiloride-insensitive, Gd3+- and/or FFA-sensitive (Wehner et al., 2003a; Wehner et al., 2006). Amiloride-sensitive HICC currents were originally observed in rat hepatocytes and shown to be involved in RVI (Wehner et al., 1995). Molecular correlates for this type of HICC in hepatocytes and HepG2 cells have been suggested to be several members of ENaC (Böhmer and Wehner, 2001; Plettenberg et al., 2008; Bondarava et al., 2009). In addition to δENaC, recently TRPM2 and TRPM5 were also suggested to be implicated in the molecular architecture of HICC in HepG2 (Koos et al., 2018). Gd3+-sensitive amiloride-insensitive HICC currents involved in RVI were first found in airway epithelial cells (Chan and Nelson, 1992), and were shown to be also sensitive to FFA in mouse cortical collecting duct M-1 cells (Volk et al., 1995). The molecular entity of this type of HICC was identified as TRPM2-∆C plus CD38 in HeLa cells, and the RVI event was shown to be inhibited by knockdown of TRPM2 or CD38 (Numata et al., 2012). The molecular mechanism of TRPM2-mediated RVI is schematically depicted in Figure 5A.
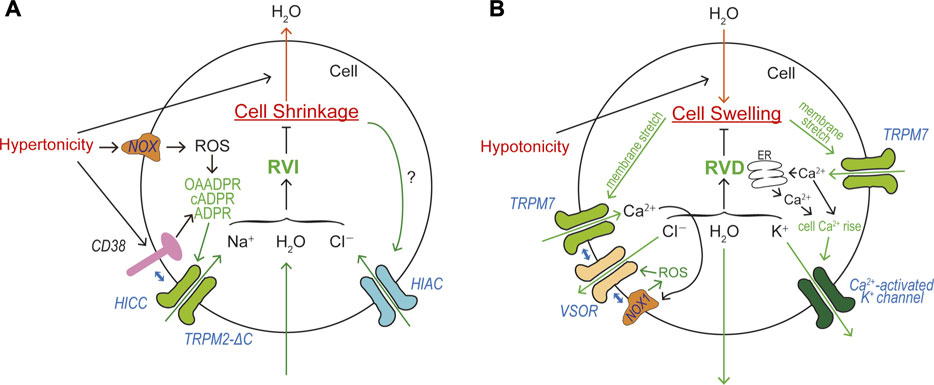
FIGURE 5. Roles of TRPM2 and TRPM7 in cell volume regulation (CVR). (A) Roles of HICC (TRPM2-∆C plus CD38) and HIAC in CVR attained after cell shrinkage, RVI. Depolarization caused by HICC activation drives Cl− influx via the HIAC pore. Resultant NaCl influx drives water influx leading to RVI. (B) Roles of TRPM7, VSOR, and Ca2+-activated K+ channel in CVR attained after cell swelling, RVD. Hyperpolarization predominantly caused by Ca2+-activated K+ channels drives Cl− efflux via the VSOR channel pore. Resultant KCl efflux drives water efflux leading to RVD.
RVD and TRPM7
RVD is a prerequisite function for animal cells devoid of the cell wall which is covering plant cells. RVD is attained by net loss of mainly KCl (and partly intracellular small organic osmolytes). Now, activation of separate conductive pathways for K+ and Cl− (and some negatively-charged amino acids) is known to be the predominant mechanism in most mammalian cells, although electroneutral KCl transport pathways such as K+-Cl− symporter (KCC) are involved in RVD of erythrocytes (Hoffmann et al., 2009; Okada, 2004). First evidence for the fact that volume-regulatory KCl efflux is accomplished by parallel activation of K+ and Cl− channels was provided by electrophysiological approaches in epithelial Intestine 407 cells (Hazama and Okada, 1988) and T lymphocytes (Cahalan and Lewis, 1988). Since a large variety of K+ channels are installed in the plasma membrane of animal cells, swollen cells exploit some of them as volume-regulatory K+ channels depending on cell types (Wilson and Mongin, 2018). In human Intestine 407 cells, a Ca2+-activated K+ channel was shown to serve as the volume-regulatory K+ channel (Hazama and Okada, 1988) and later molecularly identified as IK1 (Wang et al., 2003). For the volume-regulatory conductive Cl− pathway, a new type of Cl− channel was discovered to be activated by cell swelling in Intestine 407 cells (Hazama and Okada, 1988; Kubo and Okada, 1992) and T lymphocytes (Cahalan and Lewis, 1988). In contrast to involvements of various volume-regulatory K+ channels, only this type of Cl− channel was shown to predominantly serve as the volume-regulatory Cl− channel in most animal cells. This ubiquitous Cl− channel has been well characterized and called the volume-sensitive outwardly rectifying anion channel: VSOR (Okada, 1997), the volume-regulated Cl− channel: VRAC (Nilius et al., 1997a), or the volume-sensitive organic osmolyte/anion channel: VSOAC (Strange and Jackson, 1995). In 2014, the core molecule of VSOR was identified as LRRC8A (Qiu et al., 2014; Voss et al., 2014), and VSOR activity was shown to additionally require at least one of its paralogs (LRRC8C, LRRC8D, and/or LRRC8E) as the subcomponent of VSOR (Voss et al., 2014).
Essential roles of intracellular Ca2+ rise in RVD were shown in many cell types (Cala et al., 1986; Grinstein et al., 1982; Hazama and Okada, 1988; Hazama and Okada, 1990a; Rothstein and Mack, 1990; Wong and Chase, 1986). In the human epithelial cells, swelling was found to induce activation of Ca2+-permeable cation channels (Y. Okada et al., 1990) and then triggers Ca2+ release from the intracellular Ca2+ store and sizable cytosolic Ca2+ rise (Hazama and Okada, 1990b), thereby stimulating volume-regulatory Ca2+-activated K+ channels. Later, the molecular identity of swelling-activated Ca2+-permeable cation channel was identified as TRPM7 (Numata et al., 2007b) which exhibits direct mechano-stress sensitivity (Numata et al., 2007a). TRPM7-mediated Ca2+ influx is thus involved in RVD (Numata et al., 2007b) by triggering activation of volume-regulatory Ca2+-activated K+ channel. This Ca2+ inflow may also be involved in VSOR activation, because VSOR was shown to be activated by ROS (Browe and Baumgarten, 2004; Shimizu et al., 2004; Varela et al., 2004) through the NOX activation regulated by a local Ca2+ rise in the immediate vicinity of open Ca2+-permeable cation channels called Ca2+ nanodomain (Akita and Okada, 2014). It is noteworthy that NOX1 does physically interact not only with LRRC8A (Choi et al., 2016) but also with LRRC8C and LRRC8D (Choi et al., 2021). Recently, TRPM7 was also demonstrated to physically interact with LRRC8A thereby playing a role as an essential regulator for VSOR expression (Numata et al., 2021). Steady-state Ca2+ influx through TRPM7 enhances molecular expression of LRRC8A mRNA. In addition, the plasmalemmal presence of TRPM7 stabilizes the plasma membrane expression of LRRC8A protein through the protein-protein interaction between LRRC8A and the C-terminal α-kinase domain of TRPM7. Collectively, TRPM7 is involved in RVD in a multiple fashion: first by mediating swelling-induced Ca2+ influx leading to cytosolic Ca2+ rise, thereby activating volume-regulatory Ca2+-activated K+ channels and inducing hyperpolarization driving Cl− efflux through any available Cl− channels; second by mediating formation of the Ca2+ nanodomain, thereby activating volume-regulatory VSOR Cl− channels; third by stimulating molecular expression of LRRC8A mRNA in swollen cells; and fourth by stabilizing plasmalemmal expression of LRRC8A through the physical interaction with LRRC8A protein. The molecular mechanism of TRPM7-mediated RVD is schematically illustrated in Figure 5B.
Roles of TRPM2 and TRPM7 in cell death
In a huge number of publications, both TRPM2 and TRPM7 have been reported to have pathophysiological relevance to cell death and tissue injury. Cell death-inducing machineries utilize not only VSOR/VRAC and ASOR/PAC anion channel activities (see Reviews: Okada et al., 2021a; Okada et al., 2021b) but also the activities of TRPM2 and TRPM7 that are sensor cation channels constitutively expressed. Cell death is classified into apoptosis and necrosis. Necrotic cell death can be distinguished to accidental and programmed necrosis, and the latter one is further sorted into pyroptosis, necroptosis, and ferroptosis. Here, we summarize how TRPM2 and TRPM7 are implicated in these cell death modes. It is noted that both types of TRPM members often exert as double-edged swords in cell death induction.
Inductive/protective roles of TRPM2 in cell death and tissue injury
Reactive oxygen species (ROS) are generated in many of pathological conditions such as ischemia/reperfusion (I/R) injury (Granger and Kvietys, 2015) and exposure to pathogenic factors leading to neurodegenerative disorders. These pathogenic factors include amyloid β (Aβ) peptide developing Parkinson’s disease (Hensley et al., 1994), methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) which is used to induce Alzheimer’s disease model (Wu et al., 2003), inflammatory cytokines, and others. ROS are well known to cause cell death and dysfunction. Pathological roles of ROS-activated TRPM2 channel in cell death have been extensively studied in various tissues and cell types including neuronal cells (see Review: Malko and Jiang, 2020). Hydrogen peroxide, a kind of ROS, has been used to mimic oxidative stress experimental models. The earliest study showed that TRPM2 (the former name, LTRPC2) activation in TRPM2-expressing HEK293T cells by H2O2 and TNFα leads to the cell death which is dependent on Ca2+ influx and is suppressed by a Ca2+ chelator and antisense oligonucleotides against TRPM2 (Hara et al., 2002). H2O2-induced cell death was also reproduced in primary neurons and was found to be attenuated by extracellular Ca2+ removal with abolishing TRPM2 activation. The effects of PARP inhibitors and downregulation of TRPM2 by siRNA confirmed an involvement of TRPM2 activation downstream of PARP activity in H2O2-induced neural death (Fonfria et al., 2005; Kaneko et al., 2006). An involvement of TRPM2 activation in H2O2-induced cell death was also evidenced by siRNA-mediated knockdown of Trpm2 in non-neuronal cells, such as immortalized mouse embryonic fibroblasts (Blenn et al., 2011) and mouse RAW264.7 macrophages (Zou et al., 2013). A dominant negative variant of TRPM2 (TRPM2-S) showed an inhibitory effect on cell death mediated by TRPM2 activation (Zhang et al., 2003), and PKC-mediated phosphorylation of TRPM2-S exhibited an inhibitory effect on TRPM2 activation and cell death (Hecquet et al., 2014). TRPM2 is reportedly involved in homeostasis of intracellular Zn2+ regulating cell death. H2O2-treatment of hippocampal neurons caused an intracellular Zn2+ increase by lysosomal dysfunction, Zn2+ release from lysosome, mitochondrial Zn2+ accumulation, mitochondrial fission, and cell death (Li et al., 2017). These H2O2-induced lysosomal/mitochondrial toxicities in neurons were attenuated by TRPM2 gene knockout and by a Zn2+-specific chelator, confirming contribution of TRPM2 and dysregulation of intracellular Zn2+ in neural cell death.
Involvements of TRPM2 in cell death induced by I/R or oxygen-glucose deprivation/reoxygenation (OGD/R) were demonstrated by TRPM2 gene knockdown or knockout in brain neurons (Jia et al., 2011; Shimizu et al., 2013; Verma et al., 2012; Ye et al., 2014) and myocardial cells (Hiroi et al., 2013). Bilirubin, which exerts as an direct activator for TRPM2, was most recently found to be released in the brain subjected to oxygen-glucose deprivation (OGD), and to aggravate brain damage in the stroke in a manner strongly sensitive to molecular perturbation of the bilirubin-binding site on TRPM2 (Liu et al., 2023).
Although TRPM2 plays inductive roles in cell death under a large variety of conditions, as summarized above, TRPM2 channel activity has also been reported to participate in protection from cell death under several specified conditions. TRPM2 was observed to protect neuroblastoma SH-SY5Y cells from H2O2-induced cell death (S. J. Chen et al., 2013) as well as against lung injury induced by lipopolysaccharide (LPS) (Di et al., 2011), I/R-induced adult heart injury (Miller et al., 2013; Miller et al., 2014; Hoffman et al., 2015), doxorubicin-induced death in breast cancer cells (Koh et al., 2015) and in SH-SY5Y cells (Hirschler-Laszkiewicz et al., 2022), and H2O2- or hyperthermia-induced tissue damage in the sea anemone (Ehrlich et al., 2022). Thus, activation of TRPM2 channels plays not only detrimental or death-inducing but also beneficial or protective roles depending on the cellular conditions.
Inductive roles of TRPM2 in apoptotic cell death
The most cases of H2O2-induced cell death in which TRPM2 is implicated are classified as apoptosis in a variety of cell types, including human monocytic U937 cells (W. Zhang et al., 2006), rat ventricular myocytes (Jiang et al., 2006), and mouse endothelial cells (Hecquet et al., 2014; Sun et al., 2012). TRPM2 was also found to play essential roles in apoptosis induction caused by stimulation with a variety of exogenous pathogens. These pathogens include TNFα in U937 cells (Zhang et al., 2006) and mouse ventricular myocytes (Roberge et al., 2014), interferon-γ (IFNγ) in mouse microglia (Akyuva et al., 2021) and human neuroblastoma SH-SY5Y cells (Güzel et al., 2021), morphine in mouse hippocampal neurons (Osmanlıoğlu et al., 2020), glyceryltrinitrate (GTN) in mouse trigeminal ganglion neurons (Yazğan and Nazıroğlu, 2021), zinc oxide nanoparticle in human brain vascular pericytes (Jiang et al., 2017), anti-malarian drug, hydroxychloroquine, in human retinal pigment epithelial cells (Ertuğrul et al., 2023), and uric acid in human AC16 cardiomyocytes (Wu et al., 2023). Essential roles of TRPM2 channel were also recently shown in apoptotic cell death induced by an anti-cancer drug, doxorubicin (DOX), in rat cardiac cells (Akyuva and Nazıroğlu, 2023; Yıldızhan et al., 2023) as well.
In the brain stroke, TRPM2 was shown to partake in I/R-induced neuronal apoptosis using TRPM2 knockout mice (Alim et al., 2013; Gelderblom et al., 2014). TRPM2 deficiency was found to be protective against such a brain injury by decreasing the ratio of synaptic NMDAR subunit (GluN2A) to extra-synaptic NMDAR subunit (GluN2B) in the hippocampus (Alim et al., 2013) and attenuating immune cell filtration into the ischemic hemisphere (Gelderblom et al., 2014). I/R- or OGD/R-induced apoptosis was also shown to involve TRPM2 activity in the mouse kidney (Gao et al., 2014) and rat pheochromocytoma PC12 cells (Pan et al., 2020). Traumatic brain injury (TBI) was found to upregulate TRPM2 expression in the rat cerebral cortex and hippocampus (Cook et al., 2010) and to cause apoptotic death in rat hippocampal neurons in a manner dependent on ROS production and sensitive to 2-APB (Yürüker et al., 2015).
TRPM2 channels have been demonstrated to play an important role in induction of apoptotic cell death associated with etiology of several major diseases, such as Alzheimer’s disease, diabetes mellitus, and Parkinson’s diseases. Accumulation of amyloid β (Aβ) is causative of Alzheimer’s disease. ROS-induced neuronal cell death following exposure to Aβ was attenuated by a PARP inhibitor and PARP siRNA (Fonfria et al., 2005). TRPM2 also takes part in Aβ-induced pathological conditions including ER-stress, synaptic loss, microglial activation, and age-related memory deficits (Ostapchenko et al., 2015). A critical role of TRPM2 activity was shown in Aβ-induced neuronal apoptosis (Çınar and Nazıroğlu, 2023; Li and Jiang, 2018). In pancreatic islet cells or β cells, activation of TRPM2 channels was found to be involved in apoptosis induced by H2O2 (Bari et al., 2009; Lange et al., 2009; Manna et al., 2015) and by diabetic stimuli, such as streptozocin (STZ) (Manna et al., 2015) and palmitate (Li et al., 2017). MPTP and its active metabolite, 1-methyl-4-phenylpyridinium ions (MPP+), are neurotoxins for dopaminergic neurons and thereby causing Parkinson’s disease (Burns et al., 1983). MPP+ was demonstrated to increase TRPM2 expression in the mouse substantia nigra (Sun et al., 2018) and induce ROS elevation and apoptosis in neuronal SH-SY5Y cells differentiated by the addition of retinoic acid for 6 days (Sun et al., 2018). Since increased TRPM2 immunoreactivity and apoptotic cell death in the rat ovary were found to be coupled to ovarian hyperstimulation syndrome (OHSS) (Şanlı et al., 2021), it is possible that TRPM2 is causatively related to apoptosis induction in this disease.
Inductive roles of TRPM2 in non-apoptotic cell death
Participation of TRPM2 channels has been reported also in non-apoptotic cell death, including aponecrosis, necrosis, and pyroptosis under certain conditions. Acetaminophen overdose was found to cause activation of TRPM2 currents and induction of necrotic cell death which exhibited some apoptotic features such as DNA fragmentation and membrane blebbing in a manner sensitive to TRPM2 knockdown and knockout (Kheradpezhouh et al., 2014). This cell death mode might be classified into aponecrosis or secondary necrosis (see Step-6 in Figure 3 depicted in Okada et al., 2019). In addition, TRPM2 was reported to be involved in bile acid-induced cell injury in pancreatic acinar cells that simultaneously exhibited both necrotic and apoptotic cell death (Fanczal et al., 2020). In mouse microglial cells, H2O2 or Zn2+, one of damage-associated molecular pattern molecules (DAMPs), was observed to elicit cytosolic Ca2+ rise and necrotic cell death, both of which were abolished by TRPM2 knockdown (Mortadza et al., 2017). In mouse mastocytoma P815 cells, farnesyl pyrophosphate (FPP), a mevalonate pathway intermediate, caused necrotic cell death with exhibiting cell swelling and membrane rupture in a manner partially sensitive to TRPM2 knockout (Chen et al., 2021). Physical and functional interactions between TRPM2 and extra-synaptic NMDA receptors were recently found to exacerbate excitotoxic (necrotic) cell death in mouse cortical neurons under ischemic brain injury (Zong et al., 2022a). Pyroptosis is a caspase-1/3-dependent programmed necrosis that exhibits NLRP3 inflammasome activation in association with inflammation (see Reviews: Green, 2019; Jiang et al., 2020; Kolbrink et al., 2020). ROS-induced TRPM2 activation was shown to be involved in NLRP3 inflammasome activation in bone marrow-derived macrophages stimulated by charged liposomes (Zhong et al., 2013), in human leukemia U937 cells exposed to high glucose (Tseng et al., 2016), in microglia stimulated with Aβ (Alawieyah Syed Mortadza et al., 2018; Aminzadeh et al., 2018), and in OGD/R-challenged PC12 cells (Pan et al., 2020). However, no study has addressed the pyroptotic cell death (due to membrane rupture) eventually induced by the N-terminus of gasdermin D/E in relation to TRPM2.
Inductive roles of TRPM7 in cell death
Since overexpression of TRPM7 in HEK293 cells was found to cause cell death (Nadler et al., 2001; Monteilh-Zoller et al., 2003; Schmitz et al., 2003), it was assumed that TRPM7 channels are somehow involved in cell death induction under pathophysiological conditions. In fact, Aarts et al. (Aarts et al., 2003), for the first time, found that TRPM7 plays an essential role in neuronal cell death caused by OGD. Prolonged OGD increased non-selective cation conductance (IOGD) in cortical neurons independently of the activation of glutamate receptor. IOGD was inhibited by a vitamin E derivative, an O2− scavenger and a NOS inhibitor suggesting OGD-induced current activation by reactive oxygen/nitrogen species (ROS/RNS). IOGD led to Ca2+-overload followed by a further increase of ROS/RNS production and cell death which are inhibited by a TRPM7 blocker, Gd3+, and by TRPM7 siRNA. Subsequently, Sun et al. (Sun et al., 2009) demonstrated that suppression of TRPM7 expression by shRNA inhibits delayed neuronal cell death induced by transient global cerebral ischemia in rat hippocampal CA1 neurons, along with protective effects on ischemia-induced neuronal dysfunction such as defect of long-term potentiation and fear/spatial memories. In addition, molecular expression of TRPM7 was found to be upregulated in the hippocampus of rats subjected to I/R and in cultured rat hippocampal neurons during reoxygenation after transient OGD (Jiang et al., 2008). Similar ischemia-induced upregulation of TRPM7 was subsequently observed in rat brain tissues (Zhan et al., 2014; Zhang et al., 2012) and in the mouse brain (Chen et al., 2015). Downregulation of TRPM7 expression was found to be coupled to acquirement of resistance to an anticancer drug, DOX, in colon cancer LoVo cells (Castiglioni et al., 2015). Moreover, TRPM7 activated by Ca2+/Mg2+ removal was found to induce Zn2+ influx, thereby resulting in Zn2+ toxicity in mouse cortical neurons (Inoue et al., 2010). In the presence of extracellular Zn2+ ions, OGD was found to lead to cell toxicity in cortical neurons in a manner sensitive to Gd3+, a TRPM7 blocker, and TRPM7 shRNA.
Inductive/protective roles of TRPM7 in apoptotic cell death
Chen X et al. (2010) found that TRPM7 downregulation suppresses cell death induced by staurosporine (STS) and DOX, which are well-known apoptosis inducers, in HEK293 cells. Consecutively, Coombes et al. (Coombes et al., 2011) reported that H2O2 induces not only PI-positive (necrotic) but also TUNEL-positive (apoptotic) cell death in primary mouse cortical neurons in a manner sensitive to treatment with EGTA or 2-APB. Later, pharmacological studies also suggested that TRPM7 mediates apoptotic neuronal death caused by brain ischemia through modulation of CaMKII, calmodulin, and calcineurin (Turlova et al., 2021). These results strongly suggest that TRPM7 is involved in apoptosis induction under specified conditions. Indeed, Desai et al. (Desai et al., 2012) concluded that TRPM7 plays an inductive role in Fas-mediated apoptosis based on the following observations. TRPM7 KO attenuated Fas-mediated cell death in primary mouse T-cell. TRPM7 gene silencing by shRNA in Jurkat T-cell suppressed TRPM7 currents and PARP cleavage induced by Fas ligand (FasL). The cell death induction by FasL was suggested to be mediated by the channel function of TRPM7 but not the kinase activity of TRPM7 because the kinase-dead TRPM7 mutant (K1646A) exhibited no effect on FasL-induced caspase-3 activation and PARP cleavage, although the kinase domain is cleaved by caspase-8/3 during Fas stimulation. However, the kinase domain of TRPM7 was suggested to facilitate OGD/R-induced apoptosis in primary mouse cortical neurons through interacting with annexin A1 (Zhao et al., 2015). Subsequently, acetaminophen-induced apoptosis in human hepatoma HEPG2 cells was found to be partially suppressed by siRNA of TRPM7, though the inhibitory effect was less prominent than siRNAs for TRPC1, TRPV1, and TRPM2 (Badr et al., 2016). Also, intra-cortical injection of shRNA for TRPM7 was shown to suppress apoptosis in the cerebral cortex of rats subjected to TBI (Xu et al., 2018). In addition, high glucose-induced neuronal apoptosis was shown to be accompanied by molecular and functional upregulation of TRPM7 and suppressed by TRPM7 siRNA (Huang et al., 2018a). Furthermore, gene silencing of Trpm7 was demonstrated to inhibit apoptosis induced by an NO donor, sodium nitroprusside (SNP), in rat chondrocytes (Ma et al., 2021).
In contrast to the inductive role of TRPM7 in apoptotic processes, several pieces of molecular evidence for its protective role against apoptotic cell death have also been reported. TRPM7 knockdown was found to increase the rate of apoptotic cell death spontaneously observed during culturing the rat basophilic leukemic mast (RBL-2H3) cells (Ng et al., 2012). In human urinary bladder cancer (BCa) cells, apoptotic cell death was observed, in vitro, to be facilitated by TRPM7 knockdown (Cao et al., 2016). In this study, injection of antitumor carvacrol was found to reduce TRPM7 activity, thereby suppressing tumor growth in the mouse bladder cancer in vivo. Sun and his collaborators recently reported that apoptotic neuronal cell death induced by a Parkinson’s disease-related neurotoxin, MPP+, is protected by TRPM7 activation induced by isoproterenol (Sun et al., 2020a) and by TRPM7 overexpression (Sun et al., 2020b) in dopaminergic differentiated neuroblastoma SH-SY5Y cells. Moreover, in their in vivo studies, treatment with the neurotoxin, MPTP, was found to cause apoptotic tissue damage together with reduction of the TRPM7 level in mouse substantia nigra pars compacta (SNpc) region (Sun et al., 2020b). Consistently, they also observed increased expression of apoptotic proteins and decreased TRPM7 levels in the samples obtained from the SNpc region of human Parkinson’s disease patients (Sun et al., 2020b). It is warranted to clarify how TRPM7 plays opposite roles in apoptotic processes depending on experimental conditions.
Inductive roles of TRPM7 in non-apoptotic cell death
TRPM7 was shown to be activated by extracellular acidification (Jiang et al., 2005; Numata and Okada, 2008a) and involved in acid-induced necrotic cell death (Numata et al., 2019). Extracellular acidification (< pH 6.0) causes a persistent cell volume increase and cell death which were inhibited by TRPM7 downregulation using siRNA in HeLa cells (Numata et al., 2019). TRPM7 D1054A, an acid-activation-deficient mutant, abolished acidification-evoked cell volume increase and cell death, suggesting acid-evoked activation of TRPM7 leads to a necrotic volume increase (NVI) followed by necrotic cell death. TRPM7 is also likely to be implicated in two types of inflammatory programmed necrosis: caspase-1/3-dependent one called pyroptosis and RIPK-dependent one called necroptosis. The involvement of TRPM7 in pyroptosis was suggested by the pharmacological observations, as follows. First, cell swelling-induced release of an inflammatory cytokine, IL-1β, from human macrophages was inhibited by TRPM7 blockers, 2-APB and SKF-96365 (Compan et al., 2012). Second, LPS-induced release of IL-1β, IL-6, and TNFα from mouse small intestinal IEC-6 cells was inhibited by another TRPM7 blocker, NS8593 (Li et al., 2022). However, it must be pointed out that these recent studies examined the roles of TRPM7 in inflammatory responses but not those in eventual necroptotic cell death as yet. On the other hand, TRPM7 was shown to be involved in necroptotic cell death induced by the treatment with a combination of TNFα, Smac mimetic, and z-VAD-FMK (TSZ) in human colonic epithelial HT29 cells (Cai et al., 2014). The shRNA-mediated knockdown protected the cells from TSZ-induced necroptotic membrane rupture and to inhibit TSZ-induced TRPM7 current activation and Ca2+ influx.
Ionic mechanisms of cell death induction/protection involving TRPM2 and TRPM7
Since cell death induction is tightly coupled to dysfunction of cell volume regulation, in which TRPM2 and TRPM7, as already described, play important roles, the ionic mechanisms of cell death induction may, at least in part, involve TRPM2 and TRPM7 channel activities in relation to altered mechanisms of the cell volume regulation.
Roles of TRPM2 and TRPM7 in ionic mechanisms leading to apoptotic cell death
Normotonic cell shrinkage, termed AVD (Maeno et al., 2000), is an earliest and prerequisite event of apoptotic cell death processes (Hasegawa et al., 2012; Maeno et al., 2000; Maeno et al., 2006a; Nukui et al., 2006; Shimizu et al., 2007). The AVD induction is attained by KCl efflux that is accomplished by activation of K+ and Cl− channels and drives water efflux (Okada et al., 2001). ROS are produced in response to I/R, OGD/R, and numbers of apoptosis-inducing chemicals and exert as the central mediators linking between apoptotic stimulants and intracellular apoptotic reactions. ROS can activate TRPM2 cation channels (Hara et al., 2002) through ADPR production (Figure 1) and VSOR/VRAC anion channels (Browe and Baumgarten, 2004; Shimizu et al., 2004; Varela et al., 2004) (Figure 6). TRPM2-mediated Ca2+ entry not only triggers apoptotic intracellular biochemical reactions (Nicotera and Orrenius, 1998; Orrenius et al., 2003; Rizzuto et al., 2003) but also stimulates Ca2+-activated K+ channels which are known to be involved in apoptosis induction (see Review: Burg et al., 2006). VSOR plays dual roles in apoptosis induction first by mediating AVD-inducing Cl− efflux and second by mediating antioxidant GSH− efflux driven by membrane hyperpolarization caused by K+ channel activation (Figure 6). TRPM7 was recently demonstrated to function as an essential regulator of VSOR by enhancing expression of LRRC8A mRNA via the mediation of Ca2+ influx and stabilizing the plasmalemmal expression of LRRC8A through the molecular interaction between LRRC8A and the C-terminal domain of TRPM7 (Numata et al., 2021). Thus, TRPM7 may play a facilitating role in apoptosis induction secondary by upregulating VSOR channel activity inducing AVD and by mediating GSH release (Figure 6).
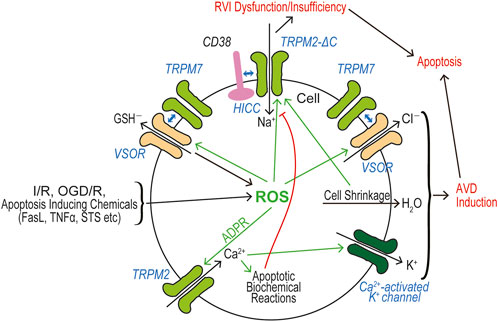
FIGURE 6. Roles of TRPM2 and TRPM7 in the ionic mechanisms leading to apoptosis induction that requisitely involves AVD induction followed by RVI dysfunction or insufficiency. Hyperpolarization predominantly caused by Ca2+-activated K+ channels drives effluxes of Cl− and GSH− via the VSOR channel pore. Resultant KCl efflux drives water efflux leading to cell shrinkage. (See the text for details.).
To eventually induce apoptotic cell death, persistent cell shrinkage is to be complemented by RVI dysfunction or insufficiency. In fact, the RVI event was shown to be inhibited during apoptotic processes (Maeno et al., 2006b), through the Akt1 inhibition caused by ROS-induced ASK1 phosphorylation (Subramanyam et al., 2010). In many cell types, RVI is known to be produced by shrinkage-induced NaCl uptake through activation of NHE, NKCC, and/or HICC (Okada et al., 2019). In HeLa cells, the main mechanism of RVI was shown to be activation of HICC (Wehner et al., 2003b), the molecular correlate of which is TRPM2-∆C plus CD38 (Numata et al., 2012). Thus, it is possible that HICC activity is downregulated by some intracellular signals produced by apoptotic biochemical reactions, such as phosphorylated ASK1 (pASK1) (Figure 6).
Roles of TRPM2 and TRPM7 in ionic mechanisms leading to necrotic cell death
Necrosis is caused by plasma membrane rupture eventually resulted from persistent cell swelling induced by a variety of insults. Necrotic cell death starts with normotonic cell swelling, called NVI (Barros et al., 2001; Okada et al., 2001), which is caused by water uptake driven by influx of osmolytes, chiefly NaCl. Persistence of necrotic cell swelling is accomplished by impairment of RVD (Okada et al., 2004).
Accidental necrosis is induced by ischemia or hypoxia (I/H) and accidental or traumatic cell injury, because these insults result in ATP depletion within the cells and the resultant extracellular acidosis as well as accumulation of glutamate released from astrocytes in the brain (Okada et al., 2019), as schematically depicted in Figure 7. ATP depletion causes suppression of Na+-K+ pump (Na/K-P) activity, thereby resulting in oncotic cell swelling due to an impairment of the pump-leak balance (P-LB) mechanism (Okada et al., 2021a; Tosteson and Hoffman, 1960). In addition, ATP depletion augments TRPM7 channel activity (Nadler et al., 2001), thereby causing the influx of cations (mainly Na+) and membrane depolarization. Acidosis also stimulates ubiquitous TRPM7 cation channels (Jiang et al., 2005; Numata and Okada, 2008b) and the neuronal amiloride-sensitive acid-sensing ion channel (ASIC) (Waldmann et al., 1997; Waldmann et al., 1999), both of which also bring about Na+ influx and membrane depolarization. In addition, acidosis activates ASOR/PAC, which is a ubiquitously expressed anion channel (Okada et al., 2021b). Membrane depolarization elicited by activation of TRPM7 and ASIC cation channels drives Cl− influx via activated ASOR anion channels. Furthermore, in brain neurons, exposure to excessive glutamate stimulates ionotropic glutamate receptors, NMDARs, which operate as non-selective cation channels and produces Na+ influx and membrane depolarization (Tymianski, 2011). TRPM2 physically interacts with and functionally augments extra-synaptic NMDARs (Zong et al., 2022a). TRPM2 is also known to be activated by a danger signal FPP (Chen et al., 2021) and H2O2 or Zn2+ (Mortadza et al., 2017). The excessive NaCl entry thus produced by these cation and anion channels drives water influx, causing cell swelling (Figure 7). Such accidental cell swelling involving TRPM2 and TRPM7 may cause necrosis and thus represent NVI induction. In accord with this view, TRPM2 knockout was shown to inhibit necrotic cell death induced by H2O2 or Zn2+ in mouse macrophages (Mortadza et al., 2017), by FPP in mouse mastrocytoma cells (Chen et al., 2021), and by ischemia in mouse neurons (Zong et al., 2022a). Also, acidotoxic necrosis was shown to be abolished by TRPM7 knockdown in HeLa cells (Numata et al., 2019). Cell swelling compelled under such ischemic, acidotoxic, and excitotoxic conditions may activate VSOR anion channels. However, operation of VSOR, the activity of which is much supported by the interaction with TRPM7, cannot serve as a pathway for volume-regulatory Cl− efflux but rather exerts as a pathway for swelling-exaggerating Cl− influx, because depolarization caused by activation of NMDAR, TRPM7, and/or TRPM2 should drive Cl− flux to the inward (but not outward) direction, thereby bringing about the RVD dysfunction (Figure 7). Thus, both TRPM2 and TRPM7 are involved in accidental necrosis by participating in the NVI induction and in the following RVD dysfunction, thus attaining persistent cell swelling due to the continuous inflows of Na+, Cl−, and osmotically obliged water (Figure 7).
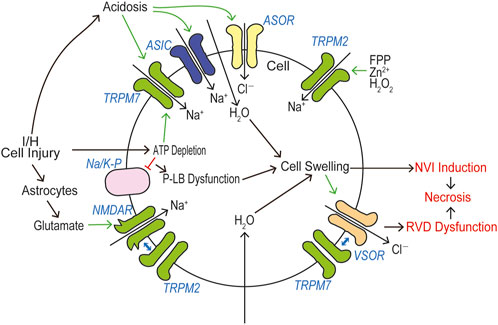
FIGURE 7. Roles of TRPM2 and TRPM7 in the ionic mechanisms leading to necrosis induction that requisitely involves NVI induction followed by RVD dysfunction. Depolarization caused by activation of TRPM7, ASIC, NMDAR, and TRPM2 drives Cl− influx via the VSOR channel pore. Resultant NaCl influx drives water influx leading to cell swelling. (See the text for details.)
As described in the preceding sections, TRPM2 and TRPM7 were molecularly proved to be involved in pyroptosis and necroptosis, respectively. Swelling- and ROS-activated VSOR/VRAC anion channels were also implicated in pyroptosis (Daniels et al., 2016; Green et al., 2018; Yang et al., 2023; Ye et al., 2021). Depletion or release ATP was observed to be coupled to necroptosis (Leist et al., 1997; Jouan-Lanhouet et al., 2012) and pyroptosis (Yang et al., 2015). Persistent, sizable cell swelling was found to be associated with necroptosis (Chen X. et al., 2016) and pyroptosis (Chen X. et al., 2016; Compan et al., 2012; Fink and Cookson, 2006; Schorn et al., 2011; Yang et al., 2023). Together, it can be assumed that the ionic mechanisms of pyroptosis and necroptotic cells death processes are, at least in part, similar to those of accidental necrosis illustrated in Figure 7. However, no study has as yet been reported as to whether TRPM2 and TRPM7 are involved in another type of programmed necrosis called ferroptosis which is caspase-independent and Fe2+-dependent.
Conclusion and perspective
TRPM2 and TRPM7 are Ca2+-permeable non-selective cation channels playing sensor roles for chemical, thermal, and mechanical stimuli. They have unique biophysical, physiological, pharmacological, and structural properties. Both activities are essentially involved as central components of the processes in cell volume regulation/dysregulation and cell death induction/protection not only by conducting cations, mainly Na+ and Ca2+, but also by interacting with other membrane-spanning proteins including LRRC8A, a core component of VSOR anion channel, NMDAR cation channels, and a cyclic ADP ribose hydrolase, CD38. Taken together, in the last section, it was sought to delve into the ionic mechanisms of cell death induction by focusing altered activities of TRPM2/TRPM7.
TRPM2 and TRPM7 have been shown to be involved in cell death associated with a large variety of diseases such as stroke, Alzheimer’s disease, Parkinson’s disease, and diabetes mellitus, as noted in the previous sections. Besides these, TRPM2 and TRPM7 were reported to be somehow related to many other diseases, although it is not clear whether these cation channels are involved in cell death processes coupled to those diseases. TRPM2 was shown to be implicated in the pathogenesis of epilepsy (Zheng et al., 2020) and atherosclerosis (Zong et al., 2022b). Also, TRPM2 was suggested to be involved in the coronavirus disease 2019 (COVID-19) (Kouhpayeh et al., 2021), based on the facts that oxidative stress plays a major role in the pathogenesis, progression, and severity of COVID-19 (Beltrán-García et al., 2020; Cecchini and Cecchini, 2020; Delgado-Roche and Mesta, 2020) and that infection of the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) is known to induce apoptosis and necroptosis (Donia and Bokhari, 2021; Li et al., 2020). TRPM7 was demonstrated to be somehow associated with causes of fibrotic diseases (Du et al., 2010; Fang et al., 2013; Yu M. et al., 2013), cancer malignancy (Yee, 2017), and multiple sclerosis (Kamermans et al., 2019). Thus, further studies are warranted to clarify how TRPM2 and TRPM7 are molecularly and causatively involved in the etiology of these diseases. In any case, TRPM2 and TRPM7 are expected to be attractive targets for future treatments and drug developments.
Author contributions
YO conceived the project and finalized the manuscript. YO, RZS, TN, and MK wrote drafts for every section with input from all authors. TN, RZS, PGM, and YO prepared figures by splitting them up. KS-N prepared references. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akyuva, Y., and Nazıroğlu, M. (2023). Silver nanoparticles potentiate antitumor and oxidant actions of cisplatin via the stimulation of TRPM2 channel in glioblastoma tumor cells. Chem. Biol. Interact. 369, 110261. doi:10.1016/j.cbi.2022.110261
Aarts, M., Iihara, K., Wei, W. L., Xiong, Z. G., Arundine, M., Cerwinski, W., et al. (2003). A key role for TRPM7 channels in anoxic neuronal death. Cell 115 (7), 863–877. doi:10.1016/s0092-8674(03)01017-1
Abiria, S. A., Krapivinsky, G., Sah, R., Santa-Cruz, A. G., Chaudhuri, D., Zhang, J., et al. (2017). TRPM7 senses oxidative stress to release Zn2+ from unique intracellular vesicles. Proc. Natl. Acad. Sci. U. S. A. 114 (30), E6079–e6088. doi:10.1073/pnas.1707380114
Abumaria, N., Li, W., and Clarkson, A. N. (2019). Role of the chanzyme TRPM7 in the nervous system in health and disease. Cell Mol. Life Sci. 76 (17), 3301–3310. doi:10.1007/s00018-019-03124-2
Akita, T., and Okada, Y. (2014). Characteristics and roles of the volume-sensitive outwardly rectifying (VSOR) anion channel in the central nervous system. Neuroscience 275, 211–231. doi:10.1016/j.neuroscience.2014.06.015
Akyuva, Y., Nazıroğlu, M., and Yıldızhan, K. (2021). Selenium prevents interferon-gamma induced activation of TRPM2 channel and inhibits inflammation, mitochondrial oxidative stress, and apoptosis in microglia. Metab. Brain Dis. 36 (2), 285–298. doi:10.1007/s11011-020-00624-0
Alawieyah Syed Mortadza, S., Sim, J. A., Neubrand, V. E., and Jiang, L. H. (2018). A critical role of TRPM2 channel in Aβ42 -induced microglial activation and generation of tumor necrosis factor-α. Glia 66 (3), 562–575. doi:10.1002/glia.23265
Alim, I., Teves, L., Li, R., Mori, Y., and Tymianski, M. (2013). Modulation of NMDAR subunit expression by TRPM2 channels regulates neuronal vulnerability to ischemic cell death. J. Neurosci. 33 (44), 17264–17277. doi:10.1523/jneurosci.1729-13.2013
Aminzadeh, M., Roghani, M., Sarfallah, A., and Riazi, G. H. (2018). TRPM2 dependence of ROS-induced NLRP3 activation in Alzheimer's disease. Int. Immunopharmacol. 54, 78–85. doi:10.1016/j.intimp.2017.10.024
Badr, H., Kozai, D., Sakaguchi, R., Numata, T., and Mori, Y. (2016). Different contribution of redox-sensitive transient receptor potential channels to acetaminophen-induced death of human hepatoma cell line. Front. Pharmacol. 7, 19. doi:10.3389/fphar.2016.00019
Balinas, C., Cabanas, H., Staines, D., and Marshall-Gradisnik, S. (2019). Transient receptor potential melastatin 2 channels are overexpressed in myalgic encephalomyelitis/chronic fatigue syndrome patients. J. Transl. Med. 17 (1), 401. doi:10.1186/s12967-019-02155-4
Bari, M. R., Akbar, S., Eweida, M., Kühn, F. J., Gustafsson, A. J., Lückhoff, A., et al. (2009). H2O2-induced Ca2+ influx and its inhibition by N-(p-amylcinnamoyl) anthranilic acid in the beta-cells: involvement of TRPM2 channels. J. Cell Mol. Med. 13 (9), 3260–3267. doi:10.1111/j.1582-4934.2009.00737.x
Barros, L. F., Hermosilla, T., and Castro, J. (2001). Necrotic volume increase and the early physiology of necrosis. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 130 (3), 401–409. doi:10.1016/s1095-6433(01)00438-x
Beck, A., Kolisek, M., Bagley, L. A., Fleig, A., and Penner, R. (2006). Nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose regulate TRPM2 channels in T lymphocytes. Faseb J. 20 (7), 962–964. doi:10.1096/fj.05-5538fje
Beesetty, P., Wieczerzak, K. B., Gibson, J. N., Kaitsuka, T., Luu, C. T., Matsushita, M., et al. (2018). Inactivation of TRPM7 kinase in mice results in enlarged spleens, reduced T-cell proliferation and diminished store-operated calcium entry. Sci. Rep. 8 (1), 3023. doi:10.1038/s41598-018-21004-w
Belrose, J. C., and Jackson, M. F. (2018). TRPM2: a candidate therapeutic target for treating neurological diseases. Acta Pharmacol. Sin. 39 (5), 722–732. doi:10.1038/aps.2018.31
Beltrán-García, J., Osca-Verdegal, R., Pallardó, F. V., Ferreres, J., Rodríguez, M., Mulet, S., et al. (2020). Oxidative stress and inflammation in COVID-19-associated sepsis: the potential role of anti-oxidant therapy in avoiding disease progression. Antioxidants (Basel) 9 (10), 936. doi:10.3390/antiox9100936
Bessac, B. F., and Fleig, A. (2007). TRPM7 channel is sensitive to osmotic gradients in human kidney cells. J. Physiol. 582 (3), 1073–1086. doi:10.1113/jphysiol.2007.130534
Bezprozvanny, I., and Tsien, R. W. (1995). Voltage-dependent blockade of diverse types of voltage-gated Ca2+ channels expressed in Xenopus oocytes by the Ca2+ channel antagonist mibefradil (Ro 40-5967). Mol. Pharmacol. 48 (3), 540–549.
Blenn, C., Wyrsch, P., Bader, J., Bollhalder, M., and Althaus, F. R. (2011). Poly(ADP-ribose)glycohydrolase is an upstream regulator of Ca2+ fluxes in oxidative cell death. Cell Mol. Life Sci. 68 (8), 1455–1466. doi:10.1007/s00018-010-0533-1
Böhmer, C., and Wehner, F. (2001). The epithelial Na+ channel (ENaC) is related to the hypertonicity-induced Na+ conductance in rat hepatocytes. FEBS Lett. 494 (1-2), 125–128. doi:10.1016/s0014-5793(01)02303-1
Bondarava, M., Li, T., Endl, E., and Wehner, F. (2009). alpha-ENaC is a functional element of the hypertonicity-induced cation channel in HepG2 cells and it mediates proliferation. Pflugers Arch. 458 (4), 675–687. doi:10.1007/s00424-009-0649-z
Bousova, K., Jirku, M., Bumba, L., Bednarova, L., Sulc, M., Franek, M., et al. (2015). PIP2 and PIP3 interact with N-terminus region of TRPM4 channel. Biophys. Chem. 205, 24–32. doi:10.1016/j.bpc.2015.06.004
Brauchi, S., Krapivinsky, G., Krapivinsky, L., and Clapham, D. E. (2008). TRPM7 facilitates cholinergic vesicle fusion with the plasma membrane. Proc. Natl. Acad. Sci. U. S. A. 105 (24), 8304–8308. doi:10.1073/pnas.0800881105
Broertjes, J., Klarenbeek, J., Habani, Y., Langeslag, M., and Jalink, K. (2019). TRPM7 residue S1269 mediates cAMP dependence of Ca2+ influx. PLoS One 14 (1), e0209563. doi:10.1371/journal.pone.0209563
Browe, D. M., and Baumgarten, C. M. (2004). Angiotensin II (AT1) receptors and NADPH oxidase regulate Cl− current elicited by beta1 integrin stretch in rabbit ventricular myocytes. J. Gen. Physiol. 124 (3), 273–287. doi:10.1085/jgp.200409040
Buelow, B., Song, Y., and Scharenberg, A. M. (2008). The Poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. J. Biol. Chem. 283 (36), 24571–24583. doi:10.1074/jbc.M802673200
Burg, E. D., Remillard, C. V., and Yuan, J. X. (2006). K+ channels in apoptosis. J. Membr. Biol. 209 (1), 3–20. doi:10.1007/s00232-005-0838-4
Burns, R. S., Chiueh, C. C., Markey, S. P., Ebert, M. H., Jacobowitz, D. M., and Kopin, I. J. (1983). A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc. Natl. Acad. Sci. U. S. A. 80 (14), 4546–4550. doi:10.1073/pnas.80.14.4546
Cahalan, M. D., and Lewis, R. S. (1988). Role of potassium and chloride channels in volume regulation by T lymphocytes. Soc. Gen. Physiol. Ser. 43, 281–301.
Cai, Z., Jitkaew, S., Zhao, J., Chiang, H. C., Choksi, S., Liu, J., et al. (2014). Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 16 (1), 55–65. doi:10.1038/ncb2883
Caiafa, P., Guastafierro, T., and Zampieri, M. (2009). Epigenetics: poly(ADP-ribosyl)ation of PARP-1 regulates genomic methylation patterns. Faseb J. 23 (3), 672–678. doi:10.1096/fj.08-123265
Cala, P. M., Mandel, L. J., and Murphy, E. (1986). Volume regulation by amphiuma red blood cells: cytosolic free Ca and alkali metal-H exchange. Am. J. Physiol. 250 (3), C423–C429. doi:10.1152/ajpcell.1986.250.3.C423
Cao, E. (2020). Structural mechanisms of transient receptor potential ion channels. J. Gen. Physiol. 152 (3), e201811998. doi:10.1085/jgp.201811998
Cao, R., Meng, Z., Liu, T., Wang, G., Qian, G., Cao, T., et al. (2016). Decreased TRPM7 inhibits activities and induces apoptosis of bladder cancer cells via ERK1/2 pathway. Oncotarget 7 (45), 72941–72960. doi:10.18632/oncotarget.12146
Castiglioni, S., Cazzaniga, A., Trapani, V., Cappadone, C., Farruggia, G., Merolle, L., et al. (2015). Magnesium homeostasis in colon carcinoma LoVo cells sensitive or resistant to doxorubicin. Sci. Rep. 5, 16538. doi:10.1038/srep16538
Cecchini, R., and Cecchini, A. L. (2020). SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med. Hypotheses 143, 110102. doi:10.1016/j.mehy.2020.110102
Chan, H. C., and Nelson, D. J. (1992). Chloride-dependent cation conductance activated during cellular shrinkage. Science 257 (5070), 669–671. doi:10.1126/science.1379742
Che, H., Yue, J., Tse, H. F., and Li, G. R. (2014). Functional TRPV and TRPM channels in human preadipocytes. Pflugers Arch. 466 (5), 947–959. doi:10.1007/s00424-013-1355-4
Chen, G. L., Zeng, B., Eastmond, S., Elsenussi, S. E., Boa, A. N., and Xu, S. Z. (2012). Pharmacological comparison of novel synthetic fenamate analogues with econazole and 2-APB on the inhibition of TRPM2 channels. Br. J. Pharmacol. 167 (6), 1232–1243. doi:10.1111/j.1476-5381.2012.02058.x
Chen, H. C., Xie, J., Zhang, Z., Su, L. T., Yue, L., and Runnels, L. W. (2010). Blockade of TRPM7 channel activity and cell death by inhibitors of 5-lipoxygenase. PLoS One 5 (6), e11161. doi:10.1371/journal.pone.0011161
Chen, J., Dou, Y., Zheng, X., Leng, T., Lu, X., Ouyang, Y., et al. (2016). TRPM7 channel inhibition mediates midazolam-induced proliferation loss in human malignant glioma. Tumour Biol. 37 (11), 14721–14731. doi:10.1007/s13277-016-5317-2
Chen, J., Zhang, X., Li, L., Ma, X., Yang, C., Liu, Z., et al. (2021). Farnesyl pyrophosphate is a new danger signal inducing acute cell death. PLoS Biol. 19 (4), e3001134. doi:10.1371/journal.pbio.3001134
Chen, S. J., Zhang, W., Tong, Q., Conrad, K., Hirschler-Laszkiewicz, I., Bayerl, M., et al. (2013). Role of TRPM2 in cell proliferation and susceptibility to oxidative stress. Am. J. Physiol. Cell Physiol. 304 (6), C548–C560. doi:10.1152/ajpcell.00069.2012
Chen, W., Xu, B., Xiao, A., Liu, L., Fang, X., Liu, R., et al. (2015). TRPM7 inhibitor carvacrol protects brain from neonatal hypoxic-ischemic injury. Mol. Brain 8, 11. doi:10.1186/s13041-015-0102-5
Chen, X., He, W. T., Hu, L., Li, J., Fang, Y., Wang, X., et al. (2016). Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 26 (9), 1007–1020. doi:10.1038/cr.2016.100
Chen, X., Numata, T., Li, M., Mori, Y., Orser, B. A., Jackson, M. F., et al. (2010). The modulation of TRPM7 currents by nafamostat mesilate depends directly upon extracellular concentrations of divalent cations. Mol. Brain 3, 38. doi:10.1186/1756-6606-3-38
Choi, H., Ettinger, N., Rohrbough, J., Dikalova, A., Nguyen, H. N., and Lamb, F. S. (2016). LRRC8A channels support TNFα-induced superoxide production by Nox1 which is required for receptor endocytosis. Free Radic. Biol. Med. 101, 413–423. doi:10.1016/j.freeradbiomed.2016.11.003
Choi, H., Rohrbough, J. C., Nguyen, H. N., Dikalova, A., and Lamb, F. S. (2021). Oxidant-resistant LRRC8A/C anion channels support superoxide production by NADPH oxidase 1. J. Physiol. 599 (12), 3013–3036. doi:10.1113/jp281577
Chokshi, R., Fruasaha, P., and Kozak, J. A. (2012a). 2-aminoethyl diphenyl borinate (2-APB) inhibits TRPM7 channels through an intracellular acidification mechanism. Channels (Austin) 6 (5), 362–369. doi:10.4161/chan.21628
Chokshi, R., Matsushita, M., and Kozak, J. A. (2012b). Detailed examination of Mg2+ and pH sensitivity of human TRPM7 channels. Am. J. Physiol. Cell Physiol. 302 (7), C1004–C1011. doi:10.1152/ajpcell.00422.2011
Chokshi, R., Matsushita, M., and Kozak, J. A. (2012c). Sensitivity of TRPM7 channels to Mg2+ characterized in cell-free patches of Jurkat T lymphocytes. Am. J. Physiol. Cell Physiol. 302 (11), C1642–C1651. doi:10.1152/ajpcell.00037.2012
Chubanov, V., Mederos y Schnitzler, M., Meißner, M., Schäfer, S., Abstiens, K., Hofmann, T., et al. (2012). Natural and synthetic modulators of SK (Kca2) potassium channels inhibit magnesium-dependent activity of the kinase-coupled cation channel TRPM7. Br. J. Pharmacol. 166 (4), 1357–1376. doi:10.1111/j.1476-5381.2012.01855.x
Çınar, R., and Nazıroğlu, M. (2023). TRPM2 channel inhibition attenuates amyloid β42-induced apoptosis and oxidative stress in the hippocampus of mice. Cell Mol. Neurobiol. 43 (3), 1335–1353. doi:10.1007/s10571-022-01253-0
Clark, K., Langeslag, M., van Leeuwen, B., Ran, L., Ryazanov, A. G., Figdor, C. G., et al. (2006). TRPM7, a novel regulator of actomyosin contractility and cell adhesion. Embo J. 25 (2), 290–301. doi:10.1038/sj.emboj.7600931
Clark, K., Middelbeek, J., Dorovkov, M. V., Figdor, C. G., Ryazanov, A. G., Lasonder, E., et al. (2008a). The alpha-kinases TRPM6 and TRPM7, but not eEF-2 kinase, phosphorylate the assembly domain of myosin IIA, IIB and IIC. FEBS Lett. 582 (20), 2993–2997. doi:10.1016/j.febslet.2008.07.043
Clark, K., Middelbeek, J., Lasonder, E., Dulyaninova, N. G., Morrice, N. A., Ryazanov, A. G., et al. (2008b). TRPM7 regulates myosin IIA filament stability and protein localization by heavy chain phosphorylation. J. Mol. Biol. 378 (4), 790–803. doi:10.1016/j.jmb.2008.02.057
Compan, V., Baroja-Mazo, A., López-Castejón, G., Gomez, A. I., Martínez, C. M., Angosto, D., et al. (2012). Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 37 (3), 487–500. doi:10.1016/j.immuni.2012.06.013
Cook, N. L., Vink, R., Helps, S. C., Manavis, J., and van den Heuvel, C. (2010). Transient receptor potential melastatin 2 expression is increased following experimental traumatic brain injury in rats. J. Mol. Neurosci. 42 (2), 192–199. doi:10.1007/s12031-010-9347-8
Coombes, E., Jiang, J., Chu, X. P., Inoue, K., Seeds, J., Branigan, D., et al. (2011). Pathophysiologically relevant levels of hydrogen peroxide induce glutamate-independent neurodegeneration that involves activation of transient receptor potential melastatin 7 channels. Antioxid. Redox Signal 14 (10), 1815–1827. doi:10.1089/ars.2010.3549
Csanády, L., and Törocsik, B. (2009). Four Ca2+ ions activate TRPM2 channels by binding in deep crevices near the pore but intracellularly of the gate. J. Gen. Physiol. 133 (2), 189–203. doi:10.1085/jgp.200810109
Daniels, M. J., Rivers-Auty, J., Schilling, T., Spencer, N. G., Watremez, W., Fasolino, V., et al. (2016). Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer's disease in rodent models. Nat. Commun. 7, 12504. doi:10.1038/ncomms12504
de Baaij, J. H., Hoenderop, J. G., and Bindels, R. J. (2015). Magnesium in man: implications for health and disease. Physiol. Rev. 95 (1), 1–46. doi:10.1152/physrev.00012.2014
Deason-Towne, F., Perraud, A. L., and Schmitz, C. (2012). Identification of Ser/Thr phosphorylation sites in the C2-domain of phospholipase C γ2 (PLCγ2) using TRPM7-kinase. Cell Signal 24 (11), 2070–2075. doi:10.1016/j.cellsig.2012.06.015
Delgado-Roche, L., and Mesta, F. (2020). Oxidative stress as key player in severe acute respiratory syndrome coronavirus (SARS-CoV) infection. Arch. Med. Res. 51 (5), 384–387. doi:10.1016/j.arcmed.2020.04.019
Demeuse, P., Penner, R., and Fleig, A. (2006). TRPM7 channel is regulated by magnesium nucleotides via its kinase domain. J. Gen. Physiol. 127 (4), 421–434. doi:10.1085/jgp.200509410
Desai, B. N., Krapivinsky, G., Navarro, B., Krapivinsky, L., Carter, B. C., Febvay, S., et al. (2012). Cleavage of TRPM7 releases the kinase domain from the ion channel and regulates its participation in Fas-induced apoptosis. Dev. Cell 22 (6), 1149–1162. doi:10.1016/j.devcel.2012.04.006
Dhakal, S., and Lee, Y. (2019). Transient receptor potential channels and metabolism. Mol. Cells 42 (8), 569–578. doi:10.14348/molcells.2019.0007
Di, A., Gao, X. P., Qian, F., Kawamura, T., Han, J., Hecquet, C., et al. (2011). The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat. Immunol. 13 (1), 29–34. doi:10.1038/ni.2171
Donia, A., and Bokhari, H. (2021). Apoptosis induced by SARS-CoV-2: can we target it? Apoptosis 26 (1-2), 7–8. doi:10.1007/s10495-021-01656-2
Dorovkov, M. V., and Ryazanov, A. G. (2004). Phosphorylation of annexin I by TRPM7 channel-kinase. J. Biol. Chem. 279 (49), 50643–50646. doi:10.1074/jbc.C400441200
Du, J., Xie, J., and Yue, L. (2009b). Modulation of TRPM2 by acidic pH and the underlying mechanisms for pH sensitivity. J. Gen. Physiol. 134 (6), 471–488. doi:10.1085/jgp.200910254
Du, J., Xie, J., and Yue, L. (2009a). Intracellular calcium activates TRPM2 and its alternative spliced isoforms. Proc. Natl. Acad. Sci. U. S. A. 106 (17), 7239–7244. doi:10.1073/pnas.0811725106
Du, J., Xie, J., Zhang, Z., Tsujikawa, H., Fusco, D., Silverman, D., et al. (2010). TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ. Res. 106 (5), 992–1003. doi:10.1161/circresaha.109.206771
Duan, J., Li, Z., Li, J., Hulse, R. E., Santa-Cruz, A., Valinsky, W. C., et al. (2018). Structure of the mammalian TRPM7, a magnesium channel required during embryonic development. Proc. Natl. Acad. Sci. U. S. A. 115 (35), E8201–e8210. doi:10.1073/pnas.1810719115
Ehrlich, W., Gahan, J. M., Rentzsch, F., and Kühn, F. J. P. (2022). TRPM2 causes sensitization to oxidative stress but attenuates high-temperature injury in the sea anemone Nematostella vectensis. J. Exp. Biol. 225 (6), jeb243717. doi:10.1242/jeb.243717
Elizondo, M. R., Arduini, B. L., Paulsen, J., MacDonald, E. L., Sabel, J. L., Henion, P. D., et al. (2005). Defective skeletogenesis with kidney stone formation in dwarf zebrafish mutant for trpm7. Curr. Biol. 15 (7), 667–671. doi:10.1016/j.cub.2005.02.050
Ertuğrul, A., Özkaya, D., and Nazıroğlu, M. (2023). Curcumin attenuates hydroxychloroquine-mediated apoptosis and oxidative stress via the inhibition of TRPM2 channel signalling pathways in a retinal pigment epithelium cell line. Graefes Arch. Clin. Exp. Ophthalmol. 2023, 1–16. doi:10.1007/s00417-023-06082-5
Esposito, E., and Cuzzocrea, S. (2009). Superoxide, NO, peroxynitrite and PARP in circulatory shock and inflammation. Front. Biosci. Landmark Ed. 14 (1), 263–296. doi:10.2741/3244
Fanczal, J., Pallagi, P., Görög, M., Diszházi, G., Almássy, J., Madácsy, T., et al. (2020). TRPM2-mediated extracellular Ca2+ entry promotes acinar cell necrosis in biliary acute pancreatitis. J. Physiol. 598 (6), 1253–1270. doi:10.1113/jp279047
Fang, L., Zhan, S., Huang, C., Cheng, X., Lv, X., Si, H., et al. (2013). TRPM7 channel regulates PDGF-BB-induced proliferation of hepatic stellate cells via PI3K and ERK pathways. Toxicol. Appl. Pharmacol. 272 (3), 713–725. doi:10.1016/j.taap.2013.08.009
Faouzi, M., Kilch, T., Horgen, F. D., Fleig, A., and Penner, R. (2017). The TRPM7 channel kinase regulates store-operated calcium entry. J. Physiol. 595 (10), 3165–3180. doi:10.1113/jp274006
Fauzee, N. J., Pan, J., and Wang, Y. L. (2010). PARP and PARG inhibitors--new therapeutic targets in cancer treatment. Pathol. Oncol. Res. 16 (4), 469–478. doi:10.1007/s12253-010-9266-6
Fink, S. L., and Cookson, B. T. (2006). Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 8 (11), 1812–1825. doi:10.1111/j.1462-5822.2006.00751.x
Fleig, A., and Penner, R. (2004). The TRPM ion channel subfamily: molecular, biophysical and functional features. Trends Pharmacol. Sci. 25 (12), 633–639. doi:10.1016/j.tips.2004.10.004
Fliegert, R., Bauche, A., Wolf Pérez, A. M., Watt, J. M., Rozewitz, M. D., Winzer, R., et al. (2017a). 2'-Deoxyadenosine 5'-diphosphoribose is an endogenous TRPM2 superagonist. Nat. Chem. Biol. 13 (9), 1036–1044. doi:10.1038/nchembio.2415
Fliegert, R., Riekehr, W. M., and Guse, A. H. (2020). Does cyclic ADP-ribose (cADPR) activate the non-selective cation channel TRPM2? Front. Immunol. 11, 2018. doi:10.3389/fimmu.2020.02018
Fliegert, R., Watt, J. M., Schöbel, A., Rozewitz, M. D., Moreau, C., Kirchberger, T., et al. (2017b). Ligand-induced activation of human TRPM2 requires the terminal ribose of ADPR and involves Arg1433 and Tyr1349. Biochem. J. 474 (13), 2159–2175. doi:10.1042/bcj20170091
Fliniaux, I., Germain, E., Farfariello, V., and Prevarskaya, N. (2018). TRPs and Ca2+ in cell death and survival. Cell Calcium 69, 4–18. doi:10.1016/j.ceca.2017.07.002
Fonfria, E., Marshall, I. C., Benham, C. D., Boyfield, I., Brown, J. D., Hill, K., et al. (2004). TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br. J. Pharmacol. 143 (1), 186–192. doi:10.1038/sj.bjp.0705914
Fonfria, E., Marshall, I. C., Boyfield, I., Skaper, S. D., Hughes, J. P., Owen, D. E., et al. (2005). Amyloid beta-peptide(1-42) and hydrogen peroxide-induced toxicity are mediated by TRPM2 in rat primary striatal cultures. J. Neurochem. 95 (3), 715–723. doi:10.1111/j.1471-4159.2005.03396.x
Franco, L., Guida, L., Bruzzone, S., Zocchi, E., Usai, C., and De Flora, A. (1998). The transmembrane glycoprotein CD38 is a catalytically active transporter responsible for generation and influx of the second messenger cyclic ADP-ribose across membranes. Faseb J. 12 (14), 1507–1520. doi:10.1096/fasebj.12.14.1507
Gao, G., Wang, W., Tadagavadi, R. K., Briley, N. E., Love, M. I., Miller, B. A., et al. (2014). TRPM2 mediates ischemic kidney injury and oxidant stress through RAC1. J. Clin. Invest. 124 (11), 4989–5001. doi:10.1172/jci76042
García-Rodríguez, S., Rosal-Vela, A., Botta, D., Cumba Garcia, L. M., Zumaquero, E., Prados-Maniviesa, V., et al. (2018). CD38 promotes pristane-induced chronic inflammation and increases susceptibility to experimental lupus by an apoptosis-driven and TRPM2-dependent mechanism. Sci. Rep. 8 (1), 3357. doi:10.1038/s41598-018-21337-6
Gattkowski, E., Johnsen, A., Bauche, A., Möckl, F., Kulow, F., Garcia Alai, M., et al. (2019). Novel CaM-binding motif in its NudT9H domain contributes to temperature sensitivity of TRPM2. Biochim. Biophys. Acta Mol. Cell Res. 1866 (7), 1162–1170. doi:10.1016/j.bbamcr.2018.12.010
Gelderblom, M., Melzer, N., Schattling, B., Göb, E., Hicking, G., Arunachalam, P., et al. (2014). Transient receptor potential melastatin subfamily member 2 cation channel regulates detrimental immune cell invasion in ischemic stroke. Stroke 45 (11), 3395–3402. doi:10.1161/strokeaha.114.005836
Granger, D. N., and Kvietys, P. R. (2015). Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. 6, 524–551. doi:10.1016/j.redox.2015.08.020
Green, D. R. (2019). The coming decade of cell death research: five riddles. Cell 177 (5), 1094–1107. doi:10.1016/j.cell.2019.04.024
Green, J. P., Yu, S., Martín-Sánchez, F., Pelegrin, P., Lopez-Castejon, G., Lawrence, C. B., et al. (2018). Chloride regulates dynamic NLRP3-dependent ASC oligomerization and inflammasome priming. Proc. Natl. Acad. Sci. U. S. A. 115 (40), E9371–e9380. doi:10.1073/pnas.1812744115
Grinstein, S., Dupre, A., and Rothstein, A. (1982). Volume regulation by human lymphocytes. Role of calcium. J. Gen. Physiol. 79 (5), 849–868. doi:10.1085/jgp.79.5.849
Grubisha, O., Rafty, L. A., Takanishi, C. L., Xu, X., Tong, L., Perraud, A. L., et al. (2006). Metabolite of SIR2 reaction modulates TRPM2 ion channel. J. Biol. Chem. 281 (20), 14057–14065. doi:10.1074/jbc.M513741200
Guan, Z., Chen, X., Fang, S., Ji, Y., Gao, Z., and Zheng, Y. (2021). CCT128930 is a novel and potent antagonist of TRPM7 channel. Biochem. Biophys. Res. Commun. 560, 132–138. doi:10.1016/j.bbrc.2021.04.119
Guida, L., Franco, L., Bruzzone, S., Sturla, L., Zocchi, E., Basile, G., et al. (2004). Concentrative influx of functionally active cyclic ADP-ribose in dimethyl sulfoxide-differentiated HL-60 cells. J. Biol. Chem. 279 (21), 22066–22075. doi:10.1074/jbc.M314137200
Guinamard, R., Simard, C., and Del Negro, C. (2013). Flufenamic acid as an ion channel modulator. Pharmacol. Ther. 138 (2), 272–284. doi:10.1016/j.pharmthera.2013.01.012
Güzel, M., Nazıroğlu, M., Akpınar, O., and Çınar, R. (2021). Interferon gamma-mediated oxidative stress induces apoptosis, neuroinflammation, zinc ion influx, and TRPM2 channel activation in neuronal cell line: modulator role of curcumin. Inflammation 44 (5), 1878–1894. doi:10.1007/s10753-021-01465-4
Gwanyanya, A., Sipido, K. R., Vereecke, J., and Mubagwa, K. (2006). ATP and PIP2 dependence of the magnesium-inhibited, TRPM7-like cation channel in cardiac myocytes. Am. J. Physiol. Cell Physiol. 291 (4), C627–C635. doi:10.1152/ajpcell.00074.2006
Hanano, T., Hara, Y., Shi, J., Morita, H., Umebayashi, C., Mori, E., et al. (2004). Involvement of TRPM7 in cell growth as a spontaneously activated Ca2+ entry pathway in human retinoblastoma cells. J. Pharmacol. Sci. 95 (4), 403–419. doi:10.1254/jphs.fp0040273
Hantute-Ghesquier, A., Haustrate, A., Prevarskaya, N., and Lehen'kyi, V. (2018). TRPM family channels in cancer. Pharm. (Basel) 11 (2), 58. doi:10.3390/ph11020058
Hara, Y., Wakamori, M., Ishii, M., Maeno, E., Nishida, M., Yoshida, T., et al. (2002). LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell 9 (1), 163–173. doi:10.1016/s1097-2765(01)00438-5
Hasegawa, Y., Shimizu, T., Takahashi, N., and Okada, Y. (2012). The apoptotic volume decrease is an upstream event of MAP kinase activation during Staurosporine-induced apoptosis in HeLa cells. Int. J. Mol. Sci. 13 (7), 9363–9379. doi:10.3390/ijms13079363
Hazama, A., and Okada, Y. (1990a). Biphasic rises in cytosolic free Ca2+ in association with activation of K+ and Cl− conductance during the regulatory volume decrease in cultured human epithelial cells. Pflugers Arch. 416 (6), 710–714. doi:10.1007/bf00370619
Hazama, A., and Okada, Y. (1988). Ca2+ sensitivity of volume-regulatory K+ and Cl− channels in cultured human epithelial cells. J. Physiol. 402, 687–702. doi:10.1113/jphysiol.1988.sp017229
Hazama, A., and Okada, Y. (1990b). Involvement of Ca2+-induced Ca2+ release in the volume regulation of human epithelial cells exposed to a hypotonic medium. Biochem. Biophys. Res. Commun. 167 (1), 287–293. doi:10.1016/0006-291x(90)91763-i
He, Y., Yao, G., Savoia, C., and Touyz, R. M. (2005). Transient receptor potential melastatin 7 ion channels regulate magnesium homeostasis in vascular smooth muscle cells: role of angiotensin II. Circ. Res. 96 (2), 207–215. doi:10.1161/01.RES.0000152967.88472.3e
Hecquet, C. M., Zhang, M., Mittal, M., Vogel, S. M., Di, A., Gao, X., et al. (2014). Cooperative interaction of trp melastatin channel transient receptor potential (TRPM2) with its splice variant TRPM2 short variant is essential for endothelial cell apoptosis. Circ. Res. 114 (3), 469–479. doi:10.1161/circresaha.114.302414
Heiner, I., Eisfeld, J., and Lückhoff, A. (2003). Role and regulation of TRP channels in neutrophil granulocytes. Cell Calcium 33 (5-6), 533–540. doi:10.1016/s0143-4160(03)00058-7
Hensley, K., Carney, J. M., Mattson, M. P., Aksenova, M., Harris, M., Wu, J. F., et al. (1994). A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to alzheimer disease. Proc. Natl. Acad. Sci. U. S. A. 91 (8), 3270–3274. doi:10.1073/pnas.91.8.3270
Hermosura, M. C., Monteilh-Zoller, M. K., Scharenberg, A. M., Penner, R., and Fleig, A. (2002). Dissociation of the store-operated calcium current I(CRAC) and the Mg-nucleotide-regulated metal ion current MagNuM. J. Physiol. 539 (2), 445–458. doi:10.1113/jphysiol.2001.013361
Higashida, H., Yuhi, T., Akther, S., Amina, S., Zhong, J., Liang, M., et al. (2018). Oxytocin release via activation of TRPM2 and CD38 in the hypothalamus during hyperthermia in mice: implication for autism spectrum disorder. Neurochem. Int. 119, 42–48. doi:10.1016/j.neuint.2017.07.009
Hill, K., Benham, C. D., McNulty, S., and Randall, A. D. (2004a). Flufenamic acid is a pH-dependent antagonist of TRPM2 channels. Neuropharmacology 47 (3), 450–460. doi:10.1016/j.neuropharm.2004.04.014
Hill, K., McNulty, S., and Randall, A. D. (2004b). Inhibition of TRPM2 channels by the antifungal agents clotrimazole and econazole. Naunyn Schmiedeb. Arch. Pharmacol. 370 (4), 227–237. doi:10.1007/s00210-004-0981-y
Hiroi, T., Wajima, T., Negoro, T., Ishii, M., Nakano, Y., Kiuchi, Y., et al. (2013). Neutrophil TRPM2 channels are implicated in the exacerbation of myocardial ischaemia/reperfusion injury. Cardiovasc Res. 97 (2), 271–281. doi:10.1093/cvr/cvs332
Hirschler-Laszkiewicz, I., Festa, F., Huang, S., Moldovan, G. L., Nicolae, C., Dhoonmoon, A., et al. (2022). The human ion channel TRPM2 modulates cell survival in neuroblastoma through E2F1 and FOXM1. Sci. Rep. 12 (1), 6311. doi:10.1038/s41598-022-10385-8
Hoffman, N. E., Miller, B. A., Wang, J., Elrod, J. W., Rajan, S., Gao, E., et al. (2015). Ca2⁺ entry via Trpm2 is essential for cardiac myocyte bioenergetics maintenance. Am. J. Physiol. Heart Circ. Physiol. 308 (6), H637–H650. doi:10.1152/ajpheart.00720.2014
Hoffmann, E. K., Lambert, I. H., and Pedersen, S. F. (2009). Physiology of cell volume regulation in vertebrates. Physiol. Rev. 89 (1), 193–277. doi:10.1152/physrev.00037.2007
Hofmann, T., Schäfer, S., Linseisen, M., Sytik, L., Gudermann, T., and Chubanov, V. (2014). Activation of TRPM7 channels by small molecules under physiological conditions. Pflugers Arch. 466 (12), 2177–2189. doi:10.1007/s00424-014-1488-0
Hu, H. Z., Gu, Q., Wang, C., Colton, C. K., Tang, J., Kinoshita-Kawada, M., et al. (2004). 2-aminoethoxydiphenyl borate is a common activator of TRPV1, TRPV2, and TRPV3. J. Biol. Chem. 279 (34), 35741–35748. doi:10.1074/jbc.M404164200
Huang, Y., Fliegert, R., Guse, A. H., Lü, W., and Du, J. (2020). A structural overview of the ion channels of the TRPM family. Cell Calcium 85, 102111. doi:10.1016/j.ceca.2019.102111
Huang, Y., Leng, T. D., Inoue, K., Yang, T., Liu, M., Horgen, F. D., et al. (2018a). TRPM7 channels play a role in high glucose-induced endoplasmic reticulum stress and neuronal cell apoptosis. J. Biol. Chem. 293 (37), 14393–14406. doi:10.1074/jbc.RA117.001032
Huang, Y., Roth, B., Lü, W., and Du, J. (2019). Ligand recognition and gating mechanism through three ligand-binding sites of human TRPM2 channel. Elife 8, e50175. doi:10.7554/eLife.50175
Huang, Y., Winkler, P. A., Sun, W., Lü, W., and Du, J. (2018b). Architecture of the TRPM2 channel and its activation mechanism by ADP-ribose and calcium. Nature 562 (7725), 145–149. doi:10.1038/s41586-018-0558-4
Huffer, K. E., Aleksandrova, A. A., Jara-Oseguera, A., Forrest, L. R., and Swartz, K. J. (2020). Global alignment and assessment of TRP channel transmembrane domain structures to explore functional mechanisms. Elife 9, e58660. doi:10.7554/eLife.58660
Ikari, A., Atomi, K., Yamazaki, Y., Sakai, H., Hayashi, H., Yamaguchi, M., et al. (2013). Hyperosmolarity-induced up-regulation of claudin-4 mediated by NADPH oxidase-dependent H2O2 production and Sp1/c-Jun cooperation. Biochim. Biophys. Acta 1833 (12), 2617–2627. doi:10.1016/j.bbamcr.2013.06.016
Inoue, H., Murayama, T., Kobayashi, T., Konishi, M., and Yokoyama, U. (2021). The zinc-binding motif of TRPM7 acts as an oxidative stress sensor to regulate its channel activity. J. Gen. Physiol. 153 (6), e202012708. doi:10.1085/jgp.202012708
Inoue, H., Murayama, T., Tashiro, M., Sakurai, T., and Konishi, M. (2014). Mg2+- and ATP-dependent inhibition of transient receptor potential melastatin 7 by oxidative stress. Free Radic. Biol. Med. 72, 257–266. doi:10.1016/j.freeradbiomed.2014.04.015
Inoue, K., Branigan, D., and Xiong, Z. G. (2010). Zinc-induced neurotoxicity mediated by transient receptor potential melastatin 7 channels. J. Biol. Chem. 285 (10), 7430–7439. doi:10.1074/jbc.M109.040485
Iordanov, I., Mihályi, C., Tóth, B., and Csanády, L. (2016). The proposed channel-enzyme transient receptor potential melastatin 2 does not possess ADP ribose hydrolase activity. Elife 5, e17600. doi:10.7554/eLife.17600
Iordanov, I., Tóth, B., Szollosi, A., and Csanády, L. (2019). Enzyme activity and selectivity filter stability of ancient TRPM2 channels were simultaneously lost in early vertebrates. Elife 8, e44556. doi:10.7554/eLife.44556
Ishii, M., Shimizu, S., Hara, Y., Hagiwara, T., Miyazaki, A., Mori, Y., et al. (2006). Intracellular-produced hydroxyl radical mediates H2O2-induced Ca2+ influx and cell death in rat beta-cell line RIN-5F. Cell Calcium 39 (6), 487–494. doi:10.1016/j.ceca.2006.01.013
Jäger, H., Dreker, T., Buck, A., Giehl, K., Gress, T., and Grissmer, S. (2004). Blockage of intermediate-conductance Ca2+-activated K+ channels inhibit human pancreatic cancer cell growth in vitro. Mol. Pharmacol. 65 (3), 630–638. doi:10.1124/mol.65.3.630
Jansen, C., Sahni, J., Suzuki, S., Horgen, F. D., Penner, R., and Fleig, A. (2016). The coiled-coil domain of zebrafish TRPM7 regulates Mg·nucleotide sensitivity. Sci. Rep. 6, 33459. doi:10.1038/srep33459
Jensen, B. S., Strobaek, D., Christophersen, P., Jorgensen, T. D., Hansen, C., Silahtaroglu, A., et al. (1998). Characterization of the cloned human intermediate-conductance Ca2+-activated K+ channel. Am. J. Physiol. 275 (3), C848–C856. doi:10.1152/ajpcell.1998.275.3.C848
Jia, J., Verma, S., Nakayama, S., Quillinan, N., Grafe, M. R., Hurn, P. D., et al. (2011). Sex differences in neuroprotection provided by inhibition of TRPM2 channels following experimental stroke. J. Cereb. Blood Flow. Metab. 31 (11), 2160–2168. doi:10.1038/jcbfm.2011.77
Jiang, H., Sha, S. H., Forge, A., and Schacht, J. (2006). Caspase-independent pathways of hair cell death induced by kanamycin in vivo. Cell Death Differ. 13 (1), 20–30. doi:10.1038/sj.cdd.4401706
Jiang, H., Tian, S. L., Zeng, Y., Li, L. L., and Shi, J. (2008). TrkA pathway(s) is involved in regulation of TRPM7 expression in hippocampal neurons subjected to ischemic-reperfusion and oxygen-glucose deprivation. Brain Res. Bull. 76 (1-2), 124–130. doi:10.1016/j.brainresbull.2008.01.013
Jiang, J., Li, M., and Yue, L. (2005). Potentiation of TRPM7 inward currents by protons. J. Gen. Physiol. 126 (2), 137–150. doi:10.1085/jgp.200409185
Jiang, M., Qi, L., Li, L., and Li, Y. (2020). The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discov. 6, 112. doi:10.1038/s41420-020-00349-0
Jiang, Q., Gao, Y., Wang, C., Tao, R., Wu, Y., Zhan, K., et al. (2017). Nitration of TRPM2 as a molecular switch induces autophagy during brain pericyte injury. Antioxid. Redox Signal 27 (16), 1297–1316. doi:10.1089/ars.2016.6873
Jouan-Lanhouet, S., Arshad, M. I., Piquet-Pellorce, C., Martin-Chouly, C., Le Moigne-Muller, G., Van Herreweghe, F., et al. (2012). TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 19 (12), 2003–2014. doi:10.1038/cdd.2012.90
Kuras, Z., Yun, Y. H., Chimote, A. A., Neumeier, L., and Conforti, L. (2012). KCa3.1 and TRPM7 channels at the uropod regulate migration of activated human T cells. PLoS One 7 (8), e43859. doi:10.1371/journal.pone.0043859
Kamermans, A., Planting, K. E., Jalink, K., van Horssen, J., and de Vries, H. E. (2019). Reactive astrocytes in multiple sclerosis impair neuronal outgrowth through TRPM7-mediated chondroitin sulfate proteoglycan production. Glia 67 (1), 68–77. doi:10.1002/glia.23526
Kaneko, S., Kawakami, S., Hara, Y., Wakamori, M., Itoh, E., Minami, T., et al. (2006). A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. J. Pharmacol. Sci. 101 (1), 66–76. doi:10.1254/jphs.fp0060128
Kashio, M., Sokabe, T., Shintaku, K., Uematsu, T., Fukuta, N., Kobayashi, N., et al. (2012). Redox signal-mediated sensitization of transient receptor potential melastatin 2 (TRPM2) to temperature affects macrophage functions. Proc. Natl. Acad. Sci. U. S. A. 109 (17), 6745–6750. doi:10.1073/pnas.1114193109
Kashio, M., and Tominaga, M. (2017). The TRPM2 channel: A thermo-sensitive metabolic sensor. Channels (Austin) 11 (5), 426–433. doi:10.1080/19336950.2017.1344801
Kerschbaum, H. H., Kozak, J. A., and Cahalan, M. D. (2003). Polyvalent cations as permeant probes of MIC and TRPM7 pores. Biophys. J. 84 (4), 2293–2305. doi:10.1016/s0006-3495(03)75035-8
Kheradpezhouh, E., Barritt, G. J., and Rychkov, G. Y. (2016). Curcumin inhibits activation of TRPM2 channels in rat hepatocytes. Redox Biol. 7, 1–7. doi:10.1016/j.redox.2015.11.001
Kheradpezhouh, E., Ma, L., Morphett, A., Barritt, G. J., and Rychkov, G. Y. (2014). TRPM2 channels mediate acetaminophen-induced liver damage. Proc. Natl. Acad. Sci. U. S. A. 111 (8), 3176–3181. doi:10.1073/pnas.1322657111
Kim, B. J. (2013). Involvement of melastatin type transient receptor potential 7 channels in ginsenoside Rd-induced apoptosis in gastric and breast cancer cells. J. Ginseng Res. 37 (2), 201–209. doi:10.5142/jgr.2013.37.201
Kim, U. H., Han, M. K., Park, B. H., Kim, H. R., and An, N. H. (1993). Function of NAD glycohydrolase in ADP-ribose uptake from NAD by human erythrocytes. Biochim. Biophys. Acta 1178 (2), 121–126. doi:10.1016/0167-4889(93)90001-6
Koh, D. W., Powell, D. P., Blake, S. D., Hoffman, J. L., Hopkins, M. M., and Feng, X. (2015). Enhanced cytotoxicity in triple-negative and estrogen receptor-positive breast adenocarcinoma cells due to inhibition of the transient receptor potential melastatin-2 channel. Oncol. Rep. 34 (3), 1589–1598. doi:10.3892/or.2015.4131
Koike, C., Numata, T., Ueda, H., Mori, Y., and Furukawa, T. (2010). TRPM1: a vertebrate TRP channel responsible for retinal on bipolar function. Cell Calcium 48 (2-3), 95–101. doi:10.1016/j.ceca.2010.08.004
Kolbrink, B., Riebeling, T., Kunzendorf, U., and Krautwald, S. (2020). Plasma membrane pores drive inflammatory cell death. Front. Cell Dev. Biol. 8, 817. doi:10.3389/fcell.2020.00817
Kolisek, M., Beck, A., Fleig, A., and Penner, R. (2005). Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol. Cell 18 (1), 61–69. doi:10.1016/j.molcel.2005.02.033
Kollewe, A., Chubanov, V., Tseung, F. T., Correia, L., Schmidt, E., Rössig, A., et al. (2021). The molecular appearance of native TRPM7 channel complexes identified by high-resolution proteomics. Elife 10, e68544. doi:10.7554/eLife.68544
Koos, B., Christmann, J., Plettenberg, S., Käding, D., Becker, J., Keteku, M., et al. (2018). Hypertonicity-induced cation channels in HepG2 cells: architecture and role in proliferation vs. apoptosis. J. Physiol. 596 (7), 1227–1241. doi:10.1113/jp275827
Kouhpayeh, S., Shariati, L., Boshtam, M., Rahimmanesh, I., Mirian, M., Esmaeili, Y., et al. (2021). The molecular basis of COVID-19 pathogenesis, conventional and nanomedicine therapy. Int. J. Mol. Sci. 22 (11), 5438. doi:10.3390/ijms22115438
Kozak, J. A., and Cahalan, M. D. (2003). MIC channels are inhibited by internal divalent cations but not ATP. Biophys. J. 84 (2), 922–927. doi:10.1016/s0006-3495(03)74909-1
Kozak, J. A., Kerschbaum, H. H., and Cahalan, M. D. (2002). Distinct properties of CRAC and MIC channels in RBL cells. J. Gen. Physiol. 120 (2), 221–235. doi:10.1085/jgp.20028601
Kozak, J. A., Matsushita, M., Nairn, A. C., and Cahalan, M. D. (2005). Charge screening by internal pH and polyvalent cations as a mechanism for activation, inhibition, and rundown of TRPM7/MIC channels. J. Gen. Physiol. 126 (5), 499–514. doi:10.1085/jgp.200509324
Kraft, R., Grimm, C., Frenzel, H., and Harteneck, C. (2006). Inhibition of TRPM2 cation channels by N-(p-amylcinnamoyl)anthranilic acid. Br. J. Pharmacol. 148 (3), 264–273. doi:10.1038/sj.bjp.0706739
Kraft, R., Grimm, C., Grosse, K., Hoffmann, A., Sauerbruch, S., Kettenmann, H., et al. (2004). Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am. J. Physiol. Cell Physiol. 286 (1), C129–C137. doi:10.1152/ajpcell.00331.2003
Krapivinsky, G., Mochida, S., Krapivinsky, L., Cibulsky, S. M., and Clapham, D. E. (2006). The TRPM7 ion channel functions in cholinergic synaptic vesicles and affects transmitter release. Neuron 52 (3), 485–496. doi:10.1016/j.neuron.2006.09.033
Kubo, M., and Okada, Y. (1992). Volume-regulatory Cl− channel currents in cultured human epithelial cells. J. Physiol. 456, 351–371. doi:10.1113/jphysiol.1992.sp019340
Kühn, F. J., Kühn, C., Winking, M., Hoffmann, D. C., and Lückhoff, A. (2016). ADP-ribose activates the TRPM2 channel from the sea anemone nematostella vectensis independently of the NUDT9H domain. PLoS One 11 (6), e0158060. doi:10.1371/journal.pone.0158060
Lang, F., Busch, G. L., Ritter, M., Völkl, H., Waldegger, S., Gulbins, E., et al. (1998). Functional significance of cell volume regulatory mechanisms. Physiol. Rev. 78 (1), 247–306. doi:10.1152/physrev.1998.78.1.247
Lange, I., Penner, R., Fleig, A., and Beck, A. (2008). Synergistic regulation of endogenous TRPM2 channels by adenine dinucleotides in primary human neutrophils. Cell Calcium 44 (6), 604–615. doi:10.1016/j.ceca.2008.05.001
Lange, I., Yamamoto, S., Partida-Sanchez, S., Mori, Y., Fleig, A., and Penner, R. (2009). TRPM2 functions as a lysosomal Ca2+-release channel in beta cells. Sci. Signal 2 (71), ra23. doi:10.1126/scisignal.2000278
Langeslag, M., Clark, K., Moolenaar, W. H., van Leeuwen, F. N., and Jalink, K. (2007). Activation of TRPM7 channels by phospholipase C-coupled receptor agonists. J. Biol. Chem. 282 (1), 232–239. doi:10.1074/jbc.M605300200
Leist, M., Single, B., Castoldi, A. F., Kühnle, S., and Nicotera, P. (1997). Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J. Exp. Med. 185 (8), 1481–1486. doi:10.1084/jem.185.8.1481
Leng, T. D., Lin, J., Sun, H. W., Zeng, Z., O'Bryant, Z., Inoue, K., et al. (2015). Local anesthetic lidocaine inhibits TRPM7 current and TRPM7-mediated zinc toxicity. CNS Neurosci. Ther. 21 (1), 32–39. doi:10.1111/cns.12318
Li, M., Du, J., Jiang, J., Ratzan, W., Su, L. T., Runnels, L. W., et al. (2007). Molecular determinants of Mg2+ and Ca2+ permeability and pH sensitivity in TRPM6 and TRPM7. J. Biol. Chem. 282 (35), 25817–25830. doi:10.1074/jbc.M608972200
Li, M., Jiang, J., and Yue, L. (2006). Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J. Gen. Physiol. 127 (5), 525–537. doi:10.1085/jgp.200609502
Li, Q., Lei, X., Liu, H., Feng, S., Cai, C., Hu, Y., et al. (2022). Transient receptor potential melastatin 7 aggravates necrotizing enterocolitis by promoting an inflammatory response in children. Transl. Pediatr. 11 (12), 2030–2039. doi:10.21037/tp-22-633
Li, S., Zhang, Y., Guan, Z., Li, H., Ye, M., Chen, X., et al. (2020). SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target Ther. 5 (1), 235. doi:10.1038/s41392-020-00334-0
Li, X., and Jiang, L. H. (2018). Multiple molecular mechanisms form a positive feedback loop driving amyloid β42 peptide-induced neurotoxicity via activation of the TRPM2 channel in hippocampal neurons. Cell Death Dis. 9 (2), 195. doi:10.1038/s41419-018-0270-1
Li, X., Yang, W., and Jiang, L. H. (2017). Alteration in intracellular Zn2+ homeostasis as a result of TRPM2 channel activation contributes to ROS-induced hippocampal neuronal death. Front. Mol. Neurosci. 10, 414. doi:10.3389/fnmol.2017.00414
Liu, H. W., Gong, L. N., Lai, K., Yu, X. F., Liu, Z. Q., Li, M. X., et al. (2023). Bilirubin gates the TRPM2 channel as a direct agonist to exacerbate ischemic brain damage. Neuron 111 (10), 1609–1625.e6. e1606. doi:10.1016/j.neuron.2023.02.022
Liu, J., Zhao, Y. J., Li, W. H., Hou, Y. N., Li, T., Zhao, Z. Y., et al. (2017). Cytosolic interaction of type III human CD38 with CIB1 modulates cellular cyclic ADP-ribose levels. Proc. Natl. Acad. Sci. U. S. A. 114 (31), 8283–8288. doi:10.1073/pnas.1703718114
Liu, Y., Mikrani, R., He, Y., Faran Ashraf Baig, M. M., Abbas, M., Naveed, M., et al. (2020). TRPM8 channels: A review of distribution and clinical role. Eur. J. Pharmacol. 882, 173312. doi:10.1016/j.ejphar.2020.173312
Liu, Y. S., Liu, Y. A., Huang, C. J., Yen, M. H., Tseng, C. T., Chien, S., et al. (2015). Mechanosensitive TRPM7 mediates shear stress and modulates osteogenic differentiation of mesenchymal stromal cells through Osterix pathway. Sci. Rep. 5, 16522. doi:10.1038/srep16522
Luo, X., Li, M., Zhan, K., Yang, W., Zhang, L., Wang, K., et al. (2018). Selective inhibition of TRPM2 channel by two novel synthesized ADPR analogues. Chem. Biol. Drug Des. 91 (2), 552–566. doi:10.1111/cbdd.13119
Ma, G., Yang, Y., Chen, Y., Wei, X., Ding, J., Zhou, R. P., et al. (2021). Blockade of TRPM7 alleviates chondrocyte apoptosis and articular cartilage damage in the adjuvant arthritis rat model through regulation of the indian hedgehog signaling pathway. Front. Pharmacol. 12, 655551. doi:10.3389/fphar.2021.655551
Maeno, E., Ishizaki, Y., Kanaseki, T., Hazama, A., and Okada, Y. (2000). Normotonic cell shrinkage because of disordered volume regulation is an early prerequisite to apoptosis. Proc. Natl. Acad. Sci. U. S. A. 97 (17), 9487–9492. doi:10.1073/pnas.140216197
Maeno, E., Shimizu, T., and Okada, Y. (2006a). Normotonic cell shrinkage induces apoptosis under extracellular low Cl conditions in human lymphoid and epithelial cells. Acta Physiol. (Oxf) 187 (1-2), 217–222. doi:10.1111/j.1748-1716.2006.01554.x
Maeno, E., Takahashi, N., and Okada, Y. (2006b). Dysfunction of regulatory volume increase is a key component of apoptosis. FEBS Lett. 580 (27), 6513–6517. doi:10.1016/j.febslet.2006.10.074
Mahbub, L., Kozlov, G., Zong, P., Lee, E. L., Tetteh, S., Nethramangalath, T., et al. (2023). Structural insights into regulation of CNNM-TRPM7 divalent cation uptake by the small GTPase ARL15. Elife 12, e86129. doi:10.7554/eLife.86129
Malko, P., and Jiang, L. H. (2020). TRPM2 channel-mediated cell death: an important mechanism linking oxidative stress-inducing pathological factors to associated pathological conditions. Redox Biol. 37, 101755. doi:10.1016/j.redox.2020.101755
Manna, P. T., Munsey, T. S., Abuarab, N., Li, F., Asipu, A., Howell, G., et al. (2015). TRPM2-mediated intracellular Zn2+ release triggers pancreatic β-cell death. Biochem. J. 466 (3), 537–546. doi:10.1042/bj20140747
Matsushita, M., Kozak, J. A., Shimizu, Y., McLachlin, D. T., Yamaguchi, H., Wei, F. Y., et al. (2005). Channel function is dissociated from the intrinsic kinase activity and autophosphorylation of TRPM7/ChaK1. J. Biol. Chem. 280 (21), 20793–20803. doi:10.1074/jbc.M413671200
McHugh, D., Flemming, R., Xu, S. Z., Perraud, A. L., and Beech, D. J. (2003). Critical intracellular Ca2+ dependence of transient receptor potential melastatin 2 (TRPM2) cation channel activation. J. Biol. Chem. 278 (13), 11002–11006. doi:10.1074/jbc.M210810200
Mei, Z. Z., Xia, R., Beech, D. J., and Jiang, L. H. (2006). Intracellular coiled-coil domain engaged in subunit interaction and assembly of melastatin-related transient receptor potential channel 2. J. Biol. Chem. 281 (50), 38748–38756. doi:10.1074/jbc.M607591200
Miller, B. A., Hoffman, N. E., Merali, S., Zhang, X. Q., Wang, J., Rajan, S., et al. (2014). TRPM2 channels protect against cardiac ischemia-reperfusion injury: role of mitochondria. J. Biol. Chem. 289 (11), 7615–7629. doi:10.1074/jbc.M113.533851
Miller, B. A., Wang, J., Hirschler-Laszkiewicz, I., Gao, E., Song, J., Zhang, X. Q., et al. (2013). The second member of transient receptor potential-melastatin channel family protects hearts from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 304 (7), H1010–H1022. doi:10.1152/ajpheart.00906.2012
Mittal, M., Urao, N., Hecquet, C. M., Zhang, M., Sudhahar, V., Gao, X. P., et al. (2015). Novel role of reactive oxygen species-activated Trp melastatin channel-2 in mediating angiogenesis and postischemic neovascularization. Arterioscler. Thromb. Vasc. Biol. 35 (4), 877–887. doi:10.1161/atvbaha.114.304802
Mittermeier, L., Demirkhanyan, L., Stadlbauer, B., Breit, A., Recordati, C., Hilgendorff, A., et al. (2019). TRPM7 is the central gatekeeper of intestinal mineral absorption essential for postnatal survival. Proc. Natl. Acad. Sci. U. S. A. 116 (10), 4706–4715. doi:10.1073/pnas.1810633116
Monteilh-Zoller, M. K., Hermosura, M. C., Nadler, M. J., Scharenberg, A. M., Penner, R., and Fleig, A. (2003). TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J. Gen. Physiol. 121 (1), 49–60. doi:10.1085/jgp.20028740
Moreau, C., Kirchberger, T., Swarbrick, J. M., Bartlett, S. J., Fliegert, R., Yorgan, T., et al. (2013). Structure-activity relationship of adenosine 5'-diphosphoribose at the transient receptor potential melastatin 2 (TRPM2) channel: rational design of antagonists. J. Med. Chem. 56 (24), 10079–10102. doi:10.1021/jm401497a
Mortadza, S. S., Sim, J. A., Stacey, M., and Jiang, L. H. (2017). Signalling mechanisms mediating Zn2+-induced TRPM2 channel activation and cell death in microglial cells. Sci. Rep. 7, 45032. doi:10.1038/srep45032
Nadezhdin, K. D., Correia, L., Narangoda, C., Patel, D. S., Neuberger, A., Gudermann, T., et al. (2023). Structural mechanisms of TRPM7 activation and inhibition. Nat. Commun. 14 (1), 2639. doi:10.1038/s41467-023-38362-3
Nadler, M. J., Hermosura, M. C., Inabe, K., Perraud, A. L., Zhu, Q., Stokes, A. J., et al. (2001). LTRPC7 is a Mg·ATP-regulated divalent cation channel required for cell viability. Nature 411 (6837), 590–595. doi:10.1038/35079092
Nadolni, W., and Zierler, S. (2018). The channel-kinase TRPM7 as novel regulator of immune system homeostasis. Cells 7 (8), 109. doi:10.3390/cells7080109
Nagamine, K., Kudoh, J., Minoshima, S., Kawasaki, K., Asakawa, S., Ito, F., et al. (1998). Molecular cloning of a novel putative Ca2+ channel protein (TRPC7) highly expressed in brain. Genomics 54 (1), 124–131. doi:10.1006/geno.1998.5551
Naziroğlu, M., and Lückhoff, A. (2008). A calcium influx pathway regulated separately by oxidative stress and ADP-ribose in TRPM2 channels: single channel events. Neurochem. Res. 33 (7), 1256–1262. doi:10.1007/s11064-007-9577-5
Naziroğlu, M. (2007). New molecular mechanisms on the activation of TRPM2 channels by oxidative stress and ADP-ribose. Neurochem. Res. 32 (11), 1990–2001. doi:10.1007/s11064-007-9386-x
Negri, S., Faris, P., Berra-Romani, R., Guerra, G., and Moccia, F. (2019). Endothelial transient receptor potential channels and vascular remodeling: extracellular Ca2+ entry for angiogenesis, arteriogenesis and vasculogenesis. Front. Physiol. 10, 1618. doi:10.3389/fphys.2019.01618
Ng, N. M., Jiang, S. P., and Lv, Z. Q. (2012). Retrovirus-mediated siRNA targeting TRPM7 gene induces apoptosis in RBL-2H3 cells. Eur. Rev. Med. Pharmacol. Sci. 16 (9), 1172–1178.
Nicotera, P., and Orrenius, S. (1998). The role of calcium in apoptosis. Cell Calcium 23 (2-3), 173–180. doi:10.1016/s0143-4160(98)90116-6
Nilius, B., Eggermont, J., Voets, T., Buyse, G., Manolopoulos, V., and Droogmans, G. (1997a). Properties of volume-regulated anion channels in mammalian cells. Prog. Biophys. Mol. Biol. 68 (1), 69–119. doi:10.1016/s0079-6107(97)00021-7
Nilius, B., Prenen, J., Kamouchi, M., Viana, F., Voets, T., and Droogmans, G. (1997b). Inhibition by mibefradil, a novel calcium channel antagonist, of Ca2+- and volume-activated Cl− channels in macrovascular endothelial cells. Br. J. Pharmacol. 121 (3), 547–555. doi:10.1038/sj.bjp.0701140
Nilius, B., Prenen, J., Vennekens, R., Hoenderop, J. G., Bindels, R. J., and Droogmans, G. (2001). Pharmacological modulation of monovalent cation currents through the epithelial Ca2+ channel ECaC1. Br. J. Pharmacol. 134 (3), 453–462. doi:10.1038/sj.bjp.0704272
Nuñez-Villena, F., Becerra, A., Echeverría, C., Briceño, N., Porras, O., Armisén, R., et al. (2011). Increased expression of the transient receptor potential melastatin 7 channel is critically involved in lipopolysaccharide-induced reactive oxygen species-mediated neuronal death. Antioxid. Redox Signal 15 (9), 2425–2438. doi:10.1089/ars.2010.3825
Nukui, M., Shimizu, T., and Okada, Y. (2006). Normotonic cell shrinkage induced by Na+ deprivation results in apoptotic cell death in human epithelial HeLa cells. J. Physiol. Sci. 56 (5), 335–339. doi:10.2170/physiolsci.RP009606
Numata, T., and Okada, Y. (2008a). Molecular determinants of sensitivity and conductivity of human TRPM7 to Mg2+ and Ca2+. Channels (Austin) 2 (4), 283–286. doi:10.4161/chan.2.4.6695
Numata, T., and Okada, Y. (2008b). Proton conductivity through the human TRPM7 channel and its molecular determinants. J. Biol. Chem. 283 (22), 15097–15103. doi:10.1074/jbc.M709261200
Numata, T., Sato, K., Christmann, J., Marx, R., Mori, Y., Okada, Y., et al. (2012). The ΔC splice-variant of TRPM2 is the hypertonicity-induced cation channel in HeLa cells, and the ecto-enzyme CD38 mediates its activation. J. Physiol. 590 (5), 1121–1138. doi:10.1113/jphysiol.2011.220947
Numata, T., Sato-Numata, K., Hermosura, M. C., Mori, Y., and Okada, Y. (2021). TRPM7 is an essential regulator for volume-sensitive outwardly rectifying anion channel. Commun. Biol. 4 (1), 599. doi:10.1038/s42003-021-02127-9
Numata, T., Sato-Numata, K., and Okada, Y. (2019). TRPM7 is involved in acid-induced necrotic cell death in a manner sensitive to progesterone in human cervical cancer cells. Physiol. Rep. 7 (13), e14157. doi:10.14814/phy2.14157
Numata, T., Shimizu, T., and Okada, Y. (2007a). Direct mechano-stress sensitivity of TRPM7 channel. Cell Physiol. Biochem. 19 (1-4), 1–8. doi:10.1159/000099187
Numata, T., Shimizu, T., and Okada, Y. (2007b). TRPM7 is a stretch- and swelling-activated cation channel involved in volume regulation in human epithelial cells. Am. J. Physiol. Cell Physiol. 292 (1), C460–C467. doi:10.1152/ajpcell.00367.2006
Numata, T., Wehner, F., and Okada, Y. (2007c). A novel inhibitor of hypertonicity-induced cation channels in HeLa cells. J. Physiol. Sci. 57 (4), 249–252. doi:10.2170/physiolsci.SC003007
Oancea, E., Wolfe, J. T., and Clapham, D. E. (2006). Functional TRPM7 channels accumulate at the plasma membrane in response to fluid flow. Circ. Res. 98 (2), 245–253. doi:10.1161/01.RES.0000200179.29375.cc
Okada, Y., Maeno, E., Shimizu, T., Dezaki, K., Wang, J., and Morishima, S. (2001). Receptor-mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD). J. Physiol. 532 (1), 3–16. doi:10.1111/j.1469-7793.2001.0003g.x
Okada, Y. (2016). Channelling frozen cells to survival after thawing: opening the door to cryo-physiology. J. Physiol. 594 (6), 1523–1524. doi:10.1113/jp271842
Okada, Y., and Hazama, A. (1989). Volume-regulatory ion channels in epithelial cells. Physiology 4 (6), 238–242. doi:10.1152/physiologyonline.1989.4.6.238
Okada, Y., Hazama, A., and Yuan, W. L. (1990). Stretch-induced activation of Ca2+-permeable ion channels is involved in the volume regulation of hypotonically swollen epithelial cells. Neurosci. Res. Suppl. 12, S5–S13. doi:10.1016/0921-8696(90)90004-m
Okada, Y. (2004). Ion channels and transporters involved in cell volume regulation and sensor mechanisms. Cell Biochem. Biophys. 41 (2), 233–258. doi:10.1385/cbb:41:2:233
Okada, Y., Maeno, E., Shimizu, T., Manabe, K., Mori, S., and Nabekura, T. (2004). Dual roles of plasmalemmal chloride channels in induction of cell death. Pflugers Arch. 448 (3), 287–295. doi:10.1007/s00424-004-1276-3
Okada, Y., Numata, T., Sato-Numata, K., Sabirov, R. Z., Liu, H., Mori, S. I., et al. (2019). Roles of volume-regulatory anion channels, VSOR and Maxi-Cl, in apoptosis, cisplatin resistance, necrosis, ischemic cell death, stroke and myocardial infarction. Curr. Top. Membr. 83, 205–283. doi:10.1016/bs.ctm.2019.03.001
Okada, Y., Sabirov, R. Z., Sato-Numata, K., and Numata, T. (2021a). Cell death induction and protection by activation of ubiquitously expressed anion/cation channels. Part 1: roles of VSOR/VRAC in cell volume regulation, release of double-edged signals and apoptotic/necrotic cell death. Front. Cell Dev. Biol. 8, 614040. doi:10.3389/fcell.2020.614040
Okada, Y., Sato-Numata, K., Sabirov, R. Z., and Numata, T. (2021b). Cell death induction and protection by activation of ubiquitously expressed anion/cation channels. Part 2: functional and molecular properties of ASOR/PAC channels and their roles in cell volume dysregulation and acidotoxic cell death. Front. Cell Dev. Biol. 9, 702317. doi:10.3389/fcell.2021.702317
Okada, Y. (1997). Volume expansion-sensing outward-rectifier Cl− channel: fresh start to the molecular identity and volume sensor. Am. J. Physiol. 273 (3), C755–C789. doi:10.1152/ajpcell.1997.273.3.C755
Orrenius, S., Zhivotovsky, B., and Nicotera, P. (2003). Regulation of cell death: the calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 4 (7), 552–565. doi:10.1038/nrm1150
Osmanlıoğlu, H., Yıldırım, M. K., Akyuva, Y., Yıldızhan, K., and Nazıroğlu, M. (2020). Morphine Induces apoptosis, inflammation, and mitochondrial oxidative stress via activation of TRPM2 channel and nitric oxide signaling pathways in the hippocampus. Mol. Neurobiol. 57 (8), 3376–3389. doi:10.1007/s12035-020-01975-6
Ostapchenko, V. G., Chen, M., Guzman, M. S., Xie, Y. F., Lavine, N., Fan, J., et al. (2015). The transient receptor potential melastatin 2 (TRPM2) channel contributes to β-amyloid oligomer-related neurotoxicity and memory impairment. J. Neurosci. 35 (45), 15157–15169. doi:10.1523/jneurosci.4081-14.2015
Pan, T., Zhu, Q. J., Xu, L. X., Ding, X., Li, J. Q., Sun, B., et al. (2020). Knocking down TRPM2 expression reduces cell injury and NLRP3 inflammasome activation in PC12 cells subjected to oxygen-glucose deprivation. Neural Regen. Res. 15 (11), 2154–2161. doi:10.4103/1673-5374.282271
Parnas, M., Peters, M., Dadon, D., Lev, S., Vertkin, I., Slutsky, I., et al. (2009). Carvacrol is a novel inhibitor of Drosophila TRPL and mammalian TRPM7 channels. Cell Calcium 45 (3), 300–309. doi:10.1016/j.ceca.2008.11.009
Partida-Sanchez, S., Gasser, A., Fliegert, R., Siebrands, C. C., Dammermann, W., Shi, G., et al. (2007). Chemotaxis of mouse bone marrow neutrophils and dendritic cells is controlled by adp-ribose, the major product generated by the CD38 enzyme reaction. J. Immunol. 179 (11), 7827–7839. doi:10.4049/jimmunol.179.11.7827
Perraud, A. L., Fleig, A., Dunn, C. A., Bagley, L. A., Launay, P., Schmitz, C., et al. (2001). ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 411 (6837), 595–599. doi:10.1038/35079100
Perraud, A. L., Schmitz, C., and Scharenberg, A. M. (2003). TRPM2 Ca2+ permeable cation channels: from gene to biological function. Cell Calcium 33 (5-6), 519–531. doi:10.1016/s0143-4160(03)00057-5
Perraud, A. L., Takanishi, C. L., Shen, B., Kang, S., Smith, M. K., Schmitz, C., et al. (2005). Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J. Biol. Chem. 280 (7), 6138–6148. doi:10.1074/jbc.M411446200
Plettenberg, S., Weiss, E. C., Lemor, R., and Wehner, F. (2008). Subunits alpha, beta and gamma of the epithelial Na+ channel (ENaC) are functionally related to the hypertonicity-induced cation channel (HICC) in rat hepatocytes. Pflugers Arch. 455 (6), 1089–1095. doi:10.1007/s00424-007-0355-7
Poblete, H., Oyarzún, I., Olivero, P., Comer, J., Zuñiga, M., Sepulveda, R. V., et al. (2015). Molecular determinants of phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) binding to transient receptor potential V1 (TRPV1) channels. J. Biol. Chem. 290 (4), 2086–2098. doi:10.1074/jbc.M114.613620
Qin, X., Yue, Z., Sun, B., Yang, W., Xie, J., Ni, E., et al. (2013). Sphingosine and FTY720 are potent inhibitors of the transient receptor potential melastatin 7 (TRPM7) channels. Br. J. Pharmacol. 168 (6), 1294–1312. doi:10.1111/bph.12012
Qiu, Z., Dubin, A. E., Mathur, J., Tu, B., Reddy, K., Miraglia, L. J., et al. (2014). SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell 157 (2), 447–458. doi:10.1016/j.cell.2014.03.024
Rah, S. Y., Kwak, J. Y., Chung, Y. J., and Kim, U. H. (2015). ADP-ribose/TRPM2-mediated Ca2+ signaling is essential for cytolytic degranulation and antitumor activity of natural killer cells. Sci. Rep. 5, 9482. doi:10.1038/srep09482
Ritter, M., Mongin, A. A., Valenti, G., and Okada, Y. (2021). Editorial: ion and water transport in cell death. Front. Cell Dev. Biol. 9, 757033. doi:10.3389/fcell.2021.757033
Rizzuto, R., Pinton, P., Ferrari, D., Chami, M., Szabadkai, G., Magalhães, P. J., et al. (2003). Calcium and apoptosis: facts and hypotheses. Oncogene 22 (53), 8619–8627. doi:10.1038/sj.onc.1207105
Roberge, S., Roussel, J., Andersson, D. C., Meli, A. C., Vidal, B., Blandel, F., et al. (2014). TNF-α-mediated caspase-8 activation induces ROS production and TRPM2 activation in adult ventricular myocytes. Cardiovasc Res. 103 (1), 90–99. doi:10.1093/cvr/cvu112
Rohács, T., Lopes, C. M., Michailidis, I., and Logothetis, D. E. (2005). PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat. Neurosci. 8 (5), 626–634. doi:10.1038/nn1451
Romagnani, A., Vettore, V., Rezzonico-Jost, T., Hampe, S., Rottoli, E., Nadolni, W., et al. (2017). TRPM7 kinase activity is essential for T cell colonization and alloreactivity in the gut. Nat. Commun. 8 (1), 1917. doi:10.1038/s41467-017-01960-z
Rössig, A., Hill, K., Nörenberg, W., Weidenbach, S., Zierler, S., Schaefer, M., et al. (2022). Pharmacological agents selectively acting on the channel moieties of TRPM6 and TRPM7. Cell Calcium 106, 102640. doi:10.1016/j.ceca.2022.102640
Rothstein, A., and Mack, E. (1990). Volume-activated K+ and Cl− pathways of dissociated epithelial cells (MDCK): role of Ca2+. Am. J. Physiol. 258 (5), C827–C834. doi:10.1152/ajpcell.1990.258.5.C827
Runnels, L. W., Yue, L., and Clapham, D. E. (2002). The TRPM7 channel is inactivated by PIP2 hydrolysis. Nat. Cell Biol. 4 (5), 329–336. doi:10.1038/ncb781
Runnels, L. W., Yue, L., and Clapham, D. E. (2001). TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science 291 (5506), 1043–1047. doi:10.1126/science.1058519
Ryazanova, L. V., Rondon, L. J., Zierler, S., Hu, Z., Galli, J., Yamaguchi, T. P., et al. (2010). TRPM7 is essential for Mg2+ homeostasis in mammals. Nat. Commun. 1, 109. doi:10.1038/ncomms1108
Şanlı, C., Atılgan, R., Kuloğlu, T., Pala, Ş., Aydın Türk, B., Keser, H. B., et al. (2021). Transient receptor potential melastatin 2 ion channel activity in ovarian hyperstimulation syndrome physiopathology. Turk J. Med. Sci. 51 (2), 787–795. doi:10.3906/sag-2005-297
Sano, Y., Inamura, K., Miyake, A., Mochizuki, S., Yokoi, H., Matsushime, H., et al. (2001). Immunocyte Ca2+ influx system mediated by LTRPC2. Science 293 (5533), 1327–1330. doi:10.1126/science.1062473
Schäfer, S., Ferioli, S., Hofmann, T., Zierler, S., Gudermann, T., and Chubanov, V. (2016). Mibefradil represents a new class of benzimidazole TRPM7 channel agonists. Pflugers Arch. 468 (4), 623–634. doi:10.1007/s00424-015-1772-7
Schlingmann, K. P., Waldegger, S., Konrad, M., Chubanov, V., and Gudermann, T. (2007). TRPM6 and TRPM7--Gatekeepers of human magnesium metabolism. Biochim. Biophys. Acta 1772 (8), 813–821. doi:10.1016/j.bbadis.2007.03.009
Schmidt, E., Narangoda, C., Nörenberg, W., Egawa, M., Rössig, A., Leonhardt, M., et al. (2022). Structural mechanism of TRPM7 channel regulation by intracellular magnesium. Cell Mol. Life Sci. 79 (5), 225. doi:10.1007/s00018-022-04192-7
Schmitz, C., Perraud, A. L., Johnson, C. O., Inabe, K., Smith, M. K., Penner, R., et al. (2003). Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell 114 (2), 191–200. doi:10.1016/s0092-8674(03)00556-7
Schorn, C., Frey, B., Lauber, K., Janko, C., Strysio, M., Keppeler, H., et al. (2011). Sodium overload and water influx activate the NALP3 inflammasome. J. Biol. Chem. 286 (1), 35–41. doi:10.1074/jbc.M110.139048
Shimizu, S., Yonezawa, R., Hagiwara, T., Yoshida, T., Takahashi, N., Hamano, S., et al. (2014). Inhibitory effects of AG490 on H2O2-induced TRPM2-mediated Ca2+ entry. Eur. J. Pharmacol. 742, 22–30. doi:10.1016/j.ejphar.2014.08.023
Shimizu, T., Dietz, R. M., Cruz-Torres, I., Strnad, F., Garske, A. K., Moreno, M., et al. (2016). Extended therapeutic window of a novel peptide inhibitor of TRPM2 channels following focal cerebral ischemia. Exp. Neurol. 283, 151–156. doi:10.1016/j.expneurol.2016.06.015
Shimizu, T., Macey, T. A., Quillinan, N., Klawitter, J., Perraud, A. L., Traystman, R. J., et al. (2013). Androgen and PARP-1 regulation of TRPM2 channels after ischemic injury. J. Cereb. Blood Flow. Metab. 33 (10), 1549–1555. doi:10.1038/jcbfm.2013.105
Shimizu, T., Maeno, E., and Okada, Y. (2007). Prerequisite role of persistent cell shrinkage in apoptosis of human epithelial cells. Sheng Li Xue Bao 59 (4), 512–516.
Shimizu, T., Numata, T., and Okada, Y. (2004). A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl− channel. Proc. Natl. Acad. Sci. U. S. A. 101 (17), 6770–6773. doi:10.1073/pnas.0401604101
Simon, F., Varela, D., and Cabello-Verrugio, C. (2013). Oxidative stress-modulated TRPM ion channels in cell dysfunction and pathological conditions in humans. Cell Signal 25 (7), 1614–1624. doi:10.1016/j.cellsig.2013.03.023
Singh, A., Hildebrand, M. E., Garcia, E., and Snutch, T. P. (2010). The transient receptor potential channel antagonist SKF96365 is a potent blocker of low-voltage-activated T-type calcium channels. Br. J. Pharmacol. 160 (6), 1464–1475. doi:10.1111/j.1476-5381.2010.00786.x
Smith, M. A., Herson, P. S., Lee, K., Pinnock, R. D., and Ashford, M. L. (2003). Hydrogen-peroxide-induced toxicity of rat striatal neurones involves activation of a non-selective cation channel. J. Physiol. 547 (2), 417–425. doi:10.1113/jphysiol.2002.034561
Song, C., Bae, Y., Jun, J., Lee, H., Kim, N. D., Lee, K. B., et al. (2017). Identification of TG100-115 as a new and potent TRPM7 kinase inhibitor, which suppresses breast cancer cell migration and invasion. Biochim. Biophys. Acta Gen. Subj. 1861 (4), 947–957. doi:10.1016/j.bbagen.2017.01.034
Song, K., Wang, H., Kamm, G. B., Pohle, J., Reis, F. C., Heppenstall, P., et al. (2016). The TRPM2 channel is a hypothalamic heat sensor that limits fever and can drive hypothermia. Science 353 (6306), 1393–1398. doi:10.1126/science.aaf7537
Starkus, J., Beck, A., Fleig, A., and Penner, R. (2007). Regulation of TRPM2 by extra- and intracellular calcium. J. Gen. Physiol. 130 (4), 427–440. doi:10.1085/jgp.200709836
Starkus, J. G., Fleig, A., and Penner, R. (2010). The calcium-permeable non-selective cation channel TRPM2 is modulated by cellular acidification. J. Physiol. 588 (8), 1227–1240. doi:10.1113/jphysiol.2010.187476
Strange, K., and Jackson, P. S. (1995). Swelling-activated organic osmolyte efflux: a new role for anion channels. Kidney Int. 48 (4), 994–1003. doi:10.1038/ki.1995.381
Su, L. T., Agapito, M. A., Li, M., Simonson, W. T., Huttenlocher, A., Habas, R., et al. (2006). TRPM7 regulates cell adhesion by controlling the calcium-dependent protease calpain. J. Biol. Chem. 281 (16), 11260–11270. doi:10.1074/jbc.M512885200
Subramanyam, M., Takahashi, N., Hasegawa, Y., Mohri, T., and Okada, Y. (2010). Inhibition of protein kinase Akt1 by apoptosis signal-regulating kinase-1 (ASK1) is involved in apoptotic inhibition of regulatory volume increase. J. Biol. Chem. 285 (9), 6109–6117. doi:10.1074/jbc.M109.072785
Sumoza-Toledo, A., and Penner, R. (2011). TRPM2: a multifunctional ion channel for calcium signalling. J. Physiol. 589 (7), 1515–1525. doi:10.1113/jphysiol.2010.201855
Sun, H. S., Jackson, M. F., Martin, L. J., Jansen, K., Teves, L., Cui, H., et al. (2009). Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat. Neurosci. 12 (10), 1300–1307. doi:10.1038/nn.2395
Sun, L., Yau, H. Y., Wong, W. Y., Li, R. A., Huang, Y., and Yao, X. (2012). Role of TRPM2 in H2O2-induced cell apoptosis in endothelial cells. PLoS One 7 (8), e43186. doi:10.1371/journal.pone.0043186
Sun, Y., Kamat, A., and Singh, B. B. (2020a). Isoproterenol-dependent activation of TRPM7 protects against neurotoxin-induced loss of neuroblastoma cells. Front. Physiol. 11, 305. doi:10.3389/fphys.2020.00305
Sun, Y., Sukumaran, P., Selvaraj, S., Cilz, N. I., Schaar, A., Lei, S., et al. (2018). TRPM2 promotes neurotoxin MPP+/MPTP-induced cell death. Mol. Neurobiol. 55 (1), 409–420. doi:10.1007/s12035-016-0338-9
Sun, Y., Sukumaran, P., and Singh, B. B. (2020b). Magnesium-induced cell survival Is dependent on TRPM7 expression and function. Mol. Neurobiol. 57 (1), 528–538. doi:10.1007/s12035-019-01713-7
Szollosi, A. (2021). Two decades of evolution of our understanding of the transient receptor potential melastatin 2 (TRPM2) cation channel. Life (Basel) 11 (5), 397. doi:10.3390/life11050397
Takasawa, S., Tohgo, A., Noguchi, N., Koguma, T., Nata, K., Sugimoto, T., et al. (1993). Synthesis and hydrolysis of cyclic ADP-ribose by human leukocyte antigen CD38 and inhibition of the hydrolysis by ATP. J. Biol. Chem. 268 (35), 26052–26054. doi:10.1016/s0021-9258(19)74275-6
Tan, C. H., and McNaughton, P. A. (2016). The TRPM2 ion channel is required for sensitivity to warmth. Nature 536 (7617), 460–463. doi:10.1038/nature19074
Thomas, G. P., Karmazyn, M., Zygmunt, A. C., Antzelevitch, C., and Narayanan, N. (1999). The antifungal antibiotic clotrimazole potently inhibits L-type calcium current in Guinea-pig ventricular myocytes. Br. J. Pharmacol. 126 (7), 1531–1533. doi:10.1038/sj.bjp.0702475
Tian, Y., Yang, T., Yu, S., Liu, C., He, M., and Hu, C. (2018). Prostaglandin E2 increases migration and proliferation of human glioblastoma cells by activating transient receptor potential melastatin 7 channels. J. Cell Mol. Med. 22 (12), 6327–6337. doi:10.1111/jcmm.13931
Toda, T., Yamamoto, S., Yonezawa, R., Mori, Y., and Shimizu, S. (2016). Inhibitory effects of Tyrphostin AG-related compounds on oxidative stress-sensitive transient receptor potential channel activation. Eur. J. Pharmacol. 786, 19–28. doi:10.1016/j.ejphar.2016.05.033
Togashi, K., Hara, Y., Tominaga, T., Higashi, T., Konishi, Y., Mori, Y., et al. (2006). TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. Embo J. 25 (9), 1804–1815. doi:10.1038/sj.emboj.7601083
Togashi, K., Inada, H., and Tominaga, M. (2008). Inhibition of the transient receptor potential cation channel TRPM2 by 2-aminoethoxydiphenyl borate (2-APB). Br. J. Pharmacol. 153 (6), 1324–1330. doi:10.1038/sj.bjp.0707675
Tong, L., and Denu, J. M. (2010). Function and metabolism of sirtuin metabolite O-acetyl-ADP-ribose. Biochim. Biophys. Acta 1804 (8), 1617–1625. doi:10.1016/j.bbapap.2010.02.007
Tong, Q., Zhang, W., Conrad, K., Mostoller, K., Cheung, J. Y., Peterson, B. Z., et al. (2006). Regulation of the transient receptor potential channel TRPM2 by the Ca2+ sensor calmodulin. J. Biol. Chem. 281 (14), 9076–9085. doi:10.1074/jbc.M510422200
Tosteson, D. C., and Hoffman, J. F. (1960). Regulation of cell volume by active cation transport in high and low potassium sheep red cells. J. Gen. Physiol. 44 (1), 169–194. doi:10.1085/jgp.44.1.169
Tóth, B., and Csanády, L. (2012). Pore collapse underlies irreversible inactivation of TRPM2 cation channel currents. Proc. Natl. Acad. Sci. U. S. A. 109 (33), 13440–13445. doi:10.1073/pnas.1204702109
Tóth, B., Iordanov, I., and Csanády, L. (2015). Ruling out pyridine dinucleotides as true TRPM2 channel activators reveals novel direct agonist ADP-ribose-2'-phosphate. J. Gen. Physiol. 145 (5), 419–430. doi:10.1085/jgp.201511377
Touyz, R. M., He, Y., Montezano, A. C., Yao, G., Chubanov, V., Gudermann, T., et al. (2006). Differential regulation of transient receptor potential melastatin 6 and 7 cation channels by ANG II in vascular smooth muscle cells from spontaneously hypertensive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 290 (1), R73–R78. doi:10.1152/ajpregu.00515.2005
Tseng, H. H., Vong, C. T., Kwan, Y. W., Lee, S. M., and Hoi, M. P. (2016). TRPM2 regulates TXNIP-mediated NLRP3 inflammasome activation via interaction with p47 phox under high glucose in human monocytic cells. Sci. Rep. 6, 35016. doi:10.1038/srep35016
Turlova, E., Bae, C. Y. J., Deurloo, M., Chen, W., Barszczyk, A., Horgen, F. D., et al. (2016). TRPM7 regulates axonal outgrowth and maturation of primary hippocampal neurons. Mol. Neurobiol. 53 (1), 595–610. doi:10.1007/s12035-014-9032-y
Turlova, E., Feng, Z. P., and Sun, H. S. (2018). The role of TRPM2 channels in neurons, glial cells and the blood-brain barrier in cerebral ischemia and hypoxia. Acta Pharmacol. Sin. 39 (5), 713–721. doi:10.1038/aps.2017.194
Turlova, E., Wong, R., Xu, B., Li, F., Du, L., Habbous, S., et al. (2021). TRPM7 mediates neuronal cell Death upstream of calcium/calmodulin-dependent protein kinase II and calcineurin mechanism in neonatal hypoxic-ischemic brain injury. Transl. Stroke Res. 12 (1), 164–184. doi:10.1007/s12975-020-00810-3
Tymianski, M. (2011). Emerging mechanisms of disrupted cellular signaling in brain ischemia. Nat. Neurosci. 14 (11), 1369–1373. doi:10.1038/nn.2951
Varela, D., Simon, F., Riveros, A., Jørgensen, F., and Stutzin, A. (2004). NAD(P)H oxidase-derived H2O2 signals chloride channel activation in cell volume regulation and cell proliferation. J. Biol. Chem. 279 (14), 13301–13304. doi:10.1074/jbc.C400020200
Vennekens, R., Mesuere, M., and Philippaert, K. (2018). TRPM5 in the battle against diabetes and obesity. Acta Physiol. (Oxf) 222 (2), e12949. doi:10.1111/apha.12949
Verma, S., Quillinan, N., Yang, Y. F., Nakayama, S., Cheng, J., Kelley, M. H., et al. (2012). TRPM2 channel activation following in vitro ischemia contributes to male hippocampal cell death. Neurosci. Lett. 530 (1), 41–46. doi:10.1016/j.neulet.2012.09.044
Viana, F., Van den Bosch, L., Missiaen, L., Vandenberghe, W., Droogmans, G., Nilius, B., et al. (1997). Mibefradil (Ro 40-5967) blocks multiple types of voltage-gated calcium channels in cultured rat spinal motoneurones. Cell Calcium 22 (4), 299–311. doi:10.1016/s0143-4160(97)90068-3
Volk, T., Frömter, E., and Korbmacher, C. (1995). Hypertonicity activates nonselective cation channels in mouse cortical collecting duct cells. Proc. Natl. Acad. Sci. U. S. A. 92 (18), 8478–8482. doi:10.1073/pnas.92.18.8478
Voss, F. K., Ullrich, F., Münch, J., Lazarow, K., Lutter, D., Mah, N., et al. (2014). Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science 344 (6184), 634–638. doi:10.1126/science.1252826
Vriens, J., and Voets, T. (2019). Heat sensing involves a TRiPlet of ion channels. Br. J. Pharmacol. 176 (20), 3893–3898. doi:10.1111/bph.14812
Waldmann, R., Champigny, G., Bassilana, F., Heurteaux, C., and Lazdunski, M. (1997). A proton-gated cation channel involved in acid-sensing. Nature 386 (6621), 173–177. doi:10.1038/386173a0
Waldmann, R., Champigny, G., Lingueglia, E., De Weille, J. R., Heurteaux, C., and Lazdunski, M. (1999). H+-gated cation channels. Ann. N. Y. Acad. Sci. 868, 67–76. doi:10.1111/j.1749-6632.1999.tb11274.x
Wang, C., Naruse, K., and Takahashi, K. (2018). Role of the TRPM4 channel in cardiovascular physiology and pathophysiology. Cells 7 (6), 62. doi:10.3390/cells7060062
Wang, H. Y., Shimizu, T., Numata, T., and Okada, Y. (2007). Role of acid-sensitive outwardly rectifying anion channels in acidosis-induced cell death in human epithelial cells. Pflugers Arch. 454 (2), 223–233. doi:10.1007/s00424-006-0193-z
Wang, J., Morishima, S., and Okada, Y. (2003). Ik channels are involved in the regulatory volume decrease in human epithelial cells. Am. J. Physiol. Cell Physiol. 284 (1), C77–C84. doi:10.1152/ajpcell.00132.2002
Wang, L., Fu, T. M., Zhou, Y., Xia, S., Greka, A., and Wu, H. (2018). Structures and gating mechanism of human TRPM2. Science 362 (6421), eaav4809. doi:10.1126/science.aav4809
Wang, L., Negro, R., and Wu, H. (2020). TRPM2, linking oxidative stress and Ca2+ permeation to NLRP3 inflammasome activation. Curr. Opin. Immunol. 62, 131–135. doi:10.1016/j.coi.2020.01.005
Wehage, E., Eisfeld, J., Heiner, I., Jüngling, E., Zitt, C., and Lückhoff, A. (2002). Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J. Biol. Chem. 277 (26), 23150–23156. doi:10.1074/jbc.M112096200
Wehner, F., Bondarava, M., ter Veld, F., Endl, E., Nürnberger, H. R., and Li, T. (2006). Hypertonicity-induced cation channels. Acta Physiol. (Oxf) 187 (1-2), 21–25. doi:10.1111/j.1748-1716.2006.01561.x
Wehner, F., Olsen, H., Tinel, H., Kinne-Saffran, E., and Kinne, R. K. (2003a). Cell volume regulation: osmolytes, osmolyte transport, and signal transduction. Rev. Physiol. Biochem. Pharmacol. 148, 1–80. doi:10.1007/s10254-003-0009-x
Wehner, F., Sauer, H., and Kinne, R. K. (1995). Hypertonic stress increases the Na+ conductance of rat hepatocytes in primary culture. J. Gen. Physiol. 105 (4), 507–535. doi:10.1085/jgp.105.4.507
Wehner, F., Shimizu, T., Sabirov, R., and Okada, Y. (2003b). Hypertonic activation of a non-selective cation conductance in HeLa cells and its contribution to cell volume regulation. FEBS Lett. 551 (1-3), 20–24. doi:10.1016/s0014-5793(03)00868-8
Wei, C., Wang, X., Chen, M., Ouyang, K., Song, L. S., and Cheng, H. (2009). Calcium flickers steer cell migration. Nature 457 (7231), 901–905. doi:10.1038/nature07577
Wilson, C. S., and Mongin, A. A. (2018). Cell volume control in healthy brain and neuropathologies. Curr. Top. Membr. 81, 385–455. doi:10.1016/bs.ctm.2018.07.006
Won, J., Vang, H., Kim, J. H., Lee, P. R., Kang, Y., and Oh, S. B. (2018). TRPM7 mediates mechanosensitivity in adult rat odontoblasts. J. Dent. Res. 97 (9), 1039–1046. doi:10.1177/0022034518759947
Wong, K. K., Banham, A. H., Yaacob, N. S., and Nur Husna, S. M. (2019). The oncogenic roles of TRPM ion channels in cancer. J. Cell Physiol. 234, 14556–14573. doi:10.1002/jcp.28168
Wong, S. M., and Chase, H. S. (1986). Role of intracellular calcium in cellular volume regulation. Am. J. Physiol. 250 (6), C841–C852. doi:10.1152/ajpcell.1986.250.6.C841
Wu, D. C., Teismann, P., Tieu, K., Vila, M., Jackson-Lewis, V., Ischiropoulos, H., et al. (2003). NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Proc. Natl. Acad. Sci. U. S. A. 100 (10), 6145–6150. doi:10.1073/pnas.0937239100
Wu, H., Dai, R., Wang, M., and Chen, C. (2023). Uric acid promotes myocardial infarction injury via activating pyrin domain-containing 3 inflammasome and reactive oxygen species/transient receptor potential melastatin 2/Ca2+ pathway. BMC Cardiovasc Disord. 23 (1), 10. doi:10.1186/s12872-023-03040-1
Wuensch, T., Thilo, F., Krueger, K., Scholze, A., Ristow, M., and Tepel, M. (2010). High glucose-induced oxidative stress increases transient receptor potential channel expression in human monocytes. Diabetes 59 (4), 844–849. doi:10.2337/db09-1100
Xia, R., Mei, Z. Z., Mao, H. J., Yang, W., Dong, L., Bradley, H., et al. (2008). Identification of pore residues engaged in determining divalent cationic permeation in transient receptor potential melastatin subtype channel 2. J. Biol. Chem. 283 (41), 27426–27432. doi:10.1074/jbc.M801049200
Xia, S., Wang, L., Fu, T. M., and Wu, H. (2019). Mechanism of TRPM2 channel gating revealed by cryo-EM. Febs J. 286 (17), 3333–3339. doi:10.1111/febs.14939
Xiao, E., Yang, H. Q., Gan, Y. H., Duan, D. H., He, L. H., Guo, Y., et al. (2015). Brief reports: TRPM7 senses mechanical stimulation inducing osteogenesis in human bone marrow mesenchymal stem cells. Stem Cells 33 (2), 615–621. doi:10.1002/stem.1858
Xie, J., Sun, B., Du, J., Yang, W., Chen, H. C., Overton, J. D., et al. (2011). Phosphatidylinositol 4,5-bisphosphate (PIP2) controls magnesium gatekeeper TRPM6 activity. Sci. Rep. 1, 146. doi:10.1038/srep00146
Xu, H. L., Liu, M. D., Yuan, X. H., and Liu, C. X. (2018). Suppression of cortical TRPM7 protein attenuates oxidative damage after traumatic brain injury via Akt/endothelial nitric oxide synthase pathway. Neurochem. Int. 112, 197–205. doi:10.1016/j.neuint.2017.07.010
Xu, S. Z., Zeng, F., Boulay, G., Grimm, C., Harteneck, C., and Beech, D. J. (2005). Block of TRPC5 channels by 2-aminoethoxydiphenyl borate: a differential, extracellular and voltage-dependent effect. Br. J. Pharmacol. 145 (4), 405–414. doi:10.1038/sj.bjp.0706197
Yamaguchi, H., Matsushita, M., Nairn, A. C., and Kuriyan, J. (2001). Crystal structure of the atypical protein kinase domain of a TRP channel with phosphotransferase activity. Mol. Cell 7 (5), 1047–1057. doi:10.1016/s1097-2765(01)00256-8
Yang, D., He, Y., Muñoz-Planillo, R., Liu, Q., and Núñez, G. (2015). Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity 43 (5), 923–932. doi:10.1016/j.immuni.2015.10.009
Yang, J., Chen, J., Del Carmen Vitery, M., Osei-Owusu, J., Chu, J., Yu, H., et al. (2019). PAC, an evolutionarily conserved membrane protein, is a proton-activated chloride channel. Science 364 (6438), 395–399. doi:10.1126/science.aav9739
Yang, T., Zhang, A., Honeggar, M., Kohan, D. E., Mizel, D., Sanders, K., et al. (2005). Hypertonic induction of COX-2 in collecting duct cells by reactive oxygen species of mitochondrial origin. J. Biol. Chem. 280 (41), 34966–34973. doi:10.1074/jbc.M502430200
Yang, W., Manna, P. T., Zou, J., Luo, J., Beech, D. J., Sivaprasadarao, A., et al. (2011). Zinc inactivates melastatin transient receptor potential 2 channels via the outer pore. J. Biol. Chem. 286 (27), 23789–23798. doi:10.1074/jbc.M111.247478
Yang, W., Zou, J., Xia, R., Vaal, M. L., Seymour, V. A., Luo, J., et al. (2010). State-dependent inhibition of TRPM2 channel by acidic pH. J. Biol. Chem. 285 (40), 30411–30418. doi:10.1074/jbc.M110.139774
Yang, X., Li, C., Liao, X., Liu, S., Li, X., Hou, X., et al. (2023). Paclitaxel induces pyroptosis by inhibiting the volume-sensitive chloride channel leucine-rich repeat-containing 8a in ovarian cancer cells. Oncol. Rep. 49 (6), 115. doi:10.3892/or.2023.8552
Yazğan, Y., and Nazıroğlu, M. (2021). Involvement of TRPM2 in the neurobiology of experimental migraine: focus on oxidative stress and apoptosis. Mol. Neurobiol. 58 (11), 5581–5601. doi:10.1007/s12035-021-02503-w
Ye, M., Yang, W., Ainscough, J. F., Hu, X. P., Li, X., Sedo, A., et al. (2014). TRPM2 channel deficiency prevents delayed cytosolic Zn2+ accumulation and CA1 pyramidal neuronal death after transient global ischemia. Cell Death Dis. 5 (11), e1541. doi:10.1038/cddis.2014.494
Ye, X., Liu, X., Wei, W., Yu, H., Jin, X., Yu, J., et al. (2021). Volume-activated chloride channels contribute to lipopolysaccharide plus nigericin-induced pyroptosis in bone marrow-derived macrophages. Biochem. Pharmacol. 193, 114791. doi:10.1016/j.bcp.2021.114791
Yee, N. S. (2017). Role of TRPM7 in cancer: potential as molecular biomarker and therapeutic target. Pharm. (Basel) 10 (2), 39. doi:10.3390/ph10020039
Yee, N. S., Kazi, A. A., and Yee, R. K. (2014). Cellular and developmental biology of TRPM7 channel-kinase: implicated roles in cancer. Cells 3 (3), 751–777. doi:10.3390/cells3030751
Yıldızhan, K., Huyut, Z., and Altındağ, F. (2023). Involvement of TRPM2 channel on doxorubicin-induced experimental cardiotoxicity model: protective role of selenium. Biol. Trace Elem. Res. 201 (5), 2458–2469. doi:10.1007/s12011-022-03377-2
Yin, Y., Wu, M., Hsu, A. L., Borschel, W. F., Borgnia, M. J., Lander, G. C., et al. (2019). Visualizing structural transitions of ligand-dependent gating of the TRPM2 channel. Nat. Commun. 10 (1), 3740. doi:10.1038/s41467-019-11733-5
Yu, H., Zhang, Z., Lis, A., Penner, R., and Fleig, A. (2013). TRPM7 is regulated by halides through its kinase domain. Cell Mol. Life Sci. 70 (15), 2757–2771. doi:10.1007/s00018-013-1284-6
Yu, M., Huang, C., Huang, Y., Wu, X., Li, X., and Li, J. (2013). Inhibition of TRPM7 channels prevents proliferation and differentiation of human lung fibroblasts. Inflamm. Res. 62 (11), 961–970. doi:10.1007/s00011-013-0653-9
Yu, P., Liu, Z., Yu, X., Ye, P., Liu, H., Xue, X., et al. (2019). Direct gating of the TRPM2 channel by cADPR via specific interactions with the ADPR binding pocket. Cell Rep. 27 (12), 3684–3695. e3684. doi:10.1016/j.celrep.2019.05.067
Yu, W., Jiang, L. H., Zheng, Y., Hu, X., Luo, J., and Yang, W. (2014). Inactivation of TRPM2 channels by extracellular divalent copper. PLoS One 9 (11), e112071. doi:10.1371/journal.pone.0112071
Yu, X., Xie, Y., Zhang, X., Ma, C., Liu, L., Zhen, W., et al. (2021). Structural and functional basis of the selectivity filter as a gate in human TRPM2 channel. Cell Rep. 37 (7), 110025. doi:10.1016/j.celrep.2021.110025
Yürüker, V., Nazıroğlu, M., and Şenol, N. (2015). Reduction in traumatic brain injury-induced oxidative stress, apoptosis, and calcium entry in rat hippocampus by melatonin: possible involvement of TRPM2 channels. Metab. Brain Dis. 30 (1), 223–231. doi:10.1007/s11011-014-9623-3
Zeng, B., Chen, G. L., and Xu, S. Z. (2012). Divalent copper is a potent extracellular blocker for TRPM2 channel. Biochem. Biophys. Res. Commun. 424 (2), 279–284. doi:10.1016/j.bbrc.2012.06.107
Zhan, H. Q., Li, P. F., Li, S. Y., Yan, F. L., Yang, J., Liu, R. L., et al. (2014). Influence of lactuside B on the expression of AQP4 and TRPM7 mRNAs in the cerebral cortex after cerebral ischemia injury. Eur. Rev. Med. Pharmacol. Sci. 18 (8), 1151–1157.
Zhang, H., Yu, P., Lin, H., Jin, Z., Zhao, S., Zhang, Y., et al. (2021). The discovery of novel ACA derivatives as specific TRPM2 inhibitors that reduce ischemic injury both in vitro and in vivo. J. Med. Chem. 64 (7), 3976–3996. doi:10.1021/acs.jmedchem.0c02129
Zhang, W., Chu, X., Tong, Q., Cheung, J. Y., Conrad, K., Masker, K., et al. (2003). A novel TRPM2 isoform inhibits calcium influx and susceptibility to cell death. J. Biol. Chem. 278 (18), 16222–16229. doi:10.1074/jbc.M300298200
Zhang, W., Hirschler-Laszkiewicz, I., Tong, Q., Conrad, K., Sun, S. C., Penn, L., et al. (2006). TRPM2 is an ion channel that modulates hematopoietic cell death through activation of caspases and PARP cleavage. Am. J. Physiol. Cell Physiol. 290 (4), C1146–C1159. doi:10.1152/ajpcell.00205.2005
Zhang, W., Tong, Q., Conrad, K., Wozney, J., Cheung, J. Y., and Miller, B. A. (2007). Regulation of TRP channel TRPM2 by the tyrosine phosphatase PTPL1. Am. J. Physiol. Cell Physiol. 292 (5), C1746–C1758. doi:10.1152/ajpcell.00569.2006
Zhang, X., Chen, Q., Wang, Y., Peng, W., and Cai, H. (2014). Effects of curcumin on ion channels and transporters. Front. Physiol. 5, 94. doi:10.3389/fphys.2014.00094
Zhang, Y., Zhou, L., Zhang, X., Bai, J., Shi, M., and Zhao, G. (2012). Ginsenoside-Rd attenuates TRPM7 and ASIC1a but promotes ASIC2a expression in rats after focal cerebral ischemia. Neurol. Sci. 33 (5), 1125–1131. doi:10.1007/s10072-011-0916-6
Zhang, Z., Tóth, B., Szollosi, A., Chen, J., and Csanády, L. (2018). Structure of a TRPM2 channel in complex with Ca2+ explains unique gating regulation. Elife 7, e36409. doi:10.7554/eLife.36409
Zhao, R., Afthinos, A., Zhu, T., Mistriotis, P., Li, Y., Serra, S. A., et al. (2019). Cell sensing and decision-making in confinement: the role of TRPM7 in a tug of war between hydraulic pressure and cross-sectional area. Sci. Adv. 5 (7), eaaw7243. doi:10.1126/sciadv.aaw7243
Zhao, Y. J., Lam, C. M., and Lee, H. C. (2012). The membrane-bound enzyme CD38 exists in two opposing orientations. Sci. Signal 5 (241), ra67. doi:10.1126/scisignal.2002700
Zhao, Y., Wang, J., Jiang, H., Yu, Z., Li, X., and Shi, J. (2015). Following OGD/R, annexin 1 nuclear translocation and subsequent induction of apoptosis in neurons are assisted by myosin IIA in a TRPM7 kinase-dependent manner. Mol. Neurobiol. 51 (2), 729–742. doi:10.1007/s12035-014-8781-y
Zhelay, T., Wieczerzak, K. B., Beesetty, P., Alter, G. M., Matsushita, M., and Kozak, J. A. (2018). Depletion of plasma membrane-associated phosphoinositides mimics inhibition of TRPM7 channels by cytosolic Mg2+, spermine, and pH. J. Biol. Chem. 293 (47), 18151–18167. doi:10.1074/jbc.RA118.004066
Zheng, Q., Zhu, T., Hu, H., Zhao, Y., Ying, Y., Luo, X., et al. (2020). TRPM2 ion channel is involved in the aggravation of cognitive impairment and down regulation of epilepsy threshold in pentylenetetrazole-induced kindling mice. Brain Res. Bull. 155, 48–60. doi:10.1016/j.brainresbull.2019.11.018
Zhong, Z., Zhai, Y., Liang, S., Mori, Y., Han, R., Sutterwala, F. S., et al. (2013). TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat. Commun. 4, 1611. doi:10.1038/ncomms2608
Zierler, S., Yao, G., Zhang, Z., Kuo, W. C., Pörzgen, P., Penner, R., et al. (2011). Waixenicin A inhibits cell proliferation through magnesium-dependent block of transient receptor potential melastatin 7 (TRPM7) channels. J. Biol. Chem. 286 (45), 39328–39335. doi:10.1074/jbc.M111.264341
Zocchi, E., Franco, L., Guida, L., Benatti, U., Bargellesi, A., Malavasi, F., et al. (1993). A single protein immunologically identified as CD38 displays NAD+ glycohydrolase, ADP-ribosyl cyclase and cyclic ADP-ribose hydrolase activities at the outer surface of human erythrocytes. Biochem. Biophys. Res. Commun. 196 (3), 1459–1465. doi:10.1006/bbrc.1993.2416
Zong, P., Feng, J., Yue, Z., Li, Y., Wu, G., Sun, B., et al. (2022a). Functional coupling of TRPM2 and extrasynaptic NMDARs exacerbates excitotoxicity in ischemic brain injury. Neuron 110 (12), 1944–1958.e8. e1948. doi:10.1016/j.neuron.2022.03.021
Zong, P., Feng, J., Yue, Z., Yu, A. S., Vacher, J., Jellison, E. R., et al. (2022b). TRPM2 deficiency in mice protects against atherosclerosis by inhibiting TRPM2-CD36 inflammatory axis in macrophages. Nat. Cardiovasc Res. 1 (4), 344–360. doi:10.1038/s44161-022-00027-7
Zou, J., Ainscough, J. F., Yang, W., Sedo, A., Yu, S. P., Mei, Z. Z., et al. (2013). A differential role of macrophage TRPM2 channels in Ca2⁺ signaling and cell death in early responses to H₂O₂. Am. J. Physiol. Cell Physiol. 305 (1), C61–C69. doi:10.1152/ajpcell.00390.2012
Keywords: cell volume regulation, apoptosis, necrosis, pyroptosis, oxidative cell death, acidotoxic cell death, TRPM2, TRPM7
Citation: Okada Y, Numata T, Sabirov RZ, Kashio M, Merzlyak PG and Sato-Numata K (2023) Cell death induction and protection by activation of ubiquitously expressed anion/cation channels. Part 3: the roles and properties of TRPM2 and TRPM7. Front. Cell Dev. Biol. 11:1246955. doi: 10.3389/fcell.2023.1246955
Received: 25 June 2023; Accepted: 15 September 2023;
Published: 29 September 2023.
Edited by:
Markus Ritter, Paracelsus Medical University, AustriaReviewed by:
Alexander A. Mongin, Albany Medical College, United StatesSusanna Zierler, Johannes Kepler University of Linz, Austria
Copyright © 2023 Okada, Numata, Sabirov, Kashio, Merzlyak and Sato-Numata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yasunobu Okada, b2thZGFAbmlwcy5hYy5qcA==