 Giulia Della Chiara
Giulia Della Chiara Carlos Jiménez
Carlos Jiménez Mohit Virdi
Mohit Virdi Nicola Crosetto1,2,3
Nicola Crosetto1,2,3- 1Human Technopole, Milan, Italy
- 2Department of Microbiology, Tumor and Cell Biology, Karolinska Institutet, Solna, Sweden
- 3Science for Life Laboratory, Solna, Sweden
Eukaryotic genomes are spatially organized inside the cell nucleus, forming a threedimensional (3D) architecture that allows for spatial separation of nuclear processes and for controlled expression of genes required for cell identity specification and tissue homeostasis. Hence, it is of no surprise that mis-regulation of genome architecture through rearrangements of the linear genome sequence or epigenetic perturbations are often linked to aberrant gene expression programs in tumor cells. Increasing research efforts have shed light into the causes and consequences of alterations of 3D genome organization. In this review, we summarize the current knowledge on how 3D genome architecture is dysregulated in cancer, with a focus on enhancer highjacking events and their contribution to tumorigenesis. Studying the functional effects of genome architecture perturbations on gene expression in cancer offers a unique opportunity for a deeper understanding of tumor biology and sets the basis for the discovery of novel therapeutic targets.
Principles of 3D genome organization and functions
The spatial organization of eukaryotic genomes within the cell nucleus meets the necessity to fit very long DNA molecules into relatively small nuclei with an average diameter of 10 µm (Sun et al., 2000; Piovesan et al., 2019). This physical constraint has represented an increasing challenge for eukaryotes during evolution as genomes became larger and more complex (Fedoroff, 2012). As a result, cells have evolved mechanisms of genome folding and compaction through DNA interactions with both structural and regulatory proteins and with RNA (Cao et al., 2021). The discovery that the genome is non-randomly spatially organized dates back to the observation that, in interphase nuclei, chromosomes form discrete territories, which are non-randomly arranged with respect to the nuclear lamina and each other (Cremer and Cremer, 2010; Crosetto and Bienko, 2020). Following the advent of high-throughput chromosome conformation capture (Hi-C) (Lieberman-Aiden et al., 2009) and of high-throughput fluorescence in situ hybridization (FISH) techniques that allow for direct visualization of genomic regions as short as few kilobases (kb) across thousands of nuclei (Bouwman et al., 2022), as well as super resolution live-cell imaging (SRLCI) approaches that measure dynamics of chromatin contacts in space and time (Gabriele et al., 2022; Mach et al., 2022), our understanding of the principles of three dimensional (3D) genome folding has dramatically expanded. These methods allow us to study 3D genome organization across structural domains at different levels within the nucleus. At a coarse-grain level of organization, the genome is folded into so-called A/B compartments, which segregate euchromatin and actively transcribed elements (A compartments) away from heterochromatin and silenced regions (B compartments) (Lieberman-Aiden et al., 2009). Embedded in these compartments, we find megabase-sized topologically associating domains (TADs) (Dixon et al., 2012; Nora et al., 2012), representing DNA regions that preferentially interact with each other over the rest of the genome, whose boundaries are well labelled by the binding of CCCTC-binding factor (CTCF) or other insulator proteins. At a lower level of 3D genome architecture, relatively short-range chromatin loops bridging distal genomic regions, including enhancers and promoters, occupy TADs and sub-TADs domains, contributing to gene expression modulation (Popay and Dixon, 2022).
Regulation of gene transcription is accomplished through the integration of events at regulatory elements, that are proximal (promoters) and distal (enhancers) to gene transcription start sites (TSSs). More than 40 years after their discovery, enhancers are considered to play a central role in the spatio-temporal control of gene transcription, underlying human development and homeostasis (Banerji et al., 1981; Gasperini et al., 2020). Enhancers are short non-coding stretches of nucleosomes free DNA sequences that act as positive regulators of transcription via their ability to bind key proteins—transcription factors (TFs) and architectural proteins (i.e., CTCF, cohesins, mediators) — and complexes that control the expression of a target gene, in a location, distance- and orientation-independent manner (Banerji et al., 1981). Enhancers’ function is affected by chromatin context (i.e., histone-specific modifications of surrounding nucleosomes–H3K4me1 and H3K27Ac) (Della Chiara et al., 2021) and cell-specific TFs or stimuli in the environment in which they reside (Heinz et al., 2015). Indeed, the recruitment of specific TFs and chromatin mobility contributes to loops formation between enhancers and their target promoters, brought in close proximity in the 3D space. The mechanism and factors underlying enhancer-promoter (E-P) communication continue to be a subject of study, especially due to seemingly transient and dynamic nature of enhancers activation (Uyehara and Apostolou, 2023). Certainly, the advent of genetic engineering, single-cell methods, SRLCI approaches and 3D chromosome conformation capture-based technologies and their continuous improvement both on wet and in silico analysis, have pushed forward a deeper understanding of genome-wide mapping of E-P contacts, aiming at assigning one or multiple target genes to each enhancer, in time and space [for more details see (Brandão et al., 2021; Chen et al., 2023; Uyehara and Apostolou, 2023; Zhang et al., 2023)]. For example, intragenic enhancer at the PVT1 locus gene interacts strongly with the PVT1 promoter and weakly with the upstream MYC promoter gene. The inhibition of PVT1 promoter increases MYC gene expression and vice versa. These transcriptional alterations are thought to be due to chromatin contact changes between these two promoters and the intragenic enhancer, highlighting how different configurations of 3D genome can affect E-P contact strength and specificity (Cho et al., 2018). More recently, two live-imaging studies focusing on the sox2 and eve loci, in mouse embryonic stem cells (Alexander et al., 2019) and drosophila embryos (Chen et al., 2018), respectively, reached opposing conclusions on the correlation between the E-P proximity and gene transcription. These results highlight our incomplete understanding of the E-P communication and its relationship to gene expression. Needles to say, perturbations to 3D chromatin organization could be responsible for enhancer dysfunctions, resulting in the activation of alternative gene programs. These mechanisms are central to sustain many pathological conditions, including tumors.
Genome architecture dysregulation in cancer
A variety of genetic and epigenetic mechanisms can disrupt 3D genome architecture and, in turn, cause aberrant gene expression in cancer cells, as summarized in Figure 1. Among genetic mechanisms, single-nucleotide variants (SNVs) or small insertions and deletions (indels) affecting the binding motifs or the structure of proteins implicated in establishing and/or maintaining the 3D genome structure—such as CTCF and cohesin—have the potential to rewire the 3D genome and gene expression, thus contributing to the cancer phenotype (Weisch et al., 2023). CTCF/cohesin-binding sites (CBS) are mutational hotspots in cancer (found primarily in gastrointestinal cancers), and somatic alterations to CBSs have been linked to altered expression of nearby gene (Katainen et al., 2015; Guo et al., 2018), likely due to altered CTCF-mediated looping. Moreover, topological stress at CBSs can create structural variants (SVs) (Canela et al., 2017), which in addition to SNVs, can have dramatic effects on 3D genome architecture and consequently gene expression. Among these two genomic rearrengements, SVs are the most common in various cancer types (Akdemir et al., 2020). They are a class of mutations encompassing losses, gains and rearrangements (inversions, duplications, or translocations) of DNA segments, ranging from few nucleotides to entire chromosomal arms (Currall et al., 2013), generated by improper repair of DNA double strand breaks (DSBs) (Aaltonen et al., 2020; Rheinbay et al., 2020). The analysis of 2,658 cancer samples originating from 38 tumor types from the International Cancer Genome Consortium (ICGC)/The Cancer Genome Atlas (TCGA) and Pan-Cancer Analysis of the Whole Genomes (PCAWG), has implicated SVs as tumor drivers in ∼62% of cases. Particularly, solid tumors seem to exhibit extremely high numbers of somatic SVs (253-310 SVs on average) (Li et al., 2020; Cosenza et al., 2022), revealing huge heterogeneity across patients, even within the same cancer type. Among all the SV types, deletions are the most common (26% of all SVs in the PCAWG dataset), and occur in the majority of tumors (Cosenza et al., 2022).
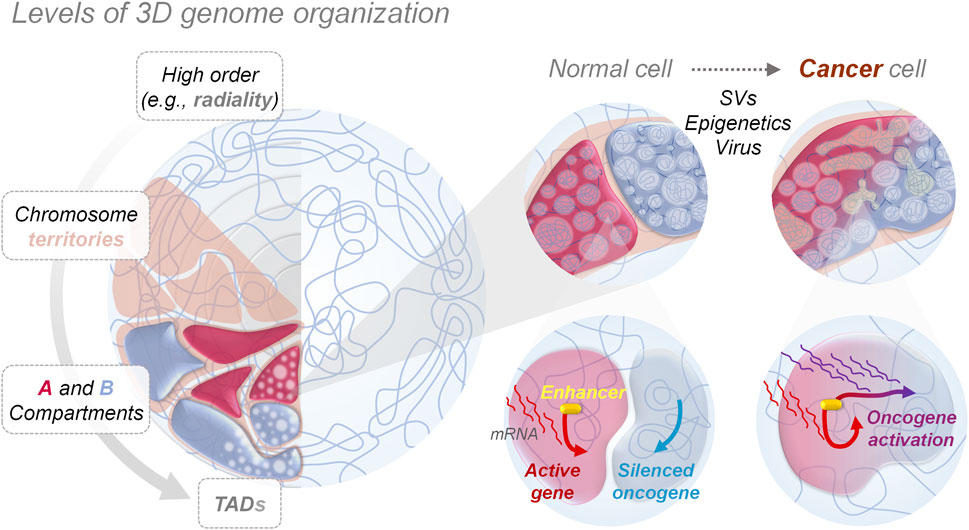
FIGURE 1. 3D genome organization and aberrant genes activation. Nucleus in the left panel shows how genomes are orderly folded forming compartments at different levels, from big chromosome territories to smaller topologically associating domains (TAD), allowing the physical separation and performance of nuclear functions, such as gene expression programming. Zooms in the right panel show how, in cancer cells, genome mis-compartmentalization occurs at multiple levels, either for genetic (e.g., structural variants—SVs), epigenetic or environmental (e.g., viral infections, chemical agents) causes, playing a crucial role in aberrant gene expression, by altering the interaction between genes and regulatory elements like enhancers, thereby promoting and allowing tumorigenesis.
Cancer-associated SVs can disrupt the physiological 3D genome structure by causing A/B compartments switching (Dubois et al., 2022)or TAD reorganization (shuffling, fusion or neo-formation) (Lupiáñez et al., 2015; San Martin et al., 2021; Xu Z. et al., 2022), resulting in the ectopic activation of oncogenes or inhibition of tumor-suppressing genes (Liu et al., 2023). That is the case for several primary and metastatic prostate cancer cell lines, where SVs-mediated DNA loci transition from the inactive to active compartment and vice versa can cause the fusion of TMPRSS2-ERG genes - a marker used for prostate cancer malignancy stratification - (San Martin et al., 2021), and the activation of numerous genes linked to carcinogenesis (i.e., WNT5, TMPRS, and CDK44) (San Martin et al., 2021) (Table 1). Another example of SVs-mediated A/B compartments switching is observed in multiple myeloma, where translocation of the histone lysine methyltransferase gene NSD2 from chromosome 4 to 14 highly increases its expression, leading to pervasive methylation of H3K36, with close TADs structure alteration and oncogenic genes pathways activation (Lhoumaud et al., 2019). One of the major consequences of SVs-mediated changes to 3D chromatin folding stems from repositioning (spatially or along the linear genome) of non-coding DNA regulatory elements (i.e., enhancers), affecting their interactions with target genes. This is because the specificity exhibited by enhancers is non-autonomous: if moved to a new locus they will generally activate any gene in their vicinity. This event called ‘enhancer hijacking’ has been increasingly identified as a driver of oncogenic behavior of structural variants (Ibrahim and Mundlos, 2020). Thus, determining how distant genomic regulatory elements, such as enhancers and promoters, communicate, is essential to any detailed understanding of transcriptional control in healthy and neoplastic development processes.
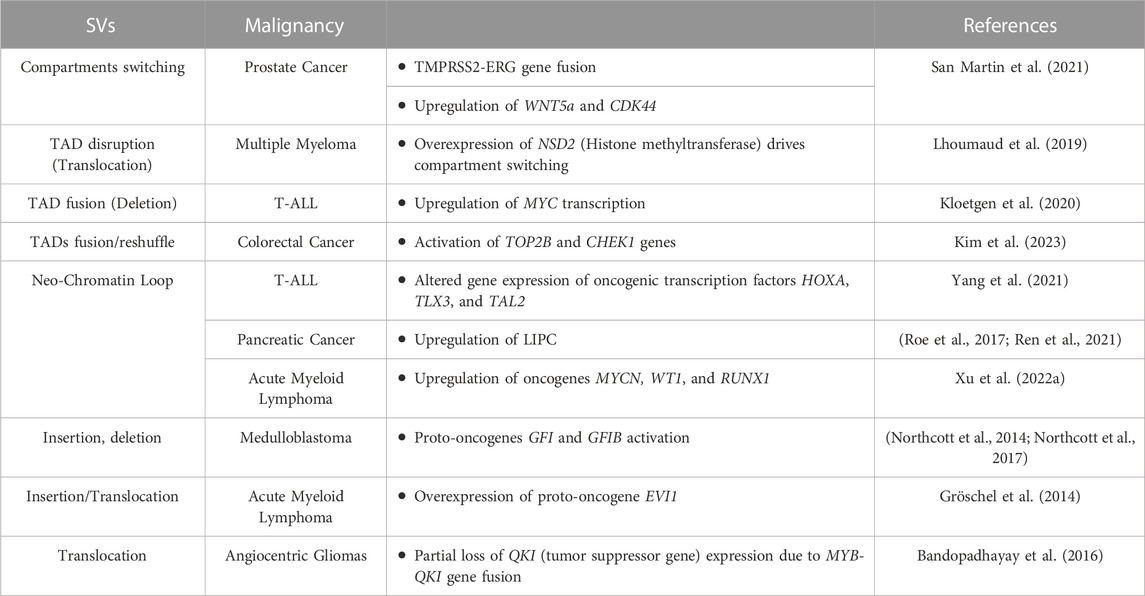
TABLE 1. Genomic rearrangements (SVs) induced enhancers hijacking events and their effect on gene expression alteration in different tumor types.
DNA regulatory elements dysfunction in cancer: enhancer hijacking
In cancer, SVs such as chromosomal translocations as well as inversions and duplications, can lead to the formation of de novo E-P contacts, leading in turn to the activation of proto-oncogenes in a process called enhancer hijacking. In this process, there are two possible mechanisms: the sequence of an active enhancer is transferred closer to a different promoter (along the linear genome) reinforcing or activating its transcription. Alternatively, SVs can trigger a fusion between contiguous TADs, leading to the emergence of long-range contacts between a promoter of a tumor-driving gene from one TAD, and an enhancer from the second TAD that normally serves other gene(s) (Bienko, 2022; Sidiropoulos et al., 2022). Enhancers hijacking has been described in several tumor types such as medulloblastoma, pancreatic and thyroid cancers, multiple myeloma, mantle cell lymphoma, and several leukemias, causing dysregulation/activation of several proto-oncogenes (Gröschel et al., 2014; Bandopadhayay et al., 2016; Northcott et al., 2017; Ren et al., 2021; Rico et al., 2022) (Table 1). For example, in T-cell acute lymphoblastic leukemia (T-ALL) deletion of a TAD boundary fuses the neighboring TADs resulting in the abnormal interaction of the MYC gene promoter with the BDME gene enhancer (Kloetgen et al., 2020), inducing MYC transcription upregulation. Moreover, formation of new chromatin loops leads to novel E-P contacts that alter expression of oncogenic transcription factors HOXA, TLX3, and TAL2 (Yang et al., 2021), contributing to T-ALL pathogenesis. Similarly, de novo chromatin loops formation creating unwanted promoter-enhancer interactions, causes upregulation of LIPC in pancreatic cancer metastasis (Roe et al., 2017; Ren et al., 2021) or upregulation of oncogenes such as MYCN, WT1, and RUNX1 in acute myeloid lymphoma (Xu J. et al., 2022). In medulloblastoma brain tumors, enhancer hijacking, caused by SVs, is an efficient mechanism driving the activation of proto-oncogenes GFI1 and GFI1B, moving their coding-sequences close to active enhancers (Northcott et al., 2014; Northcott et al., 2017). Recently, also in colorectal cancer (CRC) patients, SVs-mediated 3D genome dysregulation was found to cause super enhancers (SE) elements hijacking, leading to the hyper-activation of TOP2B and CHEK1 genes. These genes are linked to genome instability and DNA repair mechanisms and their high expression is thought to sustain tumor cells survival and proliferation, making these SE-hijacking events tumor promoting, representing new potential therapeutic targets (Kim et al., 2023). Also in gastric cancers, an integrated paired-end NanoChIP-seq (PeNChIP-seq) combined with whole genome sequencing (WGS) approach revealed new tumor-associated regulatory SVs-mediated enhancers hijacking, in regions associated with both simple and complex genomic rearrangements. These genetic and epigenetic alterations were found to activate CCNE2 and IGF2 oncogenes in 8% and 4% cancer patients respectively, further highlighting how enhancers hijacking could help in the identification of novel actionable tumor targets (Ooi et al., 2020).
Interestingly, new several in silico tools have recently emerged to identify enhancer hijacking in cancer genomes. For example, the NeoLoopFinder (Wang et al., 2021) is a computational framework aimed at identifying hijacked enhancers by integrating H3K27ac ChIPseq data, DNase-seq for chromatin accessibility and RNA-seq with Hi-C matrix, SVs input list and WGS derived from several cancer cell lines. The resulting hijacked enhancers are labelled by H3K27ac peaks or DNase accessible regions and are found in anchors of the expected neo-loops connected to gene promoters. This method predicted enhancer hijacking events in 11 cancer cell lines (for details check Wang et al. (Wang et al., 2021)) and showed how genes connected to these adoptive enhancers were expressed at higher levels compared to the same genes analysed in control cell lines, further reinforcing the relevance of enhancer hijacking in cancer. Another tool for enhancer hijacking prediction is Activity By Contact or ABC (Xu Z. et al., 2022), which integrates chromatin contacts measured by Hi-C and enhancer activity detected by ChIP-seq to predict target gene expression, after enhancer hijacking events induced by SVs. Application of ABC to patient-derived tumor samples and various cancer cell lines identified multiple TAD fusion events causing highjacking of enhancers or super-enhancers, inducing the activation of MYC, TERT and CCND1 oncogenes (Xu Z. et al., 2022). Future studies will be needed to determine whether tools such as NeoLoopFinder and ABC can be used in a clinical setting to identify targetable tumor drivers activated as a result of enhancer highjacking.
Conclusions and future perspectives
The three-dimentional architecture of the genome is vital for gene expression regulation and its disruption by genetic or epigenetic alterations is emerging as an important contributor to the pathogenesis of cancer. Several mechanisms by which the 3D genome gets perturbed in cancer have been described, including mutations in proteins shaping genome architecture or in their binding motifs, rewiring of A/B compartments and TADs and enhancer hijacking. However, several major questions in this arena remain unanswered: are genomic rearrangements and consequent 3D genome architecture perturbation the only cause for enhancers hijacking? Can enhancer hijacking be caused by non-genetic mechanisms such as reactivation of transposable elements within specific genomic regions? Can specific nucleotide sequence features (i.e., Alu, GC content, etc.) affect enhancer hijacking? Are there any shared enhancers hijacking events among different tumor types? And, most importantly, can we target dysfunctional enhancers or aberrant TADs/loops for therapeutic purposes?
Answering these questions will require charting the 3D genome directly in patient-derived tumor samples and, integrating 3D genome measurements to WGS data and new in silico tools in clinical trials, will be useful to assess the diagnostic and predictive value of SVs and associated 3D genome changes. Ultimately, developing cost-effective and scalable approaches for screening the effect of drugs or genetic perturbations on 3D genome architecture will be needed to develop the first generation of 3D genome targeted therapies.
Author contributions
GD: Conceptualization, Supervision, Writing–original draft, Writing–review and editing, Visualization. CJ: Visualization, Writing–original draft. MV: Writing–original draft. NC: Supervision, Writing–review and editing. MB: Conceptualization, Funding acquisition, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was funded by the intramural funding from the Human Technopole Foundation to MB.
Acknowledgments
We thank Simona Pedrotti (MB-NC lab at Human Technopole) for critically reading the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aaltonen, L. A., Abascal, F., Abeshouse, A., Aburatani, H., Adams, D. J., Agrawal, N., et al. (2020). Pan-cancer analysis of whole genomes. Nature 578, 82–93. doi:10.1038/s41586-020-1969-6
Akdemir, K. C., Le, V. T., Chandran, S., Li, Y., Verhaak, R. G., Beroukhim, R., et al. (2020). Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nat. Genet. 52, 294–305. doi:10.1038/s41588-019-0564-y
Alexander, J. M., Guan, J., Li, B., Maliskova, L., Song, M., Shen, Y., et al. (2019). Live-cell imaging reveals enhancer-dependent Sox2 transcription in the absence of enhancer proximity. eLife 8, e41769. doi:10.7554/eLife.41769
Bandopadhayay, P., Ramkissoon, L. A., Jain, P., Bergthold, G., Wala, J., Zeid, R., et al. (2016). MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat. Genet. 48, 273–282. doi:10.1038/ng.3500
Banerji, J., Rusconi, S., and Schaffner, W. (1981). Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 27, 299–308. doi:10.1016/0092-8674(81)90413-x
Bienko, M. (2022). Dangerous RNA links pave the way to glioblastoma. Mol. Cell 82, 1783–1785. doi:10.1016/j.molcel.2022.04.029
Bouwman, B. A. M., Crosetto, N., and Bienko, M. (2022). The era of 3D and spatial genomics. Trends Genet. TIG 38, 1062–1075. doi:10.1016/j.tig.2022.05.010
Brandão, H. B., Gabriele, M., and Hansen, A. S. (2021). Tracking and interpreting long-range chromatin interactions with super-resolution live-cell imaging. Curr. Opin. Cell Biol. 70, 18–26. doi:10.1016/j.ceb.2020.11.002
Canela, A., Maman, Y., Jung, S., Wong, N., Callen, E., Day, A., et al. (2017). Genome organization drives chromosome fragility. Cell 170, 507–521. doi:10.1016/j.cell.2017.06.034
Cao, C., Cai, Z., Ye, R., Su, R., Hu, N., Zhao, H., et al. (2021). Global in situ profiling of RNA-RNA spatial interactions with RIC-seq. Nat. Protoc. 16, 2916–2946. doi:10.1038/s41596-021-00524-2
Chen, H., Levo, M., Barinov, L., Fujioka, M., Jaynes, J. B., and Gregor, T. (2018). Dynamic interplay between enhancer–promoter topology and gene activity. Nat. Genet. 50, 1296–1303. doi:10.1038/s41588-018-0175-z
Chen, L.-F., Lee, J., and Boettiger, A. (2023). Recent progress and challenges in single-cell imaging of enhancer–promoter interaction. Curr. Opin. Genet. Dev. 79, 102023. doi:10.1016/j.gde.2023.102023
Cho, S. W., Xu, J., Sun, R., Mumbach, M. R., Carter, A. C., Chen, Y. G., et al. (2018). Promoter of lncRNA gene PVT1 is a tumor-suppressor DNA boundary element. Cell 173, 1398–1412. doi:10.1016/j.cell.2018.03.068
Cosenza, M. R., Rodriguez-Martin, B., and Korbel, J. O. (2022). Structural variation in cancer: role, prevalence, and mechanisms. Annu. Rev. Genomics Hum. Genet. 23, 123–152. doi:10.1146/annurev-genom-120121-101149
Cremer, T., and Cremer, M. (2010). Chromosome territories. Cold Spring Harb. Perspect. Biol. 2, a003889. doi:10.1101/cshperspect.a003889
Crosetto, N., and Bienko, M. (2020). Radial organization in the mammalian nucleus. Front. Genet. 11, 33. doi:10.3389/fgene.2020.00033
Currall, B. B., Chiang, C., Talkowski, M. E., and Morton, C. C. (2013). Mechanisms for structural variation in the human genome. Curr. Genet. Med. Rep. 1, 81–90. doi:10.1007/s40142-013-0012-8
Della Chiara, G., Gervasoni, F., Fakiola, M., Godano, C., D'Oria, C., Azzolin, L., et al. (2021). Epigenomic landscape of human colorectal cancer unveils an aberrant core of pan-cancer enhancers orchestrated by YAP/TAZ. Nat. Commun. 12, 2340. doi:10.1038/s41467-021-22544-y
Dixon, J. R., Selvaraj, S., Yue, F., Kim, A., Li, Y., Shen, Y., et al. (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. doi:10.1038/nature11082
Dubois, F., Sidiropoulos, N., Weischenfeldt, J., and Beroukhim, R. (2022). Structural variations in cancer and the 3D genome. Nat. Rev. Cancer 22, 533–546. doi:10.1038/s41568-022-00488-9
Fedoroff, N. V. (2012). Presidential address. Transposable elements, epigenetics, and genome evolution. Science 338, 758–767. doi:10.1126/science.338.6108.758
Gabriele, M., Brandão, H. B., Grosse-Holz, S., Jha, A., Dailey, G. M., Cattoglio, C., et al. (2022). Dynamics of CTCF- and cohesin-mediated chromatin looping revealed by live-cell imaging. Science 376, 496–501. doi:10.1126/science.abn6583
Gasperini, M., Tome, J. M., and Shendure, J. (2020). Towards a comprehensive catalogue of validated and target-linked human enhancers. Nat. Rev. Genet. 21, 292–310. doi:10.1038/s41576-019-0209-0
Gröschel, S., Sanders, M. A., Hoogenboezem, R., de Wit, E., Bouwman, B. A. M., Erpelinck, C., et al. (2014). A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157, 369–381. doi:10.1016/j.cell.2014.02.019
Guo, Y. A., Chang, M. M., Huang, W., Ooi, W. F., Xing, M., Tan, P., et al. (2018). Mutation hotspots at CTCF binding sites coupled to chromosomal instability in gastrointestinal cancers. Nat. Commun. 9, 1520. doi:10.1038/s41467-018-03828-2
Heinz, S., Romanoski, C. E., Benner, C., and Glass, C. K. (2015). The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 16, 144–154. doi:10.1038/nrm3949
Ibrahim, D. M., and Mundlos, S. (2020). Three-dimensional chromatin in disease: what holds us together and what drives us apart? Curr. Opin. Cell Biol. 64, 1–9. doi:10.1016/j.ceb.2020.01.003
Katainen, R., Dave, K., Pitkänen, E., Palin, K., Kivioja, T., Välimäki, N., et al. (2015). CTCF/cohesin-binding sites are frequently mutated in cancer. Nat. Genet. 47, 818–821. doi:10.1038/ng.3335
Kim, K., Kim, M., Lee, A. J., Song, S. H., Kang, J. K., Eom, J., et al. (2023). Spatial and clonality-resolved 3D cancer genome alterations reveal enhancer-hijacking as a potential prognostic marker for colorectal cancer. Cell Rep. 42, 112778. doi:10.1016/j.celrep.2023.112778
Kloetgen, A., Thandapani, P., Ntziachristos, P., Ghebrechristos, Y., Nomikou, S., Lazaris, C., et al. (2020). Three-dimensional chromatin landscapes in T cell acute lymphoblastic leukemia. Nat. Genet. 52, 388–400. doi:10.1038/s41588-020-0602-9
Lhoumaud, P., Badri, S., Rodriguez-Hernaez, J., Sakellaropoulos, T., Sethia, G., Kloetgen, A., et al. (2019). NSD2 overexpression drives clustered chromatin and transcriptional changes in a subset of insulated domains. Nat. Commun. 10, 4843. doi:10.1038/s41467-019-12811-4
Li, Y., Roberts, N. D., Wala, J. A., Shapira, O., Schumacher, S. E., Kumar, K., et al. (2020). Patterns of somatic structural variation in human cancer genomes. Nature 578, 112–121. doi:10.1038/s41586-019-1913-9
Lieberman-Aiden, E., van Berkum, N. L., Williams, L., Imakaev, M., Ragoczy, T., Telling, A., et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. doi:10.1126/science.1181369
Liu, H., Tsai, H., Yang, M., Li, G., Bian, Q., Ding, G., et al. (2023). Three-dimensional genome structure and function. MedComm 4, e326. doi:10.1002/mco2.326
Lupiáñez, D. G., Kraft, K., Heinrich, V., Krawitz, P., Brancati, F., Klopocki, E., et al. (2015). Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025. doi:10.1016/j.cell.2015.04.004
Mach, P., Kos, P. I., Zhan, Y., Cramard, J., Gaudin, S., Tünnermann, J., et al. (2022). Cohesin and CTCF control the dynamics of chromosome folding. Nat. Genet. 54, 1907–1918. doi:10.1038/s41588-022-01232-7
Nora, E. P., Lajoie, B. R., Schulz, E. G., Giorgetti, L., Okamoto, I., Servant, N., et al. (2012). Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385. doi:10.1038/nature11049
Northcott, P. A., Buchhalter, I., Morrissy, A. S., Hovestadt, V., Weischenfeldt, J., Ehrenberger, T., et al. (2017). The whole-genome landscape of medulloblastoma subtypes. Nature 547, 311–317. doi:10.1038/nature22973
Northcott, P. A., Lee, C., Zichner, T., Stütz, A. M., Erkek, S., Kawauchi, D., et al. (2014). Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511, 428–434. doi:10.1038/nature13379
Ooi, W. F., Nargund, A. M., Lim, K. J., Zhang, S., Xing, M., Mandoli, A., et al. (2020). Integrated paired-end enhancer profiling and whole-genome sequencing reveals recurrent CCNE1 and IGF2 enhancer hijacking in primary gastric adenocarcinoma. Gut 69, 1039–1052. doi:10.1136/gutjnl-2018-317612
Piovesan, A., Pelleri, M. C., Antonaros, F., Strippoli, P., Caracausi, M., and Vitale, L. (2019). On the length, weight and GC content of the human genome. BMC Res. Notes 12, 106. doi:10.1186/s13104-019-4137-z
Popay, T. M., and Dixon, J. R. (2022). Coming full circle: on the origin and evolution of the looping model for enhancer–promoter communication. J. Biol. Chem. 298, 102117. doi:10.1016/j.jbc.2022.102117
Ren, B., Yang, J., Wang, C., Yang, G., Wang, H., Chen, Y., et al. (2021). High-resolution Hi-C maps highlight multiscale 3D epigenome reprogramming during pancreatic cancer metastasis. J. Hematol. Oncol.J Hematol. Oncol. 14, 120. doi:10.1186/s13045-021-01131-0
Rheinbay, E., Nielsen, M. M., Abascal, F., Wala, J. A., Shapira, O., Tiao, G., et al. (2020). Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 578, 102–111. doi:10.1038/s41586-020-1965-x
Rico, D., Kent, D., Karataraki, N., Mikulasova, A., Berlinguer-Palmini, R., Walker, B. A., et al. (2022). High-resolution simulations of chromatin folding at genomic rearrangements in malignant B cells provide mechanistic insights into proto-oncogene deregulation. Genome Res. 32, 1355–1366. doi:10.1101/gr.276028.121
Roe, J.-S., Hwang, C. I., Somerville, T. D. D., Milazzo, J. P., Lee, E. J., Da Silva, B., et al. (2017). Enhancer reprogramming promotes pancreatic cancer metastasis. Cell 170, 875–888. doi:10.1016/j.cell.2017.07.007
San Martin, R., Das, P., Dos Reis Marques, R., Xu, Y., Roberts, J. M., Sanders, J. T., et al. (2021). Chromosome compartmentalization alterations in prostate cancer cell lines model disease progression. J. Cell Biol. 221, e202104108. doi:10.1083/jcb.202104108
Sidiropoulos, N., Mardin, B. R., Rodríguez-González, F. G., Bochkov, I. D., Garg, S., Stütz, A. M., et al. (2022). Somatic structural variant formation is guided by and influences genome architecture. Genome Res. 32, 643–655. doi:10.1101/gr.275790.121
Sun, H. B., Shen, J., and Yokota, H. (2000). Size-Dependent positioning of human chromosomes in interphase nuclei. Biophys. J. 79, 184–190. doi:10.1016/S0006-3495(00)76282-5
Uyehara, C. M., and Apostolou, E. (2023). 3D enhancer-promoter interactions and multi-connected hubs: organizational principles and functional roles. Cell Rep. 42, 112068. doi:10.1016/j.celrep.2023.112068
Wang, X., Xu, J., Zhang, B., Hou, Y., Song, F., Lyu, H., et al. (2021). Genome-wide detection of enhancer-hijacking events from chromatin interaction data in rearranged genomes. Nat. Methods 18, 661–668. doi:10.1038/s41592-021-01164-w
Weischenfeldt, J., and Ibrahim, D. M. (2023). When 3D genome changes cause disease: the impact of structural variations in congenital disease and cancer. Curr. Opin. Genet. Dev. 80, 102048. doi:10.1016/j.gde.2023.102048
Xu, J., Song, F., Lyu, H., Kobayashi, M., Zhang, B., Zhao, Z., et al. (2022a). Subtype-specific 3D genome alteration in acute myeloid leukaemia. Nature 611, 387–398. doi:10.1038/s41586-022-05365-x
Xu, Z., Lee, D. S., Chandran, S., Le, V. T., Bump, R., Yasis, J., et al. (2022b). Structural variants drive context-dependent oncogene activation in cancer. Nature 612, 564–572. doi:10.1038/s41586-022-05504-4
Yang, L., Chen, F., Zhu, H., Chen, Y., Dong, B., Shi, M., et al. (2021). 3D genome alterations associated with dysregulated HOXA13 expression in high-risk T-lineage acute lymphoblastic leukemia. Nat. Commun. 12, 3708. doi:10.1038/s41467-021-24044-5
Keywords: enhancers, enhancers hijacking, 3D genome organization, cancer, structural variants, epigenetics, 3D genomics, spatial genomics
Citation: Della Chiara G, Jiménez C, Virdi M, Crosetto N and Bienko M (2023) Enhancers dysfunction in the 3D genome of cancer cells. Front. Cell Dev. Biol. 11:1303862. doi: 10.3389/fcell.2023.1303862
Received: 28 September 2023; Accepted: 23 October 2023;
Published: 06 November 2023.
Edited by:
Serena Ghisletti, European Institute of Oncology (IEO), ItalyReviewed by:
Michele Gabriele, Massachusetts Institute of Technology, United StatesCopyright © 2023 Della Chiara, Jiménez, Virdi, Crosetto and Bienko. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giulia Della Chiara, Z2l1bGlhLmRlbGxhY2hpYXJhQGZodC5vcmc=; Magda Bienko, bWFnZGEuYmllbmtvQGZodC5vcmc=