Abstract
Thalassemia, a prevalent single-gene inherited disorder, relies on hematopoietic stem cell or bone marrow transplantation as its definitive treatment. However, the scarcity of suitable donors and the severe complications from anemia and iron overload pose significant challenges. An immediate need exists for a therapeutic method that addresses both the illness and its associated complications. Advancements in stem cell technology and gene-editing methods, such as clustered regularly interspaced short palindromic repeats along with its associated protein (CRISPR/Cas), offer encouraging prospects for a therapy that could liberate patients from the need for ongoing blood transfusions and iron chelation treatments. The potential of genetic reprogramming using induced pluripotent stem cells (iPSCs) to address thalassemia is highly promising. Furthermore, mesenchymal stem cells (MSCs), recognized for their capacity to self-renew and differentiate into multiple lineages that include bone, cartilage, adipose tissue, and liver, demonstrate potential in alleviating several complications faced by thalassemia patients, including osteoporosis, cirrhosis, heart conditions, respiratory issues, and immune-related disorders. In this review, we synthesize and summarize relevant studies to assess the therapeutic potential and predict the curative effects of these cellular approaches.
1 Introduction
Initially identified along the Mediterranean coast, thalassemia is considered one of the most common autosomal recessive disorders stemming from single-gene inheritance (Zakaria et al., 2022). Annually, approximately 300,000 to 500,000 infants are born with serious types of homozygous thalassemia, and it is estimated that approximately 7% of the worldwide population holds the thalassemia gene (Liu et al., 2024; Tran et al., 2023; Saleemi, 2014). There are regional differences in China; thalassemia patients or carriers mainly live in the south of the Yangtze River, such as Guangxi Province, where the proportion of carriers reaches 20%–25% (Chen et al., 2022). Major thalassemia patients require blood diffusion and iron chelation therapy to survive, which has no cure other than allogeneic hematopoietic stem cell transplantation (HSCT); however, the challenge of identifying a suitably matched donor significantly restricts the accessibility of this therapeutic approach (Zakaria et al., 2022).
Stem cell therapy utilizes the natural abilities of stem cells, such as their inherent capacity for proliferation, differentiation, and self-renewal, to repair damaged cells and promote the healing of organs, providing therapeutic advantages and improving physical development (Mousaei G et al., 2022). Currently, allogeneic HSCT is the primary stem cell treatment for thalassemia and the only recognized potentially curative method for individuals with transfusion-dependent major thalassemia (Farmakis et al., 2022). Furthermore, various research efforts are investigating the combination of gene therapy and mesenchymal stem cells (MSCs) with induced pluripotent stem cells for treating thalassemia (Li et al., 2022a). Clustered regularly interspaced short palindromic repeats along with its associated protein 9 (CRISPR/Cas9) gene-edited induced pluripotent stem cells (iPSCs) from patients can have normal genes and are hopefully capable of differentiating into normal hematopoietic cells and red blood cells (Zakaria et al., 2022). MSCs, which derive from the mesoderm, exhibit significant differentiation capabilities and low levels of immunogenicity (Rudnitsky et al., 2025). MSCs are valuable for therapeutic applications due to their potential to evolve into various adult stem cells, address complications like liver cirrhosis and osteoporosis, facilitate bone healing and liver function improvement in animal studies, and exhibit minimal immunogenicity, reducing the risk of immune rejection (Chen et al., 2023; Huang et al., 2025; Yang et al., 2020). Nevertheless, the relevant research on treating thalassemia with iPSCs and MSCs remains limited. Therefore, in this review, we aim to summarize studies on the application of these stem cells in thalassemia treatment.
2 Thalassemia and its complication
2.1 Thalassemia pathogenesis
Thalassemia is considered one of the most common genetic disorders inherited in an autosomal recessive manner, distinguished by a defect in the production (resulting from mutations or deletions) of one or several globin peptide chains (α, β, γ, and δ). This alteration creates an imbalance in the hemoglobin structure, which eventually results in the transformation or destruction of red blood cells (Kattamis et al., 2022). Due to decreased normal hemoglobin levels, red blood cells have a shorter lifespan and reduced oxygen transport capacity, and may rupture when passing through the marrow or spleen, leading to hemolytic anemia (Thiagarajan et al., 2021). Thus, the patients exhibit anemic symptoms, for instance, pallor, developmental retardation, and hepatosplenomegaly, due to chronic hypoxia. These symptoms can have a remission through blood transfusion (Musallam et al., 2024).
2.2 Clinical manifestation of thalassemia
Thalassemia can result from mutations or deletions in chromosome 16 or chromosome 11, referred to as α-thalassemia and β-thalassemia, respectively; if both chromosomes are mutated, the condition is known as αβ-thalassemia. In clinical practice, γ-thalassemia, δ-thalassemia, and δβ-thalassemia (or εγδβ-thalassemia) also exist; however, they are less prevalent than α-thalassemia and β-thalassemia (Table 1) (Taher et al., 2021).
TABLE 1
| Type and genetic deficiency | Clinical manifestation | Severity | Complication | Reference |
|---|---|---|---|---|
| α-Thalassemia (α-globin gene deletion or deficiency) | a. Silent type: asymptomatic b. Mild type: mild anemia c. Intermediate type (hemoglobin H disease): moderate anemia (may present with jaundice and hepatomegaly) d. Severe type (hydrops fetalis syndrome): severe fetal anemia, generalized edema, and hepatomegaly, often leading to fetal death |
Silent and mild types are mild; intermediate type is moderate; severe type is severe | Jaundice, hepatomegaly, hydrops fetalis syndrome, cardiac enlargement, skeletal deformities, delayed growth and development, and iron overload | Musallam et al. (2024), Li et al. (2024), Winger et al. (2025), Farashi and Harteveld (2018), and Tamary et al. (1993) |
| β-Thalassemia (β-globin gene deletion or deficiency) | a. Mild type: mild anemia; b. Intermediate type: may present with jaundice, hepatomegaly, and delayed growth and development; c. Severe type: severe anemia, pale complexion, hepatomegaly, jaundice, and poor development, with typical facial features, require long-term blood transfusions | Mild type is mild; intermediate and severe types are more severe | Jaundice, hepatomegaly, delayed growth and development, skeletal deformities, heart enlargement, and iron overload | Bajwa and Basit (2025), Makis et al., 2021; Origa (2017),Cao and Galanello (2010),Dordevic et al. (2025),Langer et al. (1993), and Cappellini et al. (2014) |
| δβ-Thalassemia (δ- and β-globin gene deletion or deficiency) | Mild anemia | Relatively mild | Slight jaundice and hepatomegaly | De Simone et al. (2022) |
| γ-Thalassemia (γ -globin chain deletion or deficiency) | Usually presents with mild anemia | Relatively mild | Slight jaundice and hepatomegaly | De Simone et al. (2022) |
| δ-Thalassemia (δ gene mutation) | Mild anemia | Relatively mild | Slight jaundice and hepatomegaly | De Simone et al. (2022) |
Thalassemia types and their genetic defects, clinical manifestations, severity, and complications.
α-Thalassemia is caused by insufficient production of α-globin peptides, which arises from genes found on chromosome 16. This chromosome typically harbors four α-globin alleles, and the condition’s severity correlates with the number of affected alleles (Harewood and Azevedo, 2024). A single mutation or deletion, known as silent carrier status, is asymptomatic. When two alleles are compromised, the condition is classified as α-thalassemia minor, which is generally mild or without clinical signs. The intermedia form, known as hemoglobin H disease, occurs when three alleles are defective; it can lead to severe symptoms, with some patients requiring blood transfusions (Musallam et al., 2024). Major α-thalassemia, also known as Hb Bart’s hydrops fetalis, often results in miscarriage in the early stages of pregnancy (Tesio and Bauer, 2023). Pregnant women affected by fetal hydrops may experience a condition known as mirror syndrome, which can lead to maternal edema, proteinuria, and hypertension. Additionally, there is an increased risk of dystocia (difficult labor) and postpartum hemorrhage due to the enlarged placenta (Biswas et al., 2023). Unfortunately, because many couples are unaware of their α-thalassemia carrier status and lack access to prenatal screening, most fetuses with α-thalassemia major (ATM) are diagnosed with hydrops or other abnormalities detected through routine prenatal ultrasound examinations (Winger et al., 2025; Tamary et al., 1993). Another kind of thalassemia in hospital is β-thalassemia, and the global morbidity rate of β-thalassemia is approximately 1.5% (Cri et al., 2019). According to available statistics, approximately 4 million infants are born with β-thalassemia (Kattamis et al., 2020), which is mainly distributed in the Mediterranean and Southeast Asia, and a few β-thalassemia infants die before they are diagnosed in some areas with poor medical resources (Taher et al., 2021; Angastiniotis and Lobitz, 2019). β-Thalassemia is difficult to be detected in utero, and most patients begin to present clinical symptoms after 6 months of age. Unlike α-thalassemia, β-thalassemia is divided into three types (Zakaria et al., 2022) based on mutation in two alleles on human chromosome 11 (Origa, 2017). Minor thalassemia, known as β-thalassemia carriers, presents with either mild anemia or no clinical symptoms. In contrast, β-thalassemia intermedia involves individuals who are double heterozygotes and exhibit clinical features ranging from mild to severe, potentially resulting in moderate anemia and hepatosplenomegaly (Origa, 2017). Severe β-thalassemia, also known as Cooley’s anemia (Khan and Shaikh, 2023), is associated with the possibility of severe anemia, jaundice, hepatosplenomegaly, growth retardation, and facial skeletal deformity; these patients require regular lifelong blood transfusions, iron chelation therapy, or hematopoietic stem cell transplantation (Needs et al., 2024). Certain patients with severe β-thalassemia may not survive into their twenties due to complications like arrhythmia and heart failure, which can lead to critical deterioration from iron overload within a span of 6 months (Coates, 2014).
2.3 Current treatments of thalassemia
Various therapeutic strategies are available for the management of thalassemia, encompassing blood transfusions, iron chelation therapy, splenectomy, bone marrow transplantation, HSCT, gene editing methods, and the induced synthesis of fetal hemoglobin (Wang et al., 2023; Khan and Shaikh, 2024), and medications such as hydroxyurea (Ali et al., 2021), which prolong the life of the patient at the different way. However, some of the treatments pose some challenges that urgently need to be addressed. For example, blood transfusion and iron removal are two symptomatic treatments for major thalassemia (Hodroj et al., 2023); however, frequent or excessive transfusions can lead to iron overload as iron released from the breakdown of red blood cells may deposit in various tissues and organs, resulting in serious complication.
2.4 Complications of thalassemia
Although patients with major thalassemia receive the treatments mentioned above, they may still experience numerous complications, including thromboembolic events, organ dysfunction, endocrinopathies, cardiovascular disease, and osteoporosis (Kattamis et al., 2022; Wood, 2023; Gaudio et al., 2019). A cohort study showed that 480 of 709 patients (67.7%) with β-thalassemia have developed at least one complication and 93 β-thalassemia patients died due to heart diseases (57.0%), complications from bone marrow transplantation (10.8%), infections (8.6%), liver diseases (4.3%), cancers (3.2%), thrombus embolism (2.2%), and severe anemia (1.1%) (Forni et al., 2023). These complications are mostly caused by anemia and the occurrence of iron overload resulting from long-term and massive blood transfusions (Yousuf et al., 2022). The accumulation of iron in various organs could result in several types of organ dysfunction, potentially leading to considerable damage and failure of those organs (Wang et al., 2025a). Complications that may arise from this include arrhythmias, dysfunction of the left ventricle (Fu and Yang, 2025), fibrosis or cirrhosis (Langer et al., 1993), osteoporosis (Bhardwaj et al., 2023), kidney disorders (Demosthen et al., 2019), and abnormalities in respiratory function (Bou-Fakhredin et al., 2023). Additionally, endocrine disorders are largely related to iron accumulation in crucial glands such as the pituitary, thyroid, and adrenal glands, significantly impacting the growth and development of patients with thalassemia (Tenuta et al., 2024; Venou et al., 2024; Evangelidis et al., 2023). In addition to other complications, patients may face infections and Graft-versus-Host Disease (GVHD) (Vento et al., 2006; Akbayram et al., 2022), as well as cancer and alloimmunization, which can further threaten the life of the patient.
3 The overview of stem cell therapies for thalassemia
Stem cell treatments encompass allogeneic stem cell therapy, which involves the transplantation of the bone marrow, peripheral blood, and umbilical cord blood sourced from relatives of the patient or from a human leukocyte antigen (HLA)-matched donor (Karponi and Zogas, 2019), as well as autologous stem cell therapy, wherein the patient’s own stem cells are grafted following gene reprogramming (Srivastava and Shaji, 2017); these approaches mainly include hematopoietic stem cell transplantation, mesenchymal stem cell transplantation, and gene-edited stem cell transplantation (Figure 1; Table 2). It is meaningful to develop autologous stem cell transplantation that can correct the genetic defect.
FIGURE 1
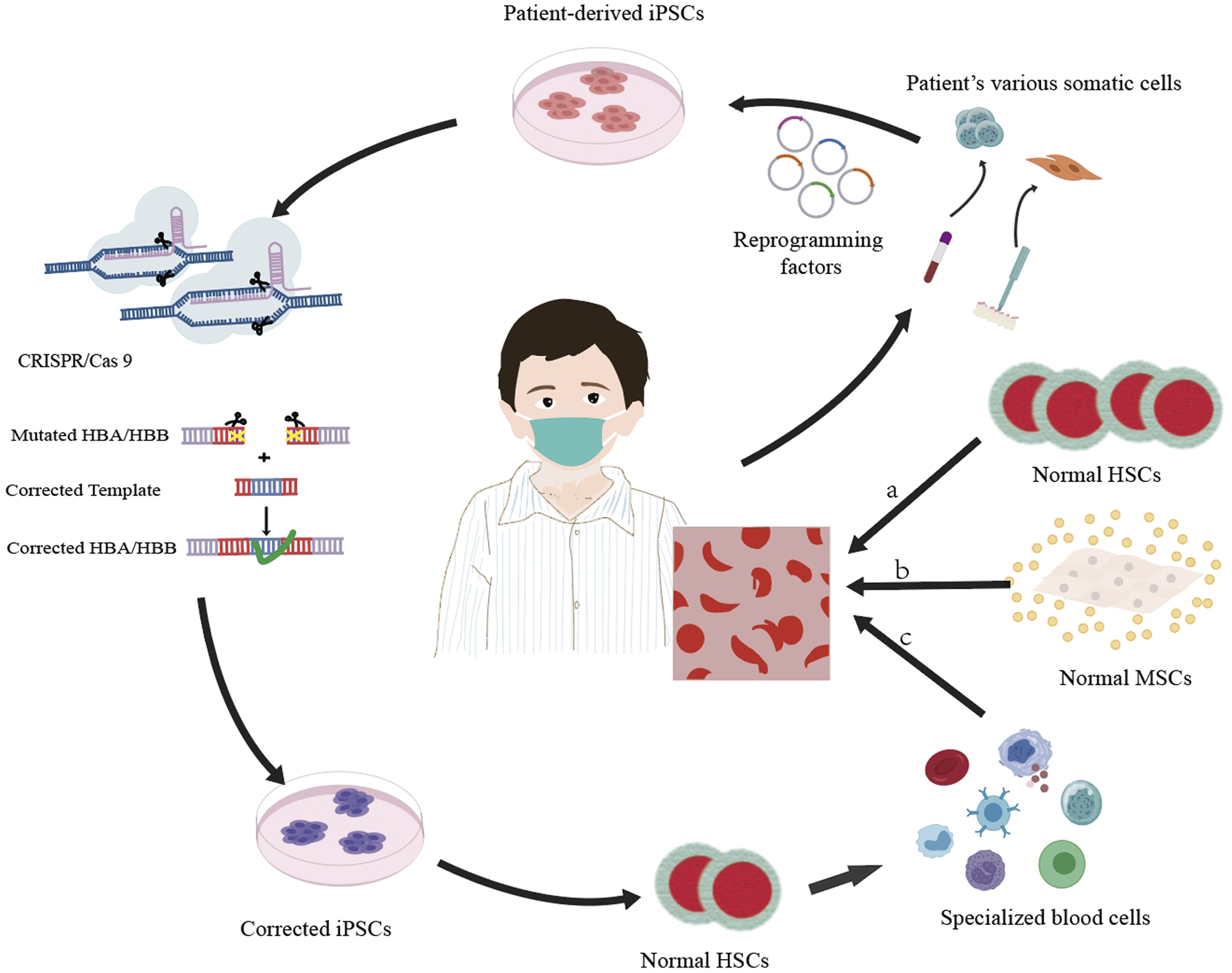
Overview of three cell therapy approaches for thalassemia treatment: a. allogeneic hematopoietic stem cell (HSC) transplantation, where healthy HSCs are transplanted into the patient to replace the defective cells; b. allogeneic mesenchymal stem cell (MSC) transplantation, which utilizes healthy MSCs to enhance the function of HSCs and may help in remedying the condition; c. by using CRISPR/Cas9 technology to modify the mutated HBA/HBB gene in the induced pluripotent stem cells (iPSCs) of the patient, differentiating these corrected iPSCs into healthy hematopoietic stem cells (HSCs), and subsequently transplanting the normal HSCs back into the patient’s body to substitute the defective cells, a targeted gene therapy approach is achieved.
TABLE 2
| Stem cell type | Advantage | Disadvantage | Reference |
|---|---|---|---|
| HSCs | a. Currently the only method capable of curing transfusion-dependent thalassemia (TDT) b. Relatively mature technology |
a. Donor limitations b. Transplant risks of GVHD. c. Toxicity of conditioning regimens |
Wood, (2023), Aljagthmi and Abdel-Aziz (2025), and Hoang et al. (2025) |
| MSCs | a. Immunomodulation, which can reduce the risk of GVHD. b. Hematopoiesis promotion c. Diverse sources: comprise bone marrow, umbilical cord blood, and adipose tissue, among others |
a. Efficacy uncertainty b. Technical challenge c. Long-term safety |
Wiewiórska-Krata et al. (2025), Terai et al. (2025), Mousavi et al. (2023), and Levy et al. (2020) |
| iPSCs | a. Personalized therapy b. Genetic modification: integrating CRISPR/Cas9 with alternative gene editing methods c. Research potential |
a. Technical complexity b. Ethical issues: involves ethical debates about embryonic stem cells c. Safety issues: gene editing may introduce off-target effects |
Hardouin et al. (2025) and Yamanaka (2020) |
Comparison of the therapies of HSCs, MSCs, and iPSCs.
3.1 The source of stem cells: stem cell bank
Stem cells have the ability to renew themselves and can be classified into two categories depending on their source (Brignier and Gewirtz, 2010), namely, embryonic and adult stem cells. Additionally, stem cells are classified into totipotent, pluripotent, multipotent, and unipotent cells, according to their differentiation potential (Wen and Wang, 2025). A zygote, which is an example of a totipotent stem cell, has the capacity to develop into a fully formed organism. In contrast, a pluripotent stem cell can differentiate into several types of tissues, but it cannot generate an entire individual (Malik and Wang, 2022). Two categories of human pluripotent stem cells exist: human embryonic stem cells (hESCs), originating from the inner cell mass of embryos, and human induced pluripotent stem cells (hiPSCs), obtained through the reprogramming of somatic cells (Tian et al., 2023). In 2006, Takahashi and Yamanaka found that fibroblasts obtained from mice could be converted into specialized cells, reflecting the three germ layers by applying four specific transcription factors: Oct4, Sox-2, Klf4, and c-Myc (Takahashi and Yamanaka, 2006); this technology involving induced pluripotent stem cells has slowly been utilized in the therapy for certain human ailments (Huang et al., 2020). A multipotent stem cell has the ability to develop into one or more specific tissues (Aprile et al., 2024). For instance, hematopoietic stem cells are capable of transforming into erythrocytes, leukocytes, and platelets, whereas mesenchymal stem cells primarily reside in connective tissue and interstitial spaces of organs (Zhang et al., 2018). Unipotent stem cell like myoblast can only differentiate into muscle cells.
Typically, stem cells used to treat thalassemia come from matched donors or the patients themselves, but technological advancements now enable the cultivation of stem cells from biological materials like umbilical cord blood under strict safety and quality regulations (Sharma et al., 2021). Stem cells can be stored in cell banks for future applications (Table 3) and are also used in research and treatment of various diseases (Stacey and Healy, 2021). At present, Zunyi Medical University has cooperated with enterprises to establish the first placental stem cell bank and seven autologous stem cell banks in China, and set up two public stem cell banks in Guizhou Province to provide perinatal stem cell storage services and store important stem cell medicine treatment needs. Ossium Health has established a cryopreserved bone marrow cell repository recovered from deceased organ vertebrae, referred to as hematopoietic progenitor cell bone marrow (Johnstone et al., 2021).
TABLE 3
| Stem cell bank | Founding time | Source | Reason for establishment | Reference |
|---|---|---|---|---|
| UK Stem Cell Bank(UKSCB) | 2003 | Approximately 100 research-grade hESC lines and several human pluripotent stem cell lines | Foster the advancement of scientific research and the clinical application of stem cell treatments | O’Shea and Abranches (2020) |
| Korea National Stem Cell Bank (KSCB) | 2005 | 17 tissue-derived adult stem cells and 228 primary genetic disease cells | Supply stem cell resources, regenerative medicine information, and hESC registry | Kim et al. (2021) |
| Karolinska Institute Human Embryonic Stem Cell Bank (KISCB) | 2002 | 60 hESC lines | Set up hESC lines for clinical use: remove xeno, chemical conditions, and ensure GMP. | Main et al. (2020) |
| Spanish National Stem Cell Bank (BNLC) | 2006 | 40 hESC lines and 171 hiPSC lines, including cord blood and adipose-derived MSCs | Advance stem cell and regenerative medicine; establish hiPSC bank from homozygous cord blood | Aran et al. (2020) |
Founding time, source, and reason for establishment of Stem Cell Banks.
3.2 Allogeneic stem cell transplantation for thalassemia
Currently, the only curative treatment for severe thalassemia is allogeneic hematopoietic stem cell transplantation (Srivastava and Shaji, 2017). This process involves utilizing hematopoietic stem cells obtained from external sources, such as the bone marrow, peripheral blood, or umbilical cord blood of relatives (Wei et al., 2024). The aim is to restore the blood and immune systems of the patient, with the success of the treatment being affected by variables including the recipient’s age, compatibility with the donor, and medical care provided prior to the transplant.
3.2.1 Bone marrow transplantation
Clinically, the initiation of bone marrow transplantation for disease treatment occurred in 1957 (Simpson and Dazzi, 2019). Bone marrow serves as a primary source of hematopoietic stem cells (Agarwal et al., 2023), and this procedure stands as the definitive treatment for thalassemia, particularly among individuals suffering from severe β-thalassemia (Ali et al., 2021). Bone marrow transplantation can help patients with thalassemia restore normal red blood cell production by providing healthy hematopoietic stem cells, thereby improving anemia symptoms and potentially achieving a cure, but it also involves inherent challenges and risks (Oikonomopoulou and Goussetis, 2021). Post-transplantation, patients might experience complications, including GVHD and hepatobiliary disorders (Dezan et al., 2023).
3.2.2 Umbilical cord blood transplantation
Umbilical cord blood contains a large number of hematopoietic stem cells, and it can be used in patients with no compatible donor (Hordyjewska et al., 2015). For more than 30 years, the transplantation of umbilical cord blood has been used in medical practices (Gupta and Wagner, 2020). Although it was initially regarded as medical waste, a thorough investigation into its biological characteristics and immunogenicity has revealed that umbilical cord blood is abundant in hematopoietic stem cells, which tend to proliferate and survive more effectively than other types of stem cells (Zhu et al., 2021a). Furthermore, it exhibits low immunogenicity, does not require strict HLA matching, causes no harm to donors, and is associated with a lower incidence of GVHD in transplant recipients (Zhu et al., 2021b). Compared to bone marrow, the matching process for cord blood demands a lesser degree of site matching, thereby making it easier to find a compatible donor (Sanchez-Petitto et al., 2023). Although umbilical cord blood transplantation has distinct benefits, it is utilized less often in clinical settings than bone marrow and peripheral blood hematopoietic stem cell transplants. This is attributable to factors such as the possibility of delayed engraftment, the risk of graft failure, elevated non-relapse mortality rates, a heightened susceptibility to infections, and the considerable expenses involved in procuring umbilical cord blood (Sirinoglu Demiriz et al., 2012; Kindwall-Keller and Ballen, 2020).
3.2.3 Peripheral blood stem cell transplantation
The use of peripheral blood stem cell transplantation (PBSCT) from a matched sibling donor has been suggested as an alternative to bone marrow transplantation to reduce the risk of transplant failure in patients with major thalassemia (Körbling et al., 2011). PBSCT is a vital treatment method for thalassemia, which involves collecting hematopoietic stem cells from a donor, who can be the patient themselves or a matched donor, and then transfusing them intravenously into the patient’s body (Angelucci et al., 2014). These stem cells migrate to the bone marrow, replacing the patient’s abnormal hematopoietic cells and restoring normal red blood cell production (Körblin et al., 2011). This method holds promise for treating thalassemia; however, it requires finding an appropriate donor and addressing complications associated with transplantation (Angelucci et al., 2014). A successful transplant can greatly enhance the patient’s quality of life, potentially decreasing or even removing the requirement for blood transfusions and iron chelation therapy. However, not all of the transplant patients can be saved; some of them die from complications after transplantation like GVHD, osteonecrosis, and infection (Kuci et al., 2020). However, the failure rate of allograft is approximately 25% (Marziali et al., 2017). Transplants of hematopoietic stem cells sourced from siblings who share identical HLA profiles show notably improved success rates when contrasted with those obtained from unrelated donors. However, it is important to note that merely 35%-40% of patients with thalassemia are able to find a sibling donor with matching HLA (O’Reilly, 1983). In addition, Jagasia demonstrated that approximately 40% HSCT of identical siblings will get GVHD, whereas 59% will get GVHD in the transplantation of the unrelated donor hematopoietic stem cells (Jagasia et al., 2012). A research indicates that approximately 15%-16% of patients experience grades II-IV acute GVHD and 4%-12% experience chronic GVHD after transplantation for thalassemia (Mahmoud et al., 2015).
3.3 Autologous stem cell transplantation and gene-editing therapy for thalassemia
Transplantation of autologous hematopoietic stem cells or therapy involving gene editing represents a novel approach that functions by modifying a patient’s own blood stem cells to correct the genetic mutation responsible for thalassemia (Zeng et al., 2023). This approach avoids donor-matching problems in allogeneic transplantation and may reduce the occurrence rate of GVHD and infectious diseases (Li and Yang, 2023). Autologous hematopoietic stem cell transplantation typically involves collecting hematopoietic stem cells from a person, using gene-editing techniques such as CRISPR-Cas9 to amend the genetic mutation, and then reinfusing the altered stem cells into the same individual (Wilkinson et al., 2021).
4 CRISPR/Cas-edited induced pluripotent stem cells
So far, gene-editing technology to treat thalassemia has been widely used in research and is considered one of the most promising therapies for diseases resulting from single-gene inheritance (Khiabani et al., 2023). Gene editing represents an advanced therapeutic approach that focuses on cultivating patients’ hematopoietic stem cells. Following this, the edited stem cells containing the corrected gene are injected back into the patients’ bodies to achieve complete healing (Ali et al., 2021). At present, gene therapy is in the experimental stage and has made some progress. Some research works have pointed out that gene-editing technologies like CRISPR/Cas 9 (Zeng et al., 2023) can accurately modify or replace defective genes and ultimately restore the function of red blood cells (Traeger-Synodinos et al., 2024). Although gene therapy offers certain advantages over other therapies, it is not suitable for all thalassemia patients, and its high cost makes it inaccessible to many patients; moreover, the therapy becomes ineffective if the corrected gene is inserted off-target (Christakopoulos et al., 2023).
4.1 CRISPR/Cas9 gene-editing technology
The CRISPR/Cas9 system was initially used by bacteria and archaea for adaptive immune responses against foreign DNA sources like plasmids and viruses. It has now become a potent instrument for gene editing (Deltcheva et al., 2011). With the introduction of CRISPR/Cas9 technology in 2012 (Jinek et al., 2012), this method has shown considerable promise in addressing genetic disorders, including thalassemia. Although CRISPR/Cas9 has limitations in off-target effects and editing efficiency, its flexibility and programmability have spurred next-generation, more precise tools like Base Editing, offering broader strategies for treating monogenic diseases such as thalassemia (Table 4) (Greco et al., 2024). The CRISPR/Cas9 system, guided by RNA, is notable for being easy to use, economical, and versatile (Anurogo et al., 2021). However, despite its broad application prospects, gene therapy raises concerns about its safety and ethical implications, particularly the risk of off-target effects, which require rigorous ethical review and regulatory oversight (Hryhorowicz et al., 2023).
TABLE 4
| Therapeutic approach | Principle | Advantage | Disadvantage | Correction of known pathogenic mutations | Safety | Reference |
|---|---|---|---|---|---|---|
| CRISPR/Cas9 gene editing |
Utilizes the CRISPR/Cas system to directly repair or insert target genes, correcting mutations | a. Precise correction of mutations b. Low risk of immune reactions c. Applicable to various cell types |
a. Unintended effects b. The efficiency of delivery and specificity for cells need additional refinement |
Suitable for cases requiring long-term stable expression of normal genes | a. Off-target effects and long-term safety b. Delivery system safety requires optimization |
Paolini Sguazzi et al. (2021), Wang et al. (2025b), Butterfield et al. (2025), Gao et al. (2025), John and Czechowicz (2025), and Ali et al. (2025) |
| Lentiviral vectors | Introduces normal genes into cells via viral vectors to replace defective genes | a. Efficient gene insertion b. Long-term stable expression c. Extensive clinical experience |
a. Potential for immune reactions b. Risk of insertional mutagenesis |
Correction of single-base mutations | a. Long-term monitoring required b. Safety relatively mature but still with risks |
Williams et al. (2025), Ottaviano and Qasim (2025), and Hackett and Crystal (2025) |
| Base editing | Chemically alters one base to another, which can be divided into two primary categories: those that target DNA and those that focus on RNA. | a. No double-strand breaks b. High efficiency for single-base conversions |
a. Lower technological maturity b. Limited scope of application |
a. Still in preclinical research stages b. Not yet widely applied clinically |
a. Long-term stability and potential side effects b. Off-target effects |
Porto et al. (2020), Komor et al. (2016), Gaudelli et al. (2017), and Rees and Liu (2018) |
Comparison of gene therapy strategies for thalassemia.
4.2 Human induced pluripotent stem cell
In 2006, Shinya Yamanaka et al. found that it was possible to reprogram adult skin fibroblasts into hiPSCs by incorporating four transcription factors: Oct4, Sox2, Klf4, and c-Myc (Takahashi and Yamanaka, 2006). iPSCs closely resemble embryonic stem cells regarding their morphological characteristics, patterns of gene expression, and capabilities for differentiation, which have been validated in various 3D cardiac tissue models (Campostrini et al., 2021). These cells exhibit remarkable self-renewal capabilities and have the potential for multipotent differentiation, rendering them a valuable resource for various applications, including regenerative medicine, drug development, and disease modeling (Poetsch et al., 2022). iPSCs derived from an individual’s own cells provide considerable medical benefits by reducing the likelihood of immune rejection. Their capacity for self-renewal and differentiation into diverse cell types positions them as essential tools in regenerative medicine, drug discovery, and disease modeling (Cerneckis et al., 2024). iPSCs possess the ability to replicate and grow indefinitely in a laboratory setting, thus offering a substantial source of cells for applications in tissue engineering and cellular therapies (Mattis and Svendsen, 2011). This property is essential for producing the required cell populations for regenerative medicine (Rowe and Daley, 2019). However, iPSCs encounter challenges including potential genetic mutations and chromosomal abnormalities arising from reprogramming, which can compromise cell quality and safety (Kavyasudha et al., 2018). Additionally, effective differentiation into specific cell types and achieving targeted differentiation with functional integration in vivo present ongoing research challenges (Lee and Son, 2021).
4.2.1 Gene-editing strategies
Technologies for gene editing offer considerable promise for utilizing iPSCs (McTague et al., 2021). For instance, CRISPR/Cas9 tools can be utilized to modify iPSCs by either knocking out genes that hinder nerve regeneration (like Nogo-A) or inserting genes that encourage nerve repair (such as the neurotrophic factor BDNF) (Hsu et al., 2019). Additionally, for allogeneic iPSCs, HLA genes can be edited to match those of the patient or to create universal donor cells, thereby reducing the risk of immune rejection (Xu et al., 2019; Song et al., 2022). It is necessary to conduct sequencing, flow cytometry and other analyses on the verified edited iPS cells to verify the accuracy of the modification and the normal differentiation ability of the cells (Maurissen and Woltjen, 2020).
4.2.2 Strategy for differentiating hiPSCs into HSCs
A fundamental approach to differentiating iPSCs into hematopoietic stem cells (HSCs) involves the creation of embryoid bodies (EBs), induction of mesoderm, formation of hematopoietic endothelium, and the process of endothelial-to-hematopoietic transition (EHT) (Ng et al., 2024). This process also encompasses the utilization of various elements, such as cytokines [interleukin (IL)-3, IL-6], dexamethasone, stem cell factor (SCF), recombinant human erythropoietin (EPO), vascular endothelial growth factor (VEGF), insulin-like growth factor I (IGF-I), Fms-like tyrosine kinase 3 (FLT3), bone morphogenetic protein 4 (BMP4), albumin, and transferrin (Lapillonne et al., 2010; Bernecker et al., 2019; Rowe et al., 2016; Oguro, 2019). In addition, retinoic acid and its precursors (such as retinol) play important roles in the mesoderm patterning stage, significantly enhancing the efficiency of iPSC differentiation into hematopoietic cells (Grace et al., 2018). However, the differentiation of iPSCs into functional HSCs still faces challenges such as cell functional stability and immunogenicity (Wattanapanitch et al., 2019).
4.3 CRISPR/Cas-edited induced pluripotent stem cell for thalassemia
iPSCs utilizing CRISPR/Cas gene-editing techniques present an innovative strategy for treating thalassemia (Wattanapanitch et al., 2018). In the context of thalassemia management, the CRISPR/Cas technology is capable of correcting genetic mutations within iPSCs, which restores their erythropoietic function and allows for the production of healthy red blood cells through in vitro cultivation and differentiation into erythroid lineages (Xie et al., 2014). These differentiated red blood cells can subsequently be reintroduced into the patient, acting as a form of cellular therapy that either supplements or replaces the malfunctioning cells, thereby alleviating the symptoms related to the genetic mutation (Ou et al., 2016). This approach is particularly advantageous as it utilizes the patient’s own cells, reducing the risk of immune rejection. Moreover, this technique could be applied to the treatment of various other genetic conditions (Li et al., 2023). The CRISPR/Cas9 technique for editing genes related to thalassemia poses numerous challenges, especially regarding safety considerations and the potential long-term consequences of this new technology, which have not yet been completely determined (Frangoul et al., 2021). Overall, using CRISPR/Cas gene-editing techniques on induced pluripotent stem cells exhibits potential as a therapy for thalassemia; nonetheless, further studies and confirmations are necessary prior to its widespread adoption in clinical applications (Zeng et al., 2023).
4.3.1 Delivery methods
The methods of delivering the CRISPR/Cas system play a vital role in the effectiveness of gene editing (Du et al., 2023). Typical methods of delivery encompass viral vectors such as adeno-associated virus (AAV) (Naso et al., 2017), adenovirus (AdV), lentivirus, Sendai virus, and retrovirus (Tong et al., 2019; Park et al., 2016), along with nonviral systems including lipid nanoparticles (Li et al., 2018; Sinclair et al., 2023). Although viral vectors offer high delivery efficiency, they are associated with risks of immune responses and insertional mutagenesis. In contrast, nonviral delivery systems are safer but less efficient (Seijas et al., 2025).
4.3.2 Editing efficiency
Editing efficiency directly impacts the therapeutic outcome. Improvements in the efficiency of the CRISPR/Cas9 system can be achieved through the optimization of sgRNA design, expression levels of Cas9 protein, and the selection of cell types (Agrawal et al., 2021). Utilizing high-precision variants of Cas9, such as eSpCas9 or SpCas9-HF1, can reduce off-target effects while also enhancing the safety of the editing procedure (Kleinstiver et al., 2021; Yin et al., 2016). In iPSCs, increased editing efficiency ensures more successful cell repair, fewer residual mutant cells, and better therapeutic efficacy, as shown by Singh et al. (2024), using p53 inhibition and pro-survival molecules to achieve over 90% CRISPR/Cas9 efficiency.
4.3.3 Differentiation into functional hematopoietic stem cells
The process of transforming corrected iPSCs into functional hematopoietic stem cells represents a crucial aspect of cellular therapy (Table 5). At present, by replicating the in vivo microenvironment that supports hematopoietic development and integrating specific cytokines (like BMP4, SCF, and TPO) with small molecular compounds (Bello et al., 2024; Jeong et al., 2020a; Kayama et al., 2024), iPSCs can be effectively guided to develop into the hematopoietic lineage. Further optimization of differentiation protocols, such as adjusting culture conditions, adding functional small molecules, and utilizing three-dimensional culture systems, holds promise for improving differentiation efficiency and cell quality (Rahman et al., 2025; Meng et al., 2014). Additionally, verifying the functionality of the differentiated cells through flow cytometry and in vivo transplantation experiments is an essential step to ensure therapeutic efficacy (Bhattacharya et al., 2014).
TABLE 5
| Disease | iPSC source | Treatment | Therapeutic effect | Reference |
|---|---|---|---|---|
| β-Thalassemia (homozygous 41/42 deletion) | β-Thal patient | CRISPR/Cas9+ iPSCs | Effectively fixes β-thal mutations in patient iPSCs | Ou et al. (2016) |
| β-Thalassemia [β17/17 (A→T) in HBB] | β-Thal patient | CRISPR/Cas9 | Edited cells show normal karyotypes, pluripotency, and no off-target. | Song et al. (2015) |
| β-Thalassemia [IVS2-654(C>T)] | β-Thal patient | CRISPR/Cas9 + piggyBac | CRISPR/Cas9 detected off-target. | Xu et al. (2015) |
| HbE mutation | β-Thal patient | CRISPR/Cas9 plasmid + ssODN | Achieves one-step HbE correction in iPSCs | Wattanapanitch (2021) |
| β-Thalassemia (a homozygous β41-42 del and heterozygous Westmead mut in HBA2) | Fetal amnion | CRISPR/Cas9+ iPSCs | Mutations fixed; hiPSCs kept normal pluripotency and could become hematopoietic progenitors | Li et al. (2022a) |
| β-Thalassemia [β17/17 (A→T) in HBB] | β-Thal patient | CRISPR/Cas9 + iPSCs | Normal karyotype, maintained pluripotency, and no off-target effects | Song et al. (2015) |
| β-Thalassemia [−28 (A>G) and the 4-bp (TCTT) del at CD41-42 in exon 2] | β-Thal patient | CRISPR/Cas9 + iPSCs + piggyBac | Seamless HBB mutation correction via HDR; no off-target effects | Xie et al. (2014) |
| β-Thalassemia [4-bp del (–TCTT) and (–CTTT) at CD41-42 mut] | β-Thal patients | CRISPR/Cas9 + ssODNs | Repaired cells had normal β-globin transcripts, low mutation load, and no off-target mutagenesis | Niu et al. (2016) |
| β-Thalassemia (CD26 G>A mut in in HBB) | HbE/β-thalassemia patient’s dermal fibroblasts with CD41/42 and CD26 mut | Cas9 + ssODNs via HDR | HBB protein restored; single CD26 allele fix normalizes β-globin in HbE/β-thalassemia | Wattanapanitch et al. (2018) |
The treatment and therapeutic effect of using iPSC to treat the thalassemia patients.
4.4 Barriers to translating research into clinical applications
4.4.1 Technical challenges and strategies for mitigation
One of the primary challenges in applying gene-editing technologies such as CRISPR/Cas9 to clinical treatments is the technical barriers associated with off-target effects (Concordet and Haeussler, 2018). Unplanned alterations to genes that are not the intended targets may heighten the risks linked to therapeutic treatments. To reduce these unintended effects, various strategies have been formulated.
High-fidelity Cas9 variants: the use of high-fidelity Cas9 variants (Zuo et al., 2022), such as eSpCas9, SpCas9-HF1, dCas9-FokI and evoCas9 (Allemailem et al., 2023; Wyvekens et al., 2015), has been extensively acknowledged in academic writings as an approach to enhance the accuracy of CRISPR/Cas9-editing systems (Naeem et al., 2020). The highly accurate version of Cas9, referred to as Hypa-Cas9, has demonstrated enhanced on-target effectiveness in human cells while minimizing off-target impacts (Chen et al., 2017).
Optimized sgRNA design: the careful design of single-guide RNA (sgRNA) improves the accuracy of gene editing and reduces off-target consequences (Doench et al., 2016). Using bioinformatic tools to predict and select sgRNA sequences with low off-target risks is an effective method to improve specificity (Concordet and Haeussler, 2018; Doench et al., 2016). Recent developments in the methods of delivering CRISPR/Cas9 for therapeutic gene editing in stem cells have been examined, emphasizing the significance of well-designed sgRNA in improving the efficiency of genome editing while minimizing off-target impacts (Lotfi et al., 2023).
Delivery approaches for CRISPR/Cas9 systems: enhancing the delivery mechanisms for the CRISPR/Cas9 system, using nonviral strategies like lipid nanoparticles (Kazemian et al., 2022) or electroporation (Hussen et al., 2024) as nonviral delivery systems, can lead to improved editing efficiency and a decrease in off-target effects.
Precise control of editing windows: introducing mutations into the deaminase allows for the narrowing of the editing window while still preserving significant editing activity (Jeong et al., 2020b). A study used adenine base editors (ABEs) to accurately correct the IVSI-110(G>A) mutation associated with beta-thalassemia, attaining a 90% efficiency in editing (Naiisseh et al., 2024). By precisely controlling the editing window, that is, performing edits during specific cell cycle stages, off-target effects can be minimized, as certain cell cycle stages are more precise in DNA damage repair mechanisms (Fichter et al., 2023).
Using these approaches allows us to more precisely modify mutations that lead to diseases, like thalassemia, while minimizing the potential for off-target effects.
4.4.2 Ethical and regulatory considerations
The translation of gene-editing technologies into clinical applications faces significant technical, ethical, and regulatory challenges (Ledford, 2020). The swift advancement of these technologies raises ethical concerns regarding safety, equity, privacy, and societal impact (Greely, 2019). The establishment of a unified global regulatory system faces challenges, as more countries become open to gene-editing technology (Sprink et al., 2022). Ethical reviews are crucial for ensuring research integrity and participant rights, and research workers must comply with laws including clinical trial regulations and data protection (Joseph et al., 2022). Public engagement and transparency are key factors to fostering understanding and discourse on these issues (Parikh et al., 2025). Overcoming these challenges is vital for advancing gene-editing technology in clinical settings (Wiley et al., 2025), potentially offering novel treatments for diseases like thalassemia.
5 Biological characteristics and function of MSCs
5.1 Biological properties
MSCs are a type of multipotent stem cell that was first discovered in the bone marrow by Friedenstein and are derived from the dental pulp, umbilical cord blood, amniotic membrane, placenta, mobilised peripheral blood, synovium and synovial fluid, endometrium, skin and muscle (Berebichez-Fridman and Montero-Olvera, 2018), which possess the ability to self-renew and differentiate into various cell types (Kolf et al., 2007). MSCs, mainly found in connective tissue and apparatus mesenchyme, are an important cell repository involved in tissue regeneration; MSCs are also a significant type of seed cell used in tissue engineering (Adil et al., 2022). MSCs proliferate rapidly and are easily isolated and cultured. These cells may be sourced from multiple origins, including bone marrow, periosteum, adipose tissue (Deo et al., 2022), umbilical cord tissue (Wu et al., 2023), dental tissue (Gan et al., 2020), and amniotic fluid (Gholami Farashah et al., 2023). Additionally, MSCs contribute to immune modulation by interacting with T cells (Kuca-Warnawin et al., 2021), B cells, NK cells (Abbasi et al., 2022), and other types of immune cells. Importantly, MSCs have little or no immunogenicity (Chen et al., 2024b) and the tolerance of immunity, which can reduce the risk of graft rejective reaction. Recently, a study found that higher-level TNF-α-induced protein 6 (TSG6) enhanced the anti-inflammatory function of CD317(+) MSCs, suggesting that CD317(+) MSCs may be a promising candidate for treating the immune-related diseases (Song et al., 2024). MSCs can repair tissues and differentiate into osteoblast, chondroblast, nerve cells, myoblast, and adipose tissues (Kim et al., 2023). Given the capabilities of MSCs, extensive research has been conducted on them, leading to their increasing implementation in clinical settings.
5.2 The possibility of MSC transplantation to treat thalassemia
A study has indicated that MSCs can enhance the homing of hematopoietic stem cells and boost their hematopoietic capacity (Aqmasheh et al., 2017). Given their role in immunological regulation, MSCs can inhibit NK cell activity and reduce T-cell proliferation by releasing specific cytokines and facilitating cell interactions, which is beneficial for minimizing rejection responses and enhancing survival rates (Johnstone et al., 2021). In patients with β-thalassemia, the MSCs in the bone marrow are functionally impaired due to iron overload and oxidative stress, leading to a decrease in their proliferation, differentiation capacity, and hematopoietic supportive function (Cri et al., 2019). The alteration of the bone marrow microenvironment in patients with thalassemia leads to a significant increase in the lipid, protein, glycogen and nucleic acid content of bone marrow mesenchymal stem cells (BM-MSCs), which is related to enhanced cell proliferation and bone marrow activity (Aksoy et al., 2012). Moreover, studies have shown that the vertebral body - adherent mesenchymal stromal cells (vBA - MSCs) extracted from donor vertebral fragments have similar characteristics to traditional bone marrow - derived mesenchymal stromal cells (BM - MSCs), but with a significantly higher abundance (Johnstone et al., 2020). Additionally, they are matched with hematopoietic progenitor cells (HPCs), which helps promote the formation of mixed chimerism, enhance peripheral immune regulatory functions, and improve the safety of transplantation (Johnstone et al., 2021).
5.3 Research on using MSCs to treat the complication of thalassemia
MSCs have shown potential therapeutic values in treating multiple complications of thalassemia, including heart disease (Szaraz et al., 2017), liver disease (Banas et al., 2007; Ortuño-Costela et al., 2025), bone destruction (Afflerbach et al., 2020), lung disease (Mansouri et al., 2019), endocrine abnormalities (Sarvari et al., 2021), and other diseases (Hoang et al., 2022) (Figure 2). First, thalassemia patients face a long-term anemic state and need regular blood transfusion, which may lead to increased heart burden, causing heart enlargement, cardiac hypertrophy, and other heart diseases (Berdoukas et al., 2015), and MSCs play a role in protecting and repairing the heart. They facilitate the regeneration and repair of injured myocardial tissue, enhancing cardiac function by differentiating into both cardiomyocytes and vascular smooth muscle cells (Ala, 2023; An et al., 2025). Second, chronic anemia and increased red blood cell destruction in individuals with thalassemia can place a greater strain on the liver, potentially leading to liver diseases such as hepatic fibrosis (Papastamataki et al., 2010). MSCs have the ability to release various growth factors and cytokines, aiding in the regeneration and repair of liver cells, minimizing liver inflammation, and enhancing liver function (Rostami et al., 2021; Wen et al., 2025). Third, patients with thalassemia may have bone destruction and osteoporosis due to chronic anemia and myelodysplasia (Bhardwaj et al., 2023), and MSCs have the ability of osteogenic differentiation, can promote the regeneration and repair of bone tissue, can increase bone density and bone strength, and can improve bone condition (Jiang et al., 2021). Fourth, thalassemia patients receiving chronic transfusions are at risk of iron overload and pulmonary fibrosis (Carnelli et al., 2003). MSCs have the potential to address these respiratory concerns by transforming into alveolar and lung epithelial cells, facilitating the repair of lung tissue, minimizing fibrosis, and enhancing overall lung performance (Doherty et al., 2024; Antoniou et al., 2010), offering a new therapeutic strategy for lung diseases associated with thalassemia. Additionally, thalassemia patients may experience endocrine system dysfunction due to long-term anemia, malnutrition, and other factors (Carsote et al., 2022). Endocrine abnormalities and MSCs can regulate the balance between the immune and endocrine systems, promote the functional recovery of endocrine glands, and improve the symptoms associated with endocrine abnormalities (Hoang et al., 2022). In addition to these challenges, individuals with thalassemia who have undergone blood transfusions, iron chelation therapy, or bone marrow transplantation might experience adverse effects resulting from these treatments (Yousuf et al., 2022). Current research on using MSCs to treat thalassemia-related complications is still in the early stages, with further exploration needed in areas such as specific treatment mechanisms, optimal administration methods, and efficacy evaluation (Li et al., 2024). Moreover, addressing the challenges related to the sourcing, preparation, and quality assurance of MSCs is essential to ensure the safety and effectiveness of these treatments (Guadix et al., 2019). In summary, MSCs demonstrate considerable potential as a therapy for various complications linked to thalassemia, and it is expected that they will provide enhanced treatment alternatives for these individuals moving forward.
FIGURE 2
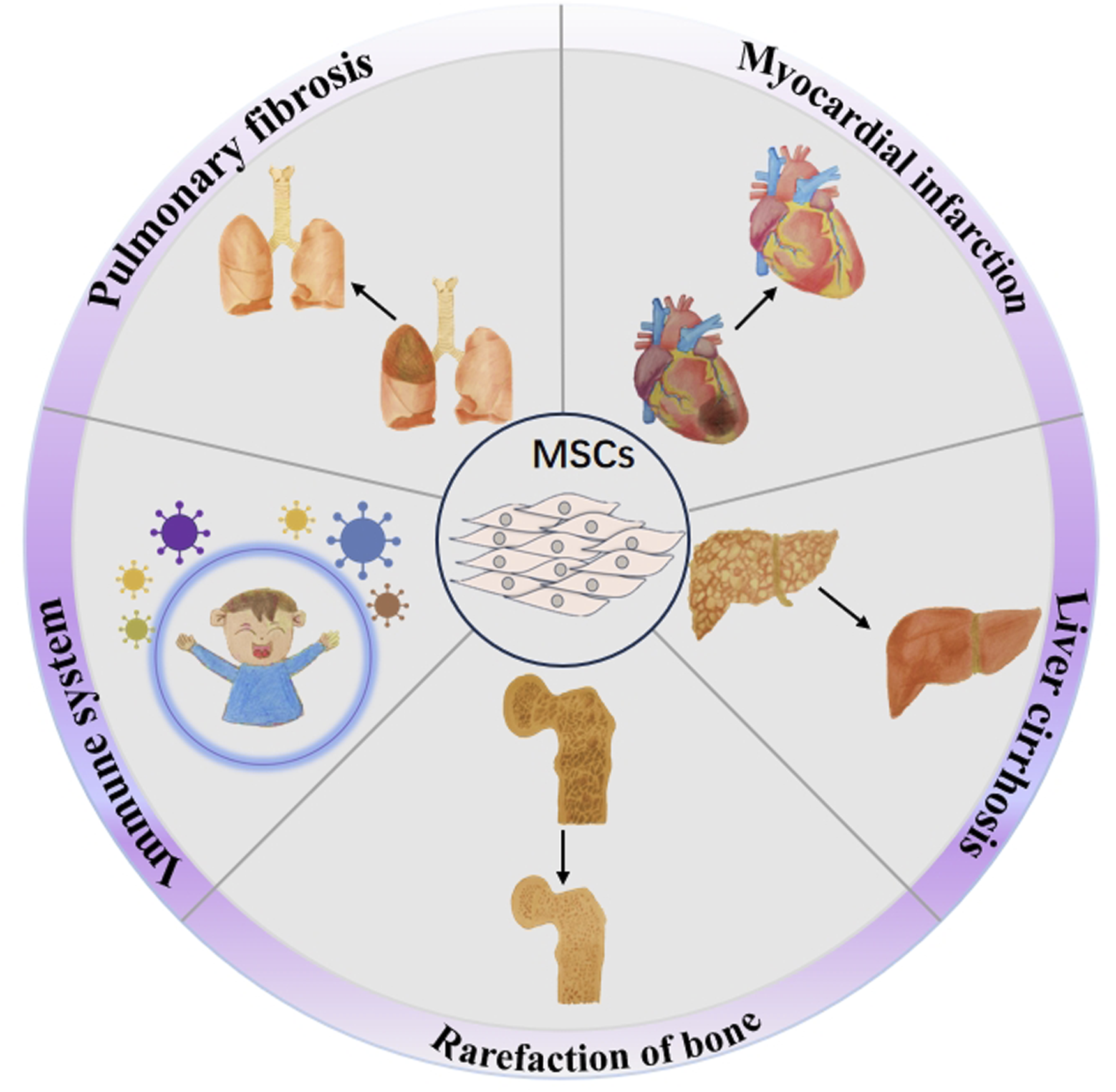
Therapeutic potential of MSCs in addressing thalassemia-related complications. The potential of MSCs to treat the complications of thalassemia including myocardial infarction, liver cirrhosis, rarefaction of bone, pulmonary fibrosis, and immune system damage.
5.4 Challenges in translational application: technical, ethical, and regulatory aspects
During the process of transplanting allogeneic MSCs into patients, challenges in technology, ethics, and regulations must be addressed (Velikova et al., 2024). Technically, it is crucial to guarantee the viability, homing ability, and long-term stability of the cells, which includes optimizing cell isolation, expansion, and cryopreservation techniques (Wang and Li, 2024). Additionally, utilizing the patient’s own MSCs after genetic modification and subsequent autologous transplantation offers a promising approach to potentially reduce the risk of immune rejection and GVHD (Večerić-Haler et al., 2022; Li et al., 2022b). Ethical considerations involve obtaining fully informed consent from patients, safeguarding patient privacy, and ensuring equitable access to treatment (Lo et al., 2009). Regulatory compliance requires adherence to international and local regulatory standards, navigating the approval process, and conducting long-term surveillance to evaluate the safety and efficacy of the therapy (Farmakis et al., 2022). Overcoming these challenges necessitates interdisciplinary collaboration, technological advancement, ethical review, and strict regulatory compliance to enhance the feasibility and acceptance of the treatment, ultimately aiming to improve therapeutic outcomes for patients with thalassemia.
6 Conclusion
6.1 The current status and challenges of stem cell therapy
Currently, in the area of stem cell therapy, the management of thalassemia mainly relies on hematopoietic stem cells, whereas MSCs and iPSCs demonstrate considerable promise for a range of uses (Muthu et al., 2022). Nonetheless, there are variations in both therapeutic effectiveness and safety among stem cells derived from different origins. For example, MSCs obtained from umbilical cord blood present advantages like reduced immunogenicity (Um et al., 2020); however, they may not exhibit the same level of cell activity and functionality as MSCs obtained from bone marrow (Shang et al., 2021). Despite their low immunogenicity, MSCs can still trigger immune responses under certain conditions, and the performance and longevity of stem cells in vivo can be affected by factors such as inflammation and hypoxia (Chen et al., 2023). Furthermore, the attributes and size of the patient cohort (including factors like age, severity of the disease, and genetic background) might also influence the comparability of the research findings (Česnik et al., 2024). The constraints of existing technologies, such as the accuracy of gene-editing instruments and inconsistencies in cell culture conditions, can also contribute to varying results (Česnik and Švajger, 2024; Mushahary et al., 2018). Although advancements have been achieved, obstacles continue to exist: iPSCs encounter problems related to epigenetic instability and the potential for tumor formation, whereas adult stem cells struggle to accomplish successful differentiation (Su et al., 2025). Ongoing assessment of long-term safety and effectiveness is essential for further verification.
6.2 The capabilities and constraints of gene-editing technology
Regarding gene-editing technology, CRISPR/Cas9 has demonstrated remarkable promise in correcting gene mutations related to thalassemia, but there are still significant differences in off-target effects and editing efficiency across different studies. Some report high editing efficiency but note potential off-target effects, whereas others manage to minimize off-target effects but still face challenges in enhancing editing efficiency (Zeng et al., 2023). These inconsistent outcomes may result from differences in experimental design, cell sources, gene-editing methods, or animal models, given that viral vectors have high delivery efficiency but are associated with risks such as immune reactions and insertional mutations, whereas nonviral delivery systems, although safer, tend to have lower efficiency (Taghdiri and Mussolino, 2024). The potential impact of the limitations of current technologies on research outcomes cannot be ignored. Despite advancements in CRISPR/Cas9 technology aimed at minimizing off-target effects, including the development of high-fidelity Cas9 variants and enhanced sgRNA designs, the total eradication of off-target effects remains a significant challenge (Guo et al., 2023).
6.3 Future directions for the development of thalassemia treatment technologies
To address these challenges, future studies should implement more standardized experimental frameworks and methods to minimize variability in outcomes while simultaneously promoting the advancement of safer and more effective gene-editing technologies and delivery systems (Lino et al., 2018). Furthermore, the significance of collaboration across multiple disciplines should not be underestimated, as integrating knowledge from gene editing, cell biology, clinical practice, and other areas can collaboratively enhance the progression of thalassemia treatment technologies (Christakopoulos et al., 2023).
Statements
Author contributions
JS: Conceptualization, Visualization, Writing – original draft, Writing – review and editing. XX: Conceptualization, Writing – original draft, Writing – review and editing. SW: Visualization, Writing – original draft. ZD: Supervision, Project administration, Writing – review and editing. PH: Supervision, Project administration, Writing – review and editing. YC: Conceptualization, Supervision, Funding acquisition, Writing – review and editing. ZH: Conceptualization, Supervision, Funding acquisition, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the National Natural Science Foundation joint fund project (U23A20498), Kweichow Moutai Hospital Research and Talent Training Fund Project (MTyk2022-01), and Guizhou Provincial Science and Technology Project (QKHCG[2024]ZD012).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. Generative AI was used for language enhancement services. Specifically, the manuscript was refined with the assistance of Kimi, an AI developed by Moonshot AI, to improve the linguistic quality and clarity of the writing.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Abbasi B. Shamsasenjan K. Ahmadi M. Beheshti S. A. Saleh M. (2022). Mesenchymal stem cells and natural killer cells interaction mechanisms and potential clinical applications. Stem Cell Res. Ther.13 (1), 97. 10.1186/s13287-022-02777-4
2
Adil A. Xu M. Haykal S. (2022). Recellularization of bioengineered scaffolds for vascular composite allotransplantation. Front. Surg.9, 843677. 10.3389/fsurg.2022.843677
3
Afflerbach A. K. Kiri M. D. Detinis T. Maoz B. M. (2020). Mesenchymal stem cells as a promising cell source for integration in novel In Vitro models. Biomolecules10 (9), 1306. 10.3390/biom10091306
4
Agarwal R. K. Dhanya R. Sedai A. Ankita K. Parmar L. Ramprakash S. et al (2023). Bone marrow quality index: a predictor of acute graft-versus-host disease in hematopoietic stem cell transplantation for thalassemia. Transpl. Cell Ther.29 (11), 711.e1–711.e6. 10.1016/j.jtct.2023.07.014
5
Agrawal S. Padmaswari M. H. Stokes A. L. Maxenberger D. Reese M. Khalil A. et al (2024). Optimizing Recombinant Cas9 Expression: Insights from E. coliBL21(DE3) Strains for Enhanced Protein Purification and Genome Editing. Biomedicines.12 (6), 1226–711.e6. 10.3390/biomedicines12061226
6
Akbayram S. Demir H. A. Kılıç A. L. Güneş A. M. Zengin E. Özmen S. et al (2022). Thalassemia-free and graft-versus-host-free survival: outcomes of hematopoietic stem cell transplantation for thalassemia major, Turkish experience. Bone Marrow Transplant.57 (5), 760–767. 10.1038/s41409-022-01613-w
7
Aksoy C. Guliyev A. Kilic E. Uckan D. Severcan F. (2012). Bone marrow mesenchymal stem cells in patients with beta thalassemia major: molecular analysis with attenuated total reflection-Fourier transform infrared spectroscopy study as a novel method. Stem Cells Dev.21 (11), 2000–2011. 10.1089/scd.2011.0444
8
Ala M. (2023). The beneficial effects of mesenchymal stem cells and their exosomes on myocardial infarction and critical considerations for enhancing their efficacy. Ageing Res. Rev.89, 101980. 10.1016/j.arr.2023.101980
9
Algeri M. Lodi M. Locatelli F. (2023). Hematopoietic stem cell transplantation in thalassemia. Hematol. Oncol. Clin. North Am.37 (2), 413–432. 10.1016/j.hoc.2022.12.009
10
Ali A. Azmat U. Khatoon A. Akbar K. Murtaza B. Ji Z. et al (2025). From gene editing to tumor eradication: the CRISPR revolution in cancer therapy. Prog. Biophys. Mol. Biol.196, 114–131. 10.1016/j.pbiomolbio.2025.04.003
11
Ali S. Mumtaz S. Shakir H. A. Khan M. Tahir H. M. Mumtaz S. et al (2021). Current status of beta-thalassemia and its treatment strategies. Mol. Genet. Genomic Med.9 (12), e1788. 10.1002/mgg3.1788
12
Aljagthmi A. A. Abdel-Aziz A. K. (2025). Hematopoietic stem cells: understanding the mechanisms to unleash the therapeutic potential of hematopoietic stem cell transplantation. Stem Cell Res. Ther.16 (1), 60. 10.1186/s13287-024-04126-z
13
Allemailem K. S. Almatroodi S. A. Almatroudi A. Alrumaihi F. Al Abdulmonem W. Al-Megrin W. A. I. et al (2023). Recent advances in genome-editing technology with CRISPR/Cas9 variants and stimuli-responsive targeting approaches within tumor cells: a future perspective of cancer management. Int. J. Mol. Sci.24 (8), 7052. 10.3390/ijms24087052
14
An C. Zhao Y. Guo L. Zhang Z. Yan C. Zhang S. et al (2025). Innovative approaches to boost mesenchymal stem cells efficacy in myocardial infarction therapy. Mater Today Bio31, 101476. 10.1016/j.mtbio.2025.101476
15
Angastiniotis M. Lobitz S. (2019). Thalassemias: an overview. Int. J. Neonatal Screen5 (1), 16. 10.3390/ijns5010016
16
Angelucci E. Matthes-Martin S. Baronciani D. Bernaudin F. Bonanomi S. Cappellini M. D. et al (2014). Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica99 (5), 811–20. 10.3324/haematol.2013.099747
17
Antoniou K. M. Papadaki H. A. Soufla G. Kastrinaki M. C. Damianaki A. Koutala H. et al (2010). Investigation of bone marrow mesenchymal stem cells (BM MSCs) involvement in idiopathic pulmonary fibrosis (IPF). Respir. Med.104 (10), 1535–1542. 10.1016/j.rmed.2010.04.015
18
Anurogo D. Yuli Prasetyo Budi N. Thi Ngo M. H. Huang Y. H. Pawitan J. A. (2021). Cell and gene therapy for anemia: hematopoietic stem cells and gene editing. Int. J. Mol. Sci.22 (12), 6275. 10.3390/ijms22126275
19
Aprile D. Patrone D. Peluso G. Galderisi U. (2024). Multipotent/pluripotent stem cell populations in stromal tissues and peripheral blood: exploring diversity, potential, and therapeutic applications. Stem Cell Res. & Ther.15 (1), 139. 10.1186/s13287-024-03752-x
20
Aran B. Lukovic D. Aguilar-Quesada R. Veiga A. (2020). Pluripotent stem cell regulation in Spain and the Spanish National Stem Cell Bank. Stem Cell Res.48, 101956. 10.1016/j.scr.2020.101956
21
Aqmasheh S. Shamsasanjan K. Akbarzadehlaleh P. Pashoutan Sarvar D. Timari H. (2017). Effects of mesenchymal stem cell derivatives on hematopoiesis and hematopoietic stem cells. Adv. Pharm. Bull.7(2), 165–177. 10.15171/apb.2017.021
22
Azvolinsky A. (2019). Molecular scissors cut in on stem cells. Nat. Med.25 (6), 864–866. 10.1038/s41591-019-0467-6
23
Bajwa H. Basit H. (2025). “Thalassemia,” in StatPearls. Treasure Island, FL: StatPearls Publishing LLC.
24
Banas A. Teratani T. Yamamoto Y. Tokuhara M. Takeshita F. Quinn G. et al (2007). Adipose tissue-derived mesenchymal stem cells as a source of human hepatocytes. Hepatology. 46(1), 219–228. 10.1002/hep.21704
25
Bello A. B. Canlas K. K. V. Kim D. Park H. Lee S. H. (2024). Stepwise dual-release microparticles of BMP-4 and SCF in induced pluripotent stem cell spheroids enhance differentiation into hematopoietic stem cells. J. Control. Release371, 386–405. 10.1016/j.jconrel.2024.06.011
26
Berdoukas V. Coates T. D. Cabantchik Z. I. Iron and oxidative stress in cardiomyopathy in thalassemia. (2015). Free Radic. Biol. Med.88(Pt A):3–9. 10.1016/j.freeradbiomed.2015.07.019
27
Berebichez-Fridman R. Montero-Olvera P. R. (2018). Sources and clinical applications of mesenchymal stem cells: State-of-the-art review. Sultan Qaboos Univ. Med. J.18 (3), e264–e277. 10.18295/squmj.2018.18.03.002
28
Bernecker C. Ackermann M. Lachmann N. Rohrhofer L. Zaehres H. Araúzo-Bravo M. J. et al (2019). Enhanced Ex Vivo generation of erythroid cells from human induced pluripotent stem cells in a simplified cell culture system with low cytokine support. Stem Cells Dev.28 (23), 1540–1551. 10.1089/scd.2019.0132
29
Bhardwaj A. Swe K. M. M. Sinha N. K. (2023). Treatment for osteoporosis in people with beta-thalassaemia. Cochrane Database Syst. Rev.5 (5), Cd010429. 10.1002/14651858.CD010429.pub3
30
Bhardwaj A. Swe K. M. M. Sinha N. K. (2023). Treatment for osteoporosis in people with beta-thalassaemia. Cochrane Database Syst. Rev.5(5), CD010429. 10.1002/14651858.CD010429.pub3
31
Bhattacharya S. Burridge P. W. Kropp E. M. Chuppa S. L. Kwok W. M. Wu J. C. et al (2014). High efficiency differentiation of human pluripotent stem cells to cardiomyocytes and characterization by flow cytometry. J. Vis. Exp.23(91), 52010. 10.3791/52010
32
Biswas S. Gomez J. Horgan R. Sibai B. M. Saad A. Powel J. E. et al (2023). Mirror syndrome: a systematic literature review. Am. J. Obstet. Gynecol. MFM5 (9), 101067. 10.1016/j.ajogmf.2023.101067
33
Bou-Fakhredin R. Motta I. Cappellini M. D. Taher A. T. (2023). Clinical complications and their management. Hematol. Oncol. Clin. North Am.37 (2), 365–378. 10.1016/j.hoc.2022.12.007
34
Brignier AC Gewirtz AM (2010). Embryonic and adult stem cell therapy. J. Allergy Clin. Immunol.125 (2), S336–44. 10.1016/j.jaci.2009.09.032
35
Butterfield G. L. Reisman S. J. Iglesias N. Gersbach C. A. (2025). Gene regulation technologies for gene and cell therapy. Mol. Ther.33 (5), 2104–2122. 10.1016/j.ymthe.2025.04.004
36
Campostrini G. Windt L. M. van Meer B. J. Bellin M. Mummery C. L. (2021). Cardiac tissues from stem cells: new routes to maturation and cardiac regeneration. Circ. Res.128 (6), 775–801. 10.1161/CIRCRESAHA.121.318183
37
Cao A. Galanello R. (2010). Beta-thalassemia. Genet. Med.12 (2), 61–76. 10.1097/GIM.0b013e3181cd68ed
38
Caocci G. La Nasa G. d'Aloja E. Vacca A. Piras E. Pintor M. et al (2011). Ethical issues of unrelated hematopoietic stem cell transplantation in adult thalassemia patients. BMC Med. Ethics12, 4. 10.1186/1472-6939-12-4
39
Cappellini M. D. Cohen A. Porter J. Taher A. Viprakasit V. (2014). Guidelines for the management of transfusion dependent thalassaemia (TDT). Nicosia, CY: Thalassaemia International Federation.
40
Carnelli V. D'Angelo E. Pecchiari M. Ligorio M. D'Angelo E. (2003). Pulmonary dysfunction in transfusion-dependent patients with thalassemia major. Am. J. Respir. Crit. Care Med.168(2), 180–184. 10.1164/rccm.200211-1292OC
41
Cerneckis J. Cai H. Shi Y. (2024). Induced pluripotent stem cells (iPSCs): molecular mechanisms of induction and applications. Signal Transduct. Target Ther.9 (1), 112. 10.1038/s41392-024-01809-0
42
Cetin B. Erendor F. Eksi Y. E. Sanlioglu A. D. Sanlioglu S. (2025). Advancing CRISPR genome editing into gene therapy clinical trials: progress and future prospects. Expert Rev. Mol. Med.27, e16. 10.1017/erm.2025.10
43
Chen A. P. Gao P. Lin L. Ashok P. He H. Ma C. et al (2024a). An improved approach to generate IL-15(+/+)/TGFβR2(-/-) iPSC-derived natural killer cells using TALEN. Cell Rep. Methods4 (9), 100857. 10.1016/j.crmeth.2024.100857
44
Chen J. S. Dagdas Y. S. Kleinstiver B. P. Welch M. M. Sousa A. A. Harrington L. B. et al (2017). Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature550 (7676), 407–410. 10.1038/nature24268
45
Chen P. Lin W. X. Li S. Q. (2022). THALASSEMIA in ASIA 2021: thalassemia in Guangxi Province, People'S Republic of China. Hemoglobin46 (1), 33–35. 10.1080/03630269.2021.2008960
46
Chen W. Lv L. Chen N. Cui E. (2023). Immunogenicity of mesenchymal stromal/stem cells. Scand. J. Immunol.97 (6), e13267. 10.1111/sji.13267
47
Chen Y. Xu Y. Chi Y. Sun T. Gao Y. Dou X. et al (2024b). Efficacy and safety of human umbilical cord-derived mesenchymal stem cells in the treatment of refractory immune thrombocytopenia: a prospective, single arm, phase I trial. Signal Transduct. Target Ther.9 (1), 102. 10.1038/s41392-024-01793-5
48
Carsote M. Vasiliu C. Trandafir A. I. Albu S. E. Dumitrascu M. C. Popa A. et al (2022). New entity-thalassemic endocrine disease: major beta-thalassemia and endocrine involvement. Diagn. (Basel). 12(8), 1921. 10.3390/diagnostics12081921
49
Česnik A. B. Švajger U. (2024). The issue of heterogeneity of MSC-based advanced therapy medicinal products-a review. Front. Cell Dev. Biol.12, 1400347. 10.3389/fcell.2024.1400347
50
Česnik A. B. Švajger U. (2024). The issue of heterogeneity of MSC-based advanced therapy medicinal products-a review. Front. Cell. Dev. Biol.12, 1400347. 10.3389/fcell.2024.1400347
51
Christakopoulos G. E. Telange R. Yen J. Weiss M. J. (2023). Gene therapy and gene editing for β-Thalassemia. Hematol. Oncol. Clin. North Am.37 (2), 433–447. 10.1016/j.hoc.2022.12.012
52
Chukwuemeka C. G. Ndubueze C. W. Kolawole A. V. Joseph J. N. Oladipo I. H. Ofoezie E. F. et al (2025). In vitro erythropoiesis: the emerging potential of induced pluripotent stem cells (iPSCs). Blood Sci.7 (1), e00215. 10.1097/BS9.0000000000000215
53
Coates T. D. (2014). Physiology and pathophysiology of iron in hemoglobin-associated diseases. Free Radic. Biol. Med.72, 23–40. 10.1016/j.freeradbiomed.2014.03.039
54
Concordet J. P. Haeussler M. (2018). CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res.46 (W1), W242–W245. 10.1093/nar/gky354
55
Crippa S. Rossella V. Aprile A. Silvestri L. Rivis S. Scaramuzza S. et al (2019). Bone marrow stromal cells from β-thalassemia patients have impaired hematopoietic supportive capacity. J. Clin. Invest129 (4), 1566–1580. 10.1172/JCI123191
56
Demirci S. Leonard A. Tisdale J. F. (2020). Hematopoietic stem cells from pluripotent stem cells: clinical potential, challenges, and future perspectives. Stem Cells Transl. Med.9 (12), 1549–1557. 10.1002/sctm.20-0247
57
Demosthenous C. Vlachaki E. Apostolou C. Eleftheriou P. Kotsiafti A. Vetsiou E. et al (2019). Beta-thalassemia: renal complications and mechanisms: a narrative review. Hematology24 (1), 426–438. 10.1080/16078454.2019.1599096
58
Deltcheva E. Chylinski K. Sharma C. M. Gonzales K. Chao Y. Pirzada Z. A. (2011). CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature471 (7340), 602–7. 10.3390/pharmaceutics14040791
59
Deo D. Marchioni M. Rao P. (2022). Mesenchymal stem/stromal cells in organ transplantation. Pharmaceutics14 (4), 791. 10.3390/pharmaceutics14040791
60
De Simone G. Quattrocchi A. Mancini B. di Masi A. Nervi C. Ascenzi P. (2022). Thalassemias: from gene to therapy. Mol. Asp. Med.84, 101028. 10.1016/j.mam.2021.101028
61
Dezan M. G. F. Cavalcante L. N. Cotrim H. P. Lyra A. C. (2023). Hepatobiliary disease after bone marrow transplant. Expert Rev. Gastroenterol. Hepatol.17 (2), 129–143. 10.1080/17474124.2023.2169671
62
Dobner J. Diecke S. Krutmann J. Prigione A. Rossi A. (2024). Reassessment of marker genes in human induced pluripotent stem cells for enhanced quality control. Nat. Commun.15 (1), 8547. 10.1038/s41467-024-52922-1
63
Doench J. G. Fusi N. Sullender M. Hegde M. Vaimberg E. W. Donovan K. F. et al Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. (2016). Nat. Biotechnol.34(2), 184–191. 10.1038/nbt.3437
64
Doench J. G. Fusi N. Sullender M. Hegde M. Vaimberg E. W. Donovan K. F. et al (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol.34 (2), 184–191. 10.1038/nbt.3437
65
Doherty D. F. Roets L. E. Dougan C. M. Brown R. R. Hawthorne I. J. O'Kane C. et al (2024). Mesenchymal stromal cells reduce inflammation and improve lung function in a mouse model of cystic fibrosis lung disease. Sci. Rep.14 (1), 30899. 10.1038/s41598-024-81276-3
66
Dordevic A. Mrakovcic-Sutic I. Pavlovic S. Ugrin M. Roganovic J. (2025). Beta thalassemia syndromes: new insights. World J. Clin. Cases13 (10), 100223. 10.12998/wjcc.v13.i10.100223
67
Du Y. Liu Y. Hu J. Peng X. Liu Z. (2023). CRISPR/Cas9 systems: delivery technologies and biomedical applications. Asian J. Pharm. Sci.18 (6), 100854. 10.1016/j.ajps.2023.100854
68
Evangelidis P. Venou T. M. Fani B. Vlachaki E. Gavriilaki E. on behalf of the International Hemoglobinopathy Research Network INHERENT (2023). Endocrinopathies in hemoglobinopathies: what is the role of iron?Int. J. Mol. Sci.24 (22), 16263. 10.3390/ijms242216263
69
Farashi S. Harteveld C. L. (2018). Molecular basis of α-thalassemia. Blood Cells Mol. Dis.70, 43–53. 10.1016/j.bcmd.2017.09.004
70
Farmakis D. Porter J. Taher A. Domenica Cappellini M. Angastiniotis M. Eleftheriou A. (2022). 2021 thalassaemia international Federation guidelines for the management of transfusion-dependent thalassemia. Hemasphere6 (8), e732. 10.1097/HS9.0000000000000732
71
Fichter K. M. Setayesh T. Malik P. (2023). Strategies for precise gene edits in mammalian cells. Mol. Ther. Nucleic Acids32, 536–552. 10.1016/j.omtn.2023.04.012
72
Forni G. L. Gianesin B. Musallam K. M. Longo F. Rosso R. Lisi R. et al (2023). Overall and complication-free survival in a large cohort of patients with β-thalassemia major followed over 50 years. Am. J. Hematol.98 (3), 381–387. 10.1002/ajh.26798
73
Frangoul H. Altshuler D. Cappellini M. D. Chen Y. S. Domm J. Eustace B. K. et al (2021). CRISPR-Cas9 gene editing for sickle cell disease and β-Thalassemia. N. Engl. J. Med.384 (3), 252–260. 10.1056/NEJMoa2031054
74
Gan L. Liu Y. Cui D. Pan Y. Zheng L. Wan M. (2020). Dental tissue-derived human mesenchymal stem cells and their potential in therapeutic application. Stem Cells Int.2020, 8864572. 10.1155/2020/8864572
75
Gao X. Zhou C. Feng Y. Ye B. Zhao Z. Qi L. et al (2025). Research progress of gene editing technology in neurological diseases. Gene962, 149534. 10.1016/j.gene.2025.149534
76
Gaudelli N. M. Komor A. C. Rees H. A. Packer M. S. Badran A. H. Bryson D. I. et al (2017). Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature551 (7681), 464–471. 10.1038/nature24644
77
Gaudio A. Morabito N. Catalano A. Rapisarda R. Xourafa A. Lasco A. (2019). Pathogenesis of thalassemia major-associated osteoporosis: a review with insights from clinical experience. J. Clin. Res. Pediatr. Endocrinol.11 (2), 110–117. 10.4274/jcrpe.galenos.2018.2018.0074
78
Gholami Farashah M. S. Mohammadi A. Javadi M. Soleimani Rad J. Shakouri S. K. Meshgi S. et al (2023). Bone marrow mesenchymal stem cells' osteogenic potential: superiority or non-superiority to other sources of mesenchymal stem cells?Cell Tissue Bank.24 (3), 663–681. 10.1007/s10561-022-10066-w
79
Gluckman E. Cappelli B. Bernaudin F. Labopin M. Volt F. Carreras J. et al (2017). Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood129 (11), 1548–1556. 10.1182/blood-2016-10-745711
80
Grace C. S. Mikkola H. K. A. Dou D. R. Calvanese V. Ronn R. E. Purton L. E. (2018). Protagonist or antagonist? The complex roles of retinoids in the regulation of hematopoietic stem cells and their specification from pluripotent stem cells. Exp. Hematol.65, 1–16. 10.1016/j.exphem.2018.06.287
81
Greco F. Cosentino M. Marino F. (2024). The Italian breakthrough in CRISPR trials for rare diseases: a focus on beta-thalassemia and sickle cell disease treatment. Front. Med. (Lausanne)11, 1356578. 10.3389/fmed.2024.1356578
82
Greely H. T. (2019). CRISPR'd babies: human germline genome editing in the 'He Jiankui affair. J. Law Biosci.6 (1), 111–183. 10.1093/jlb/lsz010
83
Guadix J. A. López-Beas J. Clares B. Soriano-Ruiz J. L. Zugaza J. L. Gálvez-Martín P. (2019). Principal criteria for evaluating the quality, safety and efficacy of hmsc-based products in clinical practice: current approaches and challenges. Pharmaceutics.11 (11), 552. 10.3390/pharmaceutics11110552
84
Guo C. Ma X. Gao F. Guo Y. (2023). Off-target effects in CRISPR/Cas9 gene editing. Front. Bioeng. Biotechnol.11, 1143157. 10.3389/fbioe.2023.1143157
85
Gupta A. O. Wagner J. E. (2020). Umbilical cord blood transplants: current status and evolving therapies. Front. Pediatr.8, 570282. 10.3389/fped.2020.570282
86
Hackett N. R. Crystal R. G. (2025). Four decades of adenovirus gene transfer vectors: history and current use. Mol. Ther.33 (5), 2192–2204. 10.1016/j.ymthe.2025.03.062
87
Hardouin G. Miccio A. Brusson M. (2025). Gene therapy for β-thalassemia: current and future options. Trends Mol. Med.31 (4), 344–358. 10.1016/j.molmed.2024.12.001
88
Harewood J. Azevedo A. M. (2024). “Alpha thalassemia,” in StatPearls. Treasure Island, FL: StatPearls Publishing LLC.
89
Hoang D. M. Pham P. T. Bach T. Q. Ngo A. T. L. Nguyen Q. T. Phan T. T. K. et al (2022). Stem cell-based therapy for human diseases. Signal Transduct. Target Ther.7 (1), 272. 10.1038/s41392-022-01134-4
90
Hoang D. M. Pham P. T. Bach T. Q. Ngo A. T. L. Nguyen Q. T. Phan T. T. K. et al (2022). Stem cell-based therapy for human diseases. Signal Transduct. Target Ther.7(1), 272. 10.1038/s41392-022-01134-4
91
Hoang V. T. Nguyen Q. T. Phan T. T. K. Pham T. H. Dinh N. T. H. Anh L. P. H. et al (2025). Tissue engineering and regenerative medicine: perspectives and challenges. MedComm (2020)6 (5), e70192. 10.1002/mco2.70192
92
Hodroj M. H. Akiki N. Bou-Fakhredin R. Taher A. T. (2023). Beta-thalassemia: is cure still a dream?Minerva Med.114 (6), 850–860. 10.23736/S0026-4806.23.08501-4
93
Hordyjewska A. Popiołek Ł. Horecka A. (2015). Characteristics of hematopoietic stem cells of umbilical cord blood. Cytotechnology67 (3), 387–96. 10.1007/s10616-014-9796-y
94
Hryhorowicz M. Lipiński D. Zeyland J. (2023). Evolution of CRISPR/Cas systems for precise genome editing. Int. J. Mol. Sci.24 (18), 14233. 10.3390/ijms241814233
95
Hsu M. N. Liao H. T. Truong V. A. Huang K. L. Yu F. J. Chen H. H. et al (2019). CRISPR-based activation of endogenous neurotrophic genes in adipose stem cell sheets to stimulate peripheral nerve regeneration. Theranostics9 (21), 6099–6111. 10.7150/thno.36790
96
Hussen B. M. Najmadden Z. B. Abdullah S. R. Rasul M. F. Mustafa S. A. Ghafouri-Fard S. et al (2024). CRISPR/Cas9 gene editing: a novel strategy for fighting drug resistance in respiratory disorders. Cell. Commun. Signal. 22(1):329. 10.1186/s12964-024-01713-8
97
Huang C. Y. Li L. H. Hsu W. T. Cheng Y. C. Nicholson M. W. Liu C. L. et al (2020). Copy number variant hotspots in Han Taiwanese population induced pluripotent stem cell lines - lessons from establishing the Taiwan human disease iPSC Consortium Bank. J. Biomed. Sci.27 (1), 92. 10.1186/s12929-020-00682-7
98
Jagasia M. Arora M. Flowers M. E. D. Chao N. J. McCarthy P. L. Cutler C. S. et al (2012). Risk factors for acute GVHD and survival after hematopoietic cell transplantation. Blood119 (1), 296–307. 10.1182/blood-2011-06-364265
99
Jeong S. An B. Kim J. H. Han H. W. Kim J. H. Heo H. R. et al (2020a). BMP4 and perivascular cells promote hematopoietic differentiation of human pluripotent stem cells in a differentiation stage-specific manner. Exp. & Mol. Med.52 (1), 56–65. 10.1038/s12276-019-0357-5
100
Jeong Y. K. Song B. Bae S. (2020b). Current status and challenges of DNA base editing tools. Mol. Ther.28 (9), 1938–1952. 10.1016/j.ymthe.2020.07.021
101
Jiang Y. Zhang P. Zhang X. Lv L. Zhou Y. (2021). Advances in mesenchymal stem cell transplantation for the treatment of osteoporosis. Cell. Prolif.54(1), e12956. 10.1111/cpr.12956
102
Jinek M. Chylinski K. Fonfara I. Hauer M. Doudna J. A. Charpentier E. (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science337 (6096), 816–821. 10.1126/science.1225829
103
John T. Czechowicz A. (2025). Clinical hematopoietic stem cell-based gene therapy. Mol. Ther.33, 2663–2678. 10.1016/j.ymthe.2025.04.029
104
Johnstone B. H. Messner F. Brandacher G. Woods E. J. (2021). A large-scale bank of organ donor bone marrow and matched mesenchymal stem cells for promoting immunomodulation and transplant tolerance. Front. Immunol.12, 622604. 10.3389/fimmu.2021.622604
105
Johnstone B. H. Miller H. M. Beck M. R. Gu D. Thirumala S. LaFontaine M. et al (2020). Identification and characterization of a large source of primary mesenchymal stem cells tightly adhered to bone surfaces of human vertebral body marrow cavities. Cytotherapy22 (11), 617–628. 10.1016/j.jcyt.2020.07.003
106
Joseph A. M. Karas M. Ramadan Y. Joubran E. Jacobs R. J. (2022). Ethical perspectives of therapeutic human genome editing from multiple and diverse viewpoints: a scoping review. Cureus14 (11), e31927. 10.7759/cureus.31927
107
Karponi G. Zogas N. (2019). Gene therapy for beta-thalassemia: updated perspectives. Appl. Clin. Genet.12, 167–180. 10.2147/TACG.S178546
108
Kattamis A. Forni G. L. Aydinok Y. Viprakasit V. (2020). Changing patterns in the epidemiology of β-thalassemia. Eur J Haematol105 (6), 692–703. 10.1111/ejh.13512
109
Kattamis A. Kwiatkowski J. L. Aydinok Y. (2022). Thalassaemia. Lancet399 (10343), 2310–2324. 10.1016/S0140-6736(22)00536-0
110
Kavyasudha C. Macrin D. ArulJothi K. N. Joseph J. P. Harishankar M. K. Devi A. (2018). Clinical applications of induced pluripotent stem cells - Stato attuale. Adv. Exp. Med. Biol.1079, 127–149. 10.1007/5584_2018_173
111
Kayama A. Eto K. (2024). Mass production of iPSC-derived platelets toward the clinical application. Regen. Ther.25, 213–219. 10.1016/j.reth.2023.12.009
112
Kazemian P. Yu S. Y. Thomson S. B. Birkenshaw A. Leavitt B. R. Ross C. J. D. (2022). Lipid-nanoparticle-based delivery of CRISPR/Cas9 genome-editing components. Mol. Pharm.19 (6), 1669–1686. 10.1021/acs.molpharmaceut.1c00916
113
Khan I. Shaikh H. (2023). “Beta thalassemia major (Cooley Anemia),” in StatPearls. Treasure Island, FL: StatPearls Publishing LLC.
114
Khiabani A. Kohansal M. H. Keshavarzi A. Shahraki H. Kooshesh M. Karimzade M. et al (2023). CRISPR/Cas9, a promising approach for the treatment of β-thalassemia: a systematic review. Mol. Genet. Genomics298 (1), 1–11. 10.1007/s00438-022-01978-z
115
Kim J. H. Jo H. Y. Ha H. Y. Kim Y. O. (2021). Korea national stem cell bank. Stem Cell Res.53, 102270. 10.1016/j.scr.2021.102270
116
Kim M. Jang H. J. Baek S. Y. Choi K. J. Han D. H. Sung J. S. (2023). Regulation of base excision repair during adipogenesis and osteogenesis of bone marrow-derived mesenchymal stem cells. Sci. Rep.13 (1), 16384. 10.1038/s41598-023-43737-z
117
Kindwall-Keller T. L. Ballen K. K. (2020). Umbilical cord blood: the promise and the uncertainty. Stem Cells Transl. Med.9 (10), 1153–1162. 10.1002/sctm.19-0288
118
Kleinstiver B. P. Prew M. S. Tsai S. Q. Topkar V. V. Nguyen N. T. Zheng Z. et al (2015). Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature523 (7561), 481–485. 10.1038/nature14592
119
Kleinstiver B. P. Pattanayak V. Prew M. S. Tsai S. Q. Nguyen N. T. Zheng Z. et al (2016). High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 529(7587), 490–495. 10.1038/nature16526
120
Komor A. C. Kim Y. B. Packer M. S. Zuris J. A. Liu D. R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature533 (7603), 420–424. 10.1038/nature17946
121
Kolf C. M. Cho E. Tuan R. S. (2007). Mesenchymal stromal cells. Biology of adult mesenchymal stem cells: regulation of niche, self-renewal and differentiation. Arthritis Res. Ther.9(1), 204. 10.1186/ar2116
122
Körbling M. Freireich E. J. (2011). Twenty-five years of peripheral blood stem cell transplantation. Blood117 (24), 6411–6416. 10.1182/blood-2010-12-322214
123
Kuca-Warnawin E. Plebańczyk M. Bonek K. Kontny E. (2021). Inhibition of allogeneic and autologous T cell proliferation by adipose-derived mesenchymal stem cells of ankylosing spondylitis patients. Stem Cells Int.2021, 6637328. 10.1155/2021/6637328
124
Kuci Z. Jordan C. Wehner S. Sörensen J. Jarisch A. Salzmann-Manrique E. et al (2020). The phenotype and functional activity of mesenchymal stromal cells in pediatric patients with non-malignant hematological diseases. Cells9 (2), 431. 10.3390/cells9020431
125
Langer A. L. Adam M. P. Feldman J. Mirzaa G. M. Pagon R. A. Wallace S. E. et al (1993). “Beta-thalassemia,” in GeneReviews (®). Editor AdamM. P., Seattle, WA: University of Washington.
126
Lapillonne H. Kobari L. Mazurier C. Tropel P. Giarratana M. C. Zanella-Cleon I. et al (2010). Red blood cell generation from human induced pluripotent stem cells: perspectives for transfusion medicine. Haematologica95 (10), 1651–1659. 10.3324/haematol.2010.023556
127
Ledford H. (2020). CRISPR gene editing in human embryos wreaks chromosomal mayhem. Nature583 (7814), 17–18. 10.1038/d41586-020-01906-4
128
Lee H. Son M. Y. (2021). Current challenges associated with the use of human induced pluripotent stem cell-derived organoids in regenerative medicine. Int. J. Stem Cells14 (1), 9–20. 10.15283/ijsc20140
129
Levy O. Kuai R. Siren E. M. J. Bhere D. Milton Y. Nissar N. et al (2020). Shattering barriers toward clinically meaningful MSC therapies. Sci. Adv.6 (30), eaba6884. 10.1126/sciadv.aba6884
130
Li L. Hu S. Chen X. (2018). Non-viral delivery systems for CRISPR/Cas9-based genome editing: challenges and opportunities. Biomaterials171, 207–218. 10.1016/j.biomaterials.2018.04.031
131
Li L. Yi H. Liu Z. Long P. Pan T. Huang Y. et al (2022a). Genetic correction of concurrent α- and β-thalassemia patient-derived pluripotent stem cells by the CRISPR-Cas9 technology. Stem Cell Res. Ther.13 (1), 102. 10.1186/s13287-022-02768-5
132
Li T. Yang Y. Qi H. Cui W. Zhang L. Fu X. et al (2023). CRISPR/Cas9 therapeutics: progress and prospects. Signal Transduct. Target Ther.8 (1), 36. 10.1038/s41392-023-01309-7
133
Li Y. Hao J. Hu Z. Yang Y. G. Zhou Q. Sun L. et al (2022b). Current status of clinical trials assessing mesenchymal stem cell therapy for graft versus host disease: a systematic review. Stem Cell Res. Ther.13 (1), 93. 10.1186/s13287-022-02751-0
134
Li Z. Yang L. (2023). Current status of producing autologous hematopoietic stem cells. Curr. Res. Transl. Med.71 (1), 103377. 10.1016/j.retram.2023.103377
135
Li Z. Yao X. Zhang J. Yang J. Ni J. Wang Y. (2024). Exploring the bone marrow micro environment in thalassemia patients: potential therapeutic alternatives. Front. Immunol.15, 1403458. 10.3389/fimmu.2024.1403458
136
Lidonnici M. R. Scaramuzza S. Ferrari G. (2023). Gene therapy for hemoglobinopathies. Hum. Gene Ther.34 (17-18), 793–807. 10.1089/hum.2023.138
137
Lino C. A. Harper J. C. Carney J. P. Timlin J. A. (2018). Delivering CRISPR: a review of the challenges and approaches. Drug Deliv.25 (1), 1234–1257. 10.1080/10717544.2018.1474964
138
Liu S. Shi J. Lin Y. Luo H. Wu Y. Yan J. et al (2024). A sandwich-type dual-mode biosensor based on graphdiyne and DNA nanoframework for ultra-sensitive detection of CD142 gene. Biosens. Bioelectron.248, 115962. 10.1016/j.bios.2023.115962
139
Lo B. Parham L. (2009). Ethical issues in stem cell research. Endocr. Rev.30(3):204–213. 10.1210/er.2008-0031
140
Lotfi M. Morshedi Rad D. Mashhadi S. S. Ashouri A. Mojarrad M. Mozaffari-Jovin S. et al (2023). Recent advances in CRISPR/Cas9 delivery approaches for therapeutic gene editing of stem cells. Stem Cell. Rev. Rep.19(8), 2576–2596. 10.1007/s12015-023-10585-3
141
Mahmoud H. K. Elhaddad A. M. Fahmy O. A. Samra M. A. Abdelfattah R. M. El-Nahass Y. H. et al (2015). Allogeneic hematopoietic stem cell transplantation for non-malignant hematological disorders. J. Adv. Res.6 (3), 449–458. 10.1016/j.jare.2014.11.001
142
Main H. Hedenskog M. Acharya G. Hovatta O. Lanner F. (2020). Karolinska institutet human embryonic stem cell bank. Stem Cell Res.45, 101810. 10.1016/j.scr.2020.101810
143
Makis A. Voskaridou E. Papassotiriou I. Hatzimichael E. (2021). Novel therapeutic advances in β-Thalassemia. Biol. (Basel)10 (6), 546. 10.3390/biology10060546
144
Malik V. Wang J. (2022). Pursuing totipotency: authentic totipotent stem cells in culture. Trends Genet.38 (7), 632–636. 10.1016/j.tig.2022.03.012
145
Mansouri N. Willis G. R. Fernandez-Gonzalez A. Reis M. Nassiri S. Mitsialis S. A. et al (2019). Mesenchymal stromal cell exosomes prevent and revert experimental pulmonary fibrosis through modulation of monocyte phenotypes. JCI Insight4 (21), e128060. 10.1172/jci.insight.128060
146
Marziali M. Isgrò A. Gaziev J. Lucarelli G. Nassiri S. Mitsialis S. A. et al (2009). Hematopoietic stem cell transplantation in thalassemia and sickle cell disease. Unicenter experience in a multi-ethnic population. Mediterr J Hematol Infect Dis1 (1), e2009027. 10.4084/MJHID.2009.027
147
Maurissen T. L. Woltjen K. (2020). Synergistic gene editing in human iPS cells via cell cycle and DNA repair modulation. Nat Commun11 (1), 2876. 10.1038/s41467-020-16643-5
148
Mattis V. B. Svendsen C. N. (2011). Induced pluripotent stem cells: a new revolution for clinical neurology?Lancet Neurology10 (4), 383–394. 10.1016/S1474-4422(11)70022-9
149
McTague A. Rossignoli G. Ferrini A. Barral S. Kurian M. A. (2021). Genome editing in iPSC-Based neural systems: from disease models to future therapeutic strategies. Front. Genome3, 630600. 10.3389/fgeed.2021.630600
150
Meng X. Leslie P. Zhang Y. Dong J. (2014). Stem cells in a three-dimensional scaffold environment. SpringerPlus3, 80. 10.1186/2193-1801-3-80
151
Mettananda S. Gibbons R. J. Higgs D. R. (2015). α-Globin as a molecular target in the treatment of β-thalassemia. Blood125 (24), 3694–3701. 10.1182/blood-2015-03-633594
152
Mousaei Ghasroldasht M. Seok J. Park H. S. Liakath Ali F. B. Al-Hendy A. (2022). Stem cell therapy: from idea to clinical practice. Int. J. Mol. Sci.23 (5), 2850. 10.3390/ijms23052850
153
Mousavi E. Khosravi A. Sedigh S. S. Mayanei S. A. T. Banakar M. Karimzadeh M. et al (2023). Exosomes derived from mesenchymal stem cells: heralding a new treatment for periodontitis?Tissue Cell82, 102070. 10.1016/j.tice.2023.102070
154
Musallam K. M. Cappellini M. D. Coates T. D. Kuo K. H. M. Al-Samkari H. Sheth S. et al (2024). Αlpha-thalassemia: a practical overview. Blood Rev.64, 101165. 10.1016/j.blre.2023.101165
155
Mushahary D. Spittler A. Kasper C. Weber V. Charwat V. (2018). Isolation, cultivation, and characterization of human mesenchymal stem cells. Cytom. A93 (1), 19–31. 10.1002/cyto.a.23242
156
Musharraf S. G. Iqbal A. Ansari S. H. Parveen S. Khan I. A. Siddiqui A. J. (2017). β-Thalassemia patients revealed a significant change of untargeted metabolites in comparison to healthy individuals. Sci. Rep.7, 42249. 10.1038/srep42249
157
Muthu S. Jeyaraman M. Kotner M. B. Jeyaraman N. Rajendran R. L. Sharma S. et al (2022). Evolution of mesenchymal stem cell therapy as an advanced therapeutic medicinal product (ATMP)-an Indian perspective. Bioengineering (Basel)9 (3), 111. 10.3390/bioengineering9030111
158
Naeem M. Majeed S. Hoque M. Z. Ahmad I. (2020). Latest developed strategies to minimize the off-target effects in CRISPR-Cas-Mediated genome editing. Cells9 (7), 1608. 10.3390/cells9071608
159
Naiisseh B. Papasavva P. L. Papaioannou N. Y. Tomazou M. Koniali L. Felekis X. et al (2024). Context base editing for splice correction of IVSI-110 β-thalassemia. Mol. Ther. Nucleic Acids35 (2), 102183. 10.1016/j.omtn.2024.102183
160
Naso M. F. Tomkowicz B. Perry W. L. Strohl W. R. (2017). Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs31 (4), 317–334. 10.1007/s40259-017-0234-5
161
Needs T. Gonzalez-Mosquera L. F. Lynch D. T. (2024). “Beta thalassemia,” in StatPearls. Treasure Island, FL: StatPearls Publishing LLC.
162
Ng E. S. Sarila G. Li J. Y. Edirisinghe H. S. Saxena R. Sun S. et al (2024). Long-term engrafting multilineage hematopoietic cells differentiated from human induced pluripotent stem cells. Nat Biotechnol.10.1038/s41587-024-02360-7
163
Niu X. He W. Song B. Ou Z. Fan D. Chen Y. et al (2016). Combining single strand oligodeoxynucleotides and CRISPR/Cas9 to correct gene mutations in β-Thalassemia-induced pluripotent stem cells. J. Biol. Chem.291 (32), 16576–16585. 10.1074/jbc.M116.719237
164
Oguro H. (2019). Generation of Hematopoietic Stem and Progenitor Cells from Human Pluripotent Stem Cells. Methods Mol Biol.2048, 245–257. 10.1007/978-1-4939-9728-2_19
165
Oikonomopoulou C. Goussetis E. (2021). HSCT remains the only cure for patients with transfusion-dependent thalassemia until gene therapy strategies are proven to be safe. Bone Marrow Transpl.56 (12), 2882–2888. 10.1038/s41409-021-01461-0
166
O’Reilly R. J. (1983). Allogenic bone marrow transplantation: current status and future directions. Blood62 (5), 941–964. 10.1182/blood.v62.5.941.941
167
Origa R. (2017). β-Thalassemia. Genet. Med.19 (6), 609–619. 10.1038/gim.2016.173
168
Ortuño-Costela M. C. Pinzani M. Vallier L. (2025). Cell therapy for liver disorders: past, present and future. Nat. Rev. Gastroenterol. Hepatol.22 (5), 329–342. 10.1038/s41575-025-01050-2
169
O'Shea O. Abranches E. (2020). UK stem cell bank. Stem Cell Res.49, 102019. 10.1016/j.scr.2020.102019
170
Ottaviano G. Qasim W. (2025). Current landscape of vector safety and genotoxicity after hematopoietic stem or immune cell gene therapy. Leukemia39, 1325–1333. 10.1038/s41375-025-02585-8
171
Ou Z. Niu X. He W. Chen Y. Song B. Xian Y. et al (2016). The combination of CRISPR/Cas9 and iPSC technologies in the gene therapy of human β-thalassemia in mice. Sci. Rep.6, 32463. 10.1038/srep32463
172
Paolini Sguazzi G. Muto V. Tartaglia M. Bertini E. Compagnucci C. (2021). Induced pluripotent stem cells (iPSCs) and gene therapy: a new era for the treatment of neurological diseases. Int. J. Mol. Sci.22 (24), 13674. 10.3390/ijms222413674
173
Papastamataki M. Delaporta P. Premetis E. Kattamis A. Ladis V. Papassotiriou I. (2010). Evaluation of liver fibrosis in patients with thalassemia: the important role of hyaluronic acid. Blood Cells, Mol. Dis.45 (3), 215–218. 10.1016/j.bcmd.2010.06.002
174
Parikh M. C. (2025). Gene editing: developments, ethical considerations, and future directions. J. Community Hosp. Intern Med. Perspect.15(1), 1–4. 10.55729/2000-9666.1445
175
Park A. Hong P. Won S. T. Thibault P. A. Vigant F. Oguntuyo K. Y. et al (2016). Sendai virus, an RNA virus with no risk of genomic integration, delivers CRISPR/Cas9 for efficient gene editing. Mol. Ther. Methods Clin. Dev.3, 16057. 10.1038/mtm.2016.57
176
Park S. H. Bao G. (2021). CRISPR/Cas9 gene editing for curing sickle cell disease. Transfus. Apher. Sci.60 (1), 103060. 10.1016/j.transci.2021.103060
177
Poetsch M. S. Strano A. Guan K. (2022). Human induced pluripotent stem cells: from cell origin, genomic stability, and epigenetic memory to translational medicine. Stem Cells40 (6), 546–555. 10.1093/stmcls/sxac020
178
Rahman M. S. Qi G. Li Q. Liu X. Bai J. Chen M. et al (2025). Three-Dimensional trilineage differentiation conditions for human induced pluripotent stem cells. Bioeng. (Basel). 12(5), 503. 10.3390/bioengineering12050503
179
Rees H. A. Liu D. R. (2018). Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet.19 (12), 770–788. 10.1038/s41576-018-0059-1
180
Rivella S. (2019). Iron metabolism under conditions of ineffective erythropoiesis in β-thalassemia. Blood133 (1), 51–58. 10.1182/blood-2018-07-815928
181
Rostami T. Kasaeian A. Maleki N. Nikbakht M. Kiumarsi A. Tavangar S. M. et al (2021). The effect of bone marrow-derived mesenchymal stem cell co-transplantation with hematopoietic stem cells on liver fibrosis alleviation and survival in patients with class III β-thalassemia major. Stem Cell Res. Ther.12 (1), 213. 10.1186/s13287-021-02242-8
182
Rowe R. G. Daley G. Q. (2019). Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet.20 (7), 377–388. 10.1038/s41576-019-0100-z
183
Rowe R. G. Mandelbaum J. Zon L. I. Daley G. Q. (2016). Engineering hematopoietic stem cells: lessons from development. Cell Stem Cell18 (6), 707–720. 10.1016/j.stem.2016.05.016
184
Rudnitsky E. Braiman A. Wolfson M. Muradian K. K. Gorbunova V. Turgeman G. et al (2025). Mesenchymal stem cells and their derivatives as potential longevity-promoting tools. Biogerontology26 (3), 96. 10.1007/s10522-025-10240-z
185
Szaraz P. Gratch Y. S. Iqbal F. Librach C. L. (2017). In vitro differentiation of human mesenchymal stem cells into functional cardiomyocyte-like cells. J. Vis. Exp. (126), 55757. 10.3791/55757
186
Saleemi S. (2014). Saudi guidelines on the diagnosis and treatment of pulmonary hypertension: pulmonary hypertension associated with hemolytic anemia. Ann. Thorac. Med.9 (Suppl. 1), S67–S73. 10.4103/1817-1737.134039
187
Sanchez-Petitto G. Rezvani K. Daher M. Rafei H. Kebriaei P. Shpall E. J. et al (2023). Umbilical cord blood transplantation: connecting its origin to its future. Stem Cells Transl. Med.12 (2), 55–71. 10.1093/stcltm/szac086
188
Santarone S. Angelini S. Natale A. Vaddinelli D. Spadano R. Casciani P. et al (2022). Survival and late effects of hematopoietic cell transplantation in patients with thalassemia major. Bone Marrow Transpl.57 (11), 1689–1697. 10.1038/s41409-022-01786-4
189
Sarvari M. Alavi-Moghadam S. Aghayan H. R. Tayanloo-Beik A. Payab M. Tootee A. et al (2021). Stem cells researches and therapies towards endocrine diseases treatment; strategies, challenges, and opportunities. J. Diabetes Metab. Disord.23(2), 1461–1467. 10.1007/s40200-020-00674-2
190
Seijas A. Cora D. Novo M. Al-Soufi W. Sánchez L. Arana Á. J. (2025). CRISPR/Cas9 delivery systems to enhance gene editing efficiency. Int. J. Mol. Sci.26(9), 4420. 10.3390/ijms26094420
191
Shang Y. Guan H. Zhou F. (2021). Biological characteristics of umbilical cord mesenchymal stem cells and its therapeutic potential for hematological disorders. Front. Cell Dev. Biol.9, 570179. 10.3389/fcell.2021.570179
192
Sharma A. Jagannath VA. Puri L. (2021). Hematopoietic stem cell transplantation for people with β-thalassaemia. Cochrane Database Syst. Rev.4(4), CD008708. 10.1002/14651858.CD008708.pub5
193
Simpson E. Dazzi F. (2019). Bone marrow transplantation 1957-2019. Front. Immunol.10, 1246. 10.3389/fimmu.2019.01246
194
Sinclair F. Begum A. A. Dai C. C. Toth I. Moyle P. M. (2023). Recent advances in the delivery and applications of nonviral CRISPR/Cas9 gene editing. Drug Deliv. Transl. Res.13 (5), 1500–1519. 10.1007/s13346-023-01320-z
195
Singh A. Babu S. Phan M. Yuan S. H. (2024). CRISPR/Cas9-Based protocol for precise genome editing in induced pluripotent stem cells. Bio Protoc.14(24), e5141. 10.21769/BioProtoc.5141
196
Sirinoglu Demiriz I. Tekgunduz E. Altuntas F. (2012). What is the most appropriate source for hematopoietic stem cell transplantation? Peripheral stem cell/bone marrow/cord blood. Bone Marrow Res.2012, 834040. 10.1155/2012/834040
197
Song B. Fan Y. He W. Zhu D. Niu X. Wang D. et al (2015). Improved hematopoietic differentiation efficiency of gene-corrected beta-thalassemia induced pluripotent stem cells by CRISPR/Cas9 system. Stem Cells Dev.24 (9), 1053–1065. 10.1089/scd.2014.0347
198
Song C. Wang L. Li Q. Liao B. Qiao W. Dong N. et al (2022). Generation of individualized immunocompatible endothelial cells from HLA-I-matched human pluripotent stem cells. Stem Cell Res Ther.13 (1), 48. 10.1186/s13287-022-02720-7
199
Song J. Ma Q. Li Y. Wang X. Chen S. Liang B. et al (2024). CD317(+) MSCs expanded with chemically defined media have enhanced immunological anti-inflammatory activities. Stem Cell Res. Ther.15 (1), 2. 10.1186/s13287-023-03618-8
200
Sprink T. Wilhelm R. Hartung F. (2022). Genome editing around the globe: an update on policies and perceptions. Plant Physiol.190 (3), 1579–1587. 10.1093/plphys/kiac359
201
Srivastava A. Shaji R. V. (2017). Cure for thalassemia major - from allogeneic hematopoietic stem cell transplantation to gene therapy. Haematologica102 (2), 214–223. 10.3324/haematol.2015.141200
202
Stacey G. N. Healy L. (2021). The International Stem Cell Banking Initiative (ISCBI). Stem Cell Res.53, 102265. 10.1016/j.scr.2021.102265
203
Su Z. Dong H. Fang X. Zhang W. Duan H. (2025). Frontier progress and translational challenges of pluripotent differentiation of stem cells. Front. Genet.16, 1583391. 10.3389/fgene.2025.1583391
204
Taghdiri M. Mussolino C. (2024). Viral and non-viral systems to deliver gene therapeutics to clinical targets. Int. J. Mol. Sci.25 (13), 7333. 10.3390/ijms25137333
205
Taher A. T. Musallam K. M. Cappellini M. D. (2021). β-Thalassemias. N. Engl. J. Med.384 (8), 727–743. 10.1056/NEJMra2021838
206
Takahashi K. Yamanaka S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell126 (4), 663–676. 10.1016/j.cell.2006.07.024
207
Tamary H. Dgany O. Adam M. P. Feldman J. Mirzaa G. M. Pagon R. A. et al (1993). “Alpha-thalassemia,” in GeneReviews (®). Editor AdamM. P., Seattle, WA: University of Washington).
208
Tenuta M. Cangiano B. Rastrelli G. Carlomagno F. Sciarra F. Sansone A. et al (2024). Iron overload disorders: growth and gonadal dysfunction in childhood and adolescence. Pediatr. Blood Cancer71 (7), e30995. 10.1002/pbc.30995
209
Terai S. Ezoe S. Mano K. Matsuzaki Y. Sato Y. Yamahara K. et al (2025). Recommendations for the safe implementation of intravenous administration of mesenchymal stromal cells. Regen. Ther.29, 171–176. 10.1016/j.reth.2025.01.024
210
Tesio N. Bauer D. E. (2023). Molecular basis and genetic modifiers of thalassemia. Hematol. Oncol. Clin. North Am.37 (2), 273–299. 10.1016/j.hoc.2022.12.001
211
Thiagarajan P. Parker C. J. Prchal J. T. (2023). How Do Red Blood Cells Die?. Front Physiol.12, 655393. 10.3389/fphys.2021.655393
212
Tian Z. Yu T. Liu J. Wang T. Higuchi A. (2023). Introduction to stem cells. Prog. Mol. Biol. Transl. Sci.199, 3–32. 10.1016/bs.pmbts.2023.02.012
213
Tong S. Moyo B. Lee C. M. Leong K. Bao G. (2019). Engineered materials for in vivo delivery of genome-editing machinery. Nat. Rev. Mater4, 726–737. 10.1038/s41578-019-0145-9
214
Traeger-Synodinos J. Vrettou C. Sofocleous C. Zurlo M. Finotti A. Gambari R. et al (2024). Impact of α-Globin gene expression and α-Globin modifiers on the phenotype of β-Thalassemia and other hemoglobinopathies: implications for patient management. Int. J. Mol. Sci.25 (6), 3400. 10.3390/ijms25063400
215
Tran D. C. Dang A. L. Hoang T. N. L. Nguyen C. T. Le T. M. P. Dinh T. N. M. et al (2023). Prevalence of thalassemia in the Vietnamese population and building a clinical decision support system for prenatal screening for thalassemia. Mediterr. J. Hematol. Infect. Dis.15 (1), e2023026. 10.4084/MJHID.2023.026
216
Um S. Ha J. Choi S. J. Oh W. Jin H. J. (2020). Prospects for the therapeutic development of umbilical cord blood-derived mesenchymal stem cells. World J. Stem Cells12 (12), 1511–1528. 10.4252/wjsc.v12.i12.1511
217
Večerić-Haler Ž. Sever M. Kojc N. Halloran P. F. Boštjančič E. Mlinšek G. et al (2022). Autologous mesenchymal stem cells for treatment of chronic active antibody-mediated kidney graft rejection: report of the phase I/II clinical trial case series. Transpl. Int.35, 10772. 10.3389/ti.2022.10772
218
Velikova T. Dekova T. Miteva D. G. (2024). Controversies regarding transplantation of mesenchymal stem cells. World J. Transpl.14(2), 90554. 10.5500/wjt.v14.i2.90554
219
Venou T. M. Barmpageorgopoulou F. Peppa M. Vlachaki E. (2024). Endocrinopathies in beta thalassemia: a narrative review. Horm. (Athens)23 (2), 205–216. 10.1007/s42000-023-00515-w
220
Vento S. Cainelli F. Cesario F. (2006). Infections and thalassaemia. Lancet Infect. Dis.6 (4), 226–233. 10.1016/S1473-3099(06)70437-6
221
Wang J. Li R. (2024). Effects, methods and limits of the cryopreservation on mesenchymal stem cells. Stem Cell Res. & Ther.15 (1), 337. 10.1186/s13287-024-03954-3
222
Wang K. C. Zheng T. Hubbard B. P. (2025b). CRISPR/Cas technologies for cancer drug discovery and treatment. Trends Pharmacol. Sci.46 (5), 437–452. 10.1016/j.tips.2025.02.009
223
Wang W. D. Hu F. Zhou D. H. Gale R. P. Lai Y. R. Yao H. X. et al (2023). Thalassaemia in China. Blood Rev.60, 101074. 10.1016/j.blre.2023.101074
224
Wattanapanitch M. (2019). Recent Updates on Induced Pluripotent Stem Cells in Hematological Disorders. Stem Cells Int. , 5171032. 10.1155/2019/5171032
225
Wattanapanitch M. (2021). Correction of hemoglobin E/Beta-Thalassemia patient-derived iPSCs using CRISPR/Cas9. Methods Mol. Biol.2211, 193–211. 10.1007/978-1-0716-0943-9_14
226
Wattanapanitch M. Damkham N. Potirat P. Trakarnsanga K. Janan M. U-Pratya Y. et al (2018). One-step genetic correction of hemoglobin E/beta-thalassemia patient-derived iPSCs by the CRISPR/Cas9 system. Stem Cell Res. Ther.9 (1), 46. 10.1186/s13287-018-0779-3
227
Wei L. Yan W. Shah W. Zhang Z. Wang M. Liu B. et al (2024). Advancements and challenges in stem cell transplantation for regenerative medicine. Heliyon10 (16), e35836. 10.1016/j.heliyon.2024.e35836
228
Wen D. Wang J. (2025). Totipotency or plenipotency: rethinking stem cell bipotentiality. Curr. Opin. Genet. Dev.92, 102342. 10.1016/j.gde.2025.102342
229
Wen Y. Li J. Mukama O. Huang R. Deng S. Li Z. (2025). New insights on mesenchymal stem cells therapy from the perspective of the pathogenesis of nonalcoholic fatty liver disease. Dig. Liver Dis.57, 1107–1118. 10.1016/j.dld.2025.03.007
230
Wiewiórska-Krata N. Foroncewicz B. Mucha K. Zagożdżon R. (2025). Cell therapies for immune-mediated disorders. Front. Med. (Lausanne)12, 1550527. 10.3389/fmed.2025.1550527
231
Wiley L. Cheek M. LaFar E. Ma X. Sekowski J. Tanguturi N. et al (2025). The ethics of human embryo editing via CRISPR-Cas9 technology: a systematic review of ethical arguments, reasons, and concerns. HEC Forum37 (2), 267–303. 10.1007/s10730-024-09538-1
232
Wilkinson A. C. Dever D. P. Baik R. Camarena J. Hsu I. Charlesworth C. T. et al (2021). Cas9-AAV6 gene correction of beta-globin in autologous HSCs improves sickle cell disease erythropoiesis in mice. Nat. Commun.12 (1), 686. 10.1038/s41467-021-20909-x
233
Williams D. A. Kohn D. B. Thrasher A. J. (2025). Ex vivo modification of hematopoietic stem and progenitor cells for gene therapy. Mol. Ther.33 (5), 2141–2153. 10.1016/j.ymthe.2025.03.058
234
Winger B. A. Ajayi A. Vichinsky E. (2025). Diagnosis and treatment of alpha thalassemia major. Hemoglobin49 (1), 3–9. 10.1080/03630269.2024.2432899
235
Wood J. C. (2023). Cardiac complications in thalassemia throughout the lifespan: victories and challenges. Ann. N. Y. Acad. Sci.1530 (1), 64–73. 10.1111/nyas.15078
236
Wu S. C. M. Zhu M. Chik S. C. C. Kwok M. Javed A. Law L. et al (2023). Adipose tissue-derived human mesenchymal stromal cells can better suppress complement lysis, engraft and inhibit acute graft-versus-host disease in mice. Stem Cell Res. Ther.14 (1), 167. 10.1186/s13287-023-03380-x
237
Wyvekens N. Topkar V. V. Khayter C. Joung J. K. Tsai S. Q. (2015). Dimeric CRISPR RNA-guided FokI-dCas9 nucleases directed by truncated gRNAs for highly specific genome editing. Hum. Gene Ther.26 (7), 425–431. 10.1089/hum.2015.084
238
Xie F. Ye L. Chang J. C. Beyer A. I. Wang J. Muench M. O. et al (2014). Seamless gene correction of β-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res.24 (9), 1526–1533. 10.1101/gr.173427.114
239
Xu P. Tong Y. Liu X. z. Wang T. t. Cheng L. Wang B. y. et al (2015). Both TALENs and CRISPR/Cas9 directly target the HBB IVS2-654 (C > T) mutation in β-thalassemia-derived iPSCs. Sci. Rep.5, 12065. 10.1038/srep12065
240
Xu H. Wang B. Ono M. Kagita A. Fujii K. Sasakawa N. et al (2019). Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility. Cell Stem Cell.24(4), 566–578. 10.1016/j.stem.2019.02.005
241
Yamanaka S. (2020). Pluripotent stem cell-based cell therapy-promise and challenges. Cell Stem Cell27 (4), 523–531. 10.1016/j.stem.2020.09.014
242
Yang X. Meng Y. Han Z. Ye F. Wei L. Zong C. (2020). Mesenchymal stem cell therapy for liver disease: full of chances and challenges. Cell Biosci.10, 123. 10.1186/s13578-020-00480-6
243
Ye L. Chang J. C. Lin C. Sun X. Yu J. Kan Y. W. (2009). Induced pluripotent stem cells offer new approach to therapy in thalassemia and sickle cell anemia and option in prenatal diagnosis in genetic diseases. Proc. Natl. Acad. Sci. U. S. A.106 (24), 9826–9830. 10.1073/pnas.0904689106
244
Yin S. Zhang M. Liu Y. Sun X. Guan Y. Chen X. et al (2023). Engineering of efficiency-enhanced Cas9 and base editors with improved gene therapy efficacies. Mol. Ther.31(3), 744–759. 10.1016/j.ymthe.2022.11.014
245
Yong J. Tao J. Wang K. Li X. Yang Y. (2025). Post-myocardial infarction cardiac remodeling: multidimensional mechanisms and clinical prospects of stem cell therapy. Stem Cell Rev. Rep.10.1007/s12015-025-10888-7
246
Yousuf R. Akter S. Wasek S. M. Sinha S. Ahmad R. Haque M. (2022). Thalassemia: a review of the challenges to the families and caregivers. Cureus14 (12), e32491. 10.7759/cureus.32491
247
Zakaria N. A. Bahar R. Abdullah W. Z. Mohamed Yusoff A. A. Shamsuddin S. Abdul Wahab R. et al (2022). Genetic manipulation strategies for β-Thalassemia: a review. Front. Pediatr.10, 901605. 10.3389/fped.2022.901605
248
Zeng S. Lei S. Qu C. Wang Y. Teng S. Huang P. (2023). CRISPR/Cas-based gene editing in therapeutic strategies for beta-thalassemia. Hum. Genet.142 (12), 1677–1703. 10.1007/s00439-023-02610-9
249
Zhang Y. Gao S. Xia J. Liu F. (2018). Hematopoietic hierarchy - an updated roadmap. Trends Cell Biol.28 (12), 976–986. 10.1016/j.tcb.2018.06.001
250
Zhu X. Tang B. Sun Z. (2021a). Umbilical cord blood transplantation: still growing and improving. Stem Cells Transl. Med.10 (Suppl. 2), S62–s74. 10.1002/sctm.20-0495
251
Zuo Z. Babu K. Ganguly C. Zolekar A. Newsom S. Rajan R. et al (2022). Rational engineering of CRISPR-Cas9 nuclease to attenuate position-dependent off-target effects. Crispr J.5 (2), 329–340. 10.1089/crispr.2021.0076
Summary
Keywords
thalassemia, gene therapy, iPSC, MSC, complication
Citation
Shu J, Xie X, Wang S, Du Z, Huang P, Chen Y and He Z (2025) CRISPR/Cas-edited iPSCs and mesenchymal stem cells: a concise review of their potential in thalassemia therapy. Front. Cell Dev. Biol. 13:1595897. doi: 10.3389/fcell.2025.1595897
Received
18 March 2025
Accepted
24 June 2025
Published
03 September 2025
Volume
13 - 2025
Edited by
Valerie Kouskoff, The University of Manchester, United Kingdom
Reviewed by
Rossella Labella, Columbia University, United States
Naseem Ahamad, The University of Texas Health Science Center at San Antonio, United States
Updates
Copyright
© 2025 Shu, Xie, Wang, Du, Huang, Chen and He.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Chen, cyz600@163.com; Zhixu He, hzx@gmc.edu.cn
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.