 Keke Mao
Keke Mao Haifeng Lv
Haifeng Lv Xiuling Li3
Xiuling Li3- 1School of Energy and Environment Science, Anhui University of Technology, Maanshan, China
- 2School of Chemistry and Materials Sciences, University of Science and Technology of China, Hefei, China
- 3School of Physical Science and Technology, Nanjing Normal University, Nanjing, China
Perfect boron nitride (BN) nanotubes are chemically inert, and hardly considered as catalysts. Nevertheless, metal wire encapsulated BN nanotubes show extraordinarily high chemical activity. We report nickel (Ni) nanowire encapsulated BN(8.0) and BN(9.0) nanotubes toward O2 activation and CO oxidization on the basis of first-principles calculations. Our results suggest that Ni wire encapsulated BN(8.0) and BN(9.0) nanotubes can easily adsorb and activate O2 molecules to form peroxo or superoxo species exothermically. Meanwhile, superoxo species are ready to react with CO molecules forming OCOO intermediate state and finally yielding CO2 molecules. Meanwhile, the rate-limiting step barrier is only 0.637 eV, implying excellent performance for CO oxidation on Ni nanowire encapsulated BN nanotubes. Furthermore, encapsulation of nickel wire improves the catalytic activity of BN nanotubes by facilitating electron transfer from Ni wire to BN nanotubes, which facilitates the adsorption of highly electronegative O2 molecules and subsequent CO oxidation. This study provides a practical and efficient strategy for activating O2 on a metal encapsulated BN nanotube toward CO oxidation.
Introduction
Low-dimensional materials have attracted a great attention for their exhibited excellent, novel properties and a wide range of potential applications (Golberg et al., 2007). The most well-known materials are two-dimensional carbon-based graphene (Geim and Novoselov, 2007) and one-dimensional carbon nanotube (Baughman et al., 2002). Boron nitride nanotube (Chopra et al., 1995; Golberg et al., 2010) is an isoelectronic analogue of carbon nanotube with boron and nitrogen alternately replacing carbon and presents ultrahigh mechanical stability as well as chemical inertness (Golberg et al., 2010) similar to carbon nanotube, but entirely different electronic properties. Carbon nanotubes (CNTs) could be metallic or semiconducting, which depends on helicities (Baughman et al., 2002). In contrast, BN nanotubes (BNNTs) are insulators independent of their chirality (Blase et al., 1994; Rubio et al., 1994) and more chemically inert (Chen Y. et al., 2004; Golberg et al., 2007) than CNTs, which render them candidates to serve as protective shields for encapsulating metallic clusters, nanowires, and nanorods.
To enhance the catalytic activity of BNNTs, a number of approaches have been conducted to manipulate BNNTs’ electronic properties and chemical activity, including electric field (Chen C.-W. et al., 2004; Attaccalite et al., 2007), strain (Wang et al., 2009), introducing defects (An et al., 2007), functionalization (Liu et al., 2020), or doping (Cho et al., 2009; Abdel Aal, 2016; Zhang et al., 2018; Jabarullah et al., 2019; Kim et al., 2019). Among them, doping is a very effective way to modulate electronic properties and chemical activities of BNNTs. For instance, the electronic property and chemical activity of BN nanomaterials, such as toward H2 (Baierle et al., 2006) and malononitrile (Li et al., 2018) molecules, might be greatly improved by nonmetal C doping. Moreover, transition metals, with their inherent magnetic moment and great chemical reactivity, are another type of commonly employed doping atoms in BNNTs. Transition metals’ doping could induce the change of BNNTs’ structure, magnetic properties (Wu and Zeng, 2006), electronic structure (Wu and Zeng, 2006; Li et al., 2011), and chemical reactivity (Soltani et al., 2012; Xie et al., 2012; Peyghan et al., 2013; Soltani et al., 2014; Zhang et al., 2018). Li et al. (2011) studied the electronic structure of 10 types of 3d transition metal (TM) atoms doped (8.0) zigzag single walled BNNTs and demonstrated that doping reduced band gap and affected conductivity of BNNTs. Moreover, modulation of chemical activity of BNNTs toward adsorption such as CO (Peyghan et al., 2013), NO (Xie et al., 2012), and NH3 (Soltani et al., 2012) is quite promising. For example, enhanced H2 molecules’ adsorption was observed on platinum-doped boron nitride nanotubes (Wu et al., 2006). However, the active sites are mainly limited to TM metals rather than the abundant B and N atoms.
According to previous report, BN nanomaterials could also provide active sites under modulation of metals, in which B atoms are always regarded as active sites. Wang et al. (2013) indicated that the adsorption of O2 molecules was boosted on BNNT (10.0) with ion Pd3-based transition metal clusters encapsulated inside. Moreover, Nigam and Majumder (2008) have shown that an inert (BN)36 cluster is activated by incorporating into Fe nanoparticles, and serves as catalysts for CO oxidation. Besides, our previous work (Mao et al., 2017) and other similar papers have presented that metal supports could effectively modulate chemical reactivity of h-BN monolayer toward O2 activation (Wasey et al., 2013; Lyalin et al., 2014) and ORR reaction (Lyalin et al., 2013). The above works demonstrate that metal can effectively modulate the chemical activity of BN nanomaterials, especially in oxidation.
In 2003, Tang et al. (2003) synthesized boron nitride nanotubes filled with Ni and NiSi2 nanowires, which shows potential applications in catalysis, especially in O2 molecules involved reaction. However, the in-depth catalytic mechanism remains unclear. How does the embedded metal wire affect the catalytic activity of BNNTs? What are the key factors in manipulating catalytic performance? Is it the same with a metal-supported h-BN monolayer? In this work, we have investigated nickel (Ni) wire encapsulated BN nanotubes toward O2 activation and benchmark reaction of CO oxidation. Distinct from extremely inert BNNTs, metal encapsulated BNNTs show great chemical reactivity. Firstly, the adsorption and dissociation of O2 molecules were investigated on Ni nanowire encapsulated (8.0) BNNT and (9.0) BNNT. Three types of adsorptions including physical adsorption, superoxo chemisorption, and peroxo chemisorption were observed on various active sites of Ni nanowire encapsulated (8.0) BNNT and (9.0) BNNT. CO oxidation starting from peroxo chemisorbed O2 molecule is always trapped in very stable CO3 intermediate state and starting from superoxo chemisorbed shows a promising CO oxidation pathway.
Computational Methods and Details
Spin polarized density functional theory (DFT) methods implemented in the Vienna ab initio simulation package (VASP) (Kresse and Hafner, 1993; Kresse and Furthmüller, 1996) were employed for all calculations. Perdew, Burke, and Ernzerhof (PBE) (Perdew et al., 1996) for the exchange–correlation functional of generalized gradient approximation and a plane wave cutoff of 400 eV were used. All atomic positions were fully relaxed using the conjugate gradient method. All structures were optimized within energy and force convergences of 10−4 eV/atom and 0.02 eV/Å, respectively. The Brillouin zone was sampled by using a 1 × 1 × 5 Gamma centered k-points mesh. A vacuum distance more than 15 Å perpendicular to the tube’s axial direction was imposed between two periodic units to avoid interactions between periodic images. The CO oxidation minimum energy path (MEP) and energy barrier was simulated by adopting climbing-image–nudged elastic band (CI-NEB) methods (Henkelman et al., 2000). The convergence criteria force of CI-NEB was set to be 0.04 eV/Å. The adsorption energy (Eads) of molecules (CO or O2 molecules) was calculated based on the following formula: Eads(mol*) = E(mol*) − E(*) − E(mol). The E(mol*) and E(*) are total energies of BN(8.0)/Ni or BN(9.0)/Ni with and without molecules adsorbed on. Meanwhile, E(mol) is the total energy of a gaseous CO or O2 molecule.
Results and Discussion
Our calculation model of metal wire encapsulated BNNT is based on Xiang et al.’s previous work (Xiang et al., 2005). The embedded metal wire is hcp Ni wire whose unit cell is composed of six atoms with ABAB staggered triangle packing. Single-walled zigzag (8.0) and (9.0) BNNTs are the chosen encapsulant in consideration of the diameters of BNNTs and adopted Ni wire model. Figure 1 presents the hybrid structures. In structure, three Ni atom are in the same plan with N atoms in zigzag (8.0) or (9.0) BNNTs. Xiang et al.’s work (Xiang et al., 2005) also calculated the formation energy of the hybrid structures. The values are 0.88 and −0.04 eV for BN(8.0)/Ni and BN(9.0)/Ni, respectively. Here, BN(8.0)/Ni and BN(9.0)/Ni denote Ni nanowire encapsulated (8.0) and (9.0) BNNTs, respectively. Negative formation energy implies that the formation of this hybrid structure is exothermic. Results indicate that the formation of BN(9.0)/Ni is more favorable in energy.
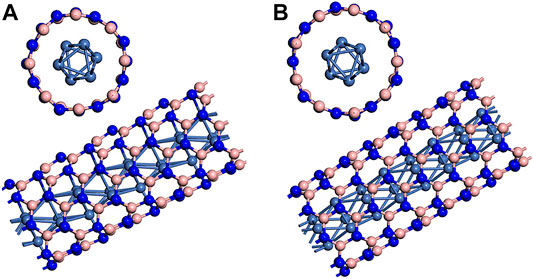
FIGURE 1. Ni nanowire encapsulated (A) (8.0) BNNT and (B) (9.0) BNNT. The pink spheres represent B atoms, the blue spheres represent N atoms, and the light blue spheres represent Ni atoms.
In order to investigate adsorption behaviors of O2 molecules on BN(8.0)/Ni and BN(9.0)/Ni, various initial adsorption structures on different adsorption sites were constructed and optimized. After optimization, three types of O2 molecules’ adsorption were observed on different adsorption sites. Here, we take BN(8.0)/Ni as an example. In type I adsorption (Supplementary Figure S1A), the O2 molecule is difficult to chemically adsorb on the site, moves away from BN(8.0)/Ni during optimization, and finally physically adsorbs on BN(8.0)/Ni. It can be seen from Supplementary Figure S1A that O–O bond length is 1.246 Å, which is very close to the bond length of 1.235 Å of a gaseous O2 molecule. This is consistent with the O2 molecule’s physical adsorption behavior. In type Ⅱ adsorption (Figure 2A, Table 1), O2 molecule superoxo chemisorbs on BN(8.0)/Ni species, with only one O atom of the O2 molecule atom chemically bonded with one B atom in the BN nanotube, as shown in Figure 2A. The adsorbed O2 molecule’s O–O bond length is elongated to 1.344 Å and the adsorption energy is −0.463 eV. The adsorbed O2 molecule possesses largely decreased local magnetic moment 0.290 and 0.523 μB, while both are 1 μB in triplet oxygen. The O atom bonded with the B atom in the BN nanotube has a lower local magnet moment, 0.290 μB. In type III adsorption (Figure 2B, Table 1), O2 molecule peroxo chemisorbs on BN(8.0)/Ni, with both two O atoms of O2 molecule bonding with B atoms in the BN nanotube, as seen in Figure 2B. The peroxo adsorbed O2 molecule has much lower adsorption energy, −0.728 eV, and much longer O–O bond length, 1.511 Å, than the superoxo chemisorbed O2 molecule’s −0.463 eV and 1.344 Å. Moreover, the peroxo adsorbed O2 molecule possesses more lower local magnetic moments of 0.008 and 0.017 μB. All these results indicate that peroxo chemisorbed O2 molecule (type III) is much more highly activated than superoxo chemisorbed O2 molecule (typeⅡ). In summary, the adsorption behaviors of O2 molecules exhibit three different adsorption types on account of different adsorption sites of BN(8.0)/Ni. Even in the same adsorption type, the adsorbed O2 molecules’ activation degree may be different. Since the adsorption sites of O2 molecules are B atoms in the BN nanotube, the activity of B atoms largely determines adsorption type and even activation degree of adsorbed O2 molecule. Our results show that B atoms’ activity is dominated by its neighboring N atoms’ distances with Ni atoms in the encapsulated Ni wire. For example, if its neighboring N atoms are on top of Ni atoms (the shortest N–Ni distance), the B atoms present the largest activity, and as a result, the adsorption type of O2 molecules are likely to be peroxo chemisorption, possessing the highest degree of activation. The adsorption behaviors of O2 molecules on metal wire encapsulated BNNTs are very different from a metal surface [Ni(111) or Cu(111)]-supported h-BN monolayer (Mao et al., 2017). On these 1 × 1 matched metal surface-supported h-BN monolayers, all the N atoms are on top of metal atoms and all the B atoms are the same, showing the same activity. Thus, the adsorbed O2 molecules exhibit the same peroxo chemisorption behavior on a metal surface-supported h-BN monolayer.
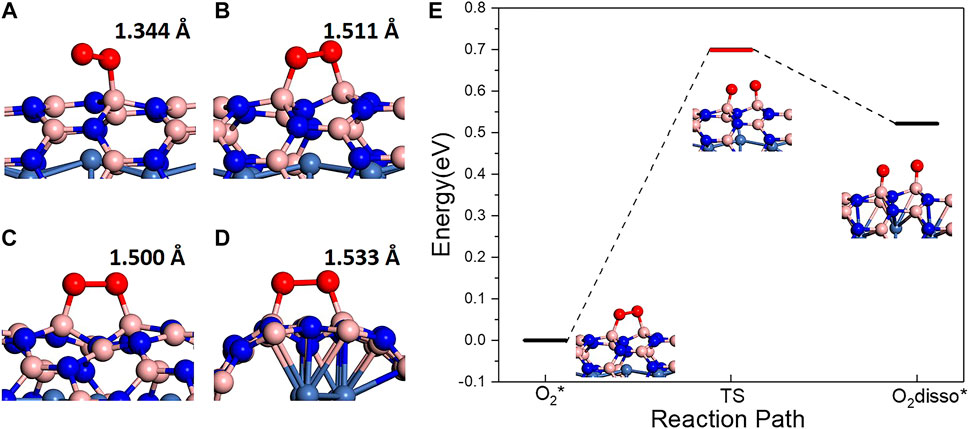
FIGURE 2. (A, B) Structures of superoxo and peroxo chemisorbed O2 molecules on BN(8.0)/Ni with the lowest adsorption energy. (C, D) Structures of peroxo chemisorbed O2 molecules on BN(9.0)/Ni with the lowest and highest adsorption energy. (E) Peroxo chemisorbed O2 molecule dissociation reaction path on BN(8.0)/Ni.
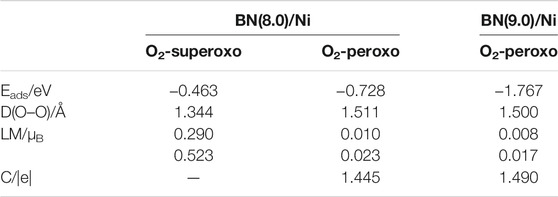
TABLE 1. The structural, electronic, and magnetic properties of adsorbed O2 molecules: superoxo chemisorbed, peroxo chemisorbed on BN(8.0)/Ni, and peroxo chemisorbed on BN(9.0)/Ni with the lowest adsorption energy. Eads is the adsorption energy. D(O–O) is the elongated O–O bonds length, LM is the local magnetic moments of two O atoms of the adsorbed O2 molecule. C is the charge transfer from BN(8.0)/Ni or BN(9.0)/Ni to adsorbed O2 molecule.
In contrast, the adsorption behaviors of O2 molecules on BN(9.0)/Ni are extraordinarily different. Firstly, we constructed 9 initial adsorption structures on different adsorption sites, in which O2 molecules are all about 2 Å away from tubes. After optimization, in eight structures, O2 molecules finally peroxo chemisorb on BN(9.0)/Ni, and in one structure, O2 molecule moved away from BN(9.0)/Ni and finally physically adsorbs on BN(9.0)/Ni (Supplementary Figure S1B). No superoxo chemisorption is observed. Interestingly, in the final physical adsorption structure, the N atom between two possible adsorption sites of B atoms is far away from encapsulated Ni atoms, and this might account for the final physical adsorption. Moreover, the degree of activation of adsorbed O2 molecules is different in the other eight O2 peroxo adsorbed structures. The adsorption energy ranges from −0.773 eV to −1.767 eV, and the O–O bond length ranges from 1.500 Å to 1.533 Å. It is worth noting that the adsorbed O2 molecule with the highest adsorption energy is the adsorbed O2 molecule perpendicular to the tube’s axial direction (Figure 2D). In this O2 adsorbed structure (Figure 2D), the adsorption energy is −0.773 eV, the elongated O–O bond length is 1.533 Å, and the formed O–B bonds length are 1.474 and 1.497 Å. Interestingly, the N atom between two active site B atoms falls down largely towards Ni atom as a result of O2 molecule’s adsorption. The N–Ni bond length changes from 2.440 Å to 1.974 Å. In the lowest adsorption energy structure (Figure 2C), the elongated O–O bond length is 1.500 Å.
Table 1 summarizes the properties of three chemisorbed O2 molecules on BN(8.0)/Ni and BN(9.0)/Ni, which have the lowest adsorption energy in superoxo chemisorption or peroxo chemisorption. It is noteworthy that the peroxo chemisorbed O2 molecule on BN(9.0)/Ni has the lowest adsorption energy of −1.767 eV, the elongated O–O bond length of 1.500 Å, and the lowest local magnetic moments of 0.008, 0.017 μB. So, the peroxo chemisorbed O2 molecule on BN(9.0)/Ni is the most highly activated in the three adsorption structures. The observed significant decrease in magnetic moments of chemisorbed O2 molecules could be attributed to charge transfer from BNNT/Ni to the adsorbed O2 molecule. Bader charge analysis indicates that the peroxo chemisorbed O2 molecule has obtained 1.445 |e| from BN(8.0)/Ni or 1.490 |e| from BN(9.0)/Ni, respectively. The gained electrons fill into the π* orbital of O2 molecules, which results in elongated O–O bond length and reduced local magnetic moments of O atoms. Furthermore, the adsorption behaviors of O2 molecules on BN(8.0)/Ni are extraordinarily different from BN(9.0)/Ni due to the different curvature of (8.0) and (9.0) BNNTs. Actually, because (9.0) BNNT has a curvature that is closer to that of an h-BN monolayer, the adsorption behaviors of O2 molecules on BN(9.0)/Ni are more similar to those of a metal-supported h-BN monolayer. In our previous work (Mao et al., 2017), we have investigated the adsorption behaviors of O2 molecules on a Ni(111) surface-supported h-BN monolayer [h-BN/Ni(111)]. On h-BN/Ni(111), the adsorption energy of the adsorbed O2 molecule is −1.709 eV, the elongated O–O bond length is 1.481 Å, and the local magnetic moments are both 0.018 μB for the two O atoms, while on BN(9.0)/Ni, they are −1.767 eV, 1.500 Å, and 0.008, 0.017 μB, respectively.
The dissociation of adsorbed O2 molecules was also investigated. On a metal-supported h-BN monolayer, the dissociation is an endothermic process, and the energy differences between the dissociated and molecular O2 are 0.960, 0.884, and 0.933 eV on h-BN/Cu(111), h-BN/Ni(111), and h-BN/Co(001) respectively. Here, on BN(8.0)/Ni and BN(9.0)/Ni, we just consider the dissociation of peroxo chemisorbed O2 molecules with the lowest adsorption energy. On BN(9.0)/Ni and BN(8.0)/Ni, the dissociation of peroxo adsorbed O2 is also endothermic by 0.962 (Supplementary Figure S1C, D) and 0.518 eV, respectively. Note that the dissociation energy has been greatly decreased on BN(8.0)/Ni compared to BN(9.0)/Ni or a metal-supported h-BN monolayer due to larger curvature in the BN(8.0) tube. Furthermore, we used the CI-NEB method to search the minimum energy path and found that the dissociation barrier is 0.700 eV (Figure 2E). Since the energy barrier 0.700 eV is low and can be satisfied at room temperature, the dissociation of adsorbed O2 on BN(8.0)/Ni is probable.
Finally, CO oxidation reaction was investigated on BNNT/Ni. In order to investigate CO oxidation reaction, coadsorption of CO and O2 molecules was firstly studied on BNNT/Ni. On both BN(8.0)/Ni and BN(9.0)/Ni, a single CO molecule is hard to be adsorbed. Interestingly, the CO molecule may adsorb on BNNT/Ni when one O2 molecule is pre-adsorbed on BNNT/Ni. In this part, we comparatively discuss the coadsorption of one pre-peroxo chemisorbed O2 molecule and another CO molecule. On both BN(8.0)/Ni and BN(9.0)/Ni, the possible CO adsorption sites are B atoms nearest to the adsorption sites of O2 molecule (another two B atoms). There are two nearest B atoms: one B atom is in the same hexatomic ring with two B atoms of the adsorption sites of the adsorbed O2 molecule (noted by the B1 site, as in Figures 3A,C), and the other is in a neighboring hexatomic ring (noted by the B2 site, as in Figures 3B,D). In Figure 3, the energy below is the adsorption energy of CO molecule after one O2 molecule pre-adsorbed. On BN(8.0)/Ni, the adsorption energy is positive, which indicates that the CO adsorptions are endothermic by 0.114 and 0.010 eV on the B1 and B2 site, respectively. On BN(9.0)/Ni, CO adsorption on the B1 site is also endothermic by 0.170 eV. However, CO adsorption on the B2 site is exothermic, releasing heat of 0.230 eV. The results indicate that CO adsorption on the B2 site of BN(9.0)/Ni is exothermic, and is the most favorable in energy.
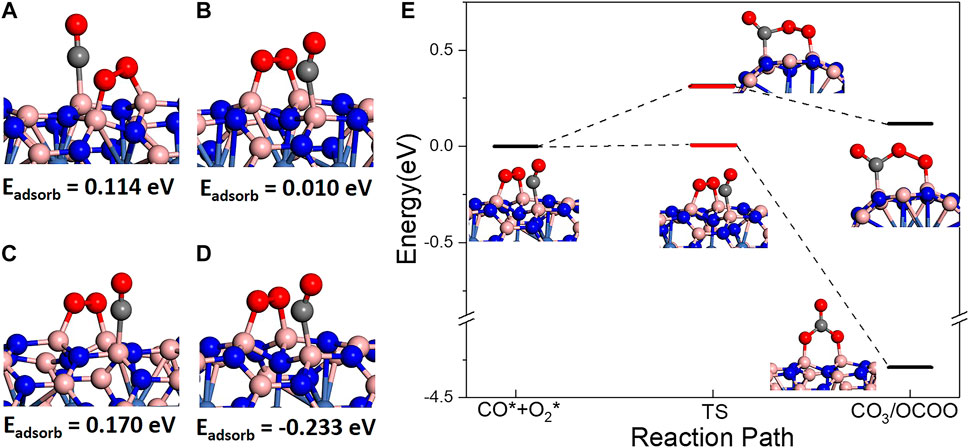
FIGURE 3. CO and O2 coadsorption structures with CO adsorbed on (A) B1 site in BN(8.0)/Ni, (B) B2 site in BN(8.0)/Ni and (C) B1 site in BN(9.0)/Ni, (D) B2 site in BN(9.0)/Ni. The values below are the adsorption energy of CO molecules after O2 pre-adsorbed. (E) Two reaction pathways of CO oxidation on BN(9.0)/Ni starting from (D) coadsorption.
Since the coadsorption on the B2 site of BN(9.0)/Ni is favorable in energy, we use it as the reactant. From this reactant, two possible reaction paths were observed. One is traditional LH reaction path. As shown in Figure 3E, coadsorbed CO and O2 molecules form OCOO intermediate state with an energy barrier of 0.317 eV, and the process is endothermic by 0.124 eV. In this path, the adsorbed O2 molecule breaks one of two O–B bonds with BN(9.0)/Ni, and rotates to the adsorbed CO molecule, finally forming an OCOO intermediate state. In the other reaction path, as presented in Figure 3E, coadsorbed CO and O2 molecules form a CO3 structure. The process is exothermic, releasing a large amount of heat of 4.293 eV, and the energy barrier is very low, only 0.008 eV. In this path, the adsorbed CO molecule jumps to the middle of the adsorbed O2 molecule, breaks the O–O bond, and finally forms the extremely stable intermediate state CO3 structure. Comparing two reaction paths, it is obvious that the CO3 formation path is much more favorable in both thermodynamics and dynamics. Afterwards, CO3 state dissociates into one gaseous CO2 molecule and one O atom adsorbed on BN(9.0)/Ni. The process is endothermic, requiring a great deal of energy of 2.130 eV. The energy barrier may be higher. In summary, although the coadsorbed CO and O2 molecules are easy to form the CO3 intermediate state, the CO3 state is very stable that the following dissociation process is hard to proceed at low temperature. As a result, CO oxidation on BN(9.0)/Ni is always trapped in a very stable CO3 state.
On BN(8.0)/Ni, since the chemical coadsorption of CO and O2 molecules is endothermic on both B1 and B2 sites, we use one peroxo chemisorbed O2 molecule and a gaseous CO molecule as the reactant. The same with BN(9.0)/Ni, two reaction paths were observed on BN(8.0)/Ni. Moreover, the CO3 intermediate state is also much more stable than OCOO because CO3’s total energy is 3.660 eV lower than OCOO, which again demonstrates that the CO3 formation process is extraordinarily favorable in thermodynamics. Supplementary Figure S2 gives the reaction path of the CO3 intermediate state formation and dissociation. The CO3 formation process is exothermic, releasing 4.780 eV heat, and the energy barrier is 0.559 eV. The following CO3 dissociation process is endothermic by 1.806 eV and the energy barrier is very high, 1.898 eV. This is very similar to reaction path on BN(9.0)/Ni. The formed CO3 intermediate state is very stable, which inhibits the following CO3 dissociation into CO2 at low temperature.
Previous work demonstrates that CO oxidation starting from the peroxo chemisorbed O2 molecule is hard to proceed at low temperature on both BN(8.0)/Ni and BN(9.0)/Ni. In this paragraph, we will discuss the reaction path starting from the superoxo chemisorbed O2 molecule on BN(8.0)/Ni (Figure 4). In the superoxo chemisorbed O2 structure, only one O atom of the adsorbed O2 molecule formed one O–B bond during the adsorption, so the adsorbed O2 will not form the CO3 intermediate state with a CO molecule. Notably, CO and O2 molecules’ co-chemisorption was not observed when the O2 molecule was superoxo pre-adsorbed. So, a superoxo chemisorbed O2 molecule and a gaseous CO molecule on BN(8.0)/Ni serve as the reactant. As seen in Figure 4, firstly the gaseous CO molecule easily reacts with the chemisorbed O2 molecule and forms OCOO intermediate state adsorbed on BN(8.0)/Ni. In this process, the energy barrier is very low, only 0.113 eV, and the reaction releases 1.345 eV heat. Then, the OCOO intermediate state dissociates into one gaseous CO2 molecule and one adsorbed O atom on BN(8.0)/Ni. The reaction releases heat of 1.866 eV and the energy barrier is low, only 0.637 eV. Finally, the remaining adsorbed O atom will react with another CO molecule and forms an adsorbed CO2 molecule and then the CO2 molecule desorbs from BN(8.0)/Ni (Supplementary Figure S3). The following two elementary reactions are both exothermic by 1.694 and 1.177 eV, respectively. The corresponding barriers are 0.291 and 0.388 eV, respectively. In all, the rate-limiting step in the whole CO oxidation is the OCOO intermediate state dissociation step with a low energy barrier of 0.637 eV. All the elementary reactions are exothermic. Thus, CO oxidation reaction on BN(8.0)/Ni starting from superoxo chemisorbed O2 molecule is easy to occur at low temperature.
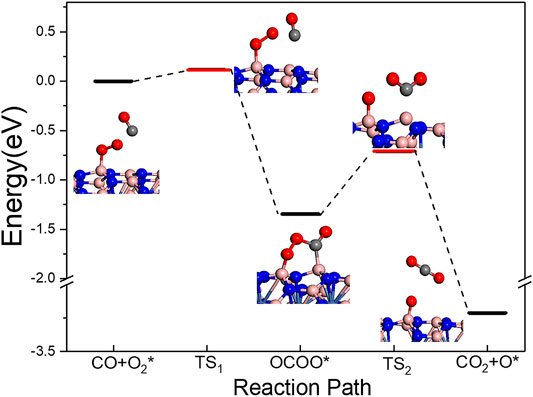
FIGURE 4. CO oxidation path starting from superoxo chemisorbed O2 molecule on BN(8.0)/Ni.
In order to further investigate the mechanism of enhanced activity of boron nitride nanotubes by encapsulation Ni wire, Bader charge analysis and partial density of states were performed. According to Bader charge analysis, BN(8.0) or BN(9.0) nanotubes in BN(8.0)/Ni or BN(9.0)/Ni obtain 2.391 or 1.815 |e| from encapsulated Ni wire. As a result, BN nanotubes became negatively charged, making it easier for highly electronegative O2 molecules to adsorb on them. Moreover, Figure 5 shows the partial density of states (PDOS) of B and N p orbitals of BN(8.0)/Ni, BN(8.0), BN(9.0)/Ni, and BN(9.0). Both BN(8.0) and BN(9.0) tubes are insulators, with band gaps of 3.607 and 3.766 eV, respectively. The highest occupied state levels come from N p orbitals, and the lowest unoccupied states are mainly derived from B p orbitals. After encapsulation by Ni wire for BN(8.0)/Ni and BN(9.0)/Ni, numerous PDOS of B and N p orbitals emerge in the gap region. In addition, the p-band center of B and N p orbitals was computed. For BN(8.0)/Ni, BN(8.0), BN(9.0)/Ni, and BN(9.0), the p-band centers of the B orbitals are −7.053, −8.795, −6.560, and −7.536 eV, respectively, while the p-band centers of the N orbitals are −7.302, −8.528, −7.441, and −7.890 eV. The results reveal that both p-band centers of B orbitals and N orbitals have shifted up toward the Fermi energy as a result of charge transfer from Ni wire to BN nanotubes, promoting the adsorption of highly electronegative O2 molecules and the following CO oxidation on BN(8.0)/Ni and BN(9.0)/Ni.
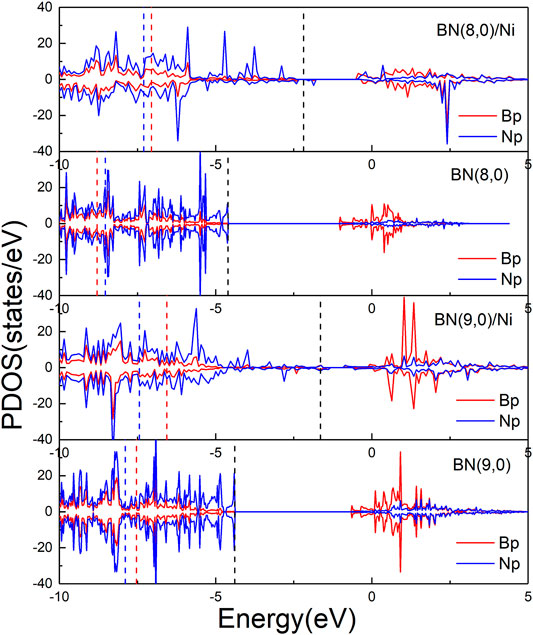
FIGURE 5. Partial density of states of B (red) and N (blue) p orbitals of BN(8.0)/Ni, BN(8.0), BN(9.0)/Ni, and BN(9.0). The black dashed line is the Fermi level of each species. The red and blue dashed lines are the p band center of each species.
Conclusion
Spin polarized density functional theory was employed to investigate the chemical activity of Ni wire encapsulated BN(8.0) and BN(9.0) nanotubes toward O2 and CO molecules. For O2 molecules, three types of adsorptions—physical adsorption and superoxo and peroxo chemisorption—were observed. The curvature of BN nanotubes and adsorption sites accounts for various types of adsorptions. On BN(8.0)/Ni, all three types of adsorptions exist. In contrast, only physical adsorption and peroxo chemisorption on BN(9.0)/Ni were observed because the curvature of BN(9.0) is closer to that of the h-BN monolayer, resulting in similar adsorption behaviors on BN(9.0)/Ni as on the metal-supported h-BN monolayer. Moreover, on BN(9.0)/Ni, the adsorbed O2 molecules are more highly activated than on BN(8.0)/Ni, showing much lower adsorption energy, longer O–O bond length, and lower possessed local magnetic moments. Furthermore, the dissociation of the peroxo chemisorbed O2 molecule on BN(8.0)/Ni is probable, whose energy barrier is only 0.70 eV. For CO oxidation, peroxo chemisorbed O2 molecules prefer to react with CO molecules, forming a very stable CO3 intermediate state that inhibits the following CO3 dissociation and CO2 production. Fortunately, superoxo chemisorbed O2 molecules on BN(8.0)/Ni are easy to react with gaseous CO molecules to form an OCOO intermediate state, and the subsequent OCOO dissociation barrier is low, only 0.637 eV, which is the rate-limiting step of the whole reaction. In all, both BN(8.0) and BN(9.0) nanotubes encapsulated by Ni nanowire have high activity toward O2 activation, and Ni nanowire encapsulated BN(8.0) has good catalytic performance in O2 dissociation and CO oxidation. In addition, according to the results of Bader charge analysis and PDOS, BN nanotubes obtain electrons from the enclosed Ni wire, causing them to become negatively charged, making it easier for highly electronegative O2 molecules to adsorb on them. It is expected that our calculations of Ni wire encapsulated BN tubes will shed light on catalytic applications of BN nanotubes.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author Contributions
KM performed DFT calculations. KM and HL wrote the manuscript. XL and JC discussed the results in the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Nos. 21803002 and 21803031).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fceng.2021.807510/full#supplementary-material
References
Abdel Aal, S. (2016). CO Catalytic Oxidation on Pt-Doped Single wall boron Nitride Nanotube: First-Principles Investigations. Surf. Sci. 644, 1–12. doi:10.1016/j.susc.2015.08.024
An, W., Wu, X., Yang, J. L., and Zeng, X. C. (2007). Adsorption and Surface Reactivity on Single-Walled Boron Nitride Nanotubes Containing Stone−Wales Defects. J. Phys. Chem. C 111 (38), 14105–14112. doi:10.1021/jp072443w
Attaccalite, C., Wirtz, L., Marini, A., and Rubio, A. (2007). Absorption of BN Nanotubes under the Influence of a Perpendicular Electric Field. Phys. Stat. Sol. (B) 244 (11), 4288–4292. doi:10.1002/pssb.200776199
Baierle, R. J., Piquini, P., Schmidt, T. M., and Fazzio, A. (2006). Hydrogen Adsorption on Carbon-Doped Boron Nitride Nanotube. J. Phys. Chem. B 110 (42), 21184–21188. doi:10.1021/jp061587s
Baughman, R. H., Zakhidov, A. A., and de Heer, W. A. (2002). Carbon Nanotubes-Tthe Route toward Applications. Science 297 (5582), 787–792. doi:10.1126/science.1060928
Blase, X., Rubio, A., Louie, S. G., and Cohen, M. L. (1994). Stability and Band Gap Constancy of Boron Nitride Nanotubes. Europhys. Lett. 28 (5), 335–340. doi:10.1209/0295-5075/28/5/007
Chen, C.-W., Lee, M.-H., and Clark, S. J. (2004a). Band gap Modification of Single-Walled Carbon Nanotube and boron Nitride Nanotube under a Transverse Electric Field. Nanotechnology 15 (12), 1837–1843. doi:10.1088/0957-4484/15/12/025
Chen, Y., Zou, J., Campbell, S. J., and Le Caer, G. (2004b). Boron Nitride Nanotubes: Pronounced Resistance to Oxidation. Appl. Phys. Lett. 84 (13), 2430–2432. doi:10.1063/1.1667278
Cho, Y. J., Kim, C. H., Kim, H. S., Park, J., Choi, H. C., Shin, H.-J., et al. (2009). Electronic Structure of Si-Doped BN Nanotubes Using X-ray Photoelectron Spectroscopy and First-Principles Calculation. Chem. Mater. 21 (1), 136–143. doi:10.1021/cm802559m
Chopra, N. G., Luyken, R. J., Cherrey, K., Crespi, V. H., Cohen, M. L., Louie, S. G., et al. (1995). Boron Nitride Nanotubes. Science 269 (5226), 966–967. doi:10.1126/science.269.5226.966
Geim, A. K., and Novoselov, K. S. (2007). The Rise of Graphene. Nat. Mater 6, 183–191. doi:10.1038/nmat1849
Golberg, D., Bando, Y., Huang, Y., Terao, T., Mitome, M., Tang, C., et al. (2010). Boron Nitride Nanotubes and Nanosheets. ACS Nano 4 (6), 2979–2993. doi:10.1021/nn1006495
Golberg, D., Bando, Y., Tang, C. C., and Zhi, C. Y. (2007). Boron Nitride Nanotubes. Adv. Mater. 19 (18), 2413–2432. doi:10.1002/adma.200700179
Henkelman, G., Uberuaga, B. P., and Jónsson, H. (2000). A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 113 (22), 9901–9904. doi:10.1063/1.1329672
Jabarullah, N. H., Razavi, R., Mohadeseh Yazdani Hamid, H., Yousif, Q. A., and Najafi, M. (2019). Potential of Ge-Adopted Boron Nitride Nanotube as Catalyst for Sulfur Dioxide Oxidation. Prot. Met. Phys. Chem. Surf. 55 (4), 671–676. doi:10.1134/s2070205119040129
Kim, J. K., Jin, C., Park, J., Iloska, M., Kim, M., Seo, D., et al. (2019). Synthesis of Boron Nitride Nanotubes Incorporated with Pd and Pt Nanoparticles for Catalytic Oxidation of Carbon Monoxide. Ind. Eng. Chem. Res. 58 (43), 20154–20161. doi:10.1021/acs.iecr.9b03954
Kresse, G., and Furthmüller, J. (1996). Efficient Iterative Schemes Forab Initiototal-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 54 (16), 11169–11186. doi:10.1103/PhysRevB.54.11169
Kresse, G., and Hafner, J. (1993). Ab Initiomolecular Dynamics for Liquid Metals. Phys. Rev. B 47 (1), 558–561. doi:10.1103/PhysRevB.47.558
Li, X.-M., Tian, W. Q., Dong, Q., Huang, X.-R., Sun, C.-C., and Jiang, L. (2011). Substitutional Doping of BN Nanotube by Transition Metal: A Density Functional Theory Simulation. Comput. Theor. Chem. 964 (1–3), 199–206. doi:10.1016/j.comptc.2010.12.026
Li, X., Lin, B., Li, H., Yu, Q., Ge, Y., Jin, X., et al. (2018). Carbon Doped Hexagonal BN as a Highly Efficient Metal-free Base Catalyst for Knoevenagel Condensation Reaction. Appl. Catal. B: Environ. 239, 254–259. doi:10.1016/j.apcatb.2018.08.021
Liu, C., Wang, C., Meng, X., Li, X., Qing, Q., Wang, X., et al. (2020). Tungsten Nitride Nanoparticles Anchored on Porous Borocarbonitride as High-Rate Anode for Lithium Ion Batteries. Chem. Eng. J. 399, 125705. doi:10.1016/j.cej.2020.125705
Lyalin, A., Nakayama, A., Uosaki, K., and Taketsugu, T. (2014). Adsorption and Catalytic Activation of the Molecular Oxygen on the Metal Supported H-BN. Top. Catal. 57 (10), 1032–1041. doi:10.1007/s11244-014-0267-7
Lyalin, A., Nakayama, A., Uosaki, K., and Taketsugu, T. (2013). Functionalization of Monolayer H-BN by a Metal Support for the Oxygen Reduction Reaction. J. Phys. Chem. C 117 (41), 21359–21370. doi:10.1021/jp406751n
Mao, K., Wu, X., and Yang, J. (2017). Enhanced Selective Oxidation of H-BN Nanosheet through a Substrate-Mediated Localized Charge Effect. Phys. Chem. Chem. Phys. 19 (6), 4435–4439. doi:10.1039/c6cp07402b
Nigam, S., and Majumder, C. (2008). CO Oxidation by BN−Fullerene Cage: Effect of Impurity on the Chemical Reactivity. ACS Nano 2 (7), 1422–1428. doi:10.1021/nn8001455
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77 (18), 3865–3868. doi:10.1103/PhysRevLett.77.3865
Peyghan, A. A., Soltani, A., Pahlevani, A. A., Kanani, Y., and Khajeh, S. (2013). A First-Principles Study of the Adsorption Behavior of CO on Al- and Ga-doped Single-Walled BN Nanotubes. Appl. Surf. Sci. 270, 25–32. doi:10.1016/j.apsusc.2012.12.008
Rubio, A., Corkill, J. L., and Cohen, M. L. (1994). Theory of Graphitic boron Nitride Nanotubes. Phys. Rev. B 49 (7), 5081–5084. doi:10.1103/PhysRevB.49.5081
Soltani, A., Baei, M. T., Ghasemi, A. S., Tazikeh Lemeski, E., and Amirabadi, K. H. (2014). Adsorption of Cyanogen Chloride over Al- and Ga-doped BN Nanotubes. Superlattices and Microstructures 75, 564–575. doi:10.1016/j.spmi.2014.07.033
Soltani, A., Raz, S. G., Rezaei, V. J., Dehno Khalaji, A., and Savar, M. (2012). Ab Initio investigation of Al- and Ga-doped Single-Walled boron Nitride Nanotubes as Ammonia Sensor. Appl. Surf. Sci. 263, 619–625. doi:10.1016/j.apsusc.2012.09.122
Tang, C., Bando, Y., Golberg, D., Ding, X., and Qi, S. (2003). Boron Nitride Nanotubes Filled with Ni and NiSi2 Nanowires In Situ. J. Phys. Chem. B 107 (27), 6539–6543. doi:10.1021/jp034310q
Wang, Q., Liu, Y.-j., and Zhao, J.-x. (2013). Theoretical Study on the Encapsulation of Pd3-Based Transition Metal Clusters inside boron Nitride Nanotubes. J. Mol. Model. 19 (3), 1143–1151. doi:10.1007/s00894-012-1662-2
Wang, Z. G., Li, Z., and Cheng, D. M. (2009). Effects of Uniaxial Strain on the Band Structure of boron Nitride Nanotubes: a First Principles Study. Eur. Phys. J. Appl. Phys. 46 (2), 20601. doi:10.1051/epjap/2009037
Wasey, A. H. M. A., Chakrabarty, S., Das, G. P., and Majumder, C. (2013). H-BN Monolayer on the Ni(111) Surface: A Potential Catalyst for Oxidation. ACS Appl. Mater. Inter. 5 (21), 10404–10408. doi:10.1021/am404321x
Wu, X., Yang, J. L., and Zeng, X. C. (2006). Adsorption of Hydrogen Molecules on the Platinum-Doped boron Nitride Nanotubes. J. Chem. Phys. 125 (4), 044704. doi:10.1063/1.2210933
Wu, X., and Zeng, X. C. (2006). Adsorption of Transition-Metal Atoms on boron Nitride Nanotube: A Density-Functional Study. J. Chem. Phys. 125 (4), 044711. doi:10.1063/1.2218841
Xiang, H. J., Yang, J., Hou, J. G., and Zhu, Q. (2005). Half-metallic Ferromagnetism in Transition-Metal Encapsulated boron Nitride Nanotubes. New J. Phys. 7 (1), 39. doi:10.1088/1367-2630/7/1/039
Xie, Y., Huo, Y.-P., and Zhang, J.-M. (2012). First-principles Study of CO and NO Adsorption on Transition Metals Doped (8,0) boron Nitride Nanotube. Appl. Surf. Sci. 258 (17), 6391–6397. doi:10.1016/j.apsusc.2012.03.048
Keywords: boron nitride nanotubes, DFT, CO oxidation, O2 activation, catalytic activity
Citation: Mao K, Lv H, Li X and Cai J (2022) Enhanced Catalytic Activity of Boron Nitride Nanotubes by Encapsulation of Nickel Wire Toward O2 Activation and CO Oxidation: A Theoretical Study. Front. Chem. Eng. 3:807510. doi: 10.3389/fceng.2021.807510
Received: 02 November 2021; Accepted: 02 December 2021;
Published: 12 January 2022.
Edited by:
Zhonglong Zhao, Inner Mongolia University, ChinaCopyright © 2022 Mao, Lv, Li and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keke Mao, bWFva2VrZUBhaHV0LmVkdS5jbg==; Jiajia Cai, Y2FpampAYWh1dC5lZHUuY24=