
 Vicky Merhej
Vicky Merhej
- Unité de Recherche en Maladies Infectieuses Tropical Emergentes, CNRS-IRD UMR 6236-198, Université de la Méditerranée, Marseille, France
Many of the definitions in microbiology are currently false. We have reviewed the great denominations of microbiology and attempted to free microorganisms from the theories of the twentieth century. The presence of compartmentation and a nucleoid in Planctomycetes clearly calls into question the accuracy of the definitions of eukaryotes and prokaryotes. Archaea are viewed as prokaryotes resembling bacteria. However, the name archaea, suggesting an archaic origin of lifestyle, is inconsistent with the lifestyle of this family. Viruses are defined as small, filterable infectious agents, but giant viruses challenge the size criteria used for the definition of a virus. Pathogenicity does not require the acquisition of virulence factors (except for toxins), and in many cases, gene loss is significantly inked to the emergence of virulence. Species classification based on 16S rRNA is useless for taxonomic purposes of human pathogens, as a 2% divergence would classify all Rickettsiae within the same species and would not identify bacteria specialized for mammal infection. The use of metagenomics helps us to understand evolution and physiology by elucidating the structure, function, and interactions of the major microbial communities, but it neglects the minority populations. Finally, Darwin’s descent with modification theory, as represented by the tree of life, no longer matches our current genomic knowledge because genomics has revealed the occurrence of de novo-created genes and the mosaic structure of genomes, the Rhizome of life is therefore more appropriate.
Introduction
Post-modern philosophy, also called the French theory (Wicks, 2003), states that the majority of theories, including scientific theories, are only based on meta-narratives expressing the influence of a culture at a given time. These scientific theories can be questioned when a change in techniques creates instability in the theory, as postulated by Karl Popper (Popper, 1959; Raoult, 2010a). In addition (and in the direction of post-modern theory), these theories can also be called into question due to an intellectual change of paradigm (Kuhn, 1962). The study of Rickettsiae has been challenging for the past few years because of the great difficulty in their handling. Moreover, the ancestors of Rickettsiae contributed to the birth of modern eukaryotes by transferring genes to the mitochondrion and the nucleus (Koonin, 2010; Renvoisé et al., 2011). During the explosion of microbial genetics, the study of Rickettsia did not benefit from the model of Escherichia coli, and rickettsiologists had to develop alternative approaches that did not include the common meta-narratives (Renvoisé et al., 2011). Among these approaches were those based on observations of the characteristics of intracellular bacteria, whose genomes and behaviors resemble those of viruses. Thus, Rickettsia has been classified as intermediate bacteria between the viruses and bacteria. Currently, the genomic revolution and “multiomics” have made it possible to analyze Rickettsia with many new tools (Bechah et al., 2010), and Rickettsia was among the species that were sequenced most quickly (Andersson et al., 1998; Ogata et al., 2001). This sequencing and generally all the work achieved by Rickettsiologists brought an important revision to the way of thinking with respect to E. coli and forced microbiologists to visualize the general theories concerning bacterial species in a different way, so several theories concerning bacteria had, or need, to be revised (Georgiades and Raoult, 2011a). In this work, our goal was to revise the overarching classifications and denominations used in microbiology. In particular, as postulated by post-modern philosophy (Lyotard, 1979; Williams, 1998), we know that the denomination of an object constrains it in its definition and that when the definition is inappropriate, one cannot conceive of the object in a reasonable way.
Definition of Eukaryotes and Prokaryotes
The word “microbe,” literally meaning “small life,” was introduced by the French surgeon Charles Sédillot in 1878 to define infinitely small living organisms (Vallery-Radot, 1885). One of the most important advances in our understanding of the living world was the realization by the French scientist Edouard Chatton that there are two major groups of organisms that he named the prokaryotic (bacteria) and the eukaryotic (organisms with nucleated cells) type (Chatton, 1925; Stanier and van Niel, 1962; Sapp, 2005). This classification was adopted by Stanier and van Niel (1962) and the prokaryote–eukaryote dichotomy was universally accepted as the natural order of things until the 1970s and the emergence of rRNA phylogenetics (Sapp, 2005). At that time, Woese achieved a comprehensive understanding of bacterial phylogeny using laborious molecular sequencing methods (Woese et al., 1975). Those data revealed two separate lineages among prokaryotes: the Archaea (Archaebacteria) and the Bacteria (Eubacteria). The prokaryote/eukaryote system was replaced by the “three domain system” and the classification of Eukarya, Archaea, and Bacteria (Woese, 1994). However, bacteria had always been defined largely in negative terms: they lacked a nucleus, compartmentation, and sexual reproduction (Sapp, 2005). This negative description is somewhat invalid because it does not define what a prokaryote is but rather what it is not (Pace, 2006). Furthermore, recent observations of Planctomycetes prove that the definitions of eukaryotes and prokaryotes are erroneous. Planctomycetes is a distinctive phylum of the domain Bacteria, in which the cells possess a different structural plan than other prokaryotes; the cells of all cultured and some uncultured species are divided into compartments by one or more membranes (Figure 1). In addition, in one particular species, Gemmata obscuriglobus, the nucleoid is enveloped in two membranes to form a nuclear body that is analogous to the structure of a eukaryotic nucleus. The existence of these organisms clearly calls into question the accuracy of the actual definitions of eukaryotes and prokaryotes (Fuerst, 1995, 2005, 2010). The nucleus of these cells likely resulted from autogenous membrane development in a prokaryote lineage (Taylor, 1976; Lake and Rivera, 1994; Glansdorff et al., 2008), most likely in Planctomycetes and the closely related Chlamydia (Ward et al., 2000; Horn et al., 2004; Figure 2). This theory has been strengthened by the discovery of nuclear envelope fold topology in Planctomycetes, which is analogous to the eukaryotic cell structure (Fuerst, 2005, 2010). Moreover, the eukaryotes all harbor mitochondria, or mitochondria-related genes, inherited from Rickettsiales (Golding and Gupta, 1995; Lang et al., 1999). Therefore, eukaryotes are younger than Rickettsia, their other ancestors are unknown, and there is no evidence that these ancestors had a nucleus. As it turns out, the three domain system, as previously defined, does not exist (Lake, 1988).
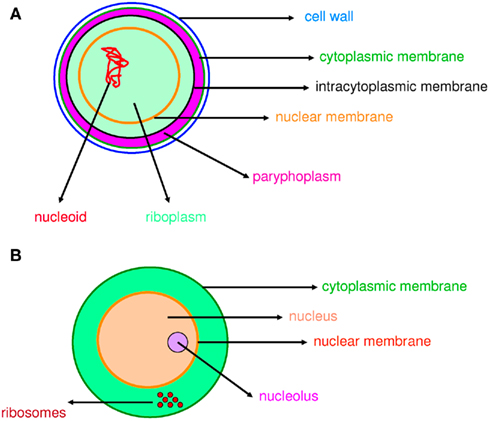
Figure 1. Compartmentation in prokaryotes and eukaryotes. Compartmentation of Gemmata obscuriglobus(A) and a eukaryotic cell (B) is comparable.
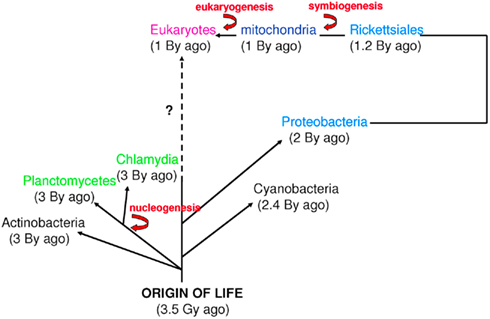
Figure 2. Time scale of eukaryogenesis and nucleogenesis. Eukaryotes are not the only species with compartmentation. First eukaryotes emerged from an endosymbiotic event. The first nucleus appeared approximately 3 billion years ago in Planctomycetes and Chlamydia. These numbers are approximations (Bromham and Penny, 2003; Cavalier-Smith, 2004; Trevors and Abel, 2004).
Archaea
When they were identified in the late 1970s based upon ribosomal sequences, Archaea were viewed as a group of archaic bacteria (Woese and Fox, 1977). Indeed, because of their capacity of methanogenesis, archaea were supposed to live in ancient organisms and received the name of “archaea.” This name is misleading as it speculates that these organisms resemble ancient cells and live in specific and “archaic like” environment. They have long been considered as extremophile bacteria that can be found in the most extreme environments (temperature, salinity, and pH). This explains the fact that archaea have not been extensively studied in clinical microbiology and their place among living organisms long went unrecognized.
Because of their archaic label, Archaea have been used as models of the early evolution of cellular life forms (Romano and Conway, 1996; Embley and Martin, 2006; Poole and Penny, 2007; Cox et al., 2008). The information processing machineries of archaea are considered ancestral forms of the more complex replication, transcription, and translation machineries of the eukaryotic cell (Gribaldo et al., 2010). Other evolutionary hypotheses about the path of life reject the archaic status of archaea. They suggested that the three domains of life evolved from a pre-cellular community containing different types of genes using a process that led to the fixation of specific subsets of genes in the ancestors of these domains (Woese, 1998). Considering evidence from molecular sequences, envelope structure, and motility mechanisms, other hypothesis suggested that the archaea evolved from Gram-positive bacteria as an adaptation to hyperthermophilic or hyperacidity (Cavalier-Smith, 2002) or in response to antibiotic selection pressure (archaea are resistant to a wide variety of antibiotics that are primarily produced by Gram-positive bacteria; Gupta, 1998a,b, 2000).
Recent results obtained using molecular approaches and metagenomic studies have changed our perspective of the nature and the diversity of archaea. Indeed, archaea were considered predominant over bacteria in all extreme environments. This is true for high-temperature environments, as only archaea can thrive at temperatures from 95 to 113°C (Huber et al., 2000). However, in all other situations, species of Bacteria and Eukarya have been found together with those of archaea (Aravalli et al., 1998; DeLong, 1998; Rothschild and Mancinelli, 2001). Novel archaea have been isolated from a variety of temperate and cold environments (Cavicchioli, 2006), agricultural and forest soils, plankton, fresh water lake sediments, and the deep waters of oceans (Schleper et al., 2005). Archaea seem to constitute a major part of global ecosystems. They were estimated to account for approximately 34% of the total marine biomass of Antarctica (DeLong, 1998) and for nearly 20% of the total marine picoplankton biomass worldwide (Karner et al., 2001). The ubiquitous abundance of archaea and their influence on biogeochemical cycles remain largely unexplored. A recent tentative to infer the ancestral conditions of life suggests that he last common ancestor of archaea has been hyperthermophilic and mesophilic species have showed adaptations to cooler environments (Groussin and Gouy, 2011).
Methanogenic bacteria play a paramount role in digestion processes. Indeed, metagenomic analysis of the gut flora in three healthy individuals established that Methanobrevibacter smithii comprised up to 11.5% of the gut microorganisms (Eckburg et al., 2005). On the contrary, while many studies using 16S rDNA sequencing confirmed the presence of M. smithii in the human gut, the prevalence was low and Methanosphaera stadtmanae was not detected in most cases (Miller and Wolin, 1982; Belay et al., 1988; Dridi et al., in revision). This contrast is due to limitations in the experimental protocols that are largely designed for the study of bacteria. Recently, in our laboratory we developed an optimized protocol for the extraction and specific PCR-based detection of M. smithii and M. stadtmanae in DNA stool samples, using specific primers (Dridi et al., 2009). Using this protocol it was demonstrated that all individuals carried methanogenic archaea with a high prevalence of M. smithii (95.5%). The application of this specific approach allowed the isolation of Methanomassiliicoccus luminyensis and its description as a new species (Dridi et al., in revision), and the same protocol can be used to identify other archaeal species (Dridi et al., 2009, 2011). It is obvious that previous methods did not allow the identification of Archaea because they were not designed for Archaea identification.
Molecular experiments and genomic approaches have suggested that the different criteria used to define archaea are not completely valid. The definition currently used for Archaea merely cloaks our lack of knowledge of this domain of life. Undoubtedly, Archaea are not a form of “archaic” bacteria, they rather represent a distinct evolutionary domain.
Bacterial Virulence Factors
It is not surprising that many people believe that bacteria that are dangerous to us are better armed than non-pathogenic bacteria. Toxins were identified in 1884 and defined as virulence factors; since, early genetic studies on bacterial virulence demonstrated that removing a certain number of genes from pathogenic bacteria decreases their capacity to infect hosts. Therefore, the conclusions of most studies on bacterial virulence, driven by anthropocentric intuition and perspective, suggested, and some still suggest that non-pathogenic bacteria lack supplementary virulence factors (Lawrence, 1999; Ochman et al., 2005).
An outstanding example of this way of thinking is the Shigella paradigm. Shigella spp. are human pathogens associated with bacillary dysentery, or shigellosis. Shigella dysenteriae causes deadly epidemics in many of the world’s poorest countries. Shigella spp. and E. coli have always been considered closely related, and they have even been placed in the same species (Pupo et al., 2000). However, most E. coli strains are commensals of the human intestine (Maurelli et al., 1998), and Shigella spp. differ from E. coli in their lack of certain phenotypic traits, such as extracellular mobility and the ability to ferment lactose and many sugars (Karaolis et al., 1994; Pupo et al., 2000). Similar to S. dysenteriae, pathogenic enteroinvasive E. coli lack lysine decarboxylase (LDC) activity. In a study by Maurelli et al. (1998), the induction of LDC expression attenuated the virulence of a transformed strain of S. flexneri. It seems plausible that Shigella evolved from the E. coli complex through the acquisition of a plasmid containing critical genes. Plasmids of Shigella spp. have been directly associated with virulence and were even named “virulence plasmids” after their discovery (Hale et al., 1983). Furthermore, actin-based motility initiated by the icsA gene has also been reported to be a virulence factor (Goldberg et al., 1994). However, virulence increased after massive gene deletions (Maurelli et al., 1998). In conclusion, S. dysenteriae was not found to have more virulence genes than related bacteria (Georgiades and Raoult, 2011a).
Many recent comparative genomics studies have demonstrated that the specialization of bacteria for the colonization of eukaryotic hosts is associated with massive gene loss (Nierman et al., 2004; Merhej et al., 2009a) and the loss of identified “virulence factors” (Audic et al., 2007). Genomic analysis has revealed that Borellia recurrentis, the agent of deadly louse-borne relapsing fever, encodes fewer putative virulence factors than Borellia duttonii (Lescot et al., 2008). Gene loss has also accompanied the evolution of pathogenic Bordetella species (Cummings et al., 2004) and gene deletions in Mycobacterium tuberculosis have resulted in a hypervirulent phenotype (Bokum et al., 2008). Finally, in a study by Audic et al. (2007), the number of putative virulence factors was found to be higher in water-dwelling bacteria than in any other categories of bacteria, including specialized pathogens (Audic et al., 2007).
One of the best examples of genome reduction can be found in the epidemic-causing Rickettsia prowazekii, which is the most dangerous rickettsial species. Genome comparisons of R. prowazekii with the less virulent R. conorii have revealed that R. prowazekii is a subset of R. conorii, with only 834 open reading frames (ORFs) compared to the 1,374 ORFs of R. conorii (Ogata et al., 2001). Although intracellular motility has been considered a virulence factor of Shigella (Goldberg and Theriot, 1995) and Listeria monocytogenes (Tilney and Portnoy, 1989; Mounier et al., 1990), R. prowazekii is completely immobile in the cytoplasm (Teysseire et al., 1992). Actin-based motility in R. conorii and R. rickettsii requires two proteins functioning together, Sca2 and RickA, suggesting that these two proteins could be virulence factors of R. rickettsii. R. typhi possesses only the Sca2 protein and is also mobile in the cytoplasm but less than R. conorii (Teysseire et al., 1992; Figure 3). However, none of these proteins are found in R. prowazekii, which lacks actin-based motility (Kleba et al., 2010). Consequently, motility is not a virulence factor per se but can be part of a virulence repertoire in some pathogens (Georgiades and Raoult, 2011a). Other studies have also demonstrated genome reduction to a lower extent in the extremely successful and fit R. africae, the agent of African tick-bite fever. In contrast with their possession of virulence factors, R. prowazekii and R. africae have the most and the least decayed genomes, respectively, among pathogenic Rickettsiae (Fournier et al., 2009). A comparison of R. africae with R. rickettsii suggested the loss of essential genes in R. rickettsii as a possible factor involved in the development of virulence (Fournier et al., 2009). In general, pathogenic Rickettsia species lack what was defined as “pathogenicity islands” and that are present in other bacterial pathogens (Hacker and Kaper, 2000). It has been suggested that plasmids contain genes encoding proteins responsible for host recognition, invasion, and pathogenicity. The presence of plasmids in Rickettsia species, however, did not show any correlation with virulence (Paddock et al., 2004; Ogata et al., 2005; Blanc et al., 2007a). The examples of Rickettsiae and Shigella spp. show that the terms “pathogenicity islands” and “virulence plasmids” are misleading. The genomic analysis of rickettsial species has revealed that the shift to pathogenicity does not require the acquisition of new genes, but in more cases, and not only in Rickettsia, gene loss seems to be implicated in the emergence of virulence (Moran, 1996; Andersson and Kurland, 1998; Andersson and Andersson, 1999; Blanc et al., 2007a; Darby et al., 2007; Merhej et al., 2009a). In a recent study in our laboratory, we demonstrated that the only features found at higher levels in extremely dangerous bacterial pathogenic species than in closely related less pathogenic species were toxins and toxin–antitoxin modules (TA; Georgiades and Raoult, 2011b).
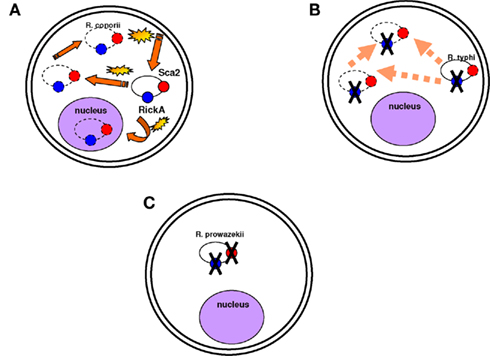
Figure 3. Motility is not necessarily a virulence factor. (A) Rickettsia conorii is mobile in the cytoplasm and in the nucleus and moves quickly. Actin-based motility is associated with the Sca2 and RickA proteins (represented in red and blue circles respectively). Yellow stars are to demonstrate that the bacterium moves fast in the cytoplasm; dotted lines are to show that the bacterium can be found anywhere in the cytoplasm and even in the nucleus. (B) Rickettsia typhi is also mobile in the cytoplasm, but it moves less quickly than R. conorii. Its mobility is associated with the Sca2 protein. (C) R. prowazekii is completely immobile in the cytoplasm. The Sca2 and RickA proteins are absent.
In conclusion, except for toxins and TA modules, which have a direct effect and are indeed virulence factors, other products named “virulence factors” are, in reality, associated with fitness in a genomic context and in a specific environment, including in tested experimental models. Comparative genomics have shown than pathogenic bacteria have smaller genomes than non-specialized bacteria. Therefore, it is not possible to say that supplementary virulence factors establish pathogenicity, but rather, the overall gene repertoire is more associated with virulence than specific genes. In a recent study, the deletion of four different gene clusters in fungi attenuated their virulence in plants, while deletion of the “divergence cluster 8–12” (region encoding effector genes with mow sequence conservation) caused a hypervirulent fungal phenotype (Schirawski et al., 2010). Under these conditions, a virulent gene repertoire is composed of both present and absent genes. The term “virulence factor” seems to be invalid, and we propose that it should be abandoned.
Phylogeny and Taxonomy
Biological dogma states that phylogeny reflects taxonomy. Indeed, the 16S rRNA sequence has been widely used for the description of many newly classified bacterial species (Rosello-Mora and Amann, 2001; Drancourt et al., 2004; Roux et al., 2004). A 16S rRNA divergence of 1–2% is considered to correspond to approximately 50 million years of divergence (Ochman et al., 1999; Ogata et al., 2001), and a cut-off of 98.7% similarity in 16S rRNA reflects a new species (Stackebrandt and Ebers, 2006). However, an accurate delineation of bacterial species cannot be guaranteed by the use of ribosomal DNA sequence identity, which often leads to misleading species definitions (Fox et al., 1992; Rosello-Mora and Amann, 2001). Bartonella henselae has two copies of 16S rRNA in some cases, which likely emerged through recombination (Sanogo et al., 2003), and these copies present a divergence higher than 1.3% (Viezens and Arvand, 2008). For several bacterial species, the presence of multiple copies of the 16S rRNA gene has been documented (Acinas et al., 2004). Although generally, these multiple copies in an organism are either identical or nearly identical, in some cases, they are divergent enough to overestimate the number of bacterial species. This overestimation can be seen in the case of Delisea pulchra strains, in which 16S rRNA gene copies were used to illustrate the effects of 16S rRNA heterogeneity in the marine bacterial community (Dahllöf et al., 2000; Adékambi et al., 2008). The use of 16S rRNA for such analysis is limited due to its inherent heterogeneity (Dahllöf et al., 2000). Moreover, using the molecular clock scale based on 16S rRNA as a species definition criterion, specialized bacteria within mammalian hosts are not defined as species (Georgiades and Raoult, 2011a). Species definition cannot be based on the percent divergence of 16S rRNA because bacteria having a divergence less than 1.3% correspond to bacterial complexes rather than species (Doolittle and Papke, 2006).
There are 9,000 validated bacterial species and 1.5 million eukaryotic species, even though the biomass of bacteria is comparable to that of eukaryotes; this suggests that use of the 16S rDNA sequence as a taxonomic tool is not adapted to the definition of species. Furthermore, genomic contents are not represented by phylogeny. In a study based on the genomic content comparison of bacteria with different lifestyles, discrepancies between taxonomy and gene content were observed (Audic et al., 2007; Merhej et al., 2009b). The phylogenomic analysis yielded a tree similar to the one produced using the 16S rDNA gene sequence. However, γ-proteobacteria appeared to be divided into three groups, confirming that these species were more similar to each other in terms of gene content than to their close phylogenetic relatives (Audic et al., 2007). Similarly, rickettsial species and relatives, such as Wolbachia and Ehrlichia species, comprise an α-proteobacterial clade characterized by small genomes; this clade is distantly related to other α-proteobacterial species with larger genomes (Moran, 2002). Phylogenetic analysis of Rickettsia species based on 16S rRNA sequences has been frequently performed; however, significant inferences about intragenus phylogeny are not possible because the sequences are almost identical (Roux and Raoult, 1997). In fact, the official molecular criteria used for the classification of a bacterial species, DNA/DNA hybridization >70%, GC content <5%, and a 16S rRNA divergence <1, 3%, cannot be applied to Rickettsia species. A 16S rRNA divergence <2% alone would classify all Rickettsiae within the same species (Fournier and Raoult, 2009). Furthermore, based on this criterion, bacteria specialized to mammalian hosts are not defined as a species (Georgiades and Raoult, 2011a). Homo sapiens emerged approximately 250,000–400,000 years ago, while the first human-specialized pathogenic bacterial species, M. tuberculosis, emerged much later, only 20,000 years ago (Wirth et al., 2008). For organisms such as archaea, bacteria, and some unicellular eukaryotes, the species and gene trees do not show much identity with each other on an evolutionary scale (Bapteste et al., 2009). This result is due to the fact that individual gene histories can be different from the history of a species. Over the past 15 years, lateral inheritance (as opposed to vertical inheritance) has been proven to be a major evolutionary force in microorganisms (Bapteste and Boucher, 2008). Examples of extensive chimerism and LGT across prokaryotes are common, and it is absolutely plausible that every gene in prokaryotes has been affected by LGT at some point in evolutionary history (Bapteste et al., 2009). With this in mind, whole gene content and present and absent genes should be taken into consideration when searching for a reliable species classification (Figure 4).
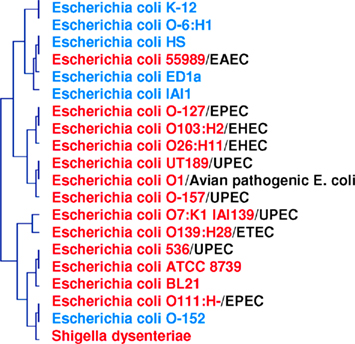
Figure 4. Phylogenomic tree based on whole gene content (present/absent genes) in pathogenic and non-pathogenic E. coli species and S. dysenteriae. Two clusters are formed: one for pathogenic species (in red), and one for non-pathogenic species (in blue). Pathogenic strains are divided into five groups: EPEC, enteropathogenic; EHEC, enterohemorrhagic; UPEC, uropathogenic; ETEC, enterotoxigenic; EAEC, enteroaggregative.
Definition of a Virus
The discovery of giant viruses with large genomes has raised many questions about virus definitions and evolution. According to Lwoff, viruses have typically been defined as “filterable infectious agents” smaller than 200 nm that are unable to undergo binary fission and have one type of nucleic acid with few protein-encoding genes (Lwoff, 1957). Giant viruses, such as mimivirus (Raoult et al., 2004, 2007) and mamavirus (La Scola et al., 2008), challenge the size criteria used for the definition of a virus. These viruses, with an icosahedral capsid diameter of nearly 400 nm, have particle sizes comparable to that of bacteria such as Mycoplasma (La Scola et al., 2003; Raoult et al., 2004). Mimivirus possesses a large double-stranded DNA genome (1,181 kb). The mimivirus genome has 1,262 putative ORFs, of which 911 are predicted to be protein-coding genes, and 298 could be associated with functional attributes (Raoult et al., 2004). Mamavirus has a slightly larger genome than mimivirus (1,200 kb), and 99% of its predicted genes are orthologous to mimivirus ORFs (Colson and Raoult, 2010). The concept of the small particle (and genome) that once defined viruses is no longer valid.
The discovery of large viruses prompted a re-evaluation of the commonly used viral isolation methods and consideration of the role played by amebae as a source of giant viruses. Because amebae ingest any particle that is larger than 100 nm, these phagocytes represent a potential source of giant viruses with chimeric repertoires (Raoult and Boyer, 2010). Indeed, another virus, Marseillevirus, has recently been isolated from amebae. It has a diameter of 250 nm and a genome size of 368,454 bp (Boyer et al., 2009). Mimivirus, Mamavirus, and Marseillevirus belong to the Mimiviridae, a family in the group of nucleo-cytoplasmic large DNA viruses (NCLDVs; Iyer et al., 2006; Boyer et al., 2009). Genomic analysis of the giant viruses showed that only 4.6 and 11.2% of the ORFs of mimivirus and marseillevirus, respectively, had homologs in the NCLDV core gene set. Hence, the majority of the genome is lineage-specific. In addition to the core genome, the gene repertoire of these ameba-associated viruses contains duplicated genes, ORFans and genes likely acquired through LGT. Indeed, a substantial proportion of the genome exhibits sequence similarities to gene homologs found in bacteria, archaea, eukaryotes, and viruses (Raoult et al., 2004). Using phylogenetic analyses, a bacterial or bacteriophage origin has been inferred for 49 genes and a eukaryotic origin for 85 genes of the marseillevirus genome (Boyer et al., 2009). Likewise, 60 genes from the mimivirus genome had reliable homologs in cellular species and seemed to be acquired from eukaryotes, especially from amebae (Moreira and Brochier-Armanet, 2008). These chimeric gene contents may have resulted from acquisitions through LGT involving the eukaryotic host (ameba) and sympatric bacteria and viruses. Amebae may serve as a genetic mixing bowl from which giant viruses may have gathered a complex set of genes, leading to large chimeric genomes (Raoult and Boyer, 2010).
The genomes of giant viruses help to elucidate their origin and early evolution. The position of viruses within the tree of life (TOL) has been a subject of disagreement. Indeed, the classification of organisms into a universal TOL based on ribosomal RNA sequences (Pace, 2006) evidently excludes viruses, which lack ribosomes. As acellular organisms, viruses were intentionally not represented with other living ribosome-encoding organisms in the TOL (Moreira and Lopez-Garcia, 2009). Like other viruses, the mimivirus genome contains genes for replication. Surprisingly, the genome of mimivirus also contains genes that code for components of translation machinery never before found in viruses, including four amino-acyl transfer RNA synthetases, peptide release factor 1, the translation elongation factor EF-TU, and translation initiation factor 1 (Raoult et al., 2004). The presence of these genomic features has triggered a reappraisal of the definition of living beings (Raoult and Forterre, 2008) and the evolutionary implication of viruses. The phylogenetic analysis of mimivirus proteins that have closely related eukaryotic homologs support the appearance of mimivirus as representing a fourth domain of life together with bacteria, archaea, and eukaryotes (Raoult et al., 2004). Indeed, there are some genes that allow tracing history, including DNA processing genes, even thought a whole, complete organism cannot be represented by a classic TOL. An additional genomic study revealed the early emergence of NCLDVs whose core genome is as ancient as the three currently accepted domains of life (Boyer et al., 2010a). These findings confirm previous hypotheses stating that viruses may be at the origin of many eukaryotic genes (Villarreal and De Filippis, 2000; Forterre, 2006) and might have contributed to nucleus formation (Bell, 2001; Takemura, 2001). Thus, the study of giant virus genomes sheds light on the origin of eukaryotes and emphasizes the possible role played by capsid-containing organisms in the evolution of ribosome-encoding organisms.
Metagenomics and Microbial Diversity
The study of many species is difficult or even impossible, mainly due to our inability to culture them in the laboratory (Zengler et al., 2005). Metagenomics, or the culture-independent genomic analysis of an assemblage of organisms, allows us to study microorganisms by deciphering their genetic information from DNA that is extracted directly from communities of environmental microorganisms. Metagenomics has revealed that the vast majority of microbial diversity has been missed using cultivation-based methods (Handelsman, 2004; Riesenfeld et al., 2004; Eckburg et al., 2005). Indeed, approximately 10 and 60% of the sequences from environmental microbial and viral metagenomes, respectively, are novel sequences; they have no significant similarity to any sequence in the GenBank non-redundant database (Tyson et al., 2004; Venter et al., 2004; Edwards and Rohwer, 2005). Thus, our knowledge has been gleaned from the relatively small number of presently culturable representatives while ignoring the “uncultured majority” (Hugenholtz et al., 1998).
Metagenomics has offered unprecedented insights into microbial diversity and sparked a revolution in the field of microbiology. Historically, microbiology has focused on single species in pure laboratory cultures; thus, the understanding of microbial communities has lagged behind the understanding of their individual members. In addition, limited information about physiology and functional roles can be gained from microbes in culture. Metagenomics is a new tool to study microbes in the complex communities in which they live and to begin to understand how these communities work. Indeed, metagenomics relies on high-throughput sequencing, which permits the isolation of large portions of genomes, providing access to protein-coding genes and biochemical pathways. Metagenomics focuses on profiling the functions encoded by a microbial community in a selected environment rather than the types of organisms producing them. Analysis of the genomic content of communities of organisms sheds light on the metabolic variability of an environment and on specific physiological functions (Eckburg et al., 2005; Dinsdale et al., 2008). Metagenomic studies of the pathogen-associated microbiome have allowed for an understanding of the role of microbial communities and their clinical implications (Gill et al., 2006; Ley et al., 2006; Turnbaugh et al., 2006; Willner et al., 2009). Information from metagenomic libraries has the ability to enrich our knowledge and has applications in many aspects of industry, therapeutics, and environmental sustainability.
Metagenomic approaches have revealed insights into environmental features with important evolutionary implications. Metagenomic functional analyses of ecosystems have revealed the correlation between geochemical conditions, metabolic capacity, and genetic diversity in microbial communities (Edwards et al., 2006; Frias-Lopez et al., 2008; Simon et al., 2009). Indeed, sequencing projects provide a means for sampling the genetic diversity of natural microbial populations by estimating the rate of recombination and have the potential to reveal much about the evolution of these populations (Johnson and Slatkin, 2009). Moreover, this gene-centric approach to environmental sequencing suggests that the functional profile predicted from environmental sequences of a community is similar to that of other communities whose environments of origin pose similar demands. These findings have provided insight into the processes of adaptation and the evolution of life on earth.
Notably, metagenomics represents a powerful tool that can be used to access the abundant biodiversity of environmental samples, but its accuracy depends on many limitations. Technical and economic constraints limit the depth of analysis necessary for obtaining a representative picture of microbial and viral communities, their metabolic profiles and their adaptation dynamics (Morgan et al., 2010). Indeed, large-scale sequencing of metagenomic DNA permits the isolation of the most predominant species in the environment, while sequences from low-abundance species may go undetected. In this way, only the most frequently represented functional genes and metabolic pathways that are relevant in a given ecosystem can be identified and assessed. However, the low-abundance species and their encoded functions could also play a critical role in the ecology and physiology of the studied environment (Piganeau and Moreau, 2007).
In conclusion, metagenomics has shown that the uncultured microbial world far outnumbers the cultured world and has emphasized the extent of our ignorance about the microbial world. Metagenomics has helped elucidate the structure, function, and interactions of microbial communities; these advances were not possible in the culture-dependent era. Metagenomics constitutes a comprehensive approach for understanding evolution and physiology.
Orphan Genes
Orphan genes constitute a class of lineage-specific genes that do not show homology to sequences in other species (Fischer and Eisenberg, 1999). They typically encode small proteins and show high non-synonymous substitution rates, but their functions are unknown (Domazet-Loso and Tautz, 2003; Daubin and Ochman, 2004). Recently, a classification of ORFans has been proposed, dividing ORFans into singletons, multipletons, and lineage ORFans (Boyer et al., 2010b). Each newly sequenced genome contains significant numbers of such genes (Toll-Riera et al., 2009). For example, of 60 fully sequenced microbial genomes, 14% of genes are species-specific orphans (Siew and Fischer, 2003), while 18% of genes in Drosophila are restricted to the Drosophila group (Zhang et al., 2007; Zhou et al., 2008). However, the origin of orphan genes remains a mystery (Merkeev and Mironov, 2008). One proposed scenario is that they derived from gene duplication events in which one copy accumulated so many sequence changes that the ancestral similarity is no longer detectable (Domazet-Loso and Tautz, 2003). It was recently proposed that such ORFans could also represent genes of viral or plasmid origin (Rocha et al., 2006), and some seem to correspond to truly new genes formed de novo through diverse mechanisms of gene evolution (Boyer et al., 2010b). This mechanism has been proposed to have made a significant contribution to the formation of novel genes in mammals, specifically in primates, in which 5.5% of orphan genes could have originated de novo from non-coding genomic regions (Toll-Riera et al., 2009). The formation of novel genes has also been described in Drosophila (Begun et al., 2006; Levine et al., 2006; Zhou et al., 2008) and Saccharomyces cerevisiae (Cai et al., 2008).
In a recent study in our laboratory, we identified a small number of gene sequences in Rickettsia species that had no match in any database and that seem to have resulted from de novo creation (Georgiades et al., 2011). Indeed, 17 rickettsial gene sequences seem to have no homologs in the NR database. The Ka/Ks ratio revealed that 15 of these sequences were either non-functional or had adopted functionality. Of course, the probability of pseudogenization or even of a possible viral origin of these genes should not be excluded, but because these genes were not found in regions with traces of active or ancient integrated extra-chromosomal elements, we strongly believe that they are novel genes (Georgiades et al., 2011).
Finally, it has been reported that new genes might be essential to an organism’s viability. In the case of Drosophila, 59 de novo genes were found to be as essential as the old genes in terms of viability. The observation of lethal phenotypes caused by the knockout of new genes suggested that de novo-created genes may integrate a vital pathway by interacting with existing genes, and this co-evolution may lead to the new gene becoming indispensable (Chen et al., 2010).
In summary, gene creation is a continuous and unsettled phenomenon, and this idea is supported by the discovery of new genes, which are permanently generated and whose identification is becoming increasingly frequent (Raoult, 2010a; Boyer et al., 2010b). De novo-created genes are evidence of life’s permanent creativity.
The Tree of Life
The TOL was used by Darwin approximately 150 years ago, as a concept to explain the evolutionary relationships between different species (Doolittle, 1999; Lawton, 2009). It has been accepted as a biological fact since (Doolittle and Bapteste, 2007). According to Darwin’s theory, namely the “descent with modification theory” (Penny, 2011), the common descent of species is demonstrated by similarities between species, while modifications driven by natural selection create differences in species that result in speciation (Doolittle and Bapteste, 2007). The TOL is therefore composed of a common ancestor, the root of the tree, species separated quickly and in a stable way, key branches, and branches containing the most recently arisen species (Raoult, 2010b,c). However, evidence acquired using comparative genomic analyses contradicts the existence of a single common ancestor for the gene repertoire of any organism. Evidence obtained through genomic analyses suggests that nearly all genes have been exchanged or recombined at some point and that there are no two genes with a similar history on the phylogenetic tree (Raoult, 2010c).
Since the late 1990s, LGT and gene loss in bacterial genomes have been recognized as much more frequent than previously proposed (Ochman et al., 2000; Lawrence, 2005; Dagan and Martin, 2007). Up to 30% of the genome-to-genome variation within a species is the result of LGT and gene loss, and homologous recombination is now thought to be the first cause of sequence divergence in many bacteria (Doolittle and Bapteste, 2007). Thus, LGT had been considered a rare phenomenon in intracellular bacteria (Audic et al., 2007) until the discovery of the mobilome in Rickettsiae, suggesting that such events were possible (Merhej and Raoult, 2010). Consequently, several further studies identified candidates for LGT in Rickettsia species (Wolf et al., 1999; Ogata et al., 2006; Blanc et al., 2007a,b; Georgiades et al., 2011). Moreover, genetic elements invade and proliferate in rickettsial genomes and eventually integrate genes into their host’s chromosomes (Merhej and Raoult, 2010). Analysis of the R. felis genome has provided evidence for gene transfers between the chromosome and the R. felis plasmid, while the plasmids themselves seem to have been acquired through conjugation (Ogata et al., 2005). The first evidence for LGT in R. bellii also indicated the role of amebae in gene exchanges; amebae constitute a melting pot in which species can exchange genetic material (Ogata et al., 2006; Moliner et al., 2010). Indeed, the genome of R. bellii contains many genes highly similar to those of intracellular bacteria of amebae, such as Legionella pneumophila and Protochlamydia amoebophila (Ogata et al., 2006). L. pneumophila has developed the ability to infect different species of amebae (Rowbotham, 1980; Fields et al., 2002). A recent study on L. pneumophila provided evidence for non-vertical inheritance: 34–57% of the genome has been involved in recombination events. In this study, LGT events between Legionella and all bacterial groups known to be present in amebae were detected (Coscolla et al., 2011). In parallel, other studies have identified eukaryotic-like genes in Legionella that are most likely of amebal origin (Lurie-Weinberger et al., 2010; Moliner et al., 2010; Schmitz-Esser et al., 2010). The most plausible scenario for the multiple phylogenetic origins of an important fraction of Legionella genes is the exchange of genetic material in the common ameba host.
These lateral transfer events do not always involve whole genes or certain gene functions. The R. felis paradigm is the first rickettsial genomic analysis in which random transfers of DNA sequences were found to occur independently of gene functions or sequence lengths (Merhej et al., 2011). The functional vision of genes and sequences often influences scientists’ analytical strategies and interpretations. Some bacterial genomes contain up to 40% of genes with no apparent function aside from duplication (selfish genes; Raoult, 2010b). Likewise, random sequences could have hybridized between species because of their sympatric lifestyle (Mayr, 1957).
In light of these post-genomic data, a post-Darwinist concept should be introduced, one that assimilates the chimerism and mosaic structure (Figure 5) of all living organisms through both non-vertical inheritance and de novo creation (Raoult, 2010c). The TOL is a biblical phrase (Penny, 2011) that matches well the desire to have classification reflecting the “natural order” that is inclusively hierarchical and goes back to a single ancestor (Doolittle, 1999). Our current genomic knowledge no longer matches with Darwin’s representation of the TOL. Species evolution looks much more like a rhizome (Deleuze and Guattari, 1976; Raoult, 2010c), reflecting all of the various origins of genomic sequences in each species (Raoult, 2010c). Every living organism has a variety of ancestors; exchanges between species are intense, and the creation of new genes is frequent and constant in all organisms. For example, the human genome is a chimera and viruses and bacterial species are also our ancestors. Retroviruses left relics in our genomes, in the same way that both HHV-6A and B viruses can integrate into human chromosomes and may be vertically transmitted in the germ line (Arbuckle et al., 2010). Trypanosoma cruzi sequences were also integrated and identified into the genomes of patients (Hecht et al., 2010). Therefore, the definition of a common ancestor should be revised and instead of referring to a single ancestor, refer to viral ancestors, bacterial ancestors, eukaryotic ancestors, and archaeal ancestors.
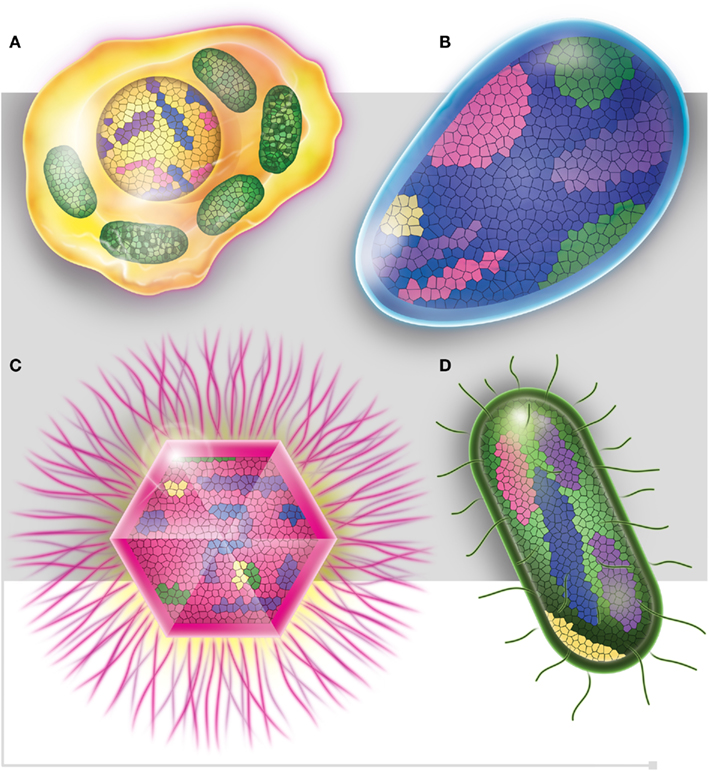
Figure 5. Each one of the four domains of life, (A) Eukaryotes (in yellow), (B) Archaea (in blue), (C) Viruses (in pink), and (D) Bacteria (in green), is represented as mosaics containing genes from all four domains. Purple squares represent ORFan genes.
Conclusion
We think that the radical approach developed by the post-modern French philosophers is useful at this time, as technology has allowed for important discoveries. From this perspective, rickettsiologists, virologists, and bacteriologists, all of whom have different points of view, can make a real contribution to their fields and to the study of the evolution of living organisms. Without the adoption of a non-traditional vision, a large proportion of living organisms, which are now within reach, will remain invisible because we will be trapped by the theories of the twentieth century. Objects are constrained by their definitions. For example, giant viruses were missed by scientists and were not identified earlier because of the misleading definitions of viruses that wanted them to be filterable and smaller than 200 nm (Lwoff, 1957) If the definitions are false, like we demonstrated for the great denominations of microbiology, objects cannot be conceived in a reasonable way and the conclusions derived from the observations of the microorganisms will be biased by misleading believes and theories.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Christelle Forzales for her help with Figure 5.
References
Acinas, S. G., Marcelino, L. A., Klepac-Ceraj, V., and Polz, M. F. (2004). Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J. Bacteriol. 186, 2629–2635.
Adékambi, T., Drancourt, M., and Raoult, D. (2008). The rpoB gene as a tool for clinical microbiologists. Trends Microbiol. 17, 37–45.
Andersson, J. O., and Andersson, S. G. (1999). Genome degradation is an ongoing process in Rickettsia. Mol. Biol. Evol. 16, 1178–1191.
Andersson, J. O., and Kurland, C. G. (1998). Reductive evolution of resident genomes. Trends Microbiol. 6, 263–268.
Andersson, S. G., Zomorodipour, A., Andersson, J. O., Sicheritz-Pontén, T., Alsmark, U. C., Podowski, R. M., Näslund, A. K., Eriksson, A. S., Winkler, H. H., and Kurland, C. G. (1998). The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 396, 133–140.
Aravalli, R. N., She, Q., and Garrett, R. A. (1998). Archaea and the new age of microorganisms. Trends Ecol. Evol. (Amst.) 13, 190–194.
Arbuckle, J. H., Medveczkya, M. M., Lukab, J., Hadleya, S. H., Luegmayrc, A., Ablashid, D., Lunde, T. C., Tolare, J., Meirleirf, K. D., Montoyag, J. G., Komaroffh, A. L., Ambrosc, P. F., and Medveczkya, P. G. (2010). The latent human herpesvirus-6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proc. Natl. Acad. Sci. U.S.A. 107, 5563–5568.
Audic, S., Robert, C., Campagna, B., Parinello, H., Claverie, J. M., Raoult, D., and Drancourt, M. (2007). Genome analysis of Minibacterium massiliensis highlights the convergent evolution of water-living bacteria. PLoS Genet. 3, e138. doi: 10.1371/journal.pgen.0030138
Bapteste, E., and Boucher, Y. (2008). Lateral gene transfer challenges principles of microbial systematics. Trends Microbiol. 16, 200–207.
Bapteste, E., O’Malley, M. A., Beiko, R. G., Ereshefsky, M., Gogarten, J. P., Franklin-Hall, L., Lapointe, F. J., Dupré, J., Dagan, T., Boucher, Y., and Martin, W. (2009). Prokaryotic evolution and the tree of life are two different things. Biol. Direct 4, 34.
Bechah, Y., El Karkouri, K., Mediannikov, O., Leroy, Q., Pelletier, N., Robert, C., Médigue, C., Mege, J. L., and Raoult, D. (2010). Genomic, proteomic and transcriptomic analysis of virulent and avirulent Rickettsia prowazekii reveals its adaptive mutation capabilities. Genome Res. 20, 655–663.
Begun, D. J., Lindfors, H. A., Thompson, M. E., and Holloway, A. K. (2006). Recently evolved genes identified from Drosophila yakuba and D. erecta accessory gland expressed sequence tags. Genetics 172, 1675–1681.
Belay, N., Johnson, R., Rajagopal, B. S., Conway de, M. E., and Daniels, L. (1988). Methanogenic bacteria from human dental plaque. Appl. Environ. Microbiol. 54, 600–603.
Bell, P. J. (2001). Viral eukaryogenesis: was the ancestor of the nucleus a complex DNA virus? J. Mol. Evol. 53, 251–256.
Blanc, G., Ogata, H., Robert, C., Audic, S., Suhre, K., Vestris, G., Claverie, J. M., and Raoult, D. (2007a). Reductive genome evolution from the mother of Rickettsia. PLoS Genet. 3, e14. doi: 10.1371/journal.pgen.0030014
Blanc, G., Ogata, H., Robert, C., Audic, S., Claverie, J. M., and Raoult, D. (2007b). Lateral gene transfer between obligate intracellular bacteria: evidence from the Rickettsia massiliae genome. Genome Res. 17, 1657–1664.
Bokum, A. M. C., Movahedzadeh, F., Frita, R., Bancroft, G. J., and Stoker, N. G. (2008). The case for hypervirulence through gene deletion in Mycobacterium tuberculosis. Cell 16, 436–441.
Boyer, M., Madoui, M. A., Gimenez, G., La Scola, B., and Raoult, D. (2010a) , Phylogenetic and phyletic studies of informational genes in genomes highlight existence of a 4 domain of life including giant viruses. PLoS ONE 5, e15530. doi: 10.1371/journal.pone.0015530
Boyer, M., Gimenez, G., Suzan-Monti, M., and Raoult, D. (2010b). Classification and determination of possible origins of ORFans through analysis of nucleocytoplasmic large DNA viruses. Intervirology 53, 310–320.
Boyer, M., Yutin, N., Pagnier, I., Barrassi, L., Fournous, G., Espinosa, L., Robert, C., Azza, S., Sun, S., Rossmann, M. G., Suzan-Monti, M., La Scola, B., Koonin, E. V., and Raoult, D. (2009). Giant Marseillevirus highlights the role of amoebae as a melting pot in emergence of chimeric microorganisms. Proc. Natl. Acad. Sci. U.S.A. 106, 21848–21853.
Cai, J., Zhao, R., Jiang, H., and Wang, W. (2008). De novo origination of a new protein-coding gene in Saccharomyces cerevisiae. Genetics 179, 487–496.
Cavalier-Smith, T. (2002). The neomuran origin of archaebacteria, the negibacterial root of the universal tree and bacterial megaclassification. Int. J. Syst. Evol. Microbiol. 52, 7–76.
Chatton, E. (1925). Pansporella perplexa. Réflexions sur la biologie et la phylogènie des protozoaires. 10e serie. Ann. Sci. Nat. Zool. 7, 1–84.
Chen, S., Zhang, Y. E., and Long, M. (2010). New genes in Drosophila quickly become essential. Science 330, 1682–1685.
Colson, P., and Raoult, D. (2010). Gene repertoire of amoeba-associated giant viruses. Intervirology 53, 330–343.
Coscolla, M., Comas, I., and Gonzales-Candelas, F. (2011). Quantifying nonvertical inheritance in the evolution of Legionella pneumophila. Mol. Biol. Evol. 28, 985–1001.
Cox, C. J., Foster, P. G., Hirt, R. P., Harris, S. R., and Embley, T. M. (2008). The archaebacterial origin of eukaryotes. Proc. Natl. Acad. Sci. U.S.A. 105, 20356–20361.
Cummings, C. A., Brinig, M. M., Lepp, P. W., van de Pas, S., and Relman, D. A. (2004). Bordetella species are distinguished by patterns of substantial gene loss and gene adaptation. J. Bacteriol. 186, 1484–1492.
Dagan, T., and Martin, W. (2007). Ancestral genome sizes specify the minimum rate of lateral gene transfer during prokaryote evolution. Proc. Natl. Acad. Sci. U.S.A. 104, 870–875.
Dahllöf, I., Baillie, H., and Kjelleberg, S. (2000). rpoB-based microbial community analysis avoids limitations inherent in 16s rRNA genes intraspecies heterogeneity. Appl. Environ. Microbiol. 66, 3376–3380.
Darby, A. C., Cho, N. H., Fuxelius, H. H., Westberg, J., and Andersson, S. G. (2007). Intracellular pathogens go extreme: genome evolution in the Rickettsiales. Trends Genet. 23, 511–520.
Daubin, V., and Ochman, H. (2004). Bacterial genomes as new gene homes: the genealogy of ORFans in E. coli. Genome Res. 14, 1036–1042.
DeLong, F. F. (1998). Everything in moderation: archea as “non-extremophiles.” Curr. Opin. Genet. Dev. 8, 649–654.
Dinsdale, E. A., Edwards, E. A., Hall, D., Angly, F., Breitbart, M., Brulc, J. M., Furlan, M., Desnues, C., Haynes, M., Li, L., McDaniel, L., Moran, M. A., Nelson, K. E., Nilsson, C., Olson, R., Paul, J., Brito, B. R., Ruan, Y., Swan, B. K., Stevens, R., Valentine, D. L., Thurber, R. V., Wegley, L., White, B. A., and Rohwer, F. (2008). Functionnal metagenomic profiling of nine biomes. Nature 452, 629–632.
Domazet-Loso, T., and Tautz, D. (2003). An evolutionary analysis of orphan genes in Drosophila. Genome Res. 13, 2213–2219.
Doolittle, W. F. (1999). Phylogenetic classification and the universal tree. Science 284, 2124–2128.
Doolittle, W. F., and Bapteste, E. (2007). Pattern pluralism and the tree of life hypothesis. Proc. Natl. Acad. Sci. U.S.A. 104, 2043–2049.
Doolittle, W. F., and Papke, R. T. (2006). Genomics and the bacterial species problem. Genome Biol. 7, 116.
Drancourt, M., Berger, P., and Raoult, D. (2004). Systematic 16S rRNA gene sequencing of atypical clinical isolates identified 27 new bacterial species associated with humans. J. Clin. Microbiol. 42, 2197–2202.
Dridi, B., Raoult, D., and Drancourt, M. (2011). Archaea as emerging organisms in complex human microbiomes. Anaerobe 17, 56–63.
Dridi, B., Henry, M., El Karkouri, K., Raoult, D., and Drancourt, M. (2009). High prevalence of Methanobrevibacter smithii and Methanosphaera stadtmanae detected in the human gut using an improved DNA detection protocol. PLoS ONE 4, e7063. doi: 10.1371/journal.pone.0007063
Eckburg, P. B., Bik, E. M., Bernstein, C. N., Purdom, E., Dethlefsen, L., Sargent, M., Gill, S. R., Nelson, K. E., and Relman, D. A. (2005). Diversity of the human intestinal microbial flora. Science 308, 1635–1638.
Edwards, R. A., Rodriguez-Brito, B., Wegley, L., Haynes, M., Breitbart, M., Peterson, D. M., Saar, M. O., Alexander, S., Alexander, E. C. Jr., and Rohwer, F. (2006). Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics 7, 57. doi: 10.1186/1471-2164-7-57
Embley, T. M., and Martin, W. (2006). Eukaryotic evolution, changes and challenges. Nature 440, 623–630.
Fields, B. S., Benson, R. F., and Besser, R. E. (2002). Legionella and Legionnaires’ disease: 25 years of investigation. Clin. Microbiol. Rev. 15, 506–526.
Fischer, D., and Eisenberg, D. (1999). Finding families for genomic ORFans. Bioinformatics 15, 759–762.
Forterre, P. (2006). The origin of viruses and their possible roles in major evolutionary transitions. Virus Res. 117, 5–16.
Fournier, P. E., Elkarkouri, K., Leroy, Q., Robert, C., Guimelli, B., Renesto, P., Socolovschi, C., Parola, P., Audic, S., and Raoult, D. (2009). Analysis of the Rickettsia africae genome reveals that virulence acquisition in Rickettsia species may be explained by genome reduction. BMC Genomics 10, 166. doi: 10.1186/1471-2164-10-166
Fournier, P. E., and Raoult, D. (2009). Current knowledge on phylogeny and taxonomy of Rickettsia spp. Ann. N. Y. Acad. Sci. 1166, 1–11.
Fox, G. E., Wisotzkey, J. D., and Jurtshuk, P. (1992). How close is close – 16S ribosomal –RNA sequence identity may no be sufficient to guarantee species identity. Int. J. Syst. Bacteriol. 42, 166–170.
Frias-Lopez, J., Shi, Y., Tyson, G. W., Coleman, M. L., Schuster, S. C., Chisholm, S. W., and DeLong, E. F. (2008). Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. U.S.A. 105, 3805–3810.
Fuerst, J. A. (1995). Planctomycetes-a phylum of emerging interest for microbial evolution and ecology. Microbiology 141, 1493–1506.
Fuerst, J. A. (2005). Intracellular compartmentation in planctomycetes. Annu. Rev. Microbiol. 59, 299–328.
Fuerst, J. A. (2010). Beyond prokaryotes and eukaryotes: planctomycetes and cell organization. Nat. Educ. 3, 44.
Georgiades, K., Merhej, V., El Karkouri, K., Raoult, D., and Pontarotti, P. (2011). Gene gain and loss events in Rickettsia and Orientia species. Biol. Direct 6, 6.
Georgiades, K., and Raoult, D. (2011a). Defining pathogenic bacterial species in the genomic era. Front. Microbiol. 1:151. doi: 10.3389/fmicb.2010.00151
Georgiades, K., and Raoult, D. (2011b). Genomes of the most dangerous epidemic bacteria have a virulence repertoire characterized by fewer genes but more toxin-antitoxin modules. PLoS ONE 6, e17962. doi: 10.1371/journal.pone.0017962
Gill, S. R., Pop, M., Deboy, R. T., Eckburg, P. B., Turnbaugh, P. J., Samuel, B. S., Gordon, J. I., Relman, D. A., Fraser-Liggett, C. M., and Nelson, K. E. (2006). Metagenomic analysis of the human distal gut microbiome. Science 312, 1355–1359.
Glansdorff, N., Xu, Y., and Labedan, B. (2008). The last common universal ancestor: emergence, constitution and genetic legacy of an elucive forerunner. Biol. Direct 3. doi: 10.1186/1745-6150-3-29
Goldberg, M. B., and Theriot, J. A. (1995). Shigella flexneri surface protein IcsA is sufficient to direct actin-based motility. Proc. Natl. Acad. Sci. U.S.A. 92, 6572–6576.
Goldberg, M. B., Theriot, J. A., and Sansonetti, P. J. (1994). Regulation of surface presentation of IcsA, a Shigella protein essential to intracellular movement and spread, is growth phase dependent. Infect. Immun. 62, 5664–5668.
Golding, G. B., and Gupta, R. S. (1995). Protein-based phylogenies support a chimeric origin for the eukaryotic genome. Mol. Biol. Evol. 12, 1–6.
Gribaldo, S., Poole, A. M., Daubin, V., Forterre, P., and Brochier-Armanet, C. (2010). The origin of eukaryotes and their relationship with the archea: are we at a phylogenomic impasse? Nat. Rev. Microbiol. 8, 743–752.
Groussin, M., and Gouy, M. (2011). Adaptation to environmental temperature is a major determinant of molecular evolutionary rates in archaea. Mol. Biol. Evol. 28, 2661–2674.
Gupta, R. S. (1998a). Protein phylogenies and signature sequences: a reappraisal of evolutionary relationships among alpha proteobacteria, eubacteria and eukaryotes. Microbiol. Mol. Biol. Rev. 62, 1435–1491.
Gupta, R. S. (1998b). What are archaebacteria: life’s third domain or monoderm prokaryotes related to gram-positive bacteria? A new proposal for the classification of prokaryotic organisms. Mol. Microbiol. 29, 695–707.
Gupta, R. S. (2000). The natural evolutionary relationships among prokaryotes. Crit. Rev. Microbiol. 26, 111–131.
Hacker, J., and Kaper, J. B. (2000). Pathogenicity islands and the evolution of microbes. Annu. Rev. Microbiol. 54, 641–679.
Hale, T. L., Sansonetti, P. J., Schad, P. A., Austin, S., and Formal, S. B. (1983). Characterization of virulence plasmids and plasmid-associated outer membrane proteins in Shigella flexneri. Infect. Immun. 40, 340–350.
Handelsman, J. (2004). Metagenomics: application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 68, 669–685.
Hecht, M. M., Nitz, N., Araujo, P. F., Sousa, A. O., de Cássia Rosa, A., Gomes, D. A., Leonardecz, D., and Teixeira, A. R. L. (2010). Inheritance of DNA transferred from American trypanosomes to human hosts. PLoS ONE 5, e9181. doi: 10.1371/journal.pone.0009181
Horn, M., Collingro, A., Schmitz-Esser, S., Beier, C. L., Purkhold, U., Fartmann, B., Brandt, P., Nyakatura, G. J., Droege, M., Frishman, D., Rattei, T., Mewes, H. W., and Wagner, M. (2004). Illuminating the evolutionary history of chlamydiae. Science 304, 728–730.
Huber, R., Huber, H., and Setter, K. O. (2000). Towards the ecology of hyperthermophiles: biotopes, new isolation strategies and novel metabolic properties. FEMS Microbiol. Rev. 24, 615–623.
Hugenholtz, P., Goebel, B. M., and Pace, N. R. (1998). Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 180, 4765–4774.
Iyer, L. M., Blaji, S., Koonin, E. V., and Aravind, L. (2006). Evolutionary genomics of nucleo-cytoplamsic large DNA viruses. Virus Res. 117, 156–184.
Johnson, P. L., and Slatkin, M. (2009). Inference of microbial recombination rates from metagenomic data. PLoS Genet. 5, e1000674. doi: 10.1371/journal.pgen.1000674
Karaolis, D. K. R., Lan, R. T., and Reeves, P. R. (1994). Sequence variation in Shigella sonnei (Sonnei), a pathogenic clone of Escherichia coli, over 4 continents and 41 years. J. Clin. Microbiol. 32, 796–802.
Karner, M. B., DeLong, E. F., and Karl, D. M. (2001). Archaeal dominance in the mesopelagic zone of the Pacific ocean. Nature 409, 507–510.
Kleba, B., Clark, T. R., Lutter, E. L., Ellson, D. W., and Hackstadt, T. (2010). Disruption of the Rickettsia rickettsii Sca2 autotransporter inhibits actin-based motility. Infect. Immun. 78, 2240–2247.
Koonin, E. V. (2010). The origin and early evolution of eukaryotes in the light of phylogenomics. Genome Biol. 11, 209.
Kuhn, S. T. (1962). The Structure of Scientific Revolutions. Chicago, IL: University of Chicago Press.
La Scola, B., Audic, S., Robert, C., Jungang, L., De, L., Drancourt, M., Birtles, R., Claverie, J. M., and Raoult, D. (2003). A giant virus in amoebae. Science 299, 2033.
La Scola, B., Desnues, C., Pagnier, I., Robert, C., Barrassi, L., Fournous, G., Merchat, M., Suzan-Monti, M., Forterre, P., Koonin, E. V., and Raoult, D. (2008). The virophage as a unique parasite of the giant mimivirus. Nature 455, 100–104.
Lake, J. A. (1988). Origin of the eukaryotic nucleus determined by rate-invariant analysis of rRNA sequences. Nature 331, 184–186.
Lake, J. A., and Rivera, M. C. (1994). Was the nucleus the first endosymbiont? Proc. Natl. Acad. Sci. U.S.A. 91, 2880–2881.
Lang, F. B., Gray, M. W., and Burger, G. (1999). Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 33, 351–397.
Lawrence, J. G. (1999). Gene transfer speciation and the evolution of bacterial genomes. Curr. Opin. Microbiol. 2, 519–523.
Lawrence, J. G. (2005). Common themes in the genome strategies of pathogens. Curr. Opin. Genet. Dev. 15, 584–588.
Lescot, M., Audic, S., Robert, C., Nguyen, T. T., Blanc, G., Cutler, S. J., Wincker, P., Couloux, A., Claverie, J. M., Raoult, D., and Drancourt, M. (2008). The genome of Borellia recurrentis, the agent of deadly louse-borne relapsing fever, is a degraded subset of tick-borne Borrelia duttonii. PLoS Genet. 4, e1000185. doi: 10.1371/journal.pgen.1000185
Levine, M. T., Jones, C. D., Kern, A. D., Lindfors, H. A., and Begun, D. J. (2006). Novel genes derived from noncoding DNA in Drosophila melanogaster are frequently X-linked and exhibit testis-biased expression. Proc. Natl. Acad. Sci. U.S.A. 103, 9935–9939.
Ley, R. E., Turnbaugh, P. J., Klein, S., and Gordon, J. I. (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023.
Lurie-Weinberger, M. N., Gomez-Valero, L., Merault, N., Glockner, G., Buchrieser, C., and Gophna, U. (2010). The origins of eukaryotic-like proteins in Legionella pneumophila. Int. J. Med. Microbiol. 7, 470–481.
Lyotard, J. F. (1979). La condition postmoderne-Rapport sur le savoir. Paris: Les éditions de minuit.
Maurelli, A. T., Fernandez, R. E., Bloch, C. A., Rode, C. K., and Fasano, A. (1998). Black holes and bacterial pathogenicity: a large genomic deletion that enhances the virulence of Shigella spp. and enteroinvasive Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 95, 3943–3948.
Mayr, E. (1957). The Species Problem. Washington, DC: American Association for the Advancement of Science.
Merhej, V., Notredame, C., Royer-Carenzi, M., Pontarotti, P., and Raoult, D. (2011). The rhizome of life: the sympatric Rickettsia felis paradigm demonstrates random transfer of sequences. Mol. Biol. Evol. (in press).
Merhej, V., and Raoult, D. (2010). Rickettsial evolution in the light of comparative genomics. Biol. Rev. Camb. Philos. Soc. 86, 379–405.
Merhej, V., Royer-Carenzi, M., Pontarotti, P., and Raoult, D. (2009a). Massive comparative genomic analysis reveals convergent evolution of specialized bacteria. Biol. Direct 4, 13.
Merhej, V., El Karkouri, K., and Raoult, D. (2009b). Whole genome-based phylogenetic analysis of Rickettsiae. Clin. Micrbiol. Infect. 2, 336–337.
Merkeev, I. V., and Mironov, A. A. (2008). Orphan genes: function, evolution and composition. Mol. Biol. (Mosk.) 42, 127–132.
Miller, T. L., and Wolin, M. J. (1982). Enumeration of Methanobrevibacter smithii in human feces. Arch. Microbiol. 131, 14–18.
Moliner, C., Fournier, P. E., and Raoult, D. (2010). Genome analysis of microorganisms living in amoebae reveals a melting pot for evolution. FEMS Microbiol. Rev. 34, 281–294.
Moran, N. A. (1996). Accelerated evolution and Muller’s rachet in endosymbiotic bacteria. Proc. Natl. Acad. Sci. U.S.A. 93, 2873–2878.
Moran, N. A. (2002). Microbial minimalism: genome reduction in bacterial pathogens. Cell 108, 583–586.
Moreira, D., and Brochier-Armanet, C. (2008). Giant viruses, giant chimeras: the multiple evolutionary histories of Mimivirus genes. BMC Evol. Biol. 8, 12. doi: 10.1186/1471-2148-8-12
Moreira, D., and Lopez-Garcia, P. (2009). Ten reasons to exclude viruses from the tree of life. Nat. Rev. Microbiol. 7, 306–311.
Morgan, J. L., Darling, A. E., and Eisen, J. A. (2010). Metagenomic sequencing of an in vitro-simulated microbial community. PLoS ONE 5, e10209. doi: 10.1371/journal.pone.0010209
Mounier, J., Ryter, A., Coquis-Rondon, M., and Sansonetti, P. J. (1990). Intracellular and cell-to-cell spread of Listeria monocytogenes involves interaction with F-actin in the enterocyte like cell line Caco-2. Infect. Immun. 58, 1048–1058.
Nierman, W. C., DeShazer, D., Kim, H. S., Tettelin, H., Nelson, K. E., Feldblyum, T., Ulrich, R. L., Ronning, C. M., Brinkac, L. M., Daugherty, S. C., Davidsen, T. D., Deboy, R. T., Dimitrov, G., Dodson, R. J., Durkin, A. S., Gwinn, M. L., Haft, D. H., Khouri, H., Kolonay, J. F., Madupu, R., Mohammoud, Y., Nelson, W. C., Radune, D., Romero, C. M., Sarria, S., Selengut, J., Shamblin, C., Sullivan, S. A., White, O., Yu, Y., Zafar, N., Zhou, L., and Fraser, C. M. (2004). Structural flexibility in the Burkholderia mallei genome. Proc. Natl. Acad. Sci. U.S.A. 101, 14146–14251.
Ochman, H., Elwyn, S., and Moran, N. A. (1999). Calibrating bacterial evolution. Proc. Natl. Acad. Sci. U.S.A. 96, 12638–12643.
Ochman, H., Lawrence, J. G., and Groisman, E. A. (2000). Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304.
Ochman, H., Lerat, E., and Daubin, V. (2005). Examining bacterial species under the specter of gene transfer and exchange. Proc. Natl. Acad. Sci. U.S.A. 102, 6595–6599.
Ogata, H., La Scola, B., Audic, S., Renesto, P., Blanc, G., Robert, C., Fournier, P. E., Claverie, J. M., and Raoult, D. (2006). Genome sequence of Rickettsia bellii illuminates the role of amoebae in gene exchanges between intracellular pathogens. PLoS Genet. 2, e76. doi: 10.1371/journal.pgen.0020076
Ogata, H., Renesto-Audiffren, P., Audic, S., Robert, C., Blanc, G., Fournier, P. E., Parinello, H., Claverie, J. M., and Raoult, D. (2005). The genome sequence of Rickettsia felis identifies the first putative conjugative plasmid in an obligate intracellular parasite. PLoS Biol. 3, e248. doi: 10.1371/journal.pbio.0030248
Ogata, H., Renesto-Audiffren, P., Fournier, P. E., Barbe, V., Samson, D., Roux, V., Cossart, P., Weissenbach, J., Claverie, J. M., and Raoult, D. (2001). Mechanisms of evolution in Rickettsia conorii and R. prowazekii. Science 293, 2093–2098.
Paddock, C. D., Sumner, J. W., Comer, J. A., Zari, S. R., Goldsmith, C. S., Goddard, J., McLellan, S. L., Tamminga, C. L., and Ohl, C. A. (2004). Rickettsia parkeri: a newly recognized cause of spotted fever rickettsiosis in the United States. Clin. Infect. Dis. 38, 805–811.
Penny, D. (2011). Darwin’s theory of descent with modification, versus the biblical tree of life. PLoS Biol. 9, e1001096. doi: 10.1371/journal.pbio.1001096
Piganeau, G., and Moreau, H. (2007). Screening the Sargasso Sea metagenome for data to investigate genome evolution in Ostreococcus (Prasinophyceae, Chlorophyta). Gene 406, 184–190.
Pupo, G. M., Lan, R. T., and Reeves, P. R. (2000). Multiple independent origins of Shigella clones of Escherichia coli and convergent evolution of many of their characteristics. Proc. Natl. Acad. Sci. U.S.A. 97, 10567–10572.
Raoult, D. (2010a). Technology-driven research will dominate hypothesis-driven research: the future of microbiology. Future Microbiol. 5, 135–137.
Raoult, D. (2010b). “L’homme, cette chimère: l’évolution selon Darwin relue et corrigée,” in Dépasser Darwin, ed. Plos (Paris: Plon V. France), 15–55.
Raoult, D., Audic, S., Robert, C., Abergel, C., Renesto, P., Ogata, H., La Scola, B., Suzan, M., and Claverie, J. M. (2004). The 1.2-megabase genome sequence of Mimivirus. Science 306, 1344–1350.
Raoult, D., and Boyer, M. (2010). Amoebae as genitors and reservoirs of giant viruses. Intervirology 53, 321–329.
Raoult, D., and Forterre, P. (2008). Redefining viruses: lessons from Mimivirus. Nat. Rev. Microbiol. 6, 315–317.
Raoult, D., La Scola, B., and Birtles, R. (2007). The discovery and characterization of Mimivirus, the largest known virus and putative pneumonia agent. Clin. Infect. Dis. 45, 95–102.
Renvoisé, A., Merhej, V., Georgiades, K., and Raoult, D. (2011). Intracellular Rickettsiales: insights into manipulators of eukaryotic cells. Trends Mol. Med. 10, 573–583.
Riesenfeld, C. S., Schloss, P. D., and Handelsman, J. (2004). Metagenomics: genomic analysis of microbial communities. Annu. Rev. Genet. 38, 525–552.
Rocha, M. T. G., Cooper, J. E., Smith, N. H., and Feil, E. J. (2006). Comparisons of dN/dS are time dependent for closely related bacterial genomes. J. Theor. Biol. 239, 226–235.
Romano, A. H., and Conway, T. (1996). Evolution of carbohydrate metabolic pathways. Res. Microbiol. 147, 448–455.
Rosello-Mora, R., and Amann, R. (2001). The species concept for prokaryotes. FEMS Microbiol. Rev. 25, 39–67.
Rothschild, L. J., and Mancinelli, R. L. (2001). Life in extreme environments. Nature 409, 1092–1101.
Roux, V., Drancourt, M., Stein, A., Riegel, P., Raoult, D., and La Scola, B. (2004). Corynebacterium species isolated from bone and joint infection identified by 16 rRNA gene sequence analysis. J. Clin. Microbiol. 42, 2231–2233.
Roux, V., and Raoult, D. (1997). Rickettsioses as paradigms of new emerging infectious diseases. Clin. Microbiol. Rev. 10, 694–719.
Rowbotham, T. J. (1980). Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae. J. Clin. Pathol. 33, 1179–1183.
Sanogo, Y. O., Zeaiter, Z., Caruso, G., Merola, F., Shpynov, S., Brouqui, P., and Raoult, D. (2003). Bartonella henselae in Ixodes ricinus ticks (Acari: Ixodida) removed from humans, Bellulo province, Italy. Emerg. Infect. Dis. 9, 329–332.
Sapp, J. (2005). The prokaryotic-eukaryotic dichotomy: meanings and mythology. Microbiol. Mol. Biol. Rev. 69, 292–305.
Schirawski, J., Mannhaupt, G., and Münch, K. (2010). Pathogenicity determinants in smut fungi revealed by genome comparison. Science 330, 1546–1548.
Schleper, C., Jurgens, G., and Jonuscheit, M. (2005). Genomic studies of uncultivated Archaea. Nat. Rev. Microbiol. 3, 479–488.
Schmitz-Esser, S., Tischler, P., Arnold, R., Montanaro, J., Wagner, M., Rattei, T., and Horn, M. (2010). The genome of the amoeba symbionts “Candidatus Amoebophilus asiaticus” reveals common mechanisms for host cell interaction among amoeba-associated bacteria. J. Bacteriol. 192, 1045–1057.
Siew, N., and Fischer, D. (2003). Analysis of singleton ORFans in fully sequenced microbial genomes. Proteins 53, 241–251.
Simon, C., Wiezer, A., Strittmatter, A. W., and Daniel, R. (2009). Phylogenetic diversity and metabolic potential revealed in a glacier ice metagenome. Appl. Environ. Microbiol. 75, 7519–7526.
Stackebrandt, E., and Ebers, J. (2006). Taxonomic parameters revisited: tarnished gold standards. Microbiol. Today 33, 152–155.
Takemura, M. (2001). Poxviruses and the origin of the eukaryotic nucleus. J. Mol. Evol. 52, 419–425.
Teysseire, N., Chiche-Portiche, C., and Raoult, D. (1992). Intracellular movements in Rickettsia conorii and R. typhi based on actin polymerization. Res. Microbiol. 143, 821–829.
Tilney, L. G., and Portnoy, D. A. (1989). Actin filaments and the growth, movement and spread of the intracellular bacterial parasite Listeria monocytogenes. J. Cell Biol. 109, 1597–1608.
Toll-Riera, M., Bosch, N., Bellora, N., Castelo, R., Armengol, L., Estivill, X., and Alba, M. M. (2009). Origin of primate orphan genes: a comparative genomics approach. Mol. Biol. Evol. 26, 603–612.
Trevors, J. T., and Abel, D. L. (2004). Chance and necessity do not explain the origin of life. Cell Biol. Int. 28, 729–739.
Turnbaugh, P. J., Ley, R. E., Mahowald, M. A., Magrini, V., Mardis, E. R., and Gordon, J. I. (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031.
Tyson, G. W., Chapman, J., Hugenholtz, P., Allen, E. E., Ram, R. J., Richerdson, P. M., Solovyev, V. V., Rubin, E. M., Rokhsar, D. S., and Banfield, J. F. (2004). Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428, 37–43.
Vallery-Radot, P. (1885). Lois Pasteur: Life and Labours, By His Son in-Law. Tr, From the French By Lady Claud Hamilton. London: Longmans, Green and Co.
Venter, J. C., Remington, K., Heidelberg, J. F., Halpern, A. L., Rusch, D., Eisen, J. A., Wu, D., Paulsen, I., Nelson, K. E., Nelson, W., Fouts, D. E., Levy, S., Knap, A. H., Lomas, M. W., Nealson, K., White, O., Hoffman, J., Parsons, R., Baden-Tillson, H., Pfannkoch, C., Rogers, Y. H., and Smith, H. O. (2004). Environmental genomes shotgun sequencing of the Sargasso Sea. Science 304, 66–74.
Viezens, J., and Arvand, M. (2008). Simultaneous presence of two different copies of the 16S rRNA gene in Bartonella henselae. Microbiology 154, 2881–2886.
Villarreal, L. P., and De Filippis, V. R. (2000). A hypothesis for DNA viruses as the origin of eukaryotic replication proteins. J. Virol. 74, 7079–7084.
Ward, N., Rainey, F., Hedlund, B., Staley, J., Ludwig, W., and Stackebrandt, E. (2000). Comparative phylogenetic analyses of members of the order Planctomycetales and the division Verrucomicrobia: 23S rRNA gene sequence analysis supports the 16S rRNA gene sequence-derived phylogeny. Int. J. Syst. Evol. Microbiol. 50, 1965–1972.
Wicks, R. (2003). Modern French Philosophy: From Existentialism to Postmodernism. Oxford: Oneworld Publications.
Willner, D., Furlan, M., Haynes, M., Schmieder, R., Angly, F. E., Silva, J., Tammadoni, S., Nosrat, B., Conrad, D., and Rohwer, F. (2009). Metagenomic analysis of respiratory tract DNA viral communities in cystic fibrosis and non-cystic fibrosis individuals. PLoS ONE 4, e7370. doi: 10.1371/journal.pone.0007370
Wirth, T., Hildebrand, F., Allix-Béguec, C., Wölbeling, F., Kubica, T., Kremer, K., van Soolingen, D., Rüsch-Gerdes, S., Locht, C., Brisse, S., Meyer, A., Supply, P., and Niemann, S. (2008). Origin, spread and demography of the Mycobacterium tuberculosis complex. PLoS Pathog. 4, e1000160. doi: 10.1371/journal.ppat.1000160
Woese, C. R. (1994). There must be a prokaryote somewhere-microbiology search for itself. Microbiol. Rev. 58, 1–9.
Woese, C. R., and Fox, G. E. (1977). Phylogenetic structure of the prokaryotic domain: the primary kingdoms. Proc. Natl. Acad. Sci. U.S.A. 74, 5088–5090.
Woese, C. R., Fox, G. E., Zablen, L., Uchida, T., Bonen, L., Pechman, K., Lewis, K. B. J., and Stahl, D. (1975). Conservation of primary structure in 16 ribosomal-RNA. Nature 254, 83–86.
Wolf, Y. I., Aravind, L., and Koonin, E. V. (1999). Rickettsiae and Chlamydiae: evidence of horizontal gene transfer and gene exchange. Trends Genet. 15, 173–175.
Zengler, K., Walcher, M., Clark, G., Haller, I., Toledo, G., Holland, T., Mathur, E. J., Woodnutt, G., Short, J. M., and Keller, M. (2005). High-throughput cultivation of microorganisms using microcapsules. Meth. Enzymol. 397, 124–130.
Zhang, G., Wang, H., Shi, J., Wang, X., Zheng, H., Wong, G. K., Clark, T., Wang, W., Wang, J., and Kang, L. (2007). Identification and characterization of insect-specific proteins by genome protein data analysis. BMC Genomics 8, 93. doi: 10.1186/1471-2164-8-93
Keywords: definitions, prokaryotes, virus, archaea, metagenomics, tree of life, bacterial virulence factors, orphan genes
Citation: Georgiades K, Merhej V and Raoult D (2011) The influence of rickettsiologists on post-modern microbiology. Front. Cell. Inf. Microbio. 1:8. doi: 10.3389/fcimb.2011.00008
Received: 05 July 2011; Accepted: 10 October 2011;
Published online: 31 October 2011.
Edited by:
D. Scott Merrell, Uniformed Services University, USAReviewed by:
Antje Flieger, Robert Koch Institute, GermanyPatrick Forterre, Université Paris-Sud, France
Copyright: © 2011 Georgiades, Merhej and Raoult. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Didier Raoult, Unité des Rickettsies URMITE, UMR CNRS 6236, IRD 198, Université de la Méditerranée, 27, Boulevard Jean Moulin, 13385 Marseille Cedex 05, France. e-mail:ZGlkaWVyLnJhb3VsdEBnbWFpbC5jb20=