 Suzana P. Salcedo1,2,3,4†
Suzana P. Salcedo1,2,3,4† María I. Marchesini5† Clara Degos1,2,3
María I. Marchesini5† Clara Degos1,2,3 Matthieu Terwagne6 Kristine Von Bargen1,2,3 Hubert Lepidi7 Claudia K. Herrmann5 Thais L. Santos Lacerda1,2,3,4 Paul R. C. Imbert4
Matthieu Terwagne6 Kristine Von Bargen1,2,3 Hubert Lepidi7 Claudia K. Herrmann5 Thais L. Santos Lacerda1,2,3,4 Paul R. C. Imbert4 Philippe Pierre1,2,3 Lena Alexopoulou1,2,3
Philippe Pierre1,2,3 Lena Alexopoulou1,2,3 Jean-Jacques Letesson6
Jean-Jacques Letesson6 Diego J. Comerci5
Diego J. Comerci5 Jean-Pierre Gorvel1,2,3*
Jean-Pierre Gorvel1,2,3*- 1Aix-Marseille Univ UM 2, Centre d'Immunologie de Marseille-Luminy, Marseille, France
- 2INSERM U 1104, Marseille, France
- 3CNRS UMR 7280, Marseille, France
- 4Bases Moléculaires et Structurales des Systèmes Infectieux, CNRS UMR 5086, Institute of Biology and Chemistry of Proteins, Université Lyon 1, Lyon, France
- 5Instituto de Investigaciones Biotecnológicas Dr. Rodolfo A. Ugalde (IIB-INTECH), Universidad Nacional de San Martín, Consejo Nacional de Investigaciones Científicas y Técnicas, San Martín, Buenos Aires, Argentina
- 6URBM, NARILIS, University of Namur (FUNDP), Namur, Belgium
- 7Laboratoire d'anatomie pathologique-neuropathologique, Aix-Marseille Université, Marseille, France
Several bacterial pathogens have TIR domain-containing proteins that contribute to their pathogenesis. We identified a second TIR-containing protein in Brucella spp. that we have designated BtpB. We show it is a potent inhibitor of TLR signaling, probably via MyD88. BtpB is a novel Brucella effector that is translocated into host cells and interferes with activation of dendritic cells. In vivo mouse studies revealed that BtpB is contributing to virulence and control of local inflammatory responses with relevance in the establishment of chronic brucellosis. Together, our results show that BtpB is a novel Brucella effector that plays a major role in the modulation of host innate immune response during infection.
Introduction
Innate immune recognition of microbial components is critical for the onset of an appropriate immune response against invading pathogens. Key contributors include the toll-like receptor (TLR)/IL-1R superfamily characterized by the presence of a conserved region designated TIR domain located in the cytosolic part of each TLR. The TIR domain is critical for protein-protein interactions between TLRs with the corresponding TIR-containing adaptors, which couple downstream protein kinases. This signaling cascade ultimately leads to activation of specific transcription factors such as nuclear factor-κB (NF-κB) and production of inflammatory mediators. Although a variety of TLR receptors have been described, in humans the most relevant for recognition of bacterial molecules are TLR2, TLR4, TLR5, and TLR9.
In addition to TLRs and their adaptors, TIR domains are present in plant resistance proteins that mediate hypersensitive responses to pathogens, as well as in a variety of bacteria, including species present in the human gut microbiota, soil bacteria and human pathogens (Spear et al., 2009; Zhang et al., 2011). Their evolutionary history is complex and their role in interaction with eukaryotic hosts remains mostly uncharacterized. Nevertheless, in a number of bacterial pathogens, bacterial TIR-containing proteins have been implicated in virulence or control of cellular responses. Salmonella enterica serovar Enteritidis TlpA is capable of reducing NF-κB activation by TLR4, IL-1R and MyD88-dependent pathways and to contribute to control of IL-1β secretion during infection (Newman et al., 2006). In the case of uropathogenic E. coli CFT073, the TIR-containing protein TcpC is able to interfere with TLR4 and TLR2 signaling by targeting MyD88 (Cirl et al., 2008) but also to inhibit TRIF- and IL-6/IL-1 dependent pathways (Yadav et al., 2010). During infection, TcpC is implicated in the control of secretion of TNF-α and IL-6 and tcpC mutants show a defect in intracellular replication in a mouse model of pyelonephritis. The Yersinia pestis TIR-containing protein YpTdp interacts with MyD88 to reduce IL-1β- and LPS-dependent signaling and to contribute to modulation of cytokine secretion during infection (Spear et al., 2012).
In the case of Brucella spp. two groups independently reported on the role of a TIR domain containing protein in control of TLR signaling (Cirl et al., 2008; Salcedo et al., 2008). Naming of this protein as Btp1 or TcpB, respectively, by two distinct laboratories has led to some confusion in the literature and has been misinterpreted by some as two independent proteins. Since neither Btp1 nor TcpB conforms to the international guidelines for bacterial nomenclature we will hereafter designate Btp1/TcpB as BtpA.
BtpA is present in B. abortus 2308 (BAB1_0279), B. abortus 9-941 (BruAb1_0274) and B. melitensis 16 M (BMEI1674) but is absent from B. suis 1330. Cirl et al. described that ectopically expressed BtpA cloned from B. melitensis 16 M is able to interfere with TLR4 and TLR2 signaling by directly interacting with MyD88. Several reports have proposed that BtpA targets the adaptor protein MAL/TIRAP (Radhakrishnan et al., 2009; Sengupta et al., 2010). Direct comparison of the in vitro interaction between BtpA and either MyD88 or TIRAP shows a stronger interaction with MyD88 (Chaudhary et al., 2011). BtpA has been shown to bind phosphoinositides at the plasma membrane (Radhakrishnan et al., 2009) but also to induce ubiquitination of TIRAP (Sengupta et al., 2010). In accordance to its modulation of TLR function, previous work from our laboratory described the role of the BtpA from B. abortus in the control of dendritic cell (DC) activation during infection (Salcedo et al., 2008). Purified BtpA was also shown to inhibit CD8+ T cell-mediated killing suggesting it may also control adaptive immune responses (Durward et al., 2012).
Here we present a novel Brucella effector with a TIR domain that we designated as BtpB. We show that BtpB efficiently inhibits TLR signaling and contributes to control of DC activation. Together, Brucella TIR-containing proteins BtpA and BtpB modulate host inflammatory responses during infection.
Results
Identification of a Second Brucella TIR Domain-Containing Protein
Analysis of the Brucella genome revealed the presence of a second TIR domain-containing protein (BAB1_0756) that we have designated BtpB (Figure 1A). We choose to continue with the Btp nomenclature to avoid any confusion with the tcpB gene necessary for conjugative transfer in Clostridium perfringens (Parsons et al., 2007). Search for conserved domains in BtpB revealed the presence of a C terminal TIR domain (aa 144-256) that belongs to the Pfam family TIR_2 (E value 1.9 e−11), a family of bacterial Toll-like receptors. TIR domains share conserved motifs called box 1 (F/Y-DAFISY), box2 (GYKLC-RD-PG) and box 3 (W residue surrounded by basic amino acids). Sequence comparison of BtpB TIR domain with the human TIR-containing proteins MAL, MyD88, TLR2 and TLR4 showed sequence similarity and conservation of box 1, essential for signaling (Rana et al., 2013) (Figure 1A).
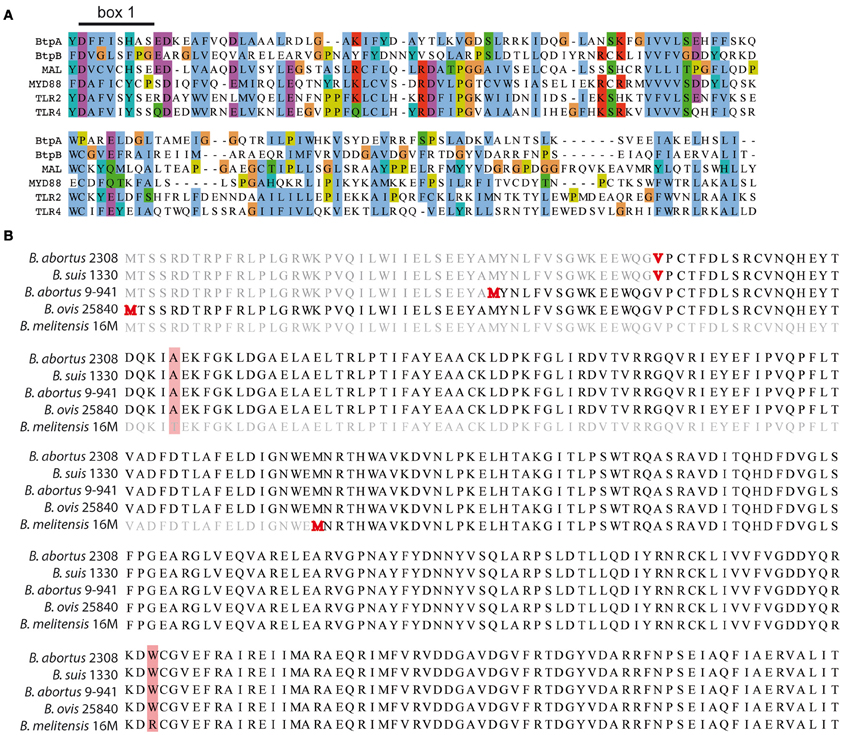
Figure 1. Identification of BtpB. (A) Identification of BtpB as bacterial member of TLR/IL-1R (TIR) family. Comparison of the predicted amino acid sequences of the TIR domain of BtpB with BtpA and the human members of the TIR family: MAL, MyD88, TLR2 and TLR4. The alignment was constructed with T-Coffee::advanced server from EMBnet (http://www.ch.embnet.org) and coloring scheme corresponds to standard ClustalX in which each residue in the alignment is assigned a color if the amino acid profile at each position meets a minimum criteria specific for the residue type. Box 1 corresponds to the signature sequence of the TLR family. (B) Alignment of BtpB amino acid sequences for B. abortus 2308 (BAB1_0756), B. suis 1330 (BR0735), B. abortus 9-941 (BruAb1_0752) and B. melitensis 16M (BME1216). The annotated starting codons (Methionine/Valine) are highlighted in red. Amino acid differences are shaded in red.
Unlike BtpA, BtpB is present in all sequenced Brucella strains, including B. suis 1330 (BR0735), B. abortus 2308 (BAB1_0756), B. abortus 9-941 (BruAb1_0752) and B. melitensis 16 M (BME1216). Alignment of the btpB sequences derived from different Brucella strains revealed 4 different annotations for the start codon (highlighted in red in Figure 1B). Analysis of the −18 to +18 nucleotides around the ATG/GTG (Kolaskar and Reddy, 1985) predicted as the most likely start codon the second methionine highlighted in Figure 1B. This open reading frame has been annotated for B. abortus 9-941 and encodes a 292 amino acid protein, BtpB (1-292). The additional annotated start codons include the first highlighted methionine, the valine (GTG) and the B. melitensis methionine resulting in proteins of either 325, 277 or 178 amino acids. None of them scored high enough to be considered as likely start codons. Comparison of all Brucella sequences available revealed only one BtpB (1-178), in B. melitensis 16 M, whereas the majority correspond to BtpB (1-292). In consequence, we decided to use in this study the BtpB (1-292).
We first investigated the ability of BtpB to interfere with TLR signaling using an in vitro NF-κB-dependent luciferase reporter system. BtpB was able to inhibit TLR2, TLR4 and TLR9 signaling (Figure 2A) even more efficiently than BtpA (Salcedo et al., 2008). This inhibition was independent on the first 114 amino acids as both BtpB (1-178) (Figure 2A), as well as, BtpB (1-292) (Figure 2B) strongly inhibited TLR signaling. BtpB was also able to inhibit flagellin-induced TLR5 signaling (Figure 2B). These results suggest that BtpB may interfere with a common molecule of these TLR pathways, such as MyD88. Consistently, BtpB did not reduce TLR3-dependent signaling which does not involve the adaptor MyD88 (Figure 2C). In addition, we observed by directed yeast two-hybrid that BtpB was able to interact with MyD88 (Figure 2D). BtpA was also able to interact with MyD88 by yeast-two hybrid as previously shown by pull-down and protein-fragment complementation assays (Cirl et al., 2008; Chaudhary et al., 2011). Neither BtpA nor BtpB interacted with any of the TLR1 to TLR10 TIR domains nor with the adaptors TIRAP or TRAM. As BtpA was previously shown to reduce TLR2 and TLR4 signaling but not TLR9 (Cirl et al., 2008; Salcedo et al., 2008) we investigated its ability to interfere with TLR5, which is also dependent on MyD88. BtpA was able to significantly reduce TLR5 signaling, following stimulation with S. typhimurium flagellin (Figure 2E).
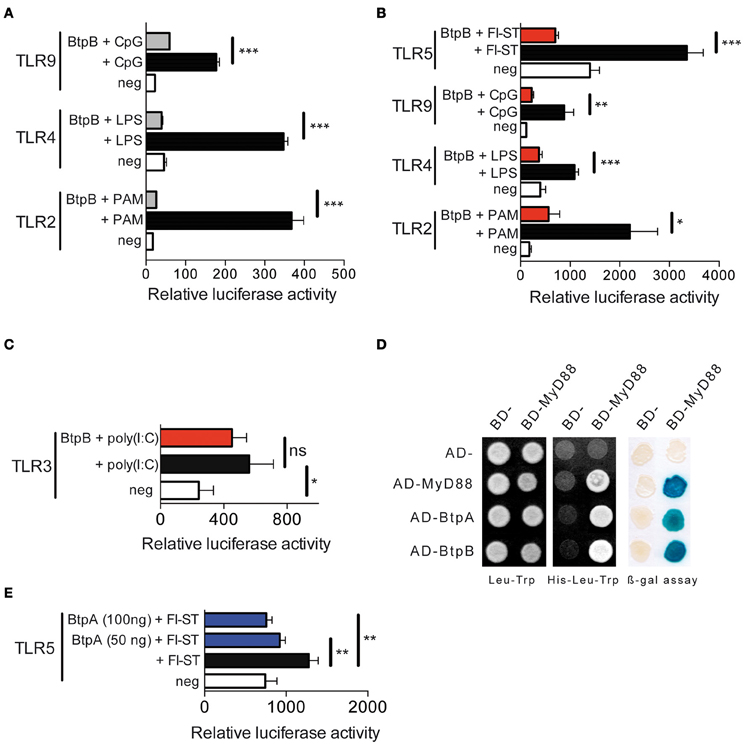
Figure 2. BtpB interferes with TLR signaling. (A) HEK293 cells were transiently transfected for 24 h with the luciferase reporter vector and either TLR2, TLR4 and TLR9, in the presence or the absence of the 178 amino acid BtpB (50 ng). Cells were then stimulated with the appropriate ligand (PAM, LPS and CpG) for 6 h before measurement of luciferase activity. White bars correspond to negative control, black bars to cells stimulated with the appropriate ligand and grey bars to cells transfected with BtpB and stimulated with the ligand. Data represent the means ± standard errors of relative luciferase activity obtained from triplicates of a representative experiment. (B) Luciferase activity in the presence or absence of the BtpB (1-292) (red bars). TLR5 was also included and stimulated with Flagellin from S. typhimurium (Fl-ST) and (C) TLR3 following stimulation with poly(I:C). (D) Yeast containing Gal4 BD- and Gal4 AD-fusion proteins were selected on synthetic medium lacking leucine (Leu) and tryptophan (Trp) (left panel). Protein interactions were identified on synthetic medium lacking histidine (His) and supplemented with 20 mM 3AT (middle panel). Growth on this medium indicates interaction between fusion proteins. The blue yeast colonies observed in the β-galactosidase expression filter assay indicate interaction between the fusion proteins (right panel). BD and AD indicate empty vectors and were used as negative controls, while MyD88 homodimerization was used as positive control. (E) Luciferase activity in cells transfected with TLR5 in the absence or presence of 100 ng and 50 ng of BtpA (275 aa). P ≤ 0.001 are denoted with ***; P ≤ 0.01 are denoted with ** and P between 0.01 and 0.05 are denoted with *.
Overall, our results show that BtpB is a potent inhibitor of TLR signaling in vitro, which may result from binding to MyD88.
BtpB is Translocated into Host Cells
In order for BtpA and BtpB to target TLR pathways they would have to be exported across the bacterial membranes and the vacuolar membrane into the host cell cytosol. To test this hypothesis we analysed the translocation of BtpA and BtpB fused at their N-terminus with the TEM-1 β-lactamase during infection of RAW macrophage-like cells. This method has been successfully used to establish translocation of several Brucella effectors, namely VceA, VceC and RicA (de Jong et al., 2008; de Barsy et al., 2011) and is traditionally carried out in live cells. RAW cells were used in order to achieve high rates of infection. VceC and VceA were included as positive controls. We could detect BtpA translocation into host cells at 4 h and 24 h after inoculation (Figures 3A,B). Translocated BtpB was detected in less than 0.5% of infected cells at 4 h and did not significantly increase at 24 h. In an attempt to try to enhance the sensitivity of this assay, we carried out the same experiments in fixed samples with observation of FRET within 15 min of fixation, which enhances the shift to 450 nm (Nothelfer et al., 2011). As in live cells, BtpA and to a lower extent BtpB were translocated into host cells at 24 h after infection (Figures 3C,D). Since the overall percentage of cells showing translocated effectors is very low with this assay, even following fixation, we analysed translocation of BtpA and BtpB fused to the adenylate cyclase CyaA (Figure 3E). In addition, we used a constitutive promoter of B. abortus bcsp31 gene to enhance expression, since this alternative approach was successfully used with the Brucella effector protein BPE123 (Marchesini et al., 2011), which was included as a positive control in our experiments. In this system, any value of cAMP bellow 1500–2000 fmol/ml corresponds to background (dotted line in Figure 3E) and is not indicative of translocation as determined after performing an exhaustive screening for the identification of Brucella abortus type IV secretion system (T4SS) substrates (Marchesini et al., 2011). We found that at 4 h after infection both BtpA and BtpB were translocated into J774.A1 macrophage-like cells (Figure 3E). Interestingly, translocation of BtpA fused with the CyaA seems to depend on the position of the tag as only BtpA with C-terminal CyaA was efficiently translocated into host cells at early stages of the infection. In contrast, for BtpB, the presence of the CyaA tag on the C-terminus reduced translocation (Figure 3E).
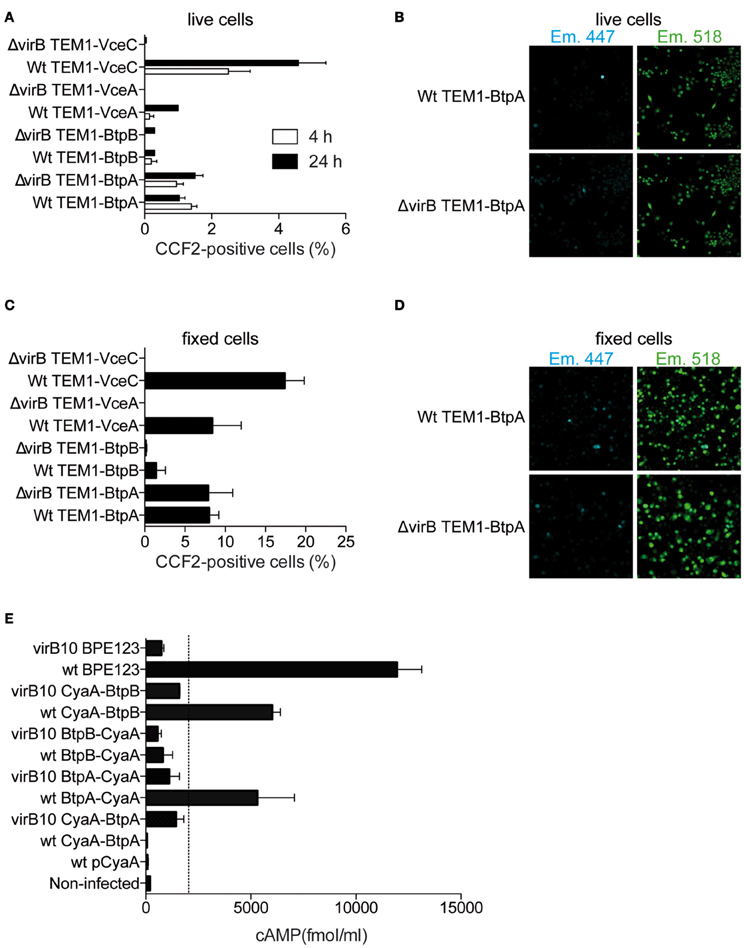
Figure 3. BtpB is translocated into host cells during infection. (A) RAW macrophages were infected with wild type (wt) or ΔvirB9 B. abortus strains carrying N-terminal TEM-1 fused VceA, VceC, BtpA, and BtpB for 4 h and 24 h. Data represents the means ± standard errors of the percentage of cells with coumarin fluorescence from 5 independent experiments. (B) Representative confocal images of RAW cells infected with either wilt-type B. abortus (wt) or Δ virB9 mutant carrying TEM-fused BtpA, at 24 h after inoculation. Appearance of blue cells is indicative of translocated TEM lactamase. (C) and (D) Analysis of TEM-1 translocation assay for fixed samples of VceA, VceC, BtpA, and BtpB 24 h after infection. (E) Intracellular cAMP levels in J774.A1 cells infected for 4 h with isogenic strains with a functional (wt) or non-functional VirB system (virB10) expressing Btp proteins fused to CyaA. Non-infected cells and a wild type strain expressing the CyaA domain alone (pCyaA) were included as negative controls. A wild type strain expressing BPE123-CyaA was included as a positive control. Means and SD are shown for one representative out of three independent experiments.
To determine if the translocation of BtpA and BtpB was dependent on the Brucella VirB T4SS, cells were infected with the virB mutant carrying either TEM- or CyaA-fused Btp proteins. We could not detect any differences between wild type and virB mutant using the TEM-1 β-lactamase assay (Figures 3A–D). In sharp contrast, translocation of BtpA-CyaA and CyaA-BtpB was clearly reduced in a virB genetic background, indicating that delivery of both proteins is dependent on the T4SS. We conclude that BtpA and BtpB are translocated into host cells and may constitute substrates for the VirB T4SS.
BtpB Replication within Murine Bone Marrow-Derived DCs
To determine the role of BtpB during infection we infected murine bone marrow-derived DCs with wild type Brucella, as well as, with a btpA, btpB or btpAbtpB mutant strains. No attenuation was observed as the btpAbtpB replicated to equivalent levels of the wild type B. abortus strain (Figure 4A). The survival curves for the single mutants overlap with that of the wild type (Figure 4A, right panel).
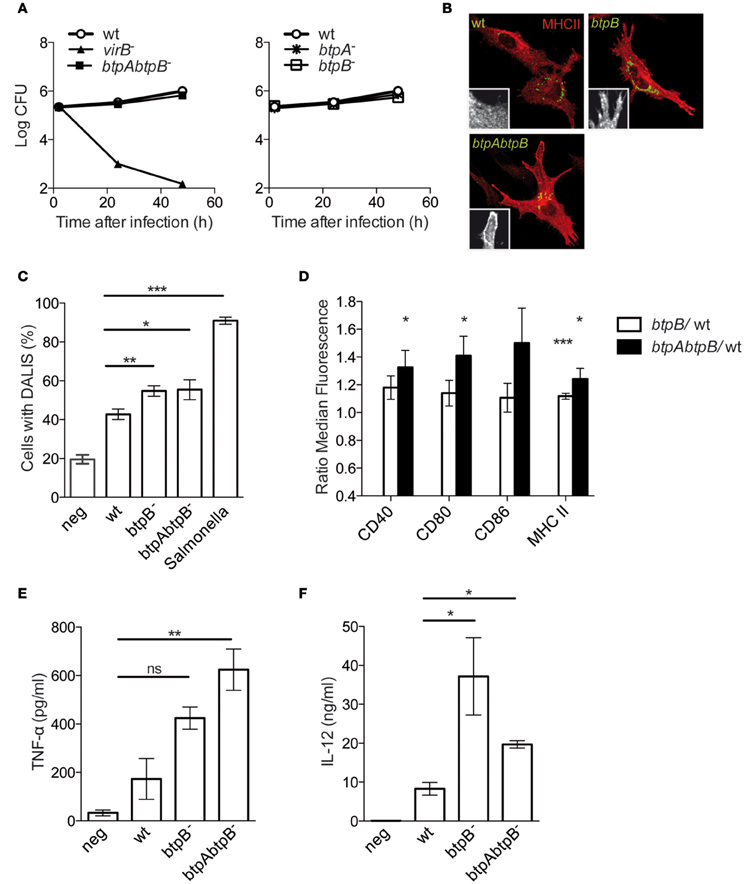
Figure 4. Role of BtpB in control of DC activation. (A) BMDCs infected with wild type B. abortus or the btpAbtpB mutant (left panel) and the single mutants (right panel) were lysed and intracellular CFUs enumerated at different times after inoculation. (B) Representative images of BMDCs infected with either the wild type, btpB or btpAbtpB mutants for 24 h. Cells were labeled for MHC class II (red) and surface expression is of a representative area is shown in zoom inlets. (C) Quantification of the percentage of DCs containing DALIS after 24 h of infection with wild type B. abortus (wt), btpB− or btpAbtpB mutant. (D) Flow cytometry of the surface expression of MHC class II, CD40, CD80 and CD86 at 24 h post-infection. Data are normalized to wt values. (E) Analysis of TNF-α and (F) IL-12 (p40/p70) secretion measured by ELISA from the supernatant of DCs 24 h after inoculation. All the results correspond to the means ± standard errors of 4 independent experiments. P ≤ 0.001 are denoted with ***; P ≤ 0.01 are denoted with ** and P between 0.01 and 0.05 are denoted with *.
As previously described for BtpA, murine DCs infected with the btpB mutant showed higher level of MHC class II surface expression and higher percentage of formation of aggresome-like induced structures (DALIS) that transiently appear during the process of activation of these immune cells (Figures 4B,C) (Lelouard et al., 2002). However, there was no additive effect of depletion of both btpA and btpB as the btpAbtpB mutant did not show an increased phenotype compared to single mutant.
Flow cytometry analysis of infected cells did not reveal a statistically significant increase in CD40, CD80 and CD86 surface expression in DCs infected with btpB mutant when compared to the wild type at 8 h post-infection. At 24 h post-infection there was a significant increase in MHC class II surface expression in DCs infected with btpB mutant relative to those infected with the wild type (Figure 4D, white bars) consistent with our microscopy observations (Figure 4B). In the case of DCs infected with btpAbtpB mutant, CD40 and CD80 co-stimulation markers were up-regulated (Figure 4D, black bars).
In terms of cytokine secretion, BtpB did not seem to be involved in the control of TNF-α secretion during infection of murine DCs (Figure 4E). The increase in TNF-α secretion observed for btpAbtpB is probably due to a lack of BtpA, previously shown to be involved in the control of secretion of this cytokine (Salcedo et al., 2008). However, an increase in the level of total IL-12 (p40/p70) secreted during infection was observed in the case of the btpB mutant compared to the wild type 24 h after infection (Figure 4F). The difference between btpB and btpAbtpB mutants is not significant. These results suggest that BtpB is contributing to the control of the inflammatory response induced in infected DCs in vitro.
BtpB Controls NF-κB Translocation in DCs
In order to analyse the effect of the BtpB effector on the early stages of DC activation, translocation of NF-κB was monitored by immunofluorescence microscopy during the course of the infection. As early as 2 h post-infection, bone marrow-derived DCs infected with the btpB mutant showed an increased translocation of NF-κB into the nucleus compared to those infected with wild type B. abortus (Figure 5). The btpB mutant phenotype was rescued by expression of BtpB from a plasmid confirming the role of BtpB in the control of NF-κB translocation into the nucleus.
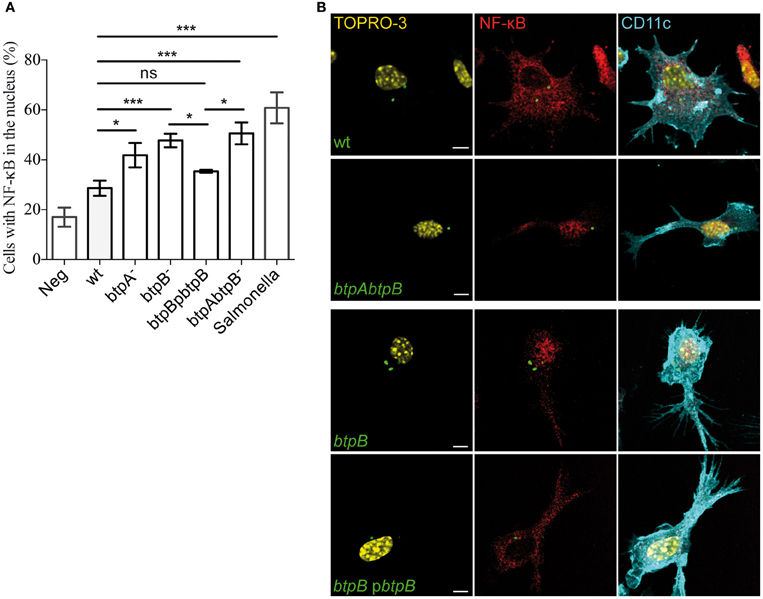
Figure 5. Modulation of NF-κB translocation to the nucleus during Brucella infection. Bone marrow-derived DCs were infected with wild type (wt) B. abortus, btpA, btpB and btpAbtpB mutants as well as btpB mutant carrying the complementing plasmid (pbtpB) for 2 h and processed for immunofluorescence confocal microscopy. Cells were labeled for CD11c (cyan) and p65 NF-κB (red). Bacteria were labeled with anti-LPS antibody followed by FITC secondary and nuclei with TOPRO3. Salmonella infected cells were used as a positive control. (A) Data corresponds to means ± standard errors of 4 independent experiments. (B) Representative images obtained by confocal microscopy are shown for DCs infected with wild type, btpB mutant, btpBpbtpB complemented strain and btpAbtpB mutant. Scale bars correspond to 5 μm. P ≤ 0.001 are denoted with ***; P ≤ 0.01 are denoted with ** and P between 0.01 and 0.05 are denoted with *.
These results confirm that BtpB has an effect on the induction of inflammatory responses during Brucella infection.
Role of BtpB in the Mouse Model of Brucellosis
To further investigate the role of BtpB during infection we carried out in vivo studies. BtpA B. melitensis mutants were previously shown to have enhanced survival in immuno-compromised Interferon Regulatory Factor-1 (IRF-1)−/− mice (Radhakrishnan et al., 2009) inoculated intra-peritoneally (i.p.), a lethality model that has been used for studying Brucella virulence (Ko et al., 2002). We therefore, inoculated IRF−/− i.p. with either btpA, btpB or btpAbtpB B. abortus mutants. Although the btpB mutant had no defect in intracellular replication in cultured cells in vitro, it showed an attenuation phenotype in IRF-1−/− mice. Mice infected with the btpB mutant survived longer than those infected with the wild type B. abortus and synergistic effect was observed for the double btpAbtpB mutant (Figure 6A, P < 0.005), despite equivalent bacterial CFU counts in the spleen at each sampling date after infection (median of 1.05 × 108 CFU/spleen for wild type versus 6.5 × 107 CFU/spleen for the btpAbtpB mutant). To better study the role of BtpB in brucellosis in vivo, we inoculated wild type BALB/c mice i.p. and enumerated the bacterial load at 30, 60, 90, and 130 days post-infection. No significant differences in bacterial CFU counts between the wild type Brucella and the btp mutants were observed at different stages of the infection (30, 60, 90, and 130 days). Data at 60 days post-infection is shown as an example (Figure 6B). We then performed histological examination of spleens obtained from wild type BALB/c mice infected with the btpA, btpB, btpAbtpB mutants or the wild type B. abortus strains to quantify granuloma formation, which usually reflects the host's ability to develop a protective immune response. No granuloma was seen in the spleen of non-infected mice. In infected mice, granulomas were detected in splenic red pulp. A significantly higher number of granulomas was observed following btpB and btpAbtpB infection (after 60 days) compared to the wild type Brucella (Figure 6C). Inflammatory granulomas showed a similar organization in all populations of infected mice and were composed mainly of macrophages and a few lymphocytes (Figure 6D). Bacteria were detected by immunohistochemistry in the spleen of mice infected with wild type B. abortus or with the btpAbtpB mutant (Figure 6E). They were seen as coarse granular immune-positive material associated with cells, which had the morphology of macrophages.
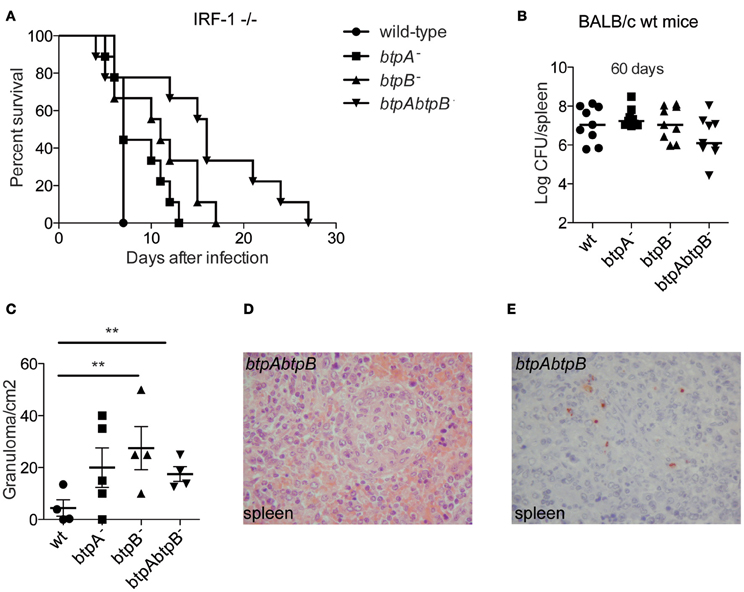
Figure 6. Role of BtpB during Brucella infection in the mouse model of brucellosis. (A) Susceptibility of IRF-1−/− to B. abortus 2308 (wt), btpA−, btpB− and btpAbtpB mutant (n = 9 per group). Infected mice were monitored daily for survival. Mice infected with btpB and tpAbtpB− survived longer than wild type Brucella infected mice (P = 0.0433 and P = 0.0152, respectively). (B) Persistence of B. abortus 2308 (wt), btpA, btpB or btpAbtpB mutants in spleens of wild type BALB/c infected mice at 60 days p.i. Each symbol represents an animal and the median values are marked by horizontal bold lines. (C) Analysis of granuloma formation in the spleens of wild type BALB/c mice infected for 60 days with wild type B. abortus, btpA, btpB or btpAbtpB mutants. Data represent means ± standard deviations of 4 or 5 mice. (D) Representative image from the spleen of a mouse infected with btpAbtpB mutant (hematoxylin-eosin, original magnification ×400). (E) Bacteria were revealed by immunostaining in the spleen of wild type BALB/c mice infected by btpAbtpB mutant of B. abortus. Macrophages present in inflammatory granulomas in the red pulp are packed with coarse immunopositive material (hemalun counterstain, original magnification ×400). P ≤ 0.01 are denoted with **.
These results are consistent with a role for BtpB in the control of inflammatory response during Brucella infection in vivo.
Discussion
Previous work from our laboratory demonstrated a role for BtpA in control of DC activation. Here we show that Brucella contains a second TIR-domain protein called BtpB that is translocated into host cells and which participates in the control of the inflammatory response during Brucella infection. In vitro, BtpB is a potent inhibitor of TLR2, TLR4, TLR5 and TLR9. Together with BtpA, BtpB contributes to the control of DC activation during infection.
Using the TEM-1 lactamase and CyaA assays we were able to detect BtpA and BtpB translocated into the cytosol as early as 4 h after infection of RAW and J774 macrophages. This is an essential step to enable BtpA and BtpB to cross the bacterial and vacuolar membranes to reach their host cellular targets during infection. It would be interesting to localize the translocated proteins during infection. We have not been able to detect neither 2HA- nor 3FLAG-tagged BtpA and BtpB by immunofluorescence microscopy. It is possible that the amounts of translocated BtpA and BtpB are too low or perhaps these proteins are quickly degraded once they reach the host cytosol. It is important to note that using the TEM-1 lactamase assay alone we were unable to detect translocation of BtpB and any differences between wild type and virB mutant in BtpA translocation. It is possible that the low sensitivity of the TEM-1 lactamase compared to the CyaA assay makes this methodology inappropriate to assess VirB dependency in the case of effectors translocated at low levels. We conclude from our results that BtpA and BtpB are likely substrates of the VirB T4SS.
In this study, we found that B. abortus lacking btpA and btpAbtpB mutants showed an increased survival time in the IRF-1 −/− mouse model, highlighting the importance of these TIR-containing proteins in virulence. Similar results were obtained for B. melitensis lacking BtpA, which is defective in systemic spread at early stages of infection (Radhakrishnan et al., 2009). However, the use of such a severely immune-compromised mouse model hampers detailed analysis of the role of these proteins in control of inflammatory responses during infection. Therefore, we proceeded with our in vivo studies using immune-competent mice. We found that absence of BtpA and/or BtpB leads to increased granuloma formation in wild type mice, probably restricting bacterial dissemination as a consequence of the inability of the mutants to modulate the inflammatory response.
Infection of DCs with B. abortus lacking BtpB revealed that this effector protein is contributing to the modulation of the inflammatory response during infection. Interestingly, significant differences were observed between BtpA and BtpB. For example, BtpA had an impact on TNF-α secretion (Salcedo et al., 2008) but not BtpB, which affected surface expression of MHC class II and co-stimulatory molecules that we had not previously seen with BtpA. These differences may be due to different kinetics of translocation and time of action of each effector or perhaps the kinetics of the cellular processes affected. These differences could also be explained by specific targeting of host pathways. Interestingly, translocation of VceC results in enhanced pro-inflammatory responses as a result of the induction of the unfolded protein response by this T4SS effector (de Jong et al., 2012). This suggests that VirB effectors can have opposing effects, resulting in either activation of host immune responses or specific inhibition of inflammatory pathways. These differences may represent host cell or tissue specificity or simply reflect different stages of disease. It is now crucial to undertake a more global analysis of the specific contribution of these effectors during infection and a better characterization of host immune responses elicited in vivo. It is also possible that some of the phenotypes observed with effectors are simply an indirect or secondary effect of their action on eukaryotic cells during infection. Defining at the molecular level the effector cellular targets and analysing their contribution during infection will hopefully shed some light on these issues.
The host interacting partner of BtpA remains controversial. BtpA has been shown to induce degradation of phosphorylated TIRAP by enhancing its poly-ubiquitination (Sengupta et al., 2010) and to efficiently block TIRAP-induced NF-κB activation (Radhakrishnan et al., 2009). Together, these studies present TIRAP as the main target of BtpA whereas other groups have shown a direct interaction with MyD88 (Cirl et al., 2008; Chaudhary et al., 2011). Although comparison of the ability of BtpA to interact with TIRAP and MyD88 revealed a stronger binding to MyD88 (Chaudhary et al., 2011), surprisingly this interaction was dependent on the Death Domain of MyD88 and not the TIR domain. By yeast-two hybrid we found that both BtpA and BtpB can interact with MyD88. In the case of BtpB this result could explain its ability to block TLRs that are dependent on MyD88 signaling but not TLR3, which is dependent on the adaptor TRIF. Although inhibition of TLR2 and TLR4 by BtpA has been described, we did not detect any inhibition of TLR9 (Salcedo et al., 2008), which would be expected if BtpA was blocking MyD88. It is possible that inhibition of TLR9 by BtpA requires higher levels of expression of BtpA and could not be detected with our assay. Consistently, BtpA interfered with TLR5 signaling which is dependent on MyD88. Further work is now required to understand the molecular mechanism by which BtpA controls TLR activation, which may involve interaction and/or competition with both MyD88 and TIRAP.
In addition to control of inflammatory responses, BtpA has been shown to interact with phosphoinositides at the plasma membrane and modulate microtubule dynamics (Radhakrishnan et al., 2009, 2011). Ectopically expressed BtpA localizes to microtubules. These could constitute important activities that may also have a consequence on control of the inflammatory response, for example by misplacing specific adaptor molecules within the cell. In addition, these data indicate that BtpA may have additional eukaryotic targets yet to be identified. It will be interesting to evaluate during infection the contribution of these different functions of BtpA described in vitro and determine if they are dependent on the TIR domain or if other domains are contributing to assigning multiple functions to this effector. Our results strongly implicate BtpB in the control of host inflammatory responses during Brucella infection. However, it is possible BtpB has additional functions as it has been described for BtpA. We are currently determining if multiple pathways are targeted by BtpB to better understand the role of this novel effector during Brucella infection.
Materials and Methods
Bacterial Strains
The bacterial strains used in this study were S. enterica serovar Typhimurium strain 12023, smooth virulent B. abortus strain 2308 (Pizarro-Cerdá et al., 1998) and the isogenic mutants virB9− (Celli et al., 2005), virB10− (Sieira et al., 2000), btpA− (Salcedo et al., 2008), btpB− (this study) and btpA− btpB− (this study). In the case of Brucella, green fluorescent protein (GFP)-expressing derivatives contain a pBBR1MCS-2 (Kovach et al., 1995) derivative expressing the gfp-mut3 gene under the control of the lac promoter. Brucella strains were grown in tryptic soy broth (TSB; Sigma-Aldrich) and Salmonella in Luria Bertani (LB) medium. For infection, we inoculated 2 ml of media for 16 h at 37°C up to an optical density (OD600 nm) of approximately 2.0 (Celli et al., 2003). Salmonella strains were cultured 16 h at 37°C with aeration to obtain stationary phase cultures.
Construction of btpB and btpAbtpB Mutants
The btpB gene (BAB1_0756) was amplified from B. abortus genomic DNA using primers 5'-acgcgacctttccggctccctt-3′ and 5′-ttcggctagacaggaatgcatg-3′ and ligated to pGem-T-Easy vector (Promega) to generate pGem-TbtpB. The plasmid was linearized with EcoRV. Linearized pGem-btpB was ligated to a fragment containing a kanamycin resistance cassette to generate pGem-TbtpB::Kan. This plasmid was electroporated into B. abortus 2308 where it is incapable of autonomous replication. Homologous recombination events were selected using kanamycin resistance (50 μg/ml) and carbenicillin sensitivity (50 μg/ml) in tryptic soy agar plates. PCR and sequencing analyses showed that the btpB wild type gene was replaced by the disrupted one. The mutant strain obtained was called btpB−. btpAbtpB− double mutant was obtained after electropration of pGem-TbtpB::Kan into btpA− mutant (Salcedo et al., 2008). Homologous recombination events were selected as previously described for btpB−single mutant. PCR and sequencing analyses confirmed that btpA and btpB wild type genes were replaced by the disrupted ones in the double mutant strain.
Construction of the btpB Complemented Strain
A DNA fragment coding for BtpB (325 aa) was amplified by PCR using primers 5′-atggatccgtggcgaatgaaccaatccgc-3′ and 5′-gcactagtctaggtgatgagggcgacgcg-3′. The PCR product was inserted by the flanking BamHI/SpeI sites (underlined) in the corresponding sites of pBBR1 MCS-4 (Kovach et al., 1995). The integrity of the construct was confirmed by sequence analysis. The plasmid was introduced into btpB− mutant by biparental mating.
Construction of Tem-1 and CyaA Fusions
DNA fragments coding for VceC, VceA, BtpA (1-275), and BtpB (1-292) were amplified by PCR, digested with XbaI and PstI and cloned into pFlagTEM1 (Raffatellu et al., 2005). Primers sequences with XbaI and PstI sites (underlined) are: VceC-Fw: 5′-tcctctagagaacgttcagagcgtccagaa-3′; VceC-Rv: 5′-aaactgcagctaattgcgggtttctcccttg-3′; VceA-Fw: 5′-tcctctagaaaaatcatcatcacggcagca-3′; VceA-Rv: 5′-aaactgcagctagttcttgggcgcgtggcc-3′; BtpA-Fw: 5′- tcctctagaagttcgtactcttctaatatt-3′; BtpA-Rv: 5′- aaactgcagtcagataagggaatgcagttc-3′; BtpB-Fw: 5′- tcctctagatacaatttatttgtttcgggc-3′; BtpB-Rv: 5′- aaactgcagctaggtgatgagggcgacgcg-3′. The integrity of all constructs was confirmed by sequence analysis. Plasmids were introduced into B. abortus 2308 or ΔvirB9/virB9− by electroporation. pFlagTEM1 encodes a copy of TEM1 β-lactamase, in which the Sec-dependent signal sequence has been deleted and replaced with a 3 × FLAG tag at the N-terminus (Raffatellu et al., 2005). Expression of the fusion proteins in Brucella was confirmed by Western blot using a mouse anti-FLAG M2 antibody (Sigma-Aldrich). To generate plasmids coding for fusions to the N-terminus of CyaA, BamHI/SpeI DNA fragments coding for BtpA (1-275) and BtpB (1-292) were obtained by PCR amplification with primers carrying BamHI/SpeI sites and ligated into the corresponding sites of pCyaA (Marchesini et al., 2011). Primers sequences with BamHI and SpeI sites (underlined) are: BtpA-Fw: 5′-atggatccatgagttcgtactcttctaata-3′; BtpA-Rv: 5′-ggactagtgataagggaatgcagttcttt-3′; BtpB-Fw 5′- atggatccatgtacaatttatttgtttcgggc-3′; BtpB-Rv: 5′-ggactagtggtgatgagggcgacgcgctc-3. To generate plasmids coding for fusions to the C-terminus of CyaA, the genes coding for BtpA (1-275) and BtpB (1-292) with flanking XbaI and SacII sites (underlined) were amplified using primers BtpA-Fw: 5′-tatctagaatgagttcgtactcttctaatattg-3′/BtpA-Rv: 5′-tccccgcggtcagataagggaatgcagttc-3′ and BtpB-Fw: 5′-tatctagaatgtacaatttatttgtttcgggct-3′/BtpB-Rv: 5′-tccccgcggctaggtgatgagggcgacgcg-3′. The DNA fragment coding for CyaA was amplified with flanking BamHI and SpeI sites (underlined) using primers 5′-cgggatccatgcagcaatcgcatcaggct-3′ and 5′-cgactagtaaggctgtcatagccggaatcctggc-3′. DNA fragments coding for CyaA and BtpA or BtpB were ligated in the corresponding sites of pDK51 under bcsp31 gene promoter as described in (Marchesini et al., 2011). The integrity of all constructs was confirmed by sequence analysis. Plasmids expressing CyaA fusion proteins were introduced in B. abortus strains by biparental mating. Expression of the fusion proteins in Brucella was confirmed by Western blot using a mouse serum raised against CyaA.
Bacterial Infection and Replication Assays
BMDCs were prepared from 6-8 week-old female C57BL/6 mice (Lelouard et al., 2002). Infections were performed at a multiplicity of infection of 30:1. Bacteria were centrifuged onto BMDCs at 400 g for 10 min at 4°C and then incubated for 30 min at 37°C with 5% CO2 atmosphere. Cells were gently washed twice with medium and then incubated for 1 h in medium supplemented with 100 μg/ml streptomycin to kill extracellular bacteria (or gentamicin for Salmonella). Thereafter, the antibiotic concentration was decreased to 20 μg/ml. Control samples were always performed by incubating cells with media only and following the exact same procedure for infection. To monitor bacterial intracellular survival, infected cells were lysed with 0.1% Triton X-100 in H2O and serial dilutions plated onto TSB agar to enumerated CFUs.
Immunofluorescence Microscopy NF-kB
Cells were fixed in 3% paraformaldehyde, pH 7.4, at room temperature for 20 min. Cells were then permeabilized for 10 min with 0.1% saponin in PBS, followed by a blocking for 1 h with 2% BSA in PBS. Primary antibodies were incubated for 1 h followed by 3 washes in PBS, 1 h incubation for secondary antibodies, 2 washes in PBS and 1 wash in water before mounting with Prolong Gold (Life technologies). Primary antibodies used: rabbit anti-p65 from Santa Cruz at 1/250, hamster anti-CD11c from BioLegend at 1/100 and cow anti-Brucella LPS antibody at 1/2000. Secondary antibodies used: goat anti-hamster Alexa 594, donkey anti-rabbit Cy3, goat anti-cow FITC, all from Jackson Immunoresearch. Nuclei were stained with TOPRO-3.
Samples were examined on a Leica SP5 laser scanning confocal microscope for image acquisition. Images of 1024 × 1024 pixels were then assembled using Adobe Photoshop 7.0. In all experiments we used an anti-CD11c antibody confirming analysis of DCs only. Quantification was always done by counting at least 100 cells in 4 independent experiments, for a total of at least 400 host cells analysed.
Flow Cytometry of Infected Cells
BMDCs were harvested 8 h or 24 h after infection and stained for 20 min at 4°C with anti CD11c APC-Cy7, anti CD40 Alexa 647, anti CD80 Pe-Cy5, anti CD86 FITC and anti MHC class II PE (all purchased at BioLegend). Cells were then washed once in 1% FCS in PBS and once in PBS. Cells were fixed for 20 min in 3% PFA at room temperature. At least 100,000 CD11c+ events were collected on flow cytometry using a FACS Canto II (Becton Dickinson) and analysis was done on FlowJo software (TreeStar).
Tem Translocation Assay
RAW cells were seeded in a 96 well plates at 1 × 104 cells/well overnight. Cells were then infected with an MOI of 500:1 by centrifugation at 4°C, 400 g for 5 min and 20 min at 37°C 5% CO2. Cells were then washed twice with DMEM and 200 μl of complete media, with gentamicin (50 μg/ml) and 1 mM of IPTG was added for 1 h. Media was replaced by 200 μl of complete DMEM, with gentamicin (10 μg/ml) and 1 mM of IPTG. At 4 or 24 h after infection cells were washed with 100 μl HBSS. 20 μl of CFF2 mix (as described by Life Technologies protocol) was then added to each well, and plate incubated for 1.5 h at room temperature in the dark. Cells were finally washed with 100 μl PBS and analysed immediately by microscopy. A total of 5000 cells were counted from 5 independent experiments in an automated manner using imageJ.
CyaA Assays
Translocation of BtpA and BtpB into host cells was assayed using the CyaA fusion approach. After infection of J774.A1 cells (MOI 250:1) for 4 h in 96-wells plates (105 cells/well), cells were gently washed five times with PBS and lysed. Intracellular cAMP levels were determined by Direct cAMP Enzyme Immunoassay Kit (Sigma, CA200) as described by the manufacturer.
Cytokine Measurement
Sandwich enzyme-linked immunosorbent assays (ELISA) from ebioscience were used to detect IL-12 (p40/p70) and TNFα from supernatants of BMDCs infected with different Brucella strains.
Luciferase Activity Assay
HEK 293 T cells were transiently transfected using Fugene (Roche) for 24 h, according to manufacturer's instructions, for a total of 0.4 μg of DNA consisting of 50 ng TLR plasmids, 200 ng of pBIIXLuc reporter plasmid, 5 ng of control Renilla luciferase (pRL-null, Promega) and 50 ng of myc-BtpA or myc-BtpB expression vectors. The total amount of DNA was kept constant by adding empty vector. Where indicated, cells were treated with E. coli LPS (1 μg/ml), Pam2CSK4 (100 ng/ml), CpG ODN1826 (1 μM), Flagellin Fl-ST (1 μg/ml) and poly(I:C) (25 μg/ml), all obtained from Invivogen, for 6 h and then cells were lysed and luciferase activity measured using Dual-Glo Luciferase Assay System (Promega). The BtpB constructs were obtained by cloning in the gateway (Life Technologies) entry vector and then cloned in pMyc. The following primers were used for BtpB (178 aa) ggggacaagtttgtacaaa
aaagcaggcttcatgaatcgtacgcactgggcg and as reverse primer ggggaccactttgtacaagaaagctgggtcctaggtgatgagggcgacgcg. For BtpB (1-292) the forward primer was ggggacaagtttgtacaaaaaagca
ggcttctacaatttatttgtttcgggc. The BtpA (1-275) primers were: ggggacaagtttgtacaaaaaagcaggcttcatgagttcgtactcttctaatatt and the reverse primer was ggggaccactttgtacaagaaagctgggtctcagat
aagggaatgcagttc.
Yeast Two-Hybrid Assay
The plasmids used for the Y2H interaction test were obtained by using the Gateway™ technique, except the pACT2 vector encoding human MyD88-Gal4 activation domain (AD) fusion that was provided by L. O'Neill. Briefly, human MyD88 and B. melitensis 16 M BtpA (BMEI1674) were amplified by PCR respectively from the pACT2-MyD88 vector and from genomic DNA with Gateway™ primers (GWMyD88F and GWMyD88R; GWbtpAF and GWbtpAR). PCR products were then separately cloned into the entry vector pDONR201 (Invitrogen Life-technologies) as previously described (Dricot et al., 2004). For btpB, the corresponding entry vector pDONR201-BMEI1216 from the ORFeome was used (Dricot et al., 2004). LR reactions were then performed as recommended by the manufacturer (Invitrogen Life-technologies) in order to clone MyD88 into pVV212 (Van Mullem et al., 2003) downstream of the Gal4 DNA-binding domain (BD), and BtpA and BtpB into pVV213 (Van Mullem et al., 2003) downstream of the Gal4 AD. Haploïd Saccharomyces cerevisiae strains Mav103 and Mav203 (Walhout and Vidal, 2001) were transformed with BD and AD fusion protein vectors respectively. Diploid yeasts carrying both plasmids were obtained by mating and selected on synthetic dextrose medium (SD) lacking leucine (leu) and tryptophan (trp) as previously described (Hallez et al., 2007). Protein interactions were assessed on medium lacking histidine (his) supplemented with 20 mM triaminotriazole (3AT). The β-galactosidase expression filter assay using the LacZ reporter gene was performed as described previously (Dozot et al., 2010). The primers used for two-hybrid constructs were: GWMyD88Fggggacaagtttgtaca
aaaaagcaggctcgcgatggctgcaggaggtcccg ;GWMyD88Rggg
gaccactttgtacaagaaagctgggtaagggcagggacaaggccttg ;GWbtpAFggggacaagtttgtacaaaaaagcaggctcgatgagttcgtactcttctaat ;GWbtpARggggaccactttgtacaagaaagctgggtagataagggaatgcagttc.
Mouse Infection Studies
Groups of 7- to 9-week-old female IRF 1−/− or BALB/c mice were intraperitoneally inoculated with 106 CFU of B. abortus strains in 0.2 ml PBS. The infected mice were housed in cages within a biosafety level 3 facility and IRF 1−/− mice were monitored daily for survival. At the indicated times post-infection, spleens from infected mice were removed and homogenized in 2 ml of PBS. Tissue homogenates were serially diluted and plated in duplicate on TSA with the appropriate antibiotic. CFU were counted after 3–4 days of incubation at 37°C.
Histological and Immunohistological Analysis
For each mouse, the spleen was removed, fixed with buffered formalin 4%, and embedded in paraffin. Serial sections (3 μm) of these specimens were obtained for routine hematoxylin-eosin and immunohistochemical investigations to assess the presence of granulomas and bacterial antigens, respectively.
Granulomas were defined as collections of ten or more macrophages within the organs. The inflammatory granulomas present in each tissue section of the spleens were counted during microscopic examination, and the total area of tissue sections was determined by quantitative image analysis as described previously (Stein et al., 2005). The results were expressed as the number of granulomas found per surface unit (i.e., square centimeters). Counts of granulomas were expressed as the mean ± the standard deviation per square centimeter and compared by using the Student t test. Immunohistochemical analysis was performed with a rabbit anti-B. abortus antibody used at a 1:1000 dilution with hemalun counterstain. The immunohistological procedure, in which an immunoperoxidase kit was used, has been described elsewhere (Leone et al., 2004). For each section, a negative control was performed with normal rabbit serum.
Statistical Analysis
Unpaired two-tailed Student's t test was carried out to determine the statistical differences between experimental data sets. P ≥ 0.05 were not considered significant; P ≤ 0.001 are denoted with ***; P ≤ 0.01 are denoted with ** and P between 0.01 and 0.05 are denoted with *. Statistical differences between IRF-1 −/− mice survival curves were determined with Mantel-Cox test.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to R. Tsolis for the pFLAG-TEM1, L. O'Neill for the pACT2 vector encoding human MyD88-Gal4 activation domain (AD) fusion and R. Jerala for the MD2 plasmid. This work was supported by the Agence National Recherche (ANR BruTir), the Centre National de la Recherche Scientifique, the Institut National de la Santé et de la Recherche Médicale and the Aix-Marseille Université. Matthieu Terwagne held a Ph.D. fellowship from the Fonds National de la Recherche Scientifique and his work was supported by an ARC convention from the French-Speaking Community of Belgium (N° 08/13–015). Paul Roger Claude Imbert held a Ph.D. fellowship from FINOVI and TLSL was funded by the ANR grant CELLPATH, awarded under the ERA-NET PathoGenoMics scheme. Diego José Comerci and María Ines Marchesini are members of the Scientific Research Career from Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET, Argentina); Claudia Karina Herrmann is a Ph.D. fellow from CONICET and this work was supported by ANPCyT PICT 2011-0253 and 2011-1485. Kristine Von Bargen was funded by the ANR BruTir and Clara Degos held a fellowship from the Aix-Marseille Université.
References
Celli, J., de Chastellier, C., Franchini, D.-M., Pizarro-Cerda, J., Moreno, E., and Gorvel, J.-P. (2003). Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J. Exp. Med. 198, 545–556. doi: 10.1084/jem.20030088
Celli, J., Salcedo, S. P., and Gorvel, J.-P. (2005). Brucella coopts the small GTPase Sar1 for intracellular replication. Proc. Natl. Acad. Sci. U.S.A. 102, 1673–1678. doi: 10.1073/pnas.0406873102
Chaudhary, A., Ganguly, K., Cabantous, S., Waldo, G. S., Micheva-Viteva, S. N., Nag, K., et al. (2011). The Brucella TIR-like protein TcpB interacts with the death domain of MyD88. Biochem. Biophys. Res. Commun. 417, 1–6.
Cirl, C., Wieser, A., Yadav, M., Duerr, S., Schubert, S., Fischer, H., et al. (2008). Subversion of Toll-like receptor signaling by a unique family of bacterial Toll/interleukin-1 receptor domain-containing proteins. Nat. Med. 14, 399–406. doi: 10.1038/nm1734
de Barsy, M., Jamet, A., Filopon, D., Nicolas, C., Laloux, G., Rual, J.-F., et al. (2011). Identification of a Brucella spp. secreted effector specifically interacting with human small GTPase Rab2. Cell Microbiol. 13, 1044–1058. doi: 10.1111/j.1462-5822.2011.01601.x
de Jong, M. F., Starr, T., Winter, M. G., den Hartigh, A. B., Child, R., Knodler, L. A., et al. (2012). Sensing of bacterial type IV secretion via the unfolded protein response. mBio 4:e00418-12. doi: 10.1128/mBio.e00418-12
de Jong, M. F., Sun, Y.-H., den Hartigh, A. B., van Dijl, J. M., and Tsolis, R. M. (2008). Identification of VceA and VceC, two members of the VjbR regulon that are translocated into macrophages by the Brucella type IV secretion system. Mol. Microbiol. 70, 1378–1396. doi: 10.1111/j.1365-2958.2008.06487.x
Dozot, M., Poncet, S., Nicolas, C., Copin, R., Bouraoui, H., Mazé, A., et al. (2010). Functional characterization of the incomplete phosphotransferase system (PTS) of the intracellular pathogen Brucella melitensis. PLoS ONE 5:e12679. doi: 10.1371/journal.pone.0012679
Dricot, A., Rual, J.-F., Lamesch, P., Bertin, N., Dupuy, D., Hao, T., et al. (2004). Generation of the Brucella melitensis ORFeome version 1.1. Genome Res. 14, 2201–2206. doi: 10.1101/gr.2456204
Durward, M., Radhakrishnan, G., Harms, J., Bareiss, C., Magnani, D., and Splitter, G. A. (2012). Active evasion of CTL mediated killing and low quality responding CD8+ T cells contribute to persistence of brucellosis. PLoS ONE 7:e34925. doi: 10.1371/journal.pone.0034925
Hallez, R., Letesson, J.-J., Vandenhaute, J., and De Bolle, X. (2007). Gateway-based destination vectors for functional analyses of bacterial ORFeomes: application to the Min system in Brucella abortus. Appl. Environ. Microbiol. 73, 1375–1379. doi: 10.1128/AEM.01873-06
Ko, J., Gendron-Fitzpatrick, A., Ficht, T. A., and Splitter, G. A. (2002). Virulence criteria for Brucella abortus strains as determined by interferon regulatory factor 1-deficient mice. Infect. Immun. 70, 7004–7012. doi: 10.1128/IAI.70.12.7004-7012.2002
Kolaskar, A. S., and Reddy, B. V. (1985). A method to locate protein coding sequences in DNA of prokaryotic systems. Nucleic Acids Res. 13, 185–194. doi: 10.1093/nar/13.1.185
Kovach, M. E., Elzer, P. H., Hill, D. S., Robertson, G. T., Farris, M. A., Roop, R. M. 2nd., et al. (1995). Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166, 175–176.
Lelouard, H., Gatti, E., Cappello, F., Gresser, O., Camosseto, V., and Pierre, P. (2002). Transient aggregation of ubiquitinated proteins during dendritic cell maturation. Nature 417, 177–182. doi: 10.1038/417177a
Leone, M., Honstettre, A., Lepidi, H., Capo, C., Bayard, F., Raoult, D., et al. (2004). Effect of sex on Coxiella burnetii infection: protective role of 17beta-estradiol. J. Infect Dis. 189, 339–345. doi: 10.1086/380798
Marchesini, M. I., Herrmann, C. K., Salcedo, S. P., Gorvel, J.-P., and Comerci, D. J. (2011). In search of Brucella abortus type IV secretion substrates: screening and identification of four proteins translocated into host cells through VirB system. Cell Microbiol. 13, 1261–1274. doi: 10.1111/j.1462-5822.2011.01618.x
Newman, R. M., Salunkhe, P., Godzik, A., and Reed, J. C. (2006). Identification and characterization of a novel bacterial virulence factor that shares homology with mammalian Toll/interleukin-1 receptor family proteins. Infect Immun. 74, 594–601. doi: 10.1128/IAI.74.1.594-601.2006
Nothelfer, K., Dias Rodrigues, C., Bobard, A., Phalipon, A., and Enninga, J. (2011). Monitoring Shigella flexnerivacuolar escape by flow cytometry. Virulence 2, 54–57. doi: 10.4161/viru.2.1.14666
Parsons, J. A., Bannam, T. L., Devenish, R. J., and Rood, J. I. (2007). TcpA, an FtsK/SpoIIIE homolog, is essential for transfer of the conjugative plasmid pCW3 in Clostridium perfringens. J. Bacteriol. 189, 7782–7790. doi: 10.1128/JB.00783-07
Pizarro-Cerdá, J., Méresse, S., Parton, R. G., van der Goot, G., Sola-Landa, A., Lopez-Goñi, I., et al. (1998). Brucella abortus transits through the autophagic pathway and replicates in the endoplasmic reticulum of nonprofessional phagocytes. Infect Immun. 66, 5711–5724.
Radhakrishnan, G. K., Harms, J. S., and Splitter, G. A. (2011). Modulation of microtubule dynamics by a TIR domain protein from the intracellular pathogen Brucella melitensis. Biochem. J. 439, 79–83. doi: 10.1042/BJ20110577
Radhakrishnan, G. K., Yu, Q., Harms, J. S., and Splitter, G. A. (2009). Brucella TIR Domain-containing protein mimics properties of the toll-like receptor adaptor protein TIRAP. J. Biol. Chem. 284, 9892–9898. doi: 10.1074/jbc.M805458200
Raffatellu, M., Sun, Y.-H., Wilson, R. P., Tran, Q. T., Chessa, D., Andrews-Polymenis, H. L., et al. (2005). Host restriction of Salmonella enterica serotype Typhi is not caused by functional alteration of SipA, SopB, or SopD. Infect Immun. 73, 7817–7826. doi: 10.1128/IAI.73.12.7817-7826.2005
Rana, R. R., Zhang, M., Spear, A. M., Atkins, H. S., and Byrne, B. (2013). Bacterial TIR-containing proteins and host innate immune system evasion. Med. Microbiol. Immunol. 202, 1–10. doi: 10.1007/s00430-012-0253-2
Salcedo, S. P., Marchesini, M. I., Lelouard, H., Fugier, E., Jolly, G., Balor, S., et al. (2008). Brucella control of dendritic cell maturation is dependent on the TIR-containing protein Btp1. PLoS Pathog. 4:e21. doi: 10.1371/journal.ppat.0040021
Sengupta, D., Koblansky, A., Gaines, J., Brown, T., West, A. P., Zhang, D., et al. (2010). Subversion of innate immune responses by Brucella through the targeted degradation of the TLR signaling adapter, MAL. J. Immunol. 184, 956–964. doi: 10.4049/jimmunol.0902008
Sieira, R., Comerci, D. J., Sánchez, D. O., and Ugalde, R. A. (2000). A homologue of an operon required for DNA transfer in Agrobacterium is required in Brucella abortus for virulence and intracellular multiplication. J. Bacteriol. 182, 4849–4855. doi: 10.1128/JB.182.17.4849-4855.2000
Spear, A. M., Loman, N. J., Atkins, H. S., and Pallen, M. J. (2009). Microbial TIR domains: not necessarily agents of subversion? Trends Microbiol. 17, 393–398. doi: 10.1016/j.tim.2009.06.005
Spear, A. M., Rana, R. R., Jenner, D. C., Flick-Smith, H. C., Oyston, P. C. F., Simpson, P., et al. (2012). A Toll/interleukin (IL)-1 receptor domain protein from Yersinia pestis interacts with mammalian IL-1/Toll-like receptor pathways but does not play a central role in the virulence of Y. pestis in a mouse model of bubonic plague. Microbiology 158, 1593–1606. doi: 10.1099/mic.0.055012-0
Stein, A., Louveau, C., Lepidi, H., Ricci, F., Baylac, P., Davoust, B., et al. (2005). Q fever pneumonia: virulence of Coxiella burnetii pathovars in a murine model of aerosol infection. Infect Immun. 73, 2469–2477. doi: 10.1128/IAI.73.4.2469-2477.2005
Van Mullem, V., Wery, M., De Bolle, X., and Vandenhaute, J. (2003). Construction of a set of Saccharomyces cerevisiae vectors designed for recombinational cloning. Yeast 20, 739–746. doi: 10.1002/yea.999
Walhout, A. J., and Vidal, M. (2001). High-throughput yeast two-hybrid assays for large-scale protein interaction mapping. Methods 24, 297–306. doi: 10.1006/meth.2001.1190
Yadav, M., Zhang, J., Fischer, H., Huang, W., Lutay, N., Cirl, C., et al. (2010). Inhibition of TIR domain signaling by TcpC: MyD88-dependent and independent effects on Escherichia coli virulence. PLoS Pathog. 6:e1001120. doi: 10.1371/journal.ppat.1001120
Keywords: Brucella, TIR domain, Btp1/BtpA, TLR, DC, NF-κB
Citation: Salcedo SP, Marchesini MI, Degos C, Terwagne M, Von Bargen K, Lepidi H, Herrmann CK, Santos Lacerda TL, Imbert PRC, Pierre P, Alexopoulou L, Letesson J-J, Comerci DJ and Gorvel J-P (2013) BtpB, a novel Brucella TIR-containing effector protein with immune modulatory functions. Front. Cell. Infect. Microbiol. 3:28. doi: 10.3389/fcimb.2013.00028
Received: 08 March 2013; Accepted: 18 June 2013;
Published online: 08 July 2013.
Edited by:
Rey Carabeo, University of Aberdeen, UKReviewed by:
Jean Celli, NIAID, NIH, USARenee M. Tsolis, University of California-Davis, USA
Nelson Gekara, Umea University, Sweden
Copyright © 2013 Salcedo, Marchesini, Degos, Terwagne, Von Bargen, Lepidi, Herrmann, Santos Lacerda, Imbert, Pierre, Alexopoulou, Letesson, Comerci and Gorvel. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Jean-Pierre Gorvel, Centre d'Immunologie de Marseille-Luminy, Parc Scientifique et Technologique de Luminy, Case 906, 13288 Marseille Cedex 09, France e-mail:Z29ydmVsQGNpbWwudW5pdi1tcnMuZnI=
†Joint-first authors.