 David S. Terman1* A. Serier2 O. Dauwalder2,3 C. Badiou2 A. Dutour4 D. Thomas2 V. Brun5 J. Bienvenu6 J. Etienne2,3 F. Vandenesch2,3
David S. Terman1* A. Serier2 O. Dauwalder2,3 C. Badiou2 A. Dutour4 D. Thomas2 V. Brun5 J. Bienvenu6 J. Etienne2,3 F. Vandenesch2,3 G. Lina2,3*
G. Lina2,3*- 1Molecular Genetics Program, Jenomic Research Institute, Carmel, CA, USA
- 2CIRI, International Center for Infectiology Research, LabEx Ecofect, Université Lyon1, Inserm U1111, Ecole Normale Supérieure de Lyon, CNRS UMR5308, Lyon, France
- 3Centre National de Références des Staphylocoques, Hospices Civils de Lyon, Bron, France
- 4Unité INSERM U590 équipe Cytokines et Cancer, Centre Léon Bérard, Lyon, France
- 5Laboratoire d'Etude de la Dynamique des Protéomes, U880 CEA/DSV/iRTSV/INSERM/UJF, Grenoble Cedex, France
- 6Laboratoire d'Immunologie, Centre de Biologie et Pathologie Sud, Hospices Civils de Lyon, Chemin du Grand Revoyet, Pierre Benite, France
The egcSEs comprise five genetically linked staphylococcal enterotoxins, SEG, SEI, SElM, SElN, and SElO and two pseudotoxins which constitute an operon present in up to 80% of Staphylococcus aureus isolates. A preparation containing these proteins was recently used to treat advanced lung cancer with pleural effusion. We investigated the hypothesis that egcSEs induce nitric oxide (NO) and associated cytokine production and that these agents may be involved in tumoricidal effects against a broad panel of clinically relevant human tumor cells. Preliminary studies showed that egcSEs and SEA activated T cells (range: 11–25%) in a concentration dependent manner. Peripheral blood mononuclear cells (PBMCs) stimulated with equimolar quantities of egcSEs expressed NO synthase and generated robust levels of nitrite (range: 200–250 μM), a breakdown product of NO; this reaction was inhibited by NG-monomethyl-L-arginine (L-NMMA) (0.3 mM), an NO synthase antagonist. Cell free supernatants (CSFs) of all egcSE-stimulated PBMCs were also equally effective in inducing concentration dependent tumor cell apoptosis in a broad panel of human tumor cells. The latter effect was due in part to the generation of NO and TNF-α since it was significantly abolished by L-NMMA, anti-TNF-α antibodies, respectively, and a combination thereof. A hierarchy of tumor cell sensitivity to these CFSs was as follows: lung carcinoma > osteogenic sarcoma > melanoma > breast carcinoma >neuroblastoma. Notably, SEG induced robust activation of NO/TNFα-dependent tumor cell apoptosis comparable to the other egcSEs and SEA despite TNF-α and IFN-γ levels that were 2 and 8 fold lower, respectively, than the other egcSEs and SEA. Thus, egcSEs produced by S. aureus induce NO synthase and the increased NO formation together with TNF-α appear to contribute to egcSE-mediated apoptosis against a broad panel of human tumor cells.
Introduction
Staphylococcus aureus produces a broad range of exoproteins, including staphylococcal enterotoxins and staphylococcal-like enterotoxins (SEs and SEls; respectively). To date, 23 different SEs have been described: they are designated SE A to X. All these toxins share superantigenic properties by stimulating a large proportion of T cells after binding to the major histocompatibility complex (MHC) class II molecule and crosslinking specific vβ regions of the T-cell receptor (TCR). This interaction results in polyclonal T-cell activation and massive secretion of cytokines such as interleukin-2 (IL)-2, interferon gamma (IFN-γ), tumor necrosis factor alpha (TNF-α), and nitric oxide (NO) (Marrack and Kappler, 1990). Several members of this group have been implicated in the pathogenesis of toxic shock syndrome and food poisoning, and have shown anti-tumor activity in animal models (Bohach, 2006; Terman et al., 2006). The egcSEs comprise five genetically linked staphylococcal enterotoxins, SEG, SEI, SElM, SElN and SElO and two pseudotoxins which constitute an operon present in up to 80% of S. aureus isolates (Jarraud et al., 2001; Becker et al., 2003). The egcSEs are structurally homologous and phylogenetically related to classic SEA-E and exhibit unique vβ signatures (Jarraud et al., 2001). Despite their prevalence and broad distribution, human serum levels of neutralizing antibodies directed against the egcSEs are significantly lower than those directed to the classic SEs (Holtfreter et al., 2004). This has been ascribed to defective mRNA transcription and impaired extracellular secretion (Grumann et al., 2008; Xu and McCormick, 2012). Interestingly, septicemia associated with the egcSEs has been reported to be less severe clinically than that linked to the classic SEs (Ferry et al., 2008).
Nitric Oxide (NO) is a pleiotropic molecule that mediates a broad spectrum of biologic functions including vasodilatation, neurotransmission, and immune defense (Moncada and Higgs, 1993; Bogdan, 2001). NO is produced by mammalian cells from one of the NG-guanidino nitrogens of L-arginine, in a reaction catalyzed by a NADPH-dependent dioxygenase and referred to as NO synthase (Kwon et al., 1990). The latter can exist in at least two distinct isoforms the first of which is a calcium-dependent NO synthase present mainly in neuronal cells (Bredt and Snyder, 1990) and vascular endothelial cells (Förstermann et al., 1991). The second enzyme is a calcium-independent inducible NO synthase found in macrophages (Marletta et al., 1988), hepatocytes (Billiar, 1990), endothelial cells (Radomski et al., 1990), and smooth muscle cells (Busse and Mülsch, 1990) after activation by bacterial lipopolysaccharide (LPS) or cytokines. NO from inducible NO synthase is responsible for killing microbial pathogens and tumor cells by activated macrophages (Hibbs et al., 1987, 1988; Nathan and Hibbs, 1991) and is further involved in the pathogenesis of LPS- or cytokine-induced hypotension and shock (Thiemermann and Vane, 1990). Tumor-associated NO, produced by tumor cells and/or host cells that permeate tumors, exerts both inhibitory and activating effects on carcinogenesis, tumor growth, angiogenesis, and metastases that appear to be concentration dependent. For example, activated macrophages and endothelial cells may produce cytotoxic levels of NO in vitro and prevent tumor growth and metastasis, presumably by killing tumor cells arrested or passaging through blood vessels or sinusoids (Hibbs et al., 1987; Xie and Fidler, 1998; Bogdan, 2001). Over-production of endogenous NO by tumor cells is auto-cytotoxic and suppresses tumor growth and metastasis (Hibbs et al., 1987; Xie and Fidler, 1998; Shi et al., 1999; Motterlini et al., 2000; Fukumura et al., 2006; Ma et al., 2010). Conversely, low levels of NOS II expression appear to promote tumor progression by interdicting tumor cell apoptosis (Billiar, 1990), altering blood vessel formation and tumor vasomotor tone (Busse and Mülsch, 1990; Radomski et al., 1990). The net effect of NO on tumor growth or apoptosis depends not only on its source and relative levels but also on the degree of activation of HIF1-α, heme oxygenase-1 (HO-1) and VEGF in the tumor microenvironment (Tamir and Tannenbaum, 1996; Ambs et al., 1997; Fukumura and Jain, 1998; Wink et al., 1998; Xie and Fidler, 1998).
Superantigens have been shown to induce tumor cell cytotoxicity in vitro and in vivo using several mechanisms. These include superantigen dependent cellular cytotoxicity (SDCC) wherein SAgs efficiently bind MHC class II-positive tumor cells and subsequently trigger human T cell proliferation and differentiation into cytotoxic T cells that kill tumor cells in a perforin/granzyme dependent manner (Dohlsten et al., 1995). In addition, MHCII deficient tumor cells are activated by selected SEs to express CD-54 which costimulates T cells in a vβ specific manner (Lamphear et al., 1998). T cell activation under these conditions may also be enhanced via a newly recognized B7- domain present in selected SEs which interfaces with T cell costimulatory receptor CD28 (Arad et al., 2011). Moreover, SAgs activated T cells and monocytes also produce various cytolytic cytokines notably IFN-γ, TNF-α, IL-2 which alone or together with nitrous oxide can induce cytotoxicity in both MHCII+ and MHCII- tumor cells (Fast et al., 1991; Dohlsten et al., 1993). Whereas SDCC is effective only against MHCII+ tumor cells (largely B cell lymphomas), cytokines and NO are able to kill MHCII− carcinomas, fibrosarcomas and mastocytomas (Fast et al., 1991; Dohlsten et al., 1993, 1995).
Together with neutralizing antibodies against superantigens, the very same cytokines that mediate tumoricidal effects are considered to be the major factors underlying the constitutional and hemodynamic toxicity of the SEs in vivo (Bette et al., 1993; Miethke et al., 1993; Giantonio et al., 1997; Cheng et al., 2004). In the last decade, modification of SEA's MHCII binding affinity has led to reduced toxicity in vivo (Abrahmsen et al., 1995; Erlandsson et al., 2003). Newer versions of SEA devoid of epitopes for preexisting neutralizing antibodies are presently being tested in clinical trials against renal cell carcinoma. Importantly, naturally occurring antibodies against the egcSEs were found in sera of less than 5% of human sera compared to 50–80% for the classic SEs (Holtfreter et al., 2004).
Here, we examined the ability of egcSEs to generate tumoricidal molecules from PMBCs. In addition, we identified such products in cell free supernatants and investigated their ability to induce cytotoxicity against a broad panel of human tumor cells. These findings unveil an efficient NO and cytokine dependent tumor killing mechanism conserved in egcSEs in the presence of diverse levels of T cell activation and TH-1 cytokines.
Materials and Methods
Preparation of Recombinant SEs
SEA, SEG, SEI, SElM, SElN and SElO were produced in Escherichia coli M15 as His-tagged recombinant toxins and purified by affinity chromatography on a nickel affinity column according to the supplier's instructions (New England Biolabs, Ipswich, USA) as previously described (Thomas et al., 2009). Protein purity was verified by SDS-PAGE. LPS was removed from toxin solutions by affinity chromatography (Detoxi-GEL endotoxin Gel®, Pierce Rockford, USA). The QCL-1000 Limulus amebocyte lysate assay® (Cambrex-BioWhittaker, Walkersville, USA) showed that the endotoxin content of the recombinant SAg solutions was less than 0.005 units/mL.
Tumor Cells
Laryngeal squamous cell carcinoma cell line Hep-2 and human non-small cell lung adenocarcinoma CRL5800 were obtained from cell library (IFR128, Lyon, France). Osteogenic sarcoma CRL1547, human breast cancer cell line MDA-MB-549, human neuroblastoma cell line SK-N-BE and human melanoma PLA-OD were a gift from Raphael Rousseau (Centre Leon Berard, Lyon, France). They were cultured in DMEM (Gibco, Invitrogen Corporation, Cergy Pontoise, France) supplemented with 10% fetal calf serum (BioWest, Paris, France), 100 U/mL penicillin and 100 μg/mL streptomycin. For SK-N-BE cells, the medium was supplemented with 1% non-essential amino acids (Gibco, Invitrogen Corporation, Cergy Pontoise, France).
Isolation of Human Mononuclear Cells
Blood packs were obtained from healthy donors through a convention with Etablissement Français du Sang after a written informed consent and according to Declaration of Helsinki principles. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Paque Plus® density gradient centrifugation (GE Healthcare Life Science, Orsay, France) and were washed with Ca- and Mg- free PBS. Cell viability was measured with the trypan blue exclusion test (>98%). The cells were washed in RPMI 1640 medium (Gibco, Invitrogen Corporation, Cergy Pontoise, France).
T Cell Activation
T cell activation with various SEs was assayed by measuring surface CD69 expression. Briefly, PBMCs (106 cells/mL) were incubated with SEA, SEG, SEI, SElM, SElN or SElO (1 pg/mL to 10 ng/mL) in Eagle's minimum essential medium (EMEN) containing 10% heat-inactivated FCS (Gibco Invitrogen, Paisley, UK) for 24 h at 37°C in humidified air with 5% CO2. EMEN and 100 μg/mL of phytohaemagglutinin (PHA) (Sigma-Aldrich, Saint Quentin Fallavier, France) were used as negative and positive control, respectively. Activated PBMCs were incubated with a mixture of anti-CD3 conjugated to cyanin-5-PE (Dako, Glostrup, Denmark) and anti-CD69 conjugated to PE (Beckman Coulter, Miami, FL). The cells were then analyzed with a FACScan® flow cytometer (BD Biosciences, San Jose, CA), and the results were expressed as the percentage of CD3+ lymphocytes expressing CD69.
Cytotoxicity Assays
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cytotoxic assay was performed to investigate the effect of stimulated PBMCs supernatants on cell viability (Mosmann, 2003). Tumor cells, 105 cells/well, were seeded in 96-well plates and incubated with 10%, 20%, 50% or 100% of supernatants from SE-stimulated or unstimulated PBMCs. After 1–4 days, 10 μL of MTT solution (5 mg/mL) (Invitrogen Corporation, Cergy Pontoise, France) was added to culture wells and plates were incubated for 3 h at 37°C. Supernatant was removed and 100 μL of 0.04 N HCl in isopropanol was added to each well before reading optical density at 540 nm with an ELISA-Reader (Bio-Rad, Marne la Coquette, France). In some experiments, 100 μg/mL PHA was used as positive control for T cell activation. Cell toxicity data are expressed as percent of the mean value obtained for untreated cells.
Annexin V-FITC/Propidium Iodide (PI) Staining
Tumor cells, 105 cells/well, were seeded in 96-well plates and incubated with 10% of SEs-stimulated PBMCs supernatant. After 4, 12, and 24 h, cells were harvested, washed with serum-containing medium and centrifuged at 3000 rpm for 5 min. The supernatant was discarded and the pellet was resuspended in 500 μL of 1× binding buffer. The sample solution was incubated with 1 μL of 5× FITC-conjugated annexin V (Abcam, Paris, France) and 1 μL of propidium iodide (PI) (Becton Dickinson, Le Pont de Claix, France) in the dark for 10 min at room temperature. The samples were analyzed using FACScanto II® flowcytometer (Becton Dickinson, Le Pont de Claix, France). Data analysis was performed with the FacsDiva® software (Becton Dickinson, Le Pont de Claix, France).
Nitric oxide (NO) assay
NO production was assessed by measuring the accumulated levels of nitrite in the supernatant with Griess reagent as previously described (Xie et al., 1995). Briefly, 100 μL of Griess reagent (1% sulfanilamide, 0.1% naphthylethylenediamine dihydrochloride, and 2.5% H3PO4) was added to 300 μL of the cell culture supernatant for 30 min at room temperature. Optical densities were read on a spectrophotometer at 548 nm. The values of NO concentration in the culture samples were obtained from standard curve of sodium nitrite solutions. For NO inhibition assays, PBMCs were incubated with or without a NO synthase inhibitor, L-NG-monomethyl arginine citrate (L-NMMA) (Sigma-Aldrich, Saint-Quentin Fallavier, France), for 2 h prior to stimulation by SEs (final concentration 3 mM). NO was quantified in supernatants as described above. Toxicity was analyzed by annexin-V and PI measurements as described above.
Cytokine assays
Levels of the cytokines IL-2, IL-4, IL-10, IL-12-p70, IL-17, IFN-γ, TNF-α, and Granulocyte Macrophage-Colony Stimulating Factor (GM-CSF) were measured in supernatants of PBMCs (106 cells/mL) incubated with 1 pg/mL of SEA, SEG, SEI, SElM, SElN or SElO in EMEM containing 10% heat-inactivated FCS (Gibco Invitrogen, Paisley, UK) for 24 h at 37°C in humidified air with 5% CO2, using Milliplex® kits based on Luminex® technology (Millipore, Molsheim, France). EMEM and 100 μg/mL PHA were the negative and positive control, respectively.
Neutralizing antibodies against TNF-α
PBMC supernatants were incubated with and without 30 μg/mL of mouse neutralizing polyclonal anti-TNF-α antibody (Abcys, Paris, France) for 18 h at 22°C before carrying out the cytotoxic assays described above. Recombinant TNF-α, (Abcam, Paris, France), 50 ng/mL was used as positive control.
Statistical analysis
The statistical analyses were based on Student t-test or Wilcoxon test for non-parametric analysis. The level of statistical significance was set at 0.05. The tests were carried out with SPSS Statistics® version 19 software (IBM France, Bois Colombes, France).
Results
egcSEs Induce T Cell CD69 Expression
CD69, a C-type lectin, disulfide linked homodimer is the earliest T cell surface activation receptor appearing even before cytokine production (Sutkowski and Huber, 2003). It is activated by superantigens, cytokines, and PHA. Thus, we investigated the ability of SEA and our recombinant egcSEs to activate CD69 expression in T cells obtained from 3 healthy donors. We noted a hierarchy of T cell activation as follows: SEA > SEI >SEG>SEM>SEO>SEN. Overall, SEA activated a larger number of T cells (26%) than all of egcSEs (p = 0.005). Among the egcSEs, SEI and SEG were the most effective T cell stimulants, activating 19 and 23% of resting T cells respectively, levels that were significantly higher than the SEM, SEN, and SEO (p = 0.019) (Figure 1). These findings are consistent with the T cell activation by egcSEs demonstrated previously using a T cell proliferation assay (Grumann et al., 2008).
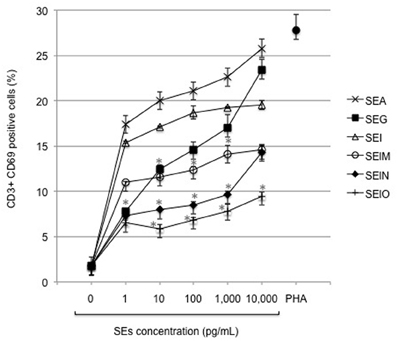
Figure 1. Expression of early T cell surface marker CD69 after incubation of human PBMCs with SEA and egcSEs. CD69 expression was measured on T lymphocytes (CD3+) after 24 h of incubation with PBMCs (106 cells/mL) in presence of SEA, SEG, SEI, SElM, SElN, or SElO (0–10 ng/mL). PHA (100 μg/mL) was used as positive control. Results are shown as the mean ± SEM for each point (n = 3 independent experiments). Asterisks indicate statistical significance compared to SEA.
Supernatants from egcSE-Activated PBMCs Kill a Broad Sampling of Human Tumor Cells
We examined the ability of supernatants from egcSE- and SEA-activated PBMCs to kill a broad sampling of human tumor cell lines. In preliminary experiments, Hep-2 squamous carcinoma cells were incubated for 96 h with various dilutions of supernatants of PBMCs that had been stimulated for 24–96 h with each egcSE, SEA or PHA. All supernatants from SE-stimulated PBMCs showed significant time and dose dependent cytotoxicity for each incubation time exceeding that of the control supernatant from unstimulated control PBMCs (p < 0.001) (Figure 2) (data 50% and 100% not shown). Supernatants from PBMCs incubated with SEs for 72 h consistently produced greater Hep-2 cell cytotoxicity than those supernatants similarly incubated for 48 h (p < 0.001). Thus, for subsequent experiments using other tumor cell lines we elected to use supernatants from PBMCs incubated with SEs for 72 hours.
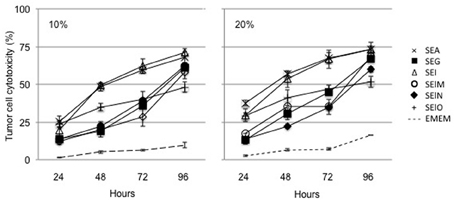
Figure 2. Cytotoxicty of SEA and egcSE-stimulated PBMCs against Hep2 squamous cell carcinoma cells. Supernatants from unstimulated PBMCs, SEA-, SEG-, SEI-, SElM-, SElN-, or SElO-stimulated PBMCs were added at 10% or 20% vol/vol to Hep2 squamous cell carcinoma cell culture. After 1–4 days, cell viability was evaluated by MTT asays. Results are shown as the mean ± SEM for each point (n = 3 independent experiments).
Having shown that supernatants from both egcSEs- and SEA-stimulated PBMCs were cytotoxic for Hep2 squamous cell carcinoma cells, we determined whether the cytotoxicity of these supernatants could also be demonstrated in broad sampling of the major human tumor histotologic types which included lung and breast carcinoma, melanoma, neuroblastoma, and osteogenic sarcoma cells. All egcSEs and SEA supernatants were equally effective in inducing significant concentration dependent cytotoxicity against all five human tumor cell lines compared to the unstimulated control (p < 0.005) (Figure 3). A hierarchy of sensitivity of the tumors to the cytotoxicity of the SE-stimulated PBMC supernatants is as follows: lung carcinoma > osteogenic sarcoma > melanoma > breast carcinoma > neuroblastona (Figure 3).
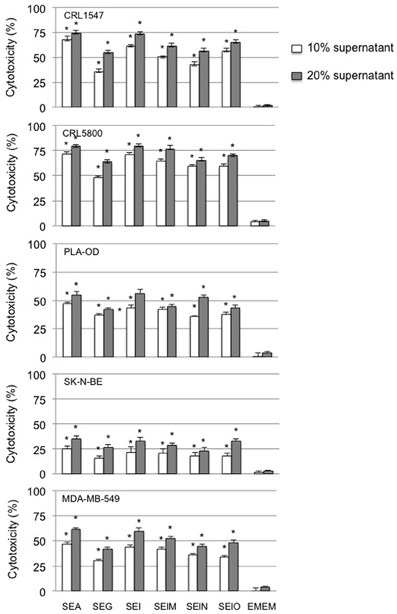
Figure 3. Cytotoxicty of SEA and egcSE-stimulated PBMCs vs. a broad panel of human tumor cell lines is shown. Cytotoxicity of 72 h supernatants from unstimulated PBMCs, SEA-, SEG-, SEI-, SElM-, SElN-, or SElO-stimulated PBMCs against human non-small cell lung adenocarcinoma CRL5800, osteogenic sarcoma CRL1547, human breast cancer cell line MDA-MB-549, human neuroblastoma cell line SK-N-BE and human melanoma PLA-OD were examined as described in Figure 2. Results are mean ± SEM (n = 3 independent experiments). Asterisks indicate statistical significance compared to the untreated PBMC control values at 10 or 20% concentrations.
By contrast, we did not observe any direct toxic effect of 1 pg to 10 ng/mL of SEA and egcSE's on any the tumor cell lines tested herein (data not shown). To determine how tumor cell lines died upon addition of SE-activated PBMC supernatants, Annexin V, and PI staining was performed on Hep-2 tumor cells treated with 10% supernatants from SEA and egcSE-stimulated PBMCs. The percentage of annexinV+ cells was 10–45 fold higher than the percentage of IP+ annexinV− cells at levels identical to those observed with unstimulated PBMC supernatants. These results suggest that upon addition of SE activated PBMC supernatants, cell lines died mainly by apoptosis.
Nitrous Oxide Generation from egcSE-Activated PBMCs
Next we determined whether NO could be induced by egcSE-activated PBMCs and mediate a tumoricidal response. PBMCs stimulation by egcSEs and SEA was associated with a significant increase in nitrite production (p < 0.001) with no difference between toxins (p > 0.05) (Figure 4). As expected, the addition of nitrous oxide synthase inhibitor L-NMMA to PBMCs inhibited NO induction by all toxins (p < 0.001) with no significant differences in the degree of inhibition.
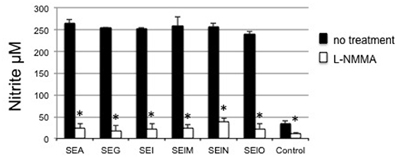
Figure 4. Nitrite generation from SEs-stimulated PBMCs. NO was quantified with Griess reagent in the supernatant of PBMCs (106 cells/mL) incubated for 24 h in a presence of EMEM, SEA, SEG, SEI, SElM, SElN, or SElO, with or without NOS inhibitor, L-NMMA (300 μM). The results are mean ± SD (n = 3 independent experiments). Asterisks indicate statistical significance compared to values obtained without the NOS inhibitor.
Nitrous Oxide Synthase Inhibition or Neutralizing Anti-TNF-α Individually or Combined Reduce(s) Tumor Cell Cytotoxicity of Supernatants from egcSE-Stimulated PBMCs
Having shown that both egcSEs and SEA induced NO production by PBMCs, we determined whether the tumor cell cytotoxicity of the supernatants could be attenuated by the addition of nitrous oxide synthase inhibitor L-NMMA. Supernatants from 10% egcSE- and SEA-stimulated PBMCs induced tumor cell cytotoxicity. In all cases, tumor cell apoptosis was confirmed by annexin V-FITC/PI staining (Figure 5). Exposure of PBMCs to L-NMMA, a competitive inhibitor of NOS, before incubation with egcSEs or SEA induced a significant decrease in tumor cell cytotoxicity of all supernatants (range: p = 0.01 for SEA to p = 0001 for SEO) with the sole exception of SEN (p = 0.85) (Figure 5). Notably, supernatants from SEG and SEO-stimulated PBMCs showed more significant reductions in tumor cytotoxicity (p = 0.002 and p = 0.001, respectively) than SEA supernatants (p = 0.01) (Figure 5). We further determined whether tumor cell cytotoxicity of all SE supernatants could be attenuated by the addition of anti-TNF-α to L-NMMA. The combined treatments significantly reduced the cytotoxicity of SEA (p = 0.005) and three of the 5 egcSEs, namely SEG (p = 0.009), SEI (p = 0.013) and SEO (p = 0.001) (Figure 5). Notably, the inhibitory effects of L-NMMA and anti-TNF-α did not completely abolish the tumor cell cytotoxicity suggesting that additional tumoricidal factors are operative in this system.
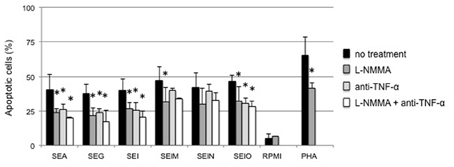
Figure 5. Anti- TNF-α and NO inhibitor, alone and in combination inhibit Hep-2 tumor cells cytotoxicity induced by 10% supernatants from SEA and egcSE-stimulated PBMCs. Hep-2 tumor cells cytotoxicity induced by 10% supernatants from SEA and egcSE-stimulated PBMCs before (black) or after treatment with L-NMMA (dark gray) or anti-TNF-α antibody (light gray) or a combination of L-NMMA plus anti-TNF-α antibody was analyzed by flow cytomerter using FITC-conjugated annexin-V and propidium iodide (IP) staining. The results are expressed as the mean ± SEM (n = 3 independent experiments). Asterisks indicate statistical significance compared to values obtained before treatment with L-NMMA or anti-TNF-α antibody.
Cytokine Profiles of Supernatants Induced by SE-Activated PBMCs
Levels of cytokines TNF-α, IFN-y, IL-2, IL-4, IL-10, IL-17, and GM-CSF produced after stimulation of PBMCs from six healthy donors by egcSEs and SEA were measured. The absolute cytokine levels induced by each egcSE and canonical SEA are shown in Table 1. Cytokine levels of egcSEs I, M, N, and SEA-activated PBMCs were not significantly different [except for IL-4 and IL-10 after stimulation with SEO (p > 0.05)]. SEA induced significantly higher levels of cytokines than SEG with mean cytokine values 5, 8, 2, 6, 7, and 2 fold higher than SEG for GM-CSF, INF-γ, IL-2, IL-4, IL-10, IL-17, and TNFα, respectively (range p = 0.01–0.04, except for TNF-α).
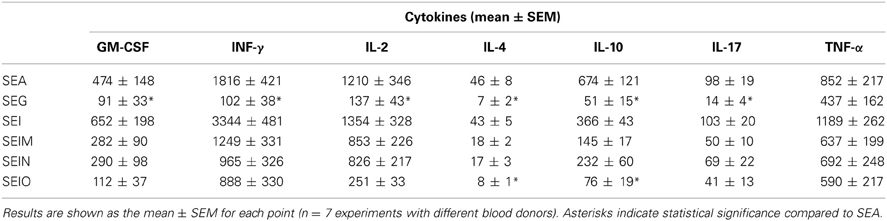
Table 1. Level of cytokines in supernatants of PBMC stimulated by SEA, SEG, SEI, SElM, SElN, and SElO.
Discussion
Our results demonstrate that egcSEs activate 11–21% or resting T cells and that cell free supernatants (CFSs) from egcSE-stimulated PBMCs induced NO synthase activation and robust generation of NO along with TH-I TH-2 cytokines. Such CFSs from all egcSEs induced an equal degree of annexin positive apoptosis in a broad panel of clinically relevant human tumor cells with a hierarchy of sensitivity: lung carcinoma > osteogenic sarcoma > melanoma > breast carcinoma > neuroblastoma. The apoptotic effect of the egcSE CSFs appears to be mediated in part by NO and TNF-α since NO synthase inhibitor L-NMMA and anti-TNF-α antibodies significantly inhibited the tumor cell cytotoxicity. Moreover, all egcSE CFSs with the exception of SEG contained substantial levels of additional TH-1 cytokines such as IFN-γ that could contribute to the tumor cell cytotoxicity.
Hibbs et al., was the first to demonstrate that NO could inhibit tumor cell growth and/or induce tumor cell death by activated macrophages (Hibbs et al., 1987). Subsequently, NO-mediated tumor cell cytotoxicity has been demonstrated by a variety of immune cells including natural killer cells, T-cells, and endothelial cells (Albina and Reichner, 1998). Fast et al. showed that SEB and TSST-1-induced NO and TNF-α derived from activated macrophages could promote cytotoxicity of murine fibrosarcoma and mastocytoma cells (Fast et al., 1991). Herein, we extend these findings by showing that NO from egcSE-stimulated PBMCs contribute to the cytotoxicity of human tumor cells.
NO donors such as L-arginine from both endogenous and exogenous sources have been shown to exert an inhibitory action on the proliferation of tumor cells, such as breast cancer, mastocytoma, neuroblastoma, epidermoid carcinoma, pheochromocytoma, colon carcinoma, and pancreatic carcinoma cells in vitro (Maragos et al., 1993; Estrada et al., 1997; Buga et al., 1998; Gansauge et al., 1998; Pervin et al., 2001; Murillo-Carretero et al., 2002; Ruano et al., 2003; Tesei et al., 2003; Ciani et al., 2004; Huguenin et al., 2004; Bal-Price et al., 2006). The stimulatory or inhibiting behavior of NO appears to be related to the distinct concentrations of NO attained under different experimental conditions. For instance, iNOS-generated NO present in excess of 300 nmol/L promotes DNA damage, gene mutation and apoptosis via increased phosphorylation of p53 and expression of MKP-1, inhibition of phosphorylation of protein kinase C (PKC), extracellular-signal-regulated protein kinase (ERK) and JUN (Pervin et al., 2003; Jones et al., 2004; Thomas et al., 2004; Ridnour et al., 2005; Fukumura et al., 2006). NO donors have been shown to induce apoptotic cell death by inhibiting NF-κ B by phosphorylation of p50 via S-nitrosylation (Marshall and Stamler, 2001), binding to iron-sulfur centers and inhibiting aconitase, complex I/complex II of the mitochondrial respiratory chain, or ribonucleotide reductase (Stuehr and Nathan, 1989; Lepoivre et al., 1990). In lower concentrations, tumor cell NO promotes tumor growth, neovascularization and invasiveness by induction of p53 mutations, upregulation of vascular endothelial growth factor resulting in neovascularization, increased vascular permeability and vasodilatation (Krischel et al., 1998; Ulibarri et al., 1999; Frank et al., 2000; Luczak et al., 2004; Wai et al., 2006). In the present study, a low dose of each egcSE induced robust nitrite concentrations of 200–250 μM suggesting that these agents may be capable of inducing a sufficient quantity of NO to exert a tumoricidal effect in vivo.
In vivo, exogenous NO or endothelial-cell derived NO in higher concentrations appears to present a tumoricidal barrier inimical to tumor cell dissemination. Selective genetic or pharmacological inhibition of eNOS or iNOS or enzymatic induction of NO deficiency in tumor cells diminishes VEGF, HO-1, and HIF1α activation and consequent tumor cell proliferation and angiogenesis (Kimura et al., 2000; Motterlini et al., 2000; Naughton et al., 2002; Kasuno, 2004). Local release of NO in endothelial cells, liver sinusoids, or pulmonary circulation causes apoptosis of the disseminated tumor cells at these sites (Fukumura et al., 1996; Wang et al., 2000, 2007; Qiu et al., 2003; Qi et al., 2004). Finally, daily intraperitoneal injections of NO-producing nitrovasodilators isosorbide mono-and dinitrate resulted in a significant decrease of the size of the primary tumor and a reduction in the number and size of spontaneous lung metastases (Pipili-Synetos et al., 1995). Thus, NO donors delivered parenterally or released from activated endothelial cells in sufficient concentration appears to be capable of inducing local tumor cell death and limiting tumor metastases.
In our study, NO and TNF-α in the CSF exhibited an additive effect in tumor cell cytotoxicity. Indeed, all of the egcSEs induced robust levels of NO and TNF-α with the exception of SEG as discussed below. TNF-α has direct effects on a variety of cell types and has been shown to work together with NO in tumor cell cytotoxicity (Laster et al., 1988; Estrada et al., 1992). NO is known to sensitize tumor cells to TNF-α-mediated apoptosis via specific disruption of the TNF-α-induced generation of hydrogen peroxide and subsequent inhibition of the NF-κ B dependent expression of anti-apoptotic genes (Schreck et al., 1992; Hong et al., 1997). Moreover, G1 arrest has been attributed to endogenous NO following activation by TNF-α, IFN-γ and IL-1 in breast and pancreatic carcinoma cells (Gansauge et al., 1998; Pervin et al., 2001). Furthermore, all of the egcSEs except SEG were also shown herein to be potent inducers of IFN-γ that could contribute to the tumor cytotoxic response.
In addition to its tumor killing properties, TNF-α has also been identified as the major cause of SE-induced toxicity in mice (Bette et al., 1993; Miethke et al., 1993). In previous cancer trials, the systemic toxicity of SEA was presumed to be related to TNF-α (Giantonio et al., 1997; Cheng et al., 2004). As shown herein, while SEG induced robust production of nitrite from PBMCs and robust tumor cell cytotoxicity, it also displayed substantially lower quantities of TNF-α and IFN-γ relative to other egcSEs. Interestingly, a supernatant from S. aureus producing SEG together with the other egcSEs used in a recent cancer trial exhibited minimal systemic toxicity (Ren et al., 2004). SEG's potent T cell activation (comparable to SEA) coupled with its significantly reduced cytokine levels (relative to SEA and the other egcSEs) is reminiscent of split T cell responses when classic peptide antigen is presented to T cells in the absence B7 costimulation (Schweitzer and Sharpe, 1998). In this context, wild type SEA has been shown to possesses an intirisic costimulatory B7-like sequence in its conserved β-strand/hinge/α-helix domains that engages and activates the T cell costimulatory CD28 homodimer resulting in T cell cytokine secretion; mutation of this sequence resulted in attenuated cytokine production (Arad et al., 2011). SEG possesses several amino acid substitutions in this conserved sequence which could alter its topographic interface with T cell CD28 resulting in reduction in cytokine levels noted herein. These findings suggest that the nature of SEG's T cell response depends not only on its affinity for MHCII but also on the strength of its intrinsic costimulatory interaction with T cell CD28. SEG's retention of NO-dependent tumor cell cytotoxicity and robust T cell activation in the presence of low cytokine levels makes it a promising model for such investigation.
Neutralizing antibodies against superantigens are considered to be the major factors underlying superantigen therapy failure. It was recently confirmed in the completed Phase II/III trial using a hybrid SEA/SEE120 superantigen (fused to a mouse monoclonal Fab targeting the 5T4 antigen on tumor cells) that SEA/SEE120 did not improve overall survival in advanced renal cell cancer (Hawkins et al., 2013). The ineffectiveness of this drug was likely due to the higher than expected levels of pre-existing anti-SEA/SEE120 antibodies because a small subgroup of patients with low levels of anti-SEA/SEE120 antibodies did apparently demonstrate longer survival. Importantly, prevalence of antibodies against egcSEs including SEG is much lower than antibodies against classical SEs (Holtfreter et al., 2004). Our results demonstrating SEG's retention of NO-dependent tumor cell cytotoxicity and robust T cell activation in the presence of low cytokine levels suggests SEG may be more appropriate for superantigen therapy in cancer.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Jacqueline Marvel, Yann Leverrier, Florence Couzon, Laure Denis and Carmen Fernandez for preliminary analysis, technical assistance and advice.
Grants
This work was supported by the European Community grant CONCORD EC 222718 and by NIH Grant Number P20 RR016454 from the INBRE Program of the National Center for Research Resources.
References
Abrahmsen, L., Dohlsten, M., Segrén, S., Björk, P., Jonsson, E., and Kalland, T. (1995). Characterization of two distinct MHC class II binding sites in the superantigen staphylococcal enterotoxin A. EMBO J. 14, 2978–2986.
Albina, J. E., and Reichner, J. S. (1998). Role of nitric oxide in mediation of macrophage cytotoxicity and apoptosis. Cancer Metastasis Rev. 17, 39–53. doi: 10.1023/A:1005904704618
Ambs, S., Hussain, S. P., and Harris, C. C. (1997). Interactive effects of nitric oxide and the p53 tumor suppressor gene in carcinogenesis and tumor progression. FASEB J. 11, 443–448.
Arad, G., Levy, R., Nasie, I., Hillman, D., Rotfogel, Z., Barash, U., et al. (2011). Binding of superantigen toxins into the CD28 homodimer interface is essential for induction of cytokine genes that mediate lethal shock. PLoS Biol. 9:e1001149. doi:10.1371/journal.pbio.1001149
Bal-Price, A., Gartlon, J., and Brown, G. C. (2006). Nitric oxide stimulates PC12 cell proliferation via cGMP and inhibits at higher concentrations mainly via energy depletion. Nitric Oxide 14, 238–246. doi: 10.1016/j.niox.2005.10.002
Becker, K., Friedrich, A. W., Lubritz, G., Weilert, M., Peters, G., and Von Eiff, C. (2003). Prevalence of genes encoding pyrogenic toxin superantigens and exfoliative toxins among strains of Staphylococcus aureus isolated from blood and nasal specimens. J. Clin. Microbiol. 41, 1434–1439. doi: 10.1128/JCM.41.4.1434-1439.2003
Bette, M., Schäfer, M. K., van Rooijen, N., Weihe, E., and Fleischer, B. (1993). Distribution and kinetics of superantigen-induced cytokine gene expression in mouse spleen. J. Exp. Med. 178, 1531–1539. doi: 10.1084/jem.178.5.1531
Billiar, T. R. (1990). Inducible cytosolic enzyme activity for the production of nitrogen oxides from L-arginine in hepatocytes. Biochem. Biophys. Res. Commun. 168, 1034–1040. doi: 10.1016/0006-291X(90)91133-D
Bogdan, C. (2001). Nitrous oxide and the immune response. Nat. Immunol. 2, 907–916. doi: 10.1038/ni1001-907
Bohach, G. A. (2006). “Staphylococcus aureus exotoxins” in Gram-Positive Pathogens, 2nd Edn., eds V. A. Fischetti, R. P., Novick, J. J. Ferreti, D. A. Portnoy, and J. I. Rood (Washington, DC: ASM Press), 464–477.
Bredt, D. S., and Snyder, S. H. (1990). Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc. Natl. Acad. Sci. U.S.A. 87, 682–685. doi: 10.1073/pnas.87.2.682
Buga, G. M., Wei, L. H., Bauer, P. M., Fukuto, J. M., and Ignarro, L. J. (1998). NG-hydroxy-L-arginine and nitric oxide inhibit Caco-2 tumor cell proliferation by distinct mechanisms. Am. J. Physiol. 275, 1256–1264.
Busse, R., and Mülsch, A. (1990). Induction of nitric oxide synthase by cytokines in vascular smooth muscle cells. FEBS Lett. 275, 87–90. doi: 10.1016/0014-5793(90)81445-T
Cheng, J. D., Babb, J. S., Langer, C., Aamdal, S., Robert, F., Engelhardt, L. R., et al. (2004). Individualized patient dosing in phase I clinical trials: the role of escalation with overdose control in PNU-214936. J. Clin. Oncol. 22, 602–609. doi: 10.1200/JCO.2004.12.034
Ciani, E., Severi, S., Contestabile, A., Bartesaghi, R., and Contestabile, A. (2004). Nitric oxide negatively regulates proliferation and promotes neuronal differentiation through N-Myc downregulation. J. Cell. Sci. 117, 4727–4737. doi: 10.1242/jcs.01348
Dohlsten, M., Lando, P. A., Björk, P., Abrahmsén, L., Ohlsson, L., Lind, P., et al. (1995). Immunotherapy of human colon cancer by antibody-targeted superantigens. Cancer Immunol. Immunother. 41, 162–168. doi: 10.1007/BF01521342
Dohlsten, M., Sundstedt, A., Björklund, M., Hedlund, G., and Kalland, T. (1993). Superantigen-induced cytokines suppress growth of human colon-carcinoma cells. Int. J. Cancer. 54, 482–488. doi: 10.1002/ijc.2910540321
Erlandsson, E., Andersson, K., Cavallin, A., Nilsson, A., Larsson-Lorek, U., Niss, U., et al. (2003). Identification of the antigenic epitopes in staphylococcal enterotoxins A and E and design of a superantigen for human cancer therapy. J. Mol. Biol. 333, 893–905. doi: 10.1016/j.jmb.2003.09.009
Estrada, C., Gómez, C., Martín, C., Moncada, S., and González, C. (1992). Nitric oxide mediates tumor necrosis factor-alpha cytotoxicity in endothelial cells. Biochem. Biophys. Res. Commun. 186, 475–482. doi: 10.1016/S0006-291X(05)80832-0
Estrada, C., Gómez, C., Martín-Nieto, J., De Frutos, T., Jiménez, A., and Villalobo, A. (1997). Nitric oxide reversibly inhibits the epidermal growth factor receptor tyrosine kinase. Biochem. J. 326, 369–376.
Fast, D. J., Shannon, B. J., Herriott, M. J., Kennedy, M. J., Rummage, J. A., and Leu, R. W. (1991). Staphylococcal exotoxins stimulate nitric oxide-dependent murine macrophage tumoricidal activity. Infect. Immun. 59, 2987–2993.
Ferry, T., Thomas, D., Perpoint, T., Lina, G., Monneret, G., Mohammedi, I., et al. (2008). Analysis of superantigenic toxin Vb T-cell signatures produced during cases of staphylococcal toxic shock syndrome and septic shock. Clin. Microbiol. Infect. 14, 546–554.
Förstermann, U., Pollock, J. S., Schmidt, H. H., Heller, M., and Murad, F. (1991). Calmodulin-dependent endothelium-derived relaxing factor/nitric oxide synthase activity is present in the particulate and cytosolic fractions of bovine aortic endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 88, 1788–1792. doi: 10.1073/pnas.88.5.1788
Frank, S., Kämpfer, H., Podda, M., Kaufmann, R., and Pfeilschifter, J. (2000). Identification of copper/zinc superoxide dismutase as a nitric oxide-regulated gene in human (HaCaT) keratinocytes: implications for keratinocyte proliferation. Biochem. J. 346, 719–728. doi: 10.1042/0264-6021:3460719
Fukumura, D., and Jain, R. K. (1998). Role of nitric oxide in angiogenesis and microcirculation in tumors. Cancer Metastasis Rev. 17, 77–89. doi: 10.1023/A:1005908805527
Fukumura, D., Kashiwagi, S., and Jain, R. K. (2006). The role of nitric oxide in tumour progression. Nat. Rev. Cancer 6, 521–534. doi: 10.1038/nrc1910
Fukumura, D., Yonei, Y., Kurose, I., Saito, H., Ohishi, T., Higuchi, H., et al. (1996). Role of nitric oxide in Kupffer cell mediated hepatoma cell cytotoxicity in vitro and ex vivo. Hepatology 24, 141–149.
Gansauge, S., Nussler, A. K., Beger, H. G., and Gansauge, F. (1998). Nitric oxide induced apoptosis in human pancreatic carcinoma cell lines is associated with G1-arrest and an increase of the cyclin-dependent kinase inhibitor p21WAF1 / CIP1. Cell Growth Differ. 9, 611–617.
Giantonio, B. J., Alpaugh, R. K., Schultz, J., McAleer, C., Newton, D. W., Shannon, B., et al. (1997). Superantigen- based immunotherapy: a phase I trial of PNU-214565, a monoclonal antibody-staphylococcal enterotoxin A recombinant fusion protein, in advanced pancreatic and colorectal cancer. J. Clin. Oncol. 15, 1994–2007.
Grumann, D., Scharf, S. S., Holtfreter, S., Kohler, C., Steil, L., Engelmann, S., et al. (2008). Immune cell activation by enterotoxin gene cluster (egc)-encoded and non-egc superantigens from Staphylococcus aureus. J. Immunol. 181, 5054–5061.
Hawkins, R., Gore, M., Shparyk, Y., Bondar, V., Gladkov, O., Ganev, T., et al. (2013). A randomized phase 2/3 study of naptumomab estafenatox plus IFN-α in advanced renal cell carcinoma. Am. Soc. Clin. Oncol. Available online at: http://www.medsci.cn/webeditor/uploadfile/201306/20130602210752381.pdf. It coorespond to the abtract page 3073.
Hibbs, J. B., Taintor, R. R., and Vavrin, Z. (1987). Macrophage cytotoxicity: role for L-arginine deiminase and imino nitrogen oxidation to nitrite. Science 235, 473–476. doi: 10.1126/science.2432665
Hibbs, J. B. Jr., Taintor, R. R., Vavrin, Z., and Rachlin, E. M. (1988). Nitric oxide: a cytotoxic activated macrophage effector molecule. Biochem. Biophys. Res. Commun. 157, 87–94. doi: 10.1016/S0006-291X(88)80015-9
Holtfreter, S., Bauer, K., Thomas, D., Feig, C., Lorenz, V., Roschack, K., et al. (2004). egc-Encoded superantigens from Staphylococcus aureus are neutralized by human sera much less efficiently than are classical staphylococcal enterotoxins or toxic shock syndrome toxin. Infect. Immun. 72, 4061–4071. doi: 10.1128/IAI.72.7.4061-4071.2004
Hong, Y. H., Peng, H. B., La Fata, V., and Liao, J. K. (1997). Hydrogen peroxide-mediated transcriptional induction of macrophage colony stimulating factor by TGF-beta1. J. Immunol. 159, 2418–2423.
Huguenin, S., Fleury-Feith, J., Kheuang, L., Jaurand, M. C., Bolla, M., Riffaud, J. P., et al. (2004). Nitrosulindac (NCX 1102): a new nitric oxide-donating non-steroidal anti-inflammatory drug (NO-NSAID), inhibits proliferation and induces apoptosis in human prostatic epithelial cell lines. Prostate 61, 132–141. doi: 10.1002/pros.20081
Jarraud, S., Peyrat, M. A., Lim, A., Tristan, A., Bes, M., Mougel, C., et al. (2001). egc, a highly prevalent operon of enterotoxin gene, forms a putative nursery of superantigens in Staphylococcus aureus. J. Immunol. 166, 669–677.
Jones, M. K., Tsugawa, K., Tarnawski, A. S., and Baatar, D. (2004). Dual actions of nitric oxide on angiogenesis: possible roles of PKC, ERK, and AP-1. Biochem. Biophys. Res. Commun. 318, 520–528. doi: 10.1016/j.bbrc.2004.04.055
Kasuno, K. (2004). Nitric oxide induces hypoxia-inducible factor 1 activation that is dependent on MAPK and phosphatidylinositol 3-kinase signaling. J. Biol. Chem. 279, 2550–2558. doi: 10.1074/jbc.M308197200
Kimura, H., Weisz, A., Kurashima, Y., Hashimoto, K., Ogura, T., D'Acquisto, F., et al. (2000). Hypoxia response element of the human vascular endothelial growth factor gene mediates transcriptional regulation by nitric oxide: control of hypoxia-inducible factor-1 activity by nitric oxide. Blood 95, 189–197.
Krischel, V., Bruch-Gerharz, D., Suschek, C., Kröncke, K. D., Ruzicka, T., and Kolb-Bachofen, V. (1998). Biphasic effect of exogenous nitric oxide on proliferation and differentiation in skin derived keratinocytes but not fibroblasts. J. Invest. Dermatol. 111, 286–291. doi: 10.1046/j.1523-1747.1998.00268.x
Kwon, N. S., Nathan, C. F., Gilker, C., Griffith, O. W., Matthews, D. E., and Stuehr, D. J. (1990). L-citrulline production from L-arginine by macrophage nitric oxide synthase. The ureido oxygen derives from dioxygen. J. Biol. Chem. 265, 13442–13445.
Lamphear, J. G., Stevens, K. R., and Rich, R. R. (1998). Intercellular adhesion molecule-1 and leukocyte function-associated antigen-3 provide costimulation for superantigen induced T lymphocyte proliferation in the absence of a specific presenting molecule. J. Immunol. 160, 615–623.
Laster, S. M., Wood, J. G., and Gooding, L. R. (1988). Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J. Immunol. 141, 2629–2634.
Lepoivre, M., Chenais, B., Yapo, A., Lemaire, G., Thelander, L., and Tenu, J. P. (1990). Alterations of ribonucleotide reductase activity following induction of the nitrite-generating pathway in adenocarcinoma cells. J. Biol. Chem. 265, 14143–14149.
Luczak, K. G., Balcerczyk, A., Soszyñski, M., and Bartosz, G. (2004). Low concentration of oxidant and nitric oxide donors stimulate proliferation of human endothelial cells in vitro. Cell Biol. Int. 28, 483–486. doi: 10.1016/j.cellbi.2004.03.004
Ma, Q., Wang, Z., Zhang, M., Hu, H., Li, J., Zhang, D., et al. (2010). Targeting the L-Arginine-nitric oxide pathway for cancer treatment. Curr. Pharm. Des. 16, 392–410. doi: 10.2174/138161210790232121
Maragos, C. M., Wang, J. M., Hrabie, J. A., Oppenheim, J. J., and Keefer, L. K. (1993). Nitric oxide/nucleophile complexes inhibit the in vitro proliferation of A375 melanoma cells via nitric oxide release. Cancer Res. 53, 564–568.
Marletta, M. A., Yoon, P. S., Iyengar, R., Leaf, C. D., and Wishnok, J. S. (1988). Macrophage oxidation of L-arginine to nitrite and nitrate: nitric oxide is an intermediate. Biochemistry 27, 8706–8711. doi: 10.1021/bi00424a003
Marrack, P., and Kappler, J. (1990). The staphylococcal enterotoxins and their relatives. Science 248, 1066–1072.
Marshall, H. E., and Stamler, J. S. (2001). Inhibition of NF-kappa B by S-nitrosylation. Biochemistry 40, 1688–1693. doi: 10.1021/bi002239y
Miethke, T., Duschek, K., Wahl, C., Heeg, K., and Wagner, H. (1993). Pathogenesis of the toxic shock syndrome: T cell mediated lethal shock caused by the superantigen TSST-1. Eur. J. Immunol. 23, 1494–1500. doi: 10.1002/eji.1830230715
Moncada, S., and Higgs, A. (1993). The L-arginine-nitric oxide pathway. N. Engl. J. Med. 329, 2002–2012. doi: 10.1056/NEJM199312303292706
Mosmann, T. (2003). Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 65, 55–63. doi: 10.1016/0022-1759(83)90303-4
Motterlini, R., Foresti, R., Bassi, R., Calabrese, V., Clark, J. E., and Green, C. J. (2000). Endothelial heme oxygenase-1 induction by hypoxia. Modulation by inducible nitric-oxide synthase and S-nitrosothiols. J. Biol. Chem. 275, 13613–13620. doi: 10.1074/jbc.275.18.13613
Murillo-Carretero, M., Ruano, M. J., Matarredona, E. R., Villalobo, A., and Estrada, C. (2002). Antiproliferative effect of nitric oxide on epidermal growth factor-responsive human neuroblastoma cells. J. Neurochem. 83, 119–131. doi: 10.1046/j.1471-4159.2002.01116.x
Nathan, C. F., and Hibbs, J. B. (1991). Role of nitric oxide synthesis in macrophage antimicrobial activity. Curr. Opin. Immunol. 3, 65–70. doi: 10.1016/0952-7915(91)90079-G
Naughton, P., Foresti, R., Bains, S. K., Hoque, M., Green, C. J., and Motterlini, R. (2002). Induction of heme oxygenase 1 by nitrosative stress. A role for nitroxyl anion. J. Biol. Chem. 277, 40666–40674. doi: 10.1074/jbc.M203863200
Pervin, S., Singh, R., and Chaudhuri, G. (2001). Nitric oxideinduced cytostasis and cell cycle arrest of a human breast cancer cell line (MDA-MB-231): potential role of cyclin D1. Proc. Natl. Acad. Sci. U.S.A. 98, 3583–3588. doi: 10.1073/pnas.041603998
Pervin, S., Singh, R., Freije, W. A., and Chaudhuri, G. (2003). MKP-1-induced dephosphorylation of extracellular signal-regulated kinase is essential for triggering nitric oxide-induced apoptosis in human breast cancer cell lines: implications in breast cancer. Cancer Res. 63, 8853–8860.
Pipili-Synetos, E., Papageorgiou, A., Sakkoula, E., Sotiropoulou, G., Fotsis, T., Karakiulakis, G., et al. (1995). Inhibition of angiogenesis, tumor growth and metastasis by the NO-releasing vasodilators, isosorbide mononitrate and dinitrate. Br. J. Pharmacol. 116, 1829–1834. doi: 10.1111/j.1476-5381.1995.tb16670.x
Qi, K., Qiu, H., Rutherford, J., Zhao, Y., Nance, D. M., and Orr, F. W. (2004). Direct visualization of nitric oxide release by liver cells after the arrest of metastatic tumor cells in the hepatic microvasculature. J. Surg. Res. 119, 29–35. doi: 10.1016/j.jss.2003.09.008
Qiu, H., Orr, F. W., Jensen, D., Wang, H. H., McIntosh, A. R., Hasinoff, B. B., et al. (2003). Arrest of B16 melanoma cells in the mouse pulmonary microcirculation induces endothelial nitric oxide synthase-dependent nitric oxide release that is cytotoxic to the tumor cells. Am. J. Pathol. 162, 403–412. doi: 10.1016/S0002-9440(10)63835-7
Radomski, M. W., Palmer, R. M., and Moncada, S. (1990). Glucocorticoids inhibit the expression of an inducible, but not the constitutive, nitric oxide synthase in vascular endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 87, 10043–10047. doi: 10.1073/pnas.87.24.10043
Ren, S., Terman, D. S., Bohach, G., Silvers, A., Hansen, C., Colt, H., et al. (2004). Intrapleural staphylococcal superantigen induces resolution of malignant pleural effusions and a survival benefit in non-small cell lung cancer. Chest 126, 1529–1539. doi: 10.1378/chest.126.5.1529
Ridnour, L. A., Isenberg, J. S., Espey, M. G., Thomas, D. D., Roberts, D. D., and Wink, D. A. (2005). Nitric oxide regulates angiogenesis through a functional switch involving thrombospondin-1. Proc. Natl. Acad. Sci. U.S.A. 102, 13147–13152. doi: 10.1073/pnas.0502979102
Ruano, M. J., Hernández-Hernando, S., Jiménez, A., Estrada, C., and Villalobo, A. (2003). Nitric oxide-induced epidermal growth factordependent phosphorylations in A431 tumor cells. Eur. J. Biochem. 270, 1828–1837. doi: 10.1046/j.1432-1033.2003.03546.x
Schreck, R., Albermann, K., and Baeuerle, P. A. (1992). Nuclear factor kappa B: an oxidative stress-responsive transcription factor of eukaryotic cells (a review). Free Radic. Res. Commun. 17, 221–237. doi: 10.3109/10715769209079515
Schweitzer, A. N., and Sharpe, A. H. (1998). Studies using antigen-presenting cells lacking expression of both B7-1 (CD80) and B7-2 (CD86) show distinct requirements for B7 molecules during priming versus restimulation of Th2 but not Th1 cytokine production. J. Immunol. 161, 2762–2771.
Shi, Q., Huang, S., Jiang, W., Kutach, L. S., Ananthaswamy, H. N., and Xie, K. (1999). Direct correlation between nitric oxide synthase II inducibility and metastatic ability of UV-2237 murine fibrosarcoma cells carrying mutant p53. Cancer Res. 59, 2072–2075.
Stuehr, D. J., and Nathan, C. F. (1989). Nitric oxide. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J. Exp. Med. 169, 1543–1555. doi: 10.1084/jem.169.5.1543
Sutkowski, N., and Huber, B. T. (2003). “Assessment of Specific T-cell activation by superantigens in Methods in Molecular Biology,” in Superantigen Protocols, Vol. 24, ed T. Krakauer (Totowa, NJ: Humana Press Inc.), 185–218.
Tamir, S., and Tannenbaum, S. R. (1996). The role of nitric oxide (NO) in the carcinogenic process. Biochim. Biophys. Acta 1288, F31–F36.
Terman, D. S., Bohach, G., Vandenesch, F., Etienne, J., Lina, G., and Sahn, S. A. (2006). Staphylococcal superantigens of the enterotoxin gene cluster (egc) for treatment of stage IIIb non–small cell lung cancer with pleural effusion. Clin. Chest Med. 27, 321–334. doi: 10.1016/j.ccm.2006.01.001
Tesei, A., Ricotti, L., Ulivi, P., Medri, L., Amadori, D., and Zoli, W. (2003). NCX 4016, a nitric oxide-releasing aspirin derivative, exhibits a significant antiproliferative effect and alters cell cycle progression in human colon adenocarcinoma cell lines. Int. J. Oncol. 22, 1297–1302.
Thiemermann, C., and Vane, J. (1990). Inhibition of nitric oxide synthesis reduces the hypotension induced by bacterial lipopolysaccharides in the rat in vivo. Eur. J. Pharmacol. 182, 591–595. doi: 10.1016/0014-2999(90)90062-B
Thomas, D., Dauwalder, O., Brun, V., Badiou, C., Ferry, T., Etienne, J., et al. (2009). Staphylococcus aureus superantigens elicit redundant and extensive human Vβ patterns. Infect. Immun. 77, 2043–2050. doi: 10.1128/IAI.01388-08
Thomas, D. D., Espey, M. G., Ridnour, L. A., Hofseth, L. J., Mancardi, D., Harris, C. C., et al. (2004). Hypoxic inducible factor 1alpha, extracellular signal-regulated kinase, and p53 are regulated by distinct threshold concentrations of nitric oxide. Proc. Natl. Acad. Sci. U.S.A. 101, 8894–8899. doi: 10.1073/pnas.0400453101
Ulibarri, J. A., Mozdziak, P. E., Schultz, E., Cook, C., and Best, T. M. (1999). Nitric oxide donors, sodium nitroprusside and S-nitro-N-acetylpencillamine, stimulate myoblast proliferation in vitro. In Vitro Cell Dev. Biol. Anim. 35, 215–218. doi: 10.1007/s11626-999-0029-1
Wai, P. Y., Guo, L., Gao, C., Mi, Z., Guo, H., and Kuo, P. C. (2006). Osteopontin inhibits macrophage nitric oxide synthesis to enhance tumor proliferation. Surgery 140, 132–140. doi: 10.1016/j.surg.2006.02.005
Wang, F., Zhang, R., Xia, T., Hsu, E., Cai, Y., Gu, Z., et al. (2007). Inhibitory effects of nitric oxide on invasion of human cancer cells. Cancer Lett. 257, 274–282. doi: 10.1016/j.canlet.2007.08.001
Wang, H. H., McIntosh, A. R., Hasinoff, B. B., Rector, E. S., Ahmed, N., Nance, D. M., et al. (2000). B16 melanoma cell arrest in the mouse liver induces nitric oxide release and sinusoidal cytotoxicity: a natural hepatic defense against metastasis. Cancer Res. 60, 5862–5869.
Wink, D. A., Vodovotz, Y., Laval, J., Laval, F., Dewhirst, M. W., and Mitchell, J. B. (1998). The multifaceted roles of nitric oxide in cancer. Carcinogenesis 19, 711–721. doi: 10.1093/carcin/19.5.711
Xie, K., and Fidler, I. J. (1998). Therapy of cancer metastasis by activation of the inducible nitric oxide synthase. Cancer Metastasis Rev. 17, 55–75. doi: 10.1023/A:1005956721457
Xie, K., Huang, S., Dong, Z., Gutman, M., and Fidler, I. J. (1995). Direct correlation between expression of endogenous inducible nitric oxide synthase and regression of M5076 reticulum cell sarcoma hepatic metastases in mice treated with liposomes containing lipopeptide CGP 31362. Cancer Res. 55, 3123–3131.
Keywords: Staphylococcus aureus, egcSE superantigens, nitric oxide, tumor cell apoptosis
Citation: Terman DS, Serier A, Dauwalder O, Badiou C, Dutour A, Thomas D, Brun V, Bienvenu J, Etienne J, Vandenesch F and Lina G (2013) Staphylococcal entertotoxins of the enterotoxin gene cluster (egcSEs) induce nitric oxide- and cytokine dependent tumor cell apoptosis in a broad panel of human tumor cells. Front. Cell. Infect. Microbiol. 3:38. doi: 10.3389/fcimb.2013.00038
Received: 23 May 2013; Accepted: 10 July 2013;
Published online: 13 August 2013.
Edited by:
Martin J. McGavin, University of Western Ontario, CanadaReviewed by:
John McCormick, University of Western Ontario, CanadaVictor J. Torres, New York University School of Medicine, USA
Copyright © 2013 Terman, Serier, Dauwalder, Badiou, Dutour, Thomas, Brun, Bienvenu, Etienne, Vandenesch and Lina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David. S. Terman, Molecular Genetics Program, Jenomic Research Institute, Seventh Avenue and Lincoln Street, Building 1, PO Box 6447, Carmel, CA 93953, USA e-mail:ZHN0QHNiY2dsb2JhbC5uZXQ=;
G. Lina, Staphylococcal Pathogenesis, INSERM U1111, Domaine de la Buire, 7 rue Guillaume Paradin, 69008 Lyon, France e-mail:Z2VyYXJkLmxpbmFAY2h1LWx5b24uZnI=