 R. K. Subbarao Malireddi
R. K. Subbarao Malireddi Thirumala-Devi Kanneganti
Thirumala-Devi Kanneganti- Department of Immunology, St. Jude Children's Research Hospital, Memphis, TN, USA
Interferons (IFNs) were discovered over a half-century ago as antiviral factors. The role of type I IFNs has been studied in the pathogenesis of both acute and chronic microbial infections. Deregulated type I IFN production results in a damaging cascade of cell death, inflammation, and immunological host responses that can lead to tissue injury and disease progression. Here, we summarize the role of type I IFNs in the regulation of cell death and disease during different microbial infections, ranging from viruses and bacteria to fungal pathogens. Understanding the specific mechanisms driving type I IFN-mediated cell death and disease could aid in the development of targeted therapies.
Introduction
Interferons (IFNs) are broadly classified into three groups, which are denoted as type I, II, and III based on the specific receptor utilization for their signal transduction. The type I IFN family comprises subtypes of IFNα (13 subtypes), IFNβ, IFNω, and IFNε (Pestka et al., 2004; Hertzog and Williams, 2013). All of the type I IFNs bind to a common heterodimeric receptor, called the IFNα/β receptor (IFNAR), composed of two chains, IFNAR1 and IFNAR2, that are associated with the tyrosine kinases Tyk2 and Jak1. Activated Tyk2 and Jak1 recruit and phosphorylate several signal transducer and activator of transcription (STAT) family members (Figure 1) (Pestka et al., 2004; Platanias, 2005). Activated STAT1 forms a dimer with STAT2, leading to the recruitment of IRF9 and subsequent formation of a heterotrimeric complex called IFN-stimulated gene factor 3 (ISGF3) (Figure 1). This complex translocates to the nucleus, where it binds upstream IFN-stimulated response elements (ISRE) and activates the transcription of type I IFN-inducible genes (Pestka et al., 2004; Platanias, 2005). Type I IFNs are classically known for their antiviral immune responses; however, several studies have demonstrated that a wide range of non-viral pathogens can also induce their expression. However, the specific mechanisms and physiological consequences of IFN responses to such pathogens are poorly understood. Various studies have attributed contrasting roles and differential outcomes to type I IFNs in immune responses to diverse microbial pathogens. The ability of IFNs to regulate cell death has been known for a long time and recent studies have started to reveal the specific mechanisms involved.
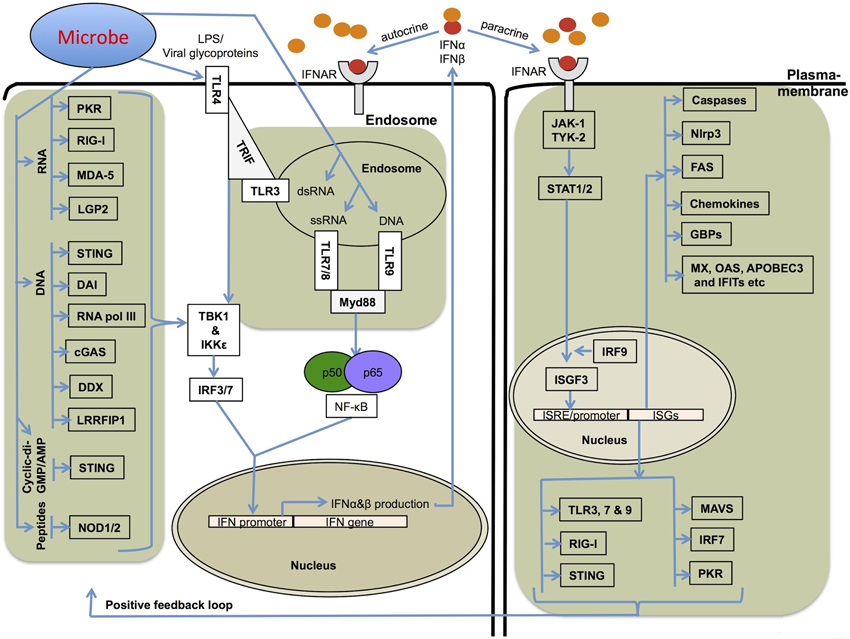
Figure 1. Mechanisms involved in type I IFN production and its receptor signaling. Several different stimuli including pathogen derived LPS, glycoproteins, RNA and DNA can induce type I IFNs (IFNα and IFNβ) via upstream pattern recognition receptors. TLRs sense the endosomally located PAMPs (pathogen associated molecular patterns) and DAMPs (damage associated molecular patterns) and recruit TRIF (TRL3 and TLR4) or Myd88 (TLR4, 7, 8, and 9), which further transfer the signals downstream to IRF3 and IRF7. RLRs, NLRs, DAI, STING, and related receptors sense nucleic acids in the cytoplasm. The RNA sensors, RIG-I, MDA-5, and LGP2, and DNA sensor STING use the adapter protein MAVS (mitochondrial antiviral signaling protein) to transfer signals to IRFs for type I IFN transcription. RIG-I and MDA-5 also sense ligands generated by RNA polymerase III from cytoplasmic DNA PAMPs. All these pathways utilize the common downstream kinases, TBK1/IKKε for activating Interferon transcription factors. Type I IFNs bind to IFNAR receptor and activate a robust transcriptional pathway through a JAK-STAT signaling pathway. The transcriptional complexes activated by type I IFN signaling bind to specific ISRE/GAS sequences and lead to the expression of several genes important for cell death, cell proliferation and immune responses.
During the course of evolution, the arms race between bacterial pathogens and host organisms has resulted in the development of virulence mechanisms by microbes and the reciprocal development of host counter strategies to efficiently defend against them. Cell death has emerged as one of the important aspects of such a race between microbes and the host, exploited by both, while the final outcome is dependent on the specific pathogen and cell types involved. The in depth molecular mechanisms of how type I IFN signaling causes differential outcomes during different microbial infections remains to be established. Here we discuss the role of type I IFNs in regulating cell death and disease in various infection models and highlight its emerging role in inflammasome activation.
Intracellular Bacteria
Listeria
Listeria monocytogenes (LM), a gram-positive intracellular bacterium that grows rapidly inside host cells, is known to potently induce type I IFN production in mouse (Havell, 1986; O'Riordan et al., 2002) and human (Reimer et al., 2007) macrophages. LM causes life-threatening infections in immunocompromised individuals and may lead to septic abortion in pregnant women (Pamer, 2004). Upon internalization by phagocytes, LM escapes the early phagosome by secreting a hemolytic toxin, Listeriolysin O (LLO) (Portnoy et al., 1988). Listeria mutants that do not express the LLO toxin fail to escape the phagosome or to induce IFN-β production. LM-induced type I IFN production is largely independent of TLR signaling and is instead mediated by cytoplasmic RLR- and STING-dependent pathways through the TBK1-IRF3 signaling axis (Ishikawa et al., 2009; Woodward et al., 2010). In addition, in vivo studies demonstrated that Irf3−/− and Ifnar−/− mice, which neither make nor respond to type I IFNs, respectively, are highly resistant to L. monocytogenes infection (Auerbuch et al., 2004; Carrero et al., 2004; O'Connell et al., 2004). Furthermore, priming with poly(I:C), a well known type I-inducing agent, results in enhanced death rate in LM-infected WT mice but not in Ifnar−/− mice (O'Connell et al., 2004), indicating a detrimental role for type I IFNs during Listeria infection. Moreover, LM-infected Ifnar−/− mice were shown to have reduced lymphocyte and splenocyte apoptosis and antibody-dependent neutralization of LLO controlled infection that resulted in reduced pathology (Auerbuch et al., 2004; Carrero et al., 2004; O'Connell et al., 2004). However, Rayamajhi et al. have proposed an alternative mechanism, where type I IFNs-dependent down-regulation of IFNγ receptor results in heightened susceptibility of wild type mice to Listeria infection (Rayamajhi et al., 2010).
Listeria is known to trigger assembly of multiple types of inflammasomes that include Aim2, Nlrc4, and Nlrp3 for caspase-1 activation in mouse macrophages. During Listeria infection, type I IFNs have significant roles in regulation of inflammasome activation and pyroptosis (Henry et al., 2007; Kim et al., 2010; Rathinam et al., 2010; Wu et al., 2010). A few studies have attempted to dissect the relative contributions of each of these inflammasomes and demonstrated that the detection of DNA by AIM2 receptor was indispensable for inflammasome activation and pyroptosis during Listeria infection in human PBMCs and mouse macrophages (Kim et al., 2010; Rathinam et al., 2010; Sauer et al., 2010). In addition, Sauer et al. demonstrated that pyroptosis was totally dependent on Aim2, while the Nlrp3 and Nlrc4 inflammasomes were dispensable for this process (Sauer et al., 2010). These reports implicate that type I IFNs play a role in the efficient induction of inflammasome activation and pyroptosis.
Legionella
Legionella pneumophila is an intracellular, gram-negative bacterial pathogen that replicates in host macrophages and causes a severe pneumonia called Legionnaires' disease. Lipmann et al. reported that L. pneumophila-infected mouse macrophages produce IFNβ in a STING- and IRF3-dependent manner (Stetson and Medzhitov, 2006a; Lippmann et al., 2011). By contrast, Monroe et al. have demonstrated that L. pneumophila RNA also stimulated a Rig-I-dependent IFN response and proposed that L. pneumophila RNA, or host RNA, rather than L. pneumophila DNA, as the primary ligand that stimulates the host IFN response (Monroe et al., 2009). IFN-αβ inhibits L. pneumophila replication in the permissive A/J or CD1 mouse macrophages (Schiavoni et al., 2004). Furthermore, Bastian et al. have reported that Legionella is controlled by IFNβ induced in human lung epithelial cells via MAVS and IRF3 (Opitz et al., 2006). Dendritic cells (DCs) and macrophages are capable of restricting L. pneumophila growth through NAIP5-dependent caspase-1 activation and cell death. However, DCs were shown to undergo a more rapid apoptosis than macrophages, leading to enhanced restriction of Legionella growth (Nogueira et al., 2009). Indeed, eliminating the pro-apoptotic proteins BAX and BAK or over-expressing the anti-apoptotic protein BCL-2 were both found to restore L. pneumophila replication in DCs (Nogueira et al., 2009). Furthermore, a sub-population of DCs, plasmacytoid DCs (pDCs) is known to express higher levels of IFNs (Liu, 2005), which can potentially contribute to the higher cell death responses to Legionella. How Legionella-induced type I IFN promotes cell death is not well understood currently, however, one possible mechanism might be that Type I IFN-dependent up regulation of pro-cell death molecules like BAK and TRAIL can potentiate apoptosis (Fuertes Marraco et al., 2011; Cohen and Prince, 2013).
Mycobacterium
Mycobacterium spp. are pathogenic intracellular bacteria that cause tuberculosis (TB) and leprosy. Human and mouse myeloid cells secrete type I IFNs in response to mycobacterial infections (Pandey et al., 2009; Berry et al., 2010; Novikov et al., 2011). Blood based profiling has identified type I IFN-induced genes as the most striking characteristic signature of active TB (Berry et al., 2010). In addition, Wu et al. have reported that several TB-induced genes have key transcription factor binding sites for STATs, IRF-1, IRF-7, and OCT-1 (Wu et al., 2012). IFN-β and its downstream genes, including interleukin-10 (IL-10), were induced in monocytes by M. leprae in vitro and were preferentially expressed in progressive lepromatous lesions (Teles et al., 2013). Manca et al. have reported that type I IFNs enhance the virulence of M. tuberculosis by suppression of Th1 type immune responses. They have also shown that treatment with purified IFN-α/β increases lung bacterial loads, resulting in reduced survival in mice (Manca et al., 2001). In addition, another study reported that treatment with exogenous type I IFN results in a striking loss of mycobacteriostatic activity in monocytes and macrophages (Bouchonnet et al., 2002). Furthermore, Mayer-Barber et al. have shown that both IL-1α and IL-1β are critical for host resistance to TB and provided evidence that type I IFNs suppressed IL-1 production (Mayer-Barber et al., 2011). However, early clinical studies suggested that type I IFNs have beneficial effects against pulmonary TB (Giosue et al., 1998; Palmero et al., 1999). Together, these studies indicate that the role of type I IFNs in mycobacterial infections is debatable and requires further research to establish a clear consensus.
Francisella
Francisella tularensis is a gram-negative bacterium and causes tularemia. F. tularensis is classified as a Class A agent (having a high potential for use as a bioweapon) by United States regulators, due to its high virulence and ability to spread rapidly. Cytosolic recognition of Francisella induces type I IFN production in an IRF3-dependent manner (Henry et al., 2007). Francisella induces Aim2 inflammasome-dependent pyroptosis, which further depends on the ability of the bacterium to induce type I IFN production (Henry et al., 2007; Fernandes-Alnemri et al., 2010). Consistent with this, Francisella-induced Aim2 inflammasome activation and IL-1β secretion are abrogated in macrophages derived from bone marrow of Irf3- and Ifnar-deficient mice (Fernandes-Alnemri et al., 2010; Jones et al., 2010; Rathinam et al., 2010). Wild-type Francisella that can escape into the cytosol induces type I IFN and Aim2 inflammasome activation and host cell death (Mariathasan et al., 2005; Henry et al., 2007; Jones et al., 2010). This observation underscores the importance of cytosolic recognition of bacteria or their components (DNA in the case of Francisella) as an important innate immune mechanism to trigger inflammasome activation. Although studies in mice certainly support an important role for Aim2 in immune responses to Francisella, its role in human monocytes seems to be less prominent due to its lack of expression and/or induction in response to type I IFNs (Gavrilin and Wewers, 2011).
Salmonella
Salmonella enterica serovar Typhimurium (S. Typhimurium) is a gram-negative, intracellular pathogen that is quickly cleared by macrophages. This pathogen is a leading cause of acute gastroenteritis worldwide, which is transmitted primarily via the consumption of contaminated food or water. Induction of rapid cell death is a virulence strategy for this pathogen and contributes to dampening host innate immune responses (Lindgren et al., 1996). Robinson et al. have reported that Salmonella exploits type I IFN signaling for eliminating macrophages to establish infection (Robinson et al., 2012). Their studies also revealed that type I IFN-induced cell death of the macrophages is mediated by the classical RIP1-RIP3 dependent necroptosis pathway (Robinson et al., 2012). Salmonella is detected by NLRP3 and NLRC4 inflammasomes resulting in casapse-1 activation and pyroptosis (Franchi et al., 2006; Miao et al., 2006; Broz et al., 2010). Broz et al. have suggested the existence of an IFN-inducible regulator of caspase-11 that is crucial for activation of non-canonical Nlrp3 inflammasome activation in mutant Salmonella- [type 3 secretion system mutant, i.e., Salmonella pathogenicity island 1(SPI-1)] infected mouse macrophages (Broz et al., 2012). Their study revealed that Salmonella induces expression and activation of caspase-11 through a Toll-like receptor 4 (TLR4)-dependent and TIR-domain containing adaptor-inducing IFN-β (TRIF)-mediated IFNβ signaling pathway. Consistent with this, Ifnar1−/− or Irf3−/−, or Stat-1−/− macrophages infected with mutant Salmonella did not process the casapase-11 or activate the non-canonical cell death pathway. Furthermore, in vivo Casp1−/− mice are more susceptible to Salmonella infection than the Casp1−/− Casp11−/− mice (Broz et al., 2012). Thus, these results indicate that caspase-11 mediated cell death results in detrimental effects to the host. Together, it is evident that Salmonella exploits type I IFN signaling to rapidly kill the immune cells to cause disease in the host.
Extracellular Bacteria
Staphylococcus
Staphylococcal infections have recently emerged as a significant problem to human health, due to the emergence of antibiotic resistant strains that cause life-threatening infections, especially in post-influenza exposures (Klevens et al., 2007; Martin et al., 2009; David and Daum, 2010). S. aureus infected mouse and human epithelial cells produce type I IFN in a STAT3-dependent manner in response to its virulent protein A (Martin et al., 2009). In mouse myeloid DCs and macrophages, TLR9 and IRF1 have important roles (Schmitz et al., 2007), while the cell wall component lipoteichoic acid (LTA) utilizes the IRF1-STAT1 axis in mouse macrophages to induce type I IFNs (Liljeroos et al., 2008). Absence of IFNAR signaling results in protection against lethal S. aureus pneumonia infection compared to wild-type control mice (Martin et al., 2009). Following recognition of S. aureus α-hemolysin, mouse macrophages undergo pyroptosis in an Nlrp3 inflammasome-dependent manner (Mariathasan et al., 2006; Craven et al., 2009). In contrast to Martin et al., a recent report demonstrated that IFNα induces phospholipid scramblase 1 (PLSCR1) in human lung epithelial cells as part of an innate protective mechanism to a bacterial pore-forming toxin (Lizak and Yarovinsky, 2012) and another study demonstrated a protective role of CpG DNA (a potent inducer of type I IFN production) in a mouse model of S. aureus pneumonia (Roquilly et al., 2010). Furthermore, Kaplan et al. have shown that phagosomal degradation and cytosolic release of intracellular ligands are essential for the induction of IFN-β in mouse and human DCs, which is required for the host defense against S. aureus during cutaneous infection in mice (Kaplan et al., 2012). Taken together, these reports suggest that type I IFNs can have both protective and detrimental roles during S. aureus infection. The disease outcome is variable and may depend on the immune status of the host, the site of infection and the specific strains causing the infection.
Streptococcus
S. pneumonia causes acute lung infections and activates type I IFN expression (Joyce et al., 2009; Parker et al., 2011). DAI (DNA-dependent activator of IFN-regulatory factors) dependent recognition of bacterial DNA is proposed to be responsible for inducing type I IFN expression through cytoplasmic DNA sensing pathway involving STING, TBK1 and IRF3-dependent signaling pathways (Parker et al., 2011). Type I IFN treatment enhances protection of mice against S. pneumoniae (Weigent et al., 1986). However, prior exposure to influenza A virus leads to increased susceptibility to bacterial infections in a type I IFN-dependent manner (Morens et al., 2008; Shahangian et al., 2009). As opposed to bacterial infection alone, type I IFNs produced during secondary infection with S. pneumonia inhibits production of chemokines like CXCL1 and CXCL2 and sensitizes hosts to secondary bacterial pneumonia (Shahangian et al., 2009).
Pseudomonas
Pseudomonas aeruginosa is a causative agent of pneumonia and infection in cystic fibrosis (CF) patients is associated with significant mortality (Zhuo et al., 2008). P. aeruginosa induces type I IFN expression predominantly through the TLR4-TRIF-IRF3 axis (Parker et al., 2012). TLR4 signaling was shown to be important for clearance of P. aeruginosa from the lungs and preventing sepsis in infected hosts (Faure et al., 2004; Ramphal et al., 2005; Skerrett et al., 2007; Cohen and Prince, 2013). Similarly, Trif and Irf3 deficiencies in mice resulted in reduced expression of type I IFN-induced chemokines including CXCL10 (IP-10) and CCL5 (RANTES) and abrogated neutrophil recruitment to the lungs leading to impaired bacterial clearance (Power et al., 2007; Carrigan et al., 2010). These results indicate a protective role for type I IFNs during P. aeruginosa infection (Roy et al., 2013). P. aeruginosa infection of mouse macrophages activates the Nlrc4 inflammasome and induces pyroptosis in a flagellin independent manner (Sutterwala et al., 2007). However, later studies demonstrated a requirement of cytosolic flagellin for Nlrc4 inflammasome activation (Miao et al., 2008; Arlehamn and Evans, 2011). Bacterial expression of specific adhesins, lipopolysaccharide, and a functional type III secretion system were all shown to be necessary to evoke apoptosis (Sutterwala et al., 2007) and the cytotoxin, ExoU-expressing P. aeruginosa strain has been shown to inhibit caspase-1 dependent pyroptosis (Sutterwala et al., 2007).
Anthrax
Bacillus anthracis is a gram-positive, aerobic bacterium that causes severe pulmonary, gastrointestinal, and cutaneous infections (Dixon et al., 1999). Production of the lethal toxin (LeTx) by this bacterium causes extensive cell death, tissue damage and systemic disease. LeTx is composed of a protective antigen (PA) and lethal factor (LF). Gold et al. found that endogenous IFNs (type I and II) inhibit the germination of B. anthracis spores, but exogenous application enhanced inflammation thereby increasing mortality (Gold et al., 2007). In addition, Walberg et al. showed that recombinant murine IFNβ or type I IFN inducers like poly(I:C) provide marked protection against “Inhalation Anthrax” (Walberg et al., 2008). B. anthracis LeTxs activate the Nlrp1b inflammasome and pyroptosis in mice (Boyden and Dietrich, 2006; Bergsbaken et al., 2009). It is recognized that macrophages from inbred mice may or may not be sensitive to B. anthracis-induced pyroptosis based on the presence of the Nlrp1b inflammasome responsive Nlrp1bS/S or non-responsive Nlrp1bR/R alleles (Moayeri et al., 2010; Terra et al., 2010). In this case, pyroptosis of macrophages is believed to counter anthrax infection since IL-1β released during this process helps to generate protective neutrophil responses. It will be essential to study the contribution of type I IFN in pyroptosis induction during B. anthracis infection and investigate whether type I IFN can promote apoptosis of neutrophils and initiate other possible detrimental effects.
Viruses
IFNs were originally discovered as antiviral molecules. Viruses are considerably smaller than other microbial pathogens but represent a major threat to human and other animal health. Extensive progress has been made in understanding the mechanisms of type I IFN production in response to viruses (Stetson and Medzhitov, 2006b; Gonzalez-Navajas et al., 2012; MacMicking, 2012). The diverse mechanisms of cellular entry and tropism of viruses are detected by different TLRs that are strategically located in different cellular compartments (Figure 1). Endosomally-located PRRs including TLR3, 7, 8, and 9 are known to trigger type I IFNs (Figure 1). TLR7 and TLR9 have particularly important roles in plasmacytoid DCs. Once the virus enters the host cytoplasm, several cytoplasmic receptors such as RIG-I-like receptors (RLRs RIG-I, MDA5 and LGP2) and NOD-like receptors (NOD1 and NOD2) monitor the cytoplasm for microbial PAMPs and initiate type I IFN production and associated immune responses (Figure 1).
Programmed cell death is a critical host defense against viruses and type I IFNs are known to be involved to this process. Multiple viruses have been discovered to encode proteins that function to subvert host-induced cell death during infection (Bowie and Unterholzner, 2008; Galluzzi et al., 2010; Kaminskyy and Zhivotovsky, 2010; Gregory et al., 2011). Death of the infected cells is detrimental to viral replication and amplification of viral progeny. However, death of the infected cells can also facilitate viral egress and enhance pathogenesis. Therefore, different viruses have evolved complex mechanisms to enhance or inhibit different forms of cell death (Kaminskyy and Zhivotovsky, 2010). Microarray based studies revealed that a large number of genes are regulated by type I IFNs and several of them are involved in cell death (Der et al., 1998; de Veer et al., 2001; Hertzog and Williams, 2013; Rusinova et al., 2013). However, the mechanisms driving cell death that involve proteins encoded by many of these genes are still awaited.
Uncontrolled chronic viral infections can result in sustained expression of type I IFNs with detrimental pathophysiological outcomes. Two recent studies reported the role of type I IFNs in viral persistence during lymphocytic choriomeningitis virus (LCMV) infection (Teijaro et al., 2013; Wilson et al., 2013). Results from these reports show that robust and acute type I IFNs secreted during early in the infection serve to control viral replication and spread by promoting apoptosis of infected cells and enhancing T cell activation. During chronic infection, prolonged expression and exposure to type I IFNs leads to lymphocyte exhaustion, and reduced viral clearance due to the presence of increased immunosuppressive molecules like IL-10 and PD-L1. Loss of circulating pDCs has been documented in chronic viral infections in mice and humans, which correlates with uncontrolled viral loads, reduced T cell counts and onset of opportunistic infections (Swiecki et al., 2011). However, the mechanism of type I IFN-dependent pDC apoptosis is not entirely clear and represents an important subject for future research.
Type I IFNs promote cell death in multiple ways. Balachandran et al. have demonstrated that type I IFN and protein kinase R (PKR) can sensitize cells to apoptosis primarily through the FADD/caspase-8 pathway (Balachandran et al., 2000). In this study, stimulation of mouse cells with IFN-α/β resulted in enhanced apoptosis and reduced viral replication. In a follow up study, Ezelle et al. showed that the poxvirus-encoded protein CrmA was able to inhibit both viral infection- and dsRNA-mediated apoptosis (Ezelle et al., 2001). Both HSV and vaccinia virus affect PKR and RNase L-mediated apoptosis pathways that are activated by dsRNA products released during viral replication (Der et al., 1997; Diaz-Guerra et al., 1997; Kibler et al., 1997). Influenza viruses cause severe lung pathology leading to lung failure and mortality. Type I IFNs have been recognized to mediate induction of pro-apoptotic TRAIL leading to excessive cell death and tissue injury (Hogner et al., 2013). Infection or treatment with type I IFN induces pro-apoptotic genes; IFN-stimulated gene 54 (ISG54) or IFN-induced gene with tetratricopeptide repeats 2 (IFIT2) that promotes apoptosis by mitochondrial-associated BCL2 family proteins (Reich, 2013).
Fungi
While type I IFNs are widely known as anti-viral factors, which are either protective or detrimental in bacterial infections, their role in fungal infections is poorly defined. Recently, two reports have shown that Candida spp. induce IFN-β in mouse bone marrow-derived DCs (BMDCs) and macrophages (Biondo et al., 2008, 2012; Bourgeois et al., 2011). Different forms of fungal glucans and mannans are sensed by a wide range of innate pattern receptors like TLRs, CLRs, dectins, and mannose receptors and initiate MyD88–mediated NF-κB and MAPK pathways or SYK-CARD9 signaling for cytokine induction and cell death (Netea et al., 2008; Brown, 2011). During fungal infections, type I IFNs are also produced by a TLR-independent pathway requiring RNA sensor MAVS and IRF3 (Inglis et al., 2010). In contrast, Del Fresno et al. have recently reported that Candida albicans induces type I IFN in DCs through a DECTIN-1, SYK-, and CARD9-dependent pathway that requires IRF5-mediated transcription but not IRF3 or IRF7 (Del Fresno et al., 2013). Jensen et al. have reported that poly(I:C)-induced or exogenously added IFNα and IFNβ treatments of macrophages suppress anti-Candida immune responses and cause death of infected mice (Worthington and Hasenclever, 1972; Jensen et al., 1992; Jensen and Balish, 1993). In addition, Ifnar−/− mice are extremely resistant to otherwise lethal Candida and Histoplasma infections (Inglis et al., 2010; Majer et al., 2012). These studies demonstrated that type I IFNs induce severe kidney damage by promoting excessive recruitment and activation of inflammatory monocytes and neutrophils. However, other reports suggest that type I IFN can be beneficial as part of the host immune response to C. albicans (Biondo et al., 2011; Del Fresno et al., 2013).
Role of Interferons In Inflammasome Activation and Pyroptosis
Type I IFNs are innate immune effector molecules with strong pro-inflammatory activities, and have been shown to contribute to the high mortality rates in septic shock syndromes (Karaghiosoff et al., 2003; Huys et al., 2009). Type I IFNs also contribute to inflammasome-dependent caspase-1 activation leading to pro-inflammatory pyroptotic cell death (Figure 2) (Anand et al., 2011; Franchi et al., 2012). There have been multiple different inflammasomes identified that sense a diverse array of microbial- and damage-associated PAMPs. These include the Naip-Nlrc4 inflammasome (Mariathasan et al., 2004; Kofoed and Vance, 2011; Zhao et al., 2011), the Nlrp1b inflammasome (Boyden and Dietrich, 2006; Masters et al., 2012), the Nlrp3 inflammasome (Kanneganti et al., 2006b; Mariathasan et al., 2006; Sutterwala et al., 2006; Anand et al., 2011), the Nlrp6 inflammasome (Elinav et al., 2011), the Nlrp12 inflammasome (Vladimer et al., 2012), the Aim2 inflammasome (Fernandes-Alnemri et al., 2010; Jones et al., 2010; Rathinam et al., 2010; Sauer et al., 2010), the RIG-I inflammasome (Poeck et al., 2010; Pothlichet et al., 2013), and the IFI16 inflammasome (Kerur et al., 2011). Inflammasome-dependent casapase-1 activation and pyroptosis are associated with the production of mature IL-1β and IL-18 cytokines, which generates a pro-inflammatory environment in host tissues (Figure 2). Inflammasome-dependent pyroptosis shares features of both apoptosis and necrosis and is tightly regulated by distinct signaling pathways.
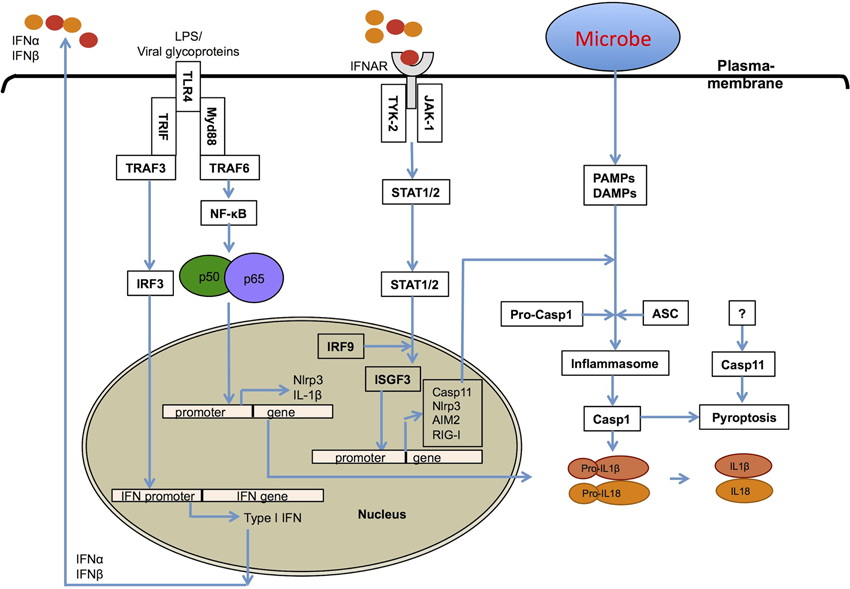
Figure 2. Role of type I interferons in inflammasome activation. Type I IFNs contribute to inflammasome activation through two different mechanisms. First, type I interferons are required for the upregulation of caspase-11, which contributes to activation of a non-canonical NLRP3 inflammasome in response to enteropathogenic bacteria, such as Citrobacter rodentium and Escherichia coli. Second, they prime the expression of inflammasome-forming NLRP3, RIG-I and AIM2 molecules for potentiating inflammasome activation.
NLRP3
NLRP3 is the most widely studied inflammasome and it requires two signals for its assembly into an active complex (Kanneganti, 2010; Anand et al., 2011). The first signal is TLR-dependent expression of NLRP3, while the second is often a damage related factor such as production of reactive oxygen species (ROS) or membrane damage (Anand et al., 2011). Activated TLRs transfer signals through two major adapters that specify the downstream signaling pathways (Takeuchi and Akira, 2010). The first one, MyD88 is required for NF-κB activation downstream of all TLRs except TLR3. The second, TRIF plays a dominant role in TLR3-dependent NF-κB activation and TLR4-mediated IRF signaling (Fitzgerald et al., 2003a,b; Sato et al., 2003; Yamamoto et al., 2003).
Recently, our lab and other groups have shown that the TLR4-TRIF axis regulates caspase-11 expression and non-canonical Nlrp3 inflammasome-mediated host defense against enteropathogens, Escherichia coli (EHEC), Citrobacter rodentium, and Salmonella Typhimurium (Kayagaki et al., 2011; Broz et al., 2012; Gurung et al., 2012; Rathinam et al., 2012). Consistent with this, Sander et al. have reported that the gram-negative bacterium E. coli induces IFN-β and activates the Nlrp3 inflammasome in the absence of virulence factors other than microbial mRNA (Sander et al., 2011). In addition, Rathinam et al. demonstrated that the IRF3-type I IFN-IFNAR-STAT-1 signaling pathway is indispensable for caspase-11 expression and activation of the Nlrp3 inflammasome and pyroptosis (Rathinam et al., 2012). Furthermore, they demonstrated that IFNβ treatment significantly increased pro-caspase-11 expression and that once induced, caspase-11 undergoes spontaneous activation. Broz et al. have observed that IFNAR-STAT-1 axis is important for caspase-11 activation, but not for its expression in Salmonella infected macrophages (Broz et al., 2012). In a subsequent review, they speculated that type I IFN-dependent expression of a yet-unidentified host molecule may trigger caspase-11 activation or that a unknown bacterial signal is required (Broz and Monack, 2013).
Two recent studies have revealed that several gram-negative bacteria, but not gram-positive bacteria, can activate the non-canonical Nlrp3 inflammasome and identified LPS as their common PAMP responsible for the activation of Caspase-11 (Hagar et al., 2013; Kayagaki et al., 2013). Both of these studies have shown that intracellular LPS is sufficient to induce the activation of caspase-11. Furthermore, these studies revealed that a sub-component of LPS, lipid A is sufficient to activate the casaspe-11-dependent non-canonical Nlrp3 inflammasome. They presented evidence that when transfected, intracellular LPS or lipid A could activate the non-canonical Nlrp3 inflammasome independent of TLR4 or the TLR4-associated lipid A-binding proteins MD1 and MD2 and even the TLR4 downstream signaling molecules TRIF and IFNAR receptor molecules. In contrast, a study by Guarda et al. reported that type I IFNs suppress Nlrp1 and Nlrp3 inflammasomes in a STAT1-dependent manner (Guarda et al., 2011). Further studies are required to clarify the role of type I IFNs and their precise mechanisms in the regulation of inflammasome activation. Of particular importance is that the intracellular sensor of lipid A is yet to be identified.
Together, regardless of the exact mechanisms, type I IFNs have clearly emerged as crucial regulators of inflammasome activation and pyroptosis (Figure 2). Physiologically, caspase-11 triggered pyroptosis is required for surveillance against bacteria that enter the cytosol, such as the sifA mutant of S. Typhimurium, an sdhA mutant of Legionella pneumophila and Burkholderia species (Aachoui et al., 2013). However, type I IFNs are known to be exploited by microbial pathogens to induce the death of immune cells and suppress host immune responses. In support of this, caspase-11-mediated cell death is responsible for pathology and detrimental effects in vivo (Wang et al., 1998; Kayagaki et al., 2011). For example, Broz et al. showed that wild type Salmonella induces caspase-11-mediated cell death in caspase-1-deficient mouse macrophages, and that caspase-11 increases the bacterial virulence and host cell susceptibility to infection (Broz et al., 2012). In addition, Salmonella also exploits type I IFN signaling to induce the RIP1-RIP3-dependent necroptotic cell death pathway to kill macrophages (Lindgren et al., 1996; Robinson et al., 2012).
RIG-I and NLRP3
Viruses are by far the best-known inducers of type I IFNs and have also been recognized to induce activation of distinct inflammasomes, including those comprised of NLRP3, AIM2, and RIG-I. The first evidence of Nlrp3 inflammasome involvement in antiviral responses showed its role in sensing of both viral RNA and its analog poly(I:C) in mouse macrophages (Kanneganti et al., 2006a,b). In vivo, Nlrp3 inflammasome activation protected mice from influenza infection (Allen et al., 2009; Thomas et al., 2009). Poeck et al. have reported that Rig-I from mouse macrophages senses cytoplasmic RNA viruses and assembles an inflammasome (Poeck et al., 2010). More recently, RIG-I was reported to induce type I IFN through a MAVS/TRIM25/RNF135 signaling axis following influenza infection, and was shown to have profound effects on NLRP3 inflammasome activation and IL-1β secretion in human lung epithelial cells (Pothlichet et al., 2013). Together, these studies demonstrate that RIG-I can itself assemble an inflammasome and also contributes toward, type I IFN-dependent potentiation of NLRP3 expression (Figure 2).
AIM2
The cytosolic bacteria Francisella novicida and Listeria both induce killing of myeloid and lymphoid cells in a manner dependent on Type I IFN signaling, an effect which has been shown to be detrimental to the host (Auerbuch et al., 2004; Carrero et al., 2004; O'Connell et al., 2004; Henry et al., 2007). Several published studies have clearly established that both Listeria (Henry et al., 2007; Warren et al., 2008; Kim et al., 2010; Tsuchiya et al., 2010; Wu et al., 2010) and Francisella (Henry and Monack, 2007; Fernandes-Alnemri et al., 2010; Jones et al., 2010; Rathinam et al., 2010) activate the Aim2 inflammasome and pyroptosis in mouse macrophages. Type I IFNs prime AIM2 expression (Kotredes and Gamero, 2013) and potentiate cytosolic bacterial DNA recognition for inflammasome activation (Fernandes-Alnemri et al., 2010). Although mice studies show that type I IFN dependent caspase-11 expression is important for the activation of the non-canonical Nlrp3 inflammasome (Kayagaki et al., 2011; Gurung et al., 2012; Rathinam et al., 2012), it is not known if it is also required for AIM2 and RIG-I mediated caspase-1 activation or cell death.
Conclusions
IFNs were the first cytokines discovered to have immune regulatory capacity. Despite their clinical application in some treatment regimens, we still do not have a complete understanding of the mechanistic effects of IFNs required to further develop treatments that capitalize their full potential therapeutic effects. A vast amount of past research has been focused on the role of IFNs as anti-viral molecules with a limited number of studies for other microbial infections. Recent studies have clearly indicated a dual role for type I IFNs both in infectious and inflammatory diseases. Despite the potential benefits, it is often challenging to manipulate type I IFNs for therapeutic purposes due to their role in regulating the expression and activation of a huge number of downstream genes, often complicating the conclusions. Part of the problem is that types I IFNs exert differential immunomodulation on diverse cell types, environments, and varying physiological conditions. The pathogen- and host-mediated counter regulatory pathways further complicate IFN-induced responses in cell death and disease.
Recently, type I IFNs were recognized as crucial regulators of non-canonical NLRP3 inflammasome activation and pyroptosis. Although the majority of literature indicates a positive role for inflammasomes in anti-microbial host defense, recent reports indicate detrimental effects due to excessive cell death, inflammation, and collateral tissue damage in vital organs (Lupfer and Kanneganti, 2012). A paradox exists, where inflammasomes intended to defend against cytoplasmic invaders by pyroptosis, which is predominantly protective in vitro, however in in vivo, if exceeds a certain level, can lead to cell and tissue damage and organ failure resulting in negative outcomes. The paradox for type-I IFNs is that their ability to inhibit microbial spread by inducing cell death is counteracted by apoptotic depletion of immune cells and inhibiting anti-microbial immune responses leading to immune suppression. Future research should explore the detailed molecular mechanisms that are responsible for type I IFN-dependent cell death and inflammasomes activation in the context of immunity and immunopathology. These findings may lead to better-targeted therapeutic interventions to treat inflammatory and infectious diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to the numerous investigators whose papers could not be cited because of space limitations. This work was supported by National Institutes of Health grants (AR056296, CA163507 and AI101935), and the American Lebanese Syrian Associated Charities (ALSAC) to Thirumala-Devi Kanneganti.
Abbreviation
IFN-I, type I interferon; IFNAR, Interferon-α/β receptor; ISRE, Interferon stimulated response element; ISGs, Interferon stimulated genes; TLR, TOLL-like receptor; NLR, NOD-like receptor.
References
Aachoui, Y., Leaf, I. A., Hagar, J. A., Fontana, M. F., Campos, C. G., Zak, D. E., et al. (2013). Caspase-11 protects against bacteria that escape the vacuole. Science 339, 975–978. doi: 10.1126/science.1230751
Allen, I. C., Scull, M. A., Moore, C. B., Holl, E. K., McElvania-TeKippe, E., Taxman, D. J., et al. (2009). The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30, 556–565. doi: 10.1016/j.immuni.2009.02.005
Anand, P. K., Malireddi, R. K., and Kanneganti, T. D. (2011). Role of the nlrp3 inflammasome in microbial infection. Front. Microbiol. 2:12. doi: 10.3389/fmicb.2011.00012
Arlehamn, C. S., and Evans, T. J. (2011). Pseudomonas aeruginosa pilin activates the inflammasome. Cell Microbiol. 13, 388–401. doi: 10.1111/j.1462-5822.2010.01541.x
Auerbuch, V., Brockstedt, D. G., Meyer-Morse, N., O'Riordan, M., and Portnoy, D. A. (2004). Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J. Exp. Med. 200, 527–533. doi: 10.1084/jem.20040976
Balachandran, S., Roberts, P. C., Kipperman, T., Bhalla, K. N., Compans, R. W., Archer, D. R., et al. (2000). Alpha/beta interferons potentiate virus-induced apoptosis through activation of the FADD/Caspase-8 death signaling pathway. J. Virol. 74, 1513–1523. doi: 10.1128/JVI.74.3.1513-1523.2000
Bergsbaken, T., Fink, S. L., and Cookson, B. T. (2009). Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109. doi: 10.1038/nrmicro2070
Berry, M. P., Graham, C. M., McNab, F. W., Xu, Z., Bloch, S. A., Oni, T., et al. (2010). An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466, 973–977. doi: 10.1038/nature09247
Biondo, C., Malara, A., Costa, A., Signorino, G., Cardile, F., Midiri, A., et al. (2012). Recognition of fungal RNA by TLR7 has a non-redundant role in host defense against experimental candidiasis. Eur. J. Immunol. 42, 2632–2643. doi: 10.1002/eji.201242532
Biondo, C., Midiri, A., Gambuzza, M., Gerace, E., Falduto, M., Galbo, R., et al. (2008). IFN-alpha/beta signaling is required for polarization of cytokine responses toward a protective type 1 pattern during experimental cryptococcosis. J. Immunol. 181, 566–573.
Biondo, C., Signorino, G., Costa, A., Midiri, A., Gerace, E., Galbo, R., et al. (2011). Recognition of yeast nucleic acids triggers a host-protective type I interferon response. Eur. J. Immunol. 41, 1969–1979. doi: 10.1002/eji.201141490
Bouchonnet, F., Boechat, N., Bonay, M., and Hance, A. J. (2002). Alpha/beta interferon impairs the ability of human macrophages to control growth of Mycobacterium bovis BCG. Infect. Immun. 70, 3020–3025. doi: 10.1128/IAI.70.6.3020-3025.2002
Bourgeois, C., Majer, O., Frohner, I. E., Lesiak-Markowicz, I., Hildering, K. S., Glaser, W., et al. (2011). Conventional dendritic cells mount a type I IFN response against Candida spp. requiring novel phagosomal TLR7-mediated IFN-beta signaling. J. Immunol. 186, 3104–3112. doi: 10.4049/jimmunol.1002599
Bowie, A. G., and Unterholzner, L. (2008). Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 8, 911–922. doi: 10.1038/nri2436
Boyden, E. D., and Dietrich, W. F. (2006). Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 38, 240–244. doi: 10.1038/ng1724
Brown, G. D. (2011). Innate antifungal immunity: the key role of phagocytes. Annu. Rev. Immunol. 29, 1–21. doi: 10.1146/annurev-immunol-030409-101229
Broz, P., and Monack, D. M. (2013). Non-canonical inflammasomes: caspase-11 activation and effector mechanisms. PLoS Pathog. 9:e1003144. doi: 10.1371/journal.ppat.1003144
Broz, P., Newton, K., Lamkanfi, M., Mariathasan, S., Dixit, V. M., and Monack, D. M. (2010). Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J. Exp. Med. 207, 1745–1755. doi: 10.1084/jem.20100257
Broz, P., Ruby, T., Belhocine, K., Bouley, D. M., Kayagaki, N., Dixit, V. M., et al. (2012). Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature 490, 288–291. doi: 10.1038/nature11419
Carrero, J. A., Calderon, B., and Unanue, E. R. (2004). Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J. Exp. Med. 200, 535–540. doi: 10.1084/jem.20040769
Carrigan, S. O., Junkins, R., Yang, Y. J., Macneil, A., Richardson, C., Johnston, B., et al. (2010). IFN regulatory factor 3 contributes to the host response during Pseudomonas aeruginosa lung infection in mice. J. Immunol. 185, 3602–3609. doi: 10.4049/jimmunol.0903429
Cohen, T. S., and Prince, A. S. (2013). Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J. Clin. Invest. 123, 1630–1637. doi: 10.1172/JCI66142
Craven, R. R., Gao, X., Allen, I. C., Gris, D., Bubeck Wardenburg, J., McElvania-Tekippe, E., et al. (2009). Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE 4:e7446. doi: 10.1371/journal.pone.0007446
David, M. Z., and Daum, R. S. (2010). Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 23, 616–687. doi: 10.1128/CMR.00081-09
Del Fresno, C., Soulat, D., Roth, S., Blazek, K., Udalova, I., Sancho, D., et al. (2013). Interferon-beta production via Dectin-1-Syk-IRF5 signaling in dendritic cells is crucial for immunity to C. albicans. Immunity 38, 1176–1186. doi: 10.1016/j.immuni.2013.05.010
Der, S. D., Yang, Y. L., Weissmann, C., and Williams, B. R. (1997). A double-stranded RNA-activated protein kinase-dependent pathway mediating stress-induced apoptosis. Proc. Natl. Acad. Sci. U.S.A. 94, 3279–3283. doi: 10.1073/pnas.94.7.3279
Der, S. D., Zhou, A., Williams, B. R., and Silverman, R. H. (1998). Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. U.S.A. 95, 15623–15628. doi: 10.1073/pnas.95.26.15623
de Veer, M. J., Holko, M., Frevel, M., Walker, E., Der, S., Paranjape, J. M., et al. (2001). Functional classification of interferon-stimulated genes identified using microarrays. J. Leukoc. Biol. 69, 912–920.
Diaz-Guerra, M., Rivas, C., and Esteban, M. (1997). Activation of the IFN-inducible enzyme RNase L causes apoptosis of animal cells. Virology 236, 354–363. doi: 10.1006/viro.1997.8719
Dixon, T. C., Meselson, M., Guillemin, J., and Hanna, P. C. (1999). Anthrax. N. Engl. J. Med. 341, 815–826. doi: 10.1056/NEJM199909093411107
Elinav, E., Strowig, T., Kau, A. L., Henao-Mejia, J., Thaiss, C. A., Booth, C. J., et al. (2011). NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757. doi: 10.1016/j.cell.2011.04.022
Ezelle, H. J., Balachandran, S., Sicheri, F., Polyak, S. J., and Barber, G. N. (2001). Analyzing the mechanisms of interferon-induced apoptosis using CrmA and hepatitis C virus NS5A. Virology 281, 124–137. doi: 10.1006/viro.2001.0815
Faure, K., Sawa, T., Ajayi, T., Fujimoto, J., Moriyama, K., Shime, N., et al. (2004). TLR4 signaling is essential for survival in acute lung injury induced by virulent Pseudomonas aeruginosa secreting type III secretory toxins. Respir. Res. 5, 1. doi: 10.1186/1465-9921-5-1
Fernandes-Alnemri, T., Yu, J. W., Juliana, C., Solorzano, L., Kang, S., Wu, J., et al. (2010). The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393. doi: 10.1038/ni.1859
Fitzgerald, K. A., McWhirter, S. M., Faia, K. L., Rowe, D. C., Latz, E., Golenbock, D. T., et al. (2003a). IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496. doi: 10.1038/ni921
Fitzgerald, K. A., Rowe, D. C., Barnes, B. J., Caffrey, D. R., Visintin, A., Latz, E., et al. (2003b). LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J. Exp. Med. 198, 1043–1055. doi: 10.1084/jem.20031023
Franchi, L., Amer, A., Body-Malapel, M., Kanneganti, T. D., Ozoren, N., Jagirdar, R., et al. (2006). Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 7, 576–582. doi: 10.1038/ni1346
Franchi, L., Munoz-Planillo, R., and Nunez, G. (2012). Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 13, 325–332. doi: 10.1038/ni.2231
Fuertes Marraco, S. A., Scott, C. L., Bouillet, P., Ives, A., Masina, S., Vremec, D., et al. (2011). Type I interferon drives dendritic cell apoptosis via multiple BH3-only proteins following activation by PolyIC in vivo. PLoS ONE 6:e20189. doi: 10.1371/journal.pone.0020189
Galluzzi, L., Kepp, O., Morselli, E., Vitale, I., Senovilla, L., Pinti, M., et al. (2010). Viral strategies for the evasion of immunogenic cell death. J. Intern. Med. 267, 526–542. doi: 10.1111/j.1365-2796.2010.02223.x
Gavrilin, M. A., and Wewers, M. D. (2011). Francisella recognition by inflammasomes: differences between mice and men. Front. Microbiol. 2:11. doi: 10.3389/fmicb.2011.00011
Giosue, S., Casarini, M., Alemanno, L., Galluccio, G., Mattia, P., Pedicelli, G., et al. (1998). Effects of aerosolized interferon-alpha in patients with pulmonary tuberculosis. Am. J. Respir. Crit. Care Med. 158, 1156–1162. doi: 10.1164/ajrccm.158.4.9803065
Gold, J. A., Hoshino, Y., Jones, M. B., Hoshino, S., Nolan, A., and Weiden, M. D. (2007). Exogenous interferon-alpha and interferon-gamma increase lethality of murine inhalational anthrax. PLoS ONE 2:e736. doi: 10.1371/journal.pone.0000736
Gonzalez-Navajas, J. M., Lee, J., David, M., and Raz, E. (2012). Immunomodulatory functions of type I interferons. Nat. Rev. Immunol. 12, 125–135. doi: 10.1038/nri3133
Gregory, S. M., Davis, B. K., West, J. A., Taxman, D. J., Matsuzawa, S., Reed, J. C., et al. (2011). Discovery of a viral NLR homolog that inhibits the inflammasome. Science 331, 330–334. doi: 10.1126/science.1199478
Guarda, G., Braun, M., Staehli, F., Tardivel, A., Mattmann, C., Forster, I., et al. (2011). Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34, 213–223. doi: 10.1016/j.immuni.2011.02.006
Gurung, P., Malireddi, R. K., Anand, P. K., Demon, D., Walle, L. V., Liu, Z., et al. (2012). Toll or interleukin-1 receptor (TIR) domain-containing adaptor inducing interferon-beta (TRIF)-mediated caspase-11 protease production integrates Toll-like receptor 4 (TLR4) protein- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J. Biol. Chem. 287, 34474–34483. doi: 10.1074/jbc.M112.401406
Hagar, J. A., Powell, D. A., Aachoui, Y., Ernst, R. K., and Miao, E. A. (2013). Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341, 1250–1253. doi: 10.1126/science.1240988
Havell, E. A. (1986). Augmented induction of interferons during Listeria monocytogenes infection. J. Infect. Dis. 153, 960–969. doi: 10.1093/infdis/153.5.960
Henry, T., Brotcke, A., Weiss, D. S., Thompson, L. J., and Monack, D. M. (2007). Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J. Exp. Med. 204, 987–994. doi: 10.1084/jem.20062665
Henry, T., and Monack, D. M. (2007). Activation of the inflammasome upon Francisella tularensis infection: interplay of innate immune pathways and virulence factors. Cell Microbiol. 9, 2543–2551. doi: 10.1111/j.1462-5822.2007.01022.x
Hertzog, P. J., and Williams, B. R. (2013). Fine tuning type i interferon responses. Cytokine Growth Factor Rev. 24, 217–225. doi: 10.1016/j.cytogfr.2013.04.002
Hogner, K., Wolff, T., Pleschka, S., Plog, S., Gruber, A. D., Kalinke, U., et al. (2013). Macrophage-expressed IFN-beta contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia. PLoS Pathog. 9:e1003188. doi: 10.1371/journal.ppat.1003188
Huys, L., Van Hauwermeiren, F., Dejager, L., Dejonckheere, E., Lienenklaus, S., Weiss, S., et al. (2009). Type I interferon drives tumor necrosis factor-induced lethal shock. J. Exp. Med. 206, 1873–1882. doi: 10.1084/jem.20090213
Inglis, D. O., Berkes, C. A., Hocking Murray, D. R., and Sil, A. (2010). Conidia but not yeast cells of the fungal pathogen Histoplasma capsulatum trigger a type I interferon innate immune response in murine macrophages. Infect. Immun. 78, 3871–3882. doi: 10.1128/IAI.00204-10
Ishikawa, H., Ma, Z., and Barber, G. N. (2009). STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792. doi: 10.1038/nature08476
Jensen, J., and Balish, E. (1993). Enhancement of susceptibility of CB-17 mice to systemic candidiasis by poly(I. C)-induced interferon. Infect. Immun. 61, 3530–3532.
Jensen, J., Vazquez-Torres, A., and Balish, E. (1992). Poly(I.C)-induced interferons enhance susceptibility of SCID mice to systemic candidiasis. Infect. Immun. 60, 4549–4557.
Jones, J. W., Kayagaki, N., Broz, P., Henry, T., Newton, K., O'Rourke, K., et al. (2010). Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. U.S.A. 107, 9771–9776. doi: 10.1073/pnas.1003738107
Joyce, E. A., Popper, S. J., and Falkow, S. (2009). Streptococcus pneumoniae nasopharyngeal colonization induces type I interferons and interferon-induced gene expression. BMC Genomics 10:404. doi: 10.1186/1471-2164-10-404
Kaminskyy, V., and Zhivotovsky, B. (2010). To kill or be killed: how viruses interact with the cell death machinery. J. Intern. Med. 267, 473–482. doi: 10.1111/j.1365-2796.2010.02222.x
Kanneganti, T. D. (2010). Central roles of NLRs and inflammasomes in viral infection. Nat. Rev. Immunol. 10, 688–698. doi: 10.1038/nri2851
Kanneganti, T. D., Body-Malapel, M., Amer, A., Park, J. H., Whitfield, J., Franchi, L., et al. (2006a). Critical role for Cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 281, 36560–36568. doi: 10.1074/jbc.M607594200
Kanneganti, T. D., Ozoren, N., Body-Malapel, M., Amer, A., Park, J. H., Franchi, L., et al. (2006b). Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440, 233–236. doi: 10.1038/nature04517
Kaplan, A., Ma, J., Kyme, P., Wolf, A. J., Becker, C. A., Tseng, C. W., et al. (2012). Failure to induce IFN-beta production during Staphylococcus aureus infection contributes to pathogenicity. J. Immunol. 189, 4537–4545. doi: 10.4049/jimmunol.1201111
Karaghiosoff, M., Steinborn, R., Kovarik, P., Kriegshauser, G., Baccarini, M., Donabauer, B., et al. (2003). Central role for type I interferons and Tyk2 in lipopolysaccharide-induced endotoxin shock. Nat. Immunol. 4, 471–477. doi: 10.1038/ni910
Kayagaki, N., Warming, S., Lamkanfi, M., Vande Walle, L., Louie, S., Dong, J., et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. doi: 10.1038/nature10558
Kayagaki, N., Wong, M. T., Stowe, I. B., Ramani, S. R., Gonzalez, L. C., Akashi-Takamura, S., et al. (2013). Non-canonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249. doi: 10.1126/science.1240248
Kerur, N., Veettil, M. V., Sharma-Walia, N., Bottero, V., Sadagopan, S., Otageri, P., et al. (2011). IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 9, 363–375. doi: 10.1016/j.chom.2011.04.008
Kibler, K. V., Shors, T., Perkins, K. B., Zeman, C. C., Banaszak, M. P., Biesterfeldt, J., et al. (1997). Double-stranded RNA is a trigger for apoptosis in vaccinia virus-infected cells. J. Virol. 71, 1992–2003.
Kim, S., Bauernfeind, F., Ablasser, A., Hartmann, G., Fitzgerald, K. A., Latz, E., et al. (2010). Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur. J. Immunol. 40, 1545–1551. doi: 10.1002/eji.201040425
Klevens, R. M., Morrison, M. A., Nadle, J., Petit, S., Gershman, K., Ray, S., et al. (2007). Invasive methicillin-resistant Staphylococcus aureus infections in the United States. Jama 298, 1763–1771. doi: 10.1001/jama.298.15.1763
Kofoed, E. M., and Vance, R. E. (2011). Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477, 592–595. doi: 10.1038/nature10394
Kotredes, K. P., and Gamero, A. M. (2013). Interferons as inducers of apoptosis in malignant cells. J. Interferon Cytokine Res. 33, 162–170. doi: 10.1089/jir.2012.0110
Liljeroos, M., Vuolteenaho, R., Rounioja, S., Henriques-Normark, B., Hallman, M., and Ojaniemi, M. (2008). Bacterial ligand of TLR2 signals Stat activation via induction of IRF1/2 and interferon-alpha production. Cell Signal 20, 1873–1881. doi: 10.1016/j.cellsig.2008.06.017
Lindgren, S. W., Stojiljkovic, I., and Heffron, F. (1996). Macrophage killing is an essential virulence mechanism of Salmonella typhimurium. Proc. Natl. Acad. Sci. U.S.A. 93, 4197–4201. doi: 10.1073/pnas.93.9.4197
Lippmann, J., Muller, H. C., Naujoks, J., Tabeling, C., Shin, S., Witzenrath, M., et al. (2011). Dissection of a type I interferon pathway in controlling bacterial intracellular infection in mice. Cell Microbiol. 13, 1668–1682. doi: 10.1111/j.1462-5822.2011.01646.x
Liu, Y. J. (2005). IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 23, 275–306. doi: 10.1146/annurev.immunol.23.021704.115633
Lizak, M., and Yarovinsky, T. O. (2012). Phospholipid scramblase 1 mediates type i interferon-induced protection against staphylococcal alpha-toxin. Cell Host Microbe 11, 70–80. doi: 10.1016/j.chom.2011.12.004
Lupfer, C. R., and Kanneganti, T. D. (2012). The role of inflammasome modulation in virulence. Virulence 3, 262–270. doi: 10.4161/viru.20266
MacMicking, J. D. (2012). Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol. 12, 367–382. doi: 10.1038/nri3210
Majer, O., Bourgeois, C., Zwolanek, F., Lassnig, C., Kerjaschki, D., Mack, M., et al. (2012). Type I interferons promote fatal immunopathology by regulating inflammatory monocytes and neutrophils during Candida infections. PLoS Pathog. 8:e1002811. doi: 10.1371/journal.ppat.1002811
Manca, C., Tsenova, L., Bergtold, A., Freeman, S., Tovey, M., Musser, J. M., et al. (2001). Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha /beta. Proc. Natl. Acad. Sci. U.S.A. 98, 5752–5757. doi: 10.1073/pnas.091096998
Mariathasan, S., Newton, K., Monack, D. M., Vucic, D., French, D. M., Lee, W. P., et al. (2004). Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218. doi: 10.1038/nature02664
Mariathasan, S., Weiss, D. S., Dixit, V. M., and Monack, D. M. (2005). Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J. Exp. Med. 202, 1043–1049. doi: 10.1084/jem.20050977
Mariathasan, S., Weiss, D. S., Newton, K., McBride, J., O'Rourke, K., Roose-Girma, M., et al. (2006). Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232. doi: 10.1038/nature04515
Martin, F. J., Gomez, M. I., Wetzel, D. M., Memmi, G., O'Seaghdha, M., Soong, G., et al. (2009). Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J. Clin. Invest. 119, 1931–1939. doi: 10.1172/JCI35879
Masters, S. L., Gerlic, M., Metcalf, D., Preston, S., Pellegrini, M., O'Donnell, J. A., et al. (2012). NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells. Immunity 37, 1009–1023. doi: 10.1016/j.immuni.2012.08.027
Mayer-Barber, K. D., Andrade, B. B., Barber, D. L., Hieny, S., Feng, C. G., Caspar, P., et al. (2011). Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity 35, 1023–1034. doi: 10.1016/j.immuni.2011.12.002
Miao, E. A., Alpuche-Aranda, C. M., Dors, M., Clark, A. E., Bader, M. W., Miller, S. I., et al. (2006). Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 7, 569–575. doi: 10.1038/ni1344
Miao, E. A., Ernst, R. K., Dors, M., Mao, D. P., and Aderem, A. (2008). Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc. Natl. Acad. Sci. U.S.A. 105, 2562–2567. doi: 10.1073/pnas.0712183105
Moayeri, M., Crown, D., Newman, Z. L., Okugawa, S., Eckhaus, M., Cataisson, C., et al. (2010). Inflammasome sensor Nlrp1b-dependent resistance to anthrax is mediated by caspase-1, IL-1 signaling and neutrophil recruitment. PLoS Pathog. 6:e1001222. doi: 10.1371/journal.ppat.1001222
Monroe, K. M., McWhirter, S. M., and Vance, R. E. (2009). Identification of host cytosolic sensors and bacterial factors regulating the type I interferon response to Legionella pneumophila. PLoS Pathog. 5:e1000665. doi: 10.1371/journal.ppat.1000665
Morens, D. M., Taubenberger, J. K., and Fauci, A. S. (2008). Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J. Infect. Dis. 198, 962–970. doi: 10.1086/591708
Netea, M. G., Brown, G. D., Kullberg, B. J., and Gow, N. A. (2008). An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. Microbiol. 6, 67–78. doi: 10.1038/nrmicro1815
Nogueira, C. V., Lindsten, T., Jamieson, A. M., Case, C. L., Shin, S., Thompson, C. B., et al. (2009). Rapid pathogen-induced apoptosis: a mechanism used by dendritic cells to limit intracellular replication of Legionella pneumophila. PLoS Pathog. 5:e1000478. doi: 10.1371/journal.ppat.1000478
Novikov, A., Cardone, M., Thompson, R., Shenderov, K., Kirschman, K. D., Mayer-Barber, K. D., et al. (2011). Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1beta production in human macrophages. J. Immunol. 187, 2540–2547. doi: 10.4049/jimmunol.1100926
O'Connell, R. M., Saha, S. K., Vaidya, S. A., Bruhn, K. W., Miranda, G. A., Zarnegar, B., et al. (2004). Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J. Exp. Med. 200, 437–445. doi: 10.1084/jem.20040712
Opitz, B., Vinzing, M., van Laak, V., Schmeck, B., Heine, G., Gunther, S., et al. (2006). Legionella pneumophila induces IFNbeta in lung epithelial cells via IPS-1 and IRF3, which also control bacterial replication. J. Biol. Chem. 281, 36173–36179. doi: 10.1074/jbc.M604638200
O'Riordan, M., Yi, C. H., Gonzales, R., Lee, K. D., and Portnoy, D. A. (2002). Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc. Natl. Acad. Sci. U.S.A. 99, 13861–13866. doi: 10.1073/pnas.202476699
Palmero, D., Eiguchi, K., Rendo, P., Castro Zorrilla, L., Abbate, E., and Gonzalez Montaner, L. J. (1999). Phase II trial of recombinant interferon-alpha2b in patients with advanced intractable multidrug-resistant pulmonary tuberculosis: long-term follow-up. Int. J. Tuberc. Lung Dis. 3, 214–218.
Pamer, E. G. (2004). Immune responses to Listeria monocytogenes. Nat. Rev. Immunol. 4, 812–823. doi: 10.1038/nri1461
Pandey, A. K., Yang, Y., Jiang, Z., Fortune, S. M., Coulombe, F., Behr, M. A., et al. (2009). NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog. 5:e1000500. doi: 10.1371/journal.ppat.1000500
Parker, D., Cohen, T. S., Alhede, M., Harfenist, B. S., Martin, F. J., and Prince, A. (2012). Induction of type I interferon signaling by Pseudomonas aeruginosa is diminished in cystic fibrosis epithelial cells. Am. J. Respir. Cell Mol. Biol. 46, 6–13. doi: 10.1165/rcmb.2011-0080OC
Parker, D., Martin, F. J., Soong, G., Harfenist, B. S., Aguilar, J. L., Ratner, A. J., et al. (2011). Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. MBio 2, e00016–e00011. doi: 10.1128/mBio.00016-11
Pestka, S., Krause, C. D., and Walter, M. R. (2004). Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 202, 8–32. doi: 10.1111/j.0105-2896.2004.00204.x
Platanias, L. C. (2005). Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386. doi: 10.1038/nri1604
Poeck, H., Bscheider, M., Gross, O., Finger, K., Roth, S., Rebsamen, M., et al. (2010). Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat. Immunol. 11, 63–69. doi: 10.1038/ni.1824
Portnoy, D. A., Jacks, P. S., and Hinrichs, D. J. (1988). Role of hemolysin for the intracellular growth of Listeria monocytogenes. J. Exp. Med. 167, 1459–1471. doi: 10.1084/jem.167.4.1459
Pothlichet, J., Meunier, I., Davis, B. K., Ting, J. P., Skamene, E., von Messling, V., et al. (2013). Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog. 9:e1003256. doi: 10.1371/journal.ppat.1003256
Power, M. R., Li, B., Yamamoto, M., Akira, S., and Lin, T. J. (2007). A role of Toll-IL-1 receptor domain-containing adaptor-inducing IFN-beta in the host response to Pseudomonas aeruginosa lung infection in mice. J. Immunol. 178, 3170–3176.
Ramphal, R., Balloy, V., Huerre, M., Si-Tahar, M., and Chignard, M. (2005). TLRs 2 and 4 are not involved in hypersusceptibility to acute Pseudomonas aeruginosa lung infections. J. Immunol. 175, 3927–3934.
Rathinam, V. A., Jiang, Z., Waggoner, S. N., Sharma, S., Cole, L. E., Waggoner, L., et al. (2010). The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402. doi: 10.1038/ni.1864
Rathinam, V. A., Vanaja, S. K., Waggoner, L., Sokolovska, A., Becker, C., Stuart, L. M., et al. (2012). TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 150, 606–619. doi: 10.1016/j.cell.2012.07.007
Rayamajhi, M., Humann, J., Penheiter, K., Andreasen, K., and Lenz, L. L. (2010). Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J. Exp. Med. 207, 327–337. doi: 10.1084/jem.20091746
Reich, N. C. (2013). A death-promoting role for ISG54/IFIT2. J. Interferon Cytokine Res. 33, 199–205. doi: 10.1089/jir.2012.0159
Reimer, T., Schweizer, M., and Jungi, T. W. (2007). Type I IFN induction in response to Listeria monocytogenes in human macrophages: evidence for a differential activation of IFN regulatory factor 3 (IRF3). J. Immunol. 179, 1166–1177.
Robinson, N., McComb, S., Mulligan, R., Dudani, R., Krishnan, L., and Sad, S. (2012). Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat. Immunol. 13, 954–962. doi: 10.1038/ni.2397
Roquilly, A., Gautreau, L., Segain, J. P., de Coppet, P., Sebille, V., Jacqueline, C., et al. (2010). CpG-ODN and MPLA prevent mortality in a murine model of post-hemorrhage-Staphyloccocus aureus pneumonia. PLoS ONE 5:e13228. doi: 10.1371/journal.pone.0013228
Roy, S., Bonfield, T., and Tartakoff, A. M. (2013). Non-apoptotic toxicity of Pseudomonas aeruginosa toward murine cells. PLoS ONE 8:e54245. doi: 10.1371/journal.pone.0054245
Rusinova, I., Forster, S., Yu, S., Kannan, A., Masse, M., Cumming, H., et al. (2013). Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 41, D1040–D1046. doi: 10.1093/nar/gks1215
Sander, L. E., Davis, M. J., Boekschoten, M. V., Amsen, D., Dascher, C. C., Ryffel, B., et al. (2011). Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature 474, 385–389. doi: 10.1038/nature10072
Sato, S., Sugiyama, M., Yamamoto, M., Watanabe, Y., Kawai, T., Takeda, K., et al. (2003). Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J. Immunol. 171, 4304–4310.
Sauer, J. D., Witte, C. E., Zemansky, J., Hanson, B., Lauer, P., and Portnoy, D. A. (2010). Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 7, 412–419. doi: 10.1016/j.chom.2010.04.004
Schiavoni, G., Mauri, C., Carlei, D., Belardelli, F., Pastoris, M. C., and Proietti, E. (2004). Type I IFN protects permissive macrophages from Legionella pneumophila infection through an IFN-gamma-independent pathway. J. Immunol. 173, 1266–1275.
Schmitz, F., Heit, A., Guggemoos, S., Krug, A., Mages, J., Schiemann, M., et al. (2007). Interferon-regulatory-factor 1 controls Toll-like receptor 9-mediated IFN-beta production in myeloid dendritic cells. Eur. J. Immunol. 37, 315–327. doi: 10.1002/eji.200636767
Shahangian, A., Chow, E. K., Tian, X., Kang, J. R., Ghaffari, A., Liu, S. Y., et al. (2009). Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Invest. 119, 1910–1920. doi: 10.1172/JCI35412
Skerrett, S. J., Wilson, C. B., Liggitt, H. D., and Hajjar, A. M. (2007). Redundant Toll-like receptor signaling in the pulmonary host response to Pseudomonas aeruginosa. Am. J. Physiol. Lung Cell Mol. Physiol. 292, L312–L322. doi: 10.1152/ajplung.00250.2006
Stetson, D. B., and Medzhitov, R. (2006a). Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103. doi: 10.1016/j.immuni.2005.12.003
Stetson, D. B., and Medzhitov, R. (2006b). Type I interferons in host defense. Immunity 25, 373–381. doi: 10.1016/j.immuni.2006.08.007
Sutterwala, F. S., Mijares, L. A., Li, L., Ogura, Y., Kazmierczak, B. I., and Flavell, R. A. (2007). Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 204, 3235–3245. doi: 10.1084/jem.20071239
Sutterwala, F. S., Ogura, Y., Szczepanik, M., Lara-Tejero, M., Lichtenberger, G. S., Grant, E. P., et al. (2006). Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 24, 317–327. doi: 10.1016/j.immuni.2006.02.004
Swiecki, M., Wang, Y., Vermi, W., Gilfillan, S., Schreiber, R. D., and Colonna, M. (2011). Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo. J. Exp. Med. 208, 2367–2374. doi: 10.1084/jem.20110654
Takeuchi, O., and Akira, S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820. doi: 10.1016/j.cell.2010.01.022
Teijaro, J. R., Ng, C., Lee, A. M., Sullivan, B. M., Sheehan, K. C., Welch, M., et al. (2013). Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340, 207–211. doi: 10.1126/science.1235214
Teles, R. M., Graeber, T. G., Krutzik, S. R., Montoya, D., Schenk, M., Lee, D. J., et al. (2013). Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science 339, 1448–1453. doi: 10.1126/science.1233665
Terra, J. K., Cote, C. K., France, B., Jenkins, A. L., Bozue, J. A., Welkos, S. L., et al. (2010). Cutting edge: resistance to Bacillus anthracis infection mediated by a lethal toxin sensitive allele of Nalp1b/Nlrp1b. J. Immunol. 184, 17–20. doi: 10.4049/jimmunol.0903114
Thomas, P. G., Dash, P., Aldridge, J. R. Jr., Ellebedy, A. H., Reynolds, C., Funk, A. J., et al. (2009). The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30, 566–575. doi: 10.1016/j.immuni.2009.02.006
Tsuchiya, K., Hara, H., Kawamura, I., Nomura, T., Yamamoto, T., Daim, S., et al. (2010). Involvement of absent in melanoma 2 in inflammasome activation in macrophages infected with Listeria monocytogenes. J. Immunol. 185, 1186–1195. doi: 10.4049/jimmunol.1001058
Vladimer, G. I., Weng, D., Paquette, S. W., Vanaja, S. K., Rathinam, V. A., Aune, M. H., et al. (2012). The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 37, 96–107. doi: 10.1016/j.immuni.2012.07.006
Walberg, K., Baron, S., Poast, J., Schwartz, B., Izotova, L., Pestka, S., et al. (2008). Interferon protects mice against inhalation anthrax. J. Interferon Cytokine Res. 28, 597–601. doi: 10.1089/jir.2007.0143
Wang, S., Miura, M., Jung, Y. K., Zhu, H., Li, E., and Yuan, J. (1998). Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 92, 501–509. doi: 10.1016/S0092-8674(00)80943-5
Warren, S. E., Mao, D. P., Rodriguez, A. E., Miao, E. A., and Aderem, A. (2008). Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J. Immunol. 180, 7558–7564.
Weigent, D. A., Huff, T. L., Peterson, J. W., Stanton, G. J., and Baron, S. (1986). Role of interferon in streptococcal infection in the mouse. Microb. Pathog. 1, 399–407. doi: 10.1016/0882-4010(86)90071-9
Wilson, E. B., Yamada, D. H., Elsaesser, H., Herskovitz, J., Deng, J., Cheng, G., et al. (2013). Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340, 202–207. doi: 10.1126/science.1235208
Woodward, J. J., Iavarone, A. T., and Portnoy, D. A. (2010). c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328, 1703–1705. doi: 10.1126/science.1189801
Worthington, M., and Hasenclever, H. (1972). Effect of an interferon stimulator, polyinosinic: polycytidylic acid, on experimental fungus infections. Infect. Immun. 5, 199–202.
Wu, J., Fernandes-Alnemri, T., and Alnemri, E. S. (2010). Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J. Clin. Immunol. 30, 693–702. doi: 10.1007/s10875-010-9425-2
Wu, K., Dong, D., Fang, H., Levillain, F., Jin, W., Mei, J., et al. (2012). An interferon-related signature in the transcriptional core response of human macrophages to Mycobacterium tuberculosis infection. PLoS ONE 7:e38367. doi: 10.1371/journal.pone.0038367
Yamamoto, M., Sato, S., Hemmi, H., Hoshino, K., Kaisho, T., Sanjo, H., et al. (2003). Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301, 640–643. doi: 10.1126/science.1087262
Zhao, Y., Yang, J., Shi, J., Gong, Y. N., Lu, Q., Xu, H., et al. (2011). The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477, 596–600. doi: 10.1038/nature10510
Keywords: type I IFN, IFNβ, NLR, caspase-1, inflammasome, cell death, Nlrp3
Citation: Malireddi RKS and Kanneganti T-D (2013) Role of type I interferons in inflammasome activation, cell death, and disease during microbial infection. Front. Cell. Infect. Microbiol. 3:77. doi: 10.3389/fcimb.2013.00077
Received: 11 August 2013; Paper pending published: 17 September 2013;
Accepted: 24 October 2013; Published online: 12 November 2013.
Edited by:
Amal O. Amer, The Ohio State University, USAReviewed by:
Mikhail A. Gavrilin, The Ohio State University, USADario S. Zamboni, Universidade de São Paulo, Brazil
Copyright © 2013 Malireddi and Kanneganti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thirumala-Devi Kanneganti, Department of Immunology, St. Jude Children's Research Hospital, MS #351, 262 Danny Thomas Place, Suite E7004, Memphis, TN 38105-2794, USA e-mail:dGhpcnVtYWxhLWRldmkua2FubmVnYW50aUBTdEp1ZGUub3Jn