 Han Ming Gan
Han Ming Gan Huan You Gan
Huan You Gan Nurul H. Ahmad3
Nurul H. Ahmad3 André O. Hudson
André O. Hudson Michael A. Savka
Michael A. Savka- 1School of Science, Monash University Malaysia, Petaling Jaya, Malaysia
- 2Genomics Facility, Monash University Malaysia, Petaling Jaya, Malaysia
- 3Thomas H. Gosnell School of Life Sciences, Rochester Institute of Technology Rochester, NY, USA
Here we report the draft genomes and annotation of four N-acyl homoserine lactone (AHL)-producing members from the family Sphingomonadaceae. Comparative genomic analyses of 62 Sphingomonadaceae genomes were performed to gain insights into the distribution of the canonical luxI/R-type quorum sensing (QS) network within this family. Forty genomes contained at least one luxR homolog while the genome of Sphingobium yanoikuyae B1 contained seven Open Reading Frames (ORFs) that have significant homology to that of luxR. Thirty-three genomes contained at least one luxI homolog while the genomes of Sphingobium sp. SYK6, Sphingobium japonicum, and Sphingobium lactosutens contained four luxI. Using phylogenetic analysis, the sphingomonad LuxR homologs formed five distinct clades with two minor clades located near the plant associated bacteria (PAB) LuxR solo clade. This work for the first time shows that 13 Sphingobium and one Sphingomonas genome(s) contain three convergently oriented genes composed of two tandem luxR genes proximal to one luxI (luxR-luxR-luxI). Interestingly, luxI solos were identified in two Sphingobium species and may represent species that contribute to AHL-based QS system by contributing AHL molecules but are unable to perceive AHLs as signals. This work provides the most comprehensive description of the luxI/R circuitry and genome-based taxonomical description of the available sphingomonad genomes to date indicating that the presence of luxR solos and luxI solos are not an uncommon feature in members of the Sphingomonadaceae family.
Introduction
Members of the Sphingomonadaceae family are Gram-negative glycosphingolipid-containing bacteria that belong to the α-4 subclass of proteobacteria (Yabuuchi et al., 1990). This family possesses a variety of metabolic capabilities that are potentially advantageous pertaining to a variety of bioremediation capabilities (White et al., 1996). Based on phylogenetic, chemotaxonomic and phenotypic observations, the Sphingomonas genus has been expanded to include three new genera, Sphingobium, Novosphingobium and Sphingopyxis (Yabuuchi et al., 1990). Recently, a fifth genus was added to include, Sphingosinicella (Maruyama et al., 2006; Geueke et al., 2007; Yoon et al., 2008; Yasir et al., 2010).
Regarding niche, sphingomonads have been isolated from a variety of terrestrial and aquatic environments, including; water supplies, respirators, blood, wounds, dialysis equipment, patients with septicemia, peritonitis, meningitis, and wound infections, soils, deep subsurface sediments, corroding copper pipes and in plants (White et al., 1996; Gan et al., 2009).
Members of the Sphingomonas genus are able to catabolize a wide range of natural recalcitrant and anthropogenic compounds including; biphenyl, naphthalenes, pyrene, furans, oestradiol, polyethylenglycols, chlorinated phenols, and various biocides such as carbofuran, 2,4-D and mecoprop (Ogramab et al., 2000; Basta et al., 2004; Stolz, 2009). It was shown that the biphenyl- and naphthalene-degrading Sphingomonas aromaticivorans F199 strain and other sphingomonads that degrade additional xenobiotic compounds contain large plasmids encoding the catabolic pathways (Romine et al., 1999; Ogramab et al., 2000; Basta et al., 2004, 2005). Evidence also supports that these replicons can only be transferred among sphingomonads (Ogramab et al., 2000; Basta et al., 2004) by conjugal transfer and that gene and gene cluster rearrangements in the plasmids occur post conjugation (Tiirola et al., 2002). The presence of multiple insertion elements in sphingomonads suggests a role in the establishment of degradative pathways and in plasmid rearrangements and differences in gene cluster localization in members of the Sphingomonadaceae family (Dogra et al., 2004; Muller et al., 2004; Thiel et al., 2005). A recent study comparing the genomes of 26 sphingomonads suggests diverse adaptations and biodegradative capabilities in this group within the phylum Alphaproteobacteria (Aylward et al., 2013). Given this complexity in niche environments, biodegradation capabilities and genome rearrangements, the whole genome sequencing of additional sphingomonads has the potential to enhance our understanding of the diversity within this group and may contribute to important biotechnological applications such as bioremediation in the future.
Quorum sensing (QS) is a system commonly employed by bacteria to monitor its cell density prior to regulating gene expression (Fuqua et al., 1994; Miller and Bassler, 2001; Waters and Bassler, 2005; Schuster et al., 2013). In one type of QS system from Gram-negative bacteria, the bacteria produce and detect chemical signals called N-acyl-homoserine lactones (AHL). These signals are produced by the enzyme AHL synthase, a member of the LuxI-type protein family. The AHL compounds are detected by a transcriptional regulator belonging to the LuxR-type family. A typical AHL-QS system contains a LuxI and a LuxR protein that are usually in a genomic context regarding proximity of the genes on the chromosome (Choudhary et al., 2013). Upon reaching a concentration threshold measured by the cell density, the AHL signal is detected by the cognate LuxR and can activate population-wide-responses leading to the coordination of gene activation or repression. In Gram-negative bacteria, AHL dependent QS regulation is used to regulate the production of diverse responses such as; the activation of virulence factors, conjugation, the production of antimicrobial metabolites, the regulation of enzyme secretion, the production of bioluminescence and the anabolism of polysaccharide production which is correlated to biofilm formation (Miller and Bassler, 2001; Fuqua and Greenberg, 2002; Waters and Bassler, 2005).
Besides the presence of the canonical luxI/luxR pairs, many bacteria contain additional luxR transcriptional regulators that are not in a genomic context regarding proximity to a luxI gene. These unpaired luxR genes have been termed solos and orphans and are homologous to QS LuxR-type transcriptional regulators in that LuxR solos contain the AHL-binding domain at the N terminus and a DNA-binding helix-turn-helix (HTH) domain at the C terminus (Fuqua, 2006; Case et al., 2008; Subramoni and Venturi, 2009; Tsai and Winans, 2010; Cude and Buchan, 2013; Gonzalez and Venturi, 2013). The solo LuxR-type transcriptional activators increase the regulatory range by responding to endogenously produced AHLs and by “listening-in” on exogenous signals produced by other bacteria. Recently, a subfamily of LuxR solos have been found that respond to plant-produced compounds and were subsequently named the plant associated bacteria (PAB) luxR solos (Ferluga et al., 2007; Zhang et al., 2007). In addition, LuxI solos were identified first by Zan et al. (2012) and have also been subsequently identified in Sulfitobacter, Ruesgeria, and Phaeobacter genera all within the Roseobacter clade (Cude and Buchan, 2013).
Members of the sphingomonads have been shown to synthesize AHL signals (D'angelo-Picard et al., 2005; Gan et al., 2009; Huang et al., 2013; Schaefer et al., 2013). Previous work by our group have, isolated, identified, sequenced and annotated the genome of an AHL-producing Novosphingobium sp. Rr 2-17 isolated from a grapevine tumor (Gan et al., 2009, 2012). Comparative genomic analysis of Rr 2-17 and five additional members from the genus Novosphingobium validated the presence of canonical luxI/luxR pairs. Furthermore, a putative luxR solo in strain PP1Y of the Novosphingobium genus was identified (Gan et al., 2013). Our initial and continuing work with a group of sphingomonads documented to degrade natural and anthropogenic compounds identified a subset of four sphingomonads capable of producing AHL QS signals. We decided to sequence their whole genomes to corroborate AHL-producing phenotype with the presence of luxI and luxR homologs in the whole genomes and more importantly to contribute molecular resources for future genetic work pertaining to microbial-based bioremediation.
Leveraging on the expansion of microbial genomics data, the additional objectives of this study are to (1) provide an updated genomic distribution of luxI/R homologs in the Sphingomonadaceae family, (2) update and validate sphingomonad taxonomy using genome-based approach, (3) provide a comprehensive LuxR phylogeny and (4) identify putative LuxR solos and LuxI solos in the currently sequenced sphingomonads.
Materials and Methods
Strains, Culture Conditions and Extract Preparation
The bacterial strains (kindly provided by Andreas Stolz, Institut fur Mikrobiologie, Universitat Stuttgart, Stuttgart, Germany) used in this work were cultured on R2A minimal agar media. To prepare extracts for the detection of AHL compounds, the four sphingomonads were grown on potato dextrose agar medium for 4 days and were resuspended in 10 mls sterile purified water. Equal volume of acidified ethyl acetate (aEtOAc) was added to the resuspended bacteria and the mixture was agitated for 3 h at 25°C with shaking at 150 rpm followed by centrifugation to separate the aqueous phase from the aEtOAc phase. Under these conditions, AHLs partition into the non-polar aEtOAC phase. The aEtOAc was aspirated off, dried in a Savant speed-vac and resuspended in aEtOAc to produce a 20-fold concentrated aEtOAc extracts. These extracts were used in AHL detection assays.
E. coli JM109, Agrobacterium tumefaciens A136 and Chromobacterium violaceum CV026 were grown in Luria-Bertani (LB). Each bacterial biosensor reporter strain used in this work is listed in Supplemental Table 1 along with its AHL receptor protein and cognate AHL signal. All media and growth conditions are as previously described by our group (Scott et al., 2006; Gan et al., 2009; Lowe et al., 2009; Savka et al., 2011).
Bioassays for AHL QS Signal Detection Using AHL-Dependent Biosensor Strains
An overnight culture of these four biosensors were grown in LB with the appropriate antibiotic and diluted 1:10 in LB and 200 μl of the diluted cell suspension was added to the round bottom tubes (12 × 50 mm) containing dried aEtOAc samples or pure AHL signals as controls. Cognate AHL signal for E. coli biosensors JM109 (pSB401) was 3-oxo-C6-HSL at 50 nM; for JM109 (pSB536) was C4-HSL at 1 μM; for JM109 (pSB1075) was 3-oxo-C12-HSL at 1 nM, unless otherwise noted. For the A. tumefaciens A136 biosensor pure C8-HSL was used at 50 nM. Tubes were incubated at 30°C with shaking for 5 to 6 h before bioluminescence was measured using a Turner Designs TD 20/20 luminometer. The TD 20/20 luminometer was adjusted to different sensitivities due to the varying responses of the JM109 series of biosensors to their cognate AHL signal. Unless noted, relative light units (RLU) measurements were made at 30.0, 39.9, 50.1, and 30.0% sensitivity for LuxR-, AhyR, LasR, and TraR-based biosensors, respectively. Luminescence is measured and given in RLU per triplicate sample. RLUs were determined with a 20-s integration period. Mean values of the RLUs were obtained with three independent biological samples.
For “T”-streak assays, the Chromobacterium violaceum colorless mutant, CV026 was used. In the presence of exogenous QS signals CVO26 produces the purple pigment violacein, indicating the presence of AHL in the sample. C. violaceum wild type strain was used as a positive control. E. coli DH5α was the negative control in the T-streak plate assays. The biosensor, controls, and samples were grown on tryptone—yeast extract medium mixed with PDA medium (1:1, v/v). Each isolate was tested at least two times using the “T”-streak bioassay. The whole cell AHL-dependent biosensor assays were performed as previously described by our group (Scott et al., 2006; Gan et al., 2009; Lowe et al., 2009; Savka et al., 2011).
Whole Genome Sequencing, Assembly and Annotation
Genomic DNA was extracted using the GenElute™ (Sigma-Aldrich, St. Louis, MO) and converted into next generation sequencing library using Nextera XT (Illumina, San Diego, CA) according to the manufacturer's instructions. Whole genome sequencing was performed using the MiSeq (Illumina, San Diego, CA) at the Monash University Malaysia Genomics Facility. The raw data for each bacterium were error-corrected and assembled using Spades v2.5 (default setting) (Bankevich et al., 2012). The generated contigs were scaffolded and gap-closed using SSPACE and GAPFiller respectively (Boetzer et al., 2011; Boetzer and Pirovano, 2012). Genome annotation was performed using Prokka and InterProScan5 (Jones et al., 2014; Seemann, 2014).
Whole Genome-Based Phylogeny Assignment
Publicly available complete and draft genome sequences (<250 contigs) from the genus Novosphingobium, Sphingomonas, Sphingopyxis, and Sphingobium were downloaded. Subsequently, gene/protein prediction was performed using Prodigal2.60 (default setting) (Hyatt et al., 2010). PhyloPhlAn was used to construct phylogenetic tree from the resulting predicted proteins based on 400 highly conserved microbial proteins (Segata et al., 2013).
Systematic Bioinformatics Identification of LuxI, LuxR and LuxR Solo Homologs
A systematic methodology for the accurate and stringent identification of LuxI, LuxR, and LuxR solo homologs is presented in Figure 1. Briefly, the predicted proteomes were scanned for protein family domain (PFAM) specifically the autoinducer synthase domain (PFAM signature: PF00765) and the autoinducer binding domain (PFAM signature: PF03472) that are universally present in reported LuxR and LuxI homologs, respectively, using profile hidden Markov models-based similarity search (E-value <1e-5). The short listed candidates were further annotated using the more time consuming but comprehensive InterProScan5. To qualify as an authentic LuxR homolog, the shortlisted protein must contain four signature LuxR homolog Interproscan identifiers e.g., IPR005143 (autoinducer binding), IPR016032 (Signal transduction response regulator, C-terminal effector), IPR011991 (Winged helix-turn-helix DNA-binding domain), and IPR000792 (Transcription regulator LuxR, C-terminal) that are universally present in functionally validated LuxR homologs. An authentic LuxI homolog on the other hand, must contain both IPR001690 (Autoinducer synthesis protein) and IPR018311 (Autoinducer synthesis, conserved site). Cognate LuxI and LuxR homologs were then manually identified based on the coordinate and close proximity of their respective protein-coding genes.
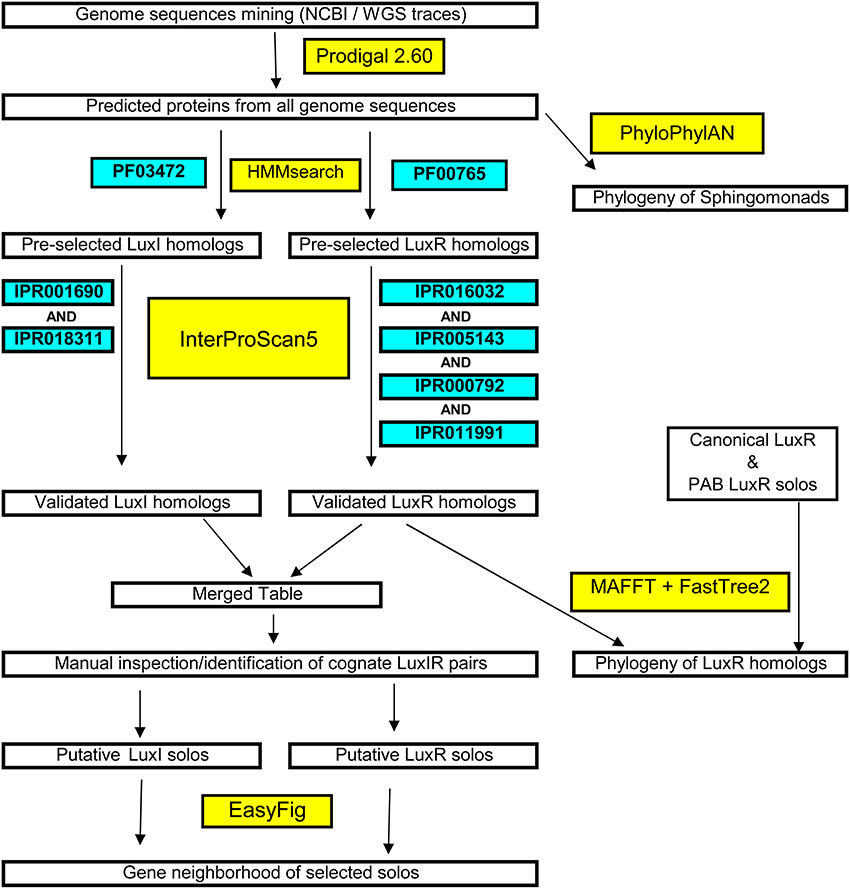
Figure 1. Flowchart of the systematic and stringent bioinformatics methodology used in this study for the identification of luxI/R in sphingomonads and large-scale phylogenetic/phylogenomic tree construction.
Maximum Likelihood Approximation of the LuxR Phylogeny
Functionally validated LuxR homologs, PAB LuxR solos and the putative sphingomonad LuxR homologs were combined and aligned with MAFFT-LINSI using the default setting (Katoh and Standley, 2013). The resulting protein alignment was then used as the input for maximum likelihood phylogenetic analysis using FastTree2 (Price et al., 2010). The constructed tree was visualized and graphically edited using FigTree (Rambaut, 2014).
Visualization of LuxR and LuxI Solos Gene Neighborhood, LuxR Homologs Alignment and Pairwise Identity Matrix Construction
Contigs containing the identified luxR solo genes were extracted from the genome, annotated with Prokka (default setting) and subsequently visualized in EasyFig (Sullivan et al., 2011). Additionally, sphingomonad LuxR homologs clustered with the PAB LuxR solos were aligned with MAFFT-LINSI (Katoh and Standley, 2013) and visualized using ALINE (Bond and Schuttelkopf, 2009). Pairwise identity matrix for selected LuxR homologs was constructed using SDT (Muhire et al., 2014).
Results
Genome Statistics of the Four Newly Sequenced Sphingomonads and their Ability to Produce AHL Signals
The genome assembly and annotation statistics of four genomes of Sphingomonas known for their biodegradation ability in addition to their isolation source and notable features are presented in Table 1. Culture extracts prepared from each of the four sphingomonads strains in this study chosen for whole genome sequencing activated at least two AHL-dependent whole cell bacterial biosensors (Supplemental Table 1). Sphingomonas paucimobilis EPA505 activated light production in the TraR-based Agrobacterium A136 and in the LasR-based E. coli JM109 (pSB1075) biosensors and activated pigment synthesis in the CviR-based Chromobacterium biosensor. The Sphingobium herbicidovorans NBRC16415, Sphingobium yanoikuyae B1 and Novosphingobium resinovorum KF1 activated light production in the TraR- and activated pigment synthesis in the CviR-based biosensors (Table 2). These results are consistent with findings by others that AHL QS signal production in members of the sphingomonad group is not uncommon (D'angelo-Picard et al., 2005; Gan et al., 2009; Huang et al., 2013; Schaefer et al., 2013).
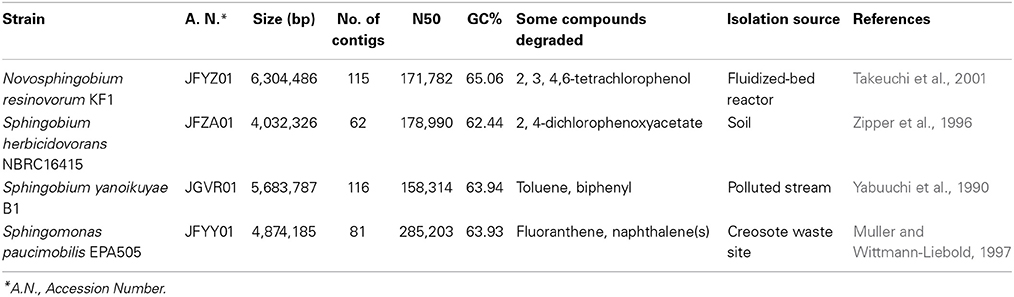
Table 1. Strain information, genome assembly and annotation statistics of the sequenced sphingomonads used in this study.
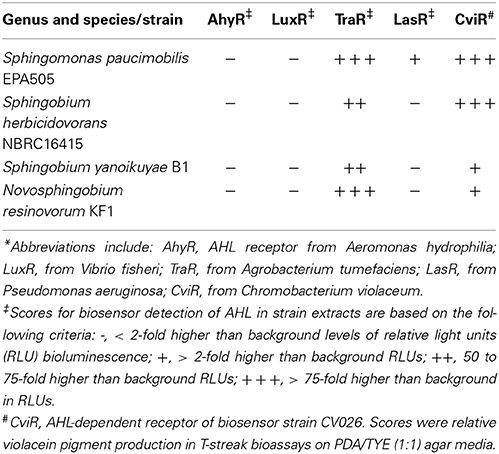
Table 2. Production of N-acyl-homoserine lactones by four newly sequenced strains of the Sphingomonas, Sphingobium and Novosphingobium group assayed by five AHL-dependent biosensor strains*.
Phylogenomic Analysis of Currently Sequenced Sphingomonad
Analysis of the currently sequenced sphingomonads (Figure 2 and see Supplemental Table 4 for accession number) indicates that there is a sequencing bias toward the genera Sphingobium, Novosphingobium, and Sphingomonas. Very recently, our group sequenced and annotated the genomes of two additional cave Sphingopyxis genomes that enabled the expansion of the taxon sampling size (Gan et al., 2014). Species from the genera Sphingobium and Novosphingobium form robust monophyletic lineages with extremely high (>90%) nodal support. Based on phylogenomic analysis, the Sphingobium clade is the sister group to the clade of Sphingopyxis and Novosphingobium. Notably, species from the genus Sphingomonas display considerable paraphyletic distributions, indicating incongruence between molecular and biochemical-based taxonomic assignment. Phylogenomic analysis also suggests that Sphingomonas sp. SKA85 and the classic Sphingomonas paucimobilis EPA505 (Nohynek et al., 1996) may have been misclassified at the genus level as evidenced by its tight clustering within the Sphingobium group.
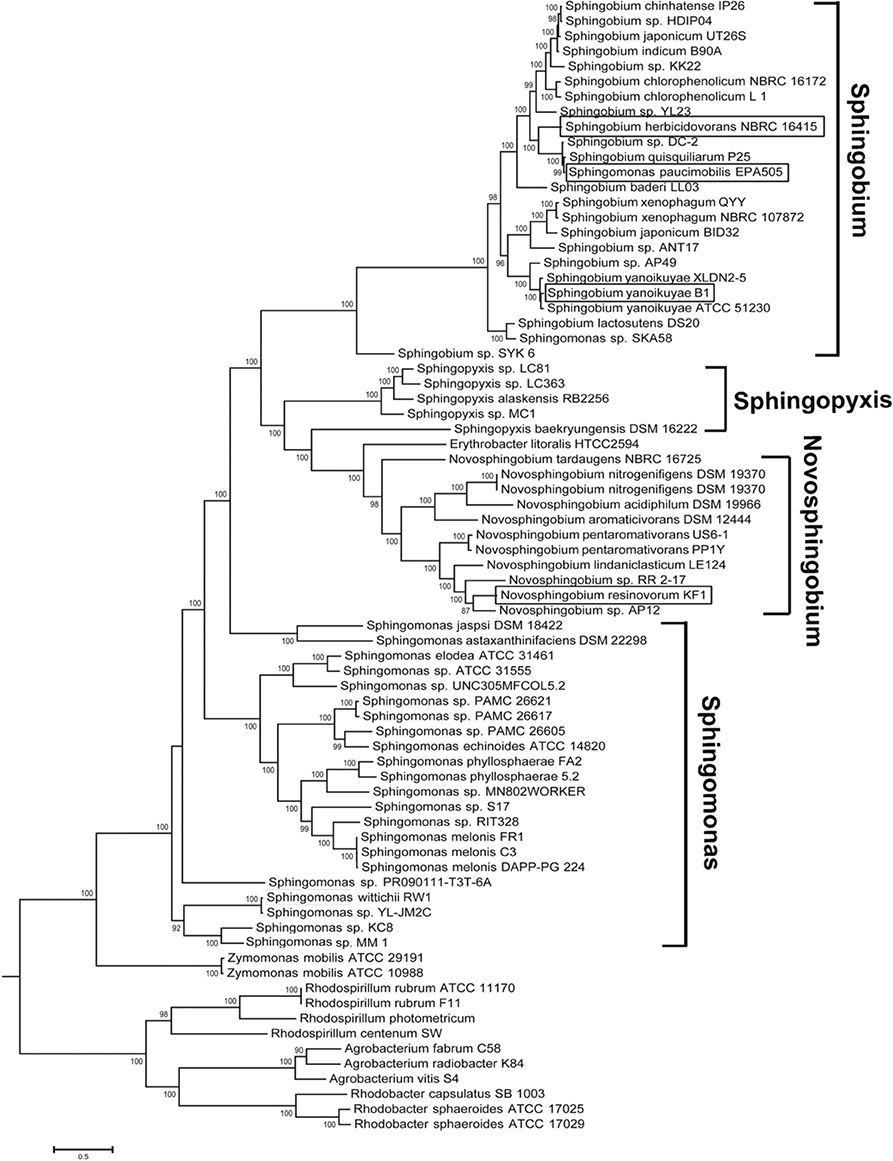
Figure 2. Phylogenomic tree depicting the evolutionary relationship of currently sequenced sphingomonads based on approximately 400 conserved single-copy genes. The four whole genomes sequenced in study were shown in rectangle boxes. Selected members from the genera Rhodospirillum, Agrobacterium and Rhodobacter were designated as outgroup. Bootstrap support of less than 50% was not shown.
The Presence and Composition of Sphingomonad LuxR and LuxI Homologs are Diverse
The analysis of 62 sphingomonads genomes provides genetic evidence that QS is a common trait within the family. 40 of the 62 genomes analyzed contain at least one putative luxI or luxR homolog with 33 of them containing at least 1 putative canonical luxI/R homolog pair (Table 3 and See Supplemental Tables 2, 3 for a complete information of the identified luxR and luxI homologs). The non-universal presence of QS genes in members of the same species e.g., Sphingobium yanoikuyae and Sphingobium xenophagum may imply that QS is a trait that is subject to purifying selection. It is also worth noting that members of the currently sequenced Sphingomonas have a relatively incomplete lux-based QS capacity as evidenced by the sparse presence of luxI and luxR homologs in this genus.
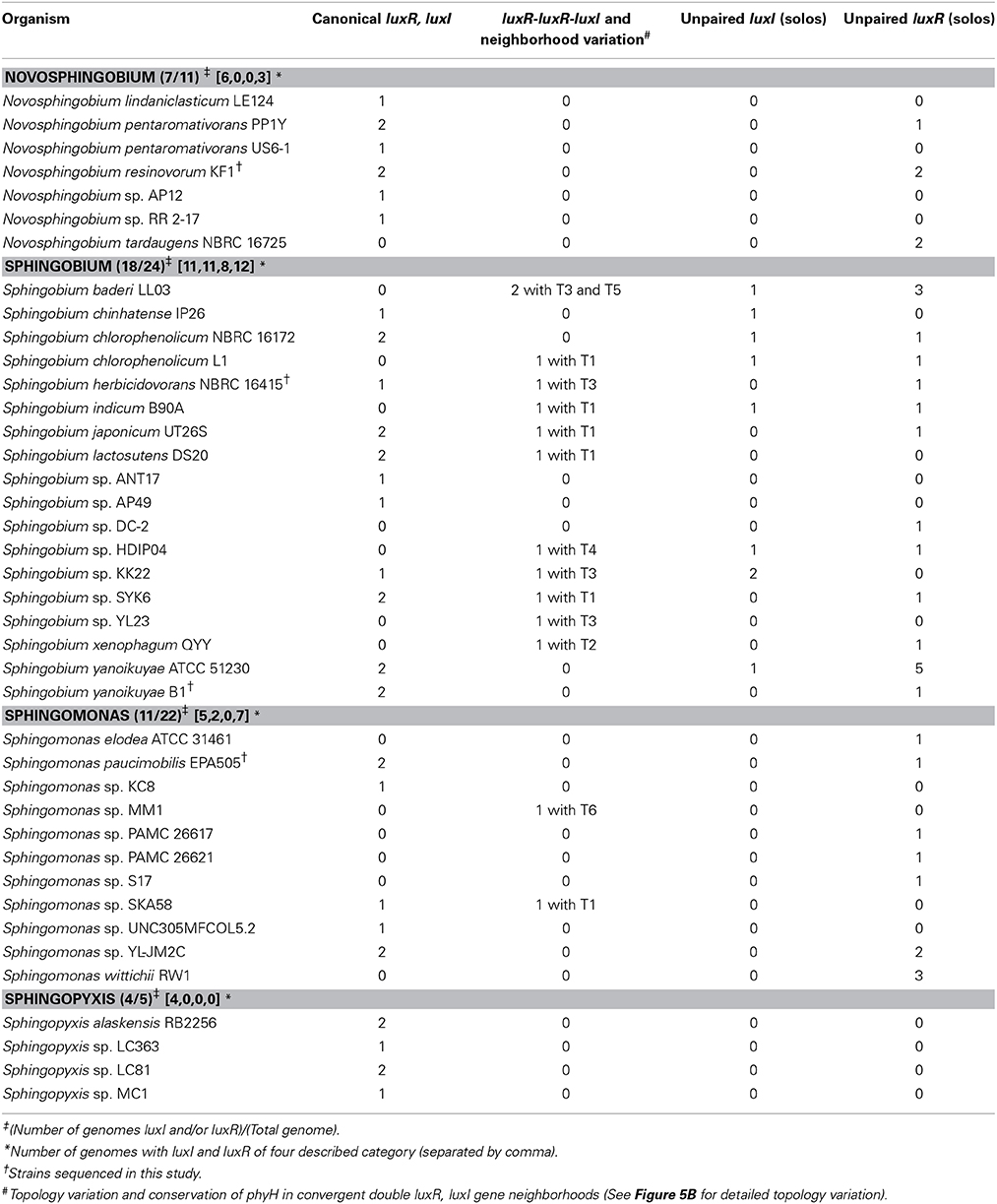
Table 3. Distribution and organization of the luxI and luxR homologs and the presence of direct double luxR-luxI topology identified in the whole genome sequences of 40 sphingomonads.
LuxR Phylogeny Reveals Diverse Origin of Sphingomonad LuxR and Supports the Monophyletic Clustering of LuxR Solos from Plant Associated Bacteria
A majority of the sphingomonad LuxR homologs form a big clade that is a sister group to the clade containing the functionally validated BjaR and RhlR (Figure 3) (Cubo et al., 1992; Lindemann et al., 2011). Consistent with previous reports, the PAB LuxR solos e.g., NesR, XagR, XccR, OryR, and PsoR (Ferluga et al., 2007; Zhang et al., 2007; Ferluga and Venturi, 2009; Chatnaparat et al., 2012; Gonzalez et al., 2013) formed a robust and well-defined monophyletic group. Based on phylogenetic clustering, six sphingomonad LuxR homologs may share a common (but distant) ancestry with the PAB LuxR solos clade. Alignment of these six putative LuxR homologs shows substitution in the highly conserved amino acid in the regulatory domain e.g., Y61W that is similarly reported in PAB LuxR solos. With the exception of a LuxR homolog from Sphingobium herbicidovorans NRBC 16415 (JFYZ01~contig3_10) that has a W57V substitution, the W57 residue was conserved in the remaining five sphingomonad LuxR homologs. Furthermore, other substitutions were observed in the conserved D70 and W85 residues for four out of the six sphingomonad LuxR homologs (Figure 4). In general, the three conserved residues in the DNA-binding domain (E178, L182, and G188) are conserved across the LuxR homologs alignment with the exception of L182I substitution in a Sphingomonas sp. S17 LuxR homolog (AFGG01~contig50_9).
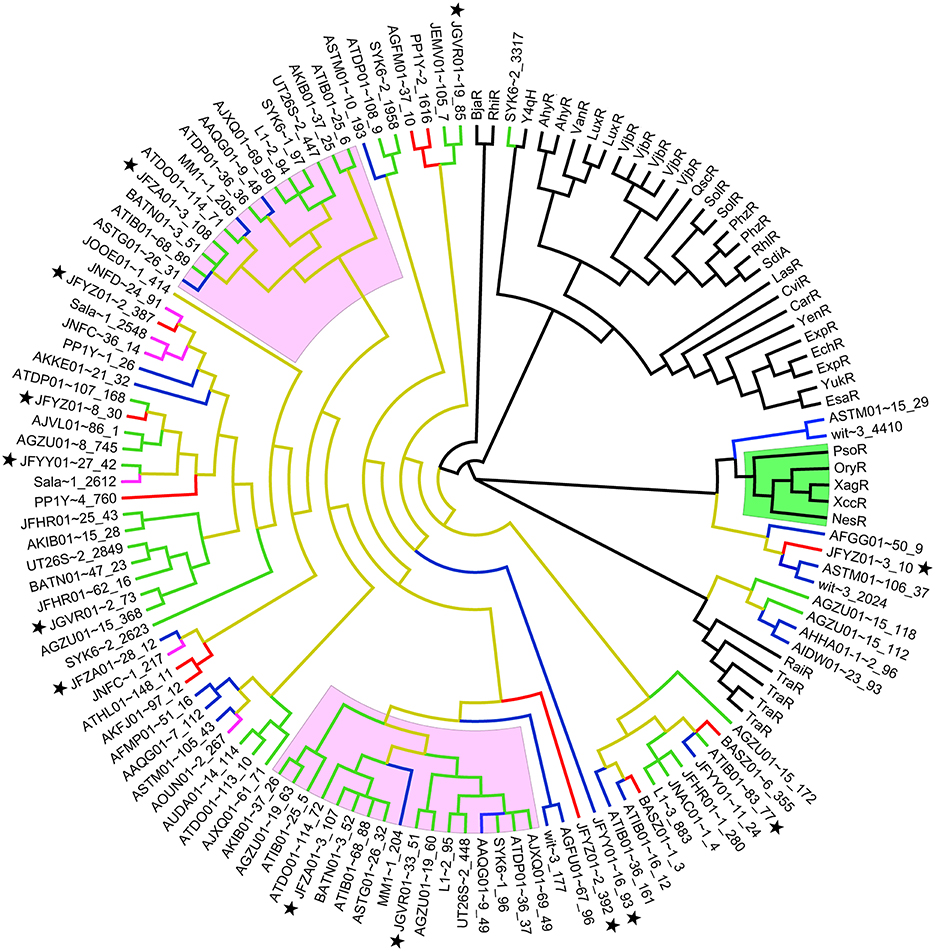
Figure 3. Unrooted phylogenetic tree of functionally validated LuxR homologs, Plant associated bacteria (PAB) LuxR Solos (Gonzalez and Venturi, 2013) and Identified sphingomonad LuxR Homologs. Clades highlighted in green and pink represent PAB LuxR solos and sphingomonad double LuxR, LuxI, respectively. Branches colored in brown, blue, red, green, and purple represent the Sphingomonadaceae, Sphingomonas, Novosphingobium, Sphingobium, and Sphingopyxis lineages, respectively. Black star next to taxa name indicates LuxR homologs from strains sequenced in this study. Accession numbers and aligned sequences are available in Supplemental Data 1.

Figure 4. Alignment of plant associated bacteria solos. LuxR solos and selected sphingomonad LuxR homologs. Number above the alignment corresponds to the residue number of the TraR protein. Regions highlighted in yellow indicate the invariant sites of canonical LuxR homologs (Fuqua and Greenberg, 2002) while variation from the conserved site was highlighted in green. The conserved sites corresponding to autoinducer binding and DNA binding were indicated by blue and purple triangles, respectively.
The Gene Neighborhood of Sphingomonad LuxR Solo and LuxR Double is not Conserved
Investigation of the genes flanking the putative luxR solos in our sequenced genomes reveals some intriguing findings (Figure 5A). In Sphingobium herbicidovorans NBRC 16415, its putative luxR solo is convergently oriented with respect to a luxI/R pair and while in N. resinovorum [contig 2], it is located four genes downstream of a luxI/R pair. Furthermore, the gene coding for a possibly truncated LuxR-like protein is located immediately downstream of the luxR solos in S. yanoikuyae and N. resinovorum (contig2) (Figure 5A), suggesting the occurrence of luxR gene duplication and/or recombination in that region. In addition to the tandem luxR duplication (luxR double) in strain NBRC16415, further analysis of the sphingomonad genomes led to the identification of additional tandem luxR duplication (Table 3 and Figure 3) with variable gene neighborhood at the 5′ end (Figure 5B).
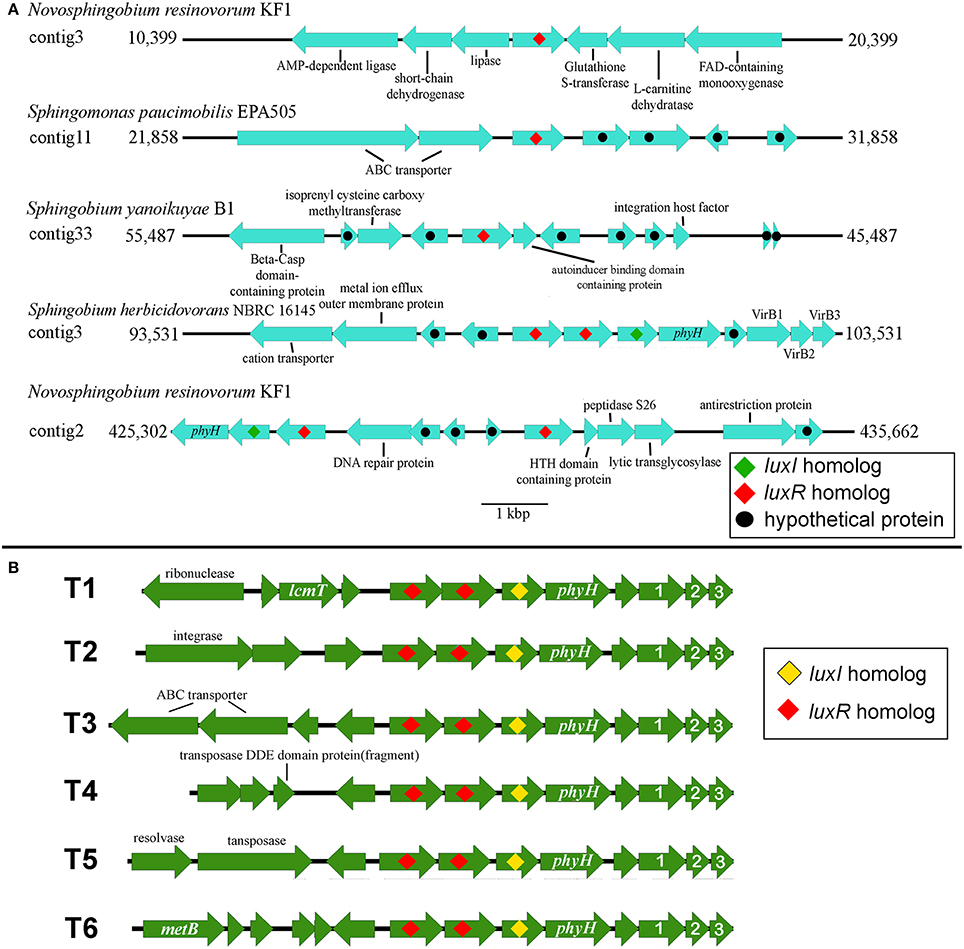
Figure 5. Gene organization of luxR Solos and luxR-luxR-luxI in the four whole genome sequenced sphingomonads. (A) EasyFig generated linear comparison of luxR solos in the genomic region of selected sphingomonads. Approximately 5000 bp of genomic region flanking the luxR solos is shown. (B) Gene orientation of the identified convergent double luxR, luxI group and its gene neighborhood variation. T1 to T6 denotes different gene neighborhoods identified in the convergent double luxR, luxI. Arrows without label represent gene coding for hypothetical protein. Please see Table 2 for topology variation present in sphingomonad genomes. The numbers “1,” “2,” and “3” represent virB1, virB2, and virB3 genes respectively. Additional abbreviations include: lcmT, Isoprenylcysteine carboxyl methyltransferase; metB, Cystathionine gamma-synthase; phyH, phytanoly dioxygenase.
Pairwise Comparison between Members of the Same Convergent Double LuxR Group Shows Considerable Sequence Divergence
The amino acid pairwise identity between members of the same LuxR double group is in the range of 50%. On the contrary, up to 94% pairwise identity could be obtained for members from different LuxR double group (Figure 6). This is consistent with the LuxR phylogenetic tree with whereby LuxR double members from the same group do not form a tight cluster with one another (Figure 3). Given that luxR double is almost exclusively observed in the genus Sphingobium, luxR double may originate from an ancient tandem gene duplication in the common ancestor of the genus Sphingobium followed by a neofunctionalization-oriented functional divergence of the luxR duplicate that was subsequently retained in several strains of the genus Sphingobium. The presence of a luxR double in a non-Sphingobium strain e.g., Sphingomonas sp. MM1 may then be attributed to horizontal gene transfer.
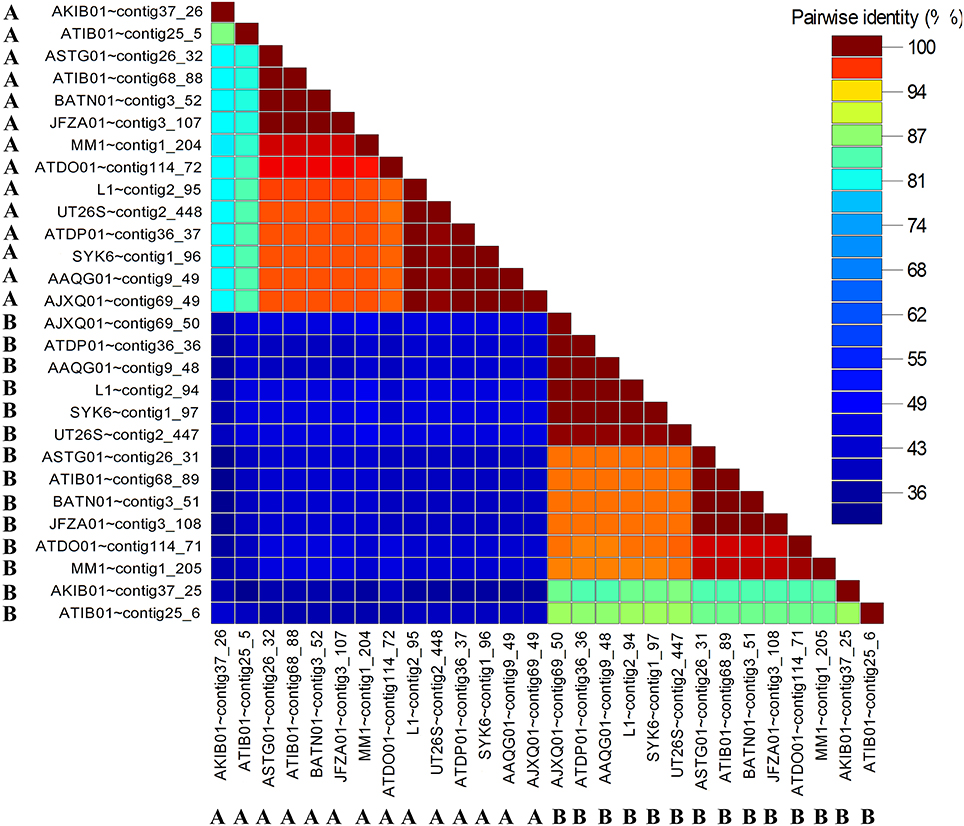
Figure 6. Pairwise identity matrix of identified convergent luxR-luxR-luxI (LuxR-A and LuxR-B) in sphingomonads. The letters A and B correspond to different partner luxRs in the luxR-luxR-luxI. The genes coding for LuxR homologs with the same symbol and color were convergently oriented with respect to each other.
Identification of LuxI Solos
Two putative luxI solos were identified in Sphingobium sp. KK2 strain that reside on different contigs and one in Sphingobium chinhatense IP26 strains (Figure 7A). A gene coding for N-terminal truncated/mutated LuxR-like protein is located immediately upstream and convergently oriented to the putative luxI solo in Sphingobium chinhatense IP26 and Sphingobium sp. KK2. Multiple sequence alignment of the three putative LuxI solos in sphingomonads with LuxI-type family proteins showed all 10 amino acid residues required for AHL synthase activity are conserved and supports that these three luxI solos encode enzymes involved in AHL synthesis (Figure 7B). Additionally, phyH gene coding for phytanoyl dioxygenase is located immediately downstream of and convergently oriented with respect to one of the luxI solos in strain KK2 which is frequently observed in several well-described luxI/R pairs (Gan et al., 2013).
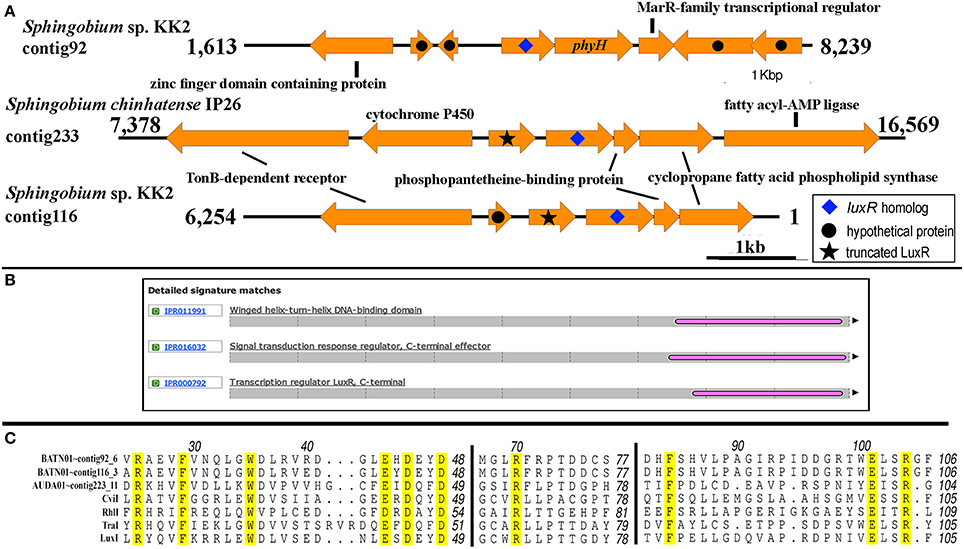
Figure 7. Genomic and genetic evidence for the presence of luxI solos in sphingomonads. (A) Gene neighborhood showing non-LuxR genes located in the vicinity of the putative LuxI genes (arrow with blue diamond). Analysis of the translated protein sequence for the gene upstream and convergently oriented to the putative luxI gene in Sphingobium chinhatense IP26 and Sphingobium sp. KK2 indicates that it may be an N-terminal truncated LuxR protein (arrow with black star). (B) A representative Interproscan analysis domain analysis of the N-terminal truncated LuxR protein noted in Figure 7A. (C) Protein alignment of the putative LuxI solos. Number above the alignment corresponds to the amino acid residue of TraI. Amino acid residues are conserved in all LuxI-type proteins (Fuqua and Greenberg, 2002) are highlighted in yellow.
Discussion
The biochemical and genetic characterization of Novosphingobium sp. Rr 2-17 isolated from grapevine tumor provided the first glimpse of QS ability in the genus Novosphingobium (Gan et al., 2009). The genome sequencing of strain Rr 2-17 and subsequent comparative genomic analysis with five additional members from the genus Novosphingobium validates the presence of luxI/R homolog(s) (Gan et al., 2012) and even more intriguingly, a luxR solo in this genus (Gan et al., 2013). Expanding from our previous study, we present four new whole genome sequences of AHL QS signal producing strains in the sphingomonad group and to our knowledge presents the most comprehensive genomic surveillance of sphingomonads for the distribution of luxI/R homologs to date. In addition, the work presents the most updated and accurate genome-based taxonomy validation of the currently sequenced sphingomonads. Although previous works provided convincing biochemical test results to support the reclassification of Sphingomonas, the constructed phylogeny based on the 16S rRNA gene failed to provide satisfactory bootstrap support particularly in the splits that separated the major genus in Sphingomonadaceae (Takeuchi et al., 2001). Our phylogenomic approach dramatically improves the bootstrap support at these major splits that highlights the presence of strong phylogenetic signal afforded by the utilization of nearly 400 universal proteins. Further, the paraphyletic clustering of the genus Sphingomonas underscores the overlooked diversity of Sphingomonas that may benefit from further sub-classification in addition to its current classification into three well-known genera e.g., Novosphingobium, Sphingopyxis, and Sphingobium and the recently proposed genus, Sphingosinicella (Takeuchi et al., 2001).
The phylogenetic clustering of sphingomonad LuxR homologs shows no evidence of phylogeny congruence i.e., inconsistent clustering of LuxR homologs from members of the same genus. Given that a majority of the sphingomonad LuxR homologs form a large clade among themselves, the incongruence with the newly constructed species phylogeny (Figure 2) can be explained by a combination of horizontal gene transfer and gene duplication within the Sphingomonadaceae family followed by speciation as proposed previously (Lerat and Moran, 2004). Interestingly, four sphingomonad LuxR homologs formed a monophyletic clade that is sister group to TraR and RaiR. The distant relationship between the this sphingomonad LuxR clade and the major sphingomonad LuxR homologs clade coupled with the localization of both traR and raiR genes on the plasmid e.g., Ti plasmid and non-symbiotic plasmid respectively (Piper et al., 1993; Gray et al., 1996; Oger and Farrand, 2002) suggest the acquisition of these four luxR homologs via plasmid-mediated horizontal gene transfer. This warrants future work focusing on the identification of plasmid-coded luxR homolog through plasmid isolation and sequencing to confirm the origin of the distant sphingomonad luxR homologs.
Five out of six of the sphingomonad LuxR homologs that are more closely related to PAB LuxR solos than the rest of the LuxR homologs (Figure 3) appear to share one of the two major signature e.g., Y61W in PAB solos (Figure 4). Recent cartography analysis of the ligand-binding sites of the LuxR homologs has demonstrated that Y61 residue is directly involved in ligand binding (in addition to W57, D70, and W85) (Covaceuszach et al., 2013). Therefore, substitution at Y61 in these specific sphingomonad LuxR homologs is a strong indicator of their inability to bind to AHL. Three dimensional structure modeling of these proteins followed by comparison of binding/active sites regarding substrate preference(s) will shed lights into the protein characteristic of these atypical sphingomonad LuxR homologs. Recently, a LuxR-homolog from Photorhabdus that has some substitutions in the conserved 9 aa residues in LuxR homologs was shown to bind to a bacterial-produced pyrone instead of AHLs or plant exudates (Brachmann et al., 2013; Brameyer et al., 2014). It should be noted that the structural-activity relationship(s) of LuxR solos is beyond the scope of this study.
The occurrence of two luxR homologs in tandem is not novel in the realm of alpha-bacteria and has been previously reported in the genus Roseobacter, noted as topology N (Cude and Buchan, 2013). However, the gene neighborhood of the double luxR in various sphingomonads is significantly different from topology N to justify the proposal of a new topology that we will coin as topology T. Topology T represents the convergently oriented luxR-luxR-luxI-phyH-X-virB1-virB2-virB3 topology whereby X denotes gene coding for hypothetical protein. It is also worth noting that one or more mobile elements are present upstream of the double luxR in three out of the six topology variants, indicating past transposition event(s) and/or transposition potential of the gene cluster.
The low pairwise identity between members of the same LuxR double group (Figure 6) support the distantly shared ancestry as observed in the LuxR phylogenetic tree (Figure 3, shaded in pink). Furthermore, the low pairwise identity between members of the same LuxR double group and retention of convergent double luxR in the genomes of several Sphingobium strains suggests that the sphingomonad LuxR duplicate has undergone sufficient functional divergence which may correlate to the evolution of the organism to be more competitive regarding niche adaptation. The presence of the complete LuxR signature domains in both members of the same convergent double LuxR group suggests the retention of the core LuxR function with perhaps dissimilar substrate range and/or DNA-binding region that warrants future protein characterization and transcription study.
In addition to harboring the newly described QS gene circuit arrangement, some members of the currently sequenced genus Sphingobium exhibit another interesting feature of QS signaling, e.g., presence of luxI solos. The assignment of LuxI solos based on the presence of signature amino acid residues in canonical LuxI homologs and the absence of unassociated luxR in the vicinity of its protein coding gene (Figure 7 and Table 3) provide strong evidence that the LuxI solos identified in both Sphingobium sp. KK2 and Sphingobium chinhatense IP26 are authentic. The presence of a gene coding for a putative N-terminal truncated LuxR-like protein immediately upstream of the luxI solo gene in strain IP26 is suggestive of the luxI solo previously being part of a functional luxI/R pair instead of having been acquired independently. However, this may not be the case for another luxI solo located in contig92 of Sphingobium sp. KK2 with more than 800 bp of an upstream non-protein coding region. Recently a detailed study of LuxR-LuxI type QS network in Ruegeria sp. KLH11 (Zan et al., 2012) confirmed the presence of a functional LuxI solo, SscI and demonstrated that SscI and a paired-LuxI homolog, SsbI, produced the same AHLs e.g., 3-OH-C14:1-HSL and 3-OH-C14-HSL that indirectly affect QS-dependent gene regulation by another LuxI/R pair homologs, SsaI/R. Given the presence of one or more luxI/R pairs in the strain KK2 and IP26 genomes, it is tempting to speculate that a similar level of QS network complexity may operate in both Sphingobium strains.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fcimb.2014.00188/abstract
References
Aylward, F. O., McDonald, B. R., Adams, S. M., Valenzuela, A., Schmidt, R. A., Goodwin, L. A., et al. (2013). Comparison of 26 sphingomonad genomes reveals diverse environmental adaptations and biodegradative capabilities. Appl. Environ. Microbiol. 79, 3724–3733. doi: 10.1128/AEM.00518-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Basta, T., Buerger, S., and Stolz, A. (2005). Structural and replicative diversity of large plasmids from sphingomonads that degrade polycyclic aromatic compounds and xenobiotics. Microbiology 151, 2025–2037. doi: 10.1099/mic.0.27965-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Basta, T., Keck, A., Klein, J., and Stolz, A. (2004). Detection and characterization of conjugative degradative plasmids in xenobiotic-degrading Sphingomonas strains. J. Bacteriol. 186, 3862–3872. doi: 10.1128/JB.186.12.3862-3872.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., and Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boetzer, M., and Pirovano, W. (2012). Toward almost closed genomes with GapFiller. Genome Biol. 13, R56. doi: 10.1186/gb-2012-13-6-r56
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bond, C. S., and Schuttelkopf, A. W. (2009). ALINE: a WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Crystallogr. D Biol. Crystallogr. 65, 510–512. doi: 10.1107/S0907444909007835
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brachmann, A. O., Brameyer, S., Kresovic, D., Hitkova, I., Kopp, Y., Manske, C., et al. (2013). Pyrones as bacterial signaling molecules. Nat. Chem. Biol. 9, 573–578. doi: 10.1038/nchembio.1295
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brameyer, S., Kresovic, D., Bode, H. B., and Heermann, R. (2014). LuxR solos in Photorhabdus species. Front. Cell. Infect. Microbiol. 4:166. doi: 10.3389/fcimb.2014.00166
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Case, R. J., Labbate, M., and Kjelleberg, S. (2008). AHL-driven quorum-sensing circuits: their frequency and function among the Proteobacteria. ISME J. 2, 345–349. doi: 10.1038/ismej.2008.13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chatnaparat, T., Prathuangwong, S., Ionescu, M., and Lindow, S. E. (2012). XagR, a LuxR homolog, contributes to the virulence of Xanthomonas axonopodis pv. glycines to soybean. Mol. Plant Microbe Interact. 25, 1104–1117. doi: 10.1094/MPMI-01-12-0008-R
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Choudhary, K. S., Hudaiberdiev, S., Gelencser, Z., Goncalves Coutinho, B., Venturi, V., and Pongor, S. (2013). The Organization of the quorum sensing luxI/R Family Genes in Burkholderia. Int. J. Mol. Sci. 14, 13727–13747. doi: 10.3390/ijms140713727
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Covaceuszach, S., Degrassi, G., Venturi, V., and Lamba, D. (2013). Structural insights into a novel interkingdom signaling circuit by cartography of the ligand-binding sites of the homologous quorum sensing LuxR-family. Int. J. Mol. Sci. 14, 20578–20596. doi: 10.3390/ijms141020578
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cubo, M. T., Economou, A., Murphy, G., Johnston, A. W., and Downie, J. A. (1992). Molecular characterization and regulation of the rhizosphere-expressed genes rhiABCR that can influence nodulation by Rhizobium leguminosarum biovar viciae. J. Bacteriol. 174, 4026–4035.
Cude, W. N., and Buchan, A. (2013). Acyl-homoserine lactone-based quorum sensing in the Roseobacter clade: complex cell-to-cell communication controls multiple physiologies. Front. Microbiol. 4:336. doi: 10.3389/fmicb.2013.00336
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
D'angelo-Picard, C., Faure, D., Penot, I., and Dessaux, Y. (2005). Diversity of N-acyl homoserine lactone-producing and -degrading bacteria in soil and tobacco rhizosphere. Environ. Microbiol. 7, 1796–1808. doi: 10.1111/j.1462-2920.2005.00886.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dogra, C., Raina, V., Pal, R., Suar, M., Lal, S., Gartemann, K. H., et al. (2004). Organization of lin genes and IS6100 among different strains of hexachlorocyclohexane-degrading Sphingomonas paucimobilis: evidence for horizontal gene transfer. J. Bacteriol. 186, 2225–2235. doi: 10.1128/JB.186.8.2225-2235.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferluga, S., Bigirimana, J., Hofte, M., and Venturi, V. (2007). A LuxR homologue of Xanthomonas oryzae pv. oryzae is required for optimal rice virulence. Mol. Plant Pathol. 8, 529–538. doi: 10.1111/j.1364-3703.2007.00415.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferluga, S., and Venturi, V. (2009). OryR is a LuxR-family protein involved in interkingdom signaling between pathogenic Xanthomonas oryzae pv. oryzae and rice. J. Bacteriol. 191, 890–897. doi: 10.1128/JB.01507-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fuqua, C., and Greenberg, E. P. (2002). Listening in on bacteria: acyl-homoserine lactone signalling. Nat. Rev. Mol. Cell Biol. 3, 685–695. doi: 10.1038/nrm907
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fuqua, C. (2006). The QscR quorum-sensing regulon of Pseudomonas aeruginosa: an orphan claims its identity. J. Bacteriol. 188, 3169–3171. doi: 10.1128/JB.188.9.3169-3171.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fuqua, W. C., Winans, S. C., and Greenberg, E. P. (1994). Quorum sensing in bacteria: the LuxR-LuxI family of cell density-responsive transcriptional regulators. J. Bacteriol. 176, 269–275.
Gan, H. M., Buckley, L., Szegedi, E., Hudson, A. O., and Savka, M. A. (2009). Identification of an rsh gene from a Novosphingobium sp. necessary for quorum-sensing signal accumulation. J. Bacteriol. 191, 2551–2560. doi: 10.1128/JB.01692-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gan, H. M., Chew, T. H., Hudson, A. O., and Savka, M. A. (2012). Genome sequence of Novosphingobium sp. strain Rr 2-17, a nopaline crown gall-associated bacterium isolated from Vitis vinifera L. grapevine. J. Bacteriol. 194, 5137–5138. doi: 10.1128/JB.01159-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gan, H. M., Hudson, A. O., Rahman, A. Y., Chan, K. G., and Savka, M. A. (2013). Comparative genomic analysis of six bacteria belonging to the genus Novosphingobium: insights into marine adaptation, cell-cell signaling and bioremediation. BMC Genomics 14:431. doi: 10.1186/1471-2164-14-431
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gan, H. Y., Gan, H. M., Tarasco, A. M., Busairi, N. I., Barton, H. A., Hudson, A. O., et al. (2014). Whole-genome sequences of five oligotrophic bacteria isolated from deep within Lechuguilla Cave, New Mexico. Genome Announc. 2:e01133-14. doi: 10.1128/genomeA.01133-14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Geueke, B., Busse, H. J., Fleischmann, T., Kampfer, P., and Kohler, H. P. (2007). Description of Sphingosinicella xenopeptidilytica sp. nov., a beta-peptide-degrading species, and emended descriptions of the genus Sphingosinicella and the species Sphingosinicella microcystinivorans. Int. J. Syst. Evol. Microbiol. 57, 107–113. doi: 10.1099/ijs.0.64509-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gonzalez, J. F., Myers, M. P., and Venturi, V. (2013). The inter-kingdom solo OryR regulator of Xanthomonas oryzae is important for motility. Mol. Plant Pathol. 14, 211–221. doi: 10.1111/j.1364-3703.2012.00843.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gonzalez, J. F., and Venturi, V. (2013). A novel widespread interkingdom signaling circuit. Trends Plant Sci. 18, 167–174. doi: 10.1016/j.tplants.2012.09.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gray, K. M., Pearson, J. P., Downie, J. A., Boboye, B. E., and Greenberg, E. P. (1996). Cell-to-cell signaling in the symbiotic nitrogen-fixing bacterium Rhizobium leguminosarum: autoinduction of a stationary phase and rhizosphere-expressed genes. J. Bacteriol. 178, 372–376.
Huang, Y., Zeng, Y., Yu, Z., Zhang, J., Feng, H., and Lin, X. (2013). In silico and experimental methods revealed highly diverse bacteria with quorum sensing and aromatics biodegradation systems–a potential broad application on bioremediation. Bioresour. Technol. 148, 311–316. doi: 10.1016/j.biortech.2013.08.155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., Mcanulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lerat, E., and Moran, N. A. (2004). The evolutionary history of quorum-sensing systems in bacteria. Mol. Biol. Evol. 21, 903–913. doi: 10.1093/molbev/msh097
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lindemann, A., Pessi, G., Schaefer, A. L., Mattmann, M. E., Christensen, Q. H., Kessler, A., et al. (2011). Isovaleryl-homoserine lactone, an unusual branched-chain quorum-sensing signal from the soybean symbiont Bradyrhizobium japonicum. Proc. Natl. Acad. Sci. U.S.A. 108, 16765–16770. doi: 10.1073/pnas.1114125108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lowe, N., Gan, H. M., Chakravartty, V., Scott, R., Szegedi, E., Burr, T. J., et al. (2009). Quorum-sensing signal production by Agrobacterium vitis strains and their tumor-inducing and tartrate-catabolic plasmids. FEMS Microbiol. Lett. 296, 102–109. doi: 10.1111/j.1574-6968.2009.01627.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maruyama, T., Park, H. D., Ozawa, K., Tanaka, Y., Sumino, T., Hamana, K., et al. (2006). Sphingosinicella microcystinivorans gen. nov., sp. nov., a microcystin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 56, 85–89. doi: 10.1099/ijs.0.63789-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miller, M. B., and Bassler, B. L. (2001). Quorum sensing in bacteria. Annu. Rev. Microbiol. 55, 165–199. doi: 10.1146/annurev.micro.55.1.165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muhire, B. M., Varsani, A., and Martin, D. P. (2014). SDT: a virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 9:e108277. doi: 10.1371/journal.pone.0108277
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muller, E. C., and Wittmann-Liebold, B. (1997). Phylogenetic relationship of organisms obtained by ribosomal protein comparison. Cell. Mol. Life Sci. 53, 34–50. doi: 10.1007/PL00000578
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Muller, T. A., Byrde, S. M., Werlen, C., Van Der Meer, J. R., and Kohler, H. P. (2004). Genetic analysis of phenoxyalkanoic acid degradation in Sphingomonas herbicidovorans MH. Appl. Environ. Microbiol. 70, 6066–6075. doi: 10.1128/AEM.70.10.6066-6075.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nohynek, L. J., Nurmiaho-Lassila, E. L., Suhonen, E. L., Busse, H. J., Mohammadi, M., Hantula, J., et al. (1996). Description of chlorophenol-degrading Pseudomonas sp. strains KF1T, KF3, and NKF1 as a new species of the genus Sphingomonas, Sphingomonas subarctica sp. nov. Int. J. Syst. Bacteriol. 46, 1042–1055. doi: 10.1099/00207713-46-4-1042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Oger, P., and Farrand, S. K. (2002). Two opines control conjugal transfer of an Agrobacterium plasmid by regulating expression of separate copies of the quorum-sensing activator gene traR. J. Bacteriol. 184, 1121–1131. doi: 10.1128/jb.184.4.1121-1131.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ogramab, A. V., Duana, Y., Trabuea, S. L., Fengc, X., Castroa, H., and Oua, L. (2000). Carbofuran degradation mediated by three related plasmid systems. FEMS Microbiol. Ecol. 32, 197–203. doi: 10.1111/j.1574-6941.2000.tb00712.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Piper, K. R., Beck Von Bodman, S., and Farrand, S. K. (1993). Conjugation factor of Agrobacterium tumefaciens regulates Ti plasmid transfer by autoinduction. Nature 362, 448–450. doi: 10.1038/362448a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490. doi: 10.1371/journal.pone.0009490
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rambaut, A. (2014). FigTree v1.4.1:Tree Figure Drawing Tool. Available online at: http://tree.bio.ed.ac.uk/software/figtree (Accessed September 18, 2014).
Romine, M. F., Fredrickson, J. K., and Li, S. M. (1999). Induction of aromatic catabolic activity in Sphingomonas aromaticivorans strain F199. J. Ind. Microbiol. Biotechnol. 23, 303–313. doi: 10.1038/sj.jim.2900750
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Savka, M. A., Le, P. T., and Burr, T. J. (2011). LasR receptor for detection of long-chain quorum-sensing signals: identification of N-acyl-homoserine lactones encoded by the avsI locus of Agrobacterium vitis. Curr. Microbiol. 62, 101–110. doi: 10.1007/s00284-010-9679-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schaefer, A. L., Lappala, C. R., Morlen, R. P., Pelletier, D. A., Lu, T. Y., Lankford, P. K., et al. (2013). luxR- and luxI-type quorum-sensing circuits are prevalent in members of the Populus deltoides microbiome. Appl. Environ. Microbiol. 79, 5745–5752. doi: 10.1128/AEM.01417-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schuster, M., Sexton, D. J., Diggle, S. P., and Greenberg, E. P. (2013). Acyl-homoserine lactone quorum sensing: from evolution to application. Annu. Rev. Microbiol. 67, 43–63. doi: 10.1146/annurev-micro-092412-155635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scott, R. A., Weil, J., Le, P. T., Williams, P., Fray, R. G., Von Bodman, S. B., et al. (2006). Long- and short-chain plant-produced bacterial N-acyl-homoserine lactones become components of phyllosphere, rhizosphere, and soil. Mol. Plant Microbe Interact. 19, 227–239. doi: 10.1094/MPMI-19-0227
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Segata, N., Bornigen, D., Morgan, X. C., and Huttenhower, C. (2013). PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 4, 2304. doi: 10.1038/ncomms3304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stolz, A. (2009). Molecular characteristics of xenobiotic-degrading sphingomonads. Appl. Microbiol. Biotechnol. 81, 793–811. doi: 10.1007/s00253-008-1752-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Subramoni, S., and Venturi, V. (2009). LuxR-family ‘solos’: bachelor sensors/regulators of signalling molecules. Microbiology 155, 1377–1385. doi: 10.1099/mic.0.026849-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Takeuchi, M., Hamana, K., and Hiraishi, A. (2001). Proposal of the genus Sphingomonas sensu stricto and three new genera, Sphingobium, Novosphingobium and Sphingopyxis, on the basis of phylogenetic and chemotaxonomic analyses. Int. J. Syst. Evol. Microbiol. 51, 1405–1417. doi: 10.1099/00207713-51-4-1405
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thiel, M., Kaschabek, S. R., Groning, J., Mau, M., and Schlomann, M. (2005). Two unusual chlorocatechol catabolic gene clusters in Sphingomonas sp. TFD44. Arch. Microbiol. 183, 80–94. doi: 10.1007/s00203-004-0748-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tiirola, M. A., Wang, H., Paulin, L., and Kulomaa, M. S. (2002). Evidence for natural horizontal transfer of the pcpB gene in the evolution of polychlorophenol-degrading sphingomonads. Appl. Environ. Microbiol. 68, 4495–4501. doi: 10.1128/AEM.68.9.4495-4501.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsai, C. S., and Winans, S. C. (2010). LuxR-type quorum-sensing regulators that are detached from common scents. Mol. Microbiol. 77, 1072–1082. doi: 10.1111/j.1365-2958.2010.07279.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Waters, C. M., and Bassler, B. L. (2005). Quorum sensing: cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 21, 319–346. doi: 10.1146/annurev.cellbio.21.012704.131001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
White, D. C., Sutton, S. D., and Ringelberg, D. B. (1996). The genus Sphingomonas: physiology and ecology. Curr. Opin. Biotechnol. 7, 301–306. doi: 10.1016/S0958-1669(96)80034-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yabuuchi, E., Yano, I., Oyaizu, H., Hashimoto, Y., Ezaki, T., and Yamamoto, H. (1990). Proposals of Sphingomonas paucimobilis gen. nov. and comb. nov., Sphingomonas parapaucimobilis sp. nov.,Sphingomonas yanoikuyae sp. nov., Sphingomonas adhaesiva sp. nov.,Sphingomonas capsulata comb. nov., and two genospecies of the genus Sphingomonas. Microbiol. Immunol. 34, 99–119. doi: 10.1111/j.1348-0421.1990.tb00996.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yasir, M., Aslam, Z., Song, G. C., Jeon, C. O., and Chung, Y. R. (2010). Sphingosinicella vermicomposti sp. nov., isolated from vermicompost, and emended description of the genus Sphingosinicella. Int. J. Syst. Evol. Microbiol. 60, 580–584. doi: 10.1099/ijs.0.010777-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yoon, J. H., Kang, S. J., Lee, J. S., Nam, S. W., Kim, W., and Oh, T. K. (2008). Sphingosinicella soli sp. nov., isolated from an alkaline soil in Korea. Int. J. Syst. Evol. Microbiol. 58, 173–177. doi: 10.1099/ijs.0.65242-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zan, J., Cicirelli, E. M., Mohamed, N. M., Sibhatu, H., Kroll, S., Choi, O., et al. (2012). A complex LuxR-LuxI type quorum sensing network in a roseobacterial marine sponge symbiont activates flagellar motility and inhibits biofilm formation. Mol. Microbiol. 85, 916–933. doi: 10.1111/j.1365-2958.2012.08149.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, L., Jia, Y., Wang, L., and Fang, R. (2007). A proline iminopeptidase gene upregulated in planta by a LuxR homologue is essential for pathogenicity of Xanthomonas campestris pv. campestris. Mol. Microbiol. 65, 121–136. doi: 10.1111/j.1365-2958.2007.05775.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zipper, C., Nickel, K., Angst, W., and Kohler, H. P. (1996). Complete microbial degradation of both enantiomers of the chiral herbicide mecoprop [(RS)-2-(4-chloro-2-methylphenoxy)propionic acid] in an enantioselective manner by Sphingomonas herbicidovorans sp. nov. Appl. Environ. Microbiol. 62, 4318–4322.
Keywords: luxI/R, luxR solos, Novosphingobium, quorum-sensing, Sphingomonadaceae, phylogenetic, whole genome sequencing
Citation: Gan HM, Gan HY, Ahmad NH, Aziz NA, Hudson AO and Savka MA (2015) Whole genome sequencing and analysis reveal insights into the genetic structure, diversity and evolutionary relatedness of luxI and luxR homologs in bacteria belonging to the Sphingomonadaceae family. Front. Cell. Infect. Microbiol. 4:188. doi: 10.3389/fcimb.2014.00188
Received: 23 September 2014; Accepted: 16 December 2014;
Published online: 08 January 2015.
Edited by:
Vittorio Venturi, International Centre for Genetic Engineering and Biotechnology, ItalyReviewed by:
Sujatha Subramoni, International Centre for Genetic Engineering and Biotechnology, ItalyAmy Schaefer, University of Washington, USA
Copyright © 2015 Gan, Gan, Ahmad, Aziz, Hudson and Savka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael A. Savka, Thomas H. Gosnell School of Life Sciences, College of Science, Rochester Institute of Technology, 85 Lomb Memorial Dr., A350 Gosnell Hall, Rochester, NY 14623, USA e-mail:bWFzc2JpQHJpdC5lZHU=