 Joshua E. Messinger
Joshua E. Messinger Emmalin Nelton
Emmalin Nelton David C. Gondek
David C. Gondek- Department of Biology, Ithaca College, Ithaca, NY, USA
Chlamydiae, obligate intracellular bacteria, cause significant human and veterinary associated diseases. Having emerged an estimated 700-million years ago, these bacteria have twice adapted to humans as a host species, causing sexually transmitted infection (C. trachomatis) and respiratory associated disease (C. pneumoniae). The principle mechanism of host cell defense against these intracellular bacteria is the induction of cell death via apoptosis. However, in the “arms race” of co-evolution, Chlamydiae have developed mechanisms to promote cell viability and inhibit cell death. Herein we examine the impact of Chlamydiae infection across multiple host species on transcription of anti-apoptotic genes. We found mostly distinct patterns of gene expression (Mcl1 and cIAPs) elicited by each pathogen-host pair indicating Chlamydiae infection across host species boundaries does not induce a universally shared host response. Understanding species specific host-pathogen interactions is paramount to deciphering how potential pathogens become emerging diseases.
Introduction
Bacterial pathogens have co-evolved with their hosts, subverting host cell biology through secreted virulence factors to maintain a niche for pathogen growth. Chlamydiae, obligate intracellular gram-negative bacterial pathogens, have evolved from a common progenitor to become infectious specialists for a variety of host species (Nunes and Gomes, 2014). Two Chlamydia species, C. trachomatis and C. pneumoniae, predominate human infection. C. trachomatis infects epithelial cells of the reproductive tract, making it the number one bacterial sexually transmitted disease while C. pneumoniae can cause respiratory and in some cases atherosclerotic disorders (Campbell and Kuo, 2004; Starnbach and Roan, 2008). Chlamydiae replicate within an intracellular compartment called the inclusion. To maintain this replicative cellular niche, Chlamydiae have evolved mechanisms to prevent host cell death, one in particular being the inhibition of apoptosis.
Apoptosis is an organized mechanism of cellular death triggered through either an extrinsic pathway (death receptor signaling) or a cellular intrinsic pathway (mitochondrial breakdown). A common feature in both these pathways is the activation of cytosolic proteases, called caspases (Thornberry and Lazebnik, 1998). While the extrinsic pathway activates caspases directly, the intrinsic pathway requires destabilization of the mitochondrial membrane, mediated by pro-apoptotic proteins such as Bax, Bak, and Bad, leading to the release of cytochrome C. The pro-apoptotic proteins can be held in check by cellular anti-apoptotic factors such as Bcl-2, Mcl-1, and cIAP. Both the intrinsic and extrinsic apoptosis pathways of apoptosis result in nuclear fragmentation and organized dispersal of cellular contents via membrane blebbing.
Intracellular bacterial infection requires host cell survival. Previous studies have shown that Chlamydia infection results in the inhibition of the host cell apoptotic response under a variety of conditions (Fan et al., 1998). Chlamydia can inhibit the cleavage of pro-apoptotic proteins (Xiao et al., 2004). Infection can also cause the upregulation of host anti-apoptotic factors such as MCL1 and IAP complexes (Rajalingam et al., 2006, 2008). However, conflicting reports exist in the literature regarding the impact of these anti-apoptotic factors. For example, Chlamydia trachomatis infection of human cells requires cIAP complexes, whereas these complexes are not essential for infection of mouse cells by this same pathogen (Ying et al., 2008).
To address this discrepancy, we took an evolutionary medicine approach. The infectious process places significant genomic pressures on both the pathogen and the host in the “arms race” of co-evolution (Red Queen Hypothesis) (Van Valen, 1973). This hypothesis captures the idea that as the host evolves mechanisms to sense infection and eliminate the invader, the pathogen is coordinately evolving mechanisms to shut down those host processes. Since the radiation of Chlamydiae has occurred over the past few million years, it is likely that each pathogen species has evolved host specific adaptations to ensure survival (Borges et al., 2012). Cellular apoptosis is a highly conserved mechanism for combating intracellular infection and Chlamydiae have been shown to repress this response in cells upon infection. However, there is no evidence that all Chlamydiae use the same anti-apoptotic mechanism to reach that outcome.
Here we show that Chlamydiae infection directly impacts host transcriptional levels of anti-apoptotic proteins. The addition of cycloheximide (CHX), a eukaryotic translational inhibitor, significantly impacts this host transcriptional profile, indicating a potential positive feedback loop of anti-apoptotic gene activation elicited by the pathogen. Utilizing three separate Chlamydiaceae (C. trachomatis-Ct, C. muridarum-Cm, C. caviae-Cc) and multiple host cell lines (human, mouse, guinea pig, monkey, and cat) we identify that each pathogen induces a unique anti-apoptotic transcriptional response dependent upon host species infection. Examining the host pathogen interactions and comparing infections will allow us to understand the boundaries for cross species infectivity. This insight is key to unraveling the evolution of new and emerging disease.
Methods
Cell Culture and Chlamydiae Stocks
McCoy (mouse fibroblast) and 104C1 (guinea pig lung fibroblast) cells were maintained in RPMI media supplemented with 10% FBS (Atlanta Biologics), L-glutamine, HEPES buffer, and β-mercaptoethanol. Vero (African green monkey kidney) and HeLa (Human endocervical) cells were maintained in Dulbecco's Modified Eagles Medium (DMEM) supplemented with 10% FBS plus additional components listed above. AK-D (Cat lung fibroblast) cells were maintained in Hams-F12 media with 10% FBS plus additional components above. All cells were grown at 37°C with 5% CO2. Chlamydia trachomatis (L2/434Bu and D/UW-3/CX), Chlamydia muridarum (MoPN), and Chalmydiophila caviae (GPIC) bacterial stocks were purified over a renografin gradient, resuspended in sodium phosphate glutamate (SPG) and stored at −80°C. To titer stocks, purified Chlamydiae were serial diluted on McCoy cells and centrifuged for 1 h at 1500RCF at 37°C. Cells were incubated for 30 h in media containing cycloheximide (CHX) (1.5 μg/mL) prior to fixation and staining. C. trachmoatis and C. muridarum strains were generously provided by the Starnbach Lab (Harvard Med) and C. caviae was purchased from ATCC.
Immunofluorescent Staining
After overnight methanol fixation, cells were rehydrated and stained with primary antibody (αHsp60—Thermo A57B9), secondary antibody (AlexaFluor A11032) and Hoechst 33258 or DAPI. Immunofluorescent stains were imaged using a Nikon Eclipse E800 UV microscope with a Nikon DS-Ri1 camera adapter. Images were analyzed with Nikon NIS-Elements D imaging software. For quantification and unbiased analysis of infectous units per milliliter images were processed using MATLAB image processing toolbox (mathworks.com). Within MATLAB, a script was written to batch convert images to grayscale, uniformly threshold the images, and then count numbers of inclusions (dots) per image. Data were exported to Excel for further analysis and quantification of infectious units per milliliter of lysate.
Recoverable Infectious Unit Assay
Confluent monolayers of McCoy, 104C1, AK-D, Vero, and HeLa cells were plated on Costar tissue culture 24 well plates for overnight growth in the conditions mentioned above. Monolayers were infected at an MOI of approximately one (1 × 105 IFU) by C. trachomatis, C. caviae, and C. muridarum in media by centrifugation at 37°C at 1500 RCF for 1 h. Media was changed and the Chlamydia were allowed to grow in the presence or absence of CHX (1.5 μg/ml) at 37°C. At 30 h post infection media was collected into a new Costar 24 well plate and cells were hypotonically lysed using sterile water. Lysates were vigorously pipetted prior to being combined with the spent media and the mixture was frozen at −80°C to ensure freeze fracture of infected cells. Cell lysates were serial diluted in SPG over confluent monolayers of McCoy cells in Costar 96-well plates and were centrifuged as described above.
Cell Death and Apoptosis Assay
Confluent monolayers of McCoy, HeLa, and 104C1 cells were plated on Costar 96-well tissue culture plates for overnight growth. Cells were infected with C. trachomatis at an MOI of approximately five, or C. caviae or C. muridarum at an MOI of approximately two in SPG using centrifugation as previously described (Coers et al., 2011). Following centrifugation, media was replaced with or without CHX (1.5 μg/ml) and allowed to incubate at 37°C for 30 h. At 36 h post infection, cells were challenged with staurosporine (104C1–25 μM, McCoy–10 μM, HeLa–2 μM) and allowed to incubate for 4 h at 37°C. Cells were then stained for immunofluorescence microscopy as described above.
Gene Expression Analysis by RT-qPCR
Genomic sequences for the following genes were obtained through the Ensembl bioinformatic database (ensemble.org). All known isoforms were analyzed and the primer binding sites that were flanking exon boundaries were determined for shared exons in all isoforms. Upon identifying exon boundaries, these sequences were entered into PRIMER3 (http://bioinfo.ut.ee/primer3/) to determine thermodynamically ideal primers for RT-qPCR(see table below) of genes previously identified as modulated by Chlamydia infection (Wahl et al., 2003; Rajalingam et al., 2006, 2008; Kun et al., 2013). Confluent monolayers of McCoy, HeLa, and Guinea Pig cells were plated in Costar six well tissue culture treated plates for overnight growth. Cells were infected at an MOI of approximately five for C. trachomatis and an MOI of approximately two for C. caviae and C. muridarum. At 30 h post infection, media was aspirated, cells washed, and lysed directly in the plate for total RNA isolation as directed by the Omega Biotech Total RNA Isolation Kit. RNA was stored at −80°C before analysis through RT-qPCR. All 1-step RT-qPCR reactions (Quanta BioSciences) were generated as directed by the manufacturer and analyzed on a Roche454 Lightcycler.
Primer Sequences Used in this Study

Caspase-3 Colorimetric Absorbance Assay
Cells were plated into six well plates (5*105 per well) for overnight growth. Cells were infected as described above. Twenty-six hours post infection cells were left either untreated or treated with staurosporine for 4 h prior to collection. Cells were probed for caspase three activity using the commercially available Caspase-3 Cellular assay kit PLUS (Enzo Life Sciences). Absorbance readings at 405 nm were made every 10 min for 6 h using a TECAN NanoQuant infinite M200 Pro plate reader.
Statistical Analysis
All experiments were independently replicated two or more times as noted in figure legends. Statistical significance was determined via One-way ANOVA and Tukey's post-hoc pairwise comparison test. P-values less than 0.05 were used to determine significant data.
Results
Chlamydiae Infection and Growth Across Host Species Boundaries
A pathogen and its host are under constant genomic pressure as the duo are locked in coevolutionary arms race of adaptation (Red Queen Hypothesis; Van Valen, 1973). Upon infection, pathogen recognition receptors (NOD/TLR) are stimulated in the cell leading to changes in gene transcription and protein activation (O'Connell et al., 2006). These pathways can elicit the production of new proteins which result in pathogen elimination and/or cell apoptosis. Chlamydiae have adapted host-specific mechanisms to subvert this cellular response by modulating the host response to infection at a transcriptional and post-translational level (Hess et al., 2001; Olive et al., 2014).
Cycloheximide (CHX) is a commonly used eukaryotic translation inhibitor that improves the growth of Chlamydia by reducing competition for intracellular amino acids (Allan and Pearce, 1983b). Thus, this treatment is able to improve the growth of poorly host adapted pathogens. A well-adapted pathogen would not need this “advantage.” Moreover, this CHX block of host cell translation could inhibit Chlamydia's ability to manipulate the infected host cell. We hypothesized that host adapted Chlamydia relies on active host cell translation to reach maximal growth potential. To test this, we examined the impact of CHX on the ability to enhance or inhibit pathogen growth across host species boundaries. We infected the following five host cell lines with each pathogen (Ct, Cc, and Cm): mouse, monkey, cat, human, and guinea pig. To compare growth potential, we took the ratio of total infectious progeny in the absence vs. presence of CHX such that a positive correlation indicates improved growth due to ongoing host translation. We found that only Cm in the guinea pig cell line was able to grow significantly better in the presence of host cell translation, compared to Ct and Cc (p < 0.05; Figure 1A).
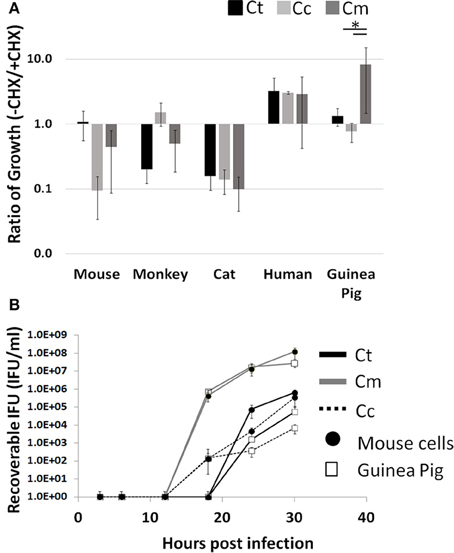
Figure 1. Enhanced growth in the presence of host cell translation—Cells were infected in 24 well plates (105 cells/well) for 30 h with or without CHX (1.5 μg/mL). Supernatants of hypotonically lysed cells were clarified by centrifugation and titered. Chlamydiae were stained with α-HSP60 to quantitate infectious units/ml (A). Presented is the ratio of (-CHX/CHX). Growth curves of Cm and Ct (MOI = 1 at 1 × 105 IFU) were determined in mouse and guinea pig cells (Gray line-Cm, solid line-Ct, dashed line-Cc open marker—guinea pig, solid marker—mouse) (B). Ct-C. trachomatis, Cc-C.caviae Cm-C.muridarum. *p < 0.05 (data are representative of two independent trials).
To determine if this high infectious load in guinea pig cells is a result of a more rapid conversion to the infectious elementary body, we compared Cm and Ct growth in guinea pig to that in mouse cells (Figure 1B). The Chlamydia IFU production during Cm infection appeared to be more rapid than Ct or Cc infection. Cm loads were similar between guinea pig and mouse cell lines across all time points. For Ct and Cc, infections of the Guinea Pig cell line typically yielded one log lower IFU at 30 h post infection, compared to McCoy cell infection. Additionally, Cm demonstrated high bacterial loads in all host species cell lines tested compared to Ct and Cc infections (Supplemental Figure 1).
Apoptosis Inhibition is a Key Determinant of Chlamydiae Growth Potential
In experiments where Chlamydiae growth potential was particularly poor, cells examined by microscopy displayed cellular blebbing and a significant debris field, indicative of apoptosis (Supplemental Figure 2C). As maintenance of host cell viability is absolutely critical for success of obligate intracellular bacteria, a host-adapted pathogen would be able to inhibit cellular apoptosis. To test this hypothesis, we focused our studies on mouse, human, and guinea pig host species. We infected each host cell species with Chlamydiae followed by treatment with staurosporine, to induce host cell apoptosis. We examined the production of active caspase three and fragmentation of nuclei in order to determine the extent of apoptosis inhibition. As shown in Figure 2, cells infected with each species of Chlamydiae were able to inhibit the activation of caspase three while uninfected cells could not. Cm exhibited the most pronounced inhibitory activity across host species boundaries, particularly in guinea pig cells, whereas Cc was the most limited in its ability to block caspase 3 activity.
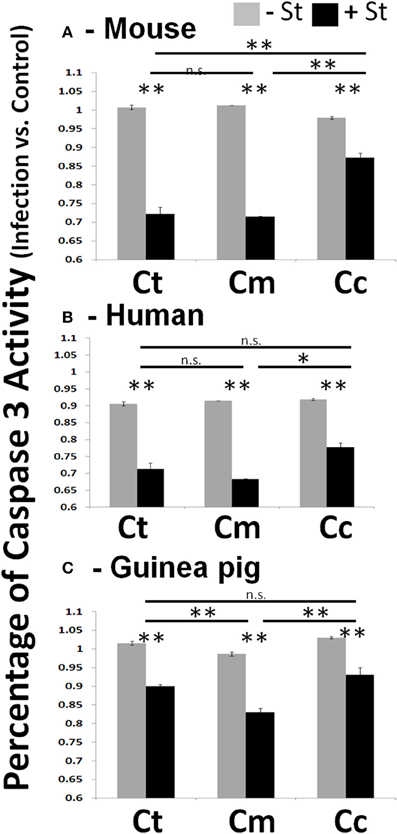
Figure 2. Chlamydiae inhibit Caspase three activation in diverse hosts—Cell lines were infected with the indicated pathogens and allowed to incubate for 26 h. Staurosporine was added for 4 h before cell lysates were collected for colorimetric analysis of caspase three activity in mouse cells (A), human cells (B), and guinea pig cells (C). Ct-C. trachomatis, Cc-C.caviae Cm-C.muridarum. **p < 0.01, *p < 0.05 (representative of greater than three independent trials).
To determine the extent to which Chlamydiae infection could impede nuclear fragmentation during apoptosis, cells were co-stained to identify both Chlamydiae inclusions and nuclear content (Figure 3). As shown in Figure 3B, when guinea pig cells are treated with staurosporine nuclear condensation occurs based on the observed increase in nuclear stain fluorescence and fragmentation of the nuclear material (p < 0.01). However, when these cells are infected with Chlamydiae the DNA fragmentation is inhibited (Figures 3C–E, p < 0.05). Cc infected cells were sparsely found on the slides due to apoptotic destruction of the cell prior to labeling leading to large variability and inability to image clusters of cells (Figure 3, Supplemental Figure 2C and data not shown). Collectively, these data indicate that Chlamydiae are able to inhibit the apoptosis of the host cell and that the level of success for the pathogen is dependent upon the species of host cell infected.
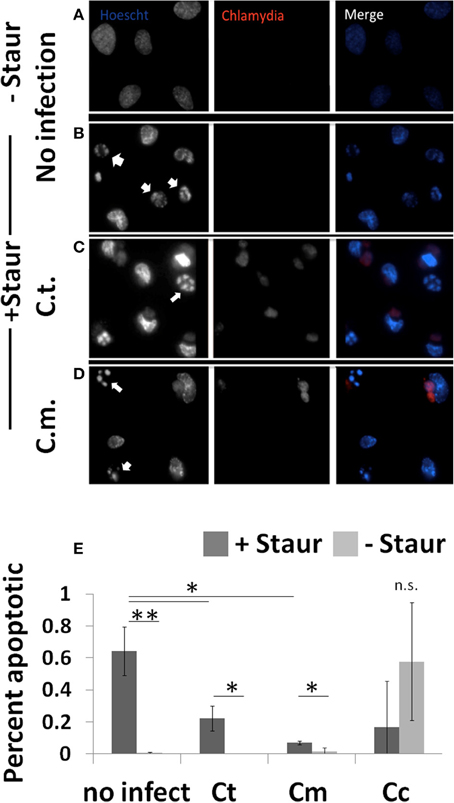
Figure 3. Chlamydiae inhibit nuclear fragmentation induced by staurosporine—Cell lines were plated on coverslips (A,B) or infected with pathogens for 26 h (C,D). Staurosporine was added for 4 h (B–D) before cells were fixed and stained to visualize Chlamydiae (αHSP-60) and DNA (Hoechst). White arrows indicate apoptotic cells. Percent apoptotic in each of the treatment conditions is quantified (E). Ct-C. trachomatis, Cc-C.caviae Cm-C.muridarum. **p < 0.01, *p < 0.05 (representative of four independent trials).
Chlamydiae Upregulate Anti-Apoptotic Genes Unique to Each Host-Pathogen Combination
We have established that Chlamydiae are capable of inhibiting apoptosis across multiple host species boundaries. However, the importance of host derived anti-apoptotic gene regulation during Chlamydiae infection has led to conflicting reports in the literature. Rajalingam et al. indicate that IAP complexes are highly upregulated in human cells following infection with Ct and these complexes are necessary for infection. Whereas Ying et al. report that Ct infection of mouse cells deficient in these IAP genes are still fully capable of inhibiting cellular apoptosis (Rajalingam et al., 2006; Ying et al., 2008). To rectify these incongruous results, we examined the regulation of anti-apoptotic transcripts in each of our host species cell lines when infected by Chlamydiae. Based on our earlier finding that the absence of CHX improves bacterial growth in certain cell lines, we decided to examine transcript levels in both the presence and absence of CHX.
As was reported by Rajalingam, we identified an upregulation of IAP genes following infection of human cells with the Ct pathogen in both the presence and absence of CHX (Figure 4A). Cm and Cc infections of human cells also follow a very similar pattern of IAP regulation except these results are dependent on CHX treatment (Figures 4B,C). In contrast to human cells, infection of mouse cells with Ct or Cm were highly specific for cIAP2 upregulation and this regulation was significantly higher in cells which were actively able to translate host proteins (Figures 4D,E). Cc infection of mouse or human cells exhbited a marginal impact on the levels of antiapoptotic transcripts (Figure 4F). Thus, the specific antiapoptotic gene transcripts (and their regulation) are completely host species dependent and are, in some cases, unique to the infecting pathogen.
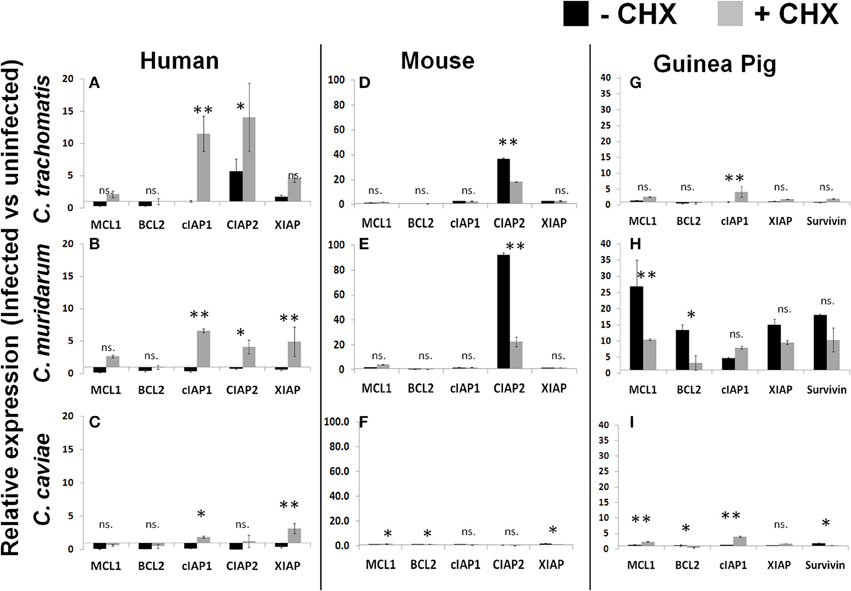
Figure 4. Infection induced transcription of anti-apoptotic factors in the presence or absence of host translation. Cell lines were plated in six well plates and infected for 30 h prior to lysis and RNA extraction. Relative expression was normalized to host GAPDH and then the ratio of infected vs. uninfected gene expression was determined in human cells (A–C), mouse cells (D–F) and guinea pig cells (G–I). **p < 0.01, *p < 0.05 (representative of three independent trials).
As described in Figure 1, Cm infection of guinea pig cells was significantly improved by the absence of CHX. When we examined the anti-apoptotic transcript expression level in these cells we found that Cm infection casued a very prominent upregulation, compared to Ct and Cc infection (Figures 4G–I). In particular, Mcl1 was very highly upregulated in guinea pig cells following Cm infection. Taken together, these data indicate that each pathogen species has a unique ability to impact the host transcription of anti-apoptotic genes. The quantity and diversity of this regulation was pathogen-species specific and the pattern of transcript expression was unique to each host species cell line.
Discussion
Chlamydiae have been co-evolving with their hosts for the last several hundred million years (Nunes and Gomes, 2014). Throughout this time the competition between pathogen and host has led to co-adaptation as the pathogen struggles to maintain its intracellular replicative niche (Nunes et al., 2008). Chlamydiae can be distinguished based on their specific amino acid requirments with a level of specificity down to identification of C. trachomatis biovars requirmeent for tryptophan(trachoma) and methionine(LGV) supplementation of culture media (Allan and Pearce, 1983a). The mechanisms for aquiring amino acids can also be very different amongst Chlamydiae. In a direct comparison to C. pneumoniae, Ct L2 was shown to be better at accessing host amino acids in the presence of a lysosomal inhibitor (Ouellette et al., 2011). The authors conclude that C. pneumoniae and Ct L2 have inherent differences in their mechanisms of amino acid acquisition. The addition of CHX provides intracellular pathogens with reduced host competition for nutrients and suppresses any de-novo protein production the host may undertake in order to rid itself of the infection (Ripa and Mårdh, 1977; Benes and McCormack, 1982; Sabet et al., 1984; Harper et al., 2000). Under these optimal conditions, in the absence of competition, Chlamydiae can readily infect across host species boundaries. We asked if this “advantage” was absolutely necessary for highly host adapted pathogens to compete with the host for nutrients and, perhaps, the pathogen could utilize the host's own protein production to its advantage. To test for different levels of human adaptation, we compared two Ct pathogens which infect at the same site but have markedly different infectious phenotypes. The invasive Ct LGV strains exhibit rapid growth kinetics and capacity to infect systemically, as compared to the genital (or ocular) strains (Miyairi et al., 2006). Based on these data, we hypothesized that the LGV strains would be better at competing with the host for nutrients and would not require CHX treatment for optimal growth. Our data support this hypothesis since the provision of “free” amino acids following CHX treatment exhibited a marginal impact on LGV growth in HeLa cells as compared to the Ct-D strain (Supplemental Figure 3). These data support a similar finding when Ct-L2 was compared to Ct-E growth in conditions ± CHX (Harper et al., 2000). Extending this study to Chlamydiae infection across multiple host species of cells, we found that only the Cm infection of guinea pig cells demonstrated a unique species specific and significant growth advantage from ongoing host cell translation, compared to Ct and Cc infection (Figure 1A). In addition, Cm infection exhibits some of the fastest growth kinetics and largest replicative burst across multiple host species cell lines (Supplemental Figure 1). From this we infer that Cm, despite being considered a mouse specific Chlamydia pathogen, is actually a generalist due to its highly permissive growth characteristics, with or without CHX treatment. In contrast, Cc infections did particularly poorly across multiple hosts indicating that it is the least well-adapted to deal with the unique biology of these divergent host species.
One main host mechanism to deny the pathogen further growth potential is the organized cellular death pathway of apoptosis. As such, the Chlamydiae pathogens have evolved mechanisms to subvert this pathway and it has been the topic of significant investigation for several years (Byrne and Ojcius, 2004). A thorough investigation of multiple Chlamydiae species had previously identified a common capacity to inhibit apoptosis in diverse tissue types of human cell lines (Greene et al., 2003). We have extended these findings and demonstrate that Chlamydiae can inhibit apoptosis across multiple host species boundaries. We used activation of caspase three, the terminal caspase in the proteolytic cascade, and nuclear fragmentation as markers of apoptosis (Thornberry and Lazebnik, 1998). As demonstrated in Figure 2, we corroborate those earlier reports in that almost all Chlamydiae species are able to inhibit host cell apoptosis. Intriguingly, Cc, the pathogen isolated from guinea pigs, was the least capable to grow within guinea pig cells. Based on light microscopy analysis, Cc infection of guinea pig cells lead to significant membrane blebbing and apoptotic phenotypes (Supplemental Figure 2). This led to particular problems in acquiring Caspase 3 activity and nuclear condensation as the cells were easily lost with gentle washing. The variability of Cc infection in Figure 3E is due to this high rate of cell loss due to combined infection and staurosporine induced apoptosis. In most situations, Cc was the least capable of down regulating the host Caspase 3 activity as compared to Ct or Cm infection within each cell line (Figure 2).
The ability of Chlamydiae to subvert host cell apoptotic responses could occur through two potential mechanisms, modulation of intrinsic host cell anti-apoptotic responses or through a direct impact on host cell biology via a type three secreted virulence factor. The latter option has been well studied in that Chlamydia post-translationally impact BH3 domain pro-apoptotic proteins even in the presence of CHX (Fan et al., 1998; Fischer et al., 2004; Pirbhai et al., 2006). However, Chlamydia infection can also cause significant changes in host cell transcription (Hess et al., 2001). These changes to host cell biology could be induced through cellular recognition of Chlamydia via pattern recognition receptors (such as TLR2) or Raf/MEK signaling pathways (Su et al., 2003; O'Connell et al., 2006). C. trachomatis has developed measures to manipulate these signaling pathways, such as modulating the host ubiquitination system necessary for NF-kB signaling (Misaghi et al., 2006). NF-kB can upregulate proteins which inhibit apoptosis (IAPs) and are critical for cell survival following Ct infection (Rajalingam et al., 2006). RNAi inhibition of these IAPs lead to a less permissive cellular niche and limited replication of Chlamydia. Additionally, signaling through the Raf/MEK pathway leads to an upregulation of anti-apoptotic protein Mcl1 and Bag-1 (Rajalingam et al., 2008; Kun et al., 2013). When Raf/MEK pathways were inhibited, Chlamydia infected cells were highly susceptible to apoptosis induction. As an alternative to manipulating cell signaling pathways, Chlamydiae could use a type three secretion system (T3SS) to directly inject the host cytosol with a bacterial protein which homes to the eukaryotic nucleus and modifies host cell transcription. Although its function is unknown, CT311 has recently been demonstrated to contain a eukaryotic nuclear localization signal (Lei et al., 2013). It is clear that infection with Chlamydiae induces significant anti-apoptotic gene transcriptional changes in a host cell which can directly impact the viability of the host cell and its resistance to apoptosis induction.
The importance of anti-apoptotic protein production during Chlamydia infection has recently been called into question. Studies with cell lines derived from mice deficient for these IAPs indicate that the proteins are dispensable for the anti-apoptotic effect (Ying et al., 2008). Discrepancies in host cell species (human/mouse) and Chlamydia type were acknowledged by both reports as confounding factors (Rajalingam et al., 2008; Ying et al., 2008). To address the host species component of these incongruous results, we examined the regulation of anti-apoptotic transcripts in various host species cell lines across multiple Chlamydiae pathogens. We identified that a single host could respond differently to each Chlamydiae pathogen (Figure 4). Differences in gene expression between hosts could be due to host species differences or cell type differences. Therefore, inter-host species comparisons are not a focus of this study. In some cases the upregulation of anti-apoptotic protein transcripts required ongoing host translation, indicating a potential positive feedback loop induced by infection. Our transcriptional profile data does support that conclusion in some cases where, in the absence of CHX, anti-apoptotic protein transcripts are significantly augmented (Figures 4D–H). We would surmise that despite the similar outcome of blocked apoptosis, each pathogen species reaches that goal via a potentially different mechanism. Intriguingly, the mouse cell line predominantly upregulated a single anti-apoptotic transcript, cIAP2, which was shown to be dispensable (Ying et al., 2008). This would indicate that, in mice, an alternative method for apoptosis inhibition could be employed.
Rather than rely solely on host cell signaling pathways for the anti-apoptotic effect, evidence indicates that Chlamydiae utilize a T3SS to inject bacterial proteins which directly impact host cell biology. Early evidence indicated that Chlamydia could induce proteolytic cleavage of pro-apoptotic BH3-only proteins (Fischer et al., 2004; Zhong et al., 2006). This proteolysis was attributed to the Chlamydia protease-like activity factor (CPAF) (Pirbhai et al., 2006; Paschen et al., 2008) However, the role of CPAF in this mechanism has been called into doubt following the development of Chlamydia genetically deficient for protease activity (Jorgensen et al., 2011; Snavely et al., 2014). Moreover, this BH3 protein cleavage mechanism was not reproducible (Rajalingam et al., 2008). The study of pro-apoptotic BH3-only protein cleavage would also benefit from an evolutionary approach to examining host-pathogen interactions and address the discrepancies in these reported systems.
Pathogens co-adapt with their hosts throughout evolution to maintain their capacity for growth and dissemination. Similar evolutionary medicine studies examining host specificity in Salmonella infection have helped to identify key virulence factors in host specificity (Bueno et al., 2008; Bäumler and Fang, 2013). For Chlamydiae, the relatively small genomes add significant evolutionary pressure due to gene loss, alternatively termed the “use it or lose it” impact of adaptation (Nunes and Gomes, 2014). These host species-specific adaptations could impair the pathogen's ability to leap across species boundaries and emerge as an infectious disease in a new host. However, data indicate one such emergence has occurred fairly recently in humans with the zoonotic acquisition C. pneumoniae, demonstrating Chlamydiae are a good model for the study of emerging infectious disease (Myers et al., 2009). The evolutionary medicine approach will be highly informative to understanding the barriers for host species-to-species pathogen transmission. The experiments in this report begin to elucidate the potential mechanisms whereby Chlamydiae are able to cross the species boundary and provide a greater understanding of the barriers to emerging infectious disease.
Author Contributions
JM, EN, CF, DG designed, performed, and analyzed the experiments for this manuscript. JM, EN, DG drafted and CF, DG revised the manuscript. All authors agree to be accountable for all aspects of the work herein.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the Ford Fund, Educational Grant Initiative, and Center for Faculty Excellence at Ithaca College for their financial support of this work. The authors would especially like to thank Dr. Ian Woods for assistance with coding the MatLab image processing scripts for image analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fcimb.2015.00096
References
Allan, I., and Pearce, J. H. (1983a). Amino acid requirements of strains of Chlamydia trachomatis and C. psittaci growing in McCoy cells: relationship with clinical syndrome and host origin. J. Gen. Microbiol. 129, 2001–2007. doi: 10.1099/00221287-129-7-2001
Allan, I., and Pearce, J. H. (1983b). Differential amino acid utilization by Chlamydia psittaci (strain guinea pig inclusion conjunctivitis) and its regulatory effect on chlamydial growth. J. Gen. Microbiol. 129, 1991–2000.
Bäumler, A., and Fang, F. C. (2013). Host specificity of bacterial pathogens. Cold Spring Harb. Perspect. Med. 3:a010041. doi: 10.1101/cshperspect.a010041
Benes, S., and McCormack, W. M. (1982). Comparison of methods for cultivation and isolation of Chlamydia trachomatis. J. Clin. Microbiol. 16, 847–50.
Borges, V., Nunes, A., Ferreira, R., Borrego, M. J., and Gomes, J. P. (2012). Directional evolution of chlamydia trachomatis towards niche-specific adaptation. J. Bacteriol. 194, 6143–6153. doi: 10.1128/JB.01291-12
Bueno, S. M., González, P. A., Carreño, L. J., Tobar, J. A., Mora, G. C., Pereda, C. J., et al. (2008). The capacity of Salmonella to survive inside dendritic cells and prevent antigen presentation to T cells is host specific. Immunology 124, 522–533. doi: 10.1111/j.1365-2567.2008.02805.x
Byrne, G. I., and Ojcius, D. M. (2004). Chlamydia and apoptosis: life and death decisions of an intracellular pathogen. Nat. Rev. Microbiol. 2, 802–808. doi: 10.1038/nrmicro1007
Campbell, L. A., and Kuo, C. (2004). Chlamydia pneumoniae – an infectious risk factor for atherosclerosis? Nat. Rev. Microbiol. 2, 23–32. doi: 10.1038/nrmicro796
Coers, J., Gondek, D. C., Olive, A. J., Rohlfing, A., Taylor, G. A., and Starnbach, M. N. (2011). Compensatory T cell responses in IRG-deficient mice prevent sustained Chlamydia trachomatis infections. PLoS Pathog. 7:e1001346. doi: 10.1371/journal.ppat.1001346
Fan, T., Lu, H., Hu, H., Shi, L., McClarty, G. A., Nance, D. M., et al. (1998). Inhibition of apoptosis in chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J. Exp. Med. 187, 487–496. doi: 10.1084/jem.187.4.487
Fischer, S. F., Vier, J., Kirschnek, S., Klos, A., Hess, S., Ying, S., et al. (2004). Chlamydia inhibit host cell apoptosis by degradation of proapoptotic BH3-only proteins. J. Exp. Med. 200, 905–916. doi: 10.1084/jem.20040402
Greene, W., Xiao, Y., Huang, Y., McClarty, G., and Zhong, G. (2003). Chlamydia-infected cells continue to undergo mitosis and resist induction of apoptosis. Infect. Immun. 72, 451–460. doi: 10.1128/IAI.72.1.451-460.2004
Harper, A., Pogson, C. I., Jones, M. L., and Pearce, J. H. (2000). Chlamydial development is adversely affected by minor changes in amino acid supply, blood plasma amino acid levels, and glucose deprivation. Infect. Immun. 68, 1457–1464. doi: 10.1128/IAI.68.3.1457-1464.2000
Hess, S., Rheinheimer, C., Tidow, F., Bartling, G., Kaps, C., Lauber, J., et al. (2001). The reprogrammed host: Chlamydia trachomatis-induced up-regulation of glycoprotein 130 cytokines, transcription factors, and antiapoptotic genes. Arthritis Rheum. 44, 2392–2401. doi: 10.1002/1529-0131(200110)44:10 < 2392::AID-ART404>3.0.CO;2-I
Jorgensen, I., Bednar, M. M., Amin, V., Davis, B. K., Ting, J. P. Y., McCafferty, D. G., et al. (2011). The Chlamydia protease CPAF regulates host and bacterial proteins to maintain pathogen vacuole integrity and promote virulence. Cell Host Microbe 10, 21–32. doi: 10.1016/j.chom.2011.06.008
Kun, D., Xiang-Lin, C., Ming, Z., and Qi, L. (2013). Chlamydia inhibit host cell apoptosis by inducing Bag-1 via the MAPK/ERK survival pathway. Apoptosis 18, 1083–1092. doi: 10.1007/s10495-013-0865-z
Lei, L., Dong, X., Li, Z., and Zhong, G. (2013). Identification of a novel nuclear localization signal sequence in Chlamydia trachomatis-secreted hypothetical protein CT311. PLoS ONE 8:e64529. doi: 10.1371/journal.pone.0064529
Misaghi, S., Balsara, Z. R., Catic, A., Spooner, E., Ploegh, H. L., and Starnbach, M. N. (2006). Chlamydia trachomatis-derived deubiquitinating enzymes in mammalian cells during infection. Mol. Microbiol. 61, 142–150. doi: 10.1111/j.1365-2958.2006.05199.x
Miyairi, I., Mahdi, O. S., Ouellette, S. P., Belland, R. J., and Byrne, G. I. (2006). Different growth rates of Chlamydia trachomatis biovars reflect pathotype. J. Infect. Dis. 194, 350–357. doi: 10.1086/505432
Myers, G. S. A., Mathews, S. A., Eppinger, M., Mitchell, C., O'Brien, K. K., White, O. R., et al. (2009). Evidence that human Chlamydia pneumoniae was zoonotically acquired. J. Bacteriol. 191, 7225–7233. doi: 10.1128/JB.00746-09
Nunes, A., and Gomes, J. P. (2014). Evolution, phylogeny, and molecular epidemiology of Chlamydia. Infect. Genet. Evol. 23, 49–64. doi: 10.1016/j.meegid.2014.01.029
Nunes, A., Nogueira, P. J., Borrego, M. J., and Gomes, J. P. (2008). Chlamydia trachomatis diversity viewed as a tissue-specific coevolutionary arms race. Genome Biol. 9:R153. doi: 10.1186/gb-2008-9-10-r153
O'Connell, C. M., Ionova, I. A., Quayle, A. J., Visintin, A., and Ingalls, R. R. (2006). Localization of TLR2 and MyD88 to Chlamydia trachomatis inclusions. evidence for signaling by intracellular TLR2 during infection with an obligate intracellular pathogen. J. Biol. Chem. 281, 1652–1659. doi: 10.1074/jbc.M510182200
Olive, A. J., Haff, M. G., Emanuele, M. J., Sack, L. M., Barker, J. R., Elledge, S. J., et al. (2014). Chlamydia trachomatis-induced alterations in the host cell proteome are required for intracellular growth. Cell Host Microbe 15, 113–124. doi: 10.1016/j.chom.2013.12.009
Ouellette, S. P., Dorsey, F. C., Moshiach, S., Cleveland, J. L., and Carabeo, R. A. (2011). Chlamydia species-dependent differences in the growth requirement for lysosomes. PLoS ONE 6:e16783. doi: 10.1371/journal.pone.0016783
Paschen, S. A., Christian, J. G., Vier, J., Schmidt, F., Walch, A., Ojcius, D. M., et al. (2008). Cytopathicity of Chlamydia is largely reproduced by expression of a single chlamydial protease. J. Cell Biol. 182, 117–127. doi: 10.1083/jcb.200804023
Pirbhai, M., Dong, F., Zhong, Y., Pan, K. Z., and Zhong, G. (2006). The secreted protease factor CPAF is responsible for degrading pro-apoptotic BH3-only proteins in Chlamydia trachomatis-infected cells. J. Biol. Chem. 281, 31495–31501. doi: 10.1074/jbc.M602796200
Rajalingam, K., Sharma, M., Lohmann, C., Oswald, M., Thieck, O., Froelich, C. J., et al. (2008). Mcl-1 is a key regulator of apoptosis resistance in Chlamydia trachomatis-infected cells. PLoS ONE 3:e3102. doi: 10.1371/journal.pone.0003102
Rajalingam, K., Sharma, M., Paland, N., Hurwitz, R., Thieck, O., Oswald, M., et al. (2006). IAP-IAP complexes required for apoptosis resistance of C. trachomatis-infected cells. PLoS Pathog. 2:e114. doi: 10.1371/journal.ppat.0020114
Ripa, K. T., and Mårdh, P. A. (1977). Cultivation of Chlamydia trachomatis in cycloheximide-treated mccoy cells. J. Clin. Microbiol. 6, 328–231.
Sabet, S. F., Simmons, J., and Caldwell, H. D. (1984). Enhancement of Chlamydia trachomatis infectious progeny by cultivation of HeLa 229 cells treated with DEAE-dextran and cycloheximide. J. Clin. Microbiol. 20, 217–222.
Snavely, E. A., Kokes, M., Dunn, J. D., Saka, H. A., Nguyen, B. D., Bastidas, R. J., et al. (2014). Reassessing the role of the secreted protease CPAF in Chlamydia trachomatis infection through genetic approaches. Pathog. Dis. 71, 336–351. doi: 10.1111/2049-632X.12179
Starnbach, M. N., and Roan, N. R. (2008). Conquering sexually transmitted diseases. Nat. Rev. Immunol. 8, 313–317. doi: 10.1038/nri2272
Su, H., McClarty, G., Dong, F., Hatch, G. M., Pan, Z. K., and Zhong, G. (2003). Activation of Raf/MEK/ERK/cPLA2 signaling pathway is essential for chlamydial acquisition of host glycerophospholipids. J. Biol. Chem. 279, 9409–9416. doi: 10.1074/jbc.M312008200
Wahl, C., Maier, S., Marre, R., and Essig, A. (2003). Chlamydia pneumoniae induces the expression of inhibitor of apoptosis 2 (c-IAP2) in a human monocytic cell line by an NF-kappaB-dependent pathway. Int. J. Med. Microbiol. 293, 377–381. doi: 10.1078/1438-4221-00274
Xiao, Y., Zhong, Y., Greene, W., Dong, F., and Zhong, G. (2004). Chlamydia trachomatis infection inhibits both Bax and Bak activation induced by staurosporine. Infect. Immun. 72, 5470–5474. doi: 10.1128/IAI.72.9.5470-5474.2004
Ying, S., Christian, J. G., Paschen, S. A., and Häcker, G. (2008). Chlamydia trachomatis can protect host cells against apoptosis in the absence of cellular Inhibitor of Apoptosis Proteins and Mcl-1. Microbes Infect. 10, 97–101. doi: 10.1016/j.micinf.2007.10.005
Keywords: apoptosis, Chlamydia, evolution, emerging disease, transcription
Citation: Messinger JE, Nelton E, Feeney C and Gondek DC (2015) Chlamydia Infection Across Host Species Boundaries Promotes Distinct Sets of Transcribed Anti-Apoptotic Factors. Front. Cell. Infect. Microbiol. 5:96. doi: 10.3389/fcimb.2015.00096
Received: 14 July 2015; Accepted: 07 December 2015;
Published: 23 December 2015.
Edited by:
Brice Rotureau, Institut Pasteur, FranceReviewed by:
Rey Carabeo, Washington State University, USAIsabel Sada, National Institute of Respiratory Diseases, Mexico
Copyright © 2015 Messinger, Nelton, Feeney and Gondek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David C. Gondek, ZGdvbmRla0BpdGhhY2EuZWR1