 Xinmiao Jia1,2†Li Yang1,2†Mengxing Dong1,2†Suting Chen3†
Xinmiao Jia1,2†Li Yang1,2†Mengxing Dong1,2†Suting Chen3† Lingna Lv3†Dandan Cao1Jing Fu1,2Tingting Yang1,2Ju Zhang1Xiangli Zhang1,2Yuanyuan Shang3Guirong Wang3Yongjie Sheng4Hairong Huang3*
Lingna Lv3†Dandan Cao1Jing Fu1,2Tingting Yang1,2Ju Zhang1Xiangli Zhang1,2Yuanyuan Shang3Guirong Wang3Yongjie Sheng4Hairong Huang3* Fei Chen1,2,5,6*
Fei Chen1,2,5,6*- 1CAS Key Laboratory of Genome Sciences & Information, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China
- 2College of Life Sciences, University of Chinese Academy of Sciences, Beijing, China
- 3National Clinical Laboratory on Tuberculosis, Beijing Key Laboratory on Drug-resistant Tuberculosis Research, Beijing Chest Hospital, Capital Medical University, Beijing Tuberculosis and Thoracic Tumor Institute, Beijing, China
- 4Key Laboratory for Molecular Enzymology and Engineering of Ministry of Education, Jilin University, Changchun, China
- 5Sino-Danish College, University of Chinese Academy of Sciences, Beijing, China
- 6Collaborative Innovation Center for Genetics and Development, Shanghai, China
Tuberculosis now exceeds HIV as the top infectious disease cause of mortality, and is caused by the Mycobacterium tuberculosis complex (MTBC). MTBC strains have highly conserved genome sequences (similarity >99%) but dramatically different phenotypes. To analyze the relationship between genotype and phenotype, we conducted the comparative genomic analysis on 12 MTBC strains representing different lineages (i.e., Mycobacterium bovis; M. bovis BCG; M. microti; M. africanum; M. tuberculosis H37Rv; M. tuberculosis H37Ra, and six M. tuberculosis clinical isolates). The analysis focused on the three aspects of pathogenicity: host association, virulence, and epitope variations. Host association analysis indicated that eight mce3 genes, two enoyl-CoA hydratases, and five PE/PPE family genes were present only in human isolates; these may have roles in host-pathogen interactions. There were 15 SNPs found on virulence factors (including five SNPs in three ESX secretion proteins) only in the Beijing strains, which might be related to their more virulent phenotype. A comparison between the virulent H37Rv and non-virulent H37Ra strains revealed three SNPs that were likely associated with the virulence attenuation of H37Ra: S219L (PhoP), A219E (MazG) and a newly identified I228M (EspK). Additionally, a comparison of animal-associated MTBC strains showed that the deletion of the first four genes (i.e., pe35, ppe68, esxB, esxA), rather than all eight genes of RD1, might play a central role in the virulence attenuation of animal isolates. Finally, by comparing epitopes among MTBC strains, we found that four epitopes were lost only in the Beijing strains; this may render them better capable of evading the human immune system, leading to enhanced virulence. Overall, our comparative genomic analysis of MTBC strains reveals the relationship between the highly conserved genotypes and the diverse phenotypes of MTBC, provides insight into pathogenic mechanisms, and facilitates the development of potential molecular targets for the prevention and treatment of tuberculosis.
Introduction
According to a 2016 World Health Organization (WHO) report, tuberculosis (TB) has surpassed HIV as the infectious disease causing the highest number of mortalities, with an estimated 1.8 million deaths and 10.4 million new TB cases worldwide in 2015 (World Health Organization, 2016). The primary cause of TB, Mycobacterium tuberculosis (Mtb), belongs to the M. tuberculosis complex (MTBC). MTBC is a genetically related group of Mycobacterium species that can cause TB in humans or other organisms (Galagan, 2014). In general, MTBC can be classified into eight main lineages (Lineage 1-8, L1-8): L1 (The Philippines and Indian Ocean), L2 (East Asia), L3 (India and East Africa), L4 (Europe and Americas), L5 (West Africa 1), L6 (West Africa 2), L7 (Ethiopia), and L8 (animal-adapted isolates). Among these, L1, L2, L3, L4, and L7 comprise Mtb. MTBC can also be divided into ancient (L1, L5-8) and modern strains (L2-4) according to the presence or absence of an M. tuberculosis specific deletion (TbD1; Brosch et al., 2002; Galagan, 2014).
After the first published genome of M. tuberculosis in 1998 (Cole et al., 1998), there were many large-scale genomic studies involving many MTBC strains (>3,500) (http://www.ncbi.nlm.nih.gov/genome/genomes). Analysis of worldwide sequencing data shows that MTBC members share more than 99% identity at the nucleotide level (Brosch et al., 2000; Rodriguez-Campos et al., 2014). Also, large genomic rearrangements are infrequent in MTBC strains (Galagan, 2014).
Although MTBC members have almost identical genome sequences/genotypes (similarity >99%) (Brosch et al., 2000), they exhibit diverse pathogenic phenotypes, which are thought to be the result of long-term co-evolution of MTBC strains with a diverse group of humans and animals (Comas et al., 2013; Galagan, 2014). Long-term geographical isolation gave rise to the accumulation of “genotype isolation,” thus resulting in different phenotypes (Ghebremichael et al., 2010).
Firstly, the MTBC members have diverse host associations. The L1 to L7 lineages mainly infect humans and rarely infect animals. Among these, the L5 and L6 lineages (M. africanum I and II) only cause human TB in West Africa (Bentley et al., 2012; Winglee et al., 2016), and the L7 lineage has only been reported in Ethiopia or in Ethiopian emigrants located in Djibouti (Firdessa et al., 2013). The other human-associated MTBC lineages (L1-4) also exhibit relatively high levels of distinct geographic distributions (Galagan, 2014). These findings suggest that the MTBC lineages have coevolved with diverse hominid ancestors since ancient times, which has lead to their diverse adaptations to specific human populations (Gagneux et al., 2006; Comas et al., 2013; Galagan, 2014). In addition, the L8 lineage (consisting of different animal-adapted MTBC isolates: M. bovis, M. bovis BCG, and M. microti, among others) has diverse animal-host adaptations, likely for the same reason (Brosch et al., 2002; Rodriguez-Campos et al., 2014). M. bovis rarely causes TB in humans but it is highly virulent for cattle (Brosch et al., 2000). Similarly, M. microti has been reported to mainly infect rodents, such as voles, and rarely infects humans (Brosch et al., 2000).
Secondly, MTBC strains have different levels of virulence [i.e., the degree of pathogenicity or the ability of the organism to invade the tissues of the host (Pirofski and Casadevall, 2012)]. The modern strains (L2-L4) are more virulent to humans than the ancient strains (L1; L5-8), and are responsible for the vast majority of today's TB cases (Brosch et al., 2002; Gagneux, 2012). Among these, the Beijing sub-lineage of the L2 lineage appears to be more virulent than the other modern isolates due to its enhanced resistance and adaptation (Ida et al., 2010).
The virulence of MTBC strains is not set in stone. For example, M. tuberculosis H37Ra, isolated by William Steenken in 1935 (Steenken and Gardner, 1946), is usually regarded as the non-virulent counterpart of the virulent H37Rv strain. Historically, both M. tuberculosis H37Ra and H37Rv are derived from the same parent strain (H37). H37Ra and H37Rv are the most common standard strains of M. tuberculosis in laboratories. Another famous example is M. bovis BCG, which was isolated from virulent M. bovis after 239 passages (Liu et al., 2009); this is the most widely used TB vaccine in the world.
Thirdly, some studies have shown a different immune response to different MTBC strains (Gagneux, 2012). It is reported that hypervirulent strains induce lower immune responses than low-pathogenic strains (Portevin et al., 2011). The distinct capacity of virulent strains to remain sequestered in host macrophages seems to be a reason for their ability to evade immune responses (Chen et al., 2006; Grace and Ernst, 2016).
In recent years, some genomic studies concerning the relationship between the genotype and phenotype of MTBC strains have been reported. A number of mycobacterial virulence genes (i.e., virulence factors) have been identified; most of these encode for cell surface proteins or lipid and fatty acid metabolism proteins (Forrellad et al., 2013). Some comparative genomic analyses between the virulent H37Rv and the non-virulent H37Ra strains revealed that the mutation of Ser219 into leucine of the PhoP virulence factor might result in the attenuation of virulence in H37Ra (Ryndak et al., 2008; Zheng et al., 2008). In addition, it is also known that the deletion of region of difference 1 (RD1) in the genome of M. bovis BCG led to its attenuation (based on a comparison with M. bovis) (Pym et al., 2002; Lewis et al., 2003).
To date, many of the comparative genomic studies only have been focused on the comparison between virulent and non-virulent MTBC strains; thus, comparative genomic studies covering the host association, virulence, and epitope variations are still lacking. This is mainly due to the fact that, on one hand, precise comparisons and analyses of almost identical MTBC genome sequences (similarity >99%) are needed to obtain precise and complete MTBC genome sequences; meanwhile, on the other hand, it is very difficult to obtain complete MTBC genome sequences using second generation sequencing technologies, such as the Illumina Hiseq platform, due to the high GC content and repetitive sequences of the PE/PPE multi-gene family.
Herein, the 12 MTBC genomes, we finished using Pacbio single-molecule real-time (SMRT) technology (Zhu et al., 2016), provided a groundwork for analyzing the relationship between the genotype and phenotype of MTBC. To obtain a precise analysis, we re-sequenced the genomes of 12 MTBC strains using an Illumina Hiseq. Based on the 12 precise and complete MTBC genomes, we performed a comparative genomic analysis looking at three aspects of pathogenicity: host association, virulence, and epitope diversity.
Materials and Methods
Isolates and Genotyping
The 12 MTBC isolates, which included six reference strains and six clinical isolates, have been previously described (Table S1; Zhu et al., 2016). Six standard reference strains were obtained from American Type Culture Collection (ATCC) specifically for the purpose of genome sequencing. The strains were grown in either Lowenstein–Jenden media or Middlebrook 7H10 media supplemented with 10% OADC (Oleic Albumin Dextrose Catalase, Becton Dickinson), glycerol, and 0.05% Tween 80. We used the VNTR-15 scheme as described in MIRU-VNTRplus (http://www.miru-vntrplus.org/) for genotyping, which uses the following markers: Mtub04; ETRC; MIRU04; MIRU40; MIRU10; MIRU16; Mtub21; QUB11b; ETRA; Mtub30; MIRU26; MIRU31; Mtub39; QUB26, and QUB4156. Each MIRU-VNTR locus was individually amplified, and electrophoresis of products on agarose gels was conducted as previously described (Fabre et al., 2004). The copy number at each locus was calculated in BioNumerics. A large sequence polymorphism (LSP) (pks15/1) was used to differentiate between the clinical TB strains using the primers shown in Table S2. A deletion of a 7-bp region in the polyketide synthase gene pks15/1 is present in the Euro-American lineage of M. tuberculosis. Whole genome SNP typing was also done using MEGA 6.06 (maximum likelihood method) as previously described (Tamura et al., 2013; Zhu et al., 2016). The results of this analysis are consistent with those of the VNTR analysis (Figure S1).
The 12 MTBC strains covered five lineages. There were two clinical L2 strains (Mtb 2242 and 2279), one clinical L3 strain (Mtb 26105), five L4 strains (three clinical strains-Mtb 22115, Mtb 22103, and Mtb 37004 and two reference strains-Mtb F1 and Mtb F28), one L6 reference strain (M. africanum 25), and three L8 reference strains (M. microti 12, M. bovis 30, and M. bovis BCG 26). Of the 12 strains, Mtb 2242, Mtb 2279, Mtb 26105, Mtb 22115, Mtb 22103, Mtb 37004, Mtb F1, Mtb F28, and M. africanum 25 are human isolates; meanwhile, M. microti 12, M. bovis 30, and M. bovis BCG 26 are animal isolates. In addition, the 12 MTBC isolates contained two homologous pairs of virulent/non-virulent strains: virulent M. tuberculosis H37Rv (Mtb F1: ATCC27294) and its non-virulent counterpart H37Ra (Mtb F28: ATCC25177), and virulent M. bovis (M. bovis 30: ATCC19210) and its non-virulent counterpart M. bovis BCG (M. bovis BCG 26: ATCC35735).
Genomic DNA Extraction, Sequencing, Correction and Re-Annotation
Genomic DNA from the 12 MTBC strains was extracted using a TIANamp Bacteria Genomic DNA Kit (Tiangen BiotechCo. Ltd., Beijing, China), and was sequenced using PacBio Single-Molecule Real-Time (SMRT) Technology as previously described (Zhu et al., 2016).
To correct the polymer errors produced during PacBio sequencing, we re-sequenced the 12 isolates using next-generation sequencing. The genomes of the 12 isolates were shotgun sequenced using an Illumina Genome Analyzer 2X. Paired-end libraries were prepared from 5 ug of isolated genomic DNA using the TruSeq DNA sample prep kit A (Illumina Inc., San Diego) according to the manufacturer's instructions. Genomic paired-end libraries were sequenced with a read length of 2 × 150 nucleotides using an Illumina GAIIx instrument according to the manufacturer's instructions. Image analysis and base calling were done in the standard Illumina pipeline. The raw Illumina sequencing reads were trimmed at a threshold of 0.01 (Phred score of 20). Filtered reads were mapped onto the genome sequences, which were assembled by the Hierarchical Genome Assembly Process (HGAP.3) algorithm in SMRT Portal (version 2.2.0) using BWA version 0.5.9 (Li and Durbin, 2010), and converted to sorted BAM format using SAMtools v0.1.9 (Li et al., 2009). The coverage ranged between 157× and 394× with an average of 255×. Pilon v1.13 (Walker et al., 2014) was then used to polish the genome sequences using these alignments, which resulted in a total of 9,493 insertions and 133 deletions. All the raw Illumina sequencing reads have also been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database (SRP064893) and the Genome Sequence Archive (GSA) of the BIG Data Center (BIGD) (PRJCA000307).
All 12 genome sequences were re-annotated with the Rapid Annotation tool in the Subsystem Technology (RAST) pipeline, which is a fully automated annotation engine for complete or draft archaeal and bacterial genomes (Aziz et al., 2008).
Genome Structure and Identification of Specific Genes and Single-Nucleotide Polymorphisms (SNPs)
The average nucleotide identity (ANI) was calculated through ANI on EzGenome (http://www.ezbiocloud.net/tools/ani). Multiple alignments of genomic sequences were performed using the Mauve multiple alignment software with the progressive alignment option (Darling et al., 2010). The output file produced by Mauve was parsed using a custom Perl script (Supplementary Data) to retrieve multiple aligned sequences for SNP loci. SNPs called in repetitive regions of the genome, which were defined as exact repetitive sequences ≥25 bp in length, and were identified using Repeat-Masker (Tarailo-Graovac and Chen, 2009), were excluded.
Gene sequences were downloaded from the RAST server after annotation. To identify specific genes, ortholog clustering was performed using orthoMCL (Li et al., 2003). Each cluster is a homology group. If the genes in a homology group covered all species in a group but no strains in other groups, we considered the genes in this group to be group specific genes.
Epitopes, Virulence Factors and PE/PPE Gene Family Distribution
Epitopes were obtained from the Immune Epitope Database (IEDB; Vita et al., 2015), and those that had been positively experimentally identified were selected and renamed. Only epitopes with 100% identical BLAT matches were considered to be the same epitope. The epitope was classified as an absence epitope if it was located in an absence gene or in a deleted/mutated region of a non-absence gene. Epitopes with the same distribution pattern were clustered into one epitope_cluster. Furthermore, BLASTn was used to search for differences between 12 genomes in terms of single nucleotide variations, insertions, and deletions in corresponding antigen genes.
Virulence genes were downloaded from the Virulence Factor Database (VFDB; Chen et al., 2005; Table S3). All of the PE/PPE genes analyzed in this study (Table S4) were based on a search of the M. tuberculosis H37Rv genes (NC_000962) from the NCBI gene database (www.ncbi.nlm.nih.gov/gene). FASTA sequences of these genes were used to search for corresponding genes in the 12 genomes using BLAST (50% coverage and 90% identity thresholds).
Results and Discussion
Conserved Genomes (Genotypes) and Diverse Phenotypes of 12 MTBC Strains
The MTBC genomes obtained by SMRT sequencing (Zhu et al., 2016) were re-sequenced to correct the homo-polymer errors using Illumina sequencing. Based on an average of 5,394,126 paired-end reads (255X coverage) (SRP064893), we resolved a total of 9,493 insertions and 133 deletions compared with the original genomes (Table S5). The precise genomes provided general information (Table 1), including the size of the genomes (4.34–4.43 Mb), the number of predicted protein-coding genes (~4,400), and the gene length (~900bp). Importantly, the ANI and SNP analyses showed that the MTBC genomes were highly conserved, as the maximum number of SNPs and the minimum ANI were 2,356 and 99.75%, respectively (Table 1). Pan-genomic analysis was further implemented and used to identified 3,761 core gene clusters (Figure S2A), which accounted for ~87% of all genes. This also demonstrates that MTBC genomes are highly conserved.
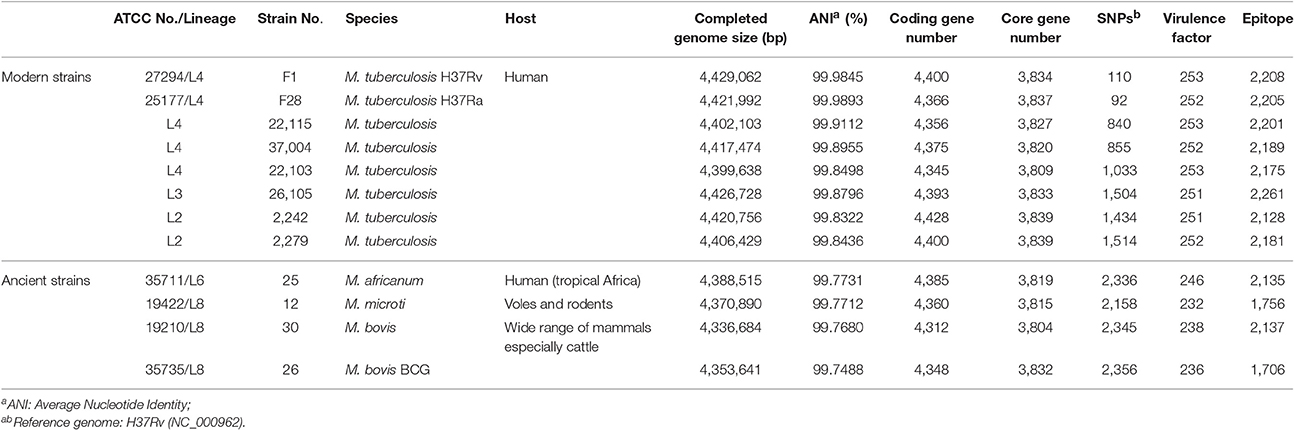
Table 1. Conserved genomes (genotypes) and diverse phenotypes of 12 MTBC strains.
The comparative genomic analysis also showed some subtle differences amongst the 12 MTBC genomes: the five L4 strains had fewer SNPs (<1,000) because the reference genome (NC_000962) belonged to the L4 lineage; the three animal-associated MTBC strains possessed more SNPs (>2,000), and the two L2 and one L3 strains all had ~1,500 SNPs (Table 1). The number of SNPs reflected the genetic distance from the reference L4 strain (Takezaki and Nei, 1996). To lower the influence of the reference, pair-wise comparisons of SNPs were conducted (Figure 1). This comparison proved that the differences in SNPs between the animal- and human-associated strains were much greater than those between intra-lineage strains. On the other hand, the analyses for the pan-genomic and pair-wise comparisons of orthologous genes (Figure S2 and Figure 1, respectively) indicated some strain-specific and lineage-specific genes.
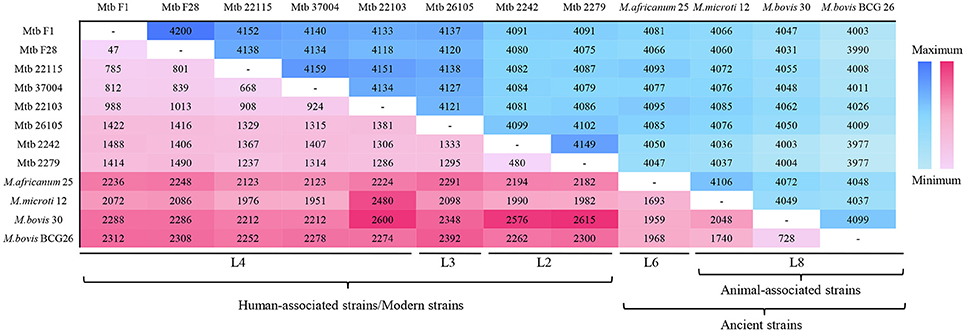
Figure 1. Pair-wise comparisons of SNPs and orthologous genes in 12 MTBC strains. The plum red region represents the SNP number, and the blue region indicates the orthologous gene number. The colors darken with increases in number.
It is well-known that genotype is a major factor influencing phenotype. Thus, the pathogenic phenotype differences amongst MTBC lineages can also be attributed to the small number of genomic variations, including the specific genes and SNPs described above. In the following sections, we investigate the correlation between these genetic variations and three essential phenotypic features (host association, virulence, and epitope variation).
Host Association Analysis
In an attempt to reveal the genetic basis for host association, we performed a comparative genomic study between the strains isolated from human (i.e., Mtb 2242, Mtb 2279, Mtb 26105, Mtb 22115, Mtb 22103, Mtb 37004, Mtb F1, and Mtb F28) and animal (i.e., M. microti 12, M. bovis 30, and M.bovis BCG 26). The average ANI was 99.76% (99.71–99.80%; Table S6), and no large genome structure variation was observed. Minor differences in genome sequences between these two groups of strains might explain the differences in host association. We found 29 genes specific to human isolates (Table 2), 16 genes specific to animal isolates (Table S7), and 579 SNPs (Table S8).
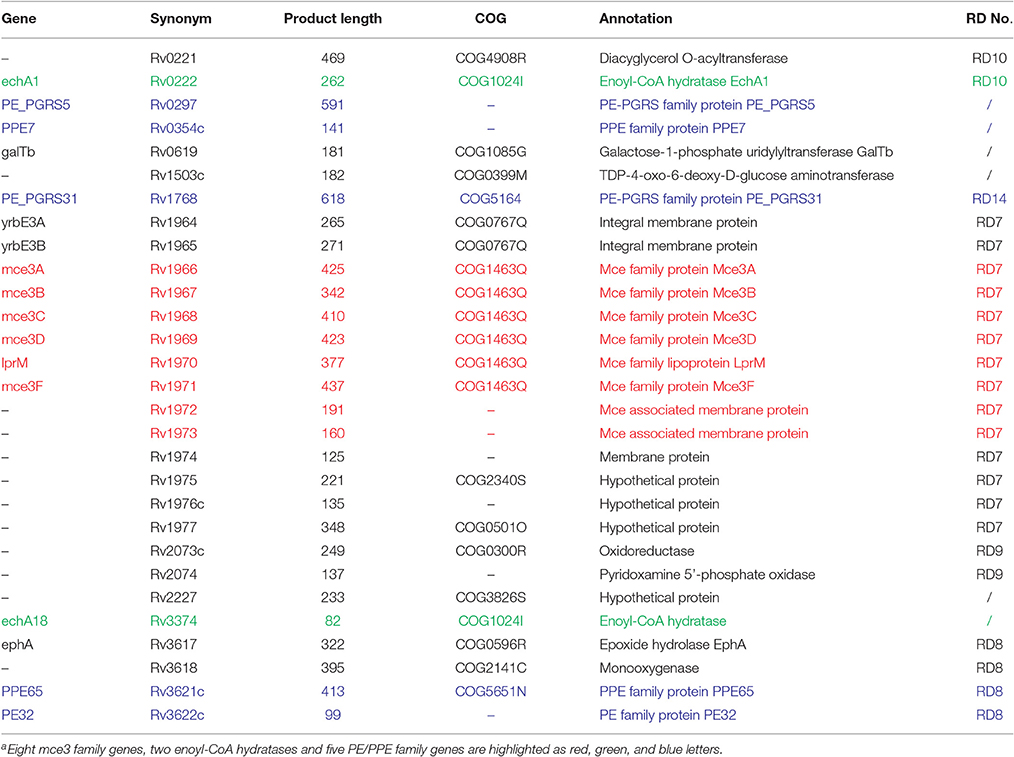
Table 2. Human-associated strain specific genesa [Reference genome: H37Rv (NC_000962)].
Interestingly, in the 29 genes specific to human isolates, 14 in region of difference 7 (RD7; Table 2) were clustered together (Figure S3). Among these, eight mammalian cell entry 3 (mce3) family genes appear to play roles in human host association of MTBC strains, since the Mce protein is an important protein family for entry and survival of bacteria in the host (Harboe et al., 2002; Ahmad et al., 2004). They are located on the cell surface and reside in the same mce3 operon of M. tuberculosis (Harboe et al., 2002). Additionally, the two genes upstream (yrbEA and yrbEB) from the eight mce3 genes and the four genes downstream (Rv1974, Rv1975, Rv1976c, and Rv1977) from the eight mce3 genes encode integral membrane proteins and signal sequences, respectively (Harboe et al., 2002). Their absence in animal isolates might be related to host association.
It is also worth noting that two enoyl-coenzyme A (CoA) hydratases (EchA1 and EchA18) were only present in human isolates (Table 2); these hydratases are key enzymes in the fatty acid β-oxidation pathway. Through β-oxidative reactions, host-cell lipids can be degraded and provide precursors for many metabolic processes, such as cell-wall synthesis (Cole et al., 1998). Therefore, the absence of enoyl-CoA hydratase in animal-associated MTBC strains might impact fatty acid metabolism and cell-wall synthesis in host cells.
In addition, five PE/PPE family genes (i.e., ppe7, ppe65, pe32, pe_pgrs5, and pe_pgrs31) were found to exist only in the human isolates (Table 2). Most of the PE/PPE proteins were localized or secreted to the cell surface; these proteins have been implicated in mycobacterial antigenic variation and host immune evasion (Akhter et al., 2012). Thus, the absence of these five PE/PPE proteins in animal isolates might lead to differences in host association.
Of the 29 genes specific to human isolates, ppe65, echA1, ephA, and Rv1977 are known antigen genes (Vita et al., 2015). The loss of these four genes might lead to changes in the immune response of animal isolates (discussed in the epitope section).
Five hundred and Seventy nine SNPs were identified between the human- and animal-isolates; 315 non-synonymous SNPs were distributed on 287 genes (Table S8A). Among them, 26 SNPs were on virulence factors and seven SNPs were located on antigen genes (Table S8B). It is likely that they influence protein function and further affect mycobacterial host association.
Genomic Analysis of Virulence Differences
Diverse MTBC lineages have different degrees of virulence (Brosch et al., 2002; Ida et al., 2010; Gagneux, 2012) even though their genomes are highly conserved. In this study, we searched for virulence factors in the 12 MTBC genomes by performing a BLAST search against 257 virulence factors collected from the VFDB (Chen et al., 2005; Table S3). Among these, 86 are experimentally validated (Table S3A), while the others are putative ones (Table S3B). The results showed that more than 80% (216: 56 experimentally validated and 160 putative) of the virulence factors were existed and conserved in all 12 strains (Figure S2B). Also, we found that the number of virulence factors in the four ancient strains was less than in the eight modern strains, which is in agreement with the more virulent phenotype of modern strains (Gagneux, 2012).
It is well known that Beijing sub-lineage strains in the L2 lineage are more virulent than the other modern isolates (Ida et al., 2010). There was no appreciable difference between Beijing sub-lineage strains (Mtb 2242 and Mtb 2279) and other modern strains (Mtb F1, Mtb F28, Mtb 22115, Mtb 37004, Mtb 22103, and Mtb 26105) in regards to the number and species of virulence factors (Table 1). We further investigated the SNPs between the Beijing and other modern strains, and 18 non-synonymous SNPs were identified (Table 3). They were then checked by performing a BLAST search against ten other Beijing strains with complete genomes from NCBI GenBank (Table S9A). 15 SNPs on 13 virulence factors (five validated and eight putative) were confirmed to exist in the 12 Beijing strains. Most noteworthy were the five SNPs located on the ESX secretion systems, which are very important for M. tuberculosis pathogenesis (Simeone et al., 2015).
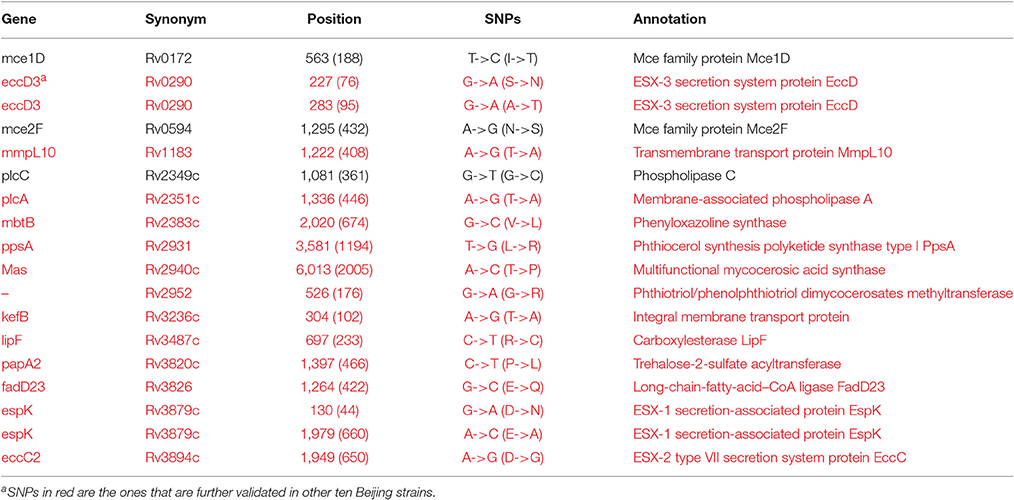
Table 3. The non-synonymous mutations between Beijing strains and other MTBC strains on virulence factors.
The two homologous pairs of virulent/non-virulent strains (i.e., H37Rv vs. H37Ra and M. bovis vs. M. bovis BCG) are well suited for virulence genetic analysis because they have the same origins (Steenken and Gardner, 1946; Liu et al., 2009). Thus, we conducted a comparative genomic analysis between virulent and non-virulent strains at the whole genome level (not limited to virulence factors).
H37Rv vs. H37Ra
H37Ra and H37Rv show very different virulence even though they share an origin. To investigate the relationship between the phenotypic differences and genetic variations, we implemented a comparative genomic study between the five H37Rv (including four online complete genomes from NCBI GenBank and Mtb H37Rv F1 genome) and two H37Ra genomes (including one online complete genome from NCBI GenBank and Mtb H37Ra F28 genome; Table S9B). ANI analysis revealed 99.98% identity between the H37Rv and H37Ra strains. No large genome structure variation was found. Four specific genes in all H37Ra strains and five missense SNPs were identified.
The four H37Ra specific genes were clustered together and located in RvD2 (H37Rv-related deletion 2) with two IS6110 transposase genes on both ends (Figure S4). Further, comparison with the other three L4 strains showed that the four genes also existed in L4 virulent strains except H37Rv; this indicates that they are not relevant to the virulence of H37Rv (Figure S4). We inferred that the four genes might have been lost through IS6110 in the long-term passage of H37Rv.
Five missense SNPs were identified between the H37Rv and H37Ra strains (Table 4). Through comparison with our six clinical virulent strains, two SNPs were eliminated. Of the other three SNPs, two of them (on PhoP and MazG) have been reported and implicated in H37Rv virulence (Ryndak et al., 2008; Lu et al., 2010). We identified a new mutation, C684G (I228M), on the validated virulence factor gene espK in H37Ra strains (Table 4). EspK is an ESX-1 secretion-associated protein that is required for virulence, growth in macrophages, and suppression of macrophage inflammatory and immune response (McLaughlin et al., 2007). Thus, this SNP might be involved in the attenuation of virulence in H37Ra.
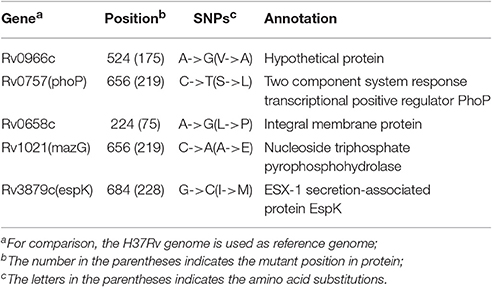
Table 4. The SNPs between non-virulent H37Ra and virulent H37Rv.
M. bovis vs. M. bovis BCG
To explore the genetic differences between M. bovis and M. bovis BCG, we performed a comparative genomic analysis between three M. bovis (including two online complete genomes from NCBI GenBank and M. bovis 30 genome) and nine M. bovis BCG genomes (including eight online complete genomes from NCBI GenBank and M. bovis BCG 26 genome; Table S9C). ANI analysis showed a 99.90% identity between M. bovis and M. bovis BCG strains. Here, we identified 28 M. bovis specific genes (Table S10A), six M. bovis BCG specific genes (Table S10B), and 308 missense SNPs (Table S11).
Eight of 28 M. bovis specific genes (i.e., pe35, ppe68, esxB, esxA, espI, eccD1, espJ, and espK) were clustered together and composed RD1 (Sreejit et al., 2014). Absence of RD1 in M. bovis BCG strains was reported to lead to attenuation of virulence (Pym et al., 2002). We further analyzed the distribution of these 8 genes in our 12 MTBC strains and found that only the first four genes (i.e., pe35, ppe68, esxB, and esxA) were absent in M. microti 12 (Figure 2). Since M. microti is a non-virulent strain, which seldom causes disease in immunocompetent individuals (Rodriguez-Campos et al., 2014), we deduced that the first four genes of RD1 (rather than RD1) might play a key role in attenuating the virulence of M. bovis BCG and M. microti strains. In addition, 13 phiRV1 phage proteins were also identified to be absent in M. bovis BCG strains. They were clustered together and composed RD3 (Table S10A). It was reported that they could sense oxygen status within the host; thus, their presence could help M. bovis adapt to environmental stress (Fan et al., 2016).
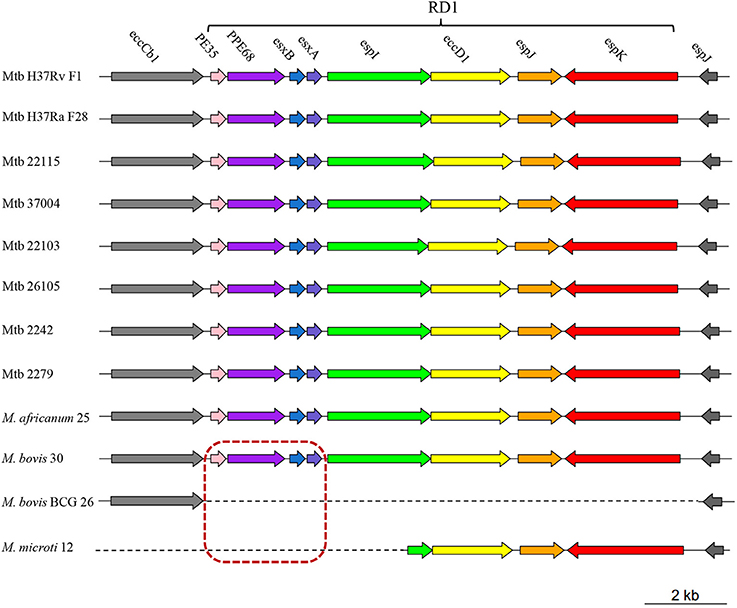
Figure 2. A schematic diagram showing the RD1 distribution in 12 MTBC strains. Different colors represent different genes on RD1 (PE35, pink; PPE68, purple; esxB, blue; esxA, slate blue; espI, green; eccD1, yellow; espJ, orange; espK, red). Genes in the red dashed box indicate the four lost genes in the attenuated M. bovis BCG 26 and M. microti 12 strains. Genes upstream (eccCb1) and downstream (espJ) of RD1 are shown in gray.
Three hundred and eight missense SNPs (on 251 genes) were identified between M. bovis and M. bovis BCG (Table S11A). Further Clusters of Orthologous Group (COG) functional analysis indicated that 19 genes (19 missense SNPs) displayed a relative enrichment in COG category I (Lipid transport and metabolism; Figure S5). MTBC strains exhibited lipid enrichment on their cell surfaces, which is reported to play a role in pathogenesis (Cole et al., 1998). Importantly, 21 of the nonsynonymous SNPs were located on 21 virulence factors (four validated and 17 putative; Table S11B). These SNPs appear to be associated with the attenuation of virulence in M. bovis BCG strains. At last, we need to emphasize that we are particularly careful about the conservation of the standard strains (obtained from ATCC), and they had not been subject to repeated in vitro cultivation and belong to the first generations of the original strains. Even so, the animal studies were not carried out due to the limitation of objective condition, all the arguments on the virulence is a speculation that need further validation.
Antigen Epitope Variations
An epitope is the part of an antigen that is recognized by the immune system, and plays a core role in immune response (https://en.wikipedia.org/wiki/Epitope). To date, epitope studies are confined to T-cells in M. tuberculosis (Comas et al., 2010; Copin et al., 2014; Lindestam Arlehamn et al., 2015). However, comparative analysis of epitopes in different lineages of MTBC strains, including T-cell and B-cell epitopes, is still lacking. In this study, we analyzed the diversity of T-cell and B-cell epitopes in 12 MTBC strains, which serves to enhance our understanding of immune response differences in diverse MTBC strains.
A total of 2,245 MTBC epitopes were downloaded from the IEDB (Vita et al., 2015), including 1,755 T-cell epitopes and 490 B-cell epitopes (Table S12). Pan-genomic analysis indicated that 1,522 epitopes were conserved in all 12 MTBC strains (Figure S2C). Among these, 1,428 epitopes possessed the same copy number.
Despite sharing the majority of both T-cell and B-cell epitopes, the 12 MTBC strains still had some differences. Two non-virulent animal-origin MTBC strains, M. bovis BCG and M. microti, had significantly less T-cell and B-cell epitopes, suggesting that these lost epitopes might be related to the non-virulence, since more than 50% of the lost epitopes (164 of 326) were located in the virulence factors (Figure 3, Table S13).
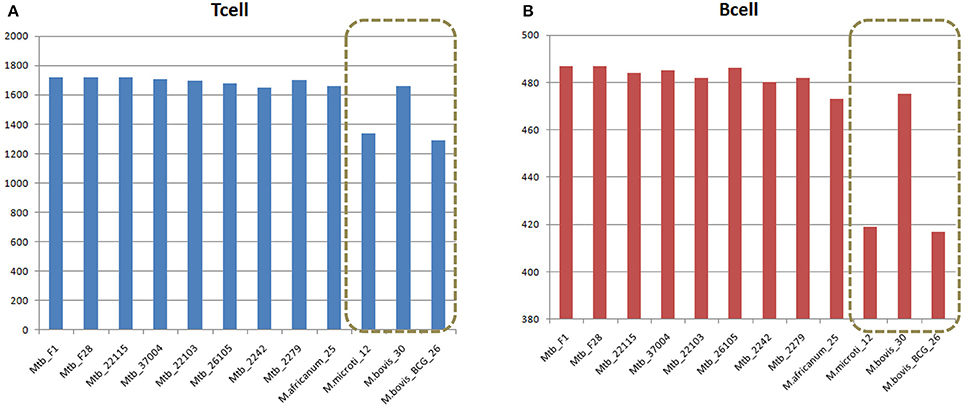
Figure 3. Comparison of (A) T cell and (B) B cell epitopes in 12 MTBC strains. Duplicate epitopes were removed, and only epitopes with 100% identical matches were considered present in the strain. The x-axis refers to the strain name, and the vertical axis indicates the number of epitopes in the corresponding strain.
Antigen Epitope Analysis between Human- and Animal-Isolates
The epitopes with the same distribution pattern in the 12 MTBC strains were clustered to further clarify the results. We then analyzed the epitope-clusters for the 12 MTBC strains (Figure 4). For T-cell epitope-clusters, we first investigated the relationship between epitopes and host association. Four epitope-clusters (Tcell_cluster_0002, 0017, 0078, and 0083: 25 epitopes) and one epitope-cluster (Tcell_cluster_0002: two epitopes) were identified to exist only in the human- and animal-associated MTBC strains, respectively (Figure 4A). In addition, the copy number of two epitope-clusters (Tcell_cluster_0015 and 0079: five epitopes) in the human isolates was higher than in the animal isolates. For B cell epitope-clusters, two epitope-clusters (Bcell_cluster_0002 and 0012: eight epitopes) only existed in human isolates, and the copy number of one epitope-cluster (Bcell_cluster_0031: one epitope) in human isolates was higher than in animal-associated ones (Figure 4B).
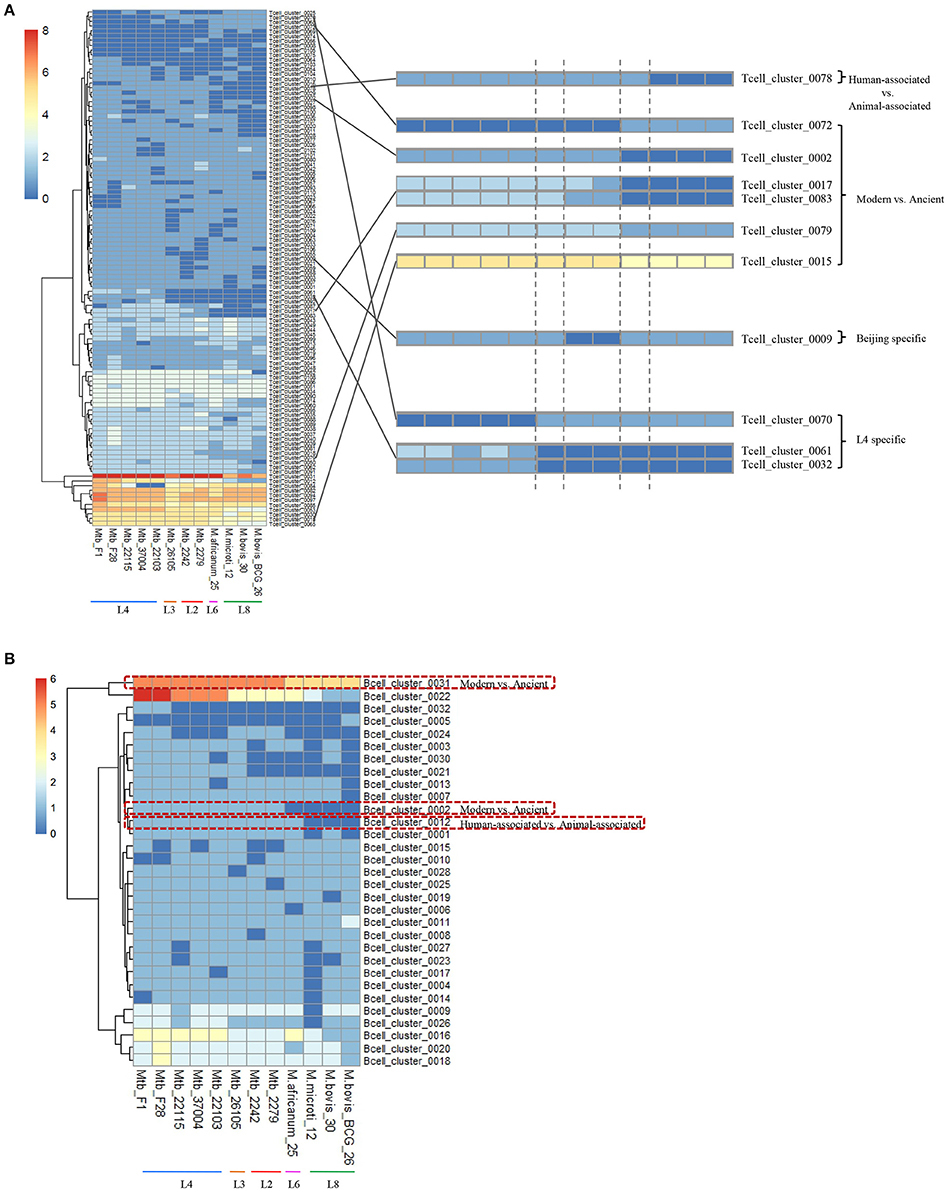
Figure 4. Distribution of differential T cell (A) and B cell (B) epitope-clusters in 12 MTBC strains. The epitope-clusters present in all 12 MTBC strains with the same copy number were excluded. Each row represents an epitope-cluster, and each column indicates a strain. The color intensity shows the copy number of each epitope-cluster. Some epitope-clusters were enlarged to highlight the differences amongst diverse MTBC lineages on the right section of figure.
To further explore the immune-genetic mechanisms, we studied the corresponding antigen genes of the previously described epitopes. The 25 human-associated strain-specific epitopes (belonging to four T-cell epitope-clusters) were located on 13 antigen genes (Figure 5). Among these, the loss of ten epitopes in animal-associated strains resulted from the deletion of the corresponding antigen genes; the loss of the other 15 epitopes was due to SNPs or Insertions or Deletions (Indels) on the relevant antigen genes (Table S14).
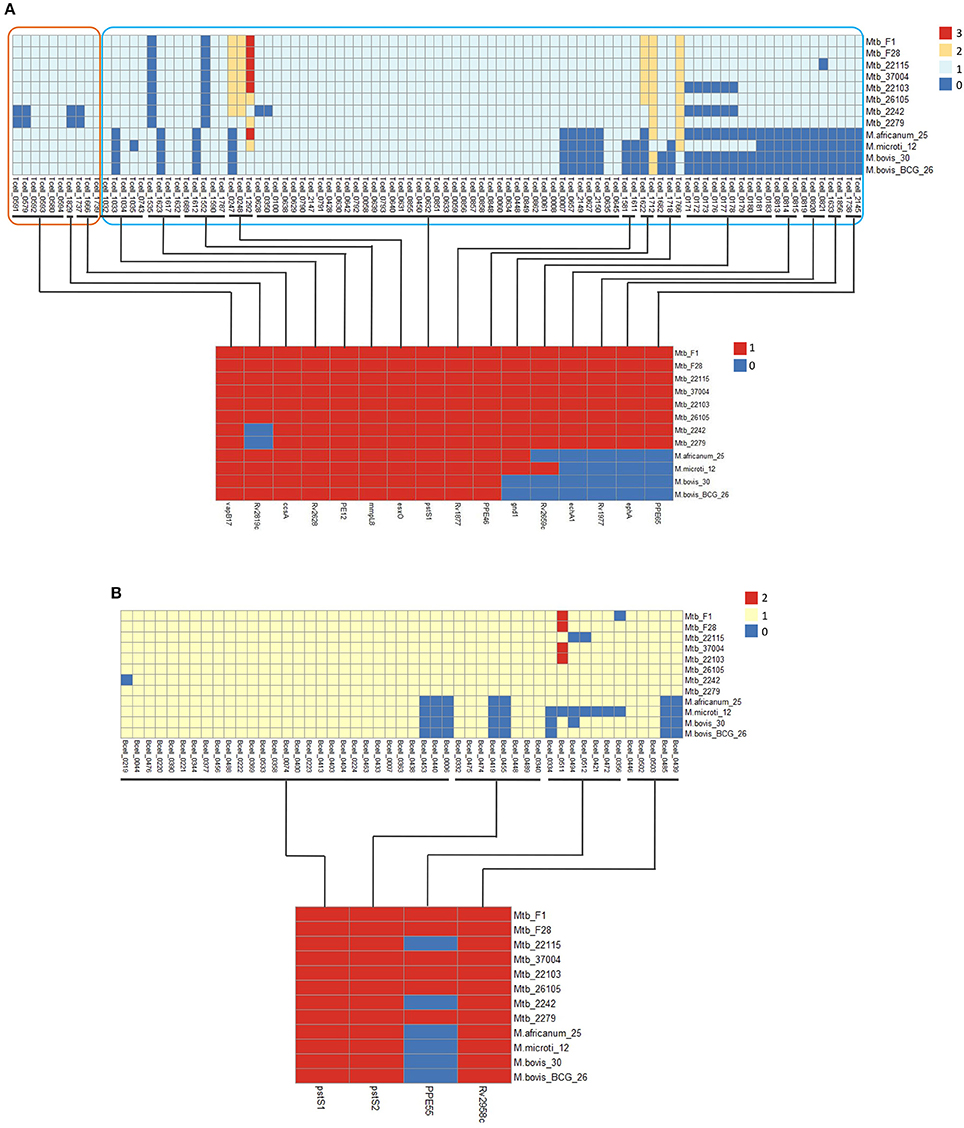
Figure 5. Distribution of some differential T-cell (A) and B-cell (B) epitopes and the corresponding antigen genes for 12 MTBC strains. The distribution of epitopes and corresponding antigens are shown at the top and bottom sections of the figure. Epitope copy number and antigen is indicated by the intensity of the color.
The deletion of antigen genes ephA (epoxide hydrolase) and ppe65 (PPE family protein), which are located in RD8, led to the loss of four epitopes (Tcell_1633 and 18566+6; Tcell_1738 and 2145) in animal-associated strains. EphA has been reported to regulate integrin-mediated T lymphocyte interactions, and its activation might lead to the down-regulation of T-cell interactions (Sharfe et al., 2008). In addition, three human-associated strain-specific epitopes (Tcell_0813, 0814, and 0815) were located on the antigen gene echA1 (described in “Host association analysis”). EchA1 was previously reported to be lost in M. bovis and M. bovis BCG strains (Brosch et al., 2002; Rosenkrands et al., 2008). Our analysis shows that echA1 is also absent from M. microti strains. In addition, our analysis indicates that the animal-associated strains lacked the antigen gene Rv1977, which resulted in the absence of epitopes (Tcell_0819-0821).
In addition to the deletion of antigens, there were 15 human-associated strains with specific epitopes attributed to SNPs and Indels in nine T cell antigen genes of animal-associated strains (Table S14). One of these was the antigen gene PstS-1, which is a lipoprotein phosphate transport receptor on the cell surface (de Araujo et al., 2014); this possessed five mutant epitopes in animal-associated strains. In our study, the five mutant epitopes were all derived from one SNP (V352A; Figure S6), which might lead to the animal-specific immune reaction.
The 25 human-associated strain-specific epitopes were further tested on ten animal-associated strains (two M. bovis and eight M. bovis BCG strains) with complete genome sequences from the NCBI (Table S9C). BLAST results confirmed our findings.
Antigen Epitope Analysis of Beijing Strains
Based on different animal experiments showing the Beijing strains to be more virulent, and to cause more histopathological changes, higher outgrowth, and increased mortality (Ida et al., 2010), the Beijing strains are considered the most pathogenic of the MTBC strains. Epitope-cluster analysis showed that Tcell_cluster_0009 (4 epitopes: Tcell_0579, 0591, 1737, and 1829) was lost only in the Beijing strains (Mtb 2242 and 2279; Figures 4, 5). Sequence alignment results revealed that the nucleotide sequence “AAACATT” from 71 to 77 of the antigen gene vapB17 mutated into “AC” in these two strains; this resulted in the absence of epitope Tcell_0579 and 0591. The “I245M” mutation on antigen CcsA led to the loss of epitope Tcell_1737. Finally the deletion of the antigen gene Rv2819c (a CRISPR type III-associated RAMP protein Csm5) led to the loss of epitope Tcell_1829 in the Beijing lineage strains. Further, verification was carried out on ten other Beijing lineage strains with complete genome sequences obtained from NCBI (Table S9A). The results confirmed the finding that these four epitopes were lost in all Beijing lineage strains. The absence of the four epitopes in all of these Beijing lineage strains might enable them to better evade the human immune system and promote growth in the hostile environment of the host cell. This could be a reason for their more virulent phenotype.
Antigen Epitope Analysis between M. bovis and M. bovis BCG Strains
To investigate the relationship between epitope variation and virulence, we compared the difference in epitopes of three virulent M. bovis strains and nine non-virulent M. bovis BCG strains (Figure S7). We determined that Tcell_cluster_A02 (149 epitopes) and Bcell_cluster_A01 (19 epitopes) were lost in all non-virulent BCG strains; meanwhile, Tcell_cluster_A06 (six epitopes) and Bcell_cluster_A04 (one epitope) were absent from all M. bovis strains. Tcell_cluster_A23 (five epitopes) had two copies in M. bovis, but only one copy in M. bovis BCG. We further examined the 13 corresponding antigen genes carrying the sequences of the above 180 epitopes (Figure S8 and Table S15). Among these, the loss of 146 epitopes (including 130 T-cell epitopes and 16 B-cell epitopes) in BCG strains resulted from the deletion of the corresponding three antigen genes (esxA, esxB, and ppe68), which were located in RD1 and are reported to be associated with the attenuation of virulence in M. bovis BCG (Lewis et al., 2003).
Conclusion
Tuberculosis now exceeds HIV as the top infectious disease killer, and is caused by the Mycobacterium tuberculosis complex (MTBC). MTBC strains possess highly similar genomes (99%) but large variations in phenotype, including host-association, virulence, and immunoreactivity. This might be the result of long-term co-evolution with diverse populations of humans and animals. Long-term geographical isolation gave rise to the accumulation of “genotype isolation,” thus resulting in different phenotypes. To analyze the relationship between genotype and phenotype, we selected 12 MTBC strains representing different lineages, re-sequenced their genomes to correct homo-polymer errors, and performed a comparative genomic analysis of the strains.
Host-association analysis indicated that eight mce3 family genes, two enoyl-CoA hydratases, and five PE/PPE family genes were present in human-associated strains but not in animal-associated strains.
Fifteen SNPs were found on virulence factors (including five SNPs in three ESX secretion-proteins) only in the Beijing strains. Comparisons between the virulent H37Rv strain and the non-virulent H37Ra strain revealed that three SNPs, S219L (PhoP), A219E (MazG), and a newly identified I228M (EspK), might lead to the attenuation of virulence in H37Ra. On the other hand, a comparison of the animal-associated MTBC strains showed that the deletion of the first four genes (i.e., pe35, ppe68, esxB, and esxA), rather than all eight RD1 genes, might play a central role in the virulence of animal-associated MTBC strains.
Finally, by comparing the epitopes of MTBC strains, we found that four epitopes were lost only in the Beijing strains. In addition, 32 T-cell epitopes and eight B-cell epitopes showed different distribution patterns in human- and animal-associated strains.
Overall, our research reveals the genetic variations leading to the differences in MTBC genotypes, and enhances our understanding of the relationship between highly conserved genome sequences/genotypes (similarity >99%) and highly different phenotypes in MTBC strains.
Accession Numbers
The SRA accession number for the sequencing data reported in this paper is SRP064893. The GAS (Genome Sequence Archive in BIG Data Center) accession number is PRJCA000307.
Author Contributions
FC, XJ, and HH designed the project. XJ, MD, and FC wrote the paper. XJ, LY, SC, LL, DC, JF, TY, JZ, XZ, YS, GW, and YS performed the experiments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Natural Science Foundation of China (31601047, 31570133, and 31600107); the “100-Talent Program” of Chinese Academy of Sciences (Y3CAS81554); Collaborative Innovation Center of Infectious diseases (PXM2015_014226_000058).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fcimb.2017.00088/full#supplementary-material
References
Ahmad, S., El-Shazly, S., Mustafa, A. S., and Al-Attiyah, R. (2004). Mammalian cell-entry proteins encoded by mce3 operon of Mycobacterium tuberculosis are expressed during natural infection in humans. Scand. J. Immunol. 60, 382–391. doi: 10.1111/j.0300-9475.2004.01490.x
Akhter, Y., Ehebauer, M. T., Mukhopadhyay, S., and Hasnain, S. E. (2012). The PE/PPE multigene family codes for virulence factors and is a possible source of mycobacterial antigenic variation: perhaps more? Biochimie 94, 110–116. doi: 10.1016/j.biochi.2011.09.026
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bentley, S. D., Comas, I., Bryant, J. M., Walker, D., Smith, N. H., Harris, S. R., et al. (2012). The Genome of Mycobacterium africanum west african 2 reveals a lineage-specific locus and genome erosion common to the M. tuberculosis complex. PLoS Negl. Trop. Dis. 6:e1552. doi: 10.1371/journal.pntd.0001552
Brosch, R., Gordon, S. V., Marmiesse, M., Brodin, P., Buchrieser, C., Eiglmeier, K., et al. (2002). A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc. Natl. Acad. Sci. U.S.A. 99, 3684–3689. doi: 10.1073/pnas.052548299
Brosch, R., Gordon, S. V., Pym, A., Eiglmeier, K., Garnier, T., and Cole, S. T. (2000). Comparative genomics of the mycobacteria. Int. J. Med. Microbiol. 290, 143–152. doi: 10.1016/S1438-4221(00)80083-1
Chen, L. H., Yang, J., Yu, J., Ya, Z. J., Sun, L. L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Chen, M. J., Gan, H. X., and Remold, H. G. (2006). A mechanism of virulence: virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J. Immunol. 176, 3707–3716. doi: 10.4049/jimmunol.176.6.3707
Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., et al. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. doi: 10.1038/31159
Comas, I., Chakravartti, J., Small, P. M., Galagan, J., Niemann, S., Kremer, K., et al. (2010). Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat. Genet. 42, 498–503. doi: 10.1038/ng.590
Comas, I., Coscolla, M., Luo, T., Borrell, S., Holt, K. E., Kato-Maeda, M., et al. (2013). Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat. Genet. 45, 1176–1182. doi: 10.1038/ng.2744
Copin, R., Coscolla, M., Efstathiadis, E., Gagneux, S., and Ernst, J. D. (2014). Impact of in vitro evolution on antigenic diversity of Mycobacterium bovis bacillus Calmette-Guerin (BCG). Vaccine 32, 5998–6004. doi: 10.1016/j.vaccine.2014.07.113
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5:e11147. doi: 10.1371/journal.pone.0011147
de Araujo, L. S., Mello, F. C. D., da Silva, N. D. M., Leung, J. A. M., Machado, S. M. A., Sardella, I. G., et al. (2014). Evaluation of gamma interferon immune response elicited by the newly constructed PstS-1(285-374): CFP10 fusion protein to detect Mycobacterium tuberculosis infection. Clin. Vaccine Immunol. 21, 552–560. doi: 10.1128/CVI.00726-13
Fabre, M., Koeck, J. L., Le Fleche, P., Simon, F., Herve, V., Vergnaud, G., et al. (2004). High genetic diversity revealed by variable-number tandem repeat genotyping and analysis of hsp65 gene polymorphism in a large collection of “Mycobacterium canettii” strains indicates that the M. tuberculosis complex is a recently emerged clone of “M. canettii.” J. Clin. Microbiol. 42, 3248–3255. doi: 10.1128/JCM.42.7.3248-3255.2004
Fan, X., Abd Alla, A. A., and Xie, J. (2016). Distribution and function of prophage phiRv1 and phiRv2 among Mycobacterium tuberculosis complex. J. Biomol. Struct. Dyn. 34, 233–238. doi: 10.1080/07391102.2015.1022602
Firdessa, R., Berg, S., Hailu, E., Schelling, E., Gumi, B., Erenso, G., et al. (2013). Mycobacterial lineages causing pulmonary and extrapulmonary tuberculosis, Ethiopia. Emerg. Infect. Dis. 19, 460–463. doi: 10.3201/eid1903.120256
Forrellad, M. A., Klepp, L. I., Gioffre, A., Sabio y Garcia, J., Morbidoni, H. R., de la Paz Santangelo, M., et al. (2013). Virulence factors of the Mycobacterium tuberculosis complex. Virulence 4, 3–66. doi: 10.4161/viru.22329
Gagneux, S. (2012). Host-pathogen coevolution in human tuberculosis. Phil. Trans. R. Soc. B 367, 850–859. doi: 10.1098/rstb.2011.0316
Gagneux, S., DeRiemer, K., Van, T., Kato-Maeda, M., de Jong, B. C., Narayanan, S., et al. (2006). Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 103, 2869–2873. doi: 10.1073/pnas.0511240103
Galagan, J. E. (2014). Genomic insights into tuberculosis. Nat. Rev. Genet. 15, 307–320. doi: 10.1038/nrg3664
Ghebremichael, S., Groenheit, R., Pennhag, A., Koivula, T., Andersson, E., Bruchfeld, J., et al. (2010). Drug resistant Mycobacterium tuberculosis of the Beijing genotype does not spread in Sweden. PLoS ONE 5:e10893. doi: 10.1371/journal.pone.0010893
Grace, P. S., and Ernst, J. D. (2016). Suboptimal antigen presentation contributes to virulence of Mycobacterium tuberculosis in vivo. J. Immunol. 196, 357–364. doi: 10.4049/jimmunol.1501494
Harboe, M., Christensen, A., Ahmad, S., Ulvund, G., Harkness, R. E., Mustafa, A. S., et al. (2002). Cross-reaction between mammalian cell entry (Mce) proteins of Mycobacterium tuberculosis. Scand. J. Immunol. 56, 580–587. doi: 10.1046/j.1365-3083.2002.01172.x
Ida, P., Reinout, V. C., and Dick, V. S. (2010). Possible underlying mechanisms for successful emergence of the Mycobacterium tuberculosis Beijing genotype strains. Lancet Infect. Dis. 10, 103–111. doi: 10.1016/S1473-3099(09)70330-5
Lewis, K. N., Liao, R. L., Guinn, K. M., Hickey, M. J., Smith, S., Behr, M. A., et al. (2003). Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette-Guerin attenuation. J. Infec. Dis. 187, 117–123. doi: 10.1086/345862
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Lindestam Arlehamn, C. S., Paul, S., Mele, F., Huang, C., Greenbaum, J. A., Vita, R., et al. (2015). Immunological consequences of intragenus conservation of Mycobacterium tuberculosis T-cell epitopes. Proc. Natl. Acad. Sci. U.S.A. 112, E147–155. doi: 10.1073/pnas.1416537112
Liu, J., Tran, V., Leung, A. S., Alexander, D. C., and Zhu, B. L. (2009). BCG vaccines: their mechanisms of attenuation and impact on safety and protective efficacy. Hum. Vaccin. 5, 70–78. doi: 10.4161/hv.5.2.7210
Lu, L. D., Sun, Q., Fan, X. Y., Zhong, Y., Yao, Y. F., and Zhao, G. P. (2010). Mycobacterial MazG is a novel NTP pyrophosphohydrolase involved in oxidative stress response. J. Biol. Chem. 285, 28076–28085. doi: 10.1074/jbc.M109.088872
McLaughlin, B., Chon, J. S., MacGurn, J. A., Carlsson, F., Cheng, T. L., Cox, J. S., et al. (2007). A mycobacterium ESX-1-secreted virulence factor with unique requirements for export. PLoS Pathog. 3:e105. doi: 10.1371/journal.ppat.0030105
Pirofski, L. A., and Casadevall, A. (2012). Q&A: What is a pathogen? a question that begs the point. BMC Biol. 10:6. doi: 10.1186/1741-7007-10-6
Portevin, D., Gagneux, S., Comas, I., and Young, D. (2011). Human macrophage responses to clinical isolates from the Mycobacterium tuberculosis complex discriminate between ancient and modern lineages. PLoS Pathog. 7:e1001307. doi: 10.1371/journal.ppat.1001307
Pym, A. S., Brodin, P., Brosch, R., Huerre, M., and Cole, S. T. (2002). Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol. Microbiol. 46, 709–717. doi: 10.1046/j.1365-2958.2002.03237.x
Rodriguez-Campos, S., Smith, N. H., Boniotti, M. B., and Aranaz, A. (2014). Overview and phylogeny of Mycobacterium tuberculosis complex organisms: implications for diagnostics and legislation of bovine tuberculosis. Res. Vet. Sci. 97, S5–S19. doi: 10.1016/j.rvsc.2014.02.009
Rosenkrands, I., Aagaard, C., Weldingh, K., Brock, I., Dziegiel, M. H., Singh, M., et al. (2008). Identification of Rv0222 from RD4 as a novel serodiagnostic target for tuberculosis. Tuberculosis 88, 335–343. doi: 10.1016/j.tube.2007.12.001
Ryndak, M., Wang, S. S., and Smith, I. (2008). PhoP, a key player in Mycobacterium tuberculosis virulence. Trends Microbiol. 16, 528–534. doi: 10.1016/j.tim.2008.08.006
Sharfe, N., Nikolic, M., Cimpeon, L., De Kratts, A. V., Freywald, A., and Roifman, C. M. (2008). EphA and ephrin-A proteins regulate integrin-mediated T lymphocyte interactions. Mol. Immunol. 45, 1208–1220. doi: 10.1016/j.molimm.2007.09.019
Simeone, R., Bottai, D., Frigui, W., Majlessi, L., and Brosch, R. (2015). ESX/type VII secretion systems of mycobacteria: insights into evolution, pathogenicity and protection. Tuberculosis 95, S150–S154. doi: 10.1016/j.tube.2015.02.019
Sreejit, G., Ahmed, A., Parveen, N., Jha, V., Valluri, V. L., Ghosh, S., et al. (2014). The ESAT-6 protein of Mycobacterium tuberculosis interacts with beta-2-microglobulin (β2M) affecting antigen presentation function of macrophage. PLoS Pathog. 10:e1004446. doi: 10.1371/journal.ppat.1004446
Steenken, W., and Gardner, L. U. (1946). History of H37 strain of tubercle bacillus. Am. Rev. Tuberc. 54, 62–66.
Takezaki, N., and Nei, M. (1996). Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA. Genetics 144, 389–399.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tarailo-Graovac, M., and Chen, N. (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. Chapter 4, Unit 4, 10. doi: 10.1002/0471250953.bi0410s25
Vita, R., Overton, J. A., Greenbaum, J. A., Ponomarenko, J., Clark, J. D., Cantrell, J. R., et al. (2015). The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 43, D405–D412. doi: 10.1093/nar/gku938
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9:e112963. doi: 10.1371/journal.pone.0112963
Winglee, K., Manson McGuire, A., Maiga, M., Abeel, T., Shea, T., Desjardins, C. A., et al. (2016). Whole genome sequencing of Mycobacterium africanum strains from Mali provides insights into the mechanisms of geographic restriction. PLoS Negl. Trop. Dis. 10:e0004332. doi: 10.1371/journal.pntd.0004332
Zheng, H. J., Lu, L. D., Wang, B. F., Pu, S. Y., Zhang, X. L., Zhu, G. F., et al. (2008). Genetic basis of virulence attenuation revealed by comparative genomic analysis of Mycobacterium tuberculosis strain H37Ra vs. H37Rv. PLoS ONE 3:e2375. doi: 10.1371/journal.pone.0002375
Keywords: Mycobacterium tuberculosis complex (MTBC), tuberculosis (TB), host association, virulence, epitope, comparative genomics, pathogenicity, PacBio
Citation: Jia X, Yang L, Dong M, Chen S, Lv L, Cao D, Fu J, Yang T, Zhang J, Zhang X, Shang Y, Wang G, Sheng Y, Huang H and Chen F (2017) The Bioinformatics Analysis of Comparative Genomics of Mycobacterium tuberculosis Complex (MTBC) Provides Insight into Dissimilarities between Intraspecific Groups Differing in Host Association, Virulence, and Epitope Diversity. Front. Cell. Infect. Microbiol. 7:88. doi: 10.3389/fcimb.2017.00088
Received: 25 November 2016; Accepted: 06 March 2017;
Published: 21 March 2017.
Edited by:
Adel M. Talaat, University of Wisconsin-Madison, USAReviewed by:
Srinand Sreevatsan, University of Minnesota, USAAndrey P. Anisimov, State Research Center for Applied Microbiology and Biotechnology, Russia
Copyright © 2017 Jia, Yang, Dong, Chen, Lv, Cao, Fu, Yang, Zhang, Zhang, Shang, Wang, Sheng, Huang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fei Chen, Y2hlbmZlaUBiaWcuYWMuY24=
Hairong Huang, aHVhbmdoYWlyb25nQHRiMTIzLm9yZw==
†These authors have contributed equally to this work.