 Maria del Mar Cendra
Maria del Mar Cendra Myron Christodoulides
Myron Christodoulides Parwez Hossain2†
Parwez Hossain2†- 1Division of Clinical and Experimental Sciences, Department of Molecular Microbiology, Faculty of Medicine, University of Southampton, Southampton, UK
- 2Eye Unit, Division of Clinical and Experimental Sciences, Faculty of Medicine, University of Southampton, Southampton, UK
Pseudomonas aeruginosa is the principal cause of bacterial keratitis worldwide and overstimulation of the innate immune system by this organism is believed to contribute significantly to sight loss. In the current study, we have used primary human corneal fibroblast (hCF) cells as an ex vivo model of corneal infection to examine the role of P. aeruginosa flagellum and type three secretion system (TTSS) in inducing inflammasome-associated molecules that trigger IL-1β and IL-18 production during the early stages of the infection. Our results show that P. aeruginosa infection stimulated the non-canonical pathway for IL-1β and IL-18 expression and pathway stimulation was influenced predominantly by the flagellum. Both IL-1β and IL-18 cytokines were expressed intracellularly during bacterial infection, but only the former was released and detected in the extracellular environment. We also investigated the signaling pathways in hCFs mediated by Toll-Like Receptor (TLR)4 and TLR5 sensing of P. aeruginosa, and our data show that the signal triggered by TLR5-flagellin sensing significantly contributed to IL-1β and IL-18 cytokine production in our model. Our study suggests that IL-18 expression is wholly dependent on extracellular flagellin sensing by TLR5, whereas IL-1β expression is also influenced by P. aeruginosa lipopolysacharide. Additionally, we demonstrate that IL-1β and IL-18 production by hCFs can be triggered by both MyD88-dependent and -independent pathways. Overall, our study provides a rationale for the development of targeted therapies, by proposing an inhibition of flagellin-PRR-signaling interactions, in order to ameliorate the inflammatory response characteristic of P. aeruginosa keratitis.
Introduction
Bacterial keratitis is a global health problem in which pathogen-induced cytotoxicity and overstimulation of the host innate inflammatory response can lead to severe visual impairment and sight lost (Taube et al., 2015). In the human eye, superficial damage and surface laceration are often associated with contact lens wear. Thus, when the corneal epithelium barrier and Bowman's membrane located between the epithelium and the stroma in the cornea are physically breached, microbes can penetrate and establish infection in the stroma. Keratitis is characterized by the production of inflammatory cytokines, the subsequent influx of polymorphonuclear leukocytes (PMNL) and extensive tissue damage. As a consequence of microbial penetration into the stroma, resident keratocytes can respond by transforming into corneal fibroblasts that exacerbate the inflammatory response (O'brien, 2003). Pseudomonas aeruginosa is the leading cause of bacterial keratitis associated with contact lens wear (Shah et al., 2011; Stapleton and Carnt, 2012).
Stimulation of the host innate immune response is initiated by pattern recognition receptors (PPR), which are immune receptors that recognize pathogen-associated molecular patterns (PAMPs) of infectious agents and trigger different types of cellular responses. Expression of cell wall lipopolysaccharide (LPS) O-antigen, the exotoxin injector type three secretion system (TTSS) and flagellum are key P. aeruginosa PAMPs (Hauser, 2011; Le Berre et al., 2011). The TTSS is a molecular machine encoded by pathogenic gram-negative bacteria, which is used to meditate interactions with eukaryotic host cells. This bacterial complex consists of a syringe-like “injectisome” that enables active translocation of bacterial proteins into the host cell cytoplasm, in order to alter eukaryotic cell biology during infection (Notti and Stebbins, 2016). Intracellular delivery of exotoxins is sensed by the host immune system: intracellular PPRs sense conserved bacterial determinants that have been internalized, e.g., components of the injectisome, thus activating a host response to infection (Puhar and Sansonetti, 2014). For P. aeruginosa, the PopB, PopD and PcrV are essential TTSS proteins for the exotoxin delivery mechanism. Possession of the TTSS has been suggested to be indispensable for PAO1 strain in promoting tissue damage and keratitis in animal models (Pier, 2007; Hauser, 2009). P. aeruginosa flagellin (FliC) is the protein product of the fliC gene, which becomes the flagellum filament when it is polymerized (Hayashi et al., 2001; Berg, 2003; Ghosh, 2004). Toll-like receptors (TLRs) are a family of PPRs and classified depending on whether they are found anchored within the cell membrane or located intracellularly (Janeway and Medzhitov, 2002; Kawasaki and Kawai, 2014). In the current study, we have focused on investigating cell membrane-associated TLR2, which senses bacterial lipoproteins; TLR4, which is activated by LPS, a classical inducer of host inflammatory processes; and TLR5, which is activated by extracellular flagellin that can subsequently be delivered within the cytosol (Kawasaki and Kawai, 2014). When stimulated, TLRs recruit TIR1 domain-containing adaptors, such as MyD882, TRIF3, TIRAP4/MAL5, or TRAM6, and activate subsequent signaling pathways that lead to transcription of NF-κβ7 and IRFs8 to orchestrate a wide range of inflammatory responses and cytokine expression (O'neill et al., 2013; Kawasaki and Kawai, 2014). P. aeruginosa has been reported to activate TRIF and MyD88 adaptor molecules in mouse macrophages, but not TIRAP (Sun et al., 2010).
Nod-like receptors (NLRs) are another family of PPRs, but they are found within the cytosol. NLRs sense intracellular PAMPS and promote the assembly of a multi-protein scaffold called the “inflammasome” (Chen et al., 2009; O'neill et al., 2013). The inflammasome recruits and activates certain caspases for maturing interleukin (IL)-1β and IL-18 cytokine pro-forms, and also induces a cell-death program called “pyroptosis” (Schroder and Tschopp, 2010; Latz et al., 2013). To date, numerous inflammasome-associated proteins have been described including NLRP91, NLRP2, NLRP3, AIM210, and NLRC114 (Schroder and Tschopp, 2010; Ozaki et al., 2015). P. aeruginosa flagellin and TTSS have been reported to be important for the intracellular assembly of the non-canonical inflammasome NLRC4 in mouse models (Sutterwala et al., 2007; Miao et al., 2010; Zhao et al., 2011).
It is generally accepted that PAMP recognition by host cell PRR leads to the production of cytokines as well as other innate immune mediator signals (Mogensen, 2009). Herein, we have studied the role of the flagellum, TTSS and LPS PAMPs in the activation of inflammasome-associated signaling molecules, and consequential IL-1β and IL-18 production by human primary stromal corneal fibroblasts (hCFs), during the early stages of P. aeruginosa infection. The rationale for studying IL-1β and IL-18 expression in our in vitro model of hCFs is based on our observation that infection of these cells with wild-type P. aeruginosa is characterized predominantly by the production of these two cytokines, with the expression of other cytokines, chemokines, and immune mediator molecules not significant (manuscript submitted elsewhere). Furthermore, we have tested the hypothesis that inhibition of intracellular signaling pathways during P. aeruginosa infection can lead to reduced pro-inflammatory cytokine expression in hCFs.
Materials and Methods
Human Primary Stromal Corneal Fibroblast (hCF)
Corneal buttons were collected aseptically from patients at the eye unit of a large hospital in the south of England. After removing the corneal epithelium, the stromal layer was dissected and digested with collagenase Type-1 (1 mg/mL; Life Technologies, Warrington, UK) for 3 h at 37°C. Digested stroma was cultured in Corneal Culture Medium (CCM) and hCF were characterized as described previously (Wong et al., 2011). Human monocytic cell line THP-1 was cultured in RPMI 1640 containing glutamine (Lonza, Biologics, Tewkesbury, UK), 1% (v/v) Penicillin-Streptomycin and 10% (v/v) decomplemented Fetal Calf Serum (dFCS; Life Technologies, Warrington, UK). Cells were cultured in a humidified incubator at 37°C and 5% (v/v) CO2.
Ethics Statement
Patients provided written informed consent to use surplus corneal tissue specimen for research via the NHS Blood Transplant Eye Retrieval Service based at Southampton General Hospital (Southampton, UK). Protocols were approved by the NRES Committee South Central-Berkshire 06/Q1602/56.
Bacterial Strains, Plasmids, and Growth Conditions
Pseudomonas aeruginosa strain PAO1 (Holloway1C Stanier131) was obtained from the National Collection of Industrial, Food and Marine Bacteria, UK. P. aeruginosa strain PA14 and PA14 ΔflgK and PA14 ΔpopB mutants were provided by George O'Toole (Dartmouth Medical School, Dartmouth, USA). PA14 ΔflgK mutant is unable to synthesize a functional flagellum and the PA14 ΔpopB mutant lacks the PopB protein, an essential protein without which the TTSS cannot deliver exotoxins within eukaryotic cells (Berg, 2003; Hauser, 2009). Green Fluorescence Protein (GFP) was inserted into PAO1, PA14 and mutant strains by transformation of the PAO1/GFP plasmid provided by Alice Prince (Columbia University, New York, USA) (Parker and Prince, 2013). To complement PA14 ΔflgK and PA14 ΔpopB mutation, the flgK and popB coding region sequences from the PA14 genome were amplified using the primers listed in Supplementary Table 1 and cloned into the HindIII and BamHI sites of the shuttle cloning vector pUCP19 (Schweizer, 1991). pUCP19-flgK and pUCP19-popB were transformed into Escherichia coli DH5α and into PA14 ΔflgK and PA14 ΔpopB strains (Irani and Rowe, 1997). Carbenicillin (Sigma-Aldrich, Poole, UK) at 100 and 300 μg/mL was added to the medium for plasmid maintenance for E. coli and P. aeruginosa, respectively. Bacterial strains were grown in Luria-Bertani broth and agar (Oxoid, Basingstoke, UK) at 37°C.
Transfection of hCFs with TLR5-siRNA (siTLR5-hCFs)
hCFs were grown to 30–50% of confluence in a 24-well plate and transfected with four TLR5 siRNA (FlexiTube GeneSolution, Qiagen, Manchester, UK) using INTERFERin® (Polyplus transfection, Illkirch-Graffenstaden, France). Mock RNA was used as a transfection control. A total of 1 nM of TLR5-siRNA per well was mixed with 100 μl of dFCS-free CCM and 2 μl of INTERFERin®. After 10 min at room temperature (RT), the transfection mix was added to the CCM (500 μl) already present in the wells and incubated for 4 h. Transfection solution was then removed and 1 mL of fresh CCM was added per monolayer and cells were incubated for 48 h. Mock transfection with an irrelevant siRNA was done to ensure that TLR5 expression remained unaltered. TLR5 silencing was checked by measuring TLR5 expression in PA14-stimulated hCFs by Real Time quantitative Polymerase Chain Reaction (RT-qPCR) and western-blot.
Expression and Purification of Recombinant (r) FliC Protein of P. aeruginosa
The fliC (flagellin) coding region of PAO1 is 100% identical to that in PA14, and it was amplified using the primers listed in Supplementary Table 1 and cloned into the NdeI and XhoI restriction sites of the T7 promoter vector pET22b(+) (Merck Millipore, Hertfordshire, UK). After checking pET22b-fliC by PCR with T7 universal oligonucleotides (Supplementary Table 1) and sequencing, the plasmid was transformed into E. coli BL21 using ampicillin (Sigma-Aldrich, Poole, UK) at 50 μg/mL to maintain the plasmid. BL21 pET22b-fliC culture was grown to an OD λ600 nm = 0.5 and induced with isopropyl-β-D-1-thiogalactopyranoside (IPTG; 1 mM) for 4 h, harvested and lysed. Cell supernatant and pellet from the lysed culture were analyzed by SDS-PAGE, which demonstrated presence of the recombinant (r)FliC in the supernatant. rFliC was purified from the soluble fraction of harvested cultures using nickel-nitrilotriacetic acid (Ni-NTA) metal-affinity chromatography (Qiagen, Manchester, UK) under native conditions, as described previously (Hung et al., 2015). Bound rFliC was eluted with 50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole buffer (pH 8.0) and elution fractions analyzed by SDS-PAGE. Pooled fractions were dialyzed against PBS, pH7.4 for 2 days. Lipopolysaccharide (LPS) content was quantified with the Pierce LAL Chromogenic Endotoxin Quantitation Kit (Thermo Fisher Scientific, Basingstoke, UK) following the manufacturer's instructions. SDS-PAGE of purified rFliC confirmed the purity and molecular mass (~49 KDa) of the protein (Supplementary Figure 1).
Infection of hCFs with P. aeruginosa Strains and Different Stimulatory Conditions
hCFs were seeded into 24-well culture plates (Greiner bio-one, Stonehouse, UK, ~105 cells/well) and infected over time with P. aeruginosa at a Multiplicity Of Infection (MOI) of 10 in dFCS-free DMEM medium (Sigma-Aldrich, Poole, UK). Stimulatory conditions included 100 ng/mL of P. aeruginosa LPS (Sigma-Aldrich, Poole, UK) and 5 μg/mL of purified P. aeruginosa rFliC protein for 6 h and when required, streptolysin-O (SLO), was used for the intracellular delivery of the molecule as described previously (Walev et al., 2001). Bacterial association to hCFs was quantified over time as described previously (Hardy et al., 2000). Briefly, monolayers were washed gently 4 times with phosphate buffered saline (PBS), pH7.4 and lysed with 250 μl of PBS containing saponin [1% (w/v); Sigma-Aldrich, Poole, UK] and dFCS (1% w/v) for 15 min. Determination of colony forming units was done by serial dilution and viable counting on agar plates.
Signaling Pathway Inhibition
Inhibition experiments were done using 50 μM of Myd88 peptide inhibitor (MyD88i, Bio-techne, Abington, UK) and 1 μM of TAK-242 (Merck Millipore), respectively (Loiarro et al., 2005; Takashima et al., 2009; Matsunaga et al., 2011). hCFs were treated with MyD88i and TAK-242 for 24 and 12 h, respectively, prior to hCFs stimulation with P. aeruginosa strains, rFliC or LPS for 6 h. Uninfected hCFs monolayers in inhibited and non-inhibited conditions were used as controls.
Total RNA Extraction and Real Time Quantitative Polymerase Chain Reaction (RT-qPCR)
In this study, total RNA was extracted from infected- and stimulated- hCF cells during different experimental conditions including hCF infection with the PAO1, PA14 wild-type and ΔflgK, ΔpopB mutant strains for 1, 3 or 6 h, and after stimulation with rFliC (5 μg/mL) and P. aeruginosa LPS (100 ng/mL) for 6 h. Total RNA was extracted from siTLR5-hCFs monolayers after 6 h of similar stimulatory and infection conditions. RNA extraction was done using the RNeasy Mini Kit (Qiagen, Manchester, UK). Control conditions were uninfected and un-stimulated hCFs or siTLR5-hCFs. RNA samples were treated with DNAse-50 (PrimerDesign, Chandler's Ford, UK) and total concentration was quantified using a NanoDrop spectrophotometer (ND-1000, NanoDrop, Thermo Fisher Scientific, Basingstoke, UK). cDNA was synthetized from 1 μg of RNA using the RT-nanoscript system (PrimerDesign, Chandler's Ford, UK) and the reverse oligonucleotides listed in Supplementary Table 1. RT-qPCRs were done with 1 μl of each cDNA in a Rotorgene-Q 5plex HRM (Qiagen, Manchester, UK) using the Power SYBR green PCR Master Mix (Life Technologies, Warrington, UK) and oligonucleotides listed in Supplementary Table 1. β-actin was used as an endogenous control and melting curves were done to ensure specific gene amplification. The 2−ΔΔCT method was used for data analyses (Livak and Schmittgen, 2001).
Protein Extraction and Western Blot Analysis
Intracellular protein analysis of TLR2, TLR4, TLR5, NLRC4, caspase-1, caspase-4, and the cytokines IL-18 and IL-1β was done after 6 h in similar infection and stimulatory conditions, including the experiments with siTLR5-hCFs cells, as for RT-qPCR experiments. hCFs infected with complemented PA14 ΔflgK pUCP19-flgK and PA14 ΔpopB pUCP19-popB strains were included for protein analyses. Intracellular mature IL-18 and IL-1β were also measured in hCFs treated with TAK-242, MyD88i and TAK-242/MyD88i and infected with PA14 and mutant strains. Cells were washed in PBS and total protein extract was obtained by using Cytobuster™ protean extraction reagent (Merck Millipore, Nottingham, UK). Total protein (5 μg) was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and electro-transferred onto polyvinylidene difluoride (PVDF) membranes (GE Healthcare, Amersham, UK). Membranes were blocked for 1 h in PBS containing 0.05% (v/v) Tween20 (PBST) and 5% (v/v) dry milk powder. Antibodies used were rabbit monoclonal anti-TLR2, -TLR4, -TLR5; rabbit polyclonal anti-NLRP3, -Caspase-4, -IL-18, and -IL-1β (Abcam, Bristol, UK); rabbit polyclonal anti-NLRC4 (Bio-techne Ltd, Abingdon, UK). All antibodies were used at the manufacturers' recommended dilutions and left overnight at 4°C. Membranes were washed three times in PBST and incubated for 1 h with goat anti-Rabbit IgG (H&L)-HRP pre-adsorbed secondary antibody (Abcam, Bristol, UK) according to the manufacturer's recommendations. Rabbit anti-β-actin polyclonal antibody (Abcam, Bristol, UK) was used as an endogenous and loading control. Immuno-detection was carried out in a Versadoc 4000 and data analyzed with Quantity One 4.6.9 software (BioRad, Langford, UK).
Cytokine Release Analysis
Extracellular IL-18 and IL-1β cytokines were quantified with the Meso Scale Discovery (MSD) electro-chemiluminescence assays (Meso Scale Diagnostics, Gaithersburg, USA.) and by western blot after protein precipitation with trichloroacetic acid (TCA). Supernatants from triplicate hCF monolayers infected with wild-type PAO1, PA14, mutant PA14 ΔflgK, and PA14 ΔpopB strains (MOI = 10) were collected after 6 h of infection.
Confocal Microscopy
For visual comparison of bacterial association with hCFs, hCF monolayers, grown on borosilicate glass cover slips, were infected for 9 h with GFP-expressing wild-type PAO1, PA14 and mutant PA14 ΔflgK and PA14 ΔpopB strains (MOI = 10). After infection, cells were washed three times with warmed DMEM medium and live-stained with Wheat Germ Agglutinin (1/1,000) for 10 min at RT. Monolayers were washed with PBS and fixed with paraformaldehyde (4% v/v) for 15 min at RT. After three more PBS washes, cells were permeabilized with PBS containing saponin 0.1% (v/v) and 10% (v/v) dFCS for 2 h at 4°C and counterstained with PBS containing DAPI (1/1,000) for 15 min at RT. hCFs monolayers were washed three times in PBS and examined with a Leica SP5 LSCM confocal microscope (Leitz).
Statistics
Data were analyzed by one-way ANOVA with Dunnett's multiple comparison test. P < 0.05 denoted significance.
Results
Pseudomonas aeruginosa Flagellum Influences Bacterial Attachment to hCFs
In preliminary experiments, hCFs were infected over time with different MOI (1-100) of P. aeruginosa. These experiments demonstrated that a MOI of 10 was optimal for infecting hCFs in order to study both logarithmic association of P. aeruginosa to hCFs and maximal gene and protein expression in intact cell monolayers (data not shown).
Adhesion patterns of P. aeruginosa wild-type bacteria and mutant strains for flagellum (ΔflgK) and TTSS (ΔpopB) to hCFs were first examined as an initial step required for cellular PRR stimulation. No significant differences (P > 0.05) in the association dynamics of wild-type PAO1 and PA14 strains and the PA14 ΔpopB mutant with hCF monolayers were observed (Figure 1A). By contrast, the PA14 ΔflgK mutant showed a significant ~1 log reduction in bacterial association with hCFs compared to the wild-type and ΔpopB strains at every time point examined (P < 0.05). The complemented PA14 ΔflgK pUCP19-flgK strain showed a similar association pattern (P > 0.05) as the wild-type PA14 strain by up to 6 h, but by 9 h there was an ~1 log divergence in adherence of the complemented strain compared with the parent strain (Figure 1A). There was no significant difference in adherence between the PA14 wild-type strain, the ΔpopB mutant and the complemented PA14 ΔpopB pUCP19-popB strain over the 9 h time period (P > 0.05; Figure 1A). Representative confocal microscopy images confirmed visually that the numbers of PA14 ΔflgK bacteria associated to hCFs monolayers were reduced compared to PA14 wild-type strain and ΔpopB mutant (Figure 1B). Bacterial growth rates were similar for all the strains (Supplementary Figure 2), except for the PA14 ΔflgK mutant, which was marginally slower between 0 and 3 h compared with the wild-type PA14 strain, although the growth rates of both were similar thereafter (P > 0.05). In addition, cytotoxicity induced by P. aeruginosa infection (MOI = 10) as determined by release of lactate dehydrogenase enzyme was not significant by 9 h (<5% observed enzyme release, data not shown). Death of the cell monolayers occurred between 9 and 24 h of infection, regardless of phenotype (data not shown).
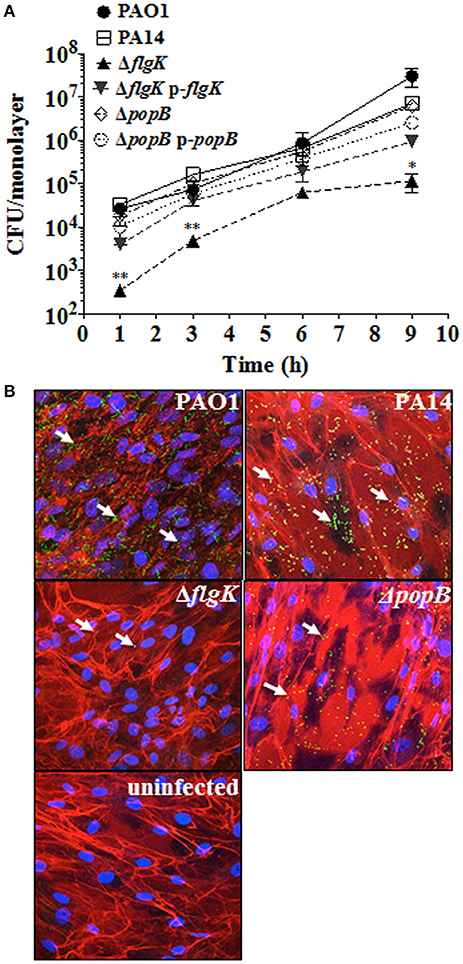
Figure 1. Expression of flagellum contributes to P. aeruginosa adhesion to hCFs. (A) Cells were infected with MOI = 10 of PAO1, PA14, ΔflgK, ΔpopB and complemented PA14 ΔflgK p-flgK, PA14 ΔpopB p-popB strains and association to hCFs monolayers was quantified by viable counting after 1, 3, 6 and 9 h. The symbols represent the means and bars the standard error of the mean (SEM) of n = 3 independent experiments. PA14 ΔflgK and PA14 ΔpopB strains carrying the cloning vector pUCP19 without insert were tested in order to exclude possible vector interferences: no differences were observed when compared with their respective mutant strains in their association capabilities (data not shown). Statistical significance was calculated with a one-way ANOVA with Dunnett's multiple comparison test and it is represented by asterisks as follows: *P < 0.05 and **P < 0.01. (B) Confocal microscopy images of GFP-expressing PAO1, PA14 and mutant strains after 9 h of infection. Arrows denote the positions of bacteria and images are representative of n = 3 independent experiments.
Pseudomonas aeruginosa Flagellum and TTSS Enhance Non-canonical Inflammasome Signaling Molecule Expression
Induction of inflammasome-associated signaling molecules in hCFs during the early stages of infection by P. aeruginosa and the effects caused by the ΔflgK and ΔpopB mutations were analyzed. Expression of the tlr2, tlr4, tlr5, nlrp3 and nlrc4 receptor genes, il-18 and il-1β cytokine genes and caspase-1 and caspase-4 genes was quantified by RT-qPCR after 1, 3 and 6 h of bacterial infection (Figure 2A). Each point in the graph represents the fold-change in expression caused by the wild-type and mutant bacteria compared against uninfected cells, using the 2−ΔΔCT method. No significant changes in expression were observed for tlr2, nlrp3 or caspase-1 (data not shown). The highest levels of gene induction were observed 6 h after infection with wild-type PAO1 and PA14 strains (Figure 2A), with significant increases at 3 h observed for il-18 and il-1β only with PA14 (P < 0.05). A one-way ANOVA with Dunnett's test was used to compare the fold-changes for these genes induced by the PA14 wild-type compared with the PAO1 wild-type, and the fold-changes induced by PA14 ΔflgK and ΔpopB mutants compared with their parental PA14 strain. At 3 h, the levels of il-18 and il-1β cytokine genes induced by PA14 wild-type infection of hCF were ~3-fold higher than the levels induced by PAO1 wild-type (P < 0.05; Figure 2A). At 6 h, PA14 induced statistically significant ~4-fold higher levels of tlr4 (P < 0.05) compared with PAO1, whereas for the other genes, the observed fold-increases induced by both strains were similar (P > 0.05). hCF infection with the PA14 ΔflgK and ΔpopB mutants, showed that at 3 h, there were significantly lower (~13-fold) levels of il-18 (P < 0.001) and il-1β (P < 0.01) cytokine gene induction, compared with the PA14 wild-type strain (Figure 2A). By 6 h, infection with the PA14 ΔflgK mutant showed significantly decreased levels (~ 5–10-fold) of tlr4, tlr5, nlrc4, casp-4, il-18, and il-1β cytokine gene expression compared with PA14 wild-type (P < 0.05; Figure 2A). By contrast, infection with the PA14 ΔpopB mutant showed significant reductions (~ 4-fold) in expression of tlr4, tlr5, nlrc4, and il-18 genes (P < 0.05) compared to PA14 wild-type, but not significantly different for casp-4 and il-1β genes (P > 0.05).
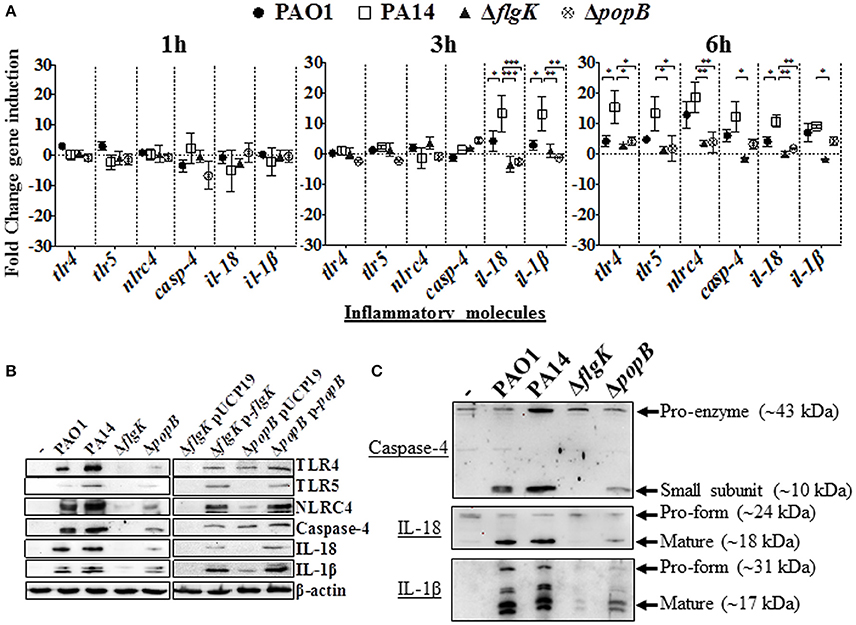
Figure 2. Inflammatory responses of human corneal fibroblast cells (hCFs) toward P. aeruginosa infection. (A) RT-qPCR of immune signaling molecules after 1, 3 and 6 h hCFs infection with PAO1 and PA14 wild-type and PA14 ΔflgK and PA14 ΔpopB mutant strains at a MOI = 10. Symbols represent the mean and bars the SEM of the log of gene expression from infected hCFs compared with uninfected controls from n = 10 independent hCF samples. Validation of the oligonucleotides used for RT-qPCR examination of gene expression in infected hCFs was done by demonstrating the same signals in THP-1 cells stimulated with 100 ng/mL of P. aeruginosa LPS (data not shown). Statistical significance was calculated with a one-way ANOVA with Dunnett's multiple comparison test and it is represented by asterisks as follows: *P < 0.05, **P < 0.01, ***P < 0.001. (B) Cells infected with MOI = 10 of PAO1 and PA14 wild-type, PA14 ΔflgK and PA14 ΔpopB mutant strains, and the complemented PA14 ΔflgK p-flgK, PA14 ΔpopB p-popB and control PA14 ΔflgK pUCP19 and PA14 ΔpopB pUCP19 strains were lysed and intracellular protein levels of TLR4, TLR5, NLRC4, active caspase-4 and matured forms of IL-18 and IL-1β were detected by western blot. Western blot image is representative of three independent experiments. (C) Western blot images show protein levels of pro- and matured forms of caspase-4, IL-18 and IL-1β in hCFs infected with PAO1 and PA14 wild-type and PA14 ΔflgK and PA14 ΔpopB mutant strains. Protein expression for uninfected hCFs is also shown. The rabbit polyclonal anti- caspase-4, -IL-18, and -IL-1β antibodies are specific for binding to the mature form of each respective protein and with less affinity for the pro-forms. Respective molecular weights of pro and mature forms are denoted with arrows. A representative image from n = 5 independent experiments is shown.
Protein levels detected by western blot were consistent with the levels of gene expression quantified by RT-qPCR. After 6 h of hCFs infection with wild-type PAO1 and PA14, significant levels of TLR4, TLR5, NLRC4, native form of caspase-4 and IL-1β and IL-18 mature-formed proteins were detected by western blot with quantification of protein densitometry ratios relative to endogenous β-actin (Figure 2B, Supplementary Table 2). Significantly lower levels of production of these molecules were induced by the PA14 ΔflgK and ΔpopB mutant strains (P < 0.05) and the percentage reductions in inflammatory molecule protein densitometry values are shown in Supplementary Table 2. In comparison with wild-type bacteria, infection with the complemented PA14 ΔflgK pUCP19-flgK and PA14 ΔpopB pUCP19-popB strains restored to varying degrees the protein levels of TLR4, TLR5, NLRC4, caspase-4 and mature IL-1β and IL-18 (Figure 2B, Supplementary Table 2). Thus, complementation of ΔflgK restored ~50, 85, 60, 40, 32, and 61% of TLR4, TLR5, NLRC4, Caspase-4, IL-18, and IL-1β protein levels, respectively (Supplementary Table 2). Complementation of ΔpopB restored ~65, 60, 62, 57 37, and 73% of TLR4, TLR5, NLRC4, Caspase-4, IL-18, and IL-1β protein levels, respectively (Supplementary Table 2).
Protein levels of the caspase-4 pro-enzyme, and IL-18 and IL-1β pro-forms were also analyzed by western blot. Caspase-4 pro-enzyme and pro-IL-18 protein expression was found to be constitutive, whereas pro-IL-1β was induced by P. aeruginosa wild-type infection, reduced by infection with the ΔpopB mutant and very weakly induced by infection with the PA14 ΔflgK mutant (Figure 2C).
hCFs Infected with P. aeruginosa Release Extracellular IL-1β but Not IL-18
The inflammatory cytokine profile of hCFs infected with wild-type P. aeruginosa is characterized predominantly by the production of IL-1β and IL-18 with the expression of other cytokines and chemokines not significant (manuscript submitted elsewhere). The early release of mature IL-1β and IL-18 was quantified by MSD-ELISA from the supernatants of hCFs infected with wild-type PAO1 and PA14 strains and isogenic PA14 ΔflgK and ΔpopB mutants after 6 h. No significant differences in IL-1β were found between PAO1 and PA14 (P > 0.05). Both P. aeruginosa wild-type strains induced ~30 pg/mL of IL-1β secretion in hCFs after 6 h of infection (Figure 3A). By contrast, a significant reduction in IL-1β secretion to levels of ~5 pg/mL was observed from hCFs infected with the PA14 ΔflgK mutant strain. These lower levels of extracellular IL-1β detected in the supernatants of hCFs infected with the PA14 ΔflgK are in agreement with the low intracellular IL-1β levels shown in Figure 2B under the same infection conditions. There were no significant differences (P > 0.05) in the levels of IL-1β secretion induced by the PA14 ΔpopB mutant (~20 pg/mL) compared to wild-type PA14 after 6 h (Figure 3A). Complementation of each mutation restored the levels of IL-1β release similar to those induced by the PA14 wild-type strain.
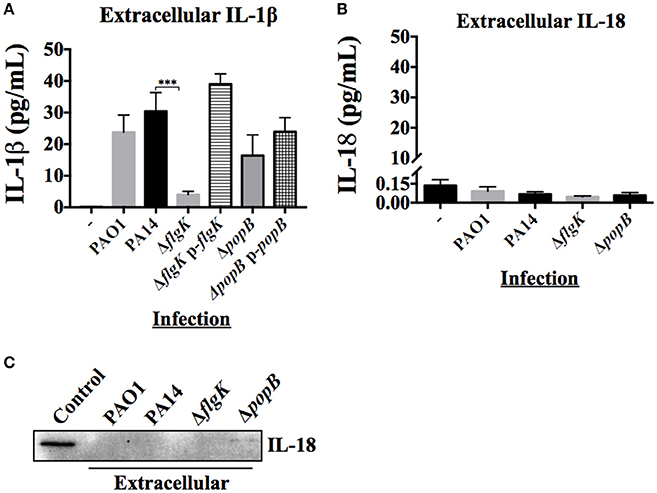
Figure 3. Extracellular levels of IL-18 and IL-1β cytokines during hCFs infection with P. aeruginosa. IL-1β and IL-18 cytokine release was measured after 6 h hCFs infection with PAO1 and PA14 wild-type and PA14 ΔflgK and PA14 ΔpopB mutant and complemented strains (MOI = 10). The columns represent the mean and error bars the SEM from n = 3 independent experiments. MSD-ELISA was used to quantify the levels of extracellular mature IL-1β (A) and IL-18 (B). Statistical significance was calculated with a one-way ANOVA with Dunnett's multiple comparison test and it is represented by asterisks as follows: ***P < 0.001. (C) Extracellular IL-18 detected by western blot in TCA-precipitated supernatants after 6 h infection of hCFs with P. aeruginosa wild-type and mutant strains. A total protein extract from hCFs infected with PA14 was used as a positive control for IL-18 detection (Control).
Measurement of IL-18 in infected hCFs supernatants revealed levels of extracellular IL-18 of ≤0.25 pg/mL for all infection conditions, which was similar to uninfected controls (P > 0.05; Figure 3B). Western blotting of IL-18 protein precipitated from infected hCF-supernatants collected after 6 h infection did not show the presence of mature IL-18 (Figure 3C), which was in agreement with the MSD-ELISA data.
Signal Pathway Inhibition in hCFs Appears to Influence IL-1β and IL-18 Expression and Release
Pathway inhibition was used to target key intracellular signaling molecules stimulated in hCFs by P. aeruginosa flagellum and TTSS that potentially could contribute to IL-18 and IL-1β production. Since our results showed similar cytokine production in hCFs caused by PAO1 and PA14 infection, only infection with PA14 was used for the subsequent inhibition experiments. Confluent monolayers were treated with MyD88i, TAK-242, and MyD88i/TAK-242 inhibitors and then infected with PA14 wild-type and PA14 ΔflgK and ΔpopB mutant strains for 6 h. MyD88i blocks the signal through the MyD88 molecule and TAK-242 interferes with the association between the TIRAP and TRAM molecules to TLRs and inhibits the consequent signaling triggered by the receptor. Although TAK-242 has been reported to have selectivity for the LPS receptor TLR4, inhibitory effect has also been seen with other TLRs including TLR5 (Loiarro et al., 2005; Takashima et al., 2009; Matsunaga et al., 2011). Intracellular IL-18 and IL-1β protein expression was examined by western blot (Figure 4A) and extracellular release of these cytokines was quantified by MSD-ELISA (Figure 4B). Treatment of hCF monolayers with MyD88i, TAK-242 alone and MyD88i/TAK-242 together, did not induce IL-18 or IL-1β production (Supplementary Figure 3).
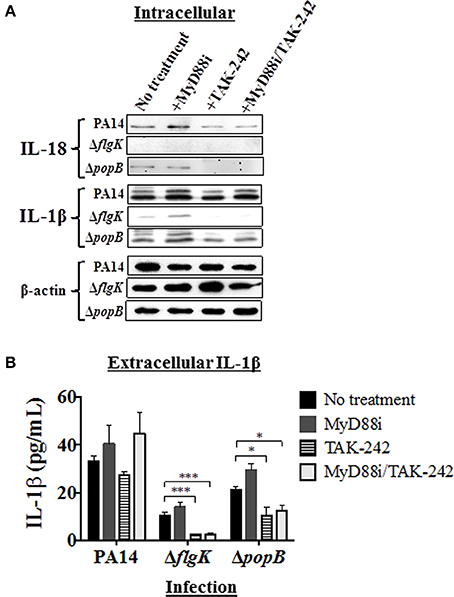
Figure 4. Effects of pathway inhibition on IL-18 and IL-1β production. Cells were treated with MyD88i, TAK-242, MyD88i/TAK-242 inhibitors and then infected with PAO1, PA14, ΔflgK and ΔpopB strains (MOI = 10). (A) Western blot was used to detect intracellular mature IL-18 and IL-1β cytokines after 6 h of bacterial infection. Detection of intracellular levels of β-actin was used as endogenous and loading control. (B) Quantification of extracellular IL-1β cytokine after 6 h of infection. The columns represent the means and the error bars the SEM of n = 3 independent experiments. Statistical significance was calculated with a one-way ANOVA with Dunnett's multiple comparison test and it is represented by asterisks as follows: *P < 0.05, ***P < 0.001.
Infection with PA14 wild-type induced intracellular IL-18 protein production and protein densitometry values (from n = 3 independent western blots) revealed no significant differences in intracellular IL-18 protein expression induced by addition of any of the inhibitors (Figure 4A and Supplementary Table 3). Infection with the PA14 ΔpopB mutant also induced mature IL-18 intracellular expression, but addition of TAK-242 (alone or in combination with MyD88i) appeared to significantly decrease its intracellular production (P < 0.01; Figure 4A and Supplementary Table 3), whereas MyD88i alone had no significant effect. PA14 ΔflgK did not induce intracellular IL-18 production, and secretory IL-18 was absent in all treated hCFs, as previously observed (Figures 3B,C).
Infection with wild-type PA14 induced intracellular IL-1β protein production, which was not inhibited by MyD88i or TAK-242 (Figure 4A and Supplementary Table 3). In cells infected with PA14 wild-type, levels of extracellular IL-1β were apparently stimulated by the addition of MyD88i or the MyD88i/TAK-242 combination, but no statistically significant differences were found compared to untreated cells (P > 0.05, Figure 4B). PA14 ΔflgK mutant induced low levels of intracellular and extracellular IL-1β in agreement with the results in Figures 2B, 3B, and were null with the addition of TAK-242, alone or in combination with MyD88i (Figures 4A,B and Supplementary Table 3B). Addition of TAK-242 also appeared to diminish intracellular and extracellular IL-1β production in hCFs infected with the PA14 ΔpopB mutant (Figures 4A,B and Supplementary Table 3).
TLR5 Silencing in hCFs Decreases IL-18 and IL-1β Expression and Release
Our results showed that infection with the PA14 ΔflgK mutant led to reduced IL-18 and IL-1β expression by hCFs. In order to demonstrate that the flagellum may be involved in inducing cytokine expression by these cells, we next tested the hypothesis that silencing the flagellum receptor TLR5 in hCFs would lead to a reduction of cytokine production regardless of bacterial adhesion. For these experiments, the flagellum filament protein FliC of P. aeruginosa was used as a control to demonstrate TLR5 stimulation. Recombinant (r) FliC was cloned and over-expressed in E. coli using the pET expression system (Supplementary Figure 1). Wild-type hCFs and hCFs that were TLR5-silenced using siRNA-TLR5 were stimulated with the PA14 wild-type, ΔflgK mutant strain and with 5 μg/mL of rFliC, and IL-18 and IL-1β gene expression was measured by RT-qPCR. Non-stimulated cells were used as a control.
Control experiments demonstrated that hCFs transfected with siRNA-TLR5 and infected with PA14 for 6 h significantly diminished TLR5 gene (P < 0.0001; Supplementary Figure 4A) and protein (Supplementary Figure 4B) expression compared to control Mock transfection. Expression of il-18 gene was reduced ~2 fold in hCF-siTLR5 compared to hCFs when stimulated with PA14 and rFliC for 6 h (Figure 5A). Infection with PA14 ΔflgK mutant did not induce IL-18 RNA expression and TLR5-silencing did not affect IL-18 expression, which is in agreement with the results shown in Figure 4A. Similarly, decreased levels of IL-1β RNA expression in hCFs-siTLR5 were found following stimulation with PA14 and rFliC for 6 h (Figure 5B). Otherwise, IL-1β RNA expression was slightly increased in hCFs-siTLR5 infected with the PA14 ΔflgK mutant compared to no infection (Figure 5B).
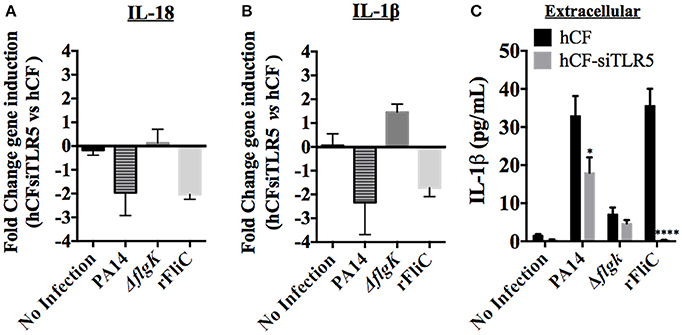
Figure 5. Effect of TLR5 silencing in hCFs on IL-18 and IL-1β expression and release. Wild-type hCFs and siTLR5-hCFs were infected with PA14 wild-type and ΔflgK mutant strain at MOI = 10, and stimulated with 5 μg/mL of rFliC for 6 h. LAL assay quantified that rFliC contained ~150 pg of LPS/mg of protein (<0.00002% w/w LPS) and RT-qPCR showed that these levels of E. coli LPS did not stimulate any gene transcription in hCFs (data not shown). Non-infected monolayers were used as a control. (A,B) Gene expression of IL-18 (A) and IL-1β (B) was measured by RT-qPCR. The bars show the fold changes in gene expression in siTLR5-hCFs compared to wild-type hCFs, which were determined using the 2−ΔΔCT method (Livak and Schmittgen, 2001). (C) Extracellular IL-1β levels measured by MSD-ELISA. rFliC was delivered intracellularly using SLO treatment. SLO treatment alone did not stimulate significant IL-1β release by hCFs (data not shown). The graph shows the levels of extracellular IL-1β quantified in the supernatants of hCFs and siTLR5-hCFs after 6 h following the different infection and stimulatory conditions. Statistical significance was calculated with a one-way ANOVA with Dunnett's multiple comparison test and it is represented by asterisks as follows: *P < 0.05, ****P < 0.0001.
Secreted cytokines (IL-18 and IL-1β) were quantified from each supernatant after 6 h of hCF and hCF-siTLR5 incubation with PA14, ΔflgK mutant and rFliC (Figure 5C). SLO-treatment was used to deliver rFliC intracellularly. As expected, no extracellular IL-18 was detected. Levels of secreted IL-1β protein were significantly reduced in supernatants of hCFs-siTLR5 compared to those detected in hCFs, both after stimulation with PA14 (P < 0.05 and ~30% of reduction). Most significantly, hCFs-siTLR5 treated with rFliC showed a reduction of IL-1β secretion of >95% (P < 0.0001) compared to wild-type cells (Figure 5C), which was similar to the data obtained by RT-qPCR (Figure 5B). No significant extracellular IL-1β secretion was induced by SLO treatment alone of hCFs (~0.7 pg/mL, data not shown).
To examine if LPS was responsible, possibly, for the IL-1β induction observed in hCFs-siTLR5 infected with the PA14 ΔflgK mutant strain (Figure 5B), IL-1β gene expression was measured in hCFs-siTLR5 treated with the TAK-242 inhibitor. Under these inhibitory conditions, signaling through TLR5 (flagellin receptor) and TLR4 (LPS receptor) should be blocked. Repression of IL-1β expression was detected in both hCFs and hCFs-siTLR5 infected with PA14 ΔflgK mutant (P < 0.05, Figure 6A) treated with TAK-242. These transcriptional results were confirmed by detecting IL-1β protein expression under the same inhibitory conditions: addition of TAK-242 blocked the little IL-1β production induced in hCFs infected with the PA14 ΔflgK mutant (Figure 6B). In order to examine inhibitory effects on TLR4 sensing of LPS, IL-1β gene expression was also measured by RT-qPCR in hCFs that were stimulated with 100 ng/mL of pure Pseudomonas LPS, with and without TAK-242, Myd88i and TAK-242/Myd88i treatments. As shown in Figure 6C, addition of either inhibitor decreased IL-1β expression in hCFs treated with pure Pseudomonas LPS, compared to untreated cells. Similar experiments were done measuring IL-18 expression, but no increases in IL-18 expression were detected in LPS-stimulated hCFs (data not shown). No extracellular IL-1β protein was detected after hCF stimulation with P. aeruginosa LPS alone or in the presence of any inhibitor (data not shown).
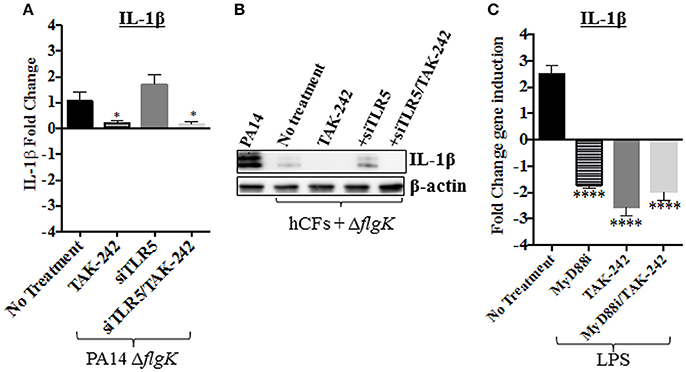
Figure 6. Effect of PA14 ΔflgK mutant LPS on IL-1β expression by hCFs. Wild-type hCFs and siTLR5-hCFs monolayers were treated with the TAK-242 inhibitor and then infected with PA14 ΔflgK mutant strain at MOI = 10 for 6 h. Un-treated and uninfected monolayers were used as control. (A) Gene expression of IL-1β was measured by RT-qPCR. The graph shows the fold changes of IL-1β expression calculated relative to the corresponding un-infected cell monolayer (hCF or siTLR5-hCF). (B) Intracellular IL-1β mature protein measured by western blot. A total protein extract from hCFs infected with PA14 was used as a positive control for IL-1β detection and intracellular levels of β-actin detected as an endogenous and loading control. (C) Gene expression of IL-1β in hCF monolayers un-treated and treated with TAK-242, Myd88i and TAK-242/Myd88i followed by stimulation with 100 ng/mL of P. aeruginosa LPS. The fold changes of IL-1β expression are calculated relative to un-stimulated monolayers. Statistical significance was calculated with a one-way ANOVA with Dunnett's multiple comparison test and it is represented by asterisks as follows: *P < 0.05, ****P < 0.0001.
Treatment of hCF Stimulated with P. aeruginosa rFliC with MyD88i and TAK-242 Inhibits IL-18 and IL-1β Expression
Thus far, our data suggest that the P. aeruginosa flagellin (FliC) appears to plays an important role in inducing IL-18 and IL-1β expression through TLR5 stimulation. We tested the hypothesis that chemical inhibition of TLR5-mediated signaling pathways reduces cytokine gene expression induced by FliC. hCF monolayers were pre-treated with MyD88i, TAK-242 and both inhibitors combined, then stimulated with rFliC for 6 h and cytokine gene expression quantified by RT-qPCR (Figure 7). Controls were hCF monolayers without chemical pre-treatment and no stimulation with rFliC.
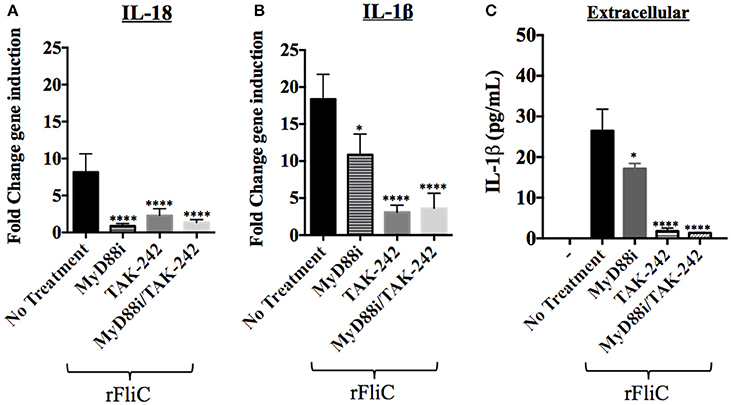
Figure 7. Effect of MyD88i, TAK-242 and MyD88i/TAK-242 inhibition on IL-18 and IL-1β expression in hCF stimulated with rFliC. IL-18 and IL-1β gene expression was measured by RT-qPCR from rFliC-stimulated hCF following treatment with MyD88i, TAK-242 and MyD88i/TAK-242 and untreated cells. The graph shows the fold changes of IL-18 (A) and IL-1β (B) gene expression calculated relative to the unstimulated and untreated hCF monolayers. The significance of the changes in gene expression shown in each inhibitory condition is compared to that measured in uninhibited hCF stimulated with P. aeruginosa rFliC. (C) Extracellular levels of IL-1β quantified from supernatants of inhibited hCFs following stimulation with rFliC. SLO-treatment was used to deliver rFliC intracellularly and no significant extracellular IL-1β was detected by treatment with SLO alone (data not shown). Unstimulated and inhibited cells were used as controls (-). Statistical significance was calculated with a one-way ANOVA with Dunnett's multiple comparison test and it is represented by asterisks as follows: *P < 0.05, ****P < 0.0001.
rFliC stimulation of hCF increased IL-18 gene expression ~10 fold compared to unstimulated cells and addition of MyD88i, TAK-242 and MyD88i/TAK-242 significantly reduced gene expression (P < 0.0001, Figure 7A). Similarly, rFliC stimulation increased IL-1β expression by ~20 fold, compared to unstimulated monolayers (Figure 7B). Inhibition with MyD88i alone reduced IL-1β gene expression by ~40% (P < 0.05), whereas treatment with TAK-242 and MyD88i/TAK-242 combined decreased IL-1β gene expression by >75% (P < 0.0001) compared to untreated cells (Figure 7B). Secreted IL-1β was quantified from supernatants of hCF stimulated with rFliC alone or in presence of the inhibitors. SLO-treatment was used to deliver the purified protein intracellularly. As shown in Figure 7C, MyD88i treatment reduced release of IL-1β by ~20%. By contrast, addition of TAK-242, alone or in combination with MyD88i, significantly reduced extracellular IL-1β release by >95% (P < 0.0001) compared to non-inhibited rFliC-stimulated hCFs.
Discussion
The major finding from our study was that flagellin-mediated TLR5 stimulation induces IL-1β and IL-18 expression in an ex vivo human model of P. aeruginosa keratitis using primary corneal fibroblasts (hCF), but that only IL-1β is released extracellularly. By contrast, the P. aeruginosa TTSS (PopB) appears to have a minor role for inducing these cytokines by hCFs. Bacterial activation of PPR and signaling molecules that lead to inflammasome activation are widely reported to induce IL-1β and IL-18 cytokines and to promote pyroptotic cell death (Lamkanfi and Dixit, 2012). In our study, expression of TLR4, TLR5, NLRC4, caspase-4, and matured IL-18 and IL-1β cytokines was induced in hCFs after infection with P. aeruginosa wild-type strains. We observed no up-regulation of TLR2 or NLRP3 in hCFs, which has also not been observed in mice infected with P. aeruginosa (Sutterwala et al., 2007; Sun et al., 2010). In addition, caspase-4, but not caspase-1, was detected in hCF cells infected with P. aeruginosa. Stimulation of caspase-4 in our human cell culture model agrees with previous studies done in mice, for which caspase-11, the murine caspase-4-homolog, was reported to be targeted by NLRC4 (Kayagaki et al., 2011). Furthermore, caspase-4 activation in human macrophages has also been detected after infection with the different Gram negative bacteria Legionella pneumophila, Yersinia pseudotuberculosis and Salmonella enterica serovar Typhimurium (Casson et al., 2015).
We observed that deletion of the flagellum (ΔflgK) resulted in lower adhesion of P. aeruginosa to hCFs, which is likely to be the main explanation for the reduced expression and activation of inflammatory-associated molecules as a consequence of lower PRR stimulation and intracellular delivery of FliC monomer. This mutant is capable of producing similar amounts of FliC as the wild-type strain, but cannot enable polymerization of FliC to produce a functional flagellum (Patankar et al., 2013). Intracellular FliC could possibly be delivered by the P. aeruginosa TTSS, which has been described already for J77A.1 macrophages infected with P. aeruginosa (Ince et al., 2015). It is possible that FliC could then bind human NAIP prior to NLRC4 stimulation, which has been reported similarly for human primary monocyte-derived macrophages infected with S. enterica serovar Typhimurium (Ince et al., 2015). In our study, the effects of deleting the TTSS PopB protein, despite showing some reduction in expression of inflammatory-associated molecules, appeared to have no effect on cytokine protein expression or release by hCFs. Therefore, our data suggest that FliC, rather than PopB, appears to play a more significant role in stimulating IL-1β and IL-18 production by hCF. Significantly, both cytokines were expressed intracellularly, but only IL-1β was released. It is possible that IL-18 Binding Protein (IL-18BP), which is constitutively expressed and binds IL-18 with high affinity and inhibits cytokine function (Dinarello et al., 2013), binds the mature IL-18 protein within hCFs and prevents cytokine release during P. aeruginosa infection in our model.
Both IL-1β and IL-18 expression by hCFs relied on TLR5 stimulation by P. aeruginosa flagellin. The inflammatory response triggered by the stimulation of TLR5 by P. aeruginosa flagellin has also been reported in human corneal epithelial cells (Zhang et al., 2003). However, stimulation through TLR4 by pure Pseudomonas LPS and LPS present on live bacteria, also appeared to contribute to inducing IL-1β, but not IL-18, expression in our model. These results confirm our observations of up-regulated TLR4 expression in hCFs stimulated with pure P. aeruginosa LPS (Wong et al., 2011), but this previous study only measured TLR4 induction after 24 h. In the early stages of infection in our model, it is possible that flagellin and LPS have dual roles in PRR activation and IL-1β production. Our data from experiments in which siTLR5-hCF knockout cells, infected with non-flagellated Pseudomonas, showed a minor induction of IL-1β expression, demonstrate that LPS probably contributes to cytokine induction. It has been reported that LPS stimulates the NLRP3 inflammasome (Guo et al., 2015). However, in our study, we found no expression of NLRP3 in hCFs infected with Pseudomonas: therefore, this may explain why we did not observe extracellular matured IL-1β production in our experiments with pure LPS. The dominant inducer appears to be the flagellin, and this conclusion is supported by studies of murine keratitis provoked by P. aeruginosa infection, wherein LPS stimulation of TLR4 occurred only in animals infected with a flagellin-mutant strain of P. aeruginosa (Sun et al., 2010).
In our study, inhibition of the signaling molecule MyD88 using MyD88i appeared to affect predominantly IL-18 expression in hCFs, rather than IL-1β. However, addition of TAK-242 significantly reduced and, in some cases, blocked hCF production of both cytokines. Activation through TLR5 is able to stimulate TRAM/TRIF molecules in addition to MyD88, e.g., as observed in human colonic epithelial cells and in murine corneal macrophages infected with P. aeruginosa (Choi et al., 2010; Sun et al., 2010). Since the inhibitory effects of TAK-242 have been also reported to occur through other TLRs (Matsunaga et al., 2011), we hypothesized that in hCFs, TLR5 could trigger signaling via stimulation of MyD88 and the TRAM-TRIF molecules for both IL-18 and IL-1β expression, with the former more dependent on signaling mediated through MyD88. However, detection of IL-18 and IL-1β expression in wild-type P. aeruginosa-infected hCF cultures that had been treated with Myd88i and TAK-242, suggests an alternative pathway for cytokine expression. In this model, IL-18 expression would be still dependent on flagellin sensing. Moreover, it is possible that this alternative mechanism could be influenced by TTSS-sensing, since only hCF infected with the PA14 ΔpopB mutant inhibited with TAK-242, showed reduced IL-1β and null IL-18 protein expression. The involvement of the respective TTSS of Yersinia pseudotuberculosis and S. typhimurium in a mechanism of NF-κβ activation in a TLR-independent manner for cytokine production has been described in murine models and also in human embryonic kidney 293T cells in vitro (Hapfelmeier et al., 2005; Auerbuch et al., 2009).
In our study, we have shown that P. aeruginosa adheres to hCFs in vitro. Bacterial adhesion appears to be a key step for enabling PAMPs stimulation of cellular PRRs. We have also observed during the time-course of infection, that P. aeruginosa can invade hCFs and reside within membrane-bound vacuoles (manuscript submitted elsewhere). It is known that both IL-18 and IL-1β require the action of the inflammasome to mature into their active forms. NLRC4 is reported to sense the intracellular rod components of Pseudomonas TTSS and FliC (after binding the hNAIP protein) (Zhao et al., 2011), both PAMPs examined in this study. Furthermore, TLR5 is a PPR receptor anchored within the mammalian cell membrane and it is likely that only specific bacterial PAMP adhesion stimulates the downstream signaling pathway. However, the possibility that the process of invasion and the presence of intracellular Pseudomonas bacteria contribute to inflammasome activation cannot be excluded entirely and potential mechanisms require further study.
Infectious Pseudomonas keratitis can lead to visual impairment as a consequence of the interactions of the bacterium with host tissue, the triggered host innate inflammatory response that leads to the characteristic influx of neutrophils, and the therapeutic drugs (antibiotics and corticosteroids) used to treat the infection (Taube et al., 2015). Activation of the innate inflammatory response and neutrophil influx are essential host defenses needed to control Pseudomonas infection, and it is possible that inhibition of these host responses may be detrimental in the early stages of infection (Rock et al., 2010). However, over-stimulation of host defenses can cause significant disease pathology (Rock and Kono, 2008). Clinically observed corneal tissue damage is likely a consequence of Pseudomonas virulence attributes and the strength of the innate inflammatory response, and is exacerbated significantly by the activities of infiltrating neutrophils. Moreover, over-stimulation of the inflammatory response can continue even when the eye is sterilized with antibiotics (O'brien, 2003), with no alleviation of tissue damage. Although topical corticosteroids are frequently used in clinical treatment, this is a non-specific attempt to reduce inflammation and can be associated with ocular complications (Taube et al., 2015). On balance, dampening the gross inflammatory response in Pseudomonas keratitis is probably more beneficial than allowing pathogen clearance through natural host defenses that induce irreversible tissue damage and permanent sight loss. In the current study, our findings show how P. aeruginosa stimulates IL-18 and IL-1β expression in hCFs during the early stages of infection and they describe an important role for bacterial flagellin. We propose, therefore, that blocking flagellin-PRR-signaling interactions could be an adjunctive approach with antibiotic chemotherapy to ameliorate the initial inflammatory response toward P. aeruginosa infection in human keratitis.
Author Contributions
MdMC, PH, and MC designed the experimental research. MdMC performed the experiments. MdMC, PH, and MC interpreted and analyzed the data. MdMC, PH, and MC wrote the manuscript.
Funding
MdMC, PH, and MC acknowledge the support from the Network for Antimicrobial Resistance and Infection Prevention (NAMRIP, University of Southampton).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank David Johnston and Laurie Lau, respectively, for help in confocal microscopy and MSD-ELISA experiments. We thank Tilman Sanchez-Elsner for guidance in establishing the siRNA experimental protocol. MdMC was a postdoctoral research fellow supported by the Royal College of Surgeons of Edinburgh, Gift of Sight, National Eye Research Centre and the British Council for Prevention of Blindness.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fcimb.2017.00130/full#supplementary-material
Footnotes
1. ^TIR: Toll/interleukin-1 receptor.
2. ^MyD88: Myeloid differentiation primary response gene 88.
3. ^TRIF: Toll/interleukin-1 receptor (TIR) domain containing adapter inducing interferon-β.
4. ^TIRAP: Toll/interleukin-1 receptor (TIR) domain containing adapter protein.
5. ^MAL: MyD88 adapter-like.
6. ^TRAM: TRIF-related adaptor molecule.
7. ^NF-κβ: Nuclear Factor Kappa-light-chain-enhancer of activated B cells.
8. ^IRF: Interferon regulatory factor.
9. ^NLRP: NLR family pyrin domain containing.
10. ^AIM2: Absent in melanoma 2.
11. ^NLRC: NLR family CARD domain containing.
References
Auerbuch, V., Golenbock, D. T., and Isberg, R. R. (2009). Innate immune recognition of Yersinia pseudotuberculosis type III secretion. PLoS Pathog. 5:e1000686. doi: 10.1371/journal.ppat.1000686
Berg, H. C. (2003). The rotary motor of bacterial flagella. Annu. Rev. Biochem. 72, 19–54. doi: 10.1146/annurev.biochem.72.121801.161737
Casson, C. N., Yu, J., Reyes, V. M., Taschuk, F. O., Yadav, A., Copenhaver, A. M., et al. (2015). Human caspase-4 mediates non-canonical inflammasome activation against gram-negative bacterial pathogens. Proc. Natl. Acad. Sci. U.S.A. 112, 6688–6693. doi: 10.1073/pnas.1421699112
Chen, G., Shaw, M. H., Kim, Y. G., and Nuñez, G. (2009). NOD-like receptors: role in innate immunity and inflammatory disease. Annu. Rev. Pathol. 4, 365–398. doi: 10.1146/annurev.pathol.4.110807.092239
Choi, Y. J., Im, E., Chung, H. K., Pothoulakis, C., and Rhee, S. H. (2010). TRIF mediates Toll-like receptor 5-induced signaling in intestinal epithelial cells. J. Biol. Chem. 285, 37570–37578. doi: 10.1074/jbc.M110.158394
Dinarello, C. A., Novick, D., Kim, S., and Kaplanski, G. (2013). Interleukin-18 and IL-18 binding protein. Front. Immunol. 4:289. doi: 10.3389/fimmu.2013.00289
Ghosh, P. (2004). Process of protein transport by the type III secretion system. Microbiol. Mol. Biol. Rev. 68, 771–795. doi: 10.1128/MMBR.68.4.771-795.2004
Guo, H., Callaway, J. B., and Ting, J. P. (2015). Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 21, 677–687. doi: 10.1038/nm.3893
Hapfelmeier, S., Stecher, B., Barthel, M., Kremer, M., Müller, A. J., Heikenwalder, M., et al. (2005). The Salmonella pathogenicity island (SPI)-2 and SPI-1 type III secretion systems allow Salmonella serovar typhimurium to trigger colitis via MyD88-dependent and MyD88-independent mechanisms. J. Immunol. 174, 1675–1685. doi: 10.4049/jimmunol.174.3.1675
Hardy, S. J., Christodoulides, M., Weller, R. O., and Heckels, J. E. (2000). Interactions of Neisseria meningitidis with cells of the human meninges. Mol. Microbiol. 36, 817–829. doi: 10.1046/j.1365-2958.2000.01923.x
Hauser, A. R. (2009). The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat. Rev. Microbiol. 7, 654–665. doi: 10.1038/nrmicro2199
Hauser, A. R. (2011). Pseudomonas aeruginosa: so many virulence factors, so little time. Crit. Care Med. 39, 2193–2194. doi: 10.1097/CCM.0b013e318221742d
Hayashi, F., Smith, K. D., Ozinsky, A., Hawn, T. R., Yi, E. C., Goodlett, D. R., et al. (2001). The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410, 1099–1103. doi: 10.1038/35074106
Hung, M. C., Humbert, M. V., Laver, J. R., Phillips, R., Heckels, J. E., and Christodoulides, M. (2015). A putative amino acid ABC transporter substrate-binding protein, NMB1612, from Neisseria meningitidis, induces murine bactericidal antibodies against meningococci expressing heterologous NMB1612 proteins. Vaccine 33, 4486–4494. doi: 10.1016/j.vaccine.2015.07.032
Ince, D., Sutterwala, F. S., and Yahr, T. L. (2015). Secretion of flagellar proteins by the Pseudomonas aeruginosa Type III secretion-injectisome system. J. Bacteriol. 197, 2003–2011. doi: 10.1128/JB.00030-15
Irani, V. R., and Rowe, J. J. (1997). Enhancement of transformation in Pseudomonas aeruginosa PAO1 by Mg2+ and heat. Biotechniques 22, 54–56.
Janeway, C. A. Jr., and Medzhitov, R. (2002). Innate immune recognition. Annu. Rev. Immunol. 20, 197–216. doi: 10.1146/annurev.immunol.20.083001.084359
Kawasaki, T., and Kawai, T. (2014). Toll-like receptor signaling pathways. Front. Immunol. 5:461. doi: 10.3389/fimmu.2014.00461
Kayagaki, N., Warming, S., Lamkanfi, M., Vande Walle, L., Louie, S., Dong, J., et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. doi: 10.1038/nature10558
Lamkanfi, M., and Dixit, V. M. (2012). Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 28, 137–161. doi: 10.1146/annurev-cellbio-101011-155745
Latz, E., Xiao, T. S., and Stutz, A. (2013). Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411. doi: 10.1038/nri3452
Le Berre, R., Nguyen, S., Nowak, E., Kipnis, E., Pierre, M., Quenee, L., et al. (2011). Relative contribution of three main virulence factors in Pseudomonas aeruginosa pneumonia. Crit. Care Med. 39, 2113–2120. doi: 10.1097/CCM.0b013e31821e899f
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Loiarro, M., Sette, C., Gallo, G., Ciacci, A., Fantò, N., Mastroianni, D., et al. (2005). Peptide-mediated interference of TIR domain dimerization in MyD88 inhibits interleukin-1-dependent activation of NF-κB. J. Biol. Chem. 280, 15809–15814. doi: 10.1074/jbc.C400613200
Matsunaga, N., Tsuchimori, N., Matsumoto, T., and Ii, M. (2011). TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol. Pharmacol. 79, 34–41. doi: 10.1124/mol.110.068064
Miao, E. A., Mao, D. P., Yudkovsky, N., Bonneau, R., Lorang, C. G., Warren, S. E., et al. (2010). Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 107, 3076–3080. doi: 10.1073/pnas.0913087107
Mogensen, T. H. (2009). Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 22, 240–273, doi: 10.1128/CMR.00046-08
Notti, R. Q., and Stebbins, C. E. (2016). The structure and function of Type III secretion systems. Microbiol. Spectr. 4, 1–18. doi: 10.1128/microbiolspec.VMBF-0004-2015
O'brien, T. P. (2003). Management of bacterial keratitis: beyond exorcism towards consideration of organism and host factors. Eye 17, 957–974. doi: 10.1038/sj.eye.6700635
O'neill, L. A., Golenbock, D., and Bowie, A. G. (2013). The history of Toll-like receptors - redefining innate immunity. Nat. Rev. Immunol. 13, 453–460. doi: 10.1038/nri3446
Ozaki, E., Campbell, M., and Doyle, S. L. (2015). Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J. Inflamm. Res. 8, 15–27. doi: 10.2147/JIR.S51250
Parker, D., and Prince, A. (2013). Epithelial uptake of flagella initiates proinflammatory signaling. PLoS ONE 8:e59932. doi: 10.1371/journal.pone.0059932
Patankar, Y. R., Lovewell, R. R., Poynter, M. E., Jyot, J., Kazmierczak, B. I., and Berwin, B. (2013). Flagellar motility is a key determinant of the magnitude of the inflammasome response to Pseudomonas aeruginosa. Infect. Immun. 81, 2043–2052. doi: 10.1128/IAI.00054-13
Pier, G. B. (2007). Pseudomonas aeruginosa lipopolysaccharide: a major virulence factor, initiator of inflammation and target for effective immunity. Int. J. Med. Microbiol. 297, 277–295. doi: 10.1016/j.ijmm.2007.03.012
Puhar, A., and Sansonetti, P. J. (2014). Type III secretion system. Curr. Biol. 24, R784–R791. doi: 10.1016/j.cub.2014.07.016
Rock, K. L., and Kono, H. (2008). The inflammatory response to cell death. Annu. Rev. Pathol. 3, 99–126. doi: 10.1146/annurev.pathmechdis.3.121806.151456
Rock, K. L., Latz, E., Ontiveros, F., and Kono, H. (2010). The sterile inflammatory response. Annu. Rev. Immunol. 28, 321–342. doi: 10.1146/annurev-immunol-030409-101311
Schroder, K., and Tschopp, J. (2010). The inflammasomes. Cell 140, 821–832. doi: 10.1016/j.cell.2010.01.040
Schweizer, H. P. (1991). Escherichia-Pseudomonas shuttle vectors derived from pUC18/19. Gene 97, 109–121. doi: 10.1016/0378-1119(91)90016-5
Shah, A., Sachdev, A., Coggon, D., and Hossain, P. (2011). Geographic variations in microbial keratitis: an analysis of the peer-reviewed literature. Br. J. Ophthalmol. 95, 762–767. doi: 10.1136/bjo.2009.169607
Stapleton, F., and Carnt, N. (2012). Contact lens-related microbial keratitis: how have epidemiology and genetics helped us with pathogenesis and prophylaxis. Eye 26, 185–193. doi: 10.1038/eye.2011.288
Sun, Y., Karmakar, M., Roy, S., Ramadan, R. T., Williams, S. R., Howell, S., et al. (2010). TLR4 and TLR5 on corneal macrophages regulate Pseudomonas aeruginosa keratitis by signaling through MyD88-dependent and -independent pathways. J. Immunol. 185, 4272–4283. doi: 10.4049/jimmunol.1000874
Sutterwala, F. S., Mijares, L. A., Li, L., Ogura, Y., Kazmierczak, B. I., and Flavell, R. A. (2007). Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 204, 3235–3245. doi: 10.1084/jem.20071239
Takashima, K., Matsunaga, N., Yoshimatsu, M., Hazeki, K., Kaisho, T., Uekata, M., et al. (2009). Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br. J. Pharmacol. 157, 1250–1262. doi: 10.1111/j.1476-5381.2009.00297.x
Taube, M. A., Del Mar Cendra, M., Elsahn, A., Christodoulides, M., and Hossain, P. (2015). Pattern recognition receptors in microbial keratitis. Eye 29, 1399–1415. doi: 10.1038/eye.2015.118
Walev, I., Bhakdi, S. C., Hofmann, F., Djonder, N., Valeva, A., Aktories, K., et al. (2001). Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc. Natl. Acad. Sci. U.S.A. 98, 3185–3190. doi: 10.1073/pnas.051429498
Wong, Y., Sethu, C., Louafi, F., and Hossain, P. (2011). Lipopolysaccharide regulation of toll-like receptor-4 and matrix metalloprotease-9 in human primary corneal fibroblasts. Invest. Ophthalmol. Vis. Sci. 52, 2796–2803. doi: 10.1167/iovs.10-5459
Zhang, J., Xu, K., Ambati, B., and Yu, F. S. (2003). Toll-like receptor 5-mediated corneal epithelial inflammatory responses to Pseudomonas aeruginosa flagellin. Invest. Ophthalmol. Vis. Sci. 44, 4247–4254. doi: 10.1167/iovs.03-0219
Keywords: Pseudomonas aeruginosa, fibroblasts, human cornea, cytokine, NLRC4, flagellin, MyD88, TLR5
Citation: Cendra MdM, Christodoulides M and Hossain P (2017) Signaling Mediated by Toll-Like Receptor 5 Sensing of Pseudomonas aeruginosa Flagellin Influences IL-1β and IL-18 Production by Primary Fibroblasts Derived from the Human Cornea. Front. Cell. Infect. Microbiol. 7:130. doi: 10.3389/fcimb.2017.00130
Received: 10 November 2016; Accepted: 29 March 2017;
Published: 19 April 2017.
Edited by:
David Dockrell, University of Edinburgh, UKReviewed by:
Gill Diamond, University of Florida, USAHelen Maria Marriott, University of Sheffield, UK
Copyright © 2017 Cendra, Christodoulides and Hossain. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Myron Christodoulides, bWM0QHNvdG9uLmFjLnVr
†These authors have contributed equally to this work.