 Philip P. Adams
Philip P. Adams Carlos Flores Avile
Carlos Flores Avile Mollie W. Jewett
Mollie W. Jewett- Division of Immunity and Pathogenesis, Burnett School of Biomedical Sciences, University of Central Florida College of Medicine, Orlando, FL, United States
Knowledge of the transcriptional responses of vector-borne pathogens at the vector-pathogen interface is critical for understanding disease transmission. Borrelia (Borreliella) burgdorferi, the causative agent of Lyme disease in the United States, is transmitted by the bite of infected Ixodes sp. ticks. It is known that B. burgdorferi has altered patterns of gene expression during tick acquisition, persistence and transmission. Recently, we and others have discovered in vitro expression of RNAs found internal, overlapping, and antisense to annotated open reading frames in the B. burgdorferi genome. However, there is a lack of molecular genetic tools for B. burgdorferi for quantitative, strand-specific, comparative analysis of these transcripts in distinct environments such as the arthropod vector. To address this need, we have developed a dual luciferase reporter system to quantify B. burgdorferi promoter activities in a strand-specific manner. We demonstrate that constitutive expression of a B. burgdorferi codon-optimized Renilla reniformis luciferase gene (rlucBb) allows normalization of the activity of a promoter of interest when fused to the B. burgdorferi codon-optimized Photinus pyralis luciferase gene (flucBb) on the same plasmid. Using the well characterized, differentially regulated, promoters for flagellin (flaBp), outer surface protein A (ospAp) and outer surface protein C (ospCp), we document the efficacy of the dual luciferase system for quantitation of promoter activities during in vitro growth and in infected ticks. Cumulatively, the dual luciferase method outlined herein is the first dual reporter system for B. burgdorferi, providing a novel and highly versatile approach for strand-specific molecular genetic analyses.
Introduction
Vector-borne illnesses account for 17% of worldwide infectious diseases, amounting to over one billion cases yearly (World Health Organization, 2016). Ticks are notorious for delivering a diversity of infectious agents to their hosts during the blood meal. Of these pathogens the Borrelia burgdorferi sensu lato complex or Borreliella genus (Adeolu and Gupta, 2014), the spirochete group that causes Lyme disease, contributes the highest incidence of arthropod-transmitted bacterial infection worldwide (Schotthoefer and Frost, 2015). Particular to the United States, Borrelia (Borreliella) burgdorferi interaction with and colonization of Ixodes species is highly specific (de Silva et al., 2009), with no other natural arthropod vector identified to date.
Newly hatched larval ticks are not colonized with B. burgdorferi, as there is currently no evidence to support transovarial transmission of the pathogen (Rollend et al., 2013). Rather, larvae can become infected by feeding on one of the numerous small vertebrates that serve as reservoirs for B. burgdorferi in nature, such as the white-footed mouse Peromyscus leucopus. Larval ticks then undergo an approximate month-long morphogenesis process and molt into nymphs. All the while, B. burgdorferi reside in the tick midgut. Like the larvae, the infected nymphs take a single blood meal from a vertebrate followed by morphogenesis to adults. During nymph feeding, B. burgdorferi migrate from the midgut to the tick salivary glands and are transmitted to the vertebrate host, maintaining the spirochete in its enzootic cycle (Radolf et al., 2012). Therefore, it has been proposed that B. burgdorferi undergoes three major tick-related events that require complex genetic regulation: acquisition, persistence, and transmission (Iyer et al., 2015; Caimano et al., 2016).
Survival of B. burgdorferi in the tick requires that the spirochete overcome a number of environmental stress conditions, such as starvation and assault from tick immune factors (Radolf et al., 2012; Caimano et al., 2016). Recently, open reading frame-based microarray analysis has provided insight into the gene expression changes that occur in the B. burgdorferi transcriptome in fed larvae, fed nymphs, and under mammalian host-like conditions in dialysis membrane chambers (Iyer et al., 2015). The unusual structure of B. burgdorferi's genome and its scarcity of characterized transcription factors, further contribute to interest in understanding the mechanisms of stress adaptation and gene regulation that the spirochete employs during its interaction with the tick vector. The B. burgdorferi segmented genome, in characterized type strain B31, is composed of an approximate 900 kb linear chromosome and 21 plasmids of size ranges 5–56 kb that include many annotated open reading frames (ORFs) of unknown function (Fraser et al., 1997; Casjens et al., 2012). A recent global examination and 5′ end mapping of the B. burgdorferi transcriptome by our laboratory has revealed that the spirochete is rich with “overlapping transcripts” where 63% of total RNA species are transcribed internal and 13% antisense to annotated open reading frames (Adams et al., 2017). Other recent RNA-seq based applications have also described the presence of these transcripts in B. burgdorferi (Arnold et al., 2016; Popitsch et al., 2017). These findings are supported by similar analyses in other bacteria, which have revealed complex transcriptomes that include a variety of antisense, intragenic, intergenic, and orphan transcripts, which in some cases represent the majority of transcript types as opposed to mRNAs for annotated open reading frames (Sharma et al., 2010; Kroger et al., 2012; Thomason et al., 2015). These discoveries drive the need for the development of new molecular genetic tools for investigating the expression patterns and functional roles of novel RNA transcripts in a strand-specific manner.
For over three decades, researchers have been isolating, expressing, and adapting bioluminescence genes for biomedical applications (de Wet et al., 1987; Lorenz et al., 1991). These techniques are based on the enzymatic (i.e., luciferase) oxidation of a substrate (i.e., luciferin) to generate light. Transcriptional reporters using bioluminescence read-outs have proven to be robust and sensitive molecular tools for investigating transcript expression (Andreu et al., 2011). Infectious disease-based research has resulted in the development of multiple luciferase systems for a variety of pathogens, and demonstrated that relative luciferase units of constitutively expressed bioluminescence reporters correlate to bacterial numbers (Andreu et al., 2011). Advanced and high-throughput adaptations for transcriptional reporters utilize multiple luciferase enzymes with unique substrates, which are compatible within the same experimental setup. In this manner, one luciferase serves as an experimental readout of promoter activity and the other as the normalization control for cell number (McNabb et al., 2005; Wright et al., 2005). A previously engineered B. burgdorferi codon-optimized Photinus pyralis (firefly) luciferase gene (Blevins et al., 2007), when fused to a constitutive promoter, has been successful for in vivo live imaging of B. burgdorferi dissemination during mouse infection (Hyde et al., 2011; Wager et al., 2015). Furthermore, this luciferase reporter has been used to characterize the promoters for a variety of annotated ORFs and novel RNAs during in vitro cultivation, in vivo mouse infection, and in infected mouse tissues ex vivo (Skare et al., 2016; Adams et al., 2017). However, this reporter plasmid is limited in that it does not contain a constitutive control reporter to allow normalization and quantitation of the data. In order to expand the utility of this approach, we engineered a dual luciferase plasmid that carries both a constitutively expressed B. burgdorferi codon-optimized Renilla reniformis (sea pansy) luciferase gene and the B. burgdorferi codon-optimized Photinus pyralis (firefly) luciferase gene driven by a promoter of interest. Luciferin, the substrate of Photinus pyralis luciferase, emits yellow-green photons (550–570 nm) of light (Marques and Esteves da Silva, 2009), whereas coelenterazine, the substrate of Renilla reniformis luciferase, produces light in the blue spectrum (470 nm) (Woo et al., 2008). Functioning on the premise that each luciferase enzyme requires unique substrates for bioluminescence readout, this approach provides a method for quantitative measurement of strand-specific transcription, in an environment of interest. It has been previously demonstrated that coelenterazine-based luciferase reporters are ineffective for in vivo live imaging detection of bacterial pathogens during murine infection (Andreu et al., 2010), despite successful in vivo applications for mammalian tumor systems (Bhaumik and Gambhir, 2002). Herein, our studies demonstrate the efficacy of the B. burgdorferi dual luciferase system for genetic studies during in vitro cultivation of spirochetes and analysis of transcriptional activity that occurs at the tick-pathogen interface, which is critical for understanding the interactions of B. burgdorferi with the tick vector for the development of novel therapeutic strategies for Lyme disease.
Materials and Methods
Bacterial Strains and Growth Conditions
B. burgdorferi clones used in this study were derived from strain B31. For genetic manipulations infectious low-passage clone A3-68Δbbe02 was utilized, which lacks cp9, lp56, and gene bbe02 on lp25 (Rego et al., 2011), and herein referred to as wild type. Spirochetes were cultivated in liquid Barbour-Stoenner-Kelly (BSK) II medium supplemented with gelatin and 6% rabbit serum (Barbour, 1984) and grown at 35°C with 2.5% CO2. Luciferase plasmids were engineered in DH5α E. coli, grown in LB broth or on LB agar plates containing 300 μg/ml spectinomycin when appropriate, and transformed into B. burgdorferi as previously described (Samuels, 1995). Transformants were selected by plating in solid BSKII medium as previously described (Rosa and Hogan, 1992), in the presence of 50 μg/ml streptomycin and/or 200 μg/ml kanamycin, when applicable. All transformants were verified by PCR to contain the plasmid content of the parent clone (Elias et al., 2002; Jewett et al., 2007).
Construction of the Dual Luciferase Plasmids
The Renilla reniformis luciferase gene (Lorenz et al., 1991) was codon-optimized for B. burgdorferi (rlucBb) with the OptimumGene™ algorithm, synthesized, and cloned into the E. coli vector pUC18 (Genscript) (Genebank accession number MF043582). All primer sequences are listed in Table 1. The rlucBb gene was PCR amplified from pUC18-rlucBb plasmid DNA using Phusion High-fidelity DNA polymerase (NEB) and primer pair 1732 and 1733. This also resulted in the addition of 27 bp of DNA from the 3′ of the flaB promoter to the 5′ of rlucBb. Concurrently, a DNA fragment containing the flaB promoter sequence with a 24 bp overhang from the 5′ of the rlucBb gene was Phusion-PCR amplified using B31 A3 genomic DNA and primer pair 1730 and 1731. The PCR fragments were ligated together by combining Gibson Assembly® Master Mix (NEB) and 0.16 pmol of each PCR fragment and incubating the reaction at 50°C for 1 h. One microliter of assembled product (flaBp-rlucBb) was Phusion-PCR amplified using primers 1730 and 1733 and the DNA fragment gel extracted using the QIAquick Gel Extraction kit (Qiagen), and cloned into pCR-Blunt using the Zero Blunt PCR cloning kit (Invitrogen) according to the manufacturer's instructions. The sequence of the flaBp-rlucBb cassette was verified by Sanger sequencing.

Table 1. Oligonucleotide primers used in this study.
The flaBp-rlucBb cassette was Phusion-PCR amplified from the pCR-Blunt flaBp-rlucBb plasmid using primer pair: 1850 and 1910, introducing BamHI and KpnI restriction sites. B. burgdorferi shuttle vectors containing the promoterless B. burgdorferi optimized Photinus pyralis luciferase gene (flucBb), flaBp-flucBb, ospAp-flucBb, or ospCp-flucBb (Blevins et al., 2007; Adams et al., 2017) were digested with BamHI and KpnI high fidelity enzymes (NEB), gel extracted using the QIAquick Gel Extraction kit (Qiagen), and ligated to the BamHI/KpnI-digested flaBp-rlucBb cassette using T4 DNA ligase (NEB), generating plasmids pCFA701, pCFA801, pCFA802, and pCFA803. All plasmid constructs were confirmed by PCR, restriction digest, and Sanger sequencing.
In vitro Dual Luciferase Assay
B. burgdorferi clones were grown to logarithmic phase (3–7 × 107 spirochetes/ml) or stationary phase (1–1.2 × 108 spirochetes/ml) in 15 ml of BSKII medium and pelleted at 3,210 × g for 10 min. Cells were washed with phosphate buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) and resuspended in 300 μl of PBS. Eighty microliters of each sample was used to measure the optical density at 600 nm (OD600) using a BioTek Synergy 4. This resulted in an average OD600 value of ~0.25 for logarithmic phase spirochetes and ~0.36 for stationary phase spirochetes. One hundred microliters of each sample was loaded into a black, solid bottom 96-well plate (Corning) and combined with 700 μM D-luciferin (Regis) in PBS or 3.5 mM water soluble native coelenterazine (NanoLight Technology) in PBS. For samples containing coelenterazine, one well was left empty, in all directions around each sample, to decrease signal overlap between samples. For determining B burgdorferi Photinus luciferase (FlucBb) and Renilla luciferase (RlucBb) sensitivity, spirochetes containing pCFA801 were grown to logarithmic phase in 15 ml of BSKII medium, cell density determined using a Petroff Hauser counting chamber, washed with PBS, and resuspended in PBS to a density of 2 × 106 cells/μl. Samples were serial diluted 10-fold and 100 μl of each dilution was loaded into a black, solid bottom 96-well plate (Corning) and combined with 700 μM D-luciferin or 3.5 mM coelenterazine. The relative luciferase units (RLUs) for FlucBb and RlucBb were determined by measuring photon emission in each well for 1 s, 10 times using the EnVision 2104 Multilabel Reader (PerkinElmer), following the addition of luciferin or coelenterazine substrate, respectively. Background relative FlucBb or RlucBb units, the average RLUs of the PBS control for either substrate, was subtracted from all experimental measurements, as appropriate. Background-subtracted relative FlucBb units were then normalized to the OD600 value or 108 background-subtracted relative RlucBb units of the same sample, when applicable (e.g., 4 × 104 FlucBb units/0.1 OD600 value = 4 × 105 relative FlucBb units/OD600; 4 × 104 FlucBb units/0.06 108 RlucBb units = 6.4 × 105 relative FlucBb units/108 RlucBb units). The limit of detection (LoD) and quantification (LoQ) for FlucBb and RlucBb were established as the average RLUs for PBS alone plus 3 or 10 standard deviations, respectively. All experiments were conducted in biological triplicate.
Ethics Statement
The University of Central Florida is accredited by the International Association for Assessment and Accreditation of Laboratory Animal Care. Protocols for all animal experiments were prepared according to the guidelines of the National Institutes of Health and were reviewed and approved by the University of Central Florida Institutional Animal Care and Use Committee.
B. burgdorferi Infection of Ticks
One week prior to inoculation, and throughout the duration of the study, mice were treated with 5 mg/ml streptomycin and 1 mg/ml Equal® sweetener in their water to maintain selection for the luciferase plasmid in the B. burgdorferi clones. Using B. burgdorferi carrying pJSB175, pCFA701, pCFA801, pCFA802, or pCFA803, groups of two 6–8 week old female C3H/HeN mice (Envigo) per clone were inoculated with 1 × 105 spirochetes per mouse 80% intraperitoneally and 20% subcutaneously. The inoculum doses were verified by colony forming unit (CFU) counts in solid BSKII medium. All inoculum were PCR verified to contain the endogenous B. burgdorferi plasmids of the parent clone as previously described (Elias et al., 2002; Jewett et al., 2007). Three weeks post inoculation mouse infection was confirmed by positive seroreactivity against B. burgdorferi protein lysate as previously described (Schwan et al., 1989; Jewett et al., 2007). Groups of approximately 200 naïve Ixodes scapularis larvae each (Centers for Disease Control, BEI resources) were fed to repletion on the B. burgdorferi infected mice (Jewett et al., 2009). Mice were further confirmed for infection by reisolation of spirochetes from bladder and joint tissues, as described (Showman et al., 2016). Larvae were analyzed for infection (Grimm et al., 2005; Jewett et al., 2009). Briefly, ticks were individually surface sterilized by sequential washes in 100 μl of 3% H2O2, 70% ethanol, and sterile H2O. Subsets of larvae were analyzed for infection by reisolation of spirochetes in BSKII medium containing RPA cocktail (60 μM rifampicin, 110 μM phosphomycin, and 2.7 μM amphotericin B), immediately post feeding to repletion. Approximately 2 weeks following feeding, additional subsets of larvae were crushed and plated in solid BSKII containing RPA cocktail and 50 μg/ml streptomycin to determine CFU counts/larva. The remaining larvae were maintained and allowed to molt into nymphs. Two groups of 10–18 infected nymphs per B. burgdorferi clone were fed to repletion on naïve 6–8 week old female C3H/HeN mice (Envigo). These mice were treated with 5 mg/ml streptomycin and 1 mg/ml Equal® sweetener in their water 1 week prior to the feeding, to help sustain the luciferase plasmids in B. burgdorferi within the feeding nymphs. Throughout the duration of the study, ticks were stored in glass desiccation jars containing saturated potassium sulfate for to maintain appropriate humidity.
In vivo Tick Dual Luciferase Assay
Approximately 2 weeks post feeding to repletion triplicate groups of 24 fed larvae or 8 fed nymphs per B. burgdorferi clone were crushed with a sterile pestle in 250 μl of PBS to generate tick extracts. For tick extracts, which were also plated for CFU counts, the ticks were first surface sterilized as described above, with a final wash in sterile PBS instead of H2O. Tick debris was allowed to settle and 100 μl of sample was removed and incubated with 700 μM D-luciferin (Regis) in PBS or 3.5 mM water soluble native coelenterazine (NanoLight Technology) in PBS. RLUs were measured as described for in vitro grown spirochetes. The limit of quantification (LoQ) for FlucBb was established as the average relative FlucBb units for PBS alone plus 10 standard deviations. The LoQ for RlucBb was established as the average relative RlucBb units for infected tick extracts with spirochetes containing pJSB175, which lacks the rlucBb gene, plus 10 standard deviations. Samples with relative FlucBb units below the LoQ were given a value of zero; whereas, samples with relative RlucBb units below the LoQ were removed from the analysis. Relative FlucBb units were normalized to 108 relative RlucBb units for each sample. One microliter of each fed nymph extract was also plated for CFUs in solid BSKII containing RPA cocktail and 50 μg/ml streptomycin.
Statistical Analysis
GraphPad Prism version 7.02 was used for all statistical analyses. One-way ANOVA was used for analysis of all luciferase assays. For statistical comparison of the relative FlucBb units normalized to OD600 or 108 relative RlucBb units, which had an extremely wide distribution (~101–107), all values were first square root transformed prior to statistical analysis. Following ANOVA, all samples were compared to the B. burgdorferi clones carrying the promoterless flucBb control plasmid pJSB161 or pCFA701 using Dunnett's multiple comparison test. To compare two groups (i.e., the same clone in logarithmic versus stationary phase) following ANOVA, Bonferroni's multiple comparison test was applied to determine significance. For association analysis, Pearson correlation coefficient (r) was determined. p ≤ 0.05 was considered statistically significant for all statistical tests.
Results
Generation of the B. burgdorferi Dual Luciferase Plasmid
The B. burgdorferi shuttle vector pJSB161 (Blevins et al., 2007) contains a promoterless B. burgdorferi codon-optimized Photinus pyralis luciferase gene (flucBb) with a BlgII restriction site upstream of a ribosome binding site (RBS) for flucBb (Figure 1A). This reporter plasmid allows a cloned promoter of interest to be analyzed for activity in a strand-specific manner via a bioluminescence detection method (Blevins et al., 2007; Skare et al., 2016; Adams et al., 2017). However, this approach does not allow for quantitative comparative analysis of promoter activity in different environments or between multiple promoters in the same environment due to the lack of an endogenous means to control for spirochete number across samples and conditions. Therefore to improve upon this technique for quantitative applications, we engineered a dual luciferase reporter system to constitutively express Renilla reniformis luciferase (rluc) (Lorenz et al., 1991), while maintaining flucBb for quantifying the activity of a promoter of interest. Codon usage in B. burgdorferi is biased (Fraser et al., 1997; Nakamura et al., 2000), as the A/T nucleotide frequency is at 71.8% across the genome (Fraser et al., 1997; Adams et al., 2017). Codon optimization has been shown to improve production and activity of non-B. burgdorferi proteins expressed in B. burgdorferi (Blevins et al., 2007; Hayes et al., 2010). Therefore to prevent rare codons interfering with the Renilla luciferase reporter, the OptimumGene™ algorithm (GenScript) was used to refine the codon adaption index (CAI) of rluc (Lorenz et al., 1991) for B. burgdorferi from 0.64 to 0.85 (where a CAI value of 1.0 indicates the highest proportion of the most abundant codons) and synthesized (GenScript). This codon-optimized rluc gene (rlucBb) (Genebank accession number MF043582) was cloned into pJSB161 (Blevins et al., 2007), for use in the dual luciferase reporter system under control of the constitutive promoter flaBp and corresponding ribosome binding site, generating pCFA701 (Figure 1B).
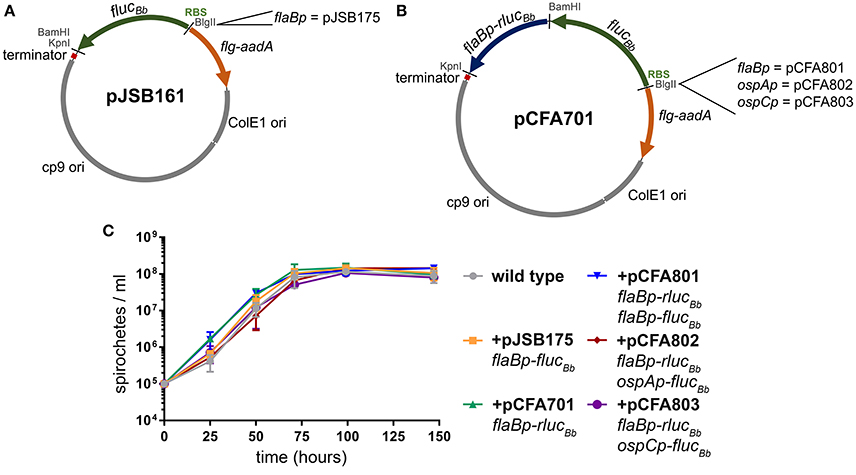
Figure 1. B. burgdorferi luciferase plasmids. All of the B. burgdorferi luciferase shuttle vectors were derived from pJSB161, which contains a Rho-independent transcription terminator sequence (terminator); ORFs 1, 2, and 3 of the B. burgdorferi cp9 replication machinery (cp9 ori); E. coli origin of replication (ColE1 ori); and the spectinomycin/streptomycin resistance cassette (flg-aadA) (Blevins et al., 2007). (A) The B. burgdorferi shuttle vector pJSB161 features a promoterless, B. burgdorferi codon optimized Photinus pyralis luciferase (flucBb), an upstream ribosome binding site (RBS) and a unique BlgII restriction site (Blevins et al., 2007). The plasmid pJSB175 was generated by addition of the flaBp promoter upstream of flucBb in pJSB161 (Blevins et al., 2007). (B) The B. burgdorferi codon optimized Renilla reniformis luciferase (rlucBb) gene under the control of the flaB promoter (flaBp-rlucBb) was added to pJSB161, generating the B. burgdorferi dual luciferase shuttle vector, pCFA701. Plasmids, pCFA801, pCFA802, and pCFA803, harbor the flaB, ospA, and ospC promoters, respectively, upstream of flucBb. (C) The density of B. burgdorferi clone A3-68Δbbe02 (wild type) alone or harboring various B. burgdorferi luciferase plasmids was assessed over a period of 144 h using a Petroff Hauser counting chamber and dark-field microscopy. The data are presented as the mean spirochete density (spirochetes/ml) ± standard deviation over time (hours).
B. burgdorferi survival in the tick vector is essential for maintenance of the pathogen in its enzootic cycle. The spirochete is known to change its transcriptional profile at different stages of tick colonization including: acquisition, persistence during the molt, and transmission to the mammalian host (Iyer et al., 2015; Caimano et al., 2016). Because of our interest in applying the dual luciferase reporter system to quantitative analysis of B. burgdorferi promoter activities in the tick, we selected three well characterized promoters with distinct patterns of expression in the tick environment for proof of principle analysis. The flagellar protein promoter, flaBp, is constitutively active (Ge et al., 1997). The promoter for outer surface protein A (ospAp) is active during in vitro culture and in the tick during acquisition and persistence. In contrast, the promoter for outer surface protein C (ospCp) is active in the feeding tick during transmission and the mammalian host during the early stages of infection (Schwan et al., 1995; Schwan and Piesman, 2000; Schwan, 2003; Srivastava and de Silva, 2008). The flaBp-rlucBb cassette was cloned into three previously constructed plasmids, each containing one of these promoters driving the expression of flucBb (Blevins et al., 2007; Adams et al., 2017), generating plasmids pCFA801, pCFA802, and pCFA803, respectively (Figure 1B). Spirochetes carrying pCFA701, pCFA801, pCFA802, or pCFA803 had no observed in vitro growth defect in BSKII medium compared to the wild type or B. burgdorferi carrying flaBp-flucBb alone (pJSB175) (Blevins et al., 2007, Figure 1C).
RlucBb Selectivity and Limit of Quantification
Photinus pyralis luciferase (Fluc) and Renilla reniformis luciferase (Rluc) are compatible for a dual reporter due to the specificity of each enzyme for distinct substrates (Bhaumik and Gambhir, 2002; McNabb et al., 2005). Therefore, we verified the selectivity of the FlucBb and RlucBb enzymes to recognize luciferin and coelenterazine, respectively. Based on our previous work using the flaBp-flucBb reporter (Adams et al., 2017), we performed these analyses with approximately 3 × 108 spirochetes harvested during log phase growth. As expected, the negative control, spirochetes not expressing flucBb or rlucBb (+pJSB161), demonstrated no significant relative luciferase units for either substrate compared to wild type. Spirochetes expressing flucBb alone (+pJSB175) demonstrated robust activity when incubated with luciferin, but no significant activity above the background of B. burgdorferi carrying pJSB161 when exposed to coelenterazine (Figure 2A). Conversely, spirochetes expressing rlucBb alone (+pCFA701) demonstrated strong activity when incubated with coelenterazine, but no significant activity above the negative control background when exposed to luciferin (Figure 2A). Spirochetes which express both flucBb and rlucBb (+pCFA801) demonstrated significant relative luciferase units compared to spirochetes containing pJSB161 for both luciferin and coelenterazine. The background relative luciferase units for wild type and negative control spirochetes exposed to coelenterazine were found to be approximately 10-fold higher than those of the same spirochetes incubated with luciferin. Collectively, these data validated the ability of the codon-optimized RlucBb enzyme to effectively oxidize coelenterazine and confirmed the specificity of the FlucBb and RlucBb enzymes for their respective substrates.
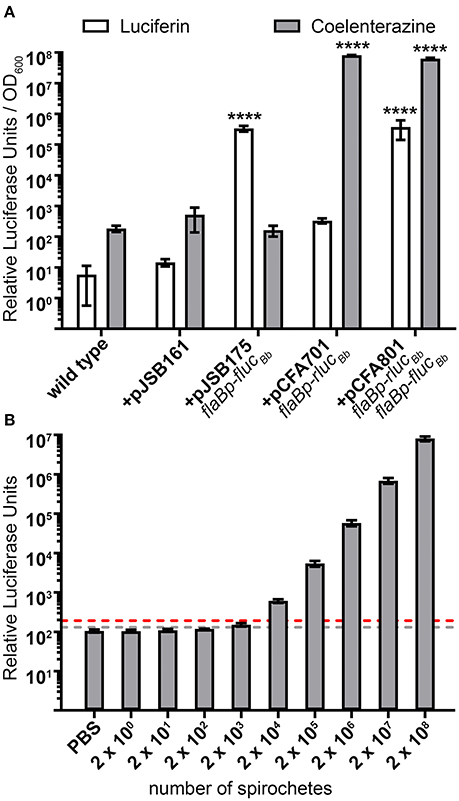
Figure 2. Selectivity and sensitivity of the dual luciferase assay in B. burgdorferi. (A) B. burgdorferi clones were grown to mid-logarithmic phase, and the in vitro luciferase assay performed with 700 μM D-luciferin or 3.5 mM coelenterazine. Relative luciferase units were normalized to optical density at 600 nm (OD600) and presented as the mean relative luciferase units/OD600 ± standard deviation for biological triplicate samples. The data were square root transformed and analyzed with a one-way ANOVA followed by Dunnett's multiple comparison test compared to B. burgdorferi containing the promoterless flucBb (+pJSB161) for each substrate. Unless indicated, means were not significantly different from the control. Significant differences are indicated with asterisks (****p ≤ 0.0001). (B) Mid-logarithmic phase grown B. burgdorferi expressing both flaBp-rlucBb and flaBp-flucBb (+pCFA801) were serial diluted from 2 × 108 to 2 × 100 spirochetes, and incubated with 3.5 mM coelenterazine. The limit of detection (LoD) was established as the mean relative luciferase units for PBS alone plus 3 standard deviations (gray dotted line). The limit of quantitation (LoQ) was established as the mean relative luciferase units for PBS alone plus 10 standard deviations (red dotted line). Data are presented as the mean relative luciferase units ± standard deviation for biological triplicate samples.
The utility of the dual luciferase reporter system not only depends on the substrate specificity of FlucBb and RlucBb, but also the sensitivity of detecting and quantifying spirochetes expressing rlucBb. The limit of detection (LoD) and limit of quantification (LoQ) were established as the number of spirochetes required to achieve relative RlucBb units greater than that of phosphate-buffered saline (PBS) alone plus three standard deviations and 10 standard deviations, respectively. Analysis of triplicate samples of 10-fold serially diluted spirochetes, 2 × 108 to 2 × 100, harvested during log phase growth and incubated with coelenterazine, demonstrated 2 × 103 spirochetes to be the lowest detectable number of flaBp-rlucBb expressing spirochetes in the assay (Figure 2B). However, the LoQ fell between 2 × 103 and 2 × 104 spirochetes. Saturation of the bioluminescence signal was never reached under the conditions examined, with a linear increase in relative RlucBb units from 2 × 103 to 2 × 108 spirochetes (y = 0.0404x; R2 = 0.9997). Extrapolating from this linear equation, the LoQ was calculated to be 4.8 × 103 spirochetes. These data indicate that a minimum of ~1 × 104 flaBp-rlucBb expressing spirochetes are needed to achieve quantifiable relative RlucBb units in the assay. Similar to what has been reported previously (Hyde et al., 2011), 2 × 103 spirochetes was also found to be the lowest detectable number of flaBp-flucBb expressing spirochetes (data not shown).
The flaBp-rlucBb Reporter is a Robust Constitutive Control for Measuring B. burgdorferi Promoter Activities during In vitro Growth
Previously, we reported quantification of in vitro active B. burgdorferi promoters by normalizing relative luciferase units (RLUs) from flucBb expressing cells to the optical density of the bacterial sample measured at 600 nm (OD600) (Adams et al., 2017). In this manner, the OD600 measurement reflects the number of spirochetes in the sample allowing normalization of RLUs across samples and assay conditions. To establish the flaBp-rlucBb reporter as an effective alternative for OD600 readings in our assay, first, relative RlucBb units were measured for all rlucBb-expressing B. burgdorferi clones and normalized to the number of spirochetes in the assay by OD600 (Figure 3A). All clones demonstrated consistent, robust relative RlucBb units, ranging from 5 × 107 to 1.68 × 108. There was no significant difference among clones except for B. burgdorferi carrying pCFA802, which demonstrated higher relative RlucBbunits/OD600 compared to all other clones as well as a difference between logarithmic and stationary phase growth. The same rlucBb-expressing clones, were also incubated with luciferin and relative FlucBb units were determined by normalizing to OD600 (Figure 3B). All flucBb promoter fusions displayed the expected relative FlucBb units/OD600, given the known expression patterns of their corresponding mRNA during logarithmic and stationary phase growth (Arnold et al., 2016). Both the flaB (+pCFA801) and ospA (+pCFA802) promoters demonstrated significant activity above the promoterless flucBb control (+pCFA701) for both logarithmic and stationary phase growth. The activity of the ospC promoter (+pCFA803) during logarithmic phase growth was no different than the promoterless flucBb control (+pCFA701). Whereas, the ospC promoter activity underwent significant induction from logarithmic to stationary phase growth (Figure 3B). Normalization of the relative FlucBb units to 108 relative RlucBb units for each clone demonstrated no difference in the trend of the data resulting from this method of analysis compared to the data resulting from FlucBb units normalized to OD600 (Figure 3B). Together these findings establish flaBp-rlucBb as an effective constitutive control reporter, whose quantitation is reflective of spirochete number and is a robust means to normalize data obtained from flucBb promoter fusions using the dual luciferase reporter system.
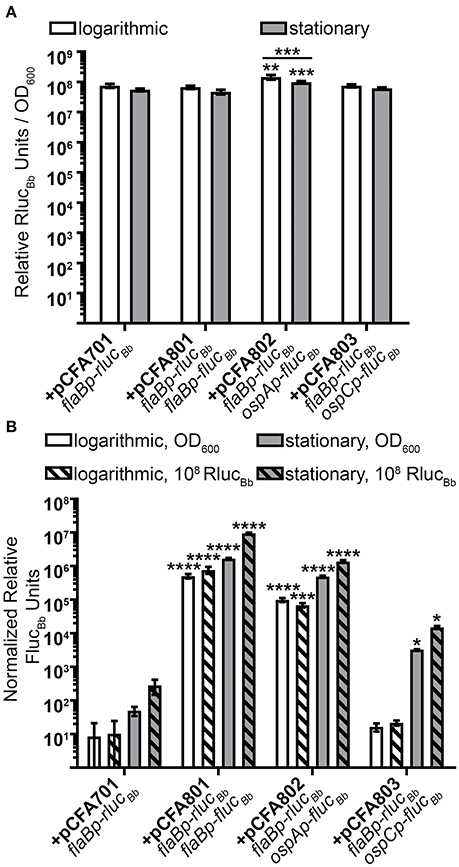
Figure 3. Dual luciferase assay with in vitro grown B. burgdorferi. Spirochetes were grown to either mid-logarithmic or stationary phase and the in vitro luciferase assay performed with 700 μM D-luciferin or 3.5 mM coelenterazine. All data are presented as mean normalized relative luciferase units ± standard deviation for biological triplicate samples. (A) Relative RlucBb units normalized to OD600. The data set for each growth condition was analyzed with a one-way ANOVA followed by Dunnett's multiple comparison test compared to B. burgdorferi expressing flaBp-rlucBb (+pCFA701) and Bonferroni's multiple comparison test to compare the same clone in the two growth phases. (B) Relative FlucBb units normalized to OD600 or 108 relative RlucBb units of the same sample. Each data set was square root transformed and analyzed with a one-way ANOVA followed by Dunnett's multiple comparison test compared to B. burgdorferi expressing flaBp-rlucBb (+pCFA701). Unless indicated, means were not significantly different from the control. Significant differences are indicated with asterisks (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001).
The Dual Luciferase Reporter System Quantifies Promoter Activities during Tick-Spirochete Interactions
Having established the dual luciferase reporter system for use with in vitro grown spirochetes, we examined the efficacy of the reporter system for measuring B. burgdorferi promoter activities in the tick vector. Naïve Ixodes scapularis larval ticks were infected with B. burgdorferi carrying the dual luciferase reporter plasmids or flaBp-flucBb, lacking rlucBb (+pJSB175) by feeding on groups of mice infected with the reporter clones via needle inoculation. Immediately following feeding to repletion, the percent of infected larvae per experimental group was determined by spirochete reisolation in BSKII medium. This analysis revealed that 60–90% of each experimental group of larvae successfully acquired B. burgdorferi upon feeding on infected mice. As an additional means to determine the percentage of infected larvae and to quantitate the number of spirochetes per tick, individual fed larvae were crushed and plated in solid medium for colony forming units (CFUs). Similar to the spirochete reisolation analysis, the groups of fed larvae were found by CFU analysis to be 66–100% infected. Moreover, although a broad range of spirochetes per tick was detected, there was no statistical difference between the average spirochete load per tick for each of the B. burgdorferi clones (Figure 4). These data suggest that all of the clones were able to colonize the ticks with the same efficiency.
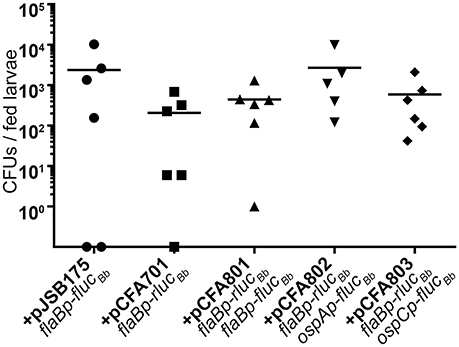
Figure 4. The dual luciferase plasmids do not affect B. burgdorferi acquisition efficiency. Groups of ~200 naïve Ixodes scapularis larvae were fed to repletion on mice infected with B. burgdorferi containing distinct luciferase plasmids. Subsets of individual fed larvae per B. burgdorferi clone were surface sterilized, crushed, and plated in solid BSKII containing RPA cocktail and 50 μg/ml streptomycin. Data points represent the number of colony forming units (CFUs) per individual fed larva. Uninfected larvae (CFU = 0) are represented as data points on the X-axis. No significant differences were detected across the data as determined with a one-way ANOVA.
Based on our quantitation of the average number of spirochetes per tick (Figure 4), we estimated that pools of 24 fed larvae would equate to approximately 104 spirochetes per sample, suggesting that the RlucBb activity would be quantifiable by our assay (Figure 2B). Therefore, 2 weeks following the blood meal, 24 fed larvae per experimental group, in triplicate, were crushed in PBS and relative FlucBb and RlucBb units measured using the luciferin and coelenterazine substrates, respectively (Table 2). The remaining fed larvae were reserved and allowed to molt into nymphs. The unfed, infected nymphs were then fed to repletion on naïve mice. Approximately 2 weeks post feeding, groups of eight fed nymphs were crushed in PBS, in triplicate, and assessed for relative FlucBb and RlucBb units (Table 2). Under the assumption that the spirochete load per fed nymph is increased approximately 10-fold compared to that of fed larvae (Jewett et al., 2007, 2009), we estimated the average spirochete load per fed nymph to be approximately 104. Therefore, a pool of eight fed nymphs was estimated to equate to approximately 8 × 104 spirochetes, which is above both the LoD and LoQ of the in vitro assay (Figure 2B). The actual LoQ for the in vivo tick assay was established using the average RlucBb units plus 10 standard deviations for tick extracts from fed ticks infected with B. burgdorferi lacking rlucBb but expressing flaBp-flucBb, (+pJSB175), rather than PBS alone. This is due to the observation that this tick extract negative control resulted in lower background relative RlucBb units compared to PBS alone (Table 2). In contrast, there was no observed difference in the background relative FlucBb units between PBS and the tick samples containing B. burgdorferi with a promoterless flucBb and expressing flaBp-rlucBb (+pCFA701) in the luciferin assay. Therefore, the LoQ for FlucBb in the in vivo tick assay was determined using the average relative FlucBb units for PBS plus 10 standard deviations. Samples that fell below the LoQ threshold for either luciferase enzyme were considered no different than background (Table 2). As expected, we detected quantifiable relative RlucBb units for all fed larvae samples, and all but two fed nymph samples (Table 2), indicating that sufficient spirochetes were present in the samples for the assay. In the pools of fed larvae only samples containing B. burgdorferi carrying flaBp-flucBb (+pCFA801) demonstrated quantifiable relative FlucBb units. The activities of ospAp and ospCp were below the LoQ for FlucBb (Table 2). In contrast, all three promoters produced quantifiable relative FlucBb units in the fed nymphs. Although one of the extracts from the fed nymphs infected with B. burgdorferi carrying ospCp-flucBb (+pCFA803) did not result in quantifiable relative FlucBb units, this sample also failed to achieve quantifiable relative RlucBb units (Table 2), indicating that the number of spirochetes in the sample was insufficient for the assay. The promoter activities of the spirochetes in the fed nymph samples were analyzed by subtracting the average relative FlucBb units of PBS from the relative FlucBb units of each sample and the average relative RlucBbunits of the infected tick extracts containing the negative control plasmid (+pJSB175) from the relative RlucBb units of each sample. Background-subtracted FlucBb units were then normalized to the respective background-subtracted relative RlucBb units, for all quantifiable values. The RlucBb-normalized promoter activities reflected the expected corresponding B. burgdorferi transcript expression pattern during the fed nymph life stage (Figure 5A, Iyer et al., 2015).
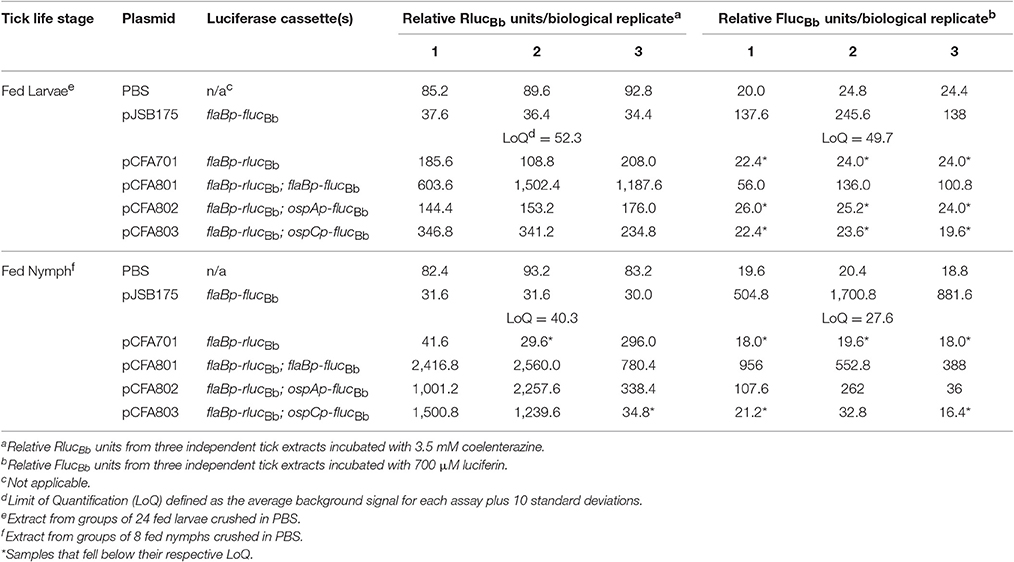
Table 2. In vivo tick dual luciferase assay.
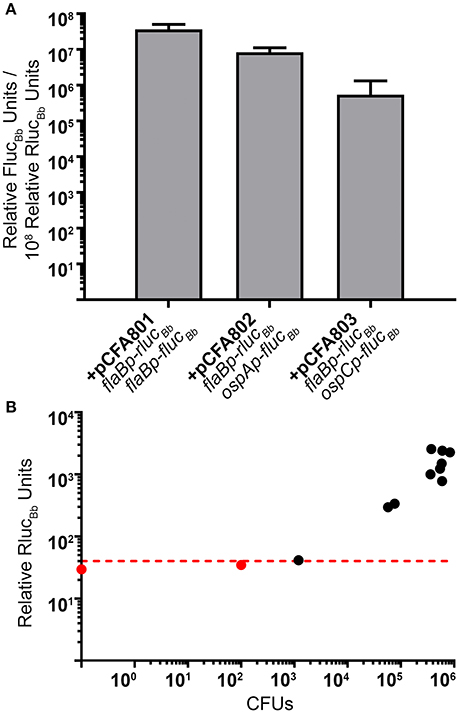
Figure 5. The dual luciferase assay quantitates known patterns of B. burgdorferi transcript expression in nymphs. (A) Groups of eight fed nymphs per B. burgdorferi clone were surface sterilized and crushed in 250 μl of PBS and the in vivo tick luciferase assay performed with 100 μl of tick extract and 700 μM D-luciferin or 3.5 mM coelenterazine. The data are presented as the mean relative FlucBb units per 108 relative RlucBb units ± standard deviation (n = 3; B. burgdorferi carrying pCFA803 n = 2). (B) Relative RlucBb luciferase activity is reflective of spirochete numbers in extracts from fed infected nymphs. 1 μl of fed nymph extract was plated for CFUs in solid BSKII containing RPA cocktail and 50 μg/ml streptomycin. The number of CFUs/100 μl of tick extract (CFUs) was plotted against the relative RlucBb units for the same extract. The red dotted line indicates the established LoQ for relative RlucBb units. Black symbols represent extracts with quantifiable relative RlucBb units and red symbols represent extracts with non-quantifiable RlucBb units. A nymph extract with no detectable CFU (CFU = 0) is represented as the data point on the Y-axis. A significant positive correlation was detected between CFU and relative RlucBb units (Pearson coefficient, r = 0.8022, p = 0.0017).
As an additional means to validate the method as well as to demonstrate that relative RlucBb units are directly reflective of spirochete numbers in the infected tick samples, a portion of each sample from the fed infected nymphs used for RlucBb and FlucBb quantitation (Table 2, Figure 5A), was plated in solid BSKII medium for determination of B. burgdorferi CFUs. The average CFUs per 100 μl of tick extract, the same volume used for the dual luciferase assay, across all clones, was found to be 3.72 × 105 spirochetes. These data support our rationalization for the use of 8 fed nymphs in the assay. Raw relative RlucBb units (Table 2) for these samples plotted against their corresponding CFU counts demonstrated a significant positive correlation (Figure 5B). Furthermore, this analysis indicated that 1.2 × 103 spirochetes are sufficient to generate relative RlucBb units above the LoQ for the in vivo tick assay, which is similar to the sensitivity we observed for the in vitro assay. Collectively, we have described a valuable new method to determine the activity of B. burgdorferi promoters of interest under in vitro growth conditions and in infected ticks. This is the first application of a dual reporter system for B. burgdorferi and, to the best of our knowledge, the first quantification of spirochete promoter activities in the tick vector.
Discussion
Promoter fusion reporter systems are elegant, simple, and powerful tools to quantitate bacterial promoter activities in environments of interest. Herein we have established a new dual luciferase reporter method using the Renilla (sea pansy) and Photinus (firefly) luciferase enzymes for measurement of B. burgdorferi promoter activities in vitro and in the feeding tick during spirochete acquisition from an infected vertebrate host and transmission to a naïve vertebrate host. We demonstrate that constitutive expression of the B. burgdorferi codon-optimized Renilla luciferase gene (rlucBb) is a specific and sensitive measurement of spirochete numbers for normalization of Photinus luciferase gene (flucBb) expression under the control of a promoter of interest.
Several reporter genes have been applied to B. burgdorferi including chloramphenicol acetyl transferase (cat) (Sohaskey et al., 1997), genes encoding a variety of fluorescent proteins (Eggers et al., 2002; Carroll et al., 2003; Schulze and Zuckert, 2006), the Photinus pyralis luciferase gene (flucBb) (Blevins et al., 2007), and lacZ encoding β-galactosidase (lacZBb) (Hayes et al., 2010). Here we describe the first use of a dual reporter system for B. burgdorferi. The combined application of the Renilla and Photinus luciferase genes has several advantages compared to other B. burgdorferi reporter systems as well as other methods of gene expression quantitation such as RT-qPCR. No sample extraction or purification is required to achieve detectable bioluminescence signals, allowing for rapid assay read out with little sample manipulation. Our data indicate that the rlucBb gene under the control of the strong, constitutive flaB promoter results in relative RlucBb units reflective of the number of live spirochetes. This allows relative RlucBb units to serve as the endogenous control against which the relative luciferase units of promoter fusions to flucBb on the same plasmid, in the same sample, can be normalized. It is even possible to measure FlucBb and RlucBb signals back-to-back in the same assay well using firefly luciferase quenching reagents, such as Stop & Glo by Promega (McNabb et al., 2005) and therefore little sample material is required. Use of optical density at 600 nm (OD600) to quantitate sample turbidity as a measure of cell number does not distinguish between live and dead cells in the sample and therefore may not accurately reflect the number of live cells that contribute to the bioluminescence signal. Furthermore, OD600 cannot be used for complex biological samples such as extracts from fed ticks. We demonstrate a significant positive correlation between relative RlucBb units and numbers of live spirochetes both in vitro and in ticks. The B. burgdorferi clone containing pCFA802 exhibited statistically different relative RlucBb units in vitro when normalized to OD600 compared to the other clones. However, the relative FlucBb units/108 relative RlucBb units for this clone followed the expected pattern of ospA expression in vitro and in nymphs. Furthermore, the relative RlucBb units for spirochetes carrying pCFA802 correlated to the number of live spirochetes in fed nymph extracts from this clone, suggesting that the observed difference may not result in a biologically significant effect. Utilizing flaBp-rlucBb as an endogenous constitutive control provides new opportunities for the development of novel high-throughput genetic screening approaches. DNA libraries engineered to drive expression of flucBb could be effectively screened for active promoters in various growth conditions of interest and relative FlucBb units normalized to relative RlucBb units. Further, the dual luciferase reporter plasmid can be manipulated to engineer FlucBb translational fusions to quantitate protein production and stability in growth conditions of interest. An additional important benefit of the dual luciferase reporter assay is the ability to quantitate the promoter activity of a transcript in a strand-specific manner. We and others have recently reported recognition of novel RNA transcripts in the B. burgdorferi genome (Arnold et al., 2016; Adams et al., 2017; Popitsch et al., 2017). Through global 5′ end mapping of the B. burgdorferi transcriptome, we have predicted promoter sequences for previously unannotated RNAs, including antisense and intragenic transcripts, and validated their activities in a variety of environments (Adams et al., 2017). Application of the dual luciferase reporter system now provides a robust means for quantitative comparative analysis of strand-specific B. burgdorferi transcription in complex regions of the genome at the tick-pathogen interface.
For the correct interpretation of molecular techniques it is important to define the lowest level of a measurement, in this case relative luciferase units, which can be reliably analyzed. The limit of detection (LoD) is the lowest amount of measurable signal above background and the limit of quantification (LoQ) signifies the lowest interpretable signal above background. Effective use of LoD and LoQ are based off the standard deviation (SD) of background readings and assume at least 95% of analyzed values are true measurements in the biological assay (Armbruster and Pry, 2008). We have stringently defined LoD as the meanbackground RLUs+ 3SD and LoQ as the meanbackground RLUs + 10SD. Thereby LoQ should be calculated for each luciferase substrate and each independent application of the B. burgdorferi dual luciferase assay to best distinguish low but quantifiable bioluminescence signals from background. It is also important to define the appropriate background controls in the context of the assay. Indeed, our studies have demonstrated that background relative RlucBb units were ~60% decreased in fed tick extracts compared to PBS alone. Therefore, extracts from fed ticks infected with B. burgdorferi lacking rlucBb expression (+pJSB175) served as the background control to calculate the LoQ for RlucBb in ticks. Conversely, this was not observed for the background relative FlucBb units for fed tick extracts and PBS alone served as the negative control for these measurements. We hypothesize that the biological matrix of the fed tick extracts contributes, in part, to alteration of the RlucBb signal by inhibiting non-specific activation of the coelenterazine substrate.
We found that not all samples with quantifiable relative RlucBb units, also had quantifiable relative FlucBb units. In some cases, the finding that a promoter fusion has non-quantifiable relative FlucBb units may accurately reflect the weak to no biological activity of that promoter in a particular environment and/or non-quantifiable relative FlucBb units may result from low numbers of spirochetes, albeit quantifiable relative RlucBb units. These challenges may be overcome by increasing the number of spirochetes used in the assay. This is evident in the data we present for the in vivo tick assay, in which the fed larvae samples for all B. burgdorferi clones achieved quantifiable relative RlucBb units; however, the clone containing flaBp-flucBb (+pCFA801), but not the clones containing ospAp-flucBb (+pCFA802) or the ospCp-flucBb (+pCFA803), produced quantifiable relative FlucBb units. This finding was not surprising for the ospC promoter, given that the ospC transcript is known to have weak to no activity in fed larvae following B. burgdorferi acquisition from infected mice. This finding was, however, unexpected for the ospA promoter, whose transcript is known to have strong activity in this environment (Caimano et al., 2015). Yet, the average number of spirochetes in the ospAp-flucBb (+pCFA802) and ospCp-flucBb (+pCFA803) containing clone extracts, as reflected by the average relative RlucBb units (1.6 × 102 and 3.1 × 102, respectively), were approximately 10-fold and 4-fold less than that of the flaBp-flucBb (+pCFA801) containing clone (1.1 × 103), suggesting that spirochete number may contribute, in part, to the non-quantifiable relative FlucBb units for these spirochetes. In contrast, the fed nymph extracts contained comparable average numbers of spirochetes regardless of the clone, as reflected by both the average relative RlucBb units (1.5 × 103 ± 380) and CFU counts (4.4 × 105 ± 2.2 × 105) and all flucBb promoter fusions achieved quantifiable relative FlucBb units. Furthermore, while it was one of our goals to measure promoter activities for spirochetes in unfed-flat nymphs post-molt, we found the luciferase signals for these samples to be below the limit of quantification of our assay. We again hypothesize that the spirochete loads in the ticks at this point in the infectious cycle may be below the number of spirochetes necessary for the assay. To examine this possibility we crushed and plated for CFU a subset of individual unfed nymphs infected with spirochetes carrying both flaBp-rlucBb and flaBp-flucBb (pCFA801). The average spirochete load was determined to be ~27 spirochetes/unfed nymph. This was approximately 10-fold lower than the average spirochete load in the fed larval ticks for the same clone (~4.4 × 102 spirochetes/fed larvae) and approximately 10,000-fold lower than that of fed nymphs (~1.6 × 105 spirochetes/fed nymph). Considering that pools of 24 fed larvae and 8 fed nymphs, and therefore ~104 and ~106 spirochetes carrying pCFA801, respectively, were used for the luciferase assays, nearly 400 up to 40,000 unfed nymphs would be required to achieve equivalent relative luciferase units. The difficulties of studying B. burgdorferi transcription in unfed nymphs was also shown by a recent microarray study, where even with an amplification step, transcript analysis in this tick life-stage was precluded (Iyer et al., 2015). RT-qPCR does remain an alternative approach for gene expression analysis in unfed nymphs, having several documented successes in determining B. burgdorferi transcript levels (Wang et al., 2002; Bykowski et al., 2007; Showman et al., 2016), albeit lacking strand specificity. It should be noted that the endogenous copies of the flaB, ospA, and ospC genes and their promoters are present in the genetic background of all of the B. burgdorferi clones that were analyzed. This raises the possibility that a reduction in FlucBb or RlucBb signals could have occurred due to titration of transcription factors away from the promoter fusions by the endogenous promoters. However, expression of flaB, ospA, and ospC are essential for survival of B. burgdorferi throughout its infectious cycle (Samuels, 2011; Sultan et al., 2013) and thus these experiments could not be conducted in the absence of these genes.
While dual fluc and rluc reporter systems have been used successfully for live imaging and quantitation of eukaryotic tumor cells in mice (Bhaumik and Gambhir, 2002), the use of Renilla luciferase and the coelenterazine substrate for live imaging of microbial infections in mice has proven challenging (Andreu et al., 2011) and few publications report exploration of the use of dual Renilla and Photinus luciferase reporters in the context of infectious disease applications. There is great interest in applying a luciferase dual reporter system to quantification of B. burgdorferi promoter activities during an active mammalian infection. We and others have demonstrated the power of the flucBb reporter for tracking B. burgdorferi dissemination and qualitative detection of promoter activities over time in live mice (Hyde et al., 2011; Chan et al., 2015; Adams et al., 2017). By extension we investigated the efficacy of the dual luciferase reporter system for live imaging applications with B. burgdorferi in infected mice. Exhaustive examination of available coelenterazine substrates including: h-Coelenterazine-SOL in vivo (NanoLight), Inject-A-Lume h-Coelenterazine (NanoLight), ViviRen™ in vivo Renilla Luciferase Substrate (Promega), and XenoLight RediJect Coelenterazine h (PerkinElmer) as well as various substrate concentrations, substrate injection methods and imaging times, resulted in no significant RlucBb signals above background (data not shown). Unlike for applications for solid cancers, use of luciferase substrates for in vivo detection of microbial pathogens relies on the substrates to be available in excess, systemically throughout the animal. Luciferin has been documented to rapidly distribute throughout the mouse (Contag et al., 1997), but the bioavailability of coelenterazine may be more limited (Luker et al., 2002). In addition, we found coelenterazine to have an extraordinary high background signal. Indeed, RlucBb signals following coelenterazine delivery were observed for mice infected with spirochetes lacking rlucBb entirely, which were not able to be overcome in mice infected with spirochetes expressing flaBp-rlucBb (data not shown). These findings are consistent with what has been reported for attempted in vivo imaging applications using coelenterazine and Mycobacterium smegmatis expressing Gaussia luciferase (Andreu et al., 2010). Rather, alternative methods of normalization may be used, such as determining spirochete loads of infected tissues immediately following FlucBb imaging (Skare et al., 2016), in instances where quantification of promoter activity during murine infection is warranted.
B. burgdorferi has been shown to colonize Ixodes scapularis via a biphasic mode of dissemination which is believed to involve complex interactions between the pathogen and the arthropod vector (Dunham-Ems et al., 2009). We are still discovering many of the mechanisms B. burgdorferi employs to survive throughout its enzootic cycle. Additionally, the recently sequenced Ixodes scapularis genome opens new areas of study for host-pathogen interactions (Gulia-Nuss et al., 2016). Successful and reliable techniques for analysis of spirochete biology in the tick are critical to drive understanding of these interactions. The dual luciferase system presented here is a simple and powerful approach for measuring transcript expression, which can be easily modified to meet the needs of the researcher and adds to the ever growing molecular genetic toolbox for investigation of B. burgdorferi transcription and gene regulation.
Author Contributions
PA and MJ conceived the study and designed experiments; PA and CF performed experiments; PA, CF, and MJ interpreted results; PA and MJ wrote the manuscript; all authors critiqued and edited the final manuscript.
Funding
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institute of Health (R01AI099094 to MJ) and the National Research Fund for Tick-Borne Diseases (MJ).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Travis Jewett, Dr. George Aranjuez, and members of the M. Jewett lab for helpful insight, discussion, and critical analysis of the work. Thanks to Chrysta Olsen, James Grant, and the UCF NAF animal care staff. Thank you Dr. Kyle Rhode for use of his plate reader, Valacia Titus and Adriana Romero for technical assistance. The authors wish to acknowledge Dr. Jon Blevins and Dr. Michael Norgard for generously providing the pJSB161 and pJSB175 vectors. The following reagent was provided by Centers for Disease Control and Prevention for distribution by BEI Resources, NIAID, NIH: Ixodes scapularis Larvae (Live), NR-44115.
References
Adams, P. P., Flores Avile, C., Popitsch, N., Bilusic, I., Schroeder, R., Lybecker, M., et al. (2017). In vivo expression technology and 5' end mapping of the Borrelia burgdorferi transcriptome identify novel RNAs expressed during mammalian infection. Nucleic Acids Res. 45, 775–792. doi: 10.1093/nar/gkw1180
Adeolu, M., and Gupta, R. S. (2014). A phylogenomic and molecular marker based proposal for the division of the genus Borrelia into two genera: the emended genus Borrelia containing only the members of the relapsing fever Borrelia, and the genus Borreliella gen. nov. containing the members of the Lyme disease Borrelia (Borrelia burgdorferi sensu lato complex). Antonie Van Leeuwenhoek 105, 1049–1072. doi: 10.1007/s10482-014-0164-x
Andreu, N., Zelmer, A., Fletcher, T., Elkington, P. T., Ward, T. H., Ripoll, J., et al. (2010). Optimisation of bioluminescent reporters for use with mycobacteria. PLoS ONE 5:e10777. doi: 10.1371/journal.pone.0010777
Andreu, N., Zelmer, A., and Wiles, S. (2011). Noninvasive biophotonic imaging for studies of infectious disease. FEMS Microbiol. Rev. 35, 360–394. doi: 10.1111/j.1574-6976.2010.00252.x
Armbruster, D. A., and Pry, T. (2008). Limit of blank, limit of detection and limit of quantitation. Clin. Biochem. Rev. 29(Suppl. 1), S49–S52.
Arnold, W. K., Savage, C. R., Brissette, C. A., Seshu, J., Livny, J., and Stevenson, B. (2016). RNA-Seq of Borrelia burgdorferi in multiple phases of growth reveals insights into the dynamics of gene expression, transcriptome architecture, and noncoding RNAs. PLoS ONE 11:e0164165. doi: 10.1371/journal.pone.0164165
Barbour, A. G. (1984). Isolation and cultivation of Lyme disease spirochetes. Yale J. Biol. Med. 57, 521–525.
Bhaumik, S., and Gambhir, S. S. (2002). Optical imaging of Renilla luciferase reporter gene expression in living mice. Proc. Natl. Acad. Sci. U.S.A. 99, 377–382. doi: 10.1073/pnas.012611099
Blevins, J. S., Revel, A. T., Smith, A. H., Bachlani, G. N., and Norgard, M. V. (2007). Adaptation of a luciferase gene reporter and lac expression system to Borrelia burgdorferi. Appl. Environ. Microbiol. 73, 1501–1513. doi: 10.1128/AEM.02454-06
Bykowski, T., Woodman, M. E., Cooley, A. E., Brissette, C. A., Brade, V., Wallich, R., et al. (2007). Coordinated expression of Borrelia burgdorferi complement regulator-acquiring surface proteins during the Lyme disease spirochete's mammal-tick infection cycle. Infect. Immun. 75, 4227–4236. doi: 10.1128/IAI.00604-07
Caimano, M. J., Drecktrah, D., Kung, F., and Samuels, D. S. (2016). Interaction of the Lyme disease spirochete with its tick vector. Cell. Microbiol. 18, 919–927. doi: 10.1111/cmi.12609
Caimano, M. J., Dunham-Ems, S., Allard, A. M., Cassera, M. B., Kenedy, M., and Radolf, J. D. (2015). Cyclic di-GMP modulates gene expression in Lyme disease spirochetes at the tick-mammal interface to promote spirochete survival during the blood meal and tick-to-mammal transmission. Infect. Immun. 83, 3043–3060. doi: 10.1128/IAI.00315-15
Carroll, J. A., Stewart, P. E., Rosa, P., Elias, A. F., and Garon, C. F. (2003). An enhanced GFP reporter system to monitor gene expression in Borrelia burgdorferi. Microbiology 149(Pt 7), 1819–1828. doi: 10.1099/mic.0.26165-0
Casjens, S. R., Mongodin, E. F., Qiu, W. G., Luft, B. J., Schutzer, S. E., Gilcrease, E. B., et al. (2012). Genome stability of Lyme disease spirochetes: comparative genomics of Borrelia burgdorferi plasmids. PLoS ONE 7:e33280. doi: 10.1371/journal.pone.0033280
Chan, K., Alter, L., Barthold, S. W., and Parveen, N. (2015). Disruption of bbe02 by insertion of a luciferase gene increases transformation efficiency of Borrelia burgdorferi and allows live imaging in lyme disease susceptible C3H mice. PLoS ONE 10:e0129532. doi: 10.1371/journal.pone.0129532
Contag, C. H., Spilman, S. D., Contag, P. R., Oshiro, M., Eames, B., Dennery, P., et al. (1997). Visualizing gene expression in living mammals using a bioluminescent reporter. Photochem. Photobiol. 66, 523–531. doi: 10.1111/j.1751-1097.1997.tb03184.x
de Silva, A. M., Tyson, K. R., and Pal, U. (2009). Molecular characterization of the tick-Borrelia interface. Front. Biosci. 14, 3051–3063. doi: 10.2741/3434
de Wet, J. R., Wood, K. V., DeLuca, M., Helinski, D. R., and Subramani, S. (1987). Firefly luciferase gene: structure and expression in mammalian cells. Mol. Cell. Biol. 7, 725–737. doi: 10.1128/MCB.7.2.725
Dunham-Ems, S. M., Caimano, M. J., Pal, U., Wolgemuth, C. W., Eggers, C. H., Balic, A., et al. (2009). Live imaging reveals a biphasic mode of dissemination of Borrelia burgdorferi within ticks. J. Clin. Invest. 119, 3652–3665. doi: 10.1172/JCI39401
Eggers, C. H., Caimano, M. J., Clawson, M. L., Miller, W. G., Samuels, D. S., and Radolf, J. D. (2002). Identification of loci critical for replication and compatibility of a Borrelia burgdorferi cp32 plasmid and use of a cp32-based shuttle vector for the expression of fluorescent reporters in the lyme disease spirochaete. Mol. Microbiol. 43, 281–295. doi: 10.1046/j.1365-2958.2002.02758.x
Elias, A. F., Stewart, P. E., Grimm, D., Caimano, M. J., Eggers, C. H., Tilly, K., et al. (2002). Clonal polymorphism of Borrelia burgdorferi strain B31 MI: implications for mutagenesis in an infectious strain background. Infect. Immun. 70, 2139–2150. doi: 10.1128/IAI.70.4.2139-2150.2002
Fraser, C. M., Casjens, S., Huang, W. M., Sutton, G. G., Clayton, R., Lathigra, R., et al. (1997). Genomic sequence of a Lyme disease spirochaete, Borrelia burgdorferi. Nature 390, 580–586. doi: 10.1038/37551
Ge, Y., Old, I. G., Girons, I. S., and Charon, N. W. (1997). The flgK motility operon of Borrelia burgdorferi is initiated by a sigma 70-like promoter. Microbiology 143(Pt 5), 1681–1690. doi: 10.1099/00221287-143-5-1681
Grimm, D., Tilly, K., Bueschel, D. M., Fisher, M. A., Policastro, P. F., Gherardini, F. C., et al. (2005). Defining plasmids required by Borrelia burgdorferi for colonization of tick vector Ixodes scapularis (Acari: Ixodidae). J. Med. Entomol. 42, 676–684.
Gulia-Nuss, M., Nuss, A. B., Meyer, J. M., Sonenshine, D. E., Roe, R. M., Waterhouse, R. M., et al. (2016). Genomic insights into the Ixodes scapularis tick vector of Lyme disease. Nat. Commun. 7:10507. doi: 10.1038/ncomms10507
Hayes, B. M., Jewett, M. W., and Rosa, P. A. (2010). A lacZ reporter system for use in Borrelia burgdorferi. Appl. Environ. Microbiol. 76, 7407–7412. doi: 10.1128/aem.01389-10
Hyde, J. A., Weening, E. H., Chang, M., Trzeciakowski, J. P., Hook, M., Cirillo, J. D., et al. (2011). Bioluminescent imaging of Borrelia burgdorferi in vivo demonstrates that the fibronectin-binding protein BBK32 is required for optimal infectivity. Mol. Microbiol. 82, 99–113. doi: 10.1111/j.1365-2958.2011.07801.x
Iyer, R., Caimano, M. J., Luthra, A., Axline, D. Jr., Corona, A., Iacobas, D. A., et al. (2015). Stage-specific global alterations in the transcriptomes of Lyme disease spirochetes during tick feeding and following mammalian host adaptation. Mol. Microbiol. 95, 509–538. doi: 10.1111/mmi.12882
Jewett, M. W., Lawrence, K. A., Bestor, A., Byram, R., Gherardini, F., and Rosa, P. A. (2009). GuaA and GuaB are essential for Borrelia burgdorferi survival in the tick-mouse infection cycle. J. Bacteriol. 191, 6231–6241. doi: 10.1128/JB.00450-09
Jewett, M. W., Lawrence, K., Bestor, A. C., Tilly, K., Grimm, D., Shaw, P., et al. (2007). The critical role of the linear plasmid lp36 in the infectious cycle of Borrelia burgdorferi. Mol. Microbiol. 64, 1358–1374. doi: 10.1111/j.1365-2958.2007.05746.x
Kroger, C., Dillon, S. C., Cameron, A. D., Papenfort, K., Sivasankaran, S. K., Hokamp, K., et al. (2012). The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc. Natl. Acad. Sci. U.S.A. 109, E1277–E1286. doi: 10.1073/pnas.1201061109
Lorenz, W. W., McCann, R. O., Longiaru, M., and Cormier, M. J. (1991). Isolation and expression of a cDNA encoding Renilla reniformis luciferase. Proc. Natl. Acad. Sci. U.S.A. 88, 4438–4442. doi: 10.1073/pnas.88.10.4438
Luker, G. D., Bardill, J. P., Prior, J. L., Pica, C. M., Piwnica-Worms, D., and Leib, D. A. (2002). Noninvasive bioluminescence imaging of herpes simplex virus type 1 infection and therapy in living mice. J. Virol. 76, 12149–12161. doi: 10.1128/JVI.76.23.12149-12161.2002
Marques, S. M., and Esteves da Silva, J. C. (2009). Firefly bioluminescence: a mechanistic approach of luciferase catalyzed reactions. IUBMB Life 61, 6–17. doi: 10.1002/iub.134
McNabb, D. S., Reed, R., and Marciniak, R. A. (2005). Dual luciferase assay system for rapid assessment of gene expression in Saccharomyces cerevisiae. Eukaryotic Cell 4, 1539–1549. doi: 10.1128/EC.4.9.1539-1549.2005
Nakamura, Y., Gojobori, T., and Ikemura, T. (2000). Codon usage tabulated from international DNA sequence databases: status for the year 2000. Nucl. Acids Res. 28:292. doi: 10.1093/nar/28.1.292
Popitsch, N., Bilusic, I., Rescheneder, P., Schroeder, R., and Lybecker, M. (2017). Temperature-dependent sRNA transcriptome of the Lyme disease spirochete. BMC Genomics 18, 28. doi: 10.1186/s12864-016-3398-3
Radolf, J. D., Caimano, M. J., Stevenson, B., and Hu, L. T. (2012). Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease spirochaetes. Nat. Rev. Microbiol. 10, 87–99. doi: 10.1038/nrmicro2714
Rego, R. O., Bestor, A., and Rosa, P. A. (2011). Defining the plasmid-borne restriction-modification systems of the Lyme disease spirochete Borrelia burgdorferi. J. Bacteriol. 193, 1161–1171. doi: 10.1128/JB.01176-10
Rollend, L., Fish, D., and Childs, J. E. (2013). Transovarial transmission of Borrelia spirochetes by Ixodes scapularis: a summary of the literature and recent observations. Ticks Tick Borne Dis. 4, 46–51. doi: 10.1016/j.ttbdis.2012.06.008
Rosa, P. A., and Hogan, D. (1992). “Colony formation by Borrelia burgdorferi in solid medium: clonal analysis of osp locus variants,” in Proceeding of the First International Conference on Tick Borne Pathogens at the Host-Vector Interface, eds U. G. Munderloh and T. J. Kurtti (St. Paul, MN: University of Minnesota), 95–103.
Samuels, D. S. (1995). “Electrotransformation of the spirochete Borrelia burgdorferi,” in Methods in Molecular Biology, ed J. A. Nickoloff (Totowa, NJ: Humana Press, Inc.), 253–259.
Samuels, D. S. (2011). Gene regulation in Borrelia burgdorferi. Annu. Rev. Microbiol. 65, 479–499. doi: 10.1146/annurev.micro.112408.134040
Schotthoefer, A. M., and Frost, H. M. (2015). Ecology and epidemiology of lyme borreliosis. Clin. Lab. Med. 35, 723–743. doi: 10.1016/j.cll.2015.08.003
Schulze, R. J., and Zuckert, W. R. (2006). Borrelia burgdorferi lipoproteins are secreted to the outer surface by default. Mol. Microbiol. 59, 1473–1484. doi: 10.1111/j.1365-2958.2006.05039.x
Schwan, T. G. (2003). Temporal regulation of outer surface proteins of the Lyme-disease spirochaete Borrelia burgdorferi. Biochem. Soc. Trans. 31(Pt 1), 108–112. doi: 10.1042/bst0310108
Schwan, T. G., Kime, K. K., Schrumpf, M. E., Coe, J. E., and Simpson, W. J. (1989). Antibody response in white-footed mice (Peromyscus leucopus) experimentally infected with the Lyme disease spirochete (Borrelia burgdorferi). Infect. Immun. 57, 3445–3451.
Schwan, T. G., and Piesman, J. (2000). Temporal changes in outer surface proteins A and C of the Lyme disease-associated spirochete, Borrelia burgdorferi, during the chain of infection in ticks and mice. J. Clin. Microbiol. 38, 382–388.
Schwan, T. G., Piesman, J., Golde, W. T., Dolan, M. C., and Rosa, P. A. (1995). Induction of an outer surface protein on Borrelia burgdorferi during tick feeding. Proc. Natl. Acad. Sci. U.S.A. 92, 2909–2913. doi: 10.1073/pnas.92.7.2909
Sharma, C. M., Hoffmann, S., Darfeuille, F., Reignier, J., Findeiss, S., Sittka, A., et al. (2010). The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464, 250–255. doi: 10.1038/nature08756
Showman, A. C., Aranjuez, G., Adams, P. P., and Jewett, M. W. (2016). Gene bb0318 is critical for the oxidative stress response and infectivity of Borrelia burgdorferi. Infect. Immun. 84, 3141–3151. doi: 10.1128/IAI.00430-16
Skare, J. T., Shaw, D. K., Trzeciakowski, J. P., and Hyde, J. A. (2016). In vivo imaging demonstrates that Borrelia burgdorferi ospC is uniquely expressed temporally and spatially throughout experimental infection. PLoS ONE 11:e0162501. doi: 10.1371/journal.pone.0162501
Sohaskey, C. D., Arnold, C., and Barbour, A. G. (1997). Analysis of promoters in Borrelia burgdorferi by use of a transiently expressed reporter gene. J. Bacteriol. 179, 6837–6842. doi: 10.1128/jb.179.21.6837-6842.1997
Srivastava, S. Y., and de Silva, A. M. (2008). Reciprocal expression of ospA and ospC in single cells of Borrelia burgdorferi. J. Bacteriol. 190, 3429–3433. doi: 10.1128/JB.00085-08
Sultan, S. Z., Manne, A., Stewart, P. E., Bestor, A., Rosa, P. A., Charon, N. W., et al. (2013). Motility is crucial for the infectious life cycle of Borrelia burgdorferi. Infect. Immun. 81, 2012–2021. doi: 10.1128/IAI.01228-12
Thomason, M. K., Bischler, T., Eisenbart, S. K., Forstner, K. U., Zhang, A., Herbig, A., et al. (2015). Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J. Bacteriol. 197, 18–28. doi: 10.1128/JB.02096-14
Wager, B., Shaw, D. K., Groshong, A. M., Blevins, J. S., and Skare, J. T. (2015). BB0744 affects tissue tropism and spatial distribution of Borrelia burgdorferi. Infect. Immun. 83, 3693–3703. doi: 10.1128/IAI.00828-15
Wang, X. G., Lin, B., Kidder, J. M., Telford, S., and Hu, L. T. (2002). Effects of environmental changes on expression of the oligopeptide permease (opp) genes of Borrelia burgdorferi. J. Bacteriol. 184, 6198–6206. doi: 10.1128/JB.184.22.6198-6206.2002
Woo, J., Howell, M. H., and von Arnim, A. G. (2008). Structure-function studies on the active site of the coelenterazine-dependent luciferase from Renilla. Protein Sci. 17, 725–735. doi: 10.1110/ps.073355508
World Health Organization (2016). Vector-Borne Diseases [Fact Sheet]. Available online at: http://www.who.int/mediacentre/factsheets/fs387/en/ (Accessed March 2017).
Keywords: Borrelia (Borreliella) burgdorferi, Lyme disease, tick-pathogen interactions, bioluminescence reporter, Photinus reniformis luciferase, Photinus pyralis luciferase
Citation: Adams PP, Flores Avile C and Jewett MW (2017) A Dual Luciferase Reporter System for B. burgdorferi Measures Transcriptional Activity during Tick-Pathogen Interactions. Front. Cell. Infect. Microbiol. 7:225. doi: 10.3389/fcimb.2017.00225
Received: 30 March 2017; Accepted: 16 May 2017;
Published: 31 May 2017.
Edited by:
Jose De La Fuente, Instituto de Investigacion en Recursos Cinegeticos, SpainReviewed by:
Janakiram Seshu, University of Texas at San Antonio, United StatesRobert D. Gilmore, Centers for Disease Control and Prevention, United States
Copyright © 2017 Adams, Flores Avile and Jewett. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mollie W. Jewett, bW9sbGllLmpld2V0dEB1Y2YuZWR1