 Abolghasem Tohidpour1*
Abolghasem Tohidpour1* Andrey V. Morgun1,2
Andrey V. Morgun1,2 Elizaveta B. Boitsova1,3
Elizaveta B. Boitsova1,3 Natalia A. Malinovskaya1
Natalia A. Malinovskaya1 Galina P. Martynova3Elena D. Khilazheva1Natalia V. Kopylevich1
Galina P. Martynova3Elena D. Khilazheva1Natalia V. Kopylevich1 Galina E. Gertsog1
Galina E. Gertsog1 Alla B. Salmina1
Alla B. Salmina1- 1Research Institute of Molecular Medicine and Pathobiochemistry, Krasnoyarsk State Medical University named after Prof. V.F. Voino-Yasenetsky, Krasnoyarsk, Russia
- 2Department of Paediatrics, Krasnoyarsk State Medical University named after Prof. V.F. Voino-Yasenetsky, Krasnoyarsk, Russia
- 3Department of Children Infectious Diseases, Krasnoyarsk State Medical University named after Prof. V.F. Voino-Yasenetsky, Krasnoyarsk, Russia
Neuroinflammation is a complex inflammatory process in the central nervous system, which is sought to play an important defensive role against various pathogens, toxins or factors that induce neurodegeneration. The onset of neurodegenerative diseases and various microbial infections are counted as stimuli that can challenge the host immune system and trigger the development of neuroinflammation. The homeostatic nature of neuroinflammation is essential to maintain the neuroplasticity. Neuroinflammation is regulated by the activity of neuronal, glial, and endothelial cells within the neurovascular unit, which serves as a “platform” for the coordinated action of pro- and anti-inflammatory mechanisms. Production of inflammatory mediators (cytokines, chemokines, reactive oxygen species) by brain resident cells or cells migrating from the peripheral blood, results in the impairment of blood-brain barrier integrity, thereby further affecting the course of local inflammation. In this review, we analyzed the most recent data on the central nervous system inflammation and focused on major mechanisms of neurovascular unit dysfunction caused by neuroinflammation and infections.
Introduction
Upon confronting different stimuli such as infectious diseases, toxins, and traumatic shocks, the host cells present an orchestrated mechanism of actions to maintain the stability of body tissues. The innate immune cells (i.e., macrophages, dendritic, and mast cells) primarily interact with antigens in non-specific pathways and stimulate tissue homeostatic (inflammatory) responses (Medzhitov, 2008). Some infectious agents can trigger intensive tissue inflammatory responses and activate the complement system. The inflammatory responses in the peripheral tissues cause dendritic cells to activate the adaptive immune system and induce some robust responses (i.e., necrosis as a result of phagocytosis). The tuned connection of the central nervous system (CNS)-immune system supports the host immune defense through various pathways such as inducing fever, pain sensitivity, and increasing the sleeping time (Maier et al., 1998).
There are substantial differences between inflammatory responses arising from the CNS and other body tissues. Perhaps the main distinction is the lack of memory T cells (adaptive immunity) in the brain parenchyma. Memory T cells present the specific antigens of invading pathogens and leaving the CNS they enter lymph nodes and initiate cellular immune responses in lymphoid tissue. In the healthy brain, parenchymal T-cells are located in the cerebrospinal fluid (CSF) but in pathological conditions they enter the CNS through the choroid plexus and meningeal veins (Charo and Ransohoff, 2006). These cells leave the CNS through the cribriform plate into the deep cervical lymph nodes (Andres et al., 1987; Goldmann et al., 2006; Ransohoff and Engelhardt, 2012; Louveau et al., 2015). On the other hand, B-cells, another group of adaptive immune cells, can enter the normal brain. However, their quantity and trafficking slightly increase after the outbreak of some pathologies such as AIDS (Anthony et al., 2003). B-cells can differentiate to plasmoblasts and produce antibodies with different functions in the CNS or exert antibody-independent activities such as cytokine production and activation of T-cells (Ransohoff et al., 2015). The brain parenchymal innate immune cells mainly consist of resident myeloid cells, astrocytes, and microglia, which enter the brain and spinal cord parenchyma during the early embryonic period (Sanes and Lichtman, 1999; Del Rio and Feller, 2006). Microglia are the key players in the physiological development of the brain (Schafer et al., 2012). However, other cell populations within the CNS, such as astrocytes, myeloid cells, and dendritic cells also contribute to the functional activities and homeostasis of the brain (Chen and Regehr, 2000; Ransohoff et al., 2015).
It is clear that the impairment of blood-brain barrier (BBB) contributes to neuroinflammation. BBB and activated microglia are components of the neurovascular unit (NVU), and their structural and functional integrity is a major factor affecting the course of neuroinflammation. Migration of immune cells, transport of cytokines and other inflammatory reactions occur due to the increased permeability of BBB and dysfunction of NVU. Therefore, coordinated activity of NVU cells (brain microvessel endothelial cells, pericytes, perivascular astrocytes, neuronal cells, etc.) is necessary to regulate local inflammation in brain tissue. For instance, the integrated neuronal activity and the stimulation of astroglial cells (so-called neuron-astrocyte metabolic coupling) cause the accumulation of extracellular lactate and H+ and affect the local inflammatory reactions in the brain. The increased level of neuronal activity also stimulates an inflammatory response in the peripheral tissues—so-called neurogenic inflammation (Figure 1; Roosterman et al., 2006; Chiu et al., 2012). Neurogenic inflammation often occurs due to pain, stress, and epileptic seizures and has a typical degree of similarity to other forms of CNS neuroinflammation (Zochodne et al., 2001; Beggs et al., 2010; Gruber-Schoffnegger et al., 2013).
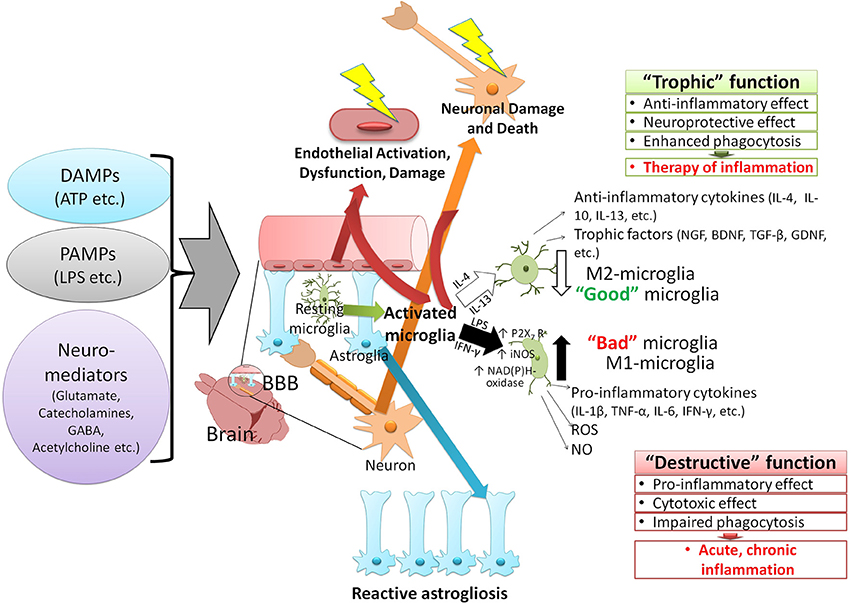
Figure 1. The paradigm of the CNS neuroinflammation. Various factors can activate the immune response of the CNS and induce neuroinflammation. These stimuli are classified into two groups: 1—pathogen-associated molecular patterns (PAMPs), which are produced by the invading microorganisms of the CNS and 2—damage-associated molecular patterns (DAMPs), molecules that are released by host due to onset of traumatic conditions or interaction with some neurotransmitters (i.e., glutamate, GABA, and acetylcholine). The immune responses to the CNS stimuli vary based on the type of stimulation but generally lead to similar outcomes such as immune adaptation, dysfunction, degeneration, and resolution. Activation of the resting microglia and converting them to two distinct phenotypes depends on various cytokines produced by surrounding cells (glia, neurons, migratory immune cells). The release of interleukins 4 and 13 (IL-4, IL-13) gives rise to M1 phenotype (anti-inflammatory) of microglia, which express inflammatory cytokines (interleukin 4, 10, and 13), cell growth factors (i.e., NGF, BDNF, TGF-β, GDNF), and exert anti-inflammatory effects. Interferon-γ (IFN-γ) and the lipopolysaccharide (LPS) of bacteria, on the other hand, activate the M2 phenotype (pro-inflammatory) of microglia. The M2 phenotype is characterized by 1—activation of purinergic receptors P2X7 subtype (activated by ATP, promoting the inflammation and destruction of cells by forming channels and pores), and 2—expression of enzymes which generate reactive oxygen and nitrogen [NAD(P)H-oxidase, iNOS], and trigger the expression of proinflammatory cytokines (IL-1β, TNF-α, IL-6, IFN- γ). Activation of microglia, especially the formation of M2 phenotype exacerbates the damage to BBB (in particular neurons and endothelial cells). Effects of these agents (PAMPs, DAMPs, neuromediators) on astroglia cause their proliferation, activation (reactive astrogliosis), and dysfunction (in particular, increased procoagulant activity and thrombosis). These CNS stimuli also cause endothelial injury, damage, and neuronal death. GABA, γ-aminobutyric acid; IL, Interleukin; NGF, nerve growth factor; BDNF, brain-derived neurotrophic factor; TGF, transforming growth factor; GDNF, glial-derived neurotrophic factors; iNOS, inducible nitric oxide synthase; TNF, tumor necrosis factor; IFN, interferon; ROS, reactive oxygen species; NO, nitric oxide.
Taken together, neuroinflammation is a form of inflammatory response within the CNS, which is significantly affected by the status of neuronal activity and BBB permeability. Moderate inflammatory responses can protect the CNS whereas an intensive inflammation aggravates the impairment of tissue homeostasis (Combes et al., 2012; Ransohoff and Brown, 2012; Skaper et al., 2012). In this review, we summarize the current knowledge on the role of BBB/NVU alterations in the development of neuroinflammation, with the emphasis on inflammatory processes caused by infectious diseases. Initially, an overview of physiologic development of NVU and BBB is made and we describe the main factors which modulate the development of neuroinflammation and other neurodegenerative disorders in the CNS. This is followed by discussing the impact of infectious diseases on inducing neuroinflammatory responses in the CNS. Finally, we unfold how neuroinflammation in the CNS is connected with genetic abnormalities of the host.
NVU/BBB Development During the Embryonic, Fetal, and After-Birth Periods
BBB formation starts in the uterus and continues during the early postnatal period through several stages as 1-vascularization and angiogenesis (the formation of the choroid plexus and overgrowth of blood vessels); 2-differentiation of cerebral endothelial cells; and 3-the maturation of cellular elements of BBB (Engelhardt and Liebner, 2014).
Vascularization and angiogenesis of BBB happen after the penetration of neuroblasts in the cranial area, where they form the perineural vascular plexus. This is followed by the proliferation of blood vessels from the perineural vascular plexus. The blood vessels grow radially into the neuroectodermal tissue and form multiple small spines that connect with the neighboring vessels. Throughout the prenatal period, vascularization is associated with the formation of a mature spatial structure and a peak of angiogenesis activity that remains stable until the early postnatal period (Ma et al., 2012).
Unlike other tissues, CNS vascularization is exclusively driven by the angiogenesis. Different factors such as vascular endothelial growth factor (VEGF), angiopoietin-1, and sonic hedgehog protein (Shh) change the phenotype of endothelial cells of the perineural vascular plexus to blood vessels that sprout into the neural tube (Lippmann et al., 2012). Differentiation of endothelial cells requires a basal membrane formed by various extracellular matrix proteins (collagen IV, fibronectin, and laminin-1). The coverage of the microvasculature by pericytes purposely determines them as the first neurovascular unit cells to physically interact with endothelial cells (Virgintino et al., 2007). Pericytes, together with neighboring neural progenitor cells and radial glia might also influence the BBB development and induce barrier properties in the brain endothelial cells (Weidenfeller et al., 2007; Daneman et al., 2010b).
During the fetal period, endothelial cells acquire the phenotypic properties of specific tissues and inhabit areas of the developing brain, containing neuroepithelial cells, radial glia, neurons, and neuroblasts (Lippmann et al., 2012). Immunophenotyping of endothelial progenitor cells showed that they express some key antigens markers such as CD31, CD34, and CD45 (Sukmawati and Tanaka, 2015). Mobilization and collection of endothelial progenitor cells in the developing and mature brain are regulated by paracrine and endocrine signals that are produced by BBB. VEGF, Insulin-like growth factor-1 (IGF-1) and angiopoietin-2 are crucial for the mobilization of endothelial progenitor cells. During embryogenesis, endothelial cells retain the important function of regulating neurogenesis by maintaining a population of the brain stem and progenitor cells using growth factors such as brain-derived neurotrophic factor (BDNF), leukemia inhibitory factor (LIF), and Platelet-derived growth factor (PDGF). These processes also occur in the mature brain during the postnatal period (Urban and Guillemot, 2014). In the embryonic brain, radial glial cells, and neuroepithelial cells interact with astrocytes and assist the maturation and maintenance of BBB. Several growth factors and signaling molecules such as angiopoietin-1, cyclic adenosine monophosphate, and basic fibroblast growth factor affect the BBB phenotype in vitro. It is clear that the BBB phenotype is influenced by the local microenvironment and is not intrinsic to brain endothelial cells themselves (Lippmann et al., 2013).
Experiments on animal models (rodents) showed that BBB formation begins about 10 days after the embryonic development, followed by the expression of transporter molecules and tight junction proteins. BBB shows transendothelial resistance, which gradually increases during the early postnatal period and corresponds to the formation of full-fledged tight junctions (Siegenthaler et al., 2013). In contrast to animal models, many fundamental features of human BBB development and maintenance remain unclear. After 5 weeks of gestation, the first vessels of the brain start to form as vesicles. During weeks 6–7, these vesicles develop to form a capillary coat in the ventricular zone (Budday et al., 2015). After 15 weeks, the vessels penetrate radially through the nervous tissue and develop into collateral vessels with increased vascular density. During weeks 20–22, the growth of horizontal branches in the lower half of the cortex is detected. This phenomenon is followed by the growth of telencephalon microvessels and the expression of claudin 5 in endothelial cells (Norman and O'Kusky, 1986; Virgintino et al., 2007).
The final step of BBB development is maturation and formation of the NVU. These events are regulated by factors such as VEGF, death receptor 6 (DR6), and tumor necrosis factor receptor superfamily member 19 (TNFRSF19; Obermeier et al., 2013). The neuroectodermal cells, perivascular astrocytes, and pericytes induce the expression of BBB-associated proteins in the brain endothelial cells, which in contact with neurons and glial cells form the BBB (Moretti et al., 2015). BBB maturation in the postnatal period is determined by the formation of stable cell-to-cell interactions within the NVU (Daneman et al., 2010a) and regulated by various humoral factors (i.e., hormones, cytokines, and neurotransmitters).
BBB Alterations in Neuroinflammation: Molecular Targets for Inflammation-Inducing Stimuli
Microglia are the primary components of the CNS innate immune system. They produce cytokines and monitor the integrity of CNS. Microglia comprise about 5–20% of brain glial cell population (Sousa et al., 2017) and are found both in the white and gray matter of the brain and spinal cord. Microglia develop early during the embryogenesis and then migrate to the CNS (Ginhoux et al., 2010). The relatively small turnover of microglia makes them sensitive to the effect of inflammatory stimuli such as trauma, stress, and age (Ajami et al., 2007; Ginhoux et al., 2010). Microglia are crucial for surveying their microenvironment (Davalos et al., 2005; Nimmerjahn et al., 2005) and transporting the inflammatory signals (Dantzer et al., 2008) to the CNS. Activated microglia respond to inflammatory signals by modifying their gene expression at the transcription level and releasing cytokines that assist in recruiting leukocytes to the CNS (Zhou et al., 2006).
The regulation of neuroinflammatory responses is often a result of cooperation between different chemokines [such as chemokine (C-C motif) ligand 2 (CCL2), chemokine (C-C motif) ligand 5 (CCL5), and chemokine (C-X-C motif) ligand 1 (CXCL1)], cytokines (such as IL-6, and TNFα), reactive species of oxygen and secondary messengers (such as prostaglandins). Most of these factors are secreted by activated microglia, which support the synaptic connections and enhance the immunological responses of CNS (Norden and Godbout, 2013; Schafer and Stevens, 2013). Other resident cells of the CNS endothelium and macrophages are also crucial for modulating the inflammatory signals (Dunn et al., 2006). Cytoskeletal rearrangement of activated microglia results in the modification of receptor patterns on their surface, which assist their migration to the source of inflammation (Russo and McGavern, 2015) to represent their macrophage-like activity on the site of injury or infection (Davalos et al., 2005). Therefore, the activated functions of microglia help to defend the CNS against the adverse effects of hostile threats while their over-activation might lead to some neuropsychiatric disorders such as depression (Norden and Godbout, 2013).
Alzheimer's disease (AD) is an example of chronic neuroinflammatory disorder. AD is associated with the activation of microglia, infiltration of the brain tissue with peripheral immune cells, and misfolding and accumulation of several essential proteins (Walter et al., 2007; Sokolova et al., 2009). These phenomena can eventually cause neuronal damage and lead to death (Bucciantini et al., 2002; Sokolova et al., 2009). Unlike in other autoimmune inflammatory diseases, the brain is the initial site of accumulation of soluble β-amyloid and phosphorylated Tau proteins which lead to AD (Van Eldik et al., 2016). Accumulation of β-amyloid causes cell toxicity, which might be counteracted by the expression of transporter molecules in the BBB to transfer the β-amyloid from the brain tissue to the blood. However, this mechanism appears to be affected in AD. Further, progression of neuroinflammation results in deposition of insoluble proteins aggregates, release of alarmins from the affected cells [i.e., HMGB1 (high mobility group box-1)] and elevated permeability of BBB leading to the migration of immune cells to the brain tissue. Alzheimer's type neurodegeneration and inflammatory responses greatly depend on metabolic alterations of neuronal and glial cells (i.e., impairment of neuron-astrocyte metabolic coupling, hypometabolism of glucose, glycolysis, and mitochondria-controlled metabolic changes in microglia) and disruption of calcium homeostasis in neuronal and astroglial cells. Such changes are linked to β-amyloid-induced hypervascularity (extensive angiogenesis) and hyperpermeability of newly-formed cerebral microvessels (Biron et al., 2011). Accumulation of β-amyloid is also related to endothelial dysfunction, vasoconstriction and regional cerebral hypoperfusion of damaged cerebral vessels in AD (Thomas et al., 1996; Niwa et al., 2000; Suter et al., 2002; Townsend et al., 2002; Smith and Greenberg, 2009). The local neuroinflammation in AD stimulates the impairment of BBB/NVU, while the loss of functional and structural integrity in the BBB promotes inflammatory alterations (Salmina et al., 2010).
In addition to the chronic inflammation, CNS can be also affected by the so-called acute neurodegeneration, which is caused by various stimuli such as stroke, head injury, and cerebral or subarachnoid hemorrhage. Some of the markers of acute neurodegeneration are the release of certain chemokines and cytokines (i.e., TNFα, IL-1β, and IL-6), and the activity of microglia. The occurrence of acute neurodegeneration has positive impacts on the coordination of the CNS function to deal with the peripheral injuries or infections and improve the behavioral and physiological responses (Imeri and Opp, 2009). During acute neurodegeneration, migration of the peripheral immune cells to the CNS is not significant, and no adverse effects such as cell atrophy or impairment of the BBB occur. It is therefore suggested that development of acute neurodegeneration assists the optimal local immune responses and provides the microenvironment that is necessary for brain recovery (Tarr et al., 2014). However, acute neuroinflammation can also associate with excessive neuronal injury, BBB leakage, and neurological deficits that hamper neuronal regeneration and recovery.
Contradictory Aspects of Neuroinflammation
The close relationship between brain and spine injuries and development of neuroinflammation unveils a cascade of modulating events in affected hosts. CNS injuries activate microglia and astrocytes, release certain chemokines and cytokines and accelerate the migration of peripheral immune cells to the CNS (Werner and Engelhard, 2007). They also cause edema, which is associated with elevated BBB permeability. Moreover, injuries to the CNS trigger post-traumatic symptoms with short-term inflammatory responses that can lead to long-term damages (David and Kroner, 2011; Woodcock and Morganti-Kossmann, 2013). The development of neuroinflammatory responses by the host immune system plays a dual role, which can be harmful or beneficial, depending on the type and extent of the stimulation. Damages to the CNS stimulate the inflammatory and repair responses of microglia and reveal their pro-inflammatory and neuroprotective roles, respectively.
The cytokine-mediated sickness behavior is an example of the positive effect of neuroinflammation. The sickness behavior is induced upon the activation of the immune system by various stimuli carried to the CNS. The corresponding inflammatory signal is processed in the NVU, brain stem, and circumventricular areas of the CNS (Laflamme et al., 1999; Hansen et al., 2001; Ching et al., 2007) which produce cytokines such as IL-1β, TNF-α, and IL-6 (Henry et al., 2009; Chen et al., 2012). These chemical messengers connect the CNS to the immune system and stimulate the development of sickness behavior, which is defined by fever, hypophagia, lethargy, listlessness, and reduced social communications (Dantzer et al., 2008). It is thought that the development of sickness behavior is evolutionarily necessary to counteract against life-threatening antigens and infections (Bluthe et al., 2000; Berg et al., 2004) without further losing the BBB integrity or transfer of peripheral immune cells to the CNS (Dantzer et al., 2008).
Other positive effects of neuroinflammation are immune conditioning (dose-dependent stimulation of the peripheral immune system using bacterial cells; DiSabato et al., 2016), developing the brain plasticity (stimulation of neurogenesis in the neurogenic niches), and assisting the repair process in the brain. On the other hand, some of the major negative impacts of neuroinflammation are injury-related hyperinflammatory responses, which trigger noradrenergic signaling (DiSabato et al., 2016) and activate inflammasomes in the effector cells. These events accelerate the aging process of the brain by activating the immunologically challenged microglia.
Altogether, it can be concluded that the acute neuroinflammation is the positive and beneficial aspect of the CNS immune response whereas the chronic state of neuroinflammation is associated with brain damage and prolonged neurological deficits.
Association of Infectious Diseases with Neuroinflammation
Activation of local inflammation often starts from the BBB endothelial cells equipped with the molecular machinery to sense bacterial and viral antigens. The first line of defense against microbial invasion comprises the antigenic recognition of a large group of conserved molecular determinants, called pathogen-associated molecular patterns (PAMPs; Hanke and Kielian, 2011) by pattern recognition receptors (PRRs). The PRRs are located on the surface or within the cytoplasm of antigen presenting dendritic cells, macrophages, or other none-immune cells. PRRs activate the innate immune responses, trigger the phagocytic pathways and directly bind to the invading microorganisms. Toll-like receptors (TLRs) are essential PRRs, containing repeats of leucine residues on the N-terminus and a highly conserved C-terminal domain so-called Toll/interleukin (IL)-1 receptor (TIR). Expressed by glial cells and neurons, TLRs are necessary for priming the adaptive immune responses and inducing the release of co-stimulatory molecules and inflammatory cytokines (Hanke and Kielian, 2011). TLRs also recognize a distinct group of host-derived molecules called danger associated molecular patterns (DAMPs), which are released upon the onset of diseases and infections that cause necrosis, apoptosis or tissue damage (Kirschning and Schumann, 2002; Kaisho and Akira, 2004; Kariko et al., 2004; Piccinini and Midwood, 2010; Hanke and Kielian, 2011). The TLR-mediated DAMPs and PAMPs recognition is followed by forming an intracellular molecular machinery (inflammasome) for caspase-dependent processing of the cytokines (IL-1, IL-18). Expression of inflammasomes by the activated glial cells stimulate the brain-leukocyte infiltration (Alfonso-Loeches et al., 2016). In particular, chronically elevated levels of IL-1 in brain tissue was shown to provoke BBB breakdown and neutrophil recruitment (Ferrari et al., 2004). Therefore, measurement of IL-1 and IL-18 levels in the CSF seems like a useful tool to evaluate the severity of the neuroinflammation, i.e., in AD (Wang et al., 2015).
The state of neurological and neurodegenerative diseases can be influenced by peripheral factors such as the gut microbiome (Dinan and Cryan, 2015). Our gastrointestinal tract is perpetually covered with a population of microbial flora (Ley et al., 2006) with the ability to cause significant impacts on the brain by releasing neurotransmitters, hormones, and neuropeptides (Selkrig et al., 2014; Wall et al., 2014). Some examples of these effects are neurodegenerative diseases, depression and autism spectrum disorder (ASD; Putignani, 2012; Mayer et al., 2014; Schroeder and Backhed, 2016; Sharon et al., 2016). Recent findings suggested that human microbiome can significantly affect brain function (Stilling et al., 2014), development (O'Mahony et al., 2017; Tognini, 2017), and neuroinflammation (Rea et al., 2016). The gut microbiota-BBB communication is evident since the gestation period: normal gut flora is required for BBB maturation, establishment of tight junctions, and BBB permeability (in mice; Braniste et al., 2014). Alterations in the composition of normal flora is associated with severe neurological disorders which affect brain development, plasticity, and cause behavioral abnormalities (El Aidy et al., 2014). Parkinson's disease (PD) is a multifactorial neurodegenerative disorder, which is associated with the accumulation of specific amyloid protein, called α-synuclein (α-Syn), a phenomenon that also occurs in other PD-related diseases such as multiple system atrophy (Brettschneider et al., 2015; Sampson et al., 2016). Patients with PD show significant gastric (intestinal) inflammation and majorly suffer from motor deficiencies. Study of a mouse model of Parkinson's disease suggested that the motor circuits dysfunctions are linked to the gastric tract abnormalities and highlighted the crucial role of gut microbial population in triggering the augmentation of α-Syn in PD (Sampson et al., 2016). Host immune responses against infectious diseases are also involved in the pathogenesis of depression and ASD (Miller and Raison, 2016). Several studies have pinpointed the association of aberrant neuroimmune responses with autism and development of depressive behavior (Buehler, 2011; McCusker and Kelley, 2013). In autism, neuroinflammation might occur in the intrauterine period of ontogenesis, and might further progress in the postnatal life in association with impaired angiogenesis and BBB hyperpermeability (Azmitia et al., 2016). In autistic patients, the loss of BBB integrity is linked to the low expression of tight junction proteins and elevated permeability of their intestinal barrier (Fiorentino et al., 2016). These findings suggest that intestinal microflora has a causative role in progression of autistic behavioral abnormalities (Diaz Heijtz, 2016).
One of the most notorious infections of CNS is caused by Mycobacterium tuberculosis. Tuberculosis (TB) creates a typical pro-inflammatory response of host immune system (Lee et al., 2009). The TB of CNS is probably the most severe type of tuberculosis and in most cases leads to death. Invasion of the CNS by M. tuberculosis is associated with formation of myddosome (see below), disruption of BBB due to cytoskeletal rearrangement in cerebral microvessel endothelial cells (Jain et al., 2006; Cervantes, 2017) and matrix metalloproteinase-mediated degradation of BBB (Green and Friedland, 2007). Tumor necrosis factor (TNF) is a proinflammatory cytokine, which is involved in priming the host immune system against M. tuberculosis infection by activating the innate immunity and maintaining the granulomas structure. Investigation of the role of TNF in the immune response against TB of the CNS has shown its protective role. Those findings also proved that neurons are essential sources of TNF production to regulate the immune response against pathogens (Francisco et al., 2015).
Some bacterial infections can develop neuroinflammation by altering the expression of endothelin-1 (ET-1; Freeman et al., 2014). ET-1 is an isoform of endothelin, a short peptide with 21 amino acid residues, which is mainly expressed by endothelial cells. ET-1 is important for maintaining the vascular homeostasis (Agapitov and Haynes, 2002; Schinelli, 2006), vascular tone and inflammation (Speciale et al., 1998; Bouallegue et al., 2007; Kohan et al., 2011). Different types of cells such as neurons, cardiomyocytes, and macrophages produce ET-1 (Freeman et al., 2014). Although ET-1 is mainly a vasoconstrictor, it also acts as a pro-inflammatory cytokine, stimulates the aggregation of platelets and induces the expression of leukocyte adhesion molecules. ET-1 also stimulates the synthesis of inflammatory mediators that cause vascular dysfunction and lead to the progression of diseases and inflammation (Teder and Noble, 2000). Several infectious diseases such as malaria (Dai et al., 2012), infection of Rickettsia conorii (Davi et al., 1995), and Chagas disease (Petkova et al., 2001) are associated with ET-1 hyper-expression. These findings indicate the potential role of infectious diseases in developing neuroinflammation by activating ET-1 as a pro-inflammatory cytokine (Freeman et al., 2014). Moreover, expression of ET-1 has been shown to increase in PD (Jain, 2014) and AD (due to the activity of β-amyloid; Palmer et al., 2012). Since overexpression of ET-1 in brain tissue mediates the breakdown of BBB (Zhang et al., 2013), degenerative disorders seen in AD and PD could be, at least partially, caused by the ET-1 hyperexpression.
Taken together, it is crucial to maintain the normal status of body microflora (microbiome), which is necessary for the development of cerebral microvessel endothelial cells. Alterations in the microbiome might provoke inflammation-mediated BBB breakdown or aberrant maturation of newly established cerebral endothelial layer. If so, the association of chronic infections with NVU impairment seen in neurodegenerative or neurodevelopmental diseases is not surprising.
LPS: A Key Player in the Development of the CNS Neuroinflammation
Repeated and minimized contact with infectious agents such as bacterial cells or their constituents can activate the peripheral immune system, thus providing immune protection, in a unique way, which might not involve stimulation of neuroinflammatory responses in the CNS. This phenomenon is called euflammation (Tarr et al., 2014; Liu et al., 2016). Euflammation alters the innate immune system, through regulating the peripheral inflammatory kinetics and controls the receptors that bind to microbial antigens. It also down-regulates the production of pro-inflammatory cytokines, inhibits the activation of brain microglia, and minimizes the development of sickness behavior in animals that received bacterial cells or lipopolysaccharide (LPS; Tarr et al., 2014). Therefore, euflammation can provide some immune protection against bacterial infections and severe toxicity by their endotoxins (Liu et al., 2016).
LPS is a major component of the cell wall structure of gram-negative bacteria and a well-described endotoxin consisting of a polysaccharide chain (varies amongst different gram-negative bacteria) and lipid A (Alexander and Rietschel, 2001). LPS endotoxins are used in modeling bacterial infections and stimulating the infection-associated inflammation via triggering TLR-4, a well-known receptor of LPS (Sandor and Buc, 2005; Rosadini and Kagan, 2017). The interaction of TLR4 with LPS triggers the formation of a macromolecular complex, so called myddosome (Rosadini and Kagan, 2017), including several proteins such as myeloid differentiation primary response gene 88 (MyD88), TIR domain-containing adaptor protein (TIRAP), and interleukin-1 receptor-associated kinase-1 (IRAK). The myddosome complex stimulates the signaling pathways that activate NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells), activation protein 1 (AP-1), and hyper-expression of several inflammatory genes (Rosadini and Kagan, 2017). TLR4 also regulates other inflammatory responses, such as the release of mediatory microRNAs which sequester LPS-induced pro-inflammatory responses in order to minimize the tissue damage caused by LPS (Molteni et al., 2016).
Nuclear binding domain NOD-like receptors (NLRs) are a main group of PRRs, which detect the bacterial cell wall components and trigger the inflammation. Nucleotide-binding oligomerization domain-containing proteins 1 and 2 (NOD-1 and NOD-2) are two major NLRs that detect bacterial peptidoglycan, induce the release of NF-κB and activate the mitogen-activated protein (MAP) kinase-dependant inflammatory responses (Elinav et al., 2011). Studying the effects of NOD-1 and NOD-2 on brain activity of mice models showed that co-activation of NOD and TLR4 stimulated the peripheral immunity and intensified LPS-activated TLR4 impact on sickness behavior and brain function (Farzi et al., 2015).
Furthermore, recent studies suggested that tolerance to LPS might trigger a late pro-inflammatory response and increase the expression of some anti-inflammatory cytokines that cause inimical injuries to the CNS (Pardon, 2015). Frequent administration of LPS triggers the hyper-expression of IL-1β, TNF-α, and IL-12 in brain but reduces the systematic expression of cytokines. The occurrence of systemic infections increases the potential of brain innate immune cells to develop tolerance and may induce or aggravate the neurodegenerative process due to the damaging effect of cytokine hyper-production (Puntener et al., 2012).
Epilepsy might be an example of an infection-mediated neuroinflammatory response, which contributes to the progression of chronic CNS disorders. Epilepsy is defined by the occurrence of unprovoked brain seizures and affected by a multitude of factors such as genetic background, CNS trauma, and infections (Vezzani and Granata, 2005). Different infections from bacteria, viruses, fungi, parasites, and prions can stimulate the CNS inflammation and induce epileptic seizures (Vezzani et al., 2016). Bacterial LPS can trigger epilepsy in mice and rat models through stimulating the secretion of cytokines, in particular, IL-1β, which is crucial in epileptogenesis (Auvin et al., 2010). Administration of LPS into the peritoneal cavity of rats induces a cyclooxygenase-2 (COX-2) dependent inflammation. COX-2 is a prostanoid-forming enzyme, which is activated during seizures (Rojas et al., 2014). The inducing effect of LPS on COX-2 indicates a higher seizure susceptibility and a more intense oxidative response during the LPS-mediated neuroinflammation (Ho et al., 2015).
Lipoteichoic acid (LTA) is a main constituent of the cell wall structure which is found in gram-positive bacteria, consisting of polyhydroxy alkane repeats. LTA assists with bonding the bacteria to the microvascular endothelial cells of brain (Sheen et al., 2010). Testing the LTA extracts on mice brain revealed that it simulated the release of interferon-γ (IFNγ), IL-6, and other cytokines. LTA was also associated with hyperexpression of circulating corticosterone and diminished expression of tight junction proteins, claudin 5 and occludin, in the brain (Mayerhofer et al., 2017). Upon the onset of bacteriolysis in blood, i.e., as a result of antibiotic therapy, LTA is released and detected by TLR2. LTA-TLR-2 triggers the secretion of inflammatory cytokines such as TNF-α and IL-1b, which ultimately damage BBB (Boveri et al., 2006).
The Impact of Infectious Diseases on the Incidence of Neurodegenerative Disorders
As mentioned above, several infectious agents could contribute to neuroinflammation and neurodegeneration. Recently, a group of pathogenic agents including Borrelia burgdorferi, Porphyromonas gingivalis, Chlamydophila pneumoniae, Helicobacter pylori, Cytomegalovirus, Herpes simplex virus type 1, Epstein-Bar virus, Human herpes virus 6, Candida glabrata, and Toxoplasma gondii have been addressed to have a significant effect on the late onset of AD in adults (LOAD; Bu et al., 2015; Lim et al., 2015; Figure 2). The late-age development of AD is purposely due to the activity of infections that initially occurred during the childhood (Khachaturian, 1985). In AD, the association of chronic infections with progressive neurodegeneration was clearly demonstrated by recent studies (Maheshwari and Eslick, 2015). Chronic spirochetal infections can induce β-amyloid accumulation in the brain, cause dementia and reproduce the clinical, pathological, and biological hallmarks of AD (Miklossy, 2011). Thus, infections by spirochete bacteria have significant potentials in developing AD (De Chiara et al., 2012; Hill et al., 2014; Maheshwari and Eslick, 2015). Different species of spirochetes, such as T. socranskii, T. pectinovorum, T. denticola, T. maltophilum, T. medium, T. amylovorum, and Borrelia burgdorferi (causing Lyme disease) are found in the brain of AD patients (Burgdorfer et al., 1982; Riviere et al., 2002). T. pallidum (the causative agent of syphilis) can reside in the brain and cause chronic infection which is associated with inflammation and dementia (Miklossy, 2015). Moreover, infection by some spirochetes such as B. burgdorferi could trigger the formation of specific granulovacuolar lesions in neurons and glial cells, which are comparable to those found in AD (Miklossy et al., 2006). AD patients are more vulnerable to infection-mediated cognitive impairment (McManus and Heneka, 2017), particularly in the case of chronic respiratory tract infections (McManus et al., 2014). Since β-amyloid shows antimicrobial properties in vitro (Welling et al., 2015; Spitzer et al., 2016), it is postulated that the excessive accumulation of β-amyloid in AD brain could reflect the response of brain neuronal cells to microbial agents. The co-morbidity of neurodegenerative and infectious diseases affects the progression of neurological disorders. For instance, the main cause of death in AD is the onset of infectious diseases such as pneumonia or urinary tract infections (Miklossy, 2015).
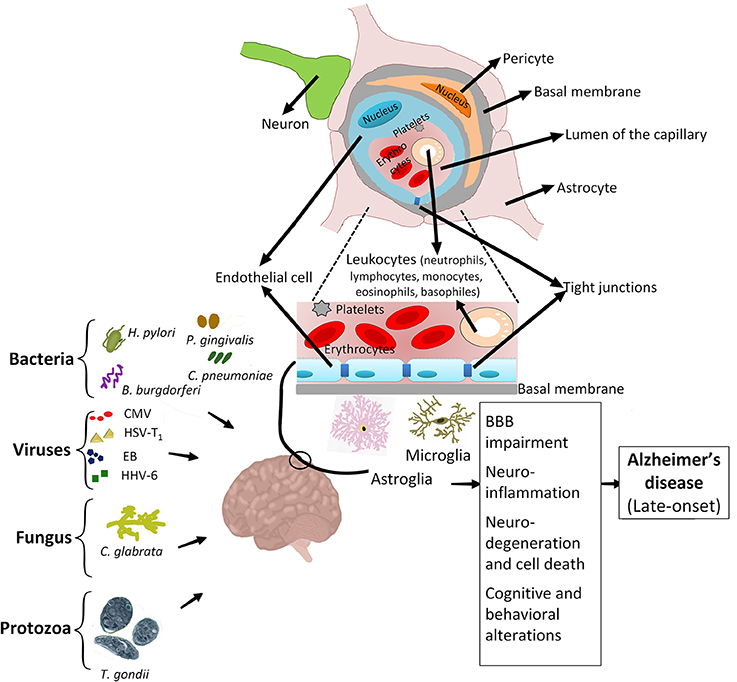
Figure 2. Association of infectious agents with Alzheimer's disease. Chronic infections caused by major infectious agents, i.e., Helicobacter pylori, various types of spirochetes, including periodontal pathogen spirochetes and Borrelia burgdorferi, Porphyromonas gingivalis, Chlamydophila pneumoniae, Cytomegalovirus, Herpes simplex virus type 1, Epstein-Bar virus, Human herpes virus 6, Candida glabrata and Toxoplasma gondii are associated with development of AD. Early life exposure to these pathogenic agents can activate the resting microglia and astroglia, trigger the migration of immune cells to the neuro-endothelial tissue, degrade cell-cell tight junctions, and cause the breakdown of BBB. These activities result in development of various side effects such as neuronal damage, neuroinflammation and ultimately predispose the adult patient to develop AD.
C. pneumoniae is an intracellular pathogenic bacterium which infects mucosal surfaces and causes respiratory infections such as community-acquired pneumonia (Grayston et al., 1990). C. pneumoniae is suggestively associated with other non-respiratory malignancies such as inflammatory arthritis, multiple sclerosis, and AD (Balin et al., 2008). The association of C. pneumoniae with pathogenesis of AD has been pinpointed in several studies (Balin et al., 2008; Shima et al., 2010). However, it is not still clear whether C. pneumoniae infection actually develops AD as there are some other studies which failed to show this relationship. Perhaps by utilizing a suitable C. pneumoniae infection model and applying standard methods which analyse homogenous sample analysis one would be able to elucidate this obscurity (Shima et al., 2010).
H. pylori is a gram-negative and spiral shaped bacterium that causes progressive and multistep inflammation of gastric lumen which, can lead to gastric cancer (adenocarcinoma; Tohidpour, 2016). H. pylori infection has been linked to high risk of AD in infected patients. Analysis of rat models of H. pylori infection showed evidence of memory and spatial learning defects as well as damage to the maturation of hippocampus dendritic spine cells (Wang et al., 2014). Such harmful effects are possibly exerted by soluble components of H. pylori surface fractions, which induce formation of Aβ42, a member of β-amyloid (Aβ) family, as main constituents of extracellular senile plaques (SP). Infection by H. pylori enhances the activity of γ-secretase, one of two enzymes, which process the amyloid precursor protein to produce mature Aβ, thus increasing levels of Aβ in brain of infected host and contributing to the development of AD (Wang et al., 2014).
The Relationship Between Viral Infections and Neuroinflammation in the CNS
The impact of viral infections on neuroinflammation develops through the interaction of CNS-associated immune responses with virulence factors of viruses, which trigger neurodegeneration and apoptosis in the brain (Amor et al., 2010; Czirr and Wyss-Coray, 2012). Viral encephalitis afflicts the CNS with various atrophies, such as loss of memory, acute CNS damage and death. The direct invasion of viruses to CNS can occur through at least five different pathways: (i) by tight junctions of brain microvascular endothelium; (ii) invading the brain microvascular endothelium and penetrating through the basolateral membranes; (iii) transfecting the migrating leukocytes to penetrate and infect the CNS; (iv) migrating through peripheral neuron to the CNS; and (v) infecting the olfactory bulb epithelium and migrating to the CNS (Miner and Diamond, 2016). Microglia and astrocytes are two main types of cells, which are affected during viral infection of the CNS. They produce pro-inflammatory cytokines and various immune-mediator molecules that activate the immune system against viruses (Ransohoff and Perry, 2009; Ransohoff and Brown, 2012; Perry and Teeling, 2013; Phares et al., 2013).
Recognition of viral pathogenic molecular patterns by host PRRs stimulates different signaling pathways and activates transcription factors such as NF-κB and interferon regulatory factor 3 (IRF3). Activated IRF3 induces the expression of interferon β (INF-β), which exerts antiviral activity by inhibiting protein synthesis of viruses. Activated NF-κB triggers the release of some anti-apoptotic proteins and pro-inflammatory cytokines with significant antiviral effects (Santoro et al., 2003; Vercammen et al., 2008). Microglia and astrocytes have been shown to actively respond to both DNA and RNA viruses [i.e., vesicular stomatitis virus (VSV), cytomegaloviruses and Sendai virus] by releasing various types of proinflammatory cytokines and chemokines such as IL-1β, TNF-α, and IL-6 (Furr and Marriott, 2012).
Viral infections also contribute to the development of AD. Viruses such as cytomegalovirus, herpesviridae, and herpes simplex type 1 (HSV-1) can escape the immune system and activate innate and adaptive immune responses via stimulating hyper-expression of pro-inflammatory cytokines with their DNA or RNA (Harris and Harris, 2015). Viral infections either directly or indirectly contribute to AD pathogenesis through various pathways such as increasing the concentration of amyloids, phosphorylating certain neuronal proteins and afflicting neurons with injury. For instance, infections with cytomegaloviruses generate a systematic population of pro-inflammatory cytokines which can pass BBB, trigger the CNS neurodegeneration and lead to AD (Lim et al., 2015). HSV-1 is usually found in the brain of infected adult hosts (Wozniak et al., 2005) and is thought to be involved in development of AD in this group of patients (Agostini et al., 2014). Primary infection with HSV-1 leads to persistent infection of sensory ganglion cells that belong to the peripheral nervous system. Various factors lead to activation of HSV-1 from the latency phase, such as stress and ultraviolet radiation (Itzhaki, 2014). Re-activated HSV-1 affects the hippocampus as well as the frontal and temporal cortices of brain, which are also being affected in AD patients' brains. The immune system is also able to reactivate HSV-1 from latency. However, results of recent experiments have suggested a protective role of humoral immunity against the damaging effect of HSV-1 in the brain of infected patients (Mancuso et al., 2014).
The ability of human immunodeficiency virus (HIV) type 1 to invade and infect the CNS is noteworthy. HIV can penetrate the CNS some weeks after the infection and induce a neuroinflammation pathway, which leads to the CNS injury (Schacker et al., 1996; Pilcher et al., 2001). It is thought that the CNS and the cerebrospinal fluid are potential niches for replication of HIV. The occurrence of genetic mutations in HIV genome leads to the evolution of HIV strains that evolve during early stages of infection and cause CNS inflammation and neurogenic damages (Dahiya et al., 2013). Although the current strategies of anti-viral therapy have drastically reduced neurocognitive impairments caused by HIV, the overall occurrence of such injuries has increased. This increase is linked to the difficulties of accessing the anti-HIV treatments in developing countries. Previous experiments revealed that brain microvascular endothelial cells are crucial for the immunity of the CNS against HIV-mediated neuroinflammation. These cells recognize HIV molecular determinants using TLR3, release antiviral compounds such as INF-β and trigger the phosphorylation of IRF3 and IRF7, activities of which control the interferon signaling pathways and confer immune defense against HIV infection (Li et al., 2013).
The neuroinflammatory disorders induced by West Nile virus (WNV) is another example of a viral infection-mediated CNS neurodegeneration. WNV is a mosquito-borne RNA virus belonging to flaviviruses (Lindenbach and Rice, 2003). WNV is a major pathogen of the CNS neurons and causes viral encephalitis. Microglia and astrocytes of CNS are both infected with WNV and develop apoptosis (Yang et al., 2002; Chambers and Diamond, 2003). A large number of viral proteins, such as envelope and capsid proteins are involved in the neuropathogenesis of WNV (Beasley et al., 2002, 2005; Lee and Lobigs, 2002). Amongst these, capsid proteins are the main viral factors causing neuroinflammation, neurotoxic effects, and apoptosis (Yang et al., 2002). Upon infection with WNV, several PRRs including MDA5 (melanoma differentiation-associated protein 5), TLR3, TLR7, and RIG-I (retinoic acid-inducible gene 1) determine the molecular patterns of WNV. This recognition leads to hyperexpression of inflammatory cytokines such as IL-1 and TNF-α, which inhibit viral replication, enhance the presentation of viral antigens and increase the migration of leukocytes (to eradicate the WNVs from the CNS; Daniels et al., 2014). Activation of PRRs by WNV also triggers the expression of IFN-α and IFN-β (type I INFs), which inhibit viral replication (Samuel and Diamond, 2005) and enhance the capability of the adaptive immune system. In vitro studies on BBB models showed that the antiviral activities of type I IFNs regulate the permeability of brain endothelial cells and thus reduce viral movements across the BBB (Daniels et al., 2014).
Two other members of flavivirus family, Zika virus (ZIKV) and Dengue virus are also linked to neuroinflammation of the CNS (Tsai et al., 2016; Roach and Alcendor, 2017). ZIKV, normally causes limited infections with symptoms such as fever, headaches, and conjunctivitis (Lum et al., 2017). However, ZIKV is also associated with brain microcephaly and ocular defects in infants, which occur during the course of pregnancy (Roach and Alcendor, 2017). Following recognition by TLR-3, infection of fetal brain cells with ZIKV impairs the neurosphere and brain organoid growth. ZIKV infection has been also found in Microglia and was shown to increase the level of cytokines such as IL-6, TNF-α, and IL-1β (Lum et al., 2017). Same as WNV and ZIKV, Dengue is also a mosquito-borne RNA flavivirus. Globally, ~2.5 billion people are at risk of infection by Dengue virus (DENV; Guabiraba and Ryffel, 2014). DENV infects around 400 million people each year causing symptoms such as fever, headache and rashes, which are referred as dengue fever stage. A minor proportion of patients who show the symptoms of the dengue fever further develop an acute form of disease called severe dengue haemorrhagic fever (DHF)/and shock syndrome (DSS). Some symptoms of DHF/DSS include gastrointestinal bleeding, renal/hepatic failure and hemorrhage which are determined by the interaction of viral virulence factors with components of host immune system. Strikingly, patients with DHF/DSS are very prone to neurological defects and CNS abnormalities. DENV can invade the CNS by transmitting though BBB and induce encephalitis (Tsai et al., 2016).
Another example of a virus, which also causes encephalitis, is Chikungunya virus (CHIKV). CHIKV is a member of encephalitogenic RNA viruses and is transmitted by mosquito bites (Long et al., 2013; Gerardin et al., 2016). The infection of CHIKV causes debilitating rheumatic diseases and inflammatory disorders which are generally non-fatal. However, in rare cases CHIKV infection can cause neurodegenerative disorders such as meningoencephalitis, myelitis, and Guillain-Barre syndrome (Gerardin et al., 2016; Brizzi, 2017).
Taken together, the onset of viral infections in the CNS can induce neuroinflammation in the glial cells of NVU (Allen et al., 2009; Kuang et al., 2009) and activate host immune responses against the invading viral pathogens. Such immune responses might eventually result in elevated local cytokines/chemokines concentrations and loss of BBB integrity.
Role of Host Factors and Genetic Alterations in Neuroinflammation
In addition to infectious agents and injuries, genetic mutations can also induce neuroinflammation. Genetic studies have elucidated the molecular mechanisms underlying the etiology and pathogenesis of various neurodegenerative disorders. Table 1 shows a list of some major immune genes whose mutations significantly induce different neurodegenerative diseases (Table 1).
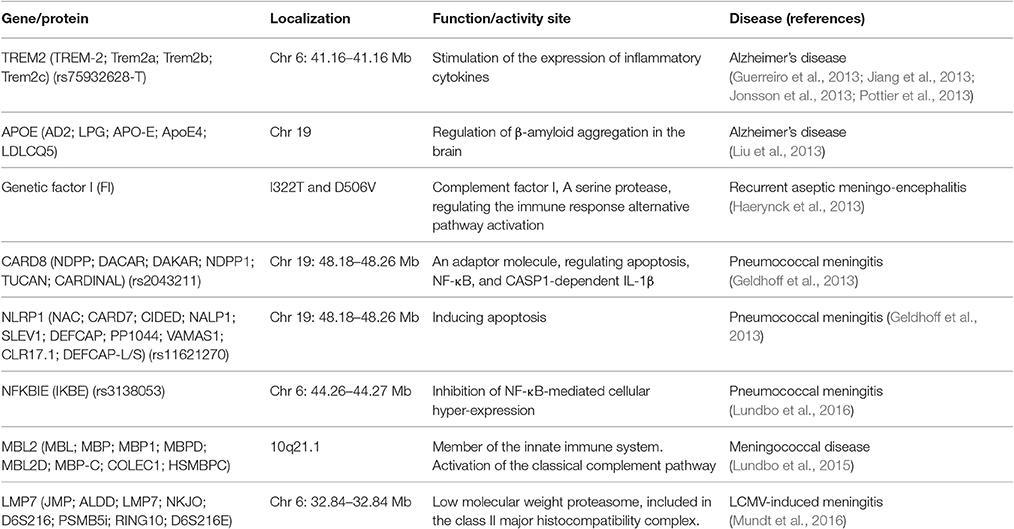
Table 1. Immune genes associated with development of neurodegenerative diseases such as AD, encephalitis, and meningitis.
Acute hemorrhagic leukoencephalitis (AHLE) is a type of acute disseminated encephalitis that often leads to death. The etiology of AHLE is thought to be associated with upper respiratory infections, mumps, and infection by Mycoplasma pneumoniae. The deficit of complement factor I (CFI) is frequently associated with recurrent pyogenic infections such as meningitis and meningoencephalitis (Floret et al., 1991; Leitao et al., 1997). CFI is a regulator of the complement alternative pathway and a major complement inhibitor. Complete deficiency of CFI results in secondary complement deficiency due to uncontrolled and spontaneous alternative pathway activation, and leads to hyper-susceptibility to infections (Nilsson et al., 2009). Broderick and co-workers described two pediatric AHLE patients of Filipino descent with partial CFI deficiency. They showed that the primary site of inflammation was in the CNS and reported symptoms such as headache, hallucinations, and reduced pupillary reaction to light. Physical examination of other patients showed that they had difficulties with speaking and suffered from fatigue. The authors identified two novel missense mutations in CFI by sequencing 13 exons of CFI and suggested that infections may trigger an uncontrolled activation of complement in brain parenchyma of predisposed individuals, leading to severe neuroinflammation and demyelination (Broderick et al., 2013). Moreover, Haerynck and co-workers studied a rare deficiency of CFI in a patient with relapsing inflammatory-mediated meningoencephalitis. They described the case of a 16-year-old patient having headaches, nausea, vomiting, neck stiffness, diplopia, and lethargy as recurrent episodes of acute aseptic meningoencephalitis. MRI of brain further approved the evidence of meningoencephalitis. Mutation analysis of the complement factor I gene showed two heterozygous mutations (I322T and D506V) that resulted in a complete CFI deficiency due to a functional CFI defect (Haerynck et al., 2013).
Clinical studies on the role of genetic abnormalities in pneumococcal meningitis showed the association of meningitis with polymorphisms in the inflammasome genes encoding caspase recruitment domain family member 8 (CARD8; SNP ID: rs2043211) and NLR family pyrin domain containing 1 (NLRP1; SNP ID: rs11621270). Genetic variations possibly influence inflammasome genes and alter the activation threshold of inflammatory responses (Geldhoff et al., 2013). Furthermore, sequencing the coding regions of 46 innate immune genes from 435 patients, Ferwerda and co-workers showed that immune susceptibility to pneumococcal meningitis is related to variations in several genes encoding the CARD8, CXCL1, NOD-2, and interleukin-1 receptor-associated kinase 4 (IRAK4; Ferwerda et al., 2016). Lundbo and co-workers also found the association of pneumococcal meningitis with the polymorphism of nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor (NFKBIE) and that the increased risk of invasive pneumococcal disease (IPD) was in the heterozygosity meningitis group for NFKBIE (Lundbo et al., 2016). Taken together, it is noteworthy to consider the impact of genetic variations and genetic abnormalities on function of certain genes, which are responsible for activity, protection, and homeostasis of NVU and the CNS. The onset of malignant mutations that disrupt the physiological functions of such genes can predispose the host to develop severe neurodegenerative disorders and/or manipulate the ability of immune system to defend against infectious agents invading the CNS.
Concluding Remarks
Neuroinflammation in the CNS has two facets: one beneficial and the other destructive. It is not yet clear whether the onset of neuroinflammation is entirely useful or causes further damages. Moreover, the genetic background of host plays a critical role in predisposing the CNS to various neuroinflammatory responses and affecting the ability of the CNS to prevent neuropsychiatric and neurodegenerative disorders.
Microbial infections can trigger the CNS-associated immune responses and cause neurodegenerative and neurodevelopmental disorders. It is well-known that there is a complicated relationship between the host normal flora and the CNS. Majority of our understanding of the microbiome effects on the CNS homeostasis or disorders are derived from the microbiome-gut-brain axis. However, microbiome from other body niches might also be able to de-regulate the CNS and induce neuroinflammatory disorders. For instance, recent studies have found the presence of RNA of α-proteobacteria in human brain (Branton et al., 2013). Some α-proteobacteria are known as pathogen of human, such as Rickettsia conorii, Rickettsia rickettsii, and Delftia acidovorans (in compromised patients) and/or have been isolated from cerebrospinal fluid (D. acidovorans; Pedersen et al., 1970). It is therefore quite possible that such niche-specific microbiomes can directly engage components of host immune system (such as TLRs or NODs to activate brain immune signaling) and trigger neuroinflammation. Therefore, it seems crucial to further determine the effect of non-gut microbiome on homeostasis of the CNS and trafficking of immune constituents. It is practical to neutralize the severity of the CNS inflammation by antibiotic therapy of infections or eradication of inflammation-stimulating effector cells. However, these strategies are unable to guarantee lessening the excessive disadvantages of neuroinflammation or prevent psychological disorders such as mood or other degenerative diseases (Xanthos and Sandkuhler, 2014). Alterations in normal composition of gut microflora can trigger some adverse brain disorders such as activation of hypothalamic pituitary adrenal (HPA) axis (Rea et al., 2016), acute brain ischemia (Singh et al., 2016), and neurodegeneration (Minter et al., 2016). BBB serves as an essential mediator of the CNS-microbiome interactions. In this context, deciphering the mechanisms by which BBB is involved in pathogenesis of the CNS neuroinflammation sounds very important. It is therefore pivotal to further study the close relationship of host normal microbiome(s) and the CNS. This would provide a better understanding of the CNS-immune system interactions to improve the treatment of the CNS injuries and reduce the CNS susceptibility to infectious agents.
Author Contributions
AT and AS wrote and edited the manuscript, and designed Figures 1, 2 and Table 1. AM and NM wrote the manuscript and designed Figures 1, 2. EB wrote the manuscript and designed Table 1. NK, EK, GM, and GG wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The study was supported by the grant #16-44-243073 provided by the RFBR and Krasnoyarsk Scientific Foundation (analysis of BBB impairment caused by infectious stimuli), and by the grant No. 10241.2016.7 of the President of Russian Federation given to Russian Leading Research Teams (analysis of neuroinflammation in chronic neurodegeneration).
References
Agapitov, A. V., and Haynes, W. G. (2002). Role of endothelin in cardiovascular disease. J. Renin Angiotensin Aldosterone Syst. 3, 1–15. doi: 10.3317/jraas.2002.001
Agostini, S., Clerici, M., and Mancuso, R. (2014). How plausible is a link between HSV-1 infection and Alzheimer's disease? Expert Rev. Anti Infect. Ther. 12, 275–278. doi: 10.1586/14787210.2014.887442
Ajami, B., Bennett, J. L., Krieger, C., Tetzlaff, W., and Rossi, F. M. (2007). Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 10, 1538–1543. doi: 10.1038/nn2014
Alexander, C., and Rietschel, E. T. (2001). Bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res. 7, 167–202. doi: 10.1179/096805101101532675
Alfonso-Loeches, S., Ureña-Peralta, J., Morillo-Bargues, M. J., Gómez-Pinedo, U., and Guerri, C. (2016). Ethanol-induced TLR4/NLRP3 neuroinflammatory response in microglial cells promotes leukocyte infiltration across the BBB. Neurochem. Res. 41, 193–209. doi: 10.1007/s11064-015-1760-5
Allen, I. C., Scull, M. A., Moore, C. B., Holl, E. K., McElvania-TeKippe, E., Taxman, D. J., et al. (2009). The NLRP3 inflammasome mediates in vivo innate immunity to Influenza A virus through recognition of viral RNA. Immunity 30, 556–565. doi: 10.1016/j.immuni.2009.02.005
Amor, S., Puentes, F., Baker, D., and van der Valk, P. (2010). Inflammation in neurodegenerative diseases. Immunology 129, 154–169. doi: 10.1111/j.1365-2567.2009.03225.x
Andres, K. H., von During, M., Muszynski, K., and Schmidt, R. F. (1987). Nerve fibres and their terminals of the dura mater encephali of the rat. Anat. Embryol. 175, 289–301. doi: 10.1007/BF00309843
Anthony, I. C., Crawford, D. H., and Bell, J. E. (2003). B lymphocytes in the normal brain: contrasts with HIV-associated lymphoid infiltrates and lymphomas. Brain 126, 1058–1067. doi: 10.1093/brain/awg118
Auvin, S., Shin, D., Mazarati, A., and Sankar, R. (2010). Inflammation induced by LPS enhances epileptogenesis in immature rat and may be partially reversed by IL1RA. Epilepsia 51(Suppl. 3), 34–38. doi: 10.1111/j.1528-1167.2010.02606.x
Azmitia, E. C., Saccomano, Z. T., Alzoobaee, M. F., Boldrini, M., and Whitaker-Azmitia, P. M. (2016). Persistent angiogenesis in the autism brain: an immunocytochemical study of postmortem cortex, brainstem and cerebellum. J. Autism Dev. Disord. 46, 1307–1318. doi: 10.1007/s10803-015-2672-6
Balin, B. J., Little, C. S., Hammond, C. J., Appelt, D. M., Whittum-Hudson, J. A., Gerard, H. C., et al. (2008). Chlamydophila pneumoniae and the etiology of late-onset Alzheimer's disease. J. Alzheimers. Dis. 13, 371–380. doi: 10.3233/JAD-2008-13403
Beasley, D. W., Li, L., Suderman, M. T., and Barrett, A. D. (2002). Mouse neuroinvasive phenotype of West Nile virus strains varies depending upon virus genotype. Virology 296, 17–23. doi: 10.1006/viro.2002.1372
Beasley, D. W., Whiteman, M. C., Zhang, S., Huang, C. Y., Schneider, B. S., Smith, D. R., et al. (2005). Envelope protein glycosylation status influences mouse neuroinvasion phenotype of genetic lineage 1 West Nile virus strains. J. Virol. 79, 8339–8347. doi: 10.1128/JVI.79.13.8339-8347.2005
Beggs, S., Liu, X. J., Kwan, C., and Salter, M. W. (2010). Peripheral nerve injury and TRPV1-expressing primary afferent C-fibers cause opening of the blood-brain barrier. Mol. Pain 6:74. doi: 10.1186/1744-8069-6-74
Berg, B. M., Godbout, J. P., Kelley, K. W., and Johnson, R. W. (2004). α-tocopherol attenuates lipopolysaccharide-induced sickness behavior in mice. Brain Behav. Immun. 18, 149–157. doi: 10.1016/S0889-1591(03)00113-2
Biron, K. E., Dickstein, D. L., Gopaul, R., and Jefferies, W. A. (2011). Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer's disease. PLoS ONE 6:e23789. doi: 10.1371/journal.pone.0023789
Bluthe, R. M., Laye, S., Michaud, B., Combe, C., Dantzer, R., and Parnet, P. (2000). Role of interleukin-1β and tumour necrosis factor-α in lipopolysaccharide-induced sickness behaviour: a study with interleukin-1 type I receptor-deficient mice. Eur. J. Neurosci. 12, 4447–4456. doi: 10.1111/j.1460-9568.2000.01348.x
Bouallegue, A., Daou, G. B., and Srivastava, A. K. (2007). Endothelin-1-induced signaling pathways in vascular smooth muscle cells. Curr. Vasc. Pharmacol. 5, 45–52. doi: 10.2174/157016107779317161
Boveri, M., Kinsner, A., Berezowski, V., Lenfant, A. M., Draing, C., Cecchelli, R., et al. (2006). Highly purified lipoteichoic acid from gram-positive bacteria induces in vitro blood-brain barrier disruption through glia activation: role of pro-inflammatory cytokines and nitric oxide. Neuroscience 137, 1193–1209. doi: 10.1016/j.neuroscience.2005.10.011
Braniste, V., Al-Asmakh, M., Kowal, C., Anuar, F., Abbaspour, A., Tóth, M., et al. (2014). The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 6:263ra158. doi: 10.1126/scitranslmed.3009759
Branton, W. G., Ellestad, K. K., Maingat, F., Wheatley, B. M., Rud, E., Warren, R. L., et al. (2013). Brain microbial populations in HIV/AIDS: α-proteobacteria predominate independent of host immune status. PLoS ONE 8:e54673. doi: 10.1371/journal.pone.0054673
Brettschneider, J., Del Tredici, K., Lee, V. M. Y., and Trojanowski, J. Q. (2015). Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci. 16, 109–120. doi: 10.1038/nrn3887
Brizzi, K. (2017). Neurologic manifestation of Chikungunya virus. Curr. Infect. Dis. Rep. 19:6. doi: 10.1007/s11908-017-0561-1
Broderick, L., Gandhi, C., Mueller, J. L., Putnam, C. D., Shayan, K., Giclas, P. C., et al. (2013). Mutations of complement factor I and potential mechanisms of neuroinflammation in acute hemorrhagic leukoencephalitis. J. Clin. Immunol. 33, 162–171. doi: 10.1007/s10875-012-9767-z
Bu, X. L., Yao, X. Q., Jiao, S. S., Zeng, F., Liu, Y. H., Xiang, Y., et al. (2015). A study on the association between infectious burden and Alzheimer's disease. Eur. J. Neurol. 22, 1519–1525. doi: 10.1111/ene.12477
Bucciantini, M., Giannoni, E., Chiti, F., Baroni, F., Formigli, L., Zurdo, J., et al. (2002). Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416, 507–511. doi: 10.1038/416507a
Budday, S., Steinmann, P., and Kuhl, E. (2015). Physical biology of human brain development. Front. Cell. Neurosci. 9:257. doi: 10.3389/fncel.2015.00257
Buehler, M. R. (2011). A proposed mechanism for autism: an aberrant neuroimmune response manifested as a psychiatric disorder. Med. Hypotheses 76, 863–870. doi: 10.1016/j.mehy.2011.02.038
Burgdorfer, W., Barbour, A. G., Hayes, S. F., Benach, J. L., Grunwaldt, E., and Davis, J. P. (1982). Lyme disease-a tick-borne spirochetosis? Science 216, 1317–1319. doi: 10.1126/science.7043737
Cervantes, J. L. (2017). MyD88 in Mycobacterium tuberculosis infection. Med. Microbiol. Immunol. 206, 187–193. doi: 10.1007/s00430-017-0495-0
Chambers, T. J., and Diamond, M. S. (2003). Pathogenesis of flavivirus encephalitis. Adv. Virus Res. 60, 273–342. doi: 10.1016/S0065-3527(03)60008-4
Charo, I. F., and Ransohoff, R. M. (2006). The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 354, 610–621. doi: 10.1056/NEJMra052723
Chen, C., and Regehr, W. G. (2000). Developmental remodeling of the retinogeniculate synapse. Neuron 28, 955–966. doi: 10.1016/S0896-6273(00)00166-5
Chen, Z., Jalabi, W., Shpargel, K. B., Farabaugh, K. T., Dutta, R., Yin, X., et al. (2012). Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J. Neurosci. 32, 11706–11715. doi: 10.1523/JNEUROSCI.0730-12.2012
Ching, S., Zhang, H., Belevych, N., He, L., Lai, W., Pu, X. A., et al. (2007). Endothelial-specific knockdown of interleukin-1 (IL-1) type 1 receptor differentially alters CNS responses to IL-1 depending on its route of administration. J. Neurosci. 27, 10476–10486. doi: 10.1523/JNEUROSCI.3357-07.2007
Chiu, I. M., von Hehn, C. A., and Woolf, C. J. (2012). Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat. Neurosci. 15, 1063–1067. doi: 10.1038/nn.3144
Combes, V., Guillemin, G. J., Chan-Ling, T., Hunt, N. H., and Grau, G. E. (2012). The crossroads of neuroinflammation in infectious diseases: endothelial cells and astrocytes. Trends Parasitol. 28, 311–319. doi: 10.1016/j.pt.2012.05.008
Czirr, E., and Wyss-Coray, T. (2012). The immunology of neurodegeneration. J. Clin. Invest. 122, 1156–1163. doi: 10.1172/JCI58656
Dahiya, S., Irish, B. P., Nonnemacher, M. R., and Wigdahl, B. (2013). Genetic variation and HIV-associated neurologic disease. Adv. Virus Res. 87, 183–240. doi: 10.1016/B978-0-12-407698-3.00006-5
Dai, M., Freeman, B., Bruno, F. P., Shikani, H. J., Tanowitz, H. B., Weiss, L. M., et al. (2012). The novel ETA receptor antagonist HJP-272 prevents cerebral microvascular hemorrhage in cerebral malaria and synergistically improves survival in combination with an artemisinin derivative. Life Sci. 91, 687–692. doi: 10.1016/j.lfs.2012.07.006
Daneman, R., Zhou, L., Agalliu, D., Cahoy, J. D., Kaushal, A., and Barres, B. A. (2010a). The mouse blood-brain barrier transcriptome: A new resource for understanding the development and function of brain endothelial cells. PLoS ONE 5:e13741. doi: 10.1371/journal.pone.0013741
Daneman, R., Zhou, L., Kebede, A. A., and Barres, B. A. (2010b). Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468, 562–566. doi: 10.1038/nature09513
Daniels, B. P., Holman, D. W., Cruz-Orengo, L., Jujjavarapu, H., Durrant, D. M., and Klein, R. S. (2014). Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. MBio 5:e01476–14. doi: 10.1128/mBio.01476-14
Dantzer, R., O'Connor, J. C., Freund, G. G., Johnson, R. W., and Kelley, K. W. (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56. doi: 10.1038/nrn2297
Davalos, D., Grutzendler, J., Yang, G., Kim, J. V., Zuo, Y., Jung, S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758. doi: 10.1038/nn1472
Davi, G., Giammarresi, C., Vigneri, S., Ganci, A., Ferri, C., Di Francesco, L., et al. (1995). Demonstration of Rickettsia conorii-induced coagulative and platelet activation in vivo in patients with mediterranean spotted fever. Thromb. Haemost. 74, 631–634.
David, S., and Kroner, A. (2011). Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 12, 388–399. doi: 10.1038/nrn3053
De Chiara, G., Marcocci, M. E., Sgarbanti, R., Civitelli, L., Ripoli, C., Piacentini, R., et al. (2012). Infectious agents and neurodegeneration. Mol. Neurobiol. 46, 614–638. doi: 10.1007/s12035-012-8320-7
Del Rio, T., and Feller, M. B. (2006). Early retinal activity and visual circuit development. Neuron 52, 221–222. doi: 10.1016/j.neuron.2006.10.001
Diaz Heijtz, R. (2016). Fetal, neonatal, and infant microbiome: Perturbations and subsequent effects on brain development and behavior. Semin. Fetal Neonatal Med. 21, 410–417. doi: 10.1016/j.siny.2016.04.012
Dinan, T. G., and Cryan, J. F. (2015). The impact of gut microbiota on brain and behaviour: implications for psychiatry. Curr. Opin. Clin. Nutr. Metab. Care 18, 552–558. doi: 10.1097/MCO.0000000000000221
DiSabato, D. J., Quan, N., and Godbout, J. P. (2016). Neuroinflammation: the devil is in the details. J. Neurochem. 139(Suppl. 2), 136–153. doi: 10.1111/jnc.13607
Dunn, A. J., Swiergiel, A. H., Zhang, H., and Quan, N. (2006). Reduced ingestion of sweetened milk induced by interleukin-1 and lipopolysaccharide is associated with induction of Cyclooxygenase-2 in brain endothelia. Neuroimmunomodulation 13, 96–104. doi: 10.1159/000096291
El Aidy, S., Dinan, T., and Cryan, J. (2014). Immune modulation of the brain-gut-microbe axis. Front. Microbiol. 5:146. doi: 10.3389/fmicb.2014.00146
Elinav, E., Strowig, T., Henao-Mejia, J., and Flavell, R. A. (2011). Regulation of the antimicrobial response by NLR proteins. Immunity 34, 665–679. doi: 10.1016/j.immuni.2011.05.007
Engelhardt, B., and Liebner, S. (2014). Novel insights into the development and maintenance of the blood-brain barrier. Cell Tissue Res. 355, 687–699. doi: 10.1007/s00441-014-1811-2
Farzi, A., Reichmann, F., Meinitzer, A., Mayerhofer, R., Jain, P., Hassan, A. M., et al. (2015). Synergistic effects of NOD1 or NOD2 and TLR4 activation on mouse sickness behavior in relation to immune and brain activity markers. Brain Behav. Immun. 44, 106–120. doi: 10.1016/j.bbi.2014.08.011
Ferrari, C. C., Depino, A. M., Prada, F., Muraro, N., Campbell, S., Podhajcer, O., et al. (2004). Reversible demyelination, blood-brain barrier breakdown, and pronounced neutrophil recruitment induced by chronic IL-1 expression in the brain. Am. J. Pathol. 165, 1827–1837. doi: 10.1016/S0002-9440(10)63438-4
Ferwerda, B., Valls Serón, M., Jongejan, A., Zwinderman, A. H., Geldhoff, M., van der Ende, A., et al. (2016). Variation of 46 innate immune genes evaluated for their contribution in pneumococcal meningitis susceptibility and outcome. EBioMed. 10, 77–84. doi: 10.1016/j.ebiom.2016.07.011
Fiorentino, M., Sapone, A., Senger, S., Camhi, S. S., Kadzielski, S. M., Buie, T. M., et al. (2016). Blood–brain barrier and intestinal epithelial barrier alterations in autism spectrum disorders. Mol. Autism 7, 49. doi: 10.1186/s13229-016-0110-z
Floret, D., Stamm, D., and Ponard, D. (1991). Increased susceptibility to infection in children with congenital deficiency of factor I. Pediatr. Infect. Dis. J. 10, 615–618. doi: 10.1097/00006454-199108000-00011
Francisco, N. M., Hsu, N. J., Keeton, R., Randall, P., Sebesho, B., Allie, N., et al. (2015). TNF-dependent regulation and activation of innate immune cells are essential for host protection against cerebral tuberculosis. J. Neuroinflam. 12, 125. doi: 10.1186/s12974-015-0345-1
Freeman, B. D., Machado, F. S., Tanowitz, H. B., and Desruisseaux, M. S. (2014). Endothelin-1 and its role in the pathogenesis of infectious diseases. Life Sci. 118, 110–119. doi: 10.1016/j.lfs.2014.04.021
Furr, S., and Marriott, I. (2012). Viral CNS infections: role of glial pattern recognition receptors in neuroinflammation. Front. Microbiol. 3:201. doi: 10.3389/fmicb.2012.00201
Geldhoff, M., Mook-Kanamori, B. B., Brouwer, M. C., Valls Seron, M., Baas, F., van der Ende, A., et al. (2013). Genetic variation in inflammasome genes is associated with outcome in bacterial meningitis. Immunogenetics 65, 9–16. doi: 10.1007/s00251-012-0653-x
Gerardin, P., Couderc, T., Bintner, M., Tournebize, P., Renouil, M., Lemant, J., et al. (2016). Chikungunya virus-associated encephalitis: a cohort study on LA Reunion island, 2005-2009. Neurology 86, 94–102. doi: 10.1212/WNL.0000000000002234
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
Goldmann, J., Kwidzinski, E., Brandt, C., Mahlo, J., Richter, D., and Bechmann, I. (2006). T cells traffic from brain to cervical lymph nodes via the cribroid plate and the nasal mucosa. J. Leukoc. Biol. 80, 797–801. doi: 10.1189/jlb.0306176
Grayston, J. T., Campbell, L. A., Kuo, C.-C., Mordhorst, C. H., Saikku, P., Thorn, D. H., et al. (1990). A new respiratory tract pathogen: Chlamydia pneumoniae strain TWAR. J. Infect. Dis. 161, 618–625. doi: 10.1093/infdis/161.4.618
Green, J. A., and Friedland, J. S. (2007). Astrocyte–leucocyte interactions and the mechanisms regulating matrix degradation in CNS tuberculosis. Biochem. Soc. Trans. 35:686. doi: 10.1042/BST0350686
Gruber-Schoffnegger, D., Drdla-Schutting, R., Honigsperger, C., Wunderbaldinger, G., Gassner, M., and Sandkuhler, J. (2013). Induction of thermal hyperalgesia and synaptic long-term potentiation in the spinal cord lamina i by TNF-α and IL-1β is mediated by glial cells. J. Neurosci. 33, 6540–6551. doi: 10.1523/JNEUROSCI.5087-12.2013
Guabiraba, R., and Ryffel, B. (2014). Dengue virus infection: Current concepts in immune mechanisms and lessons from murine models. Immunology 141, 143–156. doi: 10.1111/imm.12188
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer's disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Haerynck, F., Stordeur, P., Vandewalle, J., Van Coster, R., Bordon, V., De Baets, F., et al. (2013). Complete factor I deficiency due to dysfunctional factor I with recurrent aseptic meningo-encephalitis. J. Clin. Immunol. 33, 1293–1301. doi: 10.1007/s10875-013-9944-8
Hanke, M. L., and Kielian, T. (2011). Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin. Sci. 121, 367–387. doi: 10.1042/CS20110164
Hansen, M. K., O'Connor, K. A., Goehler, L. E., Watkins, L. R., and Maier, S. F. (2001). The contribution of the vagus nerve in interleukin-1β-induced fever is dependent on dose. Am. J. Physiol. Regul. Integr. Comp. Physiol. 280, R929–R934.
Harris, S. A., and Harris, E. A. (2015). Herpes simplex virus type 1 and other pathogens are key causative factors in sporadic Alzheimer's disease. J. Alzheimers Dis. 48, 319–353. doi: 10.3233/JAD-142853
Henry, C. J., Huang, Y., Wynne, A. M., and Godbout, J. P. (2009). Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain Behav. Immun. 23, 309–317. doi: 10.1016/j.bbi.2008.09.002
Hill, J. M., Clement, C., Pogue, A. I., Bhattacharjee, S., Zhao, Y., and Lukiw, W. J. (2014). Pathogenic microbes, the microbiome, and Alzheimer's disease (ad). Front. Aging Neurosci. 6:127. doi: 10.3389/fnagi.2014.00127
Ho, Y.-H., Lin, Y.-T., Wu, C.-W. J., Chao, Y.-M., Chang, A. Y. W., and Chan, J. Y. H. (2015). Peripheral inflammation increases seizure susceptibility via the induction of neuroinflammation and oxidative stress in the hippocampus. J. Biomed. Sci. 22, 46. doi: 10.1186/s12929-015-0157-8
Imeri, L., and Opp, M. R. (2009). How (and why) the immune system makes us sleep. Nat. Rev. Neurosci. 10, 199–210. doi: 10.1038/nrn2576
Itzhaki, R. F. (2014). Herpes simplex virus type 1 and Alzheimer's disease: Increasing evidence for a major role of the virus. Front. Aging Neurosci. 6:202. doi: 10.3389/fnagi.2014.00202
Jain, A. (2014). Endothelin-1: A potential pathological factor in Parkinson's disease?-from endoplasmic reticulum stress to beyond. J. Neurol. Sci. 344, 236–237. doi: 10.1016/j.jns.2014.06.038
Jain, S. K., Paul-Satyaseela, M., Lamichhane, G., Kim, K. S., and Bishai, W. R. (2006). Mycobacterium tuberculosis invasion and traversal across an in vitro human blood-brain barrier as a pathogenic mechanism for central nervous system tuberculosis. J. Infect. Dis. 193, 1287–1295. doi: 10.1086/502631
Jiang, T., Yu, J. T., Zhu, X. C., and Tan, L. (2013). TREM2 in Alzheimer's disease. Mol. Neurobiol. 48, 180–185. doi: 10.1007/s12035-013-8424-8
Jonsson, T., Stefansson, H., Steinberg, S., Jonsdottir, I., Jonsson, P. V., Snaedal, J., et al. (2013). Variant of TREM2 associated with the risk of Alzheimer's disease. N. Engl. J. Med. 368, 107–116. doi: 10.1056/NEJMoa1211103
Kaisho, T., and Akira, S. (2004). Pleiotropic function of toll-like receptors. Microbes Infect. 6, 1388–1394. doi: 10.1016/j.micinf.2004.08.019
Kariko, K., Ni, H., Capodici, J., Lamphier, M., and Weissman, D. (2004). mRNA is an endogenous ligand for toll-like receptor 3. J. Biol. Chem. 279, 12542–12550. doi: 10.1074/jbc.M310175200
Khachaturian, Z. S. (1985). Diagnosis of Alzheimer's disease. Arch. Neurol. 42, 1097–1105. doi: 10.1001/archneur.1985.04060100083029
Kirschning, C. J., and Schumann, R. R. (2002). TLR2: Cellular sensor for microbial and endogenous molecular patterns. Curr. Top. Microbiol. Immunol. 270, 121–144. doi: 10.1007/978-3-642-59430-4_8
Kohan, D. E., Rossi, N. F., Inscho, E. W., and Pollock, D. M. (2011). Regulation of blood pressure and salt homeostasis by endothelin. Physiol. Rev. 91, 1–77. doi: 10.1152/physrev.00060.2009
Kuang, Y., Lackay, S. N., Zhao, L., and Fu, Z. F. (2009). Role of chemokines in the enhancement of BBB permeability and inflammatory infiltration after Rabies virus infection. Virus Res. 144, 18–26. doi: 10.1016/j.virusres.2009.03.014
Laflamme, N., Lacroix, S., and Rivest, S. (1999). An essential role of interleukin-1β in mediating NF-κB activity and COX-2 transcription in cells of the blood-brain barrier in response to a systemic and localized inflammation but not during endotoxemia. J. Neurosci. 19, 10923–10930.
Lee, E., and Lobigs, M. (2002). Mechanism of virulence attenuation of glycosaminoglycan-binding variants of Japanese encephalitis virus and Murray valley encephalitis virus. J. Virol. 76, 4901–4911. doi: 10.1128/JVI.76.10.4901-4911.2002
Lee, J., Ling, C., Kosmalski, M. M., Hulseberg, P., Schreiber, H. A., Sandor, M., et al. (2009). Intracerebral Mycobacterium bovis bacilli calmette-guerin infection-induced immune responses in the CNS. J. Neuroimmunol. 213, 112–122. doi: 10.1016/j.jneuroim.2009.05.008
Leitao, M. F., Vilela, M. M., Rutz, R., Grumach, A. S., Condino-Neto, A., and Kirschfink, M. (1997). Complement factor I deficiency in a family with recurrent infections. Immunopharmacology 38, 207–213. doi: 10.1016/S0162-3109(97)00080-5
Ley, R. E., Peterson, D. A., and Gordon, J. I. (2006). Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 124, 837–848. doi: 10.1016/j.cell.2006.02.017
Li, J., Wang, Y., Wang, X., Ye, L., Zhou, Y., Persidsky, Y., et al. (2013). Immune activation of human brain microvascular endothelial cells inhibits HIV replication in macrophages. Blood 121, 2934–2942. doi: 10.1182/blood-2012-08-450353
Lim, S. L., Rodriguez-Ortiz, C. J., and Kitazawa, M. (2015). Infection, systemic inflammation, and Alzheimer's disease. Microbes Infect. 17, 549–556. doi: 10.1016/j.micinf.2015.04.004
Lindenbach, B. D., and Rice, C. M. (2003). Molecular biology of flaviviruses. Adv. Virus Res. 59, 23–61. doi: 10.1016/S0065-3527(03)59002-9
Lippmann, E. S., Al-Ahmad, A., Palecek, S. P., and Shusta, E. V. (2013). Modeling the blood-brain barrier using stem cell sources. Fluids Barriers CNS 10:2. doi: 10.1186/2045-8118-10-2
Lippmann, E. S., Azarin, S. M., Kay, J. E., Nessler, R. A., Wilson, H. K., Al-Ahmad, A., et al. (2012). Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat. Biotechnol. 30, 783–791. doi: 10.1038/nbt.2247
Liu, C. C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. doi: 10.1038/nrneurol.2012.263
Liu, X., Nemeth, D. P., Tarr, A. J., Belevych, N., Syed, Z. W., Wang, Y., et al. (2016). Euflammation attenuates peripheral inflammation-induced neuroinflammation and mitigates immune-to-brain signaling. Brain Behav. Immun. 54, 140–148. doi: 10.1016/j.bbi.2016.01.018
Long, K. M., Whitmore, A. C., Ferris, M. T., Sempowski, G. D., McGee, C., Trollinger, B., et al. (2013). Dendritic cell immunoreceptor regulates Chikungunya virus pathogenesis in mice. J. Virol. 87, 5697–5706. doi: 10.1128/JVI.01611-12
Louveau, A., Smirnov, I., Keyes, T. J., Eccles, J. D., Rouhani, S. J., Peske, J. D., et al. (2015). Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341. doi: 10.1038/nature14432
Lum, F. M., Low, D. K., Fan, Y., Tan, J. J., Lee, B., Chan, J. K., et al. (2017). Zika virus infects human fetal brain microglia and induces inflammation. Clin. Infect. Dis. 64, 914–920. doi: 10.1093/cid/ciw878
Lundbo, L. F., Harboe, Z. B., Clausen, L. N., Hollegaard, M. V., Sorensen, H. T., Hougaard, D. M., et al. (2016). Genetic variation in NFKBIE is associated with increased risk of pneumococcal meningitis in children. EBioMed. 3, 93–99. doi: 10.1016/j.ebiom.2015.11.048
Lundbo, L. F., Sorensen, H. T., Clausen, L. N., Hollegaard, M. V., Hougaard, D. M., Konradsen, H. B., et al. (2015). Mannose-binding lectin gene, MBL2, polymorphisms do not increase susceptibility to invasive meningococcal disease in a population of Danish children. Open Forum Infect. Dis. 2:ofv127. doi: 10.1093/ofid/ofv127
Ma, S., Kwon, H. J., and Huang, Z. (2012). A functional requirement for astroglia in promoting blood vessel development in the early postnatal brain. PLoS ONE 7:e48001. doi: 10.1371/journal.pone.0048001
Maheshwari, P., and Eslick, G. D. (2015). Bacterial infection and Alzheimer's disease: a meta-analysis. J. Alzheimers Dis. 43, 957–966. doi: 10.3233/JAD-140621
Maier, S. F., Goehler, L. E., Fleshner, M., and Watkins, L. R. (1998). The role of the vagus nerve in cytokine-to-brain communication. Ann. N.Y. Acad. Sci. 840, 289–300. doi: 10.1111/j.1749-6632.1998.tb09569.x
Mancuso, R., Baglio, F., Agostini, S., Cabinio, M., Lagana, M. M., Hernis, A., et al. (2014). Relationship between herpes simplex virus-1-specific antibody titers and cortical brain damage in Alzheimer's disease and amnestic mild cognitive impairment. Front. Aging Neurosci. 6:285. doi: 10.3389/fnagi.2014.00285
Mayer, E. A., Padua, D., and Tillisch, K. (2014). Altered brain-gut axis in autism: comorbidity or causative mechanisms? Bioessays 36, 933–939. doi: 10.1002/bies.201400075
Mayerhofer, R., Frohlich, E. E., Reichmann, F., Farzi, A., Kogelnik, N., Frohlich, E., et al. (2017). Diverse action of lipoteichoic acid and lipopolysaccharide on neuroinflammation, blood-brain barrier disruption, and anxiety in mice. Brain Behav. Immun. 60, 174–187. doi: 10.1016/j.bbi.2016.10.011
McCusker, R. H., and Kelley, K. W. (2013). Immune–neural connections: how the immune system's response to infectious agents influences behavior. J. Exp. Biol. 216, 84–98. doi: 10.1242/jeb.073411
McManus, R. M., and Heneka, M. T. (2017). Role of neuroinflammation in neurodegeneration: new insights. Alzheimers Res. Ther. 9, 14. doi: 10.1186/s13195-017-0241-2
McManus, R. M., Higgins, S. C., Mills, K. H. G., and Lynch, M. A. (2014). Respiratory infection promotes T cell infiltration and amyloid-β deposition in APP/PS1 mice. Neurobiol. Aging 35, 109–121. doi: 10.1016/j.neurobiolaging.2013.07.025
Medzhitov, R. (2008). Origin and physiological roles of inflammation. Nature 454, 428–435. doi: 10.1038/nature07201
Miklossy, J. (2011). Alzheimer's disease - a neurospirochetosis. Analysis of the evidence following Koch's and Hill's criteria. J. Neuroinflammation 8:90. doi: 10.1186/1742-2094-8-90
Miklossy, J. (2015). Historic evidence to support a causal relationship between spirochetal infections and Alzheimer's disease. Front. Aging Neurosci. 7:46. doi: 10.3389/fnagi.2015.00046
Miklossy, J., Kis, A., Radenovic, A., Miller, L., Forro, L., Martins, R., et al. (2006). β-amyloid deposition and Alzheimer's type changes induced by Borrelia spirochetes. Neurobiol. Aging 27, 228–236. doi: 10.1016/j.neurobiolaging.2005.01.018
Miller, A. H., and Raison, C. L. (2016). The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 16, 22–34. doi: 10.1038/nri.2015.5
Miner, J. J., and Diamond, M. S. (2016). Mechanisms of restriction of viral neuroinvasion at the blood–brain barrier. Curr. Opin. Immunol. 38, 18–23. doi: 10.1016/j.coi.2015.10.008
Minter, M. R., Zhang, C., Leone, V., Ringus, D. L., Zhang, X., Oyler-Castrillo, P., et al. (2016). Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and Amyloidosis in a murine model of Alzheimer's disease. Sci. Rep. 6:30028. doi: 10.1038/srep30028
Molteni, M., Gemma, S., and Rossetti, C. (2016). The role of toll-like receptor 4 in infectious and noninfectious inflammation. Mediators Inflamm. 2016:6978936. doi: 10.1155/2016/6978936
Moretti, R., Pansiot, J., Bettati, D., Strazielle, N., Ghersi-Egea, J. F., Damante, G., et al. (2015). Blood-brain barrier dysfunction in disorders of the developing brain. Front. Neurosci. 9:40. doi: 10.3389/fnins.2015.00040
Mundt, S., Engelhardt, B., Kirk, C. J., Groettrup, M., and Basler, M. (2016). Inhibition and deficiency of the immunoproteasome subunit LMP7 attenuates LCMV-induced meningitis. Eur. J. Immunol. 46, 104–113. doi: 10.1002/eji.201545578
Nilsson, S. C., Trouw, L. A., Renault, N., Miteva, M. A., Genel, F., Zelazko, M., et al. (2009). Genetic, molecular and functional analyses of complement factor I deficiency. Eur. J. Immunol. 39, 310–323. doi: 10.1002/eji.200838702
Nimmerjahn, A., Kirchhoff, F., and Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. doi: 10.1126/science.1110647
Niwa, K., Carlson, G. A., and Iadecola, C. (2000). Exogenous aβ1–40 reproduces cerebrovascular alterations resulting from amyloid precursor protein overexpression in mice. J. Cereb. Blood Flow Metab. 20, 1659–1668. doi: 10.1097/00004647-200012000-00005
Norden, D. M., and Godbout, J. P. (2013). Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 39, 19–34. doi: 10.1111/j.1365-2990.2012.01306.x
Norman, M. G., and O'Kusky, J. R. (1986). The growth and development of microvasculature in human cerebral cortex. J. Neuropathol. Exp. Neurol. 45, 222–232. doi: 10.1097/00005072-198605000-00003
O'Mahony, S. M., Clarke, G., Dinan, T. G., and Cryan, J. F. (2017). Early-life adversity and brain development: is the microbiome a missing piece of the puzzle? Neuroscience 342, 37–54. doi: 10.1016/j.neuroscience.2015.09.068
Obermeier, B., Daneman, R., and Ransohoff, R. M. (2013). Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 19, 1584–1596. doi: 10.1038/nm.3407
Palmer, J. C., Barker, R., Kehoe, P. G., and Love, S. (2012). Endothelin-1 is elevated in Alzheimer's disease and upregulated by amyloid-β. J. Alzheimers. Dis. 29, 853–861. doi: 10.3233/JAD-2012-111760
Pardon, M. C. (2015). Lipopolysaccharide hyporesponsiveness: protective or damaging response to the brain? Rom. J. Morphol. Embryol. 56, 903–913.
Pedersen, M. M., Marso, E., and Pickett, M. J. (1970). Nonfermentative bacilli associated with man. 3. Pathogenicity and antibiotic susceptibility. Am. J. Clin. Pathol. 54, 178–192. doi: 10.1093/ajcp/54.2.178
Perry, V. H., and Teeling, J. (2013). Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin. Immunopathol. 35, 601–612. doi: 10.1007/s00281-013-0382-8
Petkova, S. B., Huang, H., Factor, S. M., Pestell, R. G., Bouzahzah, B., Jelicks, L. A., et al. (2001). The role of endothelin in the pathogenesis of Chagas' disease. Int. J. Parasitol. 31, 499–511. doi: 10.1016/S0020-7519(01)00168-0
Phares, T. W., DiSano, K. D., Hinton, D. R., Hwang, M., Zajac, A. J., Stohlman, S. A., et al. (2013). IL-21 optimizes T cell and humoral responses in the central nervous system during viral encephalitis. J. Neuroimmunol. 263, 43–54. doi: 10.1016/j.jneuroim.2013.07.019
Piccinini, A. M., and Midwood, K. S. (2010). Dampening inflammation by modulating TLR signalling. Mediators Inflamm. 2010:672395. doi: 10.1155/2010/672395
Pilcher, C. D., Shugars, D. C., Fiscus, S. A., Miller, W. C., Menezes, P., Giner, J., et al. (2001). HIV in body fluids during primary HIV infection: implications for pathogenesis, treatment and public health. AIDS 15, 837–845. doi: 10.1097/00002030-200105040-00004
Pottier, C., Wallon, D., Rousseau, S., Rovelet-Lecrux, A., Richard, A. C., Rollin-Sillaire, A., et al. (2013). TREM2 R47H variant as a risk factor for early-onset Alzheimer's disease. J. Alzheimers. Dis. 35, 45–49. doi: 10.3233/JAD-122311
Puntener, U., Booth, S. G., Perry, V. H., and Teeling, J. L. (2012). Long-term impact of systemic bacterial infection on the cerebral vasculature and microglia. J. Neuroinflammation 9:146. doi: 10.1186/1742-2094-9-146
Putignani, L. (2012). Human gut microbiota: onset and shaping through life stages and perturbations. Front. Cell. Infect. Microbiol. 2:144. doi: 10.3389/fcimb.2012.00144
Ransohoff, R. M., and Brown, M. A. (2012). Innate immunity in the central nervous system. J. Clin. Invest. 122, 1164–1171. doi: 10.1172/JCI58644
Ransohoff, R. M., and Engelhardt, B. (2012). The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 12, 623–635. doi: 10.1038/nri3265
Ransohoff, R. M., and Perry, V. H. (2009). Microglial physiology: unique stimuli, specialized responses. Annu. Rev. Immunol. 27, 119–145. doi: 10.1146/annurev.immunol.021908.132528
Ransohoff, R. M., Schafer, D., Vincent, A., Blachere, N. E., and Bar-Or, A. (2015). Neuroinflammation: ways in which the immune system affects the brain. Neurotherapeutics 12, 896–909. doi: 10.1007/s13311-015-0385-3
Rea, K., Dinan, T. G., and Cryan, J. F. (2016). The microbiome: a key regulator of stress and neuroinflammation. Neurobiol. Stress 4, 23–33. doi: 10.1016/j.ynstr.2016.03.001
Riviere, G. R., Riviere, K. H., and Smith, K. S. (2002). Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer's disease. Oral Microbiol. Immunol. 17, 113–118. doi: 10.1046/j.0902-0055.2001.00100.x
Roach, T., and Alcendor, D. J. (2017). Zika virus infection of cellular components of the blood-retinal barriers: implications for viral associated congenital ocular disease. J. Neuroinflammation 14, 43. doi: 10.1186/s12974-017-0824-7
Rojas, A., Jiang, J., Ganesh, T., Yang, M. S., Lelutiu, N., Gueorguieva, P., et al. (2014). Cyclooxygenase-2 in epilepsy. Epilepsia 55, 17–25. doi: 10.1111/epi.12461
Roosterman, D., Goerge, T., Schneider, S. W., Bunnett, N. W., and Steinhoff, M. (2006). Neuronal control of skin function: the skin as a neuroimmunoendocrine organ. Physiol. Rev. 86, 1309–1379. doi: 10.1152/physrev.00026.2005
Rosadini, C. V., and Kagan, J. C. (2017). Early innate immune responses to bacterial LPS. Curr. Opin. Immunol. 44, 14–19. doi: 10.1016/j.coi.2016.10.005
Russo, M. V., and McGavern, D. B. (2015). Immune surveillance of the cns following infection and injury. Trends Immunol. 36, 637–650. doi: 10.1016/j.it.2015.08.002
Salmina, A. B., Inzhutova, A. I., Malinovskaya, N. A., and Petrova, M. M. (2010). Endothelial dysfunction and repair in Alzheimer-type neurodegeneration: Neuronal and glial control. J. Alzheimers Dis. 22, 17–36. doi: 10.3233/JAD-2010-091690
Sampson, T. R., Debelius, J. W., Thron, T., Janssen, S., Shastri, G. G., Ilhan, Z. E., et al. (2016). Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell 167, 1469.e1412–1480.e1412. doi: 10.1016/j.cell.2016.11.018
Samuel, M. A., and Diamond, M. S. (2005). α/β interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 79, 13350–13361. doi: 10.1128/JVI.79.21.13350-13361.2005
Sandor, F., and Buc, M. (2005). Toll-like receptors. II. Distribution and pathways involved in TLR signalling. Folia Biol. (Praha) 51, 188–197.
Sanes, J. R., and Lichtman, J. W. (1999). Development of the vertebrate neuromuscular junction. Annu. Rev. Neurosci. 22, 389–442. doi: 10.1146/annurev.neuro.22.1.389
Santoro, M., Rossi, A., and Amici, C. (2003). New EMBO member's review: NF-κB and virus infection: who controls whom. EMBO J. 22, 2552–2560. doi: 10.1093/emboj/cdg267
Schacker, T., Collier, A. C., Hughes, J., Shea, T., and Corey, L. (1996). Clinical and epidemiologic features of primary HIV infection. Ann. Intern. Med. 125, 257–264. doi: 10.7326/0003-4819-125-4-199608150-00001
Schafer, D. P., and Stevens, B. (2013). Phagocytic glial cells: Sculpting synaptic circuits in the developing nervous system. Curr. Opin. Neurobiol. 23, 1034–1040. doi: 10.1016/j.conb.2013.09.012
Schafer, D. P., Lehrman, E. K., Kautzman, A. G., Koyama, R., Mardinly, A. R., Yamasaki, R., et al. (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. doi: 10.1016/j.neuron.2012.03.026
Schinelli, S. (2006). Pharmacology and physiopathology of the brain endothelin system: an overview. Curr. Med. Chem. 13, 627–638. doi: 10.2174/092986706776055652
Schroeder, B. O., and Backhed, F. (2016). Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 22, 1079–1089. doi: 10.1038/nm.4185
Selkrig, J., Wong, P., Zhang, X., and Pettersson, S. (2014). Metabolic tinkering by the gut microbiome: Implications for brain development and function. Gut Microbes 5, 369–380. doi: 10.4161/gmic.28681
Sharon, G., Sampson, T. R., Geschwind, D. H., and Mazmanian, S. K. (2016). The central nervous system and the gut microbiome. Cell 167, 915–932. doi: 10.1016/j.cell.2016.10.027
Sheen, T. R., Ebrahimi, C. M., Hiemstra, I. H., Barlow, S. B., Peschel, A., and Doran, K. S. (2010). Penetration of the blood-brain barrier by Staphylococcus aureus: contribution of membrane-anchored lipoteichoic acid. J. Mol. Med. 88, 633–639. doi: 10.1007/s00109-010-0630-5
Shima, K., Kuhlenbaumer, G., and Rupp, J. (2010). Chlamydia pneumoniae infection and Alzheimer's disease: a connection to remember? Med. Microbiol. Immunol. 199, 283–289. doi: 10.1007/s00430-010-0162-1
Siegenthaler, J. A., Sohet, F., and Daneman, R. (2013). ‘Sealing off the CNS’: cellular and molecular regulation of blood-brain barriergenesis. Curr. Opin. Neurobiol. 23, 1057–1064. doi: 10.1016/j.conb.2013.06.006
Singh, V., Roth, S., Llovera, G., Sadler, R., Garzetti, D., Stecher, B., et al. (2016). Microbiota dysbiosis controls the neuroinflammatory response after stroke. J. Neurosci. 36, 7428. doi: 10.1523/jneurosci.1114-16.2016
Skaper, S. D., Giusti, P., and Facci, L. (2012). Microglia and mast cells: two tracks on the road to neuroinflammation. FASEB J. 26, 3103–3117. doi: 10.1096/fj.11-197194
Smith, E. E., and Greenberg, S. M. (2009). β-amyloid, blood vessels and brain function. Stroke 40, 2601–2606. doi: 10.1161/STROKEAHA.108.536839
Sokolova, A., Hill, M. D., Rahimi, F., Warden, L. A., Halliday, G. M., and Shepherd, C. E. (2009). Monocyte chemoattractant protein-1 plays a dominant role in the chronic inflammation observed in Alzheimer's disease. Brain Pathol. 19, 392–398. doi: 10.1111/j.1750-3639.2008.00188.x
Sousa, C., Biber, K., and Michelucci, A. (2017). Cellular and molecular characterization of microglia: a unique immune cell population. Front. Immunol. 8:198. doi: 10.3389/fimmu.2017.00198
Speciale, L., Roda, K., Saresella, M., Taramelli, D., and Ferrante, P. (1998). Different endothelins stimulate cytokine production by peritoneal macrophages and microglial cell line. Immunology 93, 109–114. doi: 10.1046/j.1365-2567.1998.00391.x
Spitzer, P., Condic, M., Herrmann, M., Oberstein, T. J., Scharin-Mehlmann, M., Gilbert, D. F., et al. (2016). Amyloidogenic amyloid-β-peptide variants induce microbial agglutination and exert antimicrobial activity. Sci. Rep. 6:32228. doi: 10.1038/srep32228
Stilling, R. M., Bordenstein, S. R., Dinan, T. G., and Cryan, J. F. (2014). Friends with social benefits: host-microbe interactions as a driver of brain evolution and development? Front. Cell. Infect. Microbiol. 4:147. doi: 10.3389/fcimb.2014.00147
Sukmawati, D., and Tanaka, R. (2015). Introduction to next generation of endothelial progenitor cell therapy: a promise in vascular medicine. Am. J. Transl. Res. 7, 411–421.
Suter, O.-C., Sunthorn, T., Kraftsik, R., Straubel, J., Darekar, P., Khalili, K., et al. (2002). Cerebral hypoperfusion generates cortical watershed microinfarcts in Alzheimer disease. Stroke 33, 1986. doi: 10.1161/01.str.0000024523.82311.77
Tarr, A. J., Liu, X., Reed, N. S., and Quan, N. (2014). Kinetic characteristics of euflammation: the induction of controlled inflammation without overt sickness behavior. Brain Behav. Immun. 42, 96–108. doi: 10.1016/j.bbi.2014.06.002
Teder, P., and Noble, P. W. (2000). A cytokine reborn? Endothelin-1 in pulmonary inflammation and fibrosis. Am. J. Respir. Cell Mol. Biol. 23, 7–10. doi: 10.1165/ajrcmb.23.1.f192
Thomas, T., Thomas, G., McLendon, C., Sutton, T., and Mullan, M. (1996). β-amyloid-mediated vasoactivity and vascular endothelial damage. Nature 380, 168–171. doi: 10.1038/380168a0
Tognini, P. (2017). Gut microbiota: a potential regulator of neurodevelopment. Front. Cell. Neurosci. 11:25. doi: 10.3389/fncel.2017.00025
Tohidpour, A. (2016). CagA-mediated pathogenesis of Helicobacter pylori. Microb. Pathog. 93, 44–55. doi: 10.1016/j.micpath.2016.01.005
Townsend, K. P., Obregon, D., Quadros, A., Patel, N., Volmar, C., Paris, D., et al. (2002). Proinflammatory and vasoactive effects of aβ in the cerebrovasculature. Ann. N. Y. Acad. Sci. 977, 65–76. doi: 10.1111/j.1749-6632.2002.tb04799.x
Tsai, T. T., Chen, C. L., Lin, Y. S., Chang, C. P., Tsai, C. C., Cheng, Y. L., et al. (2016). Microglia retard Dengue virus-induced acute viral encephalitis. Sci. Rep. 6:27670. doi: 10.1038/srep27670
Urban, N., and Guillemot, F. (2014). Neurogenesis in the embryonic and adult brain: same regulators, different roles. Front. Cell. Neurosci. 8:396. doi: 10.3389/fncel.2014.00396
Van Eldik, L. J., Carrillo, M. C., Cole, P. E., Feuerbach, D., Greenberg, B. D., Hendrix, J. A., et al. (2016). The roles of inflammation and immune mechanisms in Alzheimer's disease. Alzheimers Dement. Transl. Res. Clin. Interven. 2, 99–109. doi: 10.1016/j.trci.2016.05.001
Vercammen, E., Staal, J., and Beyaert, R. (2008). Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clin. Microbiol. Rev. 21, 13–25. doi: 10.1128/CMR.00022-07
Vezzani, A., and Granata, T. (2005). Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia 46, 1724–1743. doi: 10.1111/j.1528-1167.2005.00298.x
Vezzani, A., Fujinami, R. S., White, H. S., Preux, P. M., Blumcke, I., Sander, J. W., et al. (2016). Infections, inflammation and epilepsy. Acta Neuropathol. 131, 211–234. doi: 10.1007/s00401-015-1481-5
Virgintino, D., Girolamo, F., Errede, M., Capobianco, C., Robertson, D., Stallcup, W. B., et al. (2007). An intimate interplay between precocious, migrating pericytes and endothelial cells governs human fetal brain angiogenesis. Angiogenesis 10, 35–45. doi: 10.1007/s10456-006-9061-x
Wall, R., Cryan, J. F., Ross, R. P., Fitzgerald, G. F., Dinan, T. G., and Stanton, C. (2014). “Bacterial neuroactive compounds produced by psychobiotics,” in Microbial Endocrinology: The Microbiota-Gut-Brain Axis in Health and Disease, eds M. Lyte, J. F. Cryan (New York, NY: Springer New York), 221–239. doi: 10.1007/978-1-4939-0897-4_10
Walter, S., Letiembre, M., Liu, Y., Heine, H., Penke, B., Hao, W., et al. (2007). Role of the toll-like receptor 4 in neuroinflammation in Alzheimer's disease. Cell. Physiol. Biochem. 20, 947–956. doi: 10.1159/000110455
Wang, W.-Y., Tan, M.-S., Yu, J.-T., and Tan, L. (2015). Role of pro-inflammatory cytokines released from microglia in Alzheimer's disease. Ann. Transl. Med. 3, 136. doi: 10.3978/j.issn.2305-5839.2015.03.49
Wang, X.-L., Zeng, J., Feng, J., Tian, Y.-T., Liu, Y.-J., Qiu, M., et al. (2014). Helicobacter pylori filtrate impairs spatial learning and memory in rats and increases β-amyloid by enhancing expression of presenilin-2. Front. Aging Neurosci. 6:66. doi: 10.3389/fnagi.2014.00066
Weidenfeller, C., Svendsen, C. N., and Shusta, E. V. (2007). Differentiating embryonic neural progenitor cells induce blood-brain barrier properties. J. Neurochem. 101, 555–565. doi: 10.1111/j.1471-4159.2006.04394.x
Welling, M. M., Nabuurs, R. J. A., and van der Weerd, L. (2015). Potential role of antimicrobial peptides in the early onset of Alzheimer's disease. Alzheimers Dement. J. Alzheimers Assoc. 11, 51–57. doi: 10.1016/j.jalz.2013.12.020
Werner, C., and Engelhard, K. (2007). Pathophysiology of traumatic brain injury. Br. J. Anaesth. 99, 4–9. doi: 10.1093/bja/aem131
Woodcock, T., and Morganti-Kossmann, M. C. (2013). The role of markers of inflammation in traumatic brain injury. Front. Neurol. 4:18. doi: 10.3389/fneur.2013.00018
Wozniak, M. A., Shipley, S. J., Combrinck, M., Wilcock, G. K., and Itzhaki, R. F. (2005). Productive herpes simplex virus in brain of elderly normal subjects and Alzheimer's disease patients. J. Med. Virol. 75, 300–306. doi: 10.1002/jmv.20271
Xanthos, D. N., and Sandkuhler, J. (2014). Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat. Rev. Neurosci. 15, 43–53. doi: 10.1038/nrn3617
Yang, J. S., Ramanathan, M. P., Muthumani, K., Choo, A. Y., Jin, S. H., Yu, Q. C., et al. (2002). Induction of inflammation by West Nile virus capsid through the Caspase-9 apoptotic pathway. Emerg. Infect. Dis. 8, 1379–1384. doi: 10.3201/eid0812.020224
Zhang, X., Yeung, P. K. K., McAlonan, G. M., Chung, S. S. M., and Chung, S. K. (2013). Transgenic mice over-expressing endothelial endothelin-1 show cognitive deficit with blood–brain barrier breakdown after transient ischemia with long-term reperfusion. Neurobiol. Learn. Mem. 101, 46–54. doi: 10.1016/j.nlm.2013.01.002
Zhou, H., Lapointe, B. M., Clark, S. R., Zbytnuik, L., and Kubes, P. (2006). A requirement for microglial TLR4 in leukocyte recruitment into brain in response to lipopolysaccharide. J. Immunol. 177, 8103–8110. doi: 10.4049/jimmunol.177.11.8103
Keywords: infectious diseases, blood-brain barrier, immune response, neurodegeneration, brain development
Citation: Tohidpour A, Morgun AV, Boitsova EB, Malinovskaya NA, Martynova GP, Khilazheva ED, Kopylevich NV, Gertsog GE and Salmina AB (2017) Neuroinflammation and Infection: Molecular Mechanisms Associated with Dysfunction of Neurovascular Unit. Front. Cell. Infect. Microbiol. 7:276. doi: 10.3389/fcimb.2017.00276
Received: 12 April 2017; Accepted: 06 June 2017;
Published: 20 June 2017.
Edited by:
Ingar Olsen, University of Oslo, NorwayReviewed by:
Judith Miklossy, Prevention Alzheimer International Foundation, SwitzerlandFrank C. Gibson III, University of Florida, United States
Alan Paul Hudson, Wayne State University School of Medicine, United States
Copyright © 2017 Tohidpour, Morgun, Boitsova, Malinovskaya, Martynova, Khilazheva, Kopylevich, Gertsog and Salmina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Abolghasem Tohidpour, YS50b2hpZHBvdXJAZ21haWwuY29t