 Wanderson M. Silva1,2,3
Wanderson M. Silva1,2,3 Rodrigo D. De Oliveira Carvalho1Fernanda A. Dorella4
Rodrigo D. De Oliveira Carvalho1Fernanda A. Dorella4 Edson L. Folador5Gustavo H. M. F. Souza6Adriano M. C. Pimenta7
Edson L. Folador5Gustavo H. M. F. Souza6Adriano M. C. Pimenta7 Henrique C. P. Figueiredo4
Henrique C. P. Figueiredo4 Yves Le Loir2,3Artur Silva8
Yves Le Loir2,3Artur Silva8 Vasco Azevedo1*
Vasco Azevedo1*- 1Departamento de Biologia Geral, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
- 2Institut National de la Recherche Agronomique (INRA), UMR1253 Science & Technologie du Lait & de l'Oeuf (STLO), Rennes, France
- 3Agrocampus Ouest, UMR1253 Science & Technologie du Lait & de l'Oeuf (STLO), Rennes, France
- 4Escola de Veterinária, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
- 5Centro de Biotecnologia, Universidade Federal da Paraíba, João Pessoa, Brazil
- 6Waters Corporation, Waters Technologies Brazil, MS Applications Laboratory, São Paulo, Brazil
- 7Departamento de Bioquímica e Imunologia, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
- 8Instituto de Ciências Biológicas, Universidade Federal do Pará, Belém, Brazil
Corynebacterium pseudotuberculosis biovar equi is the etiologic agent of ulcerative lymphangitis. To investigate proteins that could be related to the virulence of this pathogen, we combined an experimental passage process using a murine model and high-throughput proteomics with a mass spectrometry, data-independent acquisition (LC-MSE) approach to identify and quantify the proteins released into the supernatants of strain 258_equi. To our knowledge, this approach allowed characterization of the exoproteome of a C. pseudotuberculosis equi strain for the first time. Interestingly, the recovery of this strain from infected mouse spleens induced a change in its virulence potential, and it became more virulent in a second infection challenge. Proteomic screening performed from culture supernatant of the control and recovered conditions revealed 104 proteins that were differentially expressed between the two conditions. In this context, proteomic analysis of the recovered condition detected the induction of proteins involved in bacterial pathogenesis, mainly related to iron uptake. In addition, KEGG enrichment analysis showed that ABC transporters, bacterial secretion systems and protein export pathways were significantly altered in the recovered condition. These findings show that secretion and secreted proteins are key elements in the virulence and adaptation of C. pseudotuberculosis. Collectively, bacterial pathogenesis-related proteins were identified that contribute to the processes of adherence, intracellular growth and evasion of the immune system. Moreover, this study enhances our understanding of the factors that may influence the pathogenesis of C. pseudotuberculosis.
Introduction
Corynebacterium pseudotuberculosis is a gram-positive, facultative intracellular pathogen that is globally distributed and can infect horses, cattle, sheep, goats, buffalos, and occasionally humans. C. pseudotuberculosis biovar ovis is the etiologic agent of caseous lymphadenitis in small ruminants (Dorella et al., 2006). Corynebacterium pseudotuberculosis strains belonging to biovar equi, however, cause edematous skin illness in buffalos. In horses, the infection can manifest through one of two forms: (i) externally, which usually presents with chronic ventral and pectoral lymph node abscesses and, in a more advanced stage, generates an illness denominated ulcerative lymphangitis that is characterized by ulcers of irregular shapes and sizes, or (ii) internally, which is characterized mainly by abscess formation in the lymph nodes and internal organs (kidney, liver, lung, and spleen) (Britz et al., 2014). In the United States, ulcerative lymphangitis outbreaks with large economic losses for horse farmers have been reported (Aleman et al., 1996; Foley et al., 2004; Spier, 2008). In addition, a recent study showed an increase in the case numbers of horses infected with C. pseudotuberculosis biovar equi during the last 10 years in the western region of the USA, which is being considered as an endemic area (Kilcoyne et al., 2014).
Whole genome sequencing of C. pseudotuberculosis biovar equi strain 258, isolated from a horse with ulcerative lymphangitis in Belgium, revealed the presence of pathogenic islands in its chromosome as well as genes that might contribute to its virulence, most of them coding for secreted proteins. Moreover, putative antigenic proteins were identified through reverse vaccinology (Soares et al., 2013a). Some studies have shown that extracellular proteins are related to the pathogenic process of C. pseudotuberculosis (Wilson et al., 1995; Billington et al., 2002; Pacheco et al., 2012; Seyffert et al., 2014). However, phospholipase D (Pld) exotoxin, which contributes to bacterial spread in the host, is considered the major virulence factor of this pathogen (McKean et al., 2007). In addition, some secreted factors related to the virulence of C. pseudotuberculosis have already been described, such as serine protease CP40 (Wilson et al., 1995) and two operons, fagABC and ciuABCDEF, that are involved in iron uptake (Billington et al., 2002; Ribeiro et al., 2014).
The study of host-bacteria interactions in natural hosts, such as horses, cattle or sheep, is difficult because of the underlying genetic variability among animals; it is also extremely expense and requires multiple replicates and control animals. Thus, several studies have used mice as a model for studying both the pathogenic process (Jolly, 1965; Zaki, 1966; Nieto et al., 2009) and vaccination testing against infection by C. pseudotuberculosis (Simmons et al., 1997; Lan et al., 1999; Gorman et al., 2010; Ribeiro et al., 2014; Droppa-Almeida et al., 2016). In regards to host-bacteria interactions, some work has explored the serial passage process of bacterial pathogens in vitro or in an in vivo model to identify factors that might be involved in virulence (Fernández et al., 2000, 2013; Bleich et al., 2005; Chapuis et al., 2011; Fernandez-Brando et al., 2012; Liu et al., 2015). In this study, we adopted an in vivo assay in which the strain 258_equi was experimentally inoculated in mice followed by high-throughput proteomic analysis. We screened the functional genome of 258_equi after experimental passage in the murine host by examining the proteins released into the culture supernatant of this strain using the three-phase partitioning (TPP) protocol for obtaining extracellular proteins (Paule et al., 2004) and a mass spectrometry, data-independent acquisition (LC-MSE) approach to identify and quantify the proteins (Silva et al., 2006; Pacheco et al., 2011).
Materials and Methods
Bacterial Strain and growth Conditions
Corynebacterium pseudotuberculosis biovar equi strain 258 was isolated from a horse in Belgium; this strain was cultivated under routine conditions in brain–heart infusion broth (BHI-HiMedia Laboratories Pvt. Ltd., India) at 37°C. When necessary, 1.5% agar was added to the medium for solid culture. For extracellular proteomic analyses, 258_equi was grown in a chemically defined medium (CDM) [(Na2HPO4_7H2O (12.93 g/L), KH2PO4 (2.55 g/L), NH4Cl (1 g/L), MgSO4_7H2O (0.20 g/L), CaCl2 (0.02 g/L), and 0.05% (v/v) Tween 80] with 4% (v/v) MEM Vitamins Solution (Invitrogen, Gaithersburg, MD, USA), 1% (v/v) MEM Amino Acids Solution (Invitrogen), 1% (v/v) MEM Non-Essential Amino Acids Solution (Invitrogen), and 1.2% (w/v) glucose at 37°C (Moura-Costa et al., 2002).
Experimental Infection in a murine Model
The infection parameters were performed according to Moraes et al. (2014) and Ribeiro et al. (2014). In this study, female BALB/c mice between 6- and 8-weeks-old were utilized; they were provided by the Animal Care Facility at the Biological Sciences Institute at the Federal University of Minas Gerais and were handled in accordance with the CEUA guidelines of the UFMG Ethics Committee on Animal Testing (Permit Number: CETEA 103/2011). For the bacterial passage assay, three mice were infected via intraperitoneal injection with 106 colony forming units (CFU) of strain 258_equi. Thirty-six hours after infection, the animals were sacrificed, and the spleen was aseptically removed for recovering the bacteria. Each spleen was individually macerated in a sterile saline solution (0.9% NaCl2) and seeded onto BHI agar plates for incubation at 37°C for 48 h. Subsequently, one bacterial colony of each BHI plate was isolated and cultured in BHI broth at 37°C with shaking (180 rpm) until the OD600 = 0.8. Three different stock cultures were generated and stored at −80°C in BHI broth and 10% glycerol. The recovered bacteria are referred to as Recovered (Rc), and bacteria with no previous host contact were used as the Control (Ct). For bacterial virulence assays, bacteria from the three individual frozen stocks of Rc and the Ct condition were centrifuged at 5,000 × g for 5 min and washed twice in saline solution, followed by resuspension in saline solution. Three groups of five mice were infected with bacteria from the Rc or Ct condition via intraperitoneal injection of a suspension containing 106 or 105 CFU. The animals' survival rates were calculated and represented in GraphPad Prism v.5.0 (GraphPad Software, San Diego, CA, USA) using the Kaplan-Meier survival function.
Preparation of extracellular proteins for proteome Analysis
For proteomic analysis, three independent control and recovered colonies from the three individual frozen stocks were grown in CDM to an OD600 = 0.8. The cultures were then centrifuged for 20 min at 2,700 × g. The supernatants were filtered using 0.22-μm filters, 30% (w/v) ammonium sulfate was added to the samples, and the pH of the mixtures was adjusted to 4.0. Next, 20 mL/L N-butanol was added to each sample. The samples were centrifuged for 10 min at 1,350 × g and 4°C. The interfacial precipitate was collected and resuspended in 1 mL of 20 mM Tris-HCl, pH 7.2 (Paule et al., 2004). Proteins were quantified using the Bradford assay. For label-free proteomic analysis, the protein extract was concentrated using a spin column with a 10 kDa threshold (Millipore, Billerica, MA, USA). The protein was denatured (0.1% RapiGEST SF at 60°C for 15 min) (Waters, Milford, CA, USA), reduced (10 mM DTT), alkylated (10 mM iodoacetamide) and enzymatically digested with trypsin (Promega, Sequencing Grade Modified Trypsin, Madison, WI, USA). Glycogen phosphorylase (Waters Corporation, SwissProt P00489) was added to the digests to a final concentration of 20 fmol/μl as an internal standard for normalization prior to each replicate injection. The digestion process was stopped by adding 10 μL of 5% TFA (Fluka, Buchs, Germany) (Silva et al., 2006).
Mass Spectrometry analysis, data processing and quantification
Three independent biological replicates of each experimental condition were digested, as described above, for MSE analysis. Qualitative and quantitative nanoUPLC tandem nanoESI-HDMSE (Nano Electrospray High Definition Mass Spectrometry) experiments were performed using a 1 h reversed-phase gradient from 7 to 40% (v/v) acetonitrile (with 0.1% v/v formic acid) at 500 nL.min−1 using a nanoACQUITY UPLC 2D RPxRP Technology system (Gilar et al., 2005). All analyses were performed using nano-electrospray ionization in the positive ion mode (nanoESI (+)) and a NanoLockSpray (Waters, Manchester, UK) ionization source. The mass spectrometer was calibrated with an MS/MS spectrum of human [Glu1]-Fibrinopeptide B (Glu-Fib) solution (100 fmol.mL−1) delivered through the reference sprayer of the NanoLockSpray source. The double-charged ion ([M + 2H]2+ = 785.8426) was used for initial single-point calibration, and MS/MS fragment ions of Glu-Fib were used to obtain the final instrument calibration. Multiplexed data-independent (DIA) scanning with additional specificity and selectivity for non-linear “T-wave” ion mobility (HDMSE) experiments were performed using a Synapt G2-S HDMS mass spectrometer (Waters, Manchester, UK), which was constructed to automatically switch between the application of standard MS (3 eV) and elevated collision energies HDMSE (19–45 eV) to the transfer “T-wave” CID (collision-induced dissociation) cell with argon gas.
The proteins were identified, and quantitative data were packaged using dedicated algorithms (Silva et al., 2005; Geromanos et al., 2009) and searching against a database with default parameters to account for ions (Li et al., 2009). The databases used were reversed “on-the-fly” during the database queries and appended to the original database to assess the false positive rate during identification. For proper spectra processing and database searching conditions, the ProteinLynx Global SERVER v.2.5.2 (PLGS) with IdentityE and ExpressionE informatics v.2.5.2 (Waters, Manchester, UK) was used. UniProtKB (release 2013_01) with manually reviewed annotations was used, and the search conditions were based on taxonomy (Corynebacterium pseudotuberculosis). The maximum allowed missed cleavages by trypsin was up to 1, and various modifications, including carbamidomethyl (C), N-terminal acetyl, phosphoryl (STY) and oxidation (M), were allowed. A peptide mass tolerance value of 10 ppm was used. The search threshold to accept each spectrum was the default value in the program with a false discovery rate value of 4% (Curty et al., 2014). For protein quantitation, PLGS v2.5.2 software was used with the IdentityE algorithm using Hi3 methodology and glycogen phosphorylase (muscle form; P00489) peptides were used as internal standards. The collected proteins were organized by the PLGS ExpressionE tool algorithm into a statistically significant list (p-value ≤ 0.05) that corresponded to higher or lower regulation ratios between the different groups. The calculation of the log ratio and the confidence interval was based on a Gaussian distribution model, which allows for the possibility of an uncertain peptide assignment, an incorrect assignment of data to a cluster or interference. The confidence interval of 95% was used, and the probability distribution of the measured value of a log2 ratio more than a 1.2 was more symmetric than that obtained for the direct ratio, making the results interpretations more meaningful (Levin et al., 2011). For comparing pairs of experimental groups, proteins with a differential expression log2 ratio greater than or equal to 1.2 between the two conditions were considered for higher or lower abundance level determination (Levin et al., 2011).
Bioinformatics Analysis
The proteins identified in both conditions were analyzed using the following prediction tools: SurfG+ v1.0 (Barinov et al., 2009) was used to predict subcellular localization, SecretomeP 2.0 server was used to predict proteins exported from non-classical systems (positive prediction scores greater than 0.5; Bendtsen et al., 2005a), TatP was used to predict proteins with twin-arginine signal peptides (Bendtsen et al., 2005b) and the PIPs software was used to predict the proteins in pathogenicity islands (Soares et al., 2012). Gene ontology (GO) functional annotations were generated using the COG data base (Tatusov et al., 2001). Pathway enrichment analysis of significant proteins was carried out using the Kyoto encyclopedia of genes and genomes (KEGG) database. A protein-protein interaction network was generated using Cytoscape version 2.8.3 (Shannon et al., 2003) with a spring-embedded layout.
Results
Evaluation of the virulence potential of 258_equi after passage in a murine host
In the first in vivo assay, BALB/c mice were infected with 106 CFU of bacteria that had no previous host contact and with bacteria that were recovered from mouse spleens. We observed that all of the infected animals under both the control (Ct) and recovered (Rc) conditions died within 48 h of infection (Figure 1A). These results reveal the virulence potential of 258_equi; however, in this assay, we did not observe differences in the virulence potential between the control and recovered condition. Next, the Ct and Rc condition were analyzed using a new survival assay in BALB/c mice, but a 105 CFU infection dose was used (Figure 1B). In this assay, we observed altered virulence in the Rc condition; the mice began dying in the first 10 days post-infection (40% decrease in survival rate) and mortality reached 100% in less than 20 days post-infection. For the Ct condition, while mice died during the first 10 days of infection, this early stage only resulted in 20% mortality, and mortality did not reach 100% until 23 days post-infection. Finally, we detected abscess formation in the internal organs (kidney and liver) of all animals infected with either the Ct or Rc condition in the assays using 105 CFU (data not shown). These results show that passage in a murine host affects the virulence potential of 258_equi.
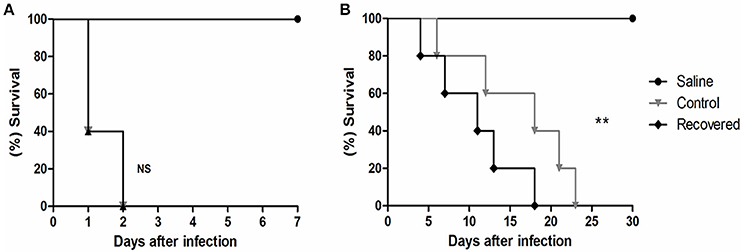
Figure 1. Survival assay of Balb/C mice infected with strain 258_equi. (A) Percent survival of BALB/c mice infected with 106 CFU of bacteria. (B) Percent survival of BALB/c mice infected with 105 CFU of bacteria. Ct, control condition and Rc, recovered condition. The mortality rates were measured daily. The results presented in (A,B) represents three independent experiments. The p-values were calculated using the log rank test. Note that infection with 105 CFU of bacteria changes the potential virulence of Rc (p = 0.0024, log-rank test) relative to Ct. NS = P > 0.05; **P < 0.01.
Overview of the exoproteome of C. pseudotuberculosis strain 258 after passage in a murine Host
After passage of 258_equi in BALB/c mice, we detected changes in its virulence potential. To assess whether this change is reflected in its proteome, considering the importance of exported proteins in bacterial infection (Hilbi et al., 2012), we used a TPP/LC-MSE approach (Pacheco et al., 2011) to compare the extracellular proteome of the control and recovered conditions. From our proteomic analysis, a total of 113 non-redundant 258_equi proteins were detected with high confidence and were identified in at least two of the three biological replicates of the two conditions tested, with an average of 17 peptides per protein and an FDR of 1%. The peptides were identified with a normal distribution of 10 ppm error for the total identified peptides (Supplementary Figure 1A). In addition, only the source fragments of peptides with a charge state of at least [M + 2H]2+ and the absence of decoys were considered to increase data quality.
The absolute quantitation of proteins present within a complex protein mixture is extremely important for understating physiological adaptations in response to biological demands promoted by environmental changes (Mallick and Kuster, 2010). To estimate the absolute abundance of identified proteins in the 258_equi exoproteome, we utilized the Hi3 method (Silva et al., 2006) where the average MS signal responses for the three most intense tryptic peptides for each protein were determined, including those of the internal standard protein glycogen phosphorylase (muscle form; P00489). All samples were normalized prior to injection using “scouting runs,” and the stoichiometry between the intensity and molarity proportion prior to the replicate runs per condition were considered. From this analysis a dynamic range of protein abundance was generated spanning three orders of magnitude (Supplementary Figure 1B). Lysozyme M1 was the most abundant protein detected. This protein, which is related to bacterial virulence, is localized in the pathogenic island Cp258PiCp02. Lysozyme M1 was also detected in a membrane shaving of a field isolate of C. pseudotuberculosis biovar ovis (Rees et al., 2015). Other proteins, such as hydrolase domain containing protein, trehalose corynomycolyl transferase B, which is involved in the cell wall synthesis, and FtsX, a protein related to division cellular, were among the most abundant proteins. All of the identified proteins on the protein abundance scale are listed in Supplementary Table 1.
For evaluating the relative differences between the core exoproteome of the Ct and Rc conditions, we used label-free quantification (Silva et al., 2005, 2014; Pacheco et al., 2011). In agreement with the PLGS analyses, 105 proteins between the Rc and Ct conditions presented significant statistical values (p < 0.05) using the ExpressionE algorithm tool (Supplementary Table 2). Differential expression was considered for proteins that were significantly different (p < 0.05) and had log2 ratios equal to or greater than a factor of 1.2, as described by Levin et al. (2011). Based on this analysis, 39 proteins were induced and 16 were down-regulated in the Rc condition (Table 1). In addition, we detected proteins exclusive to the proteome of each condition; cytochrome c nitrate reductase small subunit NrfH was detected only in the Ct condition. In only the Rc condition, two multidrug resistance proteins, the cytochrome oxidase assembly, a thioredoxin-related protein and three proteins with unknown function were identified (Supplementary Table 3).
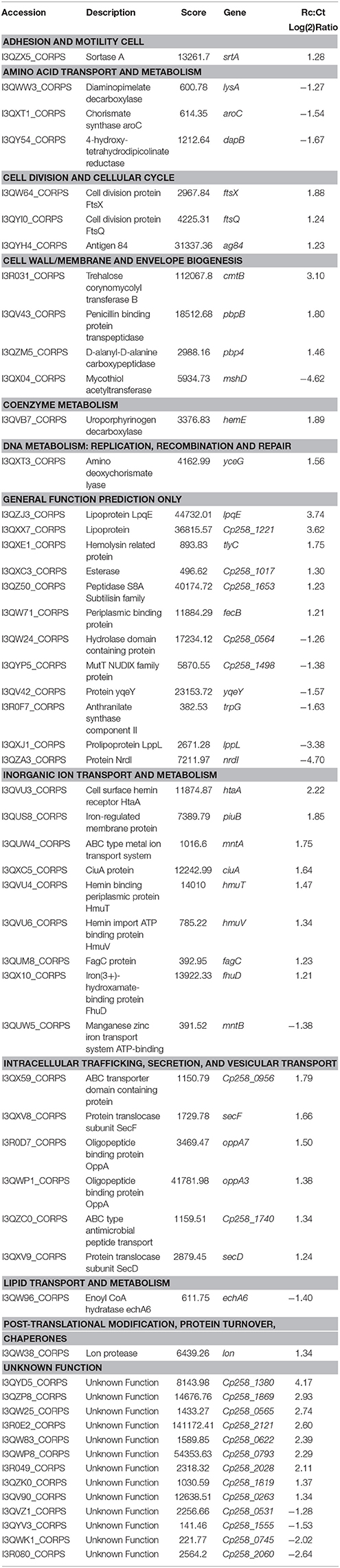
Table 1. Proteins differentially expressed between the recovered (Rc) and control (Ct) conditions.
In silico prediction of 258_equi exoprotein localization
Extracellular proteins produced by prokaryotic organisms are a subset of proteins present in the extracellular milieu, which is composed of both proteins with signal peptides that are actively secreted by classical secretion systems and proteins without signal sequences that are exported by non-classical secretion systems (Bendtsen et al., 2005a; Desvaux et al., 2009). To identify proteins that contain signal peptides and to determine their subcellular localizations, we utilized the SurfG tool (Barinov et al., 2009), which enables the classification of proteins within the following categories: cytoplasmic (CYT), membrane (MEM), potentially surface-exposed (PSE) and secreted (SEC) (Figure 2A and Supplementary Tables 2, 3). Of the total proteins identified, 66% (n = 74) presented positive predictions for signal peptides (Figure 2B). This group was composed of predicted proteins in the SEC and PSE categories. When these results were compared with the in silico data of the 258_equi genome, we had identified approximately 65% and 16% of the proteins predicted to be SEC and PSE, respectively. The proteins that did not present positive predictions for signal peptides were analyzed by SecretomeP (Bendtsen et al., 2005a) to identify proteins that could eventually be exported by non-classical secretion systems. According this analysis, nine proteins were predicted to be PSE, seven proteins were predicted to be MEM and two proteins were predicted to be CYT as they presented High SecP scores above 0.5 (Supplementary Tables 2, 3), suggesting that these proteins might be exported by a non-classical secretion system. Taken together, 86% of the 258_equi exoproteome was composed of extracellular proteins (Figure 2B). In addition, our proteomic analysis detected 17 proteins predicted to be lipoproteins (Supplementary Table 2).
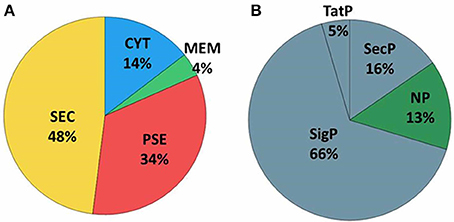
Figure 2. Prediction of the subcellular localization of the 258_equi exoproteome. (A) CYT, cytoplasmic; MEM, membrane; PSE, potentially surface-exposed; and SEC, secreted. (B) SecP, SecretomeP prediction for non-classical pathways; TatP, Tat-pathway prediction; SigP, SignalP prediction for peptide signal prediction; and NP, no prediction for SecP, TatP, or SigP.
Functional Classifications of the differentially Expressed proteins in strain 258_equi after passage in a murine Host
To evaluate the functional characteristics of the 258_equi exoproteome, we performed a Clusters of Orthologous Groups analysis (Tatusov et al., 2001). According GO analysis, the proteins were organized by clusters of orthologous groups (Figure 3A). When we evaluated each functional category, we observed that the majority of the proteins detected as induced in the Rc condition were predicted as “function unknown” and “general function only” (Table 1 and Figure 3B). These results represent a lack of knowledge regarding a protein set that might play an important role in the pathogenic process of 258_equi, and therefore, more studies are necessary to investigate the true roles of these proteins in the virulence of this strain. When we evaluated proteins with known or predicted functions, the majority of those that were more abundant in the Rc condition were related to cellular processes and signaling (Figures 3A,B). According to in silico data of the 258_equi genome, this pathogen has five iron uptake systems (Supplementary Figure 2; Soares et al., 2013a). Interestingly, in our proteomic analysis, we identified components of each of the 5 systems as more abundant in the Rc condition, including CiuA, FhuD, FagC, HmuT, HmuV, and HtaA (Table 1), suggesting that iron uptake pathways may play an important role in the pathogenesis of C. pseudotuberculosis. Moreover, we detected several proteins related to bacterial pathogenesis that contribute to processes of adherence, intracellular growth and evasion of the immune system (Table 1 and Supplementary Tables 2, 3).
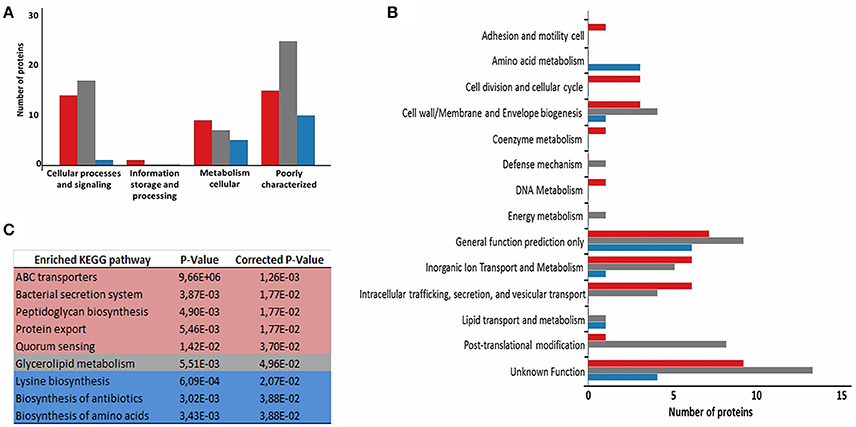
Figure 3. Functional analysis of the differentially expressed proteome between the control and recovered conditions. (A) Proteins classified by COG functional categories. (B) Categorization of differentially expressed proteins in biological processes. (C) KEGG pathway enrichment analysis of differentially expressed proteins. The colors are based on the Rc:Ct relation; red, up-regulated; gray, unchanged; and blue, down-regulated.
To identify the most relevant biological pathways of the proteins differentially expressed between the Ct and Rc conditions, we performed a KEGG enrichment analysis. Enrichment results revealed eight biological pathways with significant differences (p < 0.05). The proteins that were induced in the Rc condition are in pathways such as ABC transporters, bacterial secretion systems, peptidoglycan biosynthesis and protein export (Figure 3C). This finding confirms that secretion and secreted proteins are key elements in C. pseudotuberculosis virulence and adaptation, as suggested by previous reports that identified several secreted proteins as potential virulence factors in C. pseudotuberculosis (Pacheco et al., 2011; Silva et al., 2013; Rees et al., 2015). Most proteins perform their function in a context of networks by interacting with other proteins (Schleker et al., 2012). To evaluate the 258_equi exoproteome at the network level, we performed a protein-protein interaction analysis of the differentially expressed proteins using the Cytoscape tool. After Cytoscape analysis, the 258_equi exoproteome network was composed of 87 proteins (Figure 4). In the PPI-network, we observed enrichment clusters in heme biosynthesis and ABC transporters related to iron uptake, peptidoglycan biosynthesis and antibiotic biosynthesis. In addition, we observed that some clusters were formed by unknown proteins, which shows that these proteins may play an important role in the virulence of C. pseudotuberculosis.
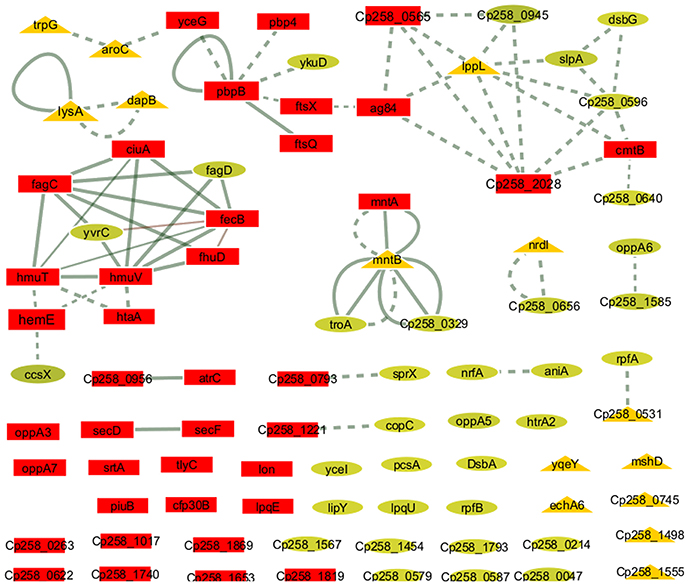
Figure 4. Protein-protein interaction network. The network nodes represent proteins, and the edges represent protein-protein associations. The node size (protein) is proportional to the amount of protein interacting (degree of interaction). Dotted line = regulatory interactions (functional), solid line, physical interactions; triangles, down-regulated proteins in the Rc condition; squares, highly induced proteins in the Rc condition; and circles, unchanged proteins.
Discussion
Here, we report a comprehensive analysis of the exoproteome of an equi isolate of C. pseudotuberculosis. The 258_equi exoproteome was composed of a high number of extracellular proteins, and a similar result was observed in a study conducted by Pacheco et al. (2011), which characterized the extracellular proteomes of C. pseudotuberculosis biovar ovis strains. The infections caused by C. pseudotuberculosis are chronic in character, and due to this, post-infection disease signs may not begin to appear until after 6 months. Necropsy is the only viable way to identify abscesses, but the cost is high. Testing several strains requires many animals and would result in high economic and ethical costs. Thus, mice are used as an alternative model for studying C. pseudotuberculosis infection, because they are relatively resistant to experimental challenge and are able to contain infection (Jolly, 1965; Zaki, 1966). In addition, mice have been shown to be efficient for the evaluation of different vaccine compounds and of humoral and cellular immune responses (Simmons et al., 1997; Lan et al., 1999; Gorman et al., 2010; Droppa-Almeida et al., 2016). Other studies have used mice to study virulence and pathogenesis, including an evaluation of hepatic disease (Nieto et al., 2009), or to study knockout strains (Moraes et al., 2014; Ribeiro et al., 2014).
In our study, the serial passage process in mice was efficient to induce changed both virulence and functional genome of 258_equi, which was showed through of proteomic analysis. In the in vivo assay, we observed changes in its virulence potential in a new infection assay with a lower infection dose. Similar results from an evaluation of the virulence potential of Shiga toxin (Stx)-producing Escherichia coli (STEC) were also observed when mice were infected with a lower dose of the recovered bacteria that had been recovered after serial passage in a murine model (Fernandez-Brando et al., 2012). Other studies have also shown that the serial passage process through in vitro or in vivo models leads to changes in the virulence potential of pathogens, including Helicobacter pylori, Escherichia coli, Xenorhabdus nematophila, Arcobacter butzleri, Salmonella enterica, and Shigella flexneri (Fernández et al., 2000, 2013; Bleich et al., 2005; Chapuis et al., 2011; Fernandez-Brando et al., 2012; Liu et al., 2015). Changes in the virulence potential of these pathogens, as well as in 258_equi, show that this strategy promotes the activation of genes related to bacterial pathogenesis.
A proteomic study conducted with Shigella flexneri after passage in an in vivo model showed the induction of important proteins that might contribute to its adaptation process during infection (Liu et al., 2015). In our proteomic analysis, we also observed changes in the 258_equi exoproteome after the recuperation process, and proteins that might play an important role in the pathogenesis of C. pseudotuberculosis were detected (Figure 5). Interestingly, when compared the proteins that were differentially induced in the Rc condition, with in silico data of the core-genome of C. pseudotuberculosis biovar equi and biovar ovis strains (Soares et al., 2013b), we observed that all proteins are present in this core-genome. This result represents a set of proteins that might be important to pathogenesis of biovar equi and biovar ovis strains. Within our proteomic repertoire, we detected predicted proteins such as lipoprotein. This class of proteins is produced by several prokaryotic organisms and then translocated across the membrane through the Sec or Tat pathway (Pugsley, 1993; Shruthi et al., 2010). Different studies have shown that these peripherally anchored membrane proteins perform an important role in the physiology, virulence and immune response of different gram-negative and gram-positive pathogens. In addition, lipoproteins are recognized as excellent vaccine targets (Nguyen and Götz, 2016). In the closely related pathogen M. tuberculosis, lipoproteins have been shown to be extremely important for virulence, contributing directly to evasion of the immune system (Su et al., 2016).
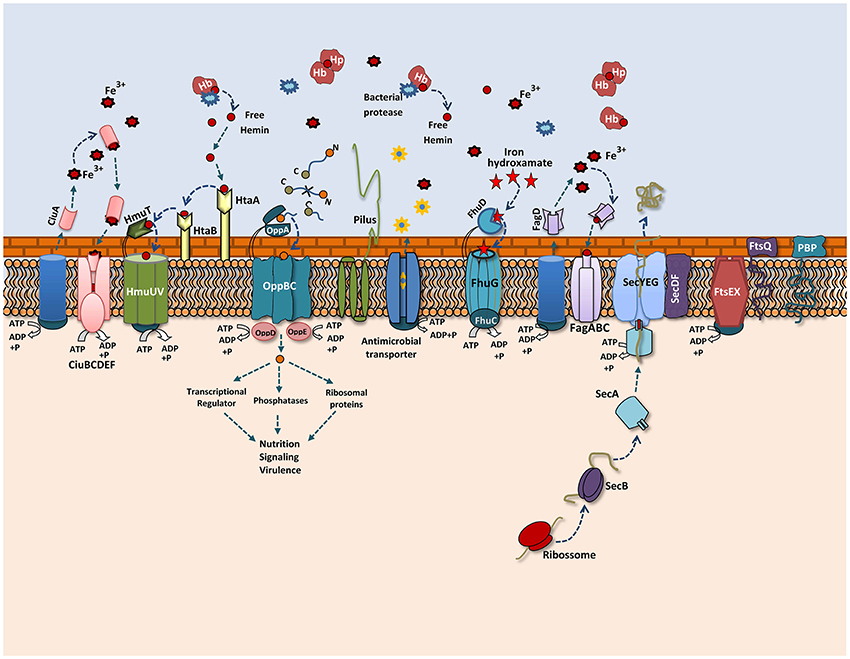
Figure 5. Overview of the 258_equi proteome after the recuperation process. A model representing the main exoproteins induced in the recovered condition, including proteins related to biogenesis of the cell wall, cellular adhesion and different secretion pathways related to iron acquisition, bacterial nutrition, efflux pumps and the Sec pathway.
Adhesion to host cells is a key determinant that contributes to bacteria–host interaction; this process is required for bacterial colonization and persistence. In vitro and in silico studies showed that C. pseudotuberculosis contains pili, and these structures play an important role in cellular adhesion (Yanagawa and Honda, 1976; Soares et al., 2013b). In C. pseudotuberculosis, spaA is a major pili gene that is encoded by the following gene cluster: srtB-spaA-srtA-spaB-spaX-spaC (Soares et al., 2013b). We found that sortase A (SrtA) was induced in the Rc supernatant. This cell surface anchored transpeptidase catalyzes the covalent attachment of precursor cell wall-attached proteins (LPXTG proteins) to the peptidoglycan. In gram-positive pathogens, such as Listeria monocytogenes (Bierne et al., 2002), Streptococcus pneumoniae (Kharat and Tomasz, 2003), and Staphylococcus aureus (Oh et al., 2006), srtA mutant strains had reduced virulence in animal infection models. We also detected important proteins related to the cell division and growth of Corynebacterium induced in the Rc condition, such as FtsQ and FtsX, which form part of the ftsXE cluster, and penicillin-binding proteins (PBPs) (Letek et al., 2008). In E. coli, the FtsEX proteins were suggested to form an ABC transporter system involved in the uptake of substrates necessary to maintain osmotic pressure during cell division (Schmidt et al., 2004; Reddy, 2007). FtsX was detected among the most abundant proteins of the 258_equi exoproteome, which suggests it is an important protein within the biology of this strain. PBPs proteins have an important role in cell-wall biosynthesis in Corynebacterium as they are essential to peptidoglycan biosynthesis. In addition, this class of proteins is a target of antibiotics (Letek et al., 2008). Antigen 84 (Ag84) was also induced in the Rc condition. Interestingly, Ag84 was also detected in a membrane shaving of an ovis strain isolated directly from the caseous nodes of a diseased animal (Rees et al., 2015). In M. tuberculosis, this protein presents antigenic characteristics and is required for growth (Sassetti et al., 2003). These proteins may have key functions in the replication and growth of C. pseudotuberculosis during the infection process.
Iron is an essential element for both the virulence and growth of several bacterial pathogens during the infection process. However, free iron is not available to the bacterial inside the host, thus several pathogens utilize different mechanisms to acquire both free iron and iron from host iron proteins (Brown and Holden, 2002). For C. pseudotuberculosis, iron acquisition is a required step in its pathogenic process (Billington et al., 2002; Ribeiro et al., 2014), and according to an in silico analysis of the 258_equi genome, this bacterium has different genetic loci associated with high-affinity iron transport systems as well as surface-associated heme-uptake pathways (Soares et al., 2012). In our proteomic analysis, we detected that specific proteins related to iron acquisition were induced in the Rc condition. Some these proteins are involved directly in the virulence of C. pseudotuberculosis, such as the FagC protein that is component of the Fag system. A study performed with strains with defective fagB(C) genes showed that these strains presented reduced virulence in goats (Billington et al., 2002).
To acquire iron inside a host, bacteria synthesize and secrete siderophores, which are low-molecular-weight iron chelators that have a high and specific affinity for ferric iron (Ellermann and Arthur, 2017). We detected the CiuA siderophore, which is localized in the operon ciuABCDE (Corynebacterium iron uptake). Studies performed with the pathogen C. diphtheria showed that Ciu is a high-affinity iron uptake system (Kunkle and Schmitt, 2005). Additionally, in a study performed with C. pseudotuberculosis, a ciuA mutated strain showed reduced virulence, demonstrating the role of this protein in the virulence of this bacterium, and was also able to protect immunized mice when they were challenged with a virulent strain (Ribeiro et al., 2014). Another siderophore detected was the FhuD siderophore, which is part of the conserved ferric hydroxamate uptake system, Fhu. The uptake of ferric ferrichrome is described in pathogens such as L. monocytogenes (Jin et al., 2006; Xiao et al., 2011), Streptococcus pyogenes (Hanks et al., 2005) and S. aureus (Sebulsky and Heinrichs, 2001). In L. monocytogenes, FhuD contributes to the uptake of ferric hydroxamate from ferrichrome, ferrichrome A and ferrioxamine B (Jin et al., 2006; Xiao et al., 2011). In S. aureus, this protein was shown to contribute both to proliferation within the blood and to the formation of renal abscesses in mice (Mishra et al., 2012). Like C. diphtheria, the genome of 258_equi also has genetic loci with genes related to heme acquisition, such as the hemin-uptake (hmu) operon HmuTUV and cell hemin specific receptors htaA, htaB and htaC. The ABC hemin transporter HmuTUV, together with cell-surface hemin receptors, is involved in heme uptake from hemoglobin (Hb), hemoglobin/haptoglobin, and myoglobin (Mb) (Kunkle and Schmitt, 2005; Allen and Schmitt, 2015). The presence of these systems shows the versatility of 258_equi in acquiring iron from different sources. Interestingly, the HtaA protein was detected to be immunoreactive in an immunoproteomic study of C. pseudotuberculosis biovar ovis (Seyffert et al., 2014). Taken together, these results indicate that, similar to other pathogens, iron acquisition likely plays an important role in 258_equi virulence as this strain uses distinct iron acquisition systems during infection. This ability to acquire iron may contribute to the increase virulence observed in the 258_equi strain.
Opp transport systems belong to the superfamily of conserved ATP-binding cassette transporters and play an important role in bacterial nutrition, signaling and virulence (Yu et al., 2014). The OppA protein, which is responsible for the uptake of peptides from the external medium, was induced in the Rc supernatant. In Mycobacterium avium, the oppA gene contributed to infections in a mouse model as well as to its viability in macrophages (Danelishvili et al., 2014). The Sec pathway is the major secretion system in several prokaryotic pathogens, components of this system was also induced in the Rc supernatant. SecDF are accessory factors from this translocation machinery and act to increase protein translocation. Different studies show that in S. aureus, the role of SecDF is related to the export of several virulence factors that contribute to parts its pathogenic process, such as adhesion, invasion and immune system evasion (Sibbald et al., 2006). In addition, SecDF belong to the resistance-nodulation-cell division (RND) family of multidrug export pumps and contribute to the resistance process against the antimicrobial effects of cathelicidins, a class of antimicrobial peptides produced by the immune system (Blodkamp et al., 2017). Similarly, proteins for antimicrobial agent resistance, such as ABC-type antimicrobial peptide transporters, which are localized in the pathogenicity island Cp258PiCp14, and efflux transporters, such as NorM, which belongs to the multidrug and toxic compound extrusion (MATE) transporter, were also detected. These data are consistent with previous in vitro studies, which showed that C. pseudotuberculosis is resistant to several classes of antimicrobial agents (Judson and Songer, 1991), and that activation of these defense pathways against antimicrobial agents might contribute to survival of this pathogen.
Conclusion
Herein, we characterized, for the first time, the exoproteome of a C. pseudotuberculosis equi isolate. In addition, we showed changes in both the virulence and proteomic profiles of 258_equi after its recovery from murine host spleens. Through a TPP/LC-MSE approach, we detected secreted virulence-associated proteins. The up-regulation of these proteins may account for the difference in virulence potential we observed in the Rc condition compared with the Ct condition. Altogether, our proteomic repertoire identified several extracellular proteins involved in key processes of bacterial pathogenesis that might contribute to the pathogenic process of C. pseudotuberculosis.
Author Contributions
WS, VA, and YL Conceived and designed the experiments. WS and FD performed in vivo experiments. WS and RD performed microbiological analyses and sample preparation for proteomic analysis. WS and GS conducted the proteomic analysis. EF performed bioinformatics analysis. AP, YL, and HF contributed substantially to data interpretation and revisions. AS, VA, and YL participated in all steps of the project as coordinators, and critically reviewed the manuscript.
Funding
The work was supported by the Brazilian Federal Agency for the Support and Evaluation of Graduate Education (CAPES), Pará Research Foundation (FAPESPA), Minas Gerais Research Foundation (FAPEMIG) and the National Council for Scientific and Technological Development (CNPq). YL is the recipient of a PVE grant (71/2013) from Programa Ciências sem Fronteiras.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank to Pará State Genomics and Proteomics Network and Waters Corporation, Brazil.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fcimb.2017.00325/full#supplementary-material
Supplementary Figure 1. (A) Two-dimensional nanoUPLC HDMSE analysis showing the distribution of fragment masses and the exact mass accuracy for 90% of the precursor ions with a 10 ppm maximum error. (B) Dynamic range based on the absolute quantitation of the proteins identified by LC-HDMSE analysis.
Supplementary Figure 2. Genomic loci encoding proteins related to iron-acquisition in 258_equi. Red genes encode proteins that were identified in our proteomic analysis.
Supplementary Table 1. Total list of identified proteins on the protein abundance scale.
Supplementary Table 2. Total list of differentially expressed proteins between the recovered and control conditions of stain 258_equi.
Supplementary Table 3. Proteins unique to the recovered and control conditions.
References
Aleman, M., Spier, S. J., Wilson, W. D., and Doherr, M. (1996). Corynebacterium pseudotuberculosis infection in horses: 538 cases (1982-1993). J. Am. Vet. Med. Assoc. 15, 804–809.
Allen, C. E., and Schmitt, M. P. (2015). Utilization of host iron sources by Corynebacterium diphtheriae: multiple hemoglobin-binding proteins are essential for the use of iron from the hemoglobin-haptoglobin complex. J. Bacteriol. 197, 553–562. doi: 10.1128/JB.02413-14
Barinov, A., Loux, V., Hammani, A., Nicolas, P., Langella, P., Ehrlich, D., et al. (2009). Prediction of surface exposed proteins in Streptococcus pyogenes, with a potential application to other Gram-positive bacteria. Proteomics 9, 61–73. doi: 10.1002/pmic.200800195
Bendtsen, J. D., Kiemer, L., Fausboll, A., and Brunak, S. (2005a). Non-classical protein secretion in bacteria. BMC Microbiol. 5:58. doi: 10.1186/1471-2180-5-58
Bendtsen, J. D., Nielsen, H., Widdick, D., Palmer, T., and Brunak, S. (2005b). Prediction of twin-arginine signal peptides. BMC Bioinformatics 2:167. doi: 10.1186/1471-2105-6-167
Bierne, H., Mazmanian, S. K., Trost, M., Pucciarelli, M. G., Liu, G., Dehoux, P., et al. (2002). Inactivation of the srtA gene in Listeria monocytogenes inhibits anchoring of surface proteins and affects virulence. Mol. Microbiol. 43, 869–881. doi: 10.1046/j.1365-2958.2002.02798.x
Billington, S. J., Esmay, P. A., Songer, J. G., and Jost, B. H. (2002). Identification and role in virulence of putative iron acquisition genes from Corynebacterium pseudotuberculosis. FEMS Microbiol. Lett. 208, 41–45. doi: 10.1111/j.1574-6968.2002.tb11058.x
Bleich, A., Köhn, I., Glage, S., Beil, W., Wagner, S., and Mähler, M. (2005). Multiple in vivo passages enhance the ability of a clinical Helicobacter pylori isolate to colonize the stomach of Mongolian gerbils and to induce gastritis. Lab. Anim. 39, 221–229. doi: 10.1258/0023677053739800
Blodkamp, S., Kadlec, K., Gutsmann, T., Quiblier, C., Naim, H. Y., and Schwarz, S. (2017). Effects of SecDF on the antimicrobial functions of cathelicidins against Staphylococcus aureus. Vet. Microbiol. 200, 52–58. doi: 10.1016/j.vetmic.2016.03.021
Britz, E., Spier, S. J., Kass, P. H., Edman, J. M., and Foley, J. E. (2014). The relationship between Corynebacterium pseudotuberculosis biovar equi phenotype with location and extent of lesions in horses. Vet. J. 200, 282–286. doi: 10.1016/j.tvjl.2014.03.009
Brown, J. S., and Holden, D. W. (2002). Iron acquisition by Gram-positive bacterial pathogens. Microb. Infect. 4, 1149–1156. doi: 10.1016/S1286-4579(02)01640-4
Chapuis, É., Pagès, S., Emelianoff, V., Givaudan, A., and Ferdy, J. B. (2011). Virulence and pathogen multiplication: a serial passage experiment in the hypervirulent bacterial insect-pathogen Xenorhabdus nematophila. PLoS ONE 31:e15872. doi: 10.1371/journal.pone.0015872
Curty, N., Kubitschek-Barreira, P. H., Neves, G. W., Gomes, D., Pizzatti, L., Abdelhay, E., et al. (2014). Discovering the infectome of human endothelial cells challenged with Aspergillus fumigatus applying a mass spectrometry label-free approach. J. Proteomics 31, 126–140. doi: 10.1016/j.jprot.2013.07.003
Danelishvili, L., Stang, B., and Bermudez, L. E. (2014). Identification of Mycobacterium avium genes expressed during in vivo infection and the role of the oligopeptide transporter OppA in virulence. Microb. Pathog. 76, 67–76. doi: 10.1016/j.micpath.2014.09.010
Desvaux, M., Hébraud, M., Talon, R., and Henderson, I. R. (2009). Secretion and subcellular localizations of bacterial proteins: a semantic awareness issue. Trends Microbiol. 17, 139–145. doi: 10.1016/j.tim.2009.01.004
Dorella, F. A., Pacheco, L. G., Oliveira, S. C., Miyoshi, A., and Azevedo, V. (2006). Corynebacterium pseudotuberculosis: microbiology, biochemical properties, pathogenesis and molecular studies of virulence. Vet. Res. 37, 201–218. doi: 10.1051/vetres:2005056
Droppa-Almeida, D., Vivas, W. L., Silva, K. K., Rezende, A. F., Simionatto, S., Meyer, R., et al. (2016). Recombinant CP40 from Corynebacterium pseudotuberculosis confers protection in mice after challenge with a virulent strain. Vaccine 17, 1091–1096. doi: 10.1016/j.vaccine.2015.12.064
Ellermann, M., and Arthur, J. C. (2017). Siderophore-mediated iron acquisition and modulation of host-bacterial interactions. Free Radic. Biol. Med. 105, 68–78. doi: 10.1016/j.freeradbiomed.2016.10.489
Fernandez-Brando, R. J., Miliwebsky, E., Mejías, M. P., Baschkier, A., Panek, C. A., Abrey-Recalde, M. J., et al. (2012). Shiga toxin-producing Escherichia coli O157: H7 shows an increased pathogenicity in mice after the passage through the gastrointestinal tract of the same host. J. Med. Microbiol. 61, 852–859. doi: 10.1099/jmm.0.041251-0
Fernández, H., Flores, S. P., Villanueva, M., Medina, G., and Carrizo, M. (2013). Enhancing adherence of Arcobacter butzleri after serial intraperitoneal passages in mice. Rev. Argent. Microbiol. 45, 75–79. doi: 10.1016/s0325-7541(13)70002-6
Fernández, H., Vivanco, T., and Eller, G. (2000). Expression of invasiveness of Campylobacter jejuni ssp. jejuni after serial intraperitoneal passages in mice. J. Vet. Med. B. Infect. Dis. Vet. Public Health 47, 635–639. doi: 10.1046/j.1439-0450.2000.00392.x
Foley, J. E., Spier, S. J., Mihalyi, J., Drazenovich, N., and Leutenegger, C. M. (2004). Molecular epidemiologic features of Corynebacterium pseudotuberculosis isolated from horses. Am. J. Vet. Res. 65, 1734–1737. doi: 10.2460/ajvr.2004.65.1734
Geromanos, S. J., Vissers, J. P., Silva, J. C., Dorschel, C. A., Li, G. Z., Gorenstein, M. V., et al. (2009). The detection, correlation, and comparison of peptide precursor and product ions from data independent LC-MS with data dependant LC-MS/MS. Proteomics 9, 1683–1695. doi: 10.1002/pmic.200800562
Gilar, M., Olivova, P., Daly, A. E., and Gebler, J. C. (2005). Two-dimensional separation of peptides using RP-RP-HPLC system with different pH in first and second separation dimensions. J. Sep. Sci. 28, 1694–1703. doi: 10.1002/jssc.200500116
Gorman, J. K., Gabriel, M., MacLachlan, N. J., Nieto, N., Foley, J., and Spier, S. (2010). Pilot immunization of mice infected with an equine strain of Corynebacterium pseudotuberculosis. Vet. Ther. 11, E1–E8.
Hanks, T. S., Liu, M., McClure, M. J., and Lei, B. (2005). ABC transporter FtsABCD of Streptococcus pyogenes mediates uptake of ferric ferrichrome. BMC Microbiol. 5:62. doi: 10.1186/1471-2180-5-62
Hilbi, H., and Haas, A. (2012). Secretive bacterial pathogens and the secretory pathway. Traffic 13, 1187–1197. doi: 10.1111/j.1600-0854.2012.01344.x
Jin, B., Newton, S. M., Shao, Y., Jiang, X., Charbit, A., and Klebba, P. E. (2006). Iron acquisition systems for ferric hydroxamates, haemin and haemoglobin in Listeria monocytogenes. Mol. Microbiol. 59, 1185–1198. doi: 10.1111/j.1365-2958.2005.05015.x
Jolly, R. D. (1965). The pathogenesis of experimental Corynebacterium ovis infection in mice. N.Z. Vet. J. 13, 141–147. doi: 10.1080/00480169.1965.33618
Judson, R., and Songer, J. G. (1991). Corynebacterium pseudotuberculosis: in vitro susceptibility to 39 antimicrobial agents. Vet. Microbiol. 27, 145–150. doi: 10.1016/0378-1135(91)90005-Z
Kharat, A. S., and Tomasz, A. (2003). Inactivation of the srtA gene affects localization of surface proteins and decreases adhesion of Streptococcus pneumoniae to human pharyngeal cells in vitro. Infect. Immun. 71, 2758–2765. doi: 10.1128/IAI.71.5.2758-2765.2003
Kilcoyne, I., Spier, S. J., Carter, C. N., Smith, J. L., Swinford, A. K., and Cohen, N. D. (2014). Frequency of Corynebacterium pseudotuberculosis infection in horses across the United States during a 10-year period. J. Am. Vet. Med. Assoc. 245, 309–314. doi: 10.2460/javma.245.3.309
Kunkle, C. A., and Schmitt, M. P. (2005). Analysis of a DtxR-regulated iron transport and siderophore biosynthesis gene cluster in Corynebacterium diphtheriae. J. Bacteriol. 187, 422–433. doi: 10.1128/JB.187.2.422-433.2005
Lan, D. T., Makino, S., Shirahata, T., Yamada, M., and Nakane, A. (1999). Tumor necrosis factor alpha and gamma interferon are required for the development of protective immunity to secondary Corynebacterium pseudotuberculosis infection in mice. J. Vet. Med. Sci. 61, 1203–1208. doi: 10.1292/jvms.61.1203
Letek, M., Fiuza, M., Ordóñez, E., Villadangos, A. F., Ramos, A., Mateos, L. M., et al. (2008). Cell growth and cell division in the rod-shaped actinomycete Corynebacterium glutamicum. Antonie Van Leeuwenhoek. 94, 99–109. doi: 10.1111/j.1574-6968.2009.01679.x
Levin, Y., Hradetzky, E., and Bahn, S. (2011). Quantification of proteins using data-independent analysis (MSE) in simple and complex samples: a systematic evaluation. Proteomics 11, 3273–3287. doi: 10.1002/pmic.201000661
Li, G. Z., Vissers, J. P., Silva, J. C., Golick, D., Gorenstein, M. V., and Geromanos, S. J. (2009). Database searching and accounting of multiplexed precursor and product ion spectra from the data independent analysis of simple and complex peptide mixtures. Proteomics 9, 1696–1719. doi: 10.1002/pmic.200800564
Liu, X., Lu, L., Liu, X., Pan, C., Feng, E., Wang, D., et al. (2015). Comparative proteomics of Shigella flexneri 2a strain using a rabbit ileal loop model reveals key proteins for bacterial adaptation in host niches. Int. J. Infect. Dis. 40, 28–33. doi: 10.1016/j.ijid.2015.09.014
Mallick, P., and Kuster, B. (2010). Proteomics: a pragmatic perspective. Nat. Biotechnol. 28, 695–709. doi: 10.1038/nbt.1658
McKean, S. C., Davies, J. K., and Moore, R. J. (2007). Expression of phospholipase D, the major virulence factor of Corynebacterium pseudotuberculosis, is regulated by multiple environmental factors and plays a role in macrophage death. Microbiology 153, 2203–2211. doi: 10.1099/mic.0.2007/005926-0
Mishra, R. P., Mariotti, P., Fiaschi, L., Nosari, S., Maccari, S., Liberatori, S., et al. (2012). Staphylococcus aureus FhuD2 is involved in the early phase of staphylococcal dissemination and generates protective immunity in mice. J. Infect. Dis. 206, 1041–1049. doi: 10.1093/infdis/jis463
Moraes, P. M., Seyffert, N., Silva, W. M., Castro, T. L., Silva, R. F., and Lima, D. D. (2014). Characterization of the opp peptide transporter of Corynebacterium pseudotuberculosis and its role in virulence and pathogenicity. Biomed. Res. Int. 2014:489782. doi: 10.1155/2014/489782
Moura-Costa, L. F., Paule, B. J. A., Freire, S. M., Nascimento, I., Schaer, R., Regis, L. F., et al. (2002). Chemically defined synthetic medium for Corynebacterium pseudotuberculosis culture. Rev. Bras. Saúde Prod. An. 3, 1–9.
Nguyen, M. T., and Götz, F. (2016). Lipoproteins of gram-positive bacteria: key players in the immune response and virulence. Microbiol. Mol. Biol. Rev. 10, 891–903. doi: 10.1128/MMBR.00028-16
Nieto, N. C., Foley, J. E., MacLachlan, N. J., Yuan, T., and Spier, S. J. (2009). Evaluation of hepatic disease in mice following intradermal inoculation with Corynebacterium pseudotuberculosis. Am. J. Vet. Res. 70, 257–262. doi: 10.2460/ajvr.70.2.257
Oh, K. B., Oh, M. N., Kim, J. G., Shin, D. S., and Shin, J. (2006). et al. inhibition of sortase-mediated Staphylococcus aureus adhesion to fibronectin via fibronectin-binding protein by sortase inhibitors. Appl. Microbiol. Biotechnol. 70, 102–106. doi: 10.1007/s00253-005-0040-8
Pacheco, L. G., Castro, T. L., Carvalho, R. D., Moraes, P. M., Dorella, F. A., Carvalho, N. B., et al. (2012). A role for sigma factor σ(E) in Corynebacterium pseudotuberculosis resistance to nitric oxide/peroxide stress. Front. Microbiol. 3:126. doi: 10.3389/fmicb.2012.00126
Pacheco, L. G., Slade, S. E., Seyffert, N., Santos, A. R., Castro, T. L., Silva, W. M., et al. (2011). A combined approach for comparative exoproteome analysis of Corynebacterium pseudotuberculosis. BMC Microbiol. 17:12. doi: 10.1186/1471-2180-11-12
Paule, B. J., Meyer, R., Moura-Costa, L. F., Bahia, R. C., Carminati, R., Regis, L. F., et al. (2004). Three-phase partitioning as an efficient method for extraction/concentration of immunoreactive excreted-secreted proteins of Corynebacterium pseudotuberculosis. Protein Expr. Purif. 34, 311–316. doi: 10.1016/j.pep.2003.12.003
Pugsley, A. P. (1993). The complete general secretory pathway in gram-negative bacteria. Microbiol. Rev. 57, 50–108.
Reddy, M. (2007). Role of FtsEX in cell division of Escherichia coli: viability of ftsEX mutants is dependent on functional sufI or high osmotic strength. J. Bacteriol. 189, 98–108. doi: 10.1128/JB.01347-06
Rees, M. A., Kleifeld, O., Crellin, P. K., Ho, B., Stinear, T. P., Smith, A. I., et al. (2015). Proteomic characterization of a natural host–pathogen interaction: repertoire of in vivo expressed bacterial and host surface-associated proteins. J. Proteome Res. 14, 120–132. doi: 10.1021/pr5010086
Ribeiro, D., Rocha, F. S., Leite, K. M., Soares, S. C., Silva, A., and Portela, R. W. (2014). An iron-acquisition-deficient mutant of Corynebacterium pseudotuberculosis efficiently protects mice against challenge. Vet. Res. 6:28. doi: 10.1186/1297-9716-45-28
Sassetti, C. M., Boyd, D. H., and Rubin, E. J. (2003). Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84. doi: 10.1046/j.1365-2958.2003.03425.x
Schleker, S., Sun, J., Raghavan, B., Srnec, M., Müller, N., Koepfinger, M., et al. (2012). The current Salmonella-host interactome. Proteomics Clin. Appl. 6, 117–133. doi: 10.1002/prca.201100083
Schmidt, K. L., Peterson, N. D., Kustusch, R. J., Wissel, M. C., Graham, B., Phillips, G. J., et al. (2004). A predicted ABC transporter, FtsEX, is needed for cell division in Escherichia coli. J. Bacteriol. 186, 785–793. doi: 10.1128/JB.186.3.785-793.2004
Sebulsky, M. T., and Heinrichs, D. E. (2001). Identification and characterization of fhuD1 and fhuD2, two genes involved in iron-hydroxamate uptake in Staphylococcus aureus. J. Bacteriol. 183, 4994–5000. doi: 10.1128/JB.183.17.4994-5000.2001
Seyffert, N., Silva, R. F., Jardin, J., Silva, W. M., Castro, T. L., and Tartaglia, N. R. (2014). Serological proteome analysis of Corynebacterium pseudotuberculosis isolated from different hosts reveals novel candidates for prophylactics to control caseous lymphadenitis. Vet. Microbiol. 7, 255–260. doi: 10.1016/j.vetmic.2014.08.024
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shruthi, H., Babu, M. M., and Sankaran, K. (2010). TAT pathway-dependent lipoproteins as a niche-based adaptation in prokaryotes, J. Mol. Evol. 70, 359–370. doi: 10.1007/s00239-010-9334-2
Sibbald, M. J., Ziebandt, A. K., Engelmann, S., Hecker, M., de Jong, A., Harmsen, H. J., et al. (2006). Mapping the pathways to staphylococcal pathogenesis by comparative secretomics. Microbiol. Mol. Biol. Rev. 70, 755–788. doi: 10.1128/MMBR.00008-06
Silva, J. C., Denny, R., Dorschel, C. A., Gorenstein, M., Kass, I. J., Li, G. Z., et al. (2005). Quantitative proteomic analysis by accurate mass retention time pairs. Anal. Chem. 1, 2187–2000. doi: 10.1021/ac048455k
Silva, J. C., Gorenstein, M. V., Li, G. Z., Vissers, J. P., and Geromanos, S. J. (2006). Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol. Cell. Proteomics 5, 144–156. doi: 10.1074/mcp.M500230-MCP200
Silva, W. M., Carvalho, R. D., Soares, S. C., Bastos, I. F., Folador, E. L., Souza, G. H., et al. (2014). Label-free proteomic analysis to confirm the predicted proteome of Corynebacterium pseudotuberculosis under nitrosative stress mediated by nitric oxide. BMC Genomics 4:1065. doi: 10.1186/1471-2164-15-1065
Silva, W. M., Seyffert, N., Santos, A. V., Castro, T. L., Pacheco, L. G., Santos, A. R., et al. (2013). Identification of 11 new exoproteins in Corynebacterium pseudotuberculosis by comparative analysis of the exoproteome. Microb. Pathog. 62, 37–42. doi: 10.1016/j.micpath.2013.05.004
Simmons, C. P., Hodgson, A. L., and Strugnell, R. A. (1997). Attenuation and vaccine potential of aroQ mutants of Corynebacterium pseudotuberculosis. Infect. Immun. 65, 3048–3056.
Soares, S. C., Abreu, V. A., Ramos, R. T., Cerdeira, L., Silva, A., et al. (2012). PIPS: pathogenicity island prediction software. PLoS ONE 7:e30848. doi: 10.1371/journal.pone.0030848
Soares, S. C., Silva, A., Trost, E., Blom, J., Ramos, R., Carneiro, A., et al. (2013b). The pan-genome of the animal pathogen Corynebacterium pseudotuberculosis reveals differences in genome plasticity between the biovar ovis and equi strains. PLoS ONE 8:e53818. doi: 10.1371/journal.pone.0053818
Soares, S. C., Trost, E., Ramos, R. T., Carneiro, A. R., Santos, A. R., Pinto, A. C., et al. (2013a). Genome sequence of Corynebacterium pseudotuberculosis biovar equi strain 258 and prediction of antigenic targets to improve biotechnological vaccine production. J. Biotechnol. 20, 135–141. doi: 10.1016/j.jbiotec.2012.11.003
Spier, S. J. (2008). Corynebacterium pseudotuberculosis infection in horses: an emerging disease associated with climate change? Equine Vet. Educ. 20, 37–39. doi: 10.2746/095777307X260106
Su, H., Zhu, S., Zhu, L., Huang, W., Wang, H., Zhang, Z., et al. (2016). Recombinant lipoprotein Rv1016c derived from Mycobacterium tuberculosis Is a TLR-2 ligand that induces macrophages apoptosis and inhibits mhc ii antigen processing. Front. Cell. Infect. Microbiol. 18:147. doi: 10.3389/fcimb.2016.00147
Tatusov, R. L., Natale, D. A., Garkavtsev, I. V., Tatusova, T. A., Shankavaram, U. T., et al. (2001). The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 29, 22–28. doi: 10.1093/nar/29.1.22
Wilson, M. J., Brandon, M. R., and Walker, J. (1995). Molecular and biochemical characterization of a protective 40-kilodalton antigen from Corynebacterium pseudotuberculosis. Infect. Immun. 63, 206–211.
Xiao, Q., Jiang, X., Moore, K. J., Shao, Y., Pi, H., Dubail, I., et al. (2011). Sortase independent and dependent systems for acquisition of haem and haemoglobin in Listeria monocytogenes. Mol. Microbiol. 80, 1581–1197. doi: 10.1111/j.1365-2958.2011.07667.x
Yanagawa, R., and Honda, E. (1976). Presence of pili in species of human and animal parasites and pathogens of the genus corynebacterium. Infect. Immun. 13, 1293–1295.
Yu, D., Pi, B., Yu, M., Wang, Y., Ruan, Z., Feng, Y., et al. (2014). Diversity and evolution of oligopeptide permease systems in staphylococcal species. Genomics 104, 8–13. doi: 10.1016/j.ygeno.2014.04.003
Keywords: Corynebacterium pseudotuberculosis, label-free proteome, ulcerative lymphangitis, bacterial proteome, bacterial virulence, serial passage
Citation: Silva WM, Carvalho RDO, Dorella FA, Folador EL, Souza GHMF, Pimenta AMC, Figueiredo HCP, Le Loir Y, Silva A and Azevedo V (2017) Quantitative Proteomic Analysis Reveals Changes in the Benchmark Corynebacterium pseudotuberculosis Biovar Equi Exoproteome after Passage in a Murine Host. Front. Cell. Infect. Microbiol. 7:325. doi: 10.3389/fcimb.2017.00325
Received: 07 April 2017; Accepted: 03 July 2017;
Published: 25 July 2017.
Edited by:
Nora Lía Padola, National University of Central Buenos Aires, ArgentinaReviewed by:
Javier Bezos, Complutense University of Madrid, SpainTravis Bourret, Creighton University, United States
Copyright © 2017 Silva, Carvalho, Dorella, Folador, Souza, Pimenta, Figueiredo, Le Loir, Silva and Azevedo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vasco Azevedo, dmFzY29AaWNiLnVmbWcuYnI=