 Sei-Jung Lee1
Sei-Jung Lee1 Young Hyun Jung2
Young Hyun Jung2 Jun Sung Kim2
Jun Sung Kim2 Hyun Jik Lee2Sang Hun Lee3,4Kyu-Ho Lee5Kyung Ku Jang6Sang Ho Choi6
Hyun Jik Lee2Sang Hun Lee3,4Kyu-Ho Lee5Kyung Ku Jang6Sang Ho Choi6 Ho Jae Han2*
Ho Jae Han2*- 1Department of Pharmaceutical Engineering, Daegu Haany University, Gyeongsan, South Korea
- 2Department of Veterinary Physiology, College of Veterinary Medicine, Research Institute for Veterinary Science and BK21 PLUS Program for Creative Veterinary Science Research Center, Seoul National University, Seoul, South Korea
- 3Medical Science Research Institute, Soonchunhyang University Seoul Hospital, Seoul, South Korea
- 4Departments of Biochemistry, Soonchunhyang University College of Medicine, Cheonan, South Korea
- 5Department of Life Science, Sogang University, Seoul, South Korea
- 6National Research Laboratory of Molecular Microbiology and Toxicology, Department of Agricultural Biotechnology, Center for Food Safety and Toxicology, Seoul National University, Seoul, South Korea
An inflammatory form of phagocyte death evoked by the Gram-negative bacterium Vibrio (V.) vulnificus (WT) is one of hallmarks to promote their colonization, but the virulence factor and infectious mechanism involved in this process remain largely unknown. Here, we identified extracellular metalloprotease VvpM as a new virulence factor and investigated the molecular mechanism of VvpM which acts during the regulation of the inflammatory form of macrophage death and bacterial colonization. Mutation of the vvpM gene appeared to play major role in the prevention of IL-1β production due to V. vulnificus infection in macrophage. However, the recombinant protein (r) VvpM caused IL-1β production coupled with necrotic cell death, which is highly susceptible to the knockdown of annexin A2 (ANXA2) located in both membrane lipid and non-lipid rafts. In lipid rafts, rVvpM recruited NOX enzymes coupled with ANXA2 to facilitate the production of ROS responsible for the epigenetic and transcriptional regulation of NF-κB in the IL-1β promoter. rVvpM acting on non-lipid rafts increased LC3 puncta formation and autophagic flux, which are required for the mRNA expression of Atg5 involved in the autophagosome formation process. The autophagy activation caused by rVvpM induced NLRP3 inflammasome-dependent caspase-1 activation in the promoting of IL-1β production. In mouse models of V. vulnificus infection, the VvpM mutant failed to elevate the level of pro-inflammatory responses closely related to IL-1β production and prevented bacterial colonization. These findings delineate VvpM efficiently regulates two pathogenic pathways that stimulate NF-κB-dependent IL-1β production and autophagy-mediated NLRP3 inflammasome via distinct spatial targeting by ANXA2.
Introduction
One of the critical activities during bacterial colonization is killing phagocytes recruited by bacterial pathogens at the inflammation site (Fink and Cookson, 2007). Vibrio vulnificus is an extremely virulent anaerobic Gram-negative marine bacterium that often causes acute inflammatory responses and the killing of phagocytes in the gut (Toma et al., 2010; Lo et al., 2011). However, the underlying cellular mechanisms involved in this process remain largely undescribed. The majority of the virulence effects of V. vulnificus are reported to be derived from secreted toxins that are encoded by cytolytic pore-forming hemolysin (VvhA) (Jeong and Satchell, 2012; Song et al., 2016), multifunctional autoprocessing repeats in the toxin (RTX) (Lee et al., 2007; Jeong and Satchell, 2012), and elastase (VvpE) (Lee et al., 2015a,b, 2016). Several other secreted and cell-associated factors have also been proposed as potential virulence determinants that are also involved in the pathogenesis of V. vulnificus. A 55-kDa zinc-metalloprotease designated as V. vulnificus VvpM is considered to be another major exoprotease that causes cytotoxic effects and an autophagic process affecting intestinal epithelial cells (Lee M. A. et al., 2014, 2015). However, it remains unclear whether VvpM is a functional virulence factor of V. vulnificus specific to the inflammatory form of phagocyte death with the ability to promote bacterial colonization.
Interaction with a distinct host plasma membrane organized into lipid rafts and non–lipid rafts has been proposed as a highly evolved bacterial infectious stratagem to manipulate a wide range of host signaling events, including the inflammatory form of phagocyte death (Manes et al., 2003; Schroeder and Hilbi, 2007). This bacterial infection-induced spatial segregation of proteins into distinct membrane phases has been shown to circumvent specific innate host defenses that are crucial for infection propagation (Manes et al., 2003; Riethmuller et al., 2006). For instance, Helicobacter pylori vacuolating toxins (Fassino et al., 2002) and the entero toxin Clostridium perfringens (van der Goot and Harder, 2001) interact with a detergent-resistant cellular membrane composed of relatively abundant cholesterol as an initial attachment platform, thereby having inflammatory and cytotoxic effects on intestinal physiological functions. Hence, identifying the functional mechanism by which pathogens can evade the immune system by exploiting host-signaling cascades via distinct membrane domains may serve as a novel target for the treatment and/or prevention of infectious diseases.
The stimulation and release of pro-inflammatory cytokines from macrophages are critical steps in the activation of an effective innate host defense and subsequently for the modulation of adaptive immune responses (Netea et al., 2010). Interleukin (IL)-1β, a pro-inflammatory cytokine, is intensely produced by activated macrophages and has a central role in the regulation of many inflammatory forms of cell death typically triggered by inflammasome-mediated caspase-1 activation during bacterial infection (Schroeder and Hilbi, 2007; Lamkanfi et al., 2011; Guo et al., 2015). Aberrant production of active IL-1β from phagocytes is functionally associated with tissue damage and chronic inflammation and therefore should be tightly controlled by the innate host defense system (Lamkanfi et al., 2011). Autophagy is an essential innate host defense mechanism against pathogens that also promotes IL-1β production (Yuan et al., 2012; Kirienko et al., 2015). Although autophagy maintains cellular homeostasis and protects the host cell from harmful stimuli, autophagic cell death occurs when the cell is overwhelmed by infection or when apoptosis is inhibited (Labbe and Saleh, 2008). Notably, it has been shown that autophagy contributes to caspase-independent macrophage death (Xu et al., 2006). This suggests that autophagic cell death of macrophages is a predominant mechanism for controlling cell viability in the absence of apoptosis. On the other hand, several studies have been conducted to determine the factors that regulate IL-1β production, including lipid rafts (Oakley et al., 2009), transcription factors (Lee et al., 2015a), and methylation status (Hashimoto et al., 2009). Specifically, a recent report showed that many enteric bacterial pathogens, such as Salmonella typhimurium (Paesold et al., 2002; Jones et al., 2008), H. pylori (Ki et al., 2008), and enteropathogenic Escherichia coli (EPEC) (Nougayrede and Donnenberg, 2004) can affect a diverse set of epigenetic factors such as DNA methylation and histone modification to regulate the selective activation or silencing of specific host genes (Takahashi, 2014). Thus, studies regarding the process leading to IL-1β production during pathogen-induced macrophage death are likely to be critical to uncovering the bacterial infection mechanisms that promote colonization.
In the present study, therefore, we investigate the molecular mechanism of VvpM action, which drives the transcriptional and epigenetic regulation of the inflammatory process and controls autophagic cell death during the production of IL-1β in RAW 264.7 murine macrophage cells.
Materials and Methods
Chemicals
Fetal bovine serum (FBS) was purchased from BioWhittaker Inc. (Walkersville, MO, USA). The following antibodies were purchased: NOX2, Rac1, and ANXA2 antibodies (BD Biosciences, Franklin Lakes, NJ, USA); NCF1 antibody (LifeSpan Biosciences, Seattle, WA, USA); p-JNK, JNK, PKCαSer 657, p-NF-κBp65, NF-κBp65, IL-1β p17, caspase-1 p10, and β-actin antibodies (Santa Cruz Biotechnology, Paso Robles, CA, USA); Horseradish peroxidase (HRP)-conjugated goat anti-rabbit and goat anti-mouse IgG antibodies (Jackson Immunoresearch, West Grove, PA, USA). 2′, 7′-dichlorofluorescein diacetate (CM-H2DCFDA) was obtained from Invitrogen (Carlsbad, CA, USA). Methyl-β-cyclodextrin (MβCD), lipopolysaccharide (LPS), adenosine 5′-triphosphate disodium salt hydrate (ATP), 3-methyladenine, bafilomycin A1, N-acetyl-l-cysteine (NAC), rotenone, and 5-azacytidine were purchased from Sigma Chemical Company (St. Louis, MO, USA). The concentrations of all of pharmacological inhibitors listed did not show any significant cytotoxic effects by themselves as confirmed by FACS analysis in each experiment. All other reagents were of the highest purity commercially available and were used as received.
Cells
RAW 264.7 murine macrophage and Caco-2 human intestinal epithelial cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured at 37°C in 5% CO2 in RPMI-1640 and DMEM containing 10% FBS and antibiotics. These cell lines have previously been used to evaluate the function of virulence factors of V. vulnificus in regulation of pro-inflammatory process and cytotoxic effects (Lee et al., 2015a; Song et al., 2016).
Host Cells Infection Protocol and IL-1β ELISA
Cells (1 × 106 cells/mL) grown to confluence in 6-well plates were incubated in serum/antibiotic-free media for 24 h. The bacterial suspensions in serum/antibiotic-free media were added to the cells at a multiplicity of infection (MOI) of 1, 5, and 10, after which the infected cells were incubated for 1 h. For IL-1β ELISA, co-culture supernatants were obtained and harvested by centrifugation at 4,000 × g for 10 min at 4°C. This was repeated once more with the resulting supernatant. The clarified supernatants were filtered through a 0.22 μM pore membrane to ensure the removal of any remaining bacterial cells and debris. The IL-1β concentration in the culture medium was quantified by an enzyme-linked immunosorbent assay (ELISA) (R&D systems, Minneapolis, MN, USA) according to the manufacturer's instructions.
Bacterial Strains, Plasmids, and Culture Media
All V. vulnificus strains (MO6-24/O; WT, MO6-24/O ΔrtxA; RTX mut, MO6-24/O vvpE; VvpE mut, MO6-24/O ΔvvhBA; VvhA mut, and MO6-24/O ΔvvpM; VvpM mut) are isogenic and naturally resistant to polymyxin B (Table 1). Unless otherwise noted, V. vulnificus strains were grown in Luria Bertani (LB) medium supplemented with 2.0% (wt/vol) NaCl (LBS) at 30°C. All media components were purchased from Difco (Difco Laboratories Inc., Detroit, MI). V. vulnificus were grown to mid-log phase (A600 = 0.500) corresponding to 2 × 108 CFU/mL and centrifuged at 6,000 × g for 5 min. The pellet was washed with phosphate buffered saline (PBS) and adjusted to desired colony-forming unit (CFU)/mL based on the A600 determined using a UV–VIS spectrophotometer (UV-1800, Shimadzu, Japan) to estimate culture density.
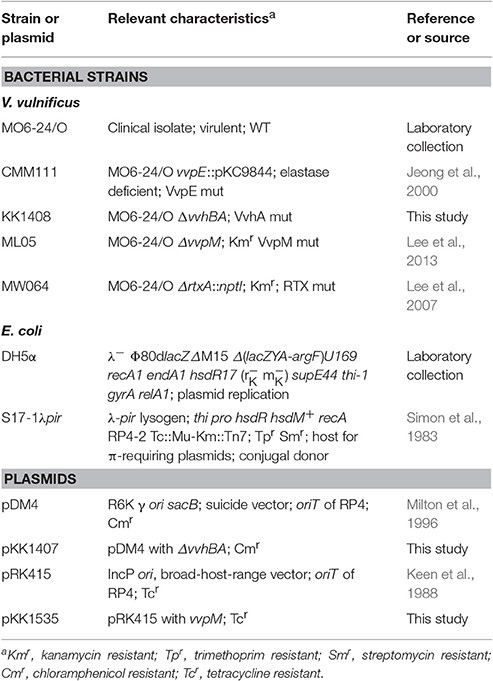
Table 1. Plasmids and bacterial strains used in this study.
Generation of the Exotoxin Mutants
Various deficient mutants in VvpM, RTX, and VvpE genes were generated by the methods as described previously (Jeong et al., 2000; Lee et al., 2007). For generation of VvhA mutant (Milton et al., 1996), the vvhBA operon was inactivated in vitro by deletion of the open reading frame (ORF) of vvhBA (998-bp of 1,935-bp) using the PCR-mediated linker-scanning mutation method as described previously (Jang et al., 2016). Briefly, pairs of primers VVHBA01-F and -R (for amplification of the 5′ amplicon) or VVHBA02-F and -R (for amplification of the 3′ amplicon) were designed and used (Table S1). The vvhBA operon with 998-bp deletion was amplified by PCR using the mixture of both amplicons as the template and VVHBA01-F and VVHBA02-R as primers. The resulting ΔvvhBA was ligated into SpeI-SphI-digested pDM4 (Milton et al., 1996) to generate pKK1407. Escherichia coli S17-1 λpir, tra strain (Simon et al., 1983) containing pKK1407 was used as a conjugal donor to either V. vulnificus MO6-24/O to generate the VvhA mutant KK1408 (Table 1).
Complementation of the VvpM Mutant and Purification of the Recombinant VvpM
To complement the vvpM mutation, an ORF of vvpM was amplified from the genomic DNA of V. vulnificus MO6-24/O by PCR with the primer pair VVPM001F and VVPM001R (Table S1) and then digested with BamHI and SacI. The amplified vvpM ORF was subcloned into the broad-host-range vector pRK415 (Keen et al., 1988) linearized with the same enzymes (Table 1) to result in pKK1535. E. coli S17-1 λ pir, tra strain (Simon et al., 1983) containing pKK1535 was used as a conjugal donor to VvpM mutant. The plasmid pKK1535 was delivered into the VvpM mutant by conjugation as described previously (Jang et al., 2016). Recombinant (r) VvpM protein (Lee M. A. et al., 2014; Lee et al., 2015a) was kindly provided by Prof. Kyu-Ho Lee (Sogang University, Korea). We have further checked the LPS contamination in rVvpM protein by using endotoxin quantitation kit (Pierce® LAL Chromogenic Endotoxin Quantitation Kit, Thermo Fisher Scientific Inc. Waltham, MA, USA). The level of endotoxin in 100 pg/mL of rVvpM was less than 0.003 EU. Thus, we suggest that rVvpM is a purified recombinant protein containing very low level of endotoxin and suitable for our experiments in this study.
RNA Isolation and Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted using the RNeasy Plus Mini Kit (Quiagen, Valencia, CA, USA). Reverse transcription (RT) was carried out with 3 μg of RNA using a Maxime RT premix kit (iNtRON Biotechnology, Sungnam, Korea). β-Actin was used as an endogenous control. The cDNA (5 μL) for Annexins family were amplified.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
The real-time quantifications of pro-inflammatory cytokines and autophagy related genes were performed using a Rotor-Gene 6000 real-time thermal cycling system (Corbett Research, New South Wales, Australia) with a QuantiMix SYBR Kit (PhileKorea Technology, Daejeon, Korea) according to the manufacturer's instructions. β-Actin was used as an endogenous control.
Flow Cytometry
Cells were synchronized in the G0/G1 phase by culture in serum-free media for 24 h before incubation of rVvpM The necrotic cell death was detected with an Annexin V and PI staining kit (BD Biosciences, Franklin Lakes, NJ) according to the manufacturer's instructions. Briefly, the cells were detached with 0.05% trypsin/EDTA and 1 × 105 cells were resuspended with Annexin V binding buffer. And then the cells were stained with Annexin V (25 μg/ml) and PI (125 ng/mL), and incubated for 15 min at room temperature in the dark. The sample was read by flow cytometry and analyzed using CXP software (Beckman Coulter, Brea, CA). Samples were gated to exclude debris (forward light scatter [FSC] area vs. side scatter-area), and then any cell doublets were excluded using FSC-area vs. FSC-width analysis.
Apoptosis/Necrosis Detection
Cell death was also detected with an Apoptosis/Necrosis Detection kit (Abcame Cambridge, MA, USA) according to the manufacturer's instructions. Briefly, the cells were treated with Apopxin Green indicator as a phosphatidylserine marker and 7-aminoactinomycin D (AAD), and were then incubated in the dark for 60 min at room temperature. After the cells were rinsed with ice-cold PBS, the level of cell death was examined using a luminometer (Victor3; Perkin-Elmer, Waltham, MA) and quantified by measuring absorbance at excitation and emission wavelengths of 490 and 525 nm for detection of Apopxin Green Indicator or at excitation and emission wavelengths of 490 and 650 nm for detection of 7-AAD.
Small Interfering (si)RNA Transfection
Cells were grown until 75% of the surface of the plate and transfected for 36 h with ON-TARGETplus siRNAs mixed by 4 different siRNAs specific for ANXA2, cav-1, PKCα, JNK, NF-κBp65, Atg5, and NLRP3 (GE Dharmacon, Lafayette, CO, USA) or non-targeting (nt) siRNA as a negative control (GE Dharmacon, Lafayette, CO, USA) with HiPerFect Transfection Reagent (QIAGEN, Valencia, CA, USA) according to the manufacturer's instructions. The siRNA efficacy for ANXA2, cav-1, PKCα, JNK, NF-κBp65, Atg5, and NLRP3 determined by Western blot was 71, 69, 76, 75, 69, 71, and 61%, respectively (Figure S1).
Detergent-Free Purification of Caveolin-Rich Membrane Fraction
Cells were washed twice with ice-cold PBS, scraped into 2 mL of 500 mM sodium carbonate (pH 11.0), transferred to a plastic tube, and homogenized with a Sonicator 250 apparatus (Branson Ultrasonic, Danbury, CT) using three 20-sec bursts. The homogenate was adjusted to 45% sucrose by the addition of 2 ml 90% sucrose prepared in 2-(N-morpholino) ethanesulfonic acid (MES)-buffered solution consisting of 25 mM MES-buffer solution (pH 6.5) and 0.15 M NaCl and placed at the bottom of an ultracentrifuge tube. A 5–35% discontinuous sucrose gradient was formed (4 ml each of 5 and 35% sucrose, both in MES-buffer solution containing 250 mM sodium carbonate) and centrifuged at 40,000 × g for 20 h in a Beckman SW41 Rotor (Beckman Coulter, Fullerton, CA). Eight fractions were collected and analyzed by 12% SDS-PAGE.
Immunofluorescence Analysis
Cells were fixed in 4% paraformaldehyde in PBS for 10 min at room temperature, permeabilized in 0.1% Triton X-100 in PBS for 5 min, and blocked in PBS containing 5% (v/v) normal goat serum (NGS) for 30 min at room temperature. Samples were then stained with primary antibody for overnight at 4°C. Following three washes with PBS, the samples were incubated with Alexa 488-conjugated goat anti-rabbit/mouse IgM (Invitrogen Co., Carlsbad, CA, USA), and counterstained with PI in PBS containing 5% (v/v) NGS for 2 h. After washing with PBS, samples were mounted on slides and visualized with an Olympus FluoView™ 300 confocal microscope with 400x objective. The expressions of LC3 in immunofluorescence image was quantified by using Image J software (NIH, Bethesda, MD), which measures the stained area per microscopic filed with consistent threshold. On the other hand, the cell numbers showing the membrane translocation of PKCα and the nuclear translocation of p-NF-κB were directly counted per random microscopic filed and converted the numbers to a percentage by multiplying by 100. Ten random fields per coverslip were counted.
Intracellular Reactive Oxygen Species (ROS) Detection
2′, 7′-dichlorofluorescein diacetate (CM-H2DCFDA) and MitoSOX™ Red Mitochondrial Superoxide Indicator (Thermo Fisher Scientific Inc.Waltham, MA, USA) were used to detect the intracellular and mitochondrial ROS production, respectively. To quantify the intracellular ROS levels, the cells treated with 10 mM CM-H2DCFDA were rinsed twice with ice-cold PBS and then scraped. A 100 μL cell suspension was loaded into a 96-well plate and examined using a luminometer (Victor3; Perkin-Elmer, MA, USA) and a fluorescent plate reader at excitation and emission wavelengths of 485 and 535 nm, respectively.
Western Blot Analysis and Subcellular Fractionation
Western blotting was performed as previously described with minor modifications (Lee S. J. et al., 2014). Cells were harvested, washed twice with PBS, and lysed with buffer (20 mM Tris [pH 7.5], 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 1 mg/mL aprotinin, and 1 mM phenylmethylsulfonylfluoride [PMSF]) for 30 min on ice. The lysates were then cleared by centrifugation (22,250 × g at 4°C for 30 min). Equal amounts of protein (20 μg) were resolved by 10~15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride (PVDF) membranes. The membranes were washed with TBST solution (10 mM Tris-HCl [pH 7.6], 150 mM NaCl, and 0.05% Tween-20), blocked with 5% skim milk for 1 h, and incubated with appropriate primary antibody at 4°C for overnight. The membrane was then washed and incubated with a horseradish peroxidase-conjugated secondary antibody for 2 h. The bands were visualized by enhanced chemiluminescence (Amersham Pharmacia Biotech Inc., Buckinghamshire, UK) and detected by using the Bio-rad ChemiDoc™ XRS+ System with images manipulated using the Image Lab™ software #170-8265 (Bio-Rad, Hercules, CA, USA). The subcellular fractionation method for the isolation of membrane and cytosolic proteins were previously reported (Cox and Emili, 2006).
DNA Binding Activity of NF-κBp65
The DNA binding activity of NF-κBp65 was determined by a NF-κBP65 transcription assay kit (Cayman chemical, Ann Arbor, MI) according to the manufacturer's instructions. Briefly, double-stranded DNA (dsDNA) sequence containing NF-κB p65 response element was incubated with 2 mg/ml nuclear extracts. DNA-bound NF-κBp65 complex was captured by an immobilized antibody specific for NF-κBp65 and detected by addition of specific secondary antibody conjugated to HRP. The absorption was measured at 450 nm.
Immunoprecipitation
Interaction of either Rac1 with ANXA2, NCF1, and caveolin-1 or ASC with NLRP3 was analyzed by immunoprecipitation and Western blotting. Cells were lysed with lysis buffer (1% Triton X-100 in 50 mM Tris–HCl pH 7.4 containing 150 mM NaCl, 5 mM EDTA, 2 mM Na3VO4, 2.5 mM Na4PO7, 100 mM NaF, 200 mM microcystin lysine–arginine, and protease inhibitors). Cell lysates (400 μg) were mixed with 10 μg of each antibodies. The samples were incubated for 4 h, mixed with Protein A/G PLUS-agarose immunoprecipitation reagent (Pierce, Rockford, IL, USA) and then incubated for an additional 12 h. The beads were washed four times, and the bound proteins were released from the beads by boiling in SDS-PAGE sample buffer for 5 min.
Chromatin Immunoprecipitation (ChIP)
ChIP was performed using the EZ-ChIP kit (EMD Millipore, Billerica, MA, USA) according to the manufacturer's protocol. Briefly, cells were treated with 1% formaldehyde for 15 min to cross-link proteins to DNA, lysed, and then sonicated. The lysate was incubated with primary antibodies overnight at 4°C. The immunocomplex was purified by incubation with 60 μL of protein G-agarose beads for 1 h and eluted for DNA purification. Quantitative real-time PCR was performed with primers for the IL-1β promoter flanking the putative NF-κBp65 binding sites. The following primer sequences of IL-1β promoter were used (sence and antisence respectively): −237 ~ −56 bp, 5′- TCCACCACGATGA CACACTT -3′ and 5′- GCTGTGAAATTTTCCCTTGG -3′. Anti-RNA polymerase II and normal mouse IgG were used as the positive and negative control for immunoprecipitation, respectively. The human IL-1β promoter sequence was found using the Eukaryotic Promoter Database. The putative binding sites were predicted using Alggen Promo software, version 3.0.2 (Messeguer et al., 2002; Farre et al., 2003).
Methylation Analysis
Genomic DNA from RAW264.7 cells was prepared with the QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA). The extracted DNA was treated with sodium bisulfite using EzWay™ DNA Methylation Detection Kit according to the manufacturer's instructions (KOMABIOTECH, Seoul, Korea). The methylation status of IL-1β gene was determined by methyl-specific PCR (MSP) analysis. We conducted MSP of IL-1β gene promoter containing the −328, −294, and −277 CpG sites due to their close distance to putative NF-κB binding sites. The following primer sequences were used (sense and antisense respectively): Methylated, 5′- TTTTAGTTTAAGTATAAGGAGGCGA-3′ and 5′-ACAC ATTCGCAAATATATCATCGTA-3′; Unmethylated, 5′-TTTTAGTTTAAGTATAAGGAGGTGA-3′ and 5′-ACACATTCACAAATATATCA TCATA -3′.
Transmission Electron Microscopy (TEM)
Cells treated with rVvpM were fixed in Karnovsky's solution and 2% OsO4 in 0.1 M cacodylate at 4°C for 2 h, respectively. The cells were rinsed with distilled water briefly and stained with 0.5% uranyl acetate en bloc solution for overnight. After dehydration in graded ethanol series, samples were infiltrated with Spurr's resin. The samples were imaged by JEM1010 (JEOL, Tokyo, Japan) transmission electron microscope operating at 80 kV.
Inflammasome RT2 Profiler PCR Array
The mouse Inflammasomes RT2 Profiler PCR Array (Qiagen, Valencia, CA, USA) was used to analyze gene expression of inflammasome components and signaling pathways in a cells treated with rVvpM for 24 h according to the manufacturer's instructions. In this array a set of optimized primer assays allows the detection of mRNA transcripts of 84 genes as well as five housekeeping genes in a 96-well plate by real-time PCR. Arrays were performed a minimum of three times for each treatment. Functional gene grouping profiles were provided as available online at http://www.sabiosciences.com/rt_pcr_product/HTML/PAMM-097Z.html#howitwork. PCR array data were analyzed using the GeneGlobe Data Analysis Center on QIAGEN's website at http://www.qiagen.com/kr/shop/genes-and-pathways/data-analysis-center-overview-page/.
Mouse Model and Colonization Assay
All animal procedures were performed following the National Institutes of Health Guidelines for the Humane Treatment of Animals, with approval from the Institutional Animal Care and Use Committee of Seoul National University (SNU-140108-4). Seven-week-old mice (n = 10) were received i.g. inoculation of boiled V. vulnificus (Cont.), V. vulnificus (WT), VvpM mutant, and VvpM complement at 1.3 × 109 CFU/mL for 16 h and sacrificed. Half of ileum tissue was collected, washed, and homogenized. The homogenates of each organ were serially diluted and spread on LB agar containing polymyxin B (100 U/mL). CFUs were normalized to grams of intestinal tissues (CFU/g) to represent superficial bacterial counts. The other half were embedded in O.C.T. compound and stored at −70°C. Samples were then cut into 6-μm-thick frozen sections.
Histologic Damage Score
Frozen sections of tissue samples were subjected to hematoxylin and eosin (H&E) staining for histological examinations. Histological parameters were determined in a blinded fashion by two experienced gastrointestinal pathologists as previously described (Clark et al., 2006). Briefly, scores were assigned as follows: 0 = no damage (normal); 1 = slight submucosal and/or lamina propria separation (mild); 2 = moderate separation of the submucosa and/or lamina propria and/or edema in the submucosa and muscular layers (moderate); 3 = severe separation of the submucosa and/or lamina propria and/or severe edema in the submucosa and muscular layers with regional villous sloughing (severe); or 4 = loss of villi and necrosis (necrosis). Intermediate scores of × 0.0 or × 0.5 were also used to more precisely assign the degree of intestinal damage.
Ileal-Ligated Mouse Model
To determine the functional role of VvpM we performed further experiment by using the ileal-ligated mouse model. Before surgery, mice were fasted for 24 h and anesthetized by intraperitoneal injection of a 2:1 mixture of Zoletil™ (20 mg/kg, Virbac Laboratories, Carros, France) and Xylazine HCl (10 mg/kg, Rompun®, Bayer, Germany). While maintaining the body temperature at 37°C using a heating pad, a small abdominal incision was made and a loop of middle ileum of intestine was isolated by silk suture (2–3 cm in length). The closed ileal loop was instilled with 100 μl of phosphate-buffered saline (PBS) containing WT, vvpM mutant, and vvpM complement at 1.3 × 109 CFU/mL for 2 h. After putting the ileal loop back into the peritoneal cavity, the cavity was closed with suture. At 2 h after the inoculation of both strains, the mice were sacrificed and the intestinal loops were removed for real-time PCR and the hematoxylin and eosin (H&E) staining.
Statistical Analysis
Results are expressed as means ± standard errors (S.E.). All experiments were analyzed by ANOVA, followed in some cases by a comparison of treatment means with a control using the Bonferroni-Dunn test. Differences were considered statistically significant at P < 0.05.
Results
V. vulnificus Metalloprotease VvpM Induces IL-1β Production Coupled with Necrotic Cell Death
V. vulnificus (WT) secretes functional virulence factors, such as multifunctional autoprocessing repeats in the toxin (RTX) (Lee et al., 2007; Jeong and Satchell, 2012), elastase (VvpE) (Lee et al., 2015a,b, 2016), cytolytic pore-forming hemolysin (VvhA) (Jeong and Satchell, 2012; Song et al., 2016), and zinc-metalloprotease (VvpM) (Lee M. A. et al., 2014; Lee et al., 2015a). RAW 264.7 murine macrophage cells were exposed to V. vulnificus (WT) and various mutants deficient in RTX, VvpE, VvhA, and VvpM genes. A robust increase in the IL-1β mRNA expression level were observed 3 h after incubation with 1 MOI of WT (Figure 1A). WT also stimulated the expression of both pro-IL-1β and cleaved IL-1β (Figure 1B), and the secretion of IL-1β into the cell media (Figure 1C). Interestingly, LPS as a positive control has similar ability with WT in the induction of IL-1β expression and the release. However, the level of IL-1β was far less evident in cells infected with the VvpM mutant as compared to cells infected with other WT mutants. Among the various pro-inflammatory cytokines, the expression of IL-1β appeared to be highly susceptible to VvpM mutant infection (Figure 1D). To determine the direct effect of VvpM, cells were exposed to various concentrations (0~200 pg/mL) of the recombinant protein (r) VvpM purified by V. vulnificus for 24 h. A significant increase in IL-1β expression was observed after 3 h of incubation with 100 pg/mL of rVvpM (Figure 1E). rVvpM preferentially increased the level of IL-1β mRNA among the mRNA amplicons of pro-inflammatory cytokines (Figure 1F). Interestingly, an increase in the active form of IL-1β (17 kDa) for 12 h (Figure 1G) and its secretion into the cell media for 24 h (Figure 1H) were also observed in a time-dependent manner after treatment with rVvpM in both RAW 264.7 cells and Caco-2 human intestinal epithelial cells. We determined the functional role of rVvpM to induce cell death by means of flow cytometric analyses over a period of 24 h (Figure 1I). rVvpM significantly induced the necrosis (an 8.5 ± 0.3-fold increase compared to a vehicle) of the cells, whereas for apoptotic cell death, a marginal effect was noted (a 1.3 ± 0.2-fold increase compared to the vehicle). We further determined the functional role of rVvpM to induce cell death by staining with annexin V for apoptotic cells, PI for necrotic cells, and CytoCalcein for live cells. Consistent with our data for flow cytometric analyses, we found that rVvpM is essential for triggering the necrotic cell death rather than the apoptosis (Figure S2). We further confirmed the necrosis-promoting effect of rVvpM using another reagent that monitors necrotic cells with 7-aminoactinomycin D. As shown in Figure S3A, we found that rVvpM shows greater stimulatory potency on necrotic cell death than on apoptotic cell death, confirming that rVvpM is essential for triggering necrotic macrophage death rather than apoptosis. In addition, the levels of IL-1β and necrotic cell death remained unchanged after treatments with identical amounts of trypsin, boiled rVvpM, or boiled WT for 12 h (Figure S3B), suggesting that the functional roles of rVvpM during the processes of IL-1β production and necrotic cell death differ from the outcome of the general proteolysis of surface proteins or contamination by the bacterial product. Thus, these results indicate that rVvpM is responsible for triggering necrotic macrophage death during V. vulnificus infection and that it evokes a pro-inflammatory response via mainly IL-1β production.
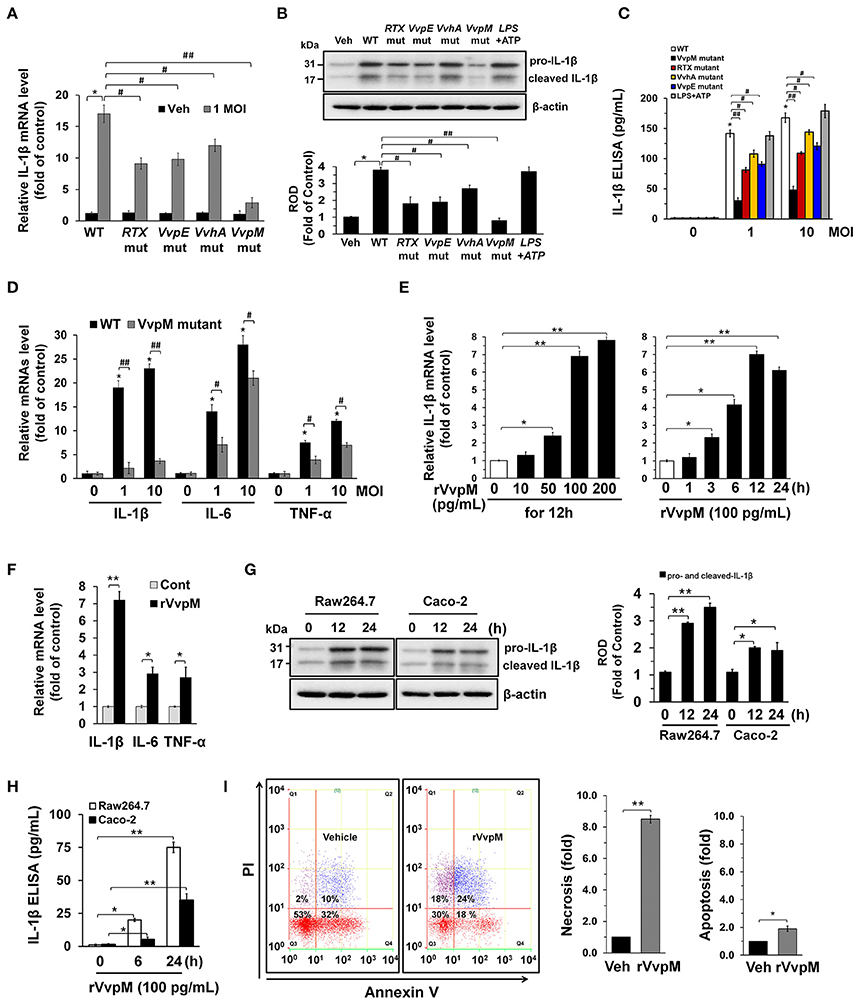
Figure 1. V. vulnificus metalloprotease VvpM induces IL-1β production coupled with necrotic cell death. The expression (A), mature form (B), and production (C) of IL-1β in RAW 264.7 murine macrophage cells infected with V. vulnificus (WT), various mutant deficients in RTX, VvpE, VvhA, and VvpM genes, and LPS (10 μg/mL) + ATP (1 mM) for 3 h are shown. Data represent the means ± S.E. (n = 5). (D) The expression of IL-1β, IL-6, and TNF-α in a cells infected with WT or VvpM mutant for 3 h is shown. n = 4. (E) Dose and time responses of rVvpM for 24 h in IL-1β mRNA expression are shown. n = 4. (F) The expression of IL-1β, IL-6, and TNF-α in a cells treated with rVvpM (100 pg/mL) for 12 h is shown. n = 3. (G) The levels of IL-1β mature form in RAW 264.7 and Caco-2 human intestinal epithelial cells treated with rVvpM (100 pg/mL) for 12 h was determined by Western blot. n = 3. (H) The level of IL-1β production in cell treated with rVvpM for 24 h was quantified by ELISA. n = 4. (I) Cells were incubated with rVvpM for 24 h. Percentages of necrosis, survival, and apoptosis were measured by using PI/Annexin V staining and flow cytometry (left panel). PI/Annexin V cells (Q1) were considered necrotic, PI/Annexin V double-positive cells (Q2) were considered late apoptotic, PI/Annexin V cells (Q3) were considered alive, and were PI/Annexin V cells (Q4) were considered early apoptotic. Quantitative analysis of the fold changes of necrotic (Q1) and apoptotic (Q2+Q4) cells by FACS analysis is shown (right panel). n = 3. *P < 0.01 vs. Veh (boiled rVvpM). (A–D) *P < 0.001 vs. Veh (boiled WT). #P < 0.01 and ##P < 0.001 vs. WT, respectively. (E–H) *P < 0.01 and **P < 0.001 vs. cells with no treatment, respectively. ROD, relative optical density.
VvpM Facilitates the Lipid Raft-Mediated Clustering of ANXA2
It is well established that annexin A2 (ANXA2) plays a critical role as a host mediator of bacterial pathogens and is essential in the pathogenesis of Enteropathogenic E. coli infections (Zobiack et al., 2002). We have previously reported that exotoxins produced by V. vulnificus contributes to their pathogenesis in multiple ways by interacting with intestinal proteins, including ANXA2 and ANXA2. However, our previous data have revealed that ANXA2, but not ANXA4 plays a critical role as a membrane mediator of V. vulnificus and that it has unique biological properties responsible for host cell death coupled with an inflammatory response (Lee et al., 2015a). We initially determined the existence of ANXA isotypes in RAW264.7 cells. ANXAs are expressed in the order of ANXA2, ANXA3, ANXA5>ANXA1, ANXA4, ANXA6 (Figure S4). The expressions of ANXA7-13 were not detected in these cell lines. Interestingly, the knockdown of ANXA2 resulted in a reduction of necrotic cell death (Figure 2A) and IL-1β production (Figure 2B), as elicited by rVvpM. In attempting to define the membrane location of functional ANXA2, our data revealed that the lipid raft markers caveolin-1 and flotillin-2 were found in mainly fraction 4, whereas ANXA2 was highly enriched both in the lipid raft parts (fraction 4) and the non-lipid raft parts (fractions 6–8) (Figure 2C). Moreover, the subunits of the NADPH oxidase (NOX) enzymes, NOX2 (gp91phox) and NCF1 (p47phox), and a cytosolic component of the NOX family, Rac1, were enriched in fraction 4. Given that caveolin-1 is constitutively expressed in lipid rafts to maintain cell homeostasis forming caveolin-1-mediated caveoli (Li et al., 2007), it is possible that individual lipid rafts attached with caveolin-1 are present in fraction 4. These individual lipid rafts were known to be dynamic microdomains and carry several membrane-bound or attached proteins or enzymes such as G-proteins, protein kinases, and NOX enzymes (Li et al., 2007). Interestingly, an increase in the level of ANXA2 together with caveolin-1, but not flotillin-2, appeared in fraction 3 after incubation with 100 pg/mL of rVvpM compared to the vehicle alone, suggesting that rVvpM enhances the recruitment of ANXA2 into a caveolin-1-enriched component. In addition, a treatment with rVvpM resulted in the translocations of NOX2, NCF1, and Rac1 into fraction 3 including the caveolin-1-enriched component. This indicate that rVvpM stimulates the movement and clustering of individual lipid rafts without flotillin-1 to form a number of lipid rafts macrodomains or platforms with aggregation or recruitment of receptors, NOX enzymes, and other proteins such as Rac1 at fraction 3. Regarding the exception of flotillin-1 move in in the cells activated by rVvpM, it reported that flotillin-1 is independently regulated and does not strictly correlate with the expression patterns of caveolin family members (Volonte et al., 1999). Indeed, it reported that the distribution patterns of flotillin 1 did not change upon cholesterol removal (Kaakinen et al., 2012). Therefore, our result in the present study indicate that rVvpM initiates the organization of rafts and the spatial distribution of caveolin which are crucially dependent on lipid raft clustering in fraction 3. The effect of rVvpM on the membrane location of ANXA2 was further visualized by staining the ANXA2 and the lipid raft marker molecule cholera toxin subunit B (CTB). As shown in Figure 2D, rVvpM significantly increased the co-localization of CTB with ANXA2. In addition to ANXA2, CTB significantly co-localized with caveolin-1 and Rac1 in the cells treated with rVvpM. Because the above approaches are qualitative at best, we also attempted to quantify the results by means of the co-immunoprecipitation of Rac1 with proteins related to the lipid rafts in the presence of rVvpM. It was noted that Rac1 co-immunoprecipitated with ANXA2, caveolin-1, and NCF1, and importantly, that these interactions were enhanced by the rVvpM treatment (Figure 2E). In good agreement with the above results, the knockdown of caveolin-1 significantly abrogated rVvpM-induced necrosis (Figure 2F) and the production of IL-1β (Figure 2G). These findings indicate that rVvpM facilitates the lipid-raft-mediated clustering of ANXA2 and NOX enzymes to trigger IL-1β production coupled with necrotic macrophage death.
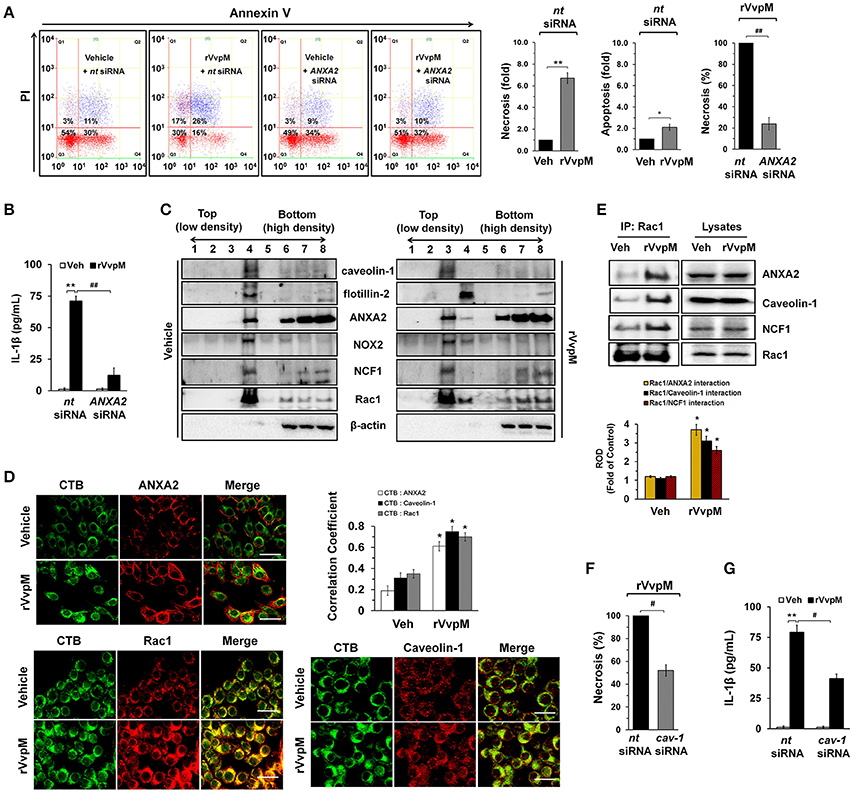
Figure 2. VvpM facilitates the lipid raft-mediated clustering of ANXA2. (A) Cells transfected with siRNAs for non-targeting (nt) control and ANXA2 were incubated with rVvpM for 24 h. FACS analysis and quantitative analysis of the percentage of necrotic (Q1) cells are shown. Data represent the means ± S.E. (n = 4). (B) The level of IL-1β protein was quantified by ELISA. n = 5. (C) Caveolin-enriched membrane fractions were prepared by discontinuous sucrose density gradient fractionation from the cell treated with rVvpM for 30 min, and the location of caveolin-1, flotillin-2, ANXA2, NOX2, NCF1, and Rac1 was determined by Western blot. n = 3. (D) The increased co-localization of CTB (green) with ANXA2 (red), caveolin-1 (red), and Rac1 (red) was determined by confocal microscopy using immunofluorescence staining. Scale bars, 100 μm (magnification, ×400). n = 3. Graphs represent Pearson's coefficient of colocalization of CTB and ANXA2, CTB and Caveolin-1, and CTB and Rac1 from 10 independent fields. *P < 0.05. (E) Rac1 co-immunoprecipitated with ANXA2, caveolin-1, and NCF1 is shown in the left side. Expression of ANXA2, caveolin-1, NCF1 and Rac1 in total cell lysates is shown in the right side. n = 4. *P < 0.01 vs. Veh (boiled rVvpM). ROD, relative optical density. (F) Cells transfected with nt siRNA and cav-1 siRNA were incubated with rVvpM for 24 h. FACS analysis and quantitative analysis of the percentage of necrotic (Q1) cells are shown; n = 4. (G) The level of IL-1β protein was quantified by ELISA. n = 5. (A,B,F,G) *P < 0.01 and **P < 0.001 vs. nt siRNA + Veh (boiled rVvpM), respectively. #P < 0.05 and ##P < 0.01 vs. nt siRNA + rVvpM, respectively.
VvpM Induces ROS Production and PKCα Phosphorylation
We subsequently determined whether rVvpM regulates the production of ROS with regard to the functional role of the lipid raft-mediated clustering of NOX enzymes. We found that rVvpM significantly increased ROS production between 10 and 60 min after incubation with 100 pg/mL of rVvpM compared to the vehicle alone (Figure 3A). The ROS production induced by rVvpM at 30 min was significantly inhibited by the knockdown of caveolin-1 and ANXA2 and by a treatment with the positive control N-acetylcysteine (NAC) as an antioxidant (Figure 3B). Despite the significant role of rVvpM in promoting autophagy induction (Lee M. A. et al., 2015), the autophagy inhibitor 3-methyladenine (3-MA) did not affect rVvpM-induced ROS production, suggesting that the signaling events related to ROS production are unrelated to the characteristics of autophagy. Moreover, we found that mitochondrial complex I inhibitor, rotenone (0.1 μM) has a very weak inhibitory effect on ROS production induced by rVvpM for 30 min. Consistently, rVvpM did not show any significant effect on mitochondiral ROS production (Figure S5). These results indicate that epithelial ROS generated by clustering of NADPH oxidase enzymes within lipid rafts facilitate the production of ROS, which results in a prominent amplification of the transmembrane signal at short time period. Importantly, a prominent increase in ROS production was also observed in cells infected with WT (Figure 3C). In contrast, while mutants deficient in VvpM and RTX significantly decreased the ability of WT to induce ROS production, macrophages infected with VvpE and VvhA mutants revealed only a modest defect. Moreover, the instances of necrotic cell death (Figure 3D) and IL-1β production (Figure 3E) induced by rVvpM were significantly blocked by a pre-treatment with NAC. These results indicate that ROS production is required for VvpM to promote IL-1β production coupled with necrotic macrophage death. ROS produced by the clustering of NADPH oxidase enzymes results in a prominent amplification of the transmembrane signal through the activation of PKC (Wu et al., 2008; Zhang et al., 2010). rVvpM significantly induced pan-PKC phosphorylation between 30 and 60 min (Figure 3F). In an experiment to identify the specific PKC isotypes, the translocation of PKCα, but not PKCδ or PKCζ, from the cytosol to the membrane compartment was observed after cells were treated with rVvpM for 30 min (Figure 3G). Importantly, the increase in PKCα phosphorylation was significantly blocked by a pretreatment with a ROS scavenger, NAC, but not by 3-MA (Figure 3H), indicating that PKCα phosphorylation is a downstream factor of ROS production induced by rVvpM acting on the membrane lipid raft, which is not affected by autophagy. The membrane expression of PKCα phosphorylation was further confirmed by immunofluorescence staining in rVvpM-treated RAW264.7 cells (Figure 3I). Not surprisingly, while WT significantly increases the level of PKCα phosphorylation, the VvpM mutant showed the most similar level to that noted in control mice compared to the other mutant forms (Figure 3J), indicating that PKCα phosphorylation mediated by ROS is a more efficient infectious mechanism of VvpM as compared to RTX. It was noted that rVvpM induces phosphorylation of PKCα at Ser 657 site responsible for PKCα autophosphorylation that leads to stabilization and maturation of the PKCα. In addition, rVvpM significantly stimulated calcium influx, which was enhanced by A23187 as a positive control (Figure S6A). Interestingly, the silencing of PKCα by PKCα siRNA significantly blocked the necrotic cell death (Figure 3K) and IL-1β production (Figure 3L) induced by rVvpM. Given that previous result showing the PKC activation also able to stimulate the ROS production via phosphorylation of NOX subunits (Cosentino-Gomes et al., 2012), we have further examined whether PKCα regulates ROS production in RAW264.7 cells treated with rVvpM. However, the knockdown of PKCα failed to regulate ROS production induced by rVvpM (Figure S6B), PKCα is downstream factor of ROS during necrotic macrophage death induced by rVvpM. Together, these results indicate that rVvpM coupling with ROS production uniquely stimulates atypical PKC activation to promote IL-1β production and necrotic macrophage death.
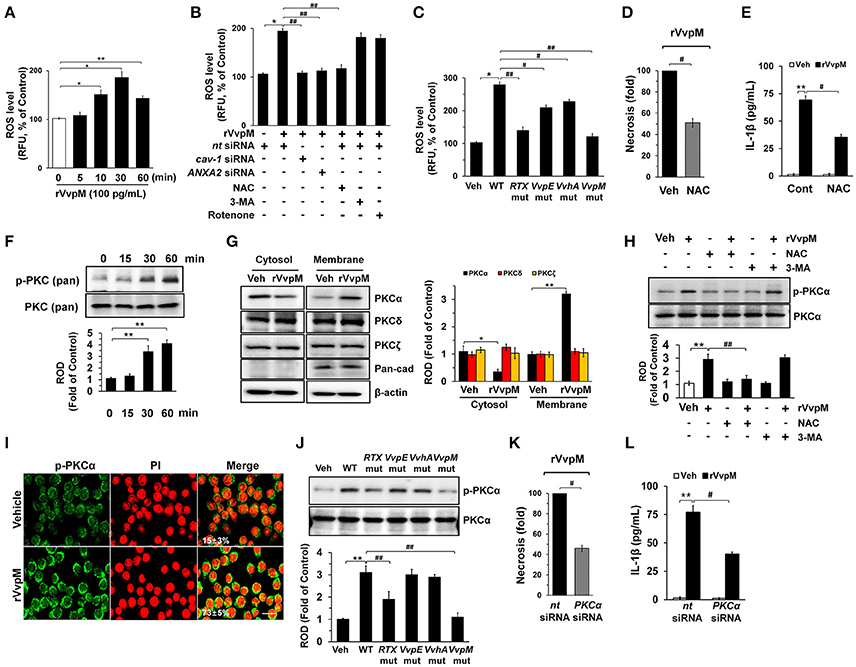
Figure 3. VvpM induces ROS production and PKCα phosphorylation. (A) Time responses of rVvpM in ROS production is shown. n = 4. (B) Cells were transfected with siRNAs for cav-1 and ANXA2 for 24 h or pre-treated with NAC (10 μM), 3-MA (10 mM), and rotenone (0.1 μM) for 30 min prior to rVvpM exposure for 30 min. n = 5. (C) Cells infected with WT or various mutant deficients for 30 min is shown. n = 4. (D) Cell were pre-treated with NAC (10 μM) for 30 min prior to rVvpM exposure for 24 h. FACS analysis and quantitative analysis of the percentage of necrotic (Q1) cells are shown. n = 4. (E) The level of IL-1β protein was quantified by ELISA. n = 5. (F) Phosphorylation of pan-PKC is shown. n = 3. (G) Membrane translocation of PKC isoforms in cells treated with rVvpM for 30 min is shown. Pan-cadherin was used as a membrane control. n = 3. (H) Phosphorylation of PKCα in cells pre-treated with NAC and 3-MA for 30 min prior to rVvpM exposure for 30 min is shown. n = 3. (I) Expression of PKCα (green) was determined by confocal microscopy. The cell numbers showing membrane translocalization of PKCα per microscopic filed were directly counted and converted to a percentage by multiplying by 100. Ten random fields per coverslip were counted. Data represent the mean ± S.E. n = 3. (J) Phosphorylation of PKCα in a cells infected with WT or various mutant deficients for 30 min is shown. n = 4. (K) Cells transfected with PKCα siRNA were incubated with rVvpM for 24 h. FACS analysis and quantitative analysis of the percentage of necrotic (Q1) cells are shown. n = 4. (G) The level of IL-1β protein was quantified by ELISA. n = 5. (A,F) *P < 0.01 and **P < 0.001 vs. cells with no treatment, respectively. (B,K,L) *P < 0.05 and **P < 0.01 vs. Veh (boiled WT), respectively. #P < 0.05 and ##P < 0.01 vs. nt siRNA + rVvpM, respectively. (C,J) *P < 0.05 and **P < 0.01 vs. Veh (boiled WT), respectively. #P < 0.05 and ##P < 0.01 vs. WT, respectively. (D,E) **P < 0.01 vs. Veh (boiled rVvpM). #P < 0.05 and ##P < 0.01 vs. Veh + rVvpM, respectively. (G,H) *P < 0.01 and **P < 0.001 vs. Veh (boiled rVvpM), respectively. ##P < 0.05 vs. rVvpM alone. ROD, relative optical density; RFU, relative fluorescence units.
VvpM Induces the Activation of JNK and NF-κBp65 to Promote IL-1β Expression
Many bacterial stimuli regulate the MAPKs and NF-κB pathways, which are interesting candidates as downstream mediators of ROS and PKC. rVvpM increased the phosphorylation of JNK between 15 and 60 min but does not affect the phosphorylation of ERK or p38 MAPK (Figure 4A), and its effect at 30 min is inhibited by the silencing of PKCα, but not by 3-MA (Figure 4B). In addition, the silencing of JNK by siRNA has a significant inhibitory effect on the necrosis (Figure 4C) and production of IL-1β (Figure 4D) in RAW264.7 cells. rVvpM also induced NF-κBp65 phosphorylation between 60 and 120 min (Figure 4E). Treatment with JNK siRNA but not 3-MA significantly blocked rVvpM-induced NF-κBp65 activation (Figure 4F). The increased accumulation of NF-κBp65 phosphorylation in the nucleus was further confirmed by immunofluorescence staining and counter-labeling with propidium iodide (PI) (Figure 4G). In agreement with this, the necrosis (Figure 4H) and production of IL-1β (Figure 4I) induced by rVvpM were significantly blocked by the knockdown of NF-κBp65. The effect of rVvpM on the activation of JNK and NF-κBp65 was further confirmed in these cells following infection with WT and various deficiency mutants (Figure 4J). WT significantly induced the activation of JNK and NF-κBp65, whereas for the VvpM mutant, only a marginal effect was noted, and the cells appeared to be highly susceptible to JNK among the various mutants tested here, although all four deficiency mutants caused significant defects in the ability of WT to activate NF-κBp65. Consistently, our results have revealed that all mutations have inhibitory effects on transcription activity of NF-κB induced by WT in RAW 264.7 and Caco-2 cells (Figures S7A,B). The data above provide important evidence that JNK-mediated NF-κBp65 phosphorylation is required for the regulation of IL-1β production and necrotic cell death as evoked by rVvpM. In addition, the phosphorylation of JNK and NF-κB induced by rVvpM was significantly inhibited by a treatment with a ROS scavenger N-acetylcysteine (NAC) (Figure S7C), suggesting that ROS induced by rVvpM plays key role in activation of PKC, JNK, and NF-κB in the promotion of necrotic macrophage death
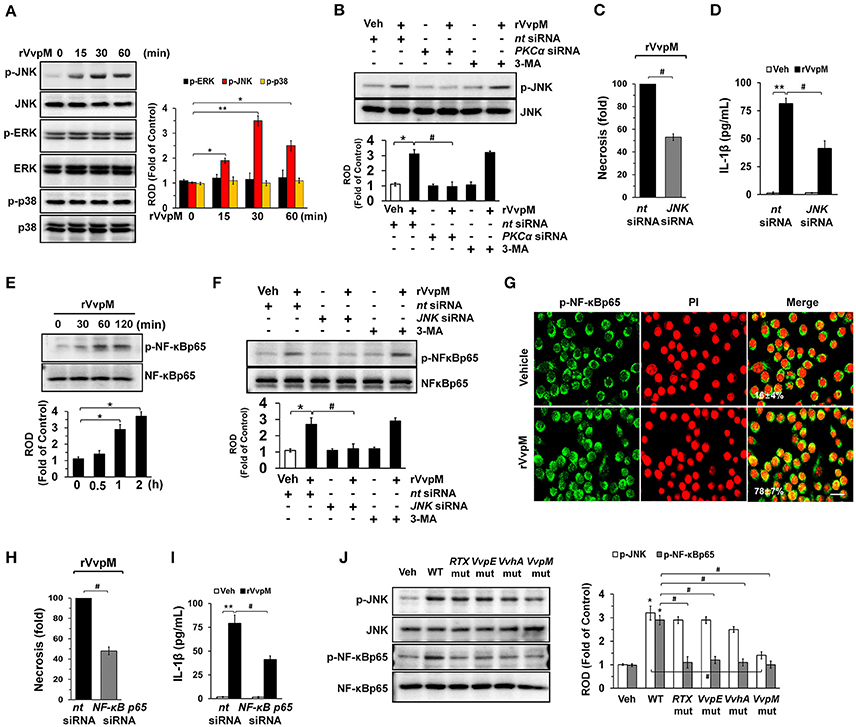
Figure 4. VvpM induces the activation of JNK and NF-κBp65 to promote IL-1β expression. (A) The effect of rVvpM on the expression of MAPK was determined by western blot. n = 3. (B) Cells were transfected with PKCα siRNA for 24 h prior to rVvpM exposure for 30 min. n = 3. (C) Cells were transfected with JNK siRNA for 24 h prior to rVvpM exposure for 24 h. FACS analysis and quantitative analysis of the percentage of necrotic (Q1) cells are shown. n = 4. (D) The level of IL-1β protein was quantified by ELISA. n = 5. (E) Time responses of rVvpM in the phosphorylation of NF-κBp65 are shown. n = 3. (F) Cells were transfected with JNK siRNA for 24 h or pre-treated with 3-MA (10 mM) for 30 min prior to rVvpM exposure for 60 min. n = 3. (G) p-NF-κBp65 (green) was detected by confocal microscopy. The cell numbers showing nuclear translocalization of p-NF-κBp65 per microscopic filed were directly counted and converted to a percentage by multiplying by 100. Ten random fields per coverslip were counted. Data represent the mean ± S.E. Scale bars, 100 μm. n = 3. (H) Cells transfected with NF-κBp65siRNA were incubated with rVvpM for 24 h. FACS analysis and quantitative analysis of the percentage of necrotic (Q1) cells are shown. n = 4. (I) The level of IL-1β protein was quantified by ELISA. n = 5. (J) Phosphorylation of JNK and NF-κBp65 in a cells infected with WT or various mutant deficients for 30 min are shown. n = 4. (A,E) *P < 0.05 and **P < 0.01 vs. cells with no treatment, respectively. (B–D,F,H,I) *P < 0.01 and **P < 0.001 vs. nt siRNA + Veh (boiled rVvpM), respectively. #P < 0.01 vs. nt siRNA + rVvpM. (J) *P < 0.01 vs. Veh (boiled WT). #P < 0.01 vs. WT, ROD; relative optical density.
To determine the role of NF-κB in the transcriptional regulation of IL-1β mRNA expression, we conducted chromatin immunoprecipitation (ChIP) assays followed by qRT-PCR in cells treated with rVvpM. With ALGGEN PROMO (Messeguer et al., 2002; Farre et al., 2003), we found that the IL-1β promoter located between the transcription start site and the point–500 bp upstream contains two putative NF-κB binding sites and six methylation sites. Test results also found a primer that includes two putative NF-κB binding sites at the region proximal −56 bp to −237 bp of the start site of the IL-1β promoter (Figure 5A). Figure 5B shows that our primer sets result in an amplicon from the anti-phospho-NF-κBp65 immunoprecipitates and, importantly, that the interaction between NF-κB and the IL-1β promoter was enhanced by a rVvpM treatment for 60 min. Interestingly, however, the level of NF-κBp65 binding to the IL-1β promoter was significantly inhibited by the silencing of ANXA2 and JNK (Figure 5B) but not by treatments with 3-MA or NLRP3 siRNA, which is a critical regulator of IL-1β production (Lamkanfi et al., 2011; Guo et al., 2015; Figure 5C). The ability of rVvpM to induce interaction between NF-κB and the IL-1β promoter is consistent with the data from a real-time PCR analysis (Figure 5D). These results indicate that rVvpM acting on the lipid raft-mediated clustering of ANXA2 transcriptionally regulates the NF-κBp65 binding to the IL-1β promoter via JNK activation. Subsequently, we postulated that rVvpM may affect methylation, which negatively regulates the interaction between NF-κB and the IL-1β promoter. We attempted to analyze the methylation status of the IL-1β gene promoter containing the −328, −294, and −277 CpG sites due to their close proximity to putative NF-κB binding sites. As shown in Figure 5E, a primer pair including these CpG sites that specifically amplified either the methylated or unmethylated form of the IL-1β promoter produced 203 bp methylated bands from vehicle-treated RAW264.7 cells, indicating that the IL-1β promoter was methylated in these alleles. However, treatment with 100 pg/mL of rVvpM for 60 min markedly inhibited the level of IL-1β promoter methylation, which was significantly blocked by the knockdown of ANXA2 and JNK and by a pre-treatment with NAC. As expected, treatment with 3-MA and NLRP3 siRNA failed to regulate the methylation status of the IL-1β gene promoter induced by rVvpM. These results were consistent with the data from a real-time PCR analysis quantified by the relative value of CpG methylation compared to an unmethylated form in the IL-1β promoter (Figure 5F). Interestingly, the levels of necrotic cell death (Figure 5G) and the production of IL-1β (Figure 5H) were markedly enhanced by a pre-treatment with the DNA methylation inhibitor 5-azacytidine (5-aza). These results suggest that the hypomethylation of the IL-1β promoter is able to affect the high level of the interaction between NF-κB and the IL-1β promoter during necrotic macrophage death as evoked by rVvpM.
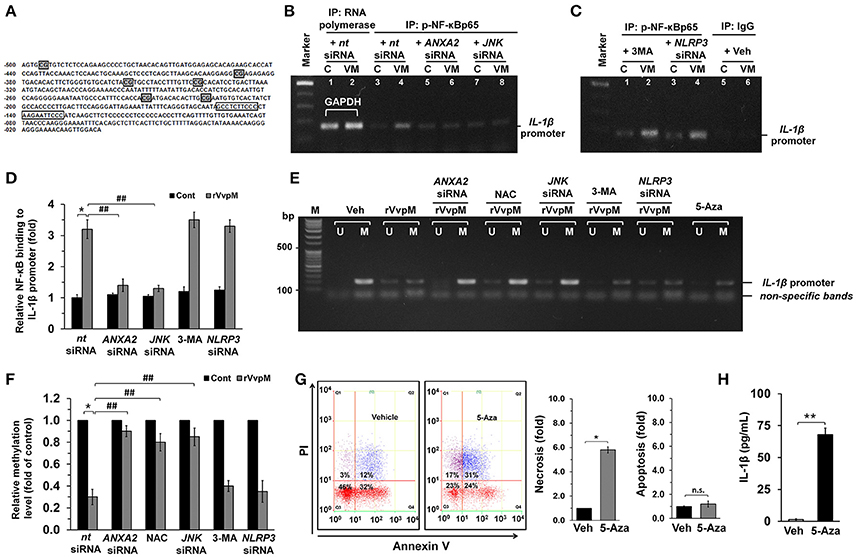
Figure 5. Regulatory effect of VvpM on the interaction of NF-κB with the IL-1β promoter. (A) The NF-κB binding sites and methylation site in the IL-1β promoter are shown. The open boxes and shaded box represent the consensus sequences of NF-κB and the methylation site, respectively. (B,C) Cells were transfected with siRNAs for ANXA2, JNK, and NLRP3 for 24 h or pre-treated with 3-MA (10 mM) for 30 min prior to rVvpM exposure for 60 min. Cells were then fixed with formaldehyde, and cell extracts were immunoprecipitated (IP) with anti-p-NF-κBp65 to detect bound IL-1β promoter DNA fragments. A representative agarose gel following RT-PCR is shown. n = 3. Normal mouse IgG or anti-RNA polymerase II antibody was used as negative or positive control for the ChIP, respectively. C, control. VM, rVvpM. (D) The level of p-NF-κBp65 binding to IL-1β promoter was quantified by qRT-PCR. Anti-RNA polymerase II antibody was used internal control. n = 3. (E) Genomic DNA of the cells transfected with siRNAs for ANXA2, JNK, and NLRP3 for 24 h or pre-treated with NAC (10 μM), 3-MA (10 mM), and 5-aza (1 μM) for 30 min prior to rVvpM exposure for 60 min was prepared. Changes in the IL-1β promoter methylation status were determined by MSP analysis. U, unmethylated form. M, methylated form. (F) The level of IL-1β methylation was quantified by qRT-PCR. Relative level of IL-1β methylation is shown, compared to the unmethylated form. n = 4. (G) Cell were treated with 5-aza (1 μM) for 24 h. FACS analysis (left panel) and quantitative analysis of the fold changes of necrotic (Q1) and apoptotic (Q2+Q4) cells by FACS analysis is shown (right panel). n = 4. (H) The level of IL-1β protein was quantified by ELISA. n = 5. (D,F) *P < 0.01 vs. nt siRNA + Veh (boiled rVvpM). ##P < 0.01 vs. nt siRNA + rVvpM. (G,H) *P < 0.01 and **P < 0.001 vs. cells with no treatment, respectively.
VvpM Induces Autophagy by Regulating Non-lipid Raft ANXA2
Despite the independent roles of autophagy and inflammasome in regulating the ROS-PKCα-JNK-NF-κBp65 pathway through the lipid-raft-mediated clustering of ANXA2, we revisited the issue of whether rVvpM has the ability to regulate the autophagic process and inflammasome formation due to the fundamental parts of these processes play role in regulating the production of IL-1β during bacterial infection. Importantly, while the expressions of LC3-II and Beclin-1 were increased by a treatment with rVvpM in a time-dependent manner, the level of p62 was attenuated (Figure 6A), indicating that rVvpM has the ability to regulate the autophagic process. We also confirmed through confocal microscopy that rVvpM increases autophagic vesicle formation, during which a significant increase in LC3 puncta positive cells was observed (Figure 6B). Comparing the functional roles of VvpM with those of other virulence factors which evoke the autophagic responses, we found that WT markedly induced the expression levels of LC3-II and Beclin-1 while inhibiting that of p62 (Figure 6C). In contrast, cells infected with the VvpM mutant among all cells infected with the mutants tested here exhibited the lowest LC3-II level similar to that of the control, suggesting that VvpM is a major virulence factor responsible for autophagy induction. Moreover, the functional effects of the VvpM mutant in LC3 activation were significantly restored by the complementation of the VvpM mutant with a functional VvpM gene (Figure S8). A unique feature of autophagosomes characterized by the peculiar double-membraned vesicle was also observed in cells treated with rVvpM by transmission electron microscopy (TEM), where rVvpM caused significant abnormalities such as swollen mitochondria while also widening the endoplasmic reticulum (ER) (Figure 6D). To confirm the enhanced autophagic flux, we used bafilomycin A1 (BafA1) which is a known inhibitor of V-ATPase (vacuolar-type H+ ATPase) that prevents the maturation of autophagic vacuoles. rVvpM significantly enhanced the accumulation of LC3-II in cells pretreated with BafA1 (Figure S9), suggesting that the lysosomal degradation pathway was not inhibited by rVvpM. Importantly, the autophagy inhibitor 3-MA significantly blocked necrotic cell death (Figure 6E) and IL-1β production (Figure 6F) as induced by rVvpM. This indicates that rVvpM differentially regulates IL-1β production through the induction of autophagic macrophage death. It was unclear as to whether the activation of two different signaling pathways for the regulation of IL-1β production reflects the selective interaction between VvpM and the lipid raft. We focused on the fact that ANXA2 was also highly enriched in the non-lipid raft parts of the cell membrane. Notably, the level of LC3-II evoked by rVvpM was markedly inhibited by the knockdown of ANXA2 with ANXA2 siRNA (Figure 6G). However, the knockdown of caveolin-1 (Figure 6H) and treatment with the lipid raft sequester MβCD failed to regulate the level of LC3-II in cells treated with rVvpM (Figure S10). These results indicate the ANXA2 in the non-lipid raft parts is functionally relevant for autophagy induction and that it distinctively regulates necrotic macrophage death and the pro-inflammatory response as a host mediator of rVvpM. To gain insight into how ANXA2 influences the autophagic process via the non-lipid raft parts, we further determined the effect of rVvpM on the expression of mRNA for induction, expansion/closure, and fusion/degradation in an autophagy system (Figure 6I). The prominent expressions of Atg5 and Atg16L1 for autophagy expansion/closure were observed following an rVvpM treatment with a trend toward increased mRNA expressions in an overall autophagy system. Intriguingly, the knockdown of Atg5 in RAW264.7 cells reduced the levels of necrotic cell death (Figure 6J) and IL-1β production (Figure 6K), suggesting the involvement of Atg5 in IL-1β production coupled with autophagic macrophage death elicited by rVvpM.
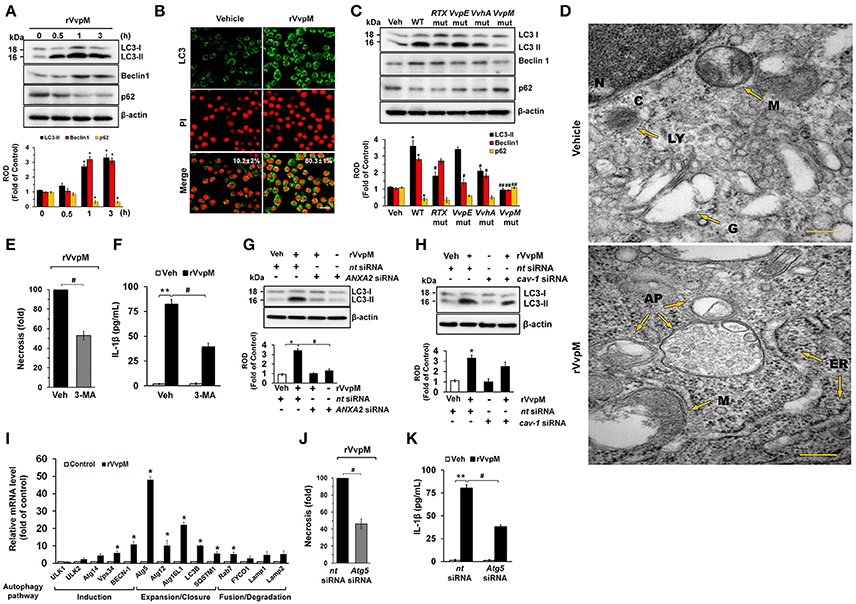
Figure 6. VvpM induces autophagy by regulating non-lipid raft ANXA2. (A) The effect of rVvpM on expressions of autophagy-related proteins, LC3, Beclin-1, and p62 were determined by Western blot. Data represent the mean ± S.E. n = 4. (B) LC3 expression (green) in cells treated with rVvpM for 3 h was visualized by confocal microscopy and quantified by using Image J program, which measures the stained area per microscopic filed with consistent threshold. Scale bars, 100 μm. n = 4. (C) The expressions of autophagy-related proteins in a cells infected with WT or various mutant deficients for 1 h are shown. n = 4. *P < 0.05 vs. Veh (boiled WT). #P < 0.05 and ##P < 0.01 vs. WT, respectively. (D) Formation of autophagic vesicles in cells treated with rVvpM for 3 h was visualized by transmission electron microscopy. Scale bars, 100 nm. n = 3. AP, autophagosome; C, cytosol; ER, endoplasmic reticulum; N, nucleus; G, golgi body; M, mitochondria. (E) Cells were pre-treated with 3-MA (10 mM) for 30 min prior to rVvpM exposure for 24 h. FACS analysis and quantitative analysis of the percentage of necrotic (Q1) cells are shown. n = 4. (F) The level of IL-1β protein was quantified by ELISA. n = 5. LC3 expression in cells transfected with siRNAs for ANXA2 (G) and cav-1 (H) for 24 h prior to rVvpM exposure for 3 h. (I) The effect of rVvpM on the expression of autophagy-related proteins was evaluated by qRT-PCR. The expression level of Atg5 and Atg16L1 was markedly increased by rVvpM treatment. n = 4. (J) Cells transfected with Atg5 siRNA were incubated with rVvpM for 24 h. FACS analysis and quantitative analysis of the percentage of necrotic (Q1) cells are shown. n = 4. (K) The level of IL-1β protein was quantified by ELISA. n = 5. (A,I) *P < 0.01 vs. cells with no treatment. (E,F) **P < 0.01 vs. Veh (boiled rVvpM). #P < 0.05 vs. Veh + rVvpM. (G,H,J,K) *P < 0.05 and **P < 0.01 vs. nt siRNA + Veh (boiled rVvpM), respectively. #P < 0.05 and ##P < 0.01 vs. nt siRNA + rVvpM, respectively. ROD, relative optical density.
VvpM Facilitates the Formation of NLRP3 Inflammasomes via Autophagy Activation
Given that RTX and VvhA produced by V. vulnificus are also known to cause the death of phagocytic cells via NLRP3-dependent caspase-1 activation in vitro and in vivo (Toma et al., 2010; Lo et al., 2011), it is important to clarify whether rVvpM regulates the expression of genes associated with the assembly, activation, and down-stream signaling of inflammasomes, which are linked to autophagy (Yuk and Jo, 2013). Our results from a RT2 Profiler PCR array showed that expression changes were detected in 13 of the 84 genes studied here after a 24 h rVvpM treatment (Figures 7A,B). Expression levels were increased in 10 genes (>two-fold, as shown by the green bar graph) but decreased in the other three genes (>two-fold, as shown by the blue bar graph). The remaining 61 genes showed no significant change in comparison with a vehicle treatment. The 10 genes showing increased expression levels were NLRP3 (NM_145827), Casp1 (caspase-1, NM_009807), Tnf (tumor necrosis factor, NM_013693), Ccl7 (chemokine ligand 7, NM_013654), Ikbkb (IKK-β, NM_010546), IL-1β, Map3k7 (TGF-β activated kinase 1, NM_172688), Mapk8 (JNK1, NM_016700), Mapk9 (JNK2, NM_016961), and Traf6 (Tnf receptor-associated factor 6, NM_009424), while those showing decreased expression levels were Ctsb (cathepsin B, NM_007798), Ccl12 (chemokine ligand 12, NM_011331), and Cxcl3 (chemokine (C-X-C motif) ligand 3, NM_203320). Among these differentially regulated genes, we identified two genes (IL-1β and NLRP3) which were highly expressed in cells treated with rVvpM compared to a treatment with a vehicle (>10-fold). Importantly, the necrotic cell death (Figure 7C) as well as the production of IL-1β (Figure 7D) induced by rVvpM were significantly abrogated by NLRP3 knockdown. These results indicate VvpM is another virulence factor of V. vulnificus to regulate NLRP3 inflammasome responsible for IL-1β production coupled with necrotic macrophage death. Consistently, our results have revealed that rVvpM has ability to induce speck formation of pyrin domain of the adaptor protein, ASC (apoptosis-associated speck-like protein containing a CARD) (Figure S11). We also found that the ASC was co-immunoprecipitated with NLRP3, and importantly, that the interaction between ASC and NLRP3 was enhanced by a treatment with rVvpM (Figure 7E). In contrast, these interactions induced by rVvpM were significantly blocked by a treatment with the autophagy inhibitor 3-MA and knockdown of Atg5 expression, suggesting that the formation of NLRP3 inflammasomes induced by rVvpM is an autophagy-dependent process. An increase in the active form of caspase-1 was also observed after 6 h of incubation with 100 pg/mL of rVvpM (Figure 7F). However, the activation of caspase-1 induced by rVvpM was markedly inhibited by transfection with siRNAs for NLRP3 (Figure 7G). Importantly, WT significantly induced the activation of caspase-1 and IL-1β maturation, whereas the VvpM mutant showed a marginal effect and appeared to be highly susceptible among the various mutants (Figure 7H). Overall, these novel findings demonstrate that VvpM facilitates the formation of NLRP3 inflammasomes via autophagy activation to regulate caspase-1-mediated IL-1β production.
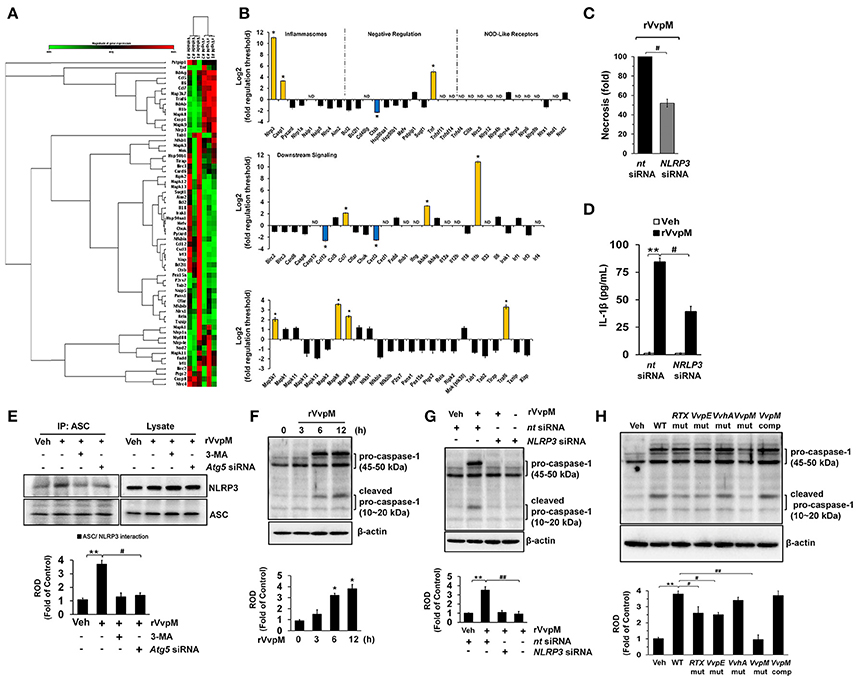
Figure 7. NLRP3 inflammasome is a downstream mediator of autophagy. (A,B) The mRNA expression levels of genes within the inflammasome pathway in cells treated with rVvpM for 24 h are shown. Expression of 84 genes involved in the inflammasome pathway assessed by RT2 Profiler PCR array, according to the manufacturer's instructions. Heat maps were generated and genes hierarchically clustered by Euclidean distance and single linkage by using the GeneGlobe Data Analysis Center on QIAGEN's website at http://www.qiagen.com/kr/shop/genes-and-pathways/data-analysis-center-overview-page/. n = 3. *P < 0.01 vs. Veh (boiled rVvpM). (C) Cells transfected with NLRP3 siRNA were incubated with rVvpM for 24 h. FACS analysis and quantitative analysis of the percentage of necrotic (Q1) cells are shown. n = 4. (D) The level of IL-1β protein was quantified by ELISA. n = 5. (E) ASC co-immunoprecipitated with NLRP3 is shown (left side). Expression of ASC and NLRP3 in total cell lysates is shown (right side). n = 4. **P < 0.01 vs. Veh (boiled rVvpM). #P < 0.05 vs. rVvpM alone. (F) The effect of rVvpM on the expression of caspase-1 was determined by western blot. n = 3. **P < 0.01 vs. cells with no treatment. (G) Cells transfected with NLRP3 siRNA were incubated with rVvpM for 6 h. n = 3. **P < 0.01 vs. nt siRNA + Veh (boiled rVvpM). ##P < 0.01 vs. nt siRNA + rVvpM. (H) The expression of caspase-1 in a cells infected with WT, various mutant deficient, or complemented VvpM mutant for 2 h are shown. n = 4. **P < 0.01 vs. Veh (boiled WT). #P < 0.05 vs. WT. (E–H) ROD, relative optical density.
VvpM Contributes to the Intestinal Colonization of V. vulnificus
To determine whether the functional role of VvpM in promoting IL-1β production coupled with macrophage cell death contributes to the growth of V. vulnificus in the gut, we further determined the level of bacterial colonization in mice inoculated intragastrically with a control, WT, and with the VvpM mutant and VvpM complementation (comp) at 1.3 × 109 CFU/mL for 16 h (Figure 8A). WT colonization in the ileum was increased by 4.6 ± 1.1 (× 106 CFU/g tissue) at 16 h. However, when mice were inoculated with the VvpM mutant, the levels were diminished by 3.3 ± 0.6 (× 106 CFU/g tissue) compared to those associated with the WT. In contrast, complementation of the VvpM mutant with a functional VvpM gene completely reversed the effect of WT on ileal colonization. It was noted that all of the mice given oral administrations of WT had survived at 18 h post-injection (data not shown). WT also induced severe inflammation of the intestine, where it caused shortened villi heights accompanied by increased numbers of inflammatory cells at 16 h infection, resulting in an increased histopathological damage score compared to that in the control mice (Figure 8B). However, the VvpM mutant significantly failed to regulate intestinal villi structures and the related inflammation. In contrast, complementation of the VvpM mutant with a functional VvpM gene restored the effect of the VvpM mutant on the histopathological damage score. These results indicate that VvpM contributes to V. vulnificus colonization, facilitating pro-inflammatory responses in vivo.
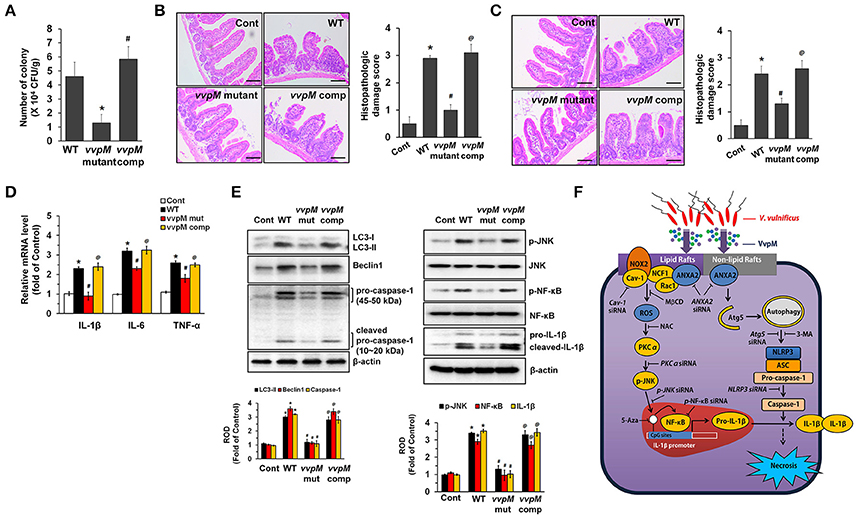
Figure 8. VvpM contributes to the intestinal colonization of V. vulnificus. (A) Mice inoculated with WT, boiled WT (Cont), VvpM mutant, and VvpM complement (comp) at 1.3 × 109 CFU/mL, and sacrificed 16 h later. Colonization activities were determined. n = 10. (B) Representative ileum tissues stained with H&E are shown (left panel). n = 10. Scale bars represent 100 μm. Average scores of histopathologic damage index from mouse ileum is shown (right panel). n = 10. *P < 0.01 vs. Cont. #P < 0.05 vs. WT. @P < 0.05 vs. VvpM mutant. (C) Closed ileal loop in mice was instilled with 100 μl of PBS containing WT, boiled WT (Cont), VvpM mutant, or VvpM complement (comp) at 1.3 × 109 CFU/mL, and sacrificed 2 h later. Representative ileum tissues stained with H&E are shown (left panel). n = 10. Scale bars represent 100 μm. Average scores of histopathologic damage index from mouse ileum is shown (right panel). n = 10. *P < 0.05 vs. Cont. #P < 0.05 vs. WT. @P < 0.05 vs. VvpM mutant. (D) The expression levels of pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α) in ileal-ligated mouse model are shown. n = 5. *P < 0.01 vs. Cont. #P < 0.05 vs. WT. @P < 0.05 vs. VvpM mutant. (E) The expression of LC3-II, Beclin 1, Caspase-1 and IL-1β, and the phosphorylation of JNK and NF-κB were determined by western blot. n = 4. *P < 0.01 vs. Cont. #P < 0.01 vs. WT. @P < 0.01 vs. VvpM mutant. (F) A hypothetical model for VvpM-induced signaling pathway, where VvpM regulates two pathogenic pathways that stimulate NF-κB-dependent IL-1β production and autophagy-mediated NLRP3 inflammasome via spatial targeting distinct ANXA2.
To eliminate the possibility that the lower inflammatory responses evoked by the VvpM mutant can simply be explained by the presence of less bacteria, we undertook an additional experiment using the ileal-ligated mouse model in which a closed ileal loop was instilled with 100 μl of PBS containing WT, the VvpM mutant, or VvpM complementation at 1.3 × 109 CFU/mL for 2 h. We anticipated that 2 h was the minimum duration for enterotoxicity and pro-inflammatory responses based on our recent report (Kim et al., 2013). There were no statistical differences between the groups treated with WT and the VvpM mutant in terms of the bacterial growth (data not shown). However, the injection of mice with the VvpM mutant at a time of 2 h failed to elevate the histopathological damage score (Figure 8C) or the expression levels of pro-inflammatory cytokines (Figure 8D) as caused by WT infection. The results in Figure 8E revealed that inoculation with WT for 2 h significantly increased the expressions of LC3-II, Beclin 1, Caspase-1 and mature IL-1β as well as the phosphorylation of JNK and NF-κB in mice, which were prevented by infection with the VvpM mutant. Notably, the functional effects of the VvpM mutant were significantly restored by the complementation of the VvpM mutant with a functional VvpM gene (Figure 8E). Thus, these results suggest that VvpM is a major virulence factor of V. vulnificus responsible for IL-1β production coupled with necrotic cell death.
Discussion
In the present study, we demonstrate that VvpM mainly targets the critical pathogenic pathway of V. vulnificus to promote IL-1β production coupled with necrotic macrophage death. A key finding of our study is that VvpM acting through ANXA2 organized into lipid rafts stimulates the ROS-mediated NF-κB pathway to result in the transcriptional induction of IL-1β, whereas ANXA2 organized into non-lipid rafts induces activation of autophagy and NLRP3 inflammasome, resulting in caspase-1-mediated IL-1β maturation and necrotic macrophage death. VvpM may stimulate IL-1β production via ANXA2, which would remain segregated from raft elements in non-lipid rafts of the macrophage, while a specific raft-associated ANXA2 in response to VvpM would additively promote IL-1β production by co-clustering with other proteins recruited to rafts to enhance their affinity for the distinct raft subtype caveolin-1. We suggest that this is one of the reasons why VvpM makes a greater contribution to IL-1β production than the contributions of all other subtypes of exotoxins produced by V. vulnificus. Hence, these findings demonstrate that ANXA2 activated by VvpM via an identical extracellular cue regulates two pathogenic pathways closely related to IL-1β production via its spatial placement within the cell. Annexins are a well-known multigene family of calcium- and phospholipid-binding protein (Gerke and Moss, 2002). Having shown that ANXA2 has an important role in inflammatory responses responsible for IL-1β production during V. vulnificus infection (Lee et al., 2015a, 2016), our current results indicate that ANXA2 is highly expressed in macrophages and that spatial targeting of ANXA2 is required for IL-1β production coupled with necrotic macrophage death. In contrast to our results, phenotypes observed in ANXA2 knockout mouse models revealed that ANXA2 functions as a mediator of the anti-inflammatory response to maintain host defense mechanisms (He et al., 2016). We believe that these opposing ANXA2 effects are, in part, due to the presence of multiple ANXA receptors that have compensatory and redundancy actions with each other, and that these functions may vary depending on cellular characteristics and stress conditions. In fact, the phenotypes of most knockout models of annexin family have also been reported to be rather subtle, and all mice strains lacking one or even two annexins are viable and develop normally (Grewal et al., 2016). Compelling evidence also supports the critical role of ANXA2 in inflammatory myopathies (Probst-Cousin et al., 2004) and in inflammatory bowel disease (Tsukamoto et al., 2013). Thus, we contend that ANXA2 functions are linked to its ability to mediate the pro-inflammatory response of macrophages during V. vulnificus infection. Given that ANXA2 is not a transmembrane protein, it is very likely unable to transmit the signal initiated by the binding of VvpM. Therefore, it is possible that ANXA2 is associated with additional proteins that transfer the signal generated by VvpM into the macrophage. Our results indicate that VvpM may utilize reversible and multiple functions of ANXA2 to transmit the signal into the cell as a unique infectious stratagem, although the exact mechanism of VvpM interactions with ANXA2 remains undescribed. Alternatively, ANXA2 may merely function as an anchor protein to facilitate the binding of VvpM to the cell surface. Indeed, the importance of ANXA2 during microbial pathogenesis is underscored by the finding that EPEC adherence induces the aggregation of lipid rafts at sites of bacterial contact, where ANXA2 is recruited to the cytoplasmic membrane surface, possibly stabilizing raft patches and their links to the actin cytoskeleton beneath adhering to EPEC (Zobiack et al., 2002). Thus, our results indicate the possibility that rVvpM influences the structure and localization of ANXA2 through lipid rafts to promote pro-inflammatory signaling in macrophages.
With regard to lipid raft signaling events, VvpM induces the transcription of the IL-1β gene through the ROS-PKCα-JNK-NF-κB pathway via ANXA2. NADPH oxidase is a critical source of cellular ROS and has been studied extensively in phagocytes as an innate host defense component (Quinn and Gauss, 2004; Li et al., 2007). Increasing evidence has suggested that lipid rafts are clustered to form a redox-signaling platform through activation of NOX2 by interacting with NCF1 and Rac1 for membrane ROS production (Kusumoto et al., 2005; Li et al., 2007). Moreover, multiple signaling processes, such as those acting through the Ca2+ and PKC/MAPK pathways, are rapidly activated in target cells through ROS production (Lee et al., 2009, 2010). In fact, H. pylori and C. perfringens have also been shown to elicit a Ca2+ response (Fasano, 2002). Moreover, ANXA2 is a Ca2+/lipid-binding protein that differs from most other Ca2+-binding proteins, and the binding of Ca2+ to ANXA2 has been shown to trigger exposure of its N-terminal region, making it available for additional interactions/activities (Rescher and Gerke, 2004). Many studies have suggested a critical role of conventional PKCα in the cell death process during EPEC or C. perfringens infections (Crane and Vezina, 2005; Monturiol-Gross et al., 2014). These previous results are consistent with our current result showing that rVvpM may induce an influx of Ca2+ upon activation of ANXA2 and PKCα, as required for ROS production to stimulate IL-1β production and necrotic macrophage death. Despite the frequent involvement of ERK and p38 MAPK in the ROS signaling pathway induced by H. pylori infection (Ki et al., 2008) and V. vulnificus VvhA (Lee et al., 2015c), our results reveal that VvpM uniquely regulates IL-1β production through activation of the JNK-mediated NF-κB pathway. These results imply a functional role of rVvpM in the determination of downstream targets. Moreover, earlier research has shown that the JNK pathway induced by ROS can influence NF-κB activation in promoting apoptotic cell death (Seo et al., 2013). Hence, it is conceivable that ROS induced by rVvpM has a potential role in promoting the NF-κB pathway through activation of PKCα/JNK cascades. While the NF-κB binding activity of the IL-1β gene promoter is a critical mechanism in the transcriptional regulation of IL-1β, the promoter methylation of cytosine residues at CpG dinucleotides is also an important post-translational modification affecting NF-κB transcription. Indeed, it was clearly shown that ROS induced by H2O2 treatment regulates histone acetyltransferases (HAT) and deacetylases (HDAC), key enzymes responsible for chromatin remodeling, suggesting that post-translational modifications affecting NF-κB are also redox-sensitive processes (Rahman et al., 2004). In the present study, we observed that the bacterial signaling caused by rVvpM acting through the ROS-PKCα-JNK-NF-κB pathway via lipid-raft-associated ANXA2 is not required for autophagy activation, ensuring that the autophagy-mediated activation of the NLRP3 inflammasome is independent of the action of lipid rafts. Thus, our data also provide important evidence that VvpM induces the transcriptional regulation of NF-κB as well as the hypomethylation of the IL-1β promoter to promote IL-1β production coupled with necrotic macrophage death.
On the other hand, our results reveal that VvpM acting through non-lipid raft ANXA2 promotes autophagic death and inflammasome-dependent IL-1β secretion in macrophages. Although the role of non-lipid rafts coupled with ANXA2 has not been identified, the non-lipid raft protein transferrin receptor critically participates in promoting bacterial signaling of Neisseriaceae and Pasteurellaceae families (Moraes et al., 2009). While autophagic cell death is an essential innate immune signaling mechanism to promote cytokine and chemokine production, and bacterial clearance mechanisms (Xu et al., 2006; Labbe and Saleh, 2008; Kirienko et al., 2015), many pathogens, including S. typhimurium, M. tuberculosis and L. monocytogenes, are reported to manipulate the autophagosome machinery for their replication and control their phagocytosis by using immune cells to facilitate their dissemination into the blood stream (Birmingham et al., 2006, 2007; Biswas et al., 2008; Cemma and Brumell, 2012). Thus, describing the processes in autophagy-related events may reveal why certain cells may be more or less susceptible to pathogen-induced cell death and may reveal novel therapeutic targets. Importantly, rVvpM acting on non-raft ANXA2 distinctively regulates autophagic cell death in macrophages via Atg5, which is critical for the formation of autophagosomes. Moreover, Atg5 covalently conjugates with Atg12 and interacts with Atg16L1 to form a dimeric complex known as the Atg16L1 complex which promotes LC3 lipidation in expanding phagophores for elongation of the autophagic membrane (Ohsumi, 2001; Geng and Klionsky, 2008). In addition to Atg5, recent research employing the ANXA2 KO mouse model has provided new insights into the biological functions of ANXA2, indicating that ANXA2 is a critical autophagy inducer for the recruitment of phosphatidylserine and phosphatidylinositides into Atg16L-positive vesicles, representing very early events in the generation of phagophores (Morozova et al., 2015). The formation of autophagophore is also related to the ability of ANXA2 to interact and organize cytoskeleton proteins including actins and actin nucleation factor Spire1 and Arp2/3 located in both non-lipid and lipid raft for inward budding (Hayes et al., 2009; Morozova et al., 2015). Thus, it is possible that non-lipid ANXA2 could binds to actin-related proteins to form the autophagophore in rVvpM-treated macrophage. Hence, the results of our present study indicate that rVvpM has unique infectious stratagems by which to initiate Atg5-dependent autophagic cell death via the non-lipid raft ANXA2 and that this bacterial signaling pathway acts in concert with the lipid raft-associated pathways in manipulating IL-1β production. Although previous reports have long considered that the autophagy pathway prevents tissue inflammation through its role in apoptotic cell corpse clearance, our results clearly indicate that VvpM mediates the inflammatory response of V. vulnificus by promoting autophagic cell death. A remaining question is how does VvpM utilize the inflammatory signaling pathway via autophagy. Given that inflammasome activation is an alternative cell death pathway associated with releasing mature pro-inflammatory cytokine IL-1β (Lamkanfi et al., 2011; Guo et al., 2015), our present results reveal that VvpM is a major virulence factor of V. vulnificus and is responsible for triggering the activation of caspase-1, which is cleaved by the assembly of the NLR protein NLRP3 and the ASC adaptor to promote IL-1β production, and, importantly, that the NLRP3 inflammasome assembly strongly depends on autophagy activation. These results are consistent with those in previous studies showing that autophagy controls inflammasome-dependent IL-1β secretion via Atg5, suggesting a role of autophagy in the positive regulation of inflammasome activation (Dupont et al., 2011).
During the early stages of the VvpM treatment (for 6 h), we observed that VvpM had the ability to promote the NLRP3 inflammasome complex and ASC speck formation without affecting protein expression levels, whereas later in the treatment (for 24 h), VvpM clearly induced expressions of NLRP3, caspase-1, and IL-1β, which are all associated with the formation of the NLRP3 inflammasome complex. These results indicate that genes related to NLRP3 inflammasomes must be transcriptionally induced by some key mediator of immunity to generate IL-1β production continually. Although we did not investigate the reasons for the unexpected contrary results regarding the up-regulation of Tnf expression and down-regulation of Ctsb, Ccl12, and Cxcl3, we did note that VvpM has the additional ability to stimulate the expressions of JNK, IKK-β, and Traf6, which are responsible for NF-κB activation (Matsumura et al., 2003; Lamkanfi et al., 2004; Sollberger et al., 2014; Lee et al., 2015c) and TGF-β activated kinase 1 (TAK1) and CCL7 expression, all of which are related to NLRP3 inflammasome activation (Inoue et al., 2012; Latz et al., 2013). These results suggest that VvpM stimulates IL-1β production coupled with necrotic macrophage death by regulating the Atg5-depednent formation of the NLRP3 inflammasome complex and by governing expressions of genes closely related to NLRP3 inflammasomes. Taken together, these findings therefore indicate that VvpM acting with non-raft ANXA2 is a major signaling regulator of autophagy in promoting NLRP3 inflammasome activation, although earlier studies found a mutual relationship between autophagy and inflammasomes (Yuk and Jo, 2013). Concerning the role of RTX in necrotic cell death, it is well-known multifunctional exotoxins of V. vulnificus and causes the necrotic cell death and/or IL-1β production through the lysis of a variety of cell types including macrophages (Toma et al., 2010) and epithelial cells (Lee et al., 2007; Kim et al., 2013). Moreover it already has been reported to play a major role in the pathogenesis of V. vulnificus (Toma et al., 2010; Jeong and Satchell, 2012; Kim et al., 2013). Given current in vitro observations that RTX is also responsible for ROS production, PKC phosphorylation and IL-1β production, it is possible that RTX induces necrotic cell death. However, it recently have shown that RTX has multiple effector domains, which catalyse the site-specific processing of the Switch I region of Ras and Rap1 (Antic et al., 2015). Thus, we suggest that the virulence mechanism of RTX during the necrotic cell death differs from the VvpM, which efficiently regulates two pathogenic pathways via distinct spatial targeting by membrane ANXA2.
Finally, in mouse models of V. vulnificus infection, our results obtained from gain- and loss-of-function approaches for VvpM revealed that the functional importance of two pathogenic pathways via ANXA2 to promote IL-1β production coupled with macrophage death may contribute to the colonization of V. vulnificus in the gut. Our results are consistent with those in a previous report showing that V. vulnificus not only induces massive inflammation, leading to the recruitment of monocytes, neutrophils and F4/80-positive macrophages (Jeong and Satchell, 2012), but that it also kills phagocytes in the gut (Toma et al., 2010; Lo et al., 2011). Concerning the role of VvpM in intestinal colonization, it is unclear whether the functional role of VvpM in facilitating V. vulnificus colonization is a direct effect of its capacity to grow in the microenvironment of the small intestine or, alternatively, a sequential process involving other cellular signaling events related to the killing of the recruited phagocytes. In agreement with our results, it was previously shown that Salmonella induces pyroptosis, leading to inflammation, which allows Salmonella to use tetrathionate respiration in the promotion of bacterial growth and colonization (Fink and Cookson, 2007). Thus, it is possible that the killing of macrophages with VvpM together with the promotion of the IL-1β production is one mechanism that would promote rapid in vivo V. vulnificus growth and colonization. We also used an ileal-ligated mouse model to investigate the actual contribution of VvpM to inflammatory responses under similar infection levels of WT, VvpM mutant, and complementation. Our data revealed that VvpM is required for the initiation of massive necrotizing inflammation, accompanying the activation of autophagy, JNK/NF-κB, caspase-1, and IL-1β. While our results indicate that the action of VvpM against infiltrated phagocytes is induction of pro-inflammatory responses, our results do not negate the contention that this toxin has a significant role in the intestinal epithelium, just as it does in Caco-2 cells and in the findings in previous reports (Lee M. A. et al., 2014; Lee et al., 2015a).
In conclusion, our results suggest that VvpM induces two different pathogenic pathways closely related to IL-1β production coupled with necrotic macrophage death and that VvpM regulates the NF-κB-dependent IL-1β production pathway via lipid-raft-dependent ANXA2 recruitment, whereas VvpM acting on non-lipid raft ANXA2 additively facilitates the activation of NLRP3 inflammasome via the Atg5-dependent autophagic pathway (Figure 8F). Given the current in vivo observations, we further suggest that VvpM is a new virulence factor responsible for regulation of the intestinal colonization of V. vulnificus. Thus, research highlighting the rVvpM signaling pathways involved in IL-1β production coupled with necrotic macrophage death may indicate potential therapeutic targets for strategic modulation of V. vulnificus infections.
Author Contributions
Study concept and design, acquisition of data, analysis and interpretation of data, statistical analysis, and drafting of the manuscript: SL and HJH. Acquisition of data, analysis and interpretation of data: YHJ, JSK, HJL, SHL, and KKJ. Technical or material support, analysis and interpretation of data: KL and SHC.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by grants to both HJH and SHC from the Agriculture, Food and Rural Affairs Research Center Support Program, Ministry of Agriculture, Food and Rural Affairs, Republic of Korea (Grant 710002-07-5).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fcimb.2017.00352/full#supplementary-material
References
Antic, I., Biancucci, M., Zhu, Y., Gius, D. R., and Satchell, K. J. (2015). Site-specific processing of Ras and Rap1 Switch I by a MARTX toxin effector domain. Nat. Commun. 6:7396. doi: 10.1038/ncomms8396
Birmingham, C. L., Canadien, V., Gouin, E., Troy, E. B., Yoshimori, T., Cossart, P., et al. (2007). Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy 3, 442–451. doi: 10.4161/auto.4450
Birmingham, C. L., Smith, A. C., Bakowski, M. A., Yoshimori, T., and Brumell, J. H. (2006). Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J. Biol. Chem. 281, 11374–11383. doi: 10.1074/jbc.M509157200
Biswas, D., Qureshi, O. S., Lee, W.-Y., Croudace, J. E., Mura, M., and Lammas, D. A. (2008). ATP-induced autophagy is associated with rapid killing of intracellular mycobacteria within human monocytes/macrophages. BMC Immunol. 9:35. doi: 10.1186/1471-2172-9-35
Cemma, M., and Brumell, J. H. (2012). Interactions of pathogenic bacteria with autophagy systems. Curr. Biol. 22, R540–R545. doi: 10.1016/j.cub.2012.06.001
Clark, J. A., Doelle, S. M., Halpern, M. D., Saunders, T. A., Holubec, H., Dvorak, K., et al. (2006). Intestinal barrier failure during experimental necrotizing enterocolitis: protective effect of EGF treatment. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G938–G949. doi: 10.1152/ajpgi.00090.2006
Cosentino-Gomes, D., Rocco-Machado, N., and Meyer-Fernandes, J. R. (2012). Cell signaling through protein kinase C oxidation and activation. Int. J. Mol. Sci. 13, 10697–10721. doi: 10.3390/ijms130910697
Cox, B., and Emili, A. (2006). Tissue subcellular fractionation and protein extraction for use in mass-spectrometry-based proteomics. Nat. Protoc. 1, 1872–1878. doi: 10.1038/nprot.2006.273
Crane, J. K., and Vezina, C. M. (2005). Externalization of host cell protein kinase C during enteropathogenic Escherichia coli infection. Cell Death Differ. 12, 115–127. doi: 10.1038/sj.cdd.4401531
Dupont, N., Jiang, S., Pilli, M., Ornatowski, W., Bhattacharya, D., and Deretic, V. (2011). Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 30, 4701–4711. doi: 10.1038/emboj.2011.398
Farre, D., Roset, R., Huerta, M., Adsuara, J. E., Rosello, L., Alba, M. M., et al. (2003). Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 31, 3651–3653. doi: 10.1093/nar/gkg605
Fasano, A. (2002). Toxins and the gut: role in human disease. Gut 50(Suppl. 3), 9–14. doi: 10.1136/gut.50.suppl_3.iii9
Fassino, S., Svrakic, D., Abbate-Daga, G., Leombruni, P., Amianto, F., Stanic, S., et al. (2002). Anorectic family dynamics: temperament and character data. Compr. Psychiatry 43, 114–120. doi: 10.1053/comp.2002.30806
Fink, S. L., and Cookson, B. T. (2007). Pyroptosis and host cell death responses during Salmonella infection. Cell. Microbiol. 9, 2562–2570. doi: 10.1111/j.1462-5822.2007.01036.x
Geng, J., and Klionsky, D. J. (2008). The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. EMBO Rep. 9, 859–864. doi: 10.1038/embor.2008.163
Gerke, V., and Moss, S. E. (2002). Annexins: from structure to function. Physiol. Rev. 82, 331–371. doi: 10.1152/physrev.00030.2001
Grewal, T., Wason, S. J., Enrich, C., and Rentero, C. (2016). Annexins—insights from knockout mice. Biol. Chem. 397, 1031–1053. doi: 10.1515/hsz-2016-0168
Guo, H., Callaway, J. B., and Ting, J. P. (2015). Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 21, 677–687. doi: 10.1038/nm.3893
Hashimoto, K., Oreffo, R. O., Gibson, M. B., Goldring, M. B., and Roach, H. I. (2009). DNA demethylation at specific CpG sites in the IL1B promoter in response to inflammatory cytokines in human articular chondrocytes. Arthritis Rheum. 60, 3303–3313. doi: 10.1002/art.24882
Hayes, M. J., Shao, D. M., Grieve, A., Levine, T., Bailly, M., and Moss, S. E. (2009). Annexin A2 at the interface between F-actin and membranes enriched in phosphatidylinositol 4,5,-bisphosphate. Biochim. Biophys. Acta 1793, 1086–1095. doi: 10.1016/j.bbamcr.2008.10.007
He, S., Li, X., Li, R., Fang, L., Sun, L., Wang, Y., et al. (2016). Annexin A2 modulates ROS and impacts inflammatory response via IL-17 signaling in polymicrobial sepsis mice. PLoS Pathog. 12:e1005743. doi: 10.1371/journal.ppat.1005743
Inoue, M., Williams, K. L., Gunn, M. D., and Shinohara, M. L. (2012). NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. U.S.A. 109, 10480–10485. doi: 10.1073/pnas.1201836109
Jang, K. K., Gil, S. Y., Lim, J. G., and Choi, S. H. (2016). Regulatory characteristics of Vibrio vulnificus gbpA gene encoding a mucin-binding protein essential for pathogenesis. J. Biol. Chem. 291, 5774–5787. doi: 10.1074/jbc.M115.685321
Jeong, H. G., and Satchell, K. J. (2012). Additive function of Vibrio vulnificus MARTXVv and VvhA cytolysins promotes rapid growth and epithelial tissue necrosis during intestinal infection. PLoS Pathog. 8:e1002581. doi: 10.1371/journal.ppat.1002581
Jeong, K. C., Jeong, H. S., Rhee, J. H., Lee, S. E., Chung, S. S., Starks, A. M., et al. (2000). Construction and phenotypic evaluation of a Vibrio vulnificus vvpE mutant for elastolytic protease. Infect. Immun. 68, 5096–5106. doi: 10.1128/IAI.68.9.5096-5106.2000
Jones, R. M., Wu, H., Wentworth, C., Luo, L., Collier-Hyams, L., and Neish, A. S. (2008). Salmonella AvrA coordinates suppression of host immune and apoptotic defenses via JNK pathway blockade. Cell Host Microbe 3, 233–244. doi: 10.1016/j.chom.2008.02.016
Kaakinen, M., Kaisto, T., Rahkila, P., and Metsikko, K. (2012). Caveolin 3, flotillin 1 and influenza virus hemagglutinin reside in distinct domains on the sarcolemma of skeletal myofibers. Biochem. Res. Int. 2012:497572. doi: 10.1155/2012/497572
Keen, N. T., Tamaki, S., Kobayashi, D., and Trollinger, D. (1988). Improved broad-host-range plasmids for DNA cloning in gram-negative bacteria. Gene 70, 191–197. doi: 10.1016/0378-1119(88)90117-5
Ki, M. R., Lee, H. R., Goo, M. J., Hong, I. H., Do, S. H., Jeong, D. H., et al. (2008). Differential regulation of ERK1/2 and p38 MAP kinases in VacA-induced apoptosis of gastric epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G635–G647. doi: 10.1152/ajpgi.00281.2007
Kim, S. M., Park, J. H., Lee, H. S., Kim, W. B., Ryu, J. M., Han, H. J., et al. (2013). LuxR homologue SmcR is essential for Vibrio vulnificus pathogenesis and biofilm detachment, and its expression is induced by host cells. Infect. Immun. 81, 3721–3730. doi: 10.1128/IAI.00561-13
Kim, Y. R., Lee, S. E., Kang, I. C., Nam, K. I., Choy, H. E., and Rhee, J. H. (2013). A bacterial RTX toxin causes programmed necrotic cell death through calcium-mediated mitochondrial dysfunction. J. Infect. Dis. 207, 1406–1415. doi: 10.1093/infdis/jis746
Kirienko, N. V., Ausubel, F. M., and Ruvkun, G. (2015). Mitophagy confers resistance to siderophore-mediated killing by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 112, 1821–1826. doi: 10.1073/pnas.1424954112
Kusumoto, K., Kawahara, T., Kuwano, Y., Teshima-Kondo, S., Morita, K., Kishi, K., et al. (2005). Ecabet sodium inhibits Helicobacter pylori lipopolysaccharide-induced activation of NADPH oxidase 1 or apoptosis of guinea pig gastric mucosal cells. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G300–G307. doi: 10.1152/ajpgi.00274.2004
Labbe, K., and Saleh, M. (2008). Cell death in the host response to infection. Cell Death Differ. 15, 1339–1349. doi: 10.1038/cdd.2008.91
Lamkanfi, M., Kalai, M., Saelens, X., Declercq, W., and Vandenabeele, P. (2004). Caspase-1 activates nuclear factor of the k-enhancer in B cells independently of its enzymatic activity. J. Biol. Chem. 279, 24785–24793. doi: 10.1074/jbc.M400985200
Lamkanfi, M., Vande Walle, L., and Kanneganti, T. D. (2011). Deregulated inflammasome signaling in disease. Immunol. Rev. 243, 163–173. doi: 10.1111/j.1600-065X.2011.01042.x
Latz, E., Xiao, T. S., and Stutz, A. (2013). Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411. doi: 10.1038/nri3452
Lee, H. J., Kim, J. A., Lee, M. A., Park, S. J., and Lee, K. H. (2013). Regulation of haemolysin (VvhA) production by ferric uptake regulator (Fur) in Vibrio vulnificus: repression of vvhA transcription by Fur and proteolysis of VvhA by Fur-repressive exoproteases. Mol. Microbiol. 88, 813–826. doi: 10.1111/mmi.12224
Lee, J. H., Kim, M. W., Kim, B. S., Kim, S. M., Lee, B. C., Kim, T. S., et al. (2007). Identification and characterization of the Vibrio vulnificus rtxA essential for cytotoxicity in vitro and virulence in mice. J. Microbiol. 45, 146–152.
Lee, M. A., Kim, J. A., Shin, M. Y., Lee, J. K., Park, S. J., and Lee, K. H. (2015). VvpM induces human cell death via multifarious modes including necroptosis and autophagy. J. Microbiol. Biotechnol. 25, 302–306. doi: 10.4014/jmb.1501.01007
Lee, M. A., Kim, J. A., Yang, Y. J., Shin, M. Y., Park, S. J., and Lee, K. H. (2014). VvpM, an extracellular metalloprotease of Vibrio vulnificus, induces apoptotic death of human cells. J. Microbiol. 52, 1036–1043. doi: 10.1007/s12275-014-4531-0
Lee, S. H., Lee, Y. J., and Han, H. J. (2010). Effect of arachidonic acid on hypoxia-induced IL-6 production in mouse ES cells: involvement of MAPKs, NF-κB, and HIF-1α. J. Cell. Physiol. 222, 574–585. doi: 10.1002/jcp.21973
Lee, S. H., Na, S. I., Heo, J. S., Kim, M. H., Kim, Y. H., Lee, M. Y., et al. (2009). Arachidonic acid release by H2O2 mediated proliferation of mouse embryonic stem cells: involvement of Ca2+/PKC and MAPKs-induced EGFR transactivation. J. Cell. Biochem. 106, 787–797. doi: 10.1002/jcb.22013
Lee, S. J., Jung, Y. H., Oh, S. Y., Jang, K. K., Lee, H. S., Choi, S. H., et al. (2015b). Vibrio vulnificus VvpE inhibits mucin 2 expression by hypermethylation via lipid raft-mediated ROS signaling in intestinal epithelial cells. Cell Death Dis. 6:e1787. doi: 10.1038/cddis.2015.152
Lee, S. J., Jung, Y. H., Oh, S. Y., Song, E. J., Choi, S. H., and Han, H. J. (2015c). Vibrio vulnificus VvhA induces NF-κB-dependent mitochondrial cell death via lipid raft-mediated ROS production in intestinal epithelial cells. Cell Death Dis. 6:1655. doi: 10.1038/cddis.2015.19
Lee, S. J., Jung, Y. H., Oh, S. Y., Yun, S. P., and Han, H. J. (2014). Melatonin enhances the human mesenchymal stem cells motility via melatonin receptor 2 coupling with Gαq in skin wound healing. J. Pineal Res. 57, 393–407. doi: 10.1111/jpi.12179
Lee, S. J., Jung, Y. H., Ryu, J. M., Jang, K. K., Choi, S. H., and Han, H. J. (2016). VvpE mediates the intestinal colonization of Vibrio vulnificus by the disruption of tight junctions. Int. J. Med. Microbiol. 306, 10–19. doi: 10.1016/j.ijmm.2015.10.006
Lee, S. J., Jung, Y. H., Song, E. J., Jang, K. K., Choi, S. H., and Han, H. J. (2015a). Vibrio vulnificus VvpE stimulates IL-1β production by the hypomethylation of the IL-1β promoter and NF-κB activation via lipid raft-dependent ANXA2 recruitment and reactive oxygen species signaling in intestinal epithelial cells. J. Immunol. 195, 2282–2293. doi: 10.4049/jimmunol.1500951
Li, P. L., Zhang, Y., and Yi, F. (2007). Lipid raft redox signaling platforms in endothelial dysfunction. Antioxid. Redox Signal. 9, 1457–1470. doi: 10.1089/ars.2007.1667
Lo, H. R., Lin, J. H., Chen, Y. H., Chen, C. L., Shao, C. P., Lai, Y. C., et al. (2011). RTX toxin enhances the survival of Vibrio vulnificus during infection by protecting the organism from phagocytosis. J. Infect. Dis. 203, 1866–1874. doi: 10.1093/infdis/jir070
Manes, S., del Real, G., and Martinez, A. C. (2003). Pathogens: raft hijackers. Nat. Rev. Immunol. 3, 557–568. doi: 10.1038/nri1129
Matsumura, T., Degawa, T., Takii, T., Hayashi, H., Okamoto, T., Inoue, J., et al. (2003). TRAF6-NF-κB pathway is essential for interleukin-1-induced TLR2 expression and its functional response to TLR2 ligand in murine hepatocytes. Immunology 109, 127–136. doi: 10.1046/j.1365-2567.2003.01627.x
Messeguer, X., Escudero, R., Farre, D., Nunez, O., Martinez, J., and Alba, M. M. (2002). PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 18, 333–334. doi: 10.1093/bioinformatics/18.2.333
Milton, D. L., O'Toole, R., Horstedt, P., and Wolf-Watz, H. (1996). Flagellin A is essential for the virulence of Vibrio anguillarum. J. Bacteriol. 178, 1310–1319. doi: 10.1128/jb.178.5.1310-1319.1996
Monturiol-Gross, L., Flores-Diaz, M., Pineda-Padilla, M. J., Castro-Castro, A. C., and Alape-Giron, A. (2014). Clostridium perfringens phospholipase C induced ROS production and cytotoxicity require PKC, MEK1 and NF-κB activation. PLoS ONE 9:e86475. doi: 10.1371/journal.pone.0086475
Moraes, T. F., Yu, R. H., Strynadka, N. C., and Schryvers, A. B. (2009). Insights into the bacterial transferrin receptor: the structure of transferrin-binding protein B from Actinobacillus pleuropneumoniae. Mol. Cell 35, 523–533. doi: 10.1016/j.molcel.2009.06.029
Morozova, K., Sridhar, S., Zolla, V., Clement, C. C., Scharf, B., Verzani, Z., et al. (2015). Annexin A2 promotes phagophore assembly by enhancing Atg16+ vesicle biogenesis and homotypic fusion. Nat. Commun. 6:5856. doi: 10.1038/ncomms7816
Netea, M. G., Simon, A., van de Veerdonk, F., Kullberg, B. J., Van der Meer, J. W., and Joosten, L. A. (2010). IL-1β processing in host defense: beyond the inflammasomes. PLoS Pathog. 6:e1000661. doi: 10.1371/journal.ppat.1000661
Nougayrede, J. P., and Donnenberg, M. S. (2004). Enteropathogenic Escherichia coli EspF is targeted to mitochondria and is required to initiate the mitochondrial death pathway. Cell. Microbiol. 6, 1097–1111. doi: 10.1111/j.1462-5822.2004.00421.x
Oakley, F. D., Smith, R. L., and Engelhardt, J. F. (2009). Lipid rafts and caveolin-1 coordinate interleukin-1β (IL-1β)-dependent activation of NF-κB by controlling endocytosis of Nox2 and IL-1β receptor 1 from the plasma membrane. J. Biol. Chem. 284, 33255–33264. doi: 10.1074/jbc.M109.042127
Ohsumi, Y. (2001). Molecular dissection of autophagy: two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2, 211–216. doi: 10.1038/35056522
Paesold, G., Guiney, D. G., Eckmann, L., and Kagnoff, M. F. (2002). Genes in the Salmonella pathogenicity island 2 and the Salmonella virulence plasmid are essential for Salmonella-induced apoptosis in intestinal epithelial cells. Cell. Microbiol. 4, 771–781. doi: 10.1046/j.1462-5822.2002.00233.x
Probst-Cousin, S., Berghoff, C., Neundorfer, B., and Heuss, D. (2004). Annexin expression in inflammatory myopathies. Muscle Nerve 30, 102–110. doi: 10.1002/mus.20077
Quinn, M. T., and Gauss, K. A. (2004). Structure and regulation of the neutrophil respiratory burst oxidase: comparison with nonphagocyte oxidases. J. Leukoc. Biol. 76, 760–781. doi: 10.1189/jlb.0404216
Rahman, I., Marwick, J., and Kirkham, P. (2004). Redox modulation of chromatin remodeling: impact on histone acetylation and deacetylation, NF-κB and pro-inflammatory gene expression. Biochem. Pharmacol. 68, 1255–1267. doi: 10.1016/j.bcp.2004.05.042
Rescher, U., and Gerke, V. (2004). Annexins–unique membrane binding proteins with diverse functions. J. Cell Sci. 117(Pt 13), 2631–2639. doi: 10.1242/jcs.01245
Riethmuller, J., Riehle, A., Grassme, H., and Gulbins, E. (2006). Membrane rafts in host-pathogen interactions. Biochim. Biophys. Acta 1758, 2139–2147. doi: 10.1016/j.bbamem.2006.07.017
Schroeder, G. N., and Hilbi, H. (2007). Cholesterol is required to trigger caspase-1 activation and macrophage apoptosis after phagosomal escape of Shigella. Cell. Microbiol. 9, 265–278. doi: 10.1111/j.1462-5822.2006.00787.x
Seo, B. N., Ryu, J. M., Yun, S. P., Jeon, J. H., Park, S. S., Oh, K. B., et al. (2013). Delphinidin prevents hypoxia-induced mouse embryonic stem cell apoptosis through reduction of intracellular reactive oxygen species-mediated activation of JNK and NF-κB, and Akt inhibition. Apoptosis 18, 811–824. doi: 10.1007/s10495-013-0838-2
Simon, R., Priefer, U., and Puhler, A. (1983). A broad host range mobilization system for in vivo genetic-engineering - transposon mutagenesis in gram-negative bacteria. Bio-Technology 1, 784–791. doi: 10.1038/nbt1183-784
Sollberger, G., Strittmatter, G. E., Garstkiewicz, M., Sand, J., and Beer, H. D. (2014). Caspase-1: the inflammasome and beyond. Innate Immun. 20, 115–125. doi: 10.1177/1753425913484374
Song, E. J., Lee, S. J., Lim, H. S., Kim, J. S., Jang, K. K., Choi, S. H., et al. (2016). Vibrio vulnificus VvhA induces autophagy-related cell death through the lipid raft-dependent c-Src/NOX signaling pathway. Sci. Rep. 6:27080. doi: 10.1038/srep27080
Takahashi, K. (2014). Influence of bacteria on epigenetic gene control. Cell. Mol. Life Sci. 71, 1045–1054. doi: 10.1007/s00018-013-1487-x
Toma, C., Higa, N., Koizumi, Y., Nakasone, N., Ogura, Y., McCoy, A. J., et al. (2010). Pathogenic Vibrio activate NLRP3 inflammasome via cytotoxins and TLR/nucleotide-binding oligomerization domain-mediated NF-KB signaling. J. Immunol. 184, 5287–5297. doi: 10.4049/jimmunol.0903536
Tsukamoto, H., Tanida, S., Ozeki, K., Ebi, M., Mizoshita, T., Shimura, T., et al. (2013). Annexin A2 regulates a disintegrin and metalloproteinase 17-mediated ectodomain shedding of pro-tumor necrosis factor-α in monocytes and colon epithelial cells. Inflamm. Bowel Dis. 19, 1365–1373. doi: 10.1097/MIB.0b013e318281f43a
van der Goot, F. G., and Harder, T. (2001). Raft membrane domains: from a liquid-ordered membrane phase to a site of pathogen attack. Semin. Immunol. 13, 89–97. doi: 10.1006/smim.2000.0300
Volonte, D., Galbiati, F., Li, S., Nishiyama, K., Okamoto, T., and Lisanti, M. P. (1999). Flotillins/cavatellins are differentially expressed in cells and tissues and form a hetero-oligomeric complex with caveolins in vivo. Characterization and epitope-mapping of a novel flotillin-1 monoclonal antibody probe. J. Biol. Chem. 274, 12702–12709. doi: 10.1074/jbc.274.18.12702
Wu, W. S., Wu, J. R., and Hu, C. T. (2008). Signal cross talks for sustained MAPK activation and cell migration: the potential role of reactive oxygen species. Cancer Metastasis Rev. 27, 303–314. doi: 10.1007/s10555-008-9112-4
Xu, Y., Kim, S. O., Li, Y., and Han, J. (2006). Autophagy contributes to caspase-independent macrophage cell death. J. Biol. Chem. 281, 19179–19187. doi: 10.1074/jbc.M513377200
Yuan, K., Huang, C., Fox, J., Laturnus, D., Carlson, E., Zhang, B., et al. (2012). Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. J. Cell Sci. 125(Pt 2), 507–515. doi: 10.1242/jcs.094573
Yuk, J. M., and Jo, E. K. (2013). Crosstalk between autophagy and inflammasomes. Mol. Cells 36, 393–399. doi: 10.1007/s10059-013-0298-0
Zhang, C., Hu, J. J., Xia, M., Boini, K. M., Brimson, C., and Li, P. L. (2010). Redox signaling via lipid raft clustering in homocysteine-induced injury of podocytes. Biochim. Biophys. Acta 1803, 482–491. doi: 10.1016/j.bbamcr.2009.12.006
Keywords: V. vulnificus, VvpM, macrophage, cell death, colonization, ANXA2, Atg5, NLRP3
Citation: Lee S-J, Jung YH, Kim JS, Lee HJ, Lee SH, Lee K-H, Jang KK, Choi SH and Han HJ (2017) A Vibrio vulnificus VvpM Induces IL-1β Production Coupled with Necrotic Macrophage Death via Distinct Spatial Targeting by ANXA2. Front. Cell. Infect. Microbiol. 7:352. doi: 10.3389/fcimb.2017.00352
Received: 27 April 2017; Accepted: 21 July 2017;
Published: 11 August 2017.
Edited by:
Margaret E. Bauer, Indiana University School of Medicine, United StatesReviewed by:
Chiou-Feng Lin, Taipei Medical University, TaiwanShivani Agarwal, Northwestern University, United States
Copyright © 2017 Lee, Jung, Kim, Lee, Lee, Lee, Jang, Choi and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ho Jae Han, aGpoYW5Ac251LmFjLmty