 Marcelo L. M. Pereira
Marcelo L. M. Pereira Claudio R. F. Marinho
Claudio R. F. Marinho Sabrina Epiphanio
Sabrina Epiphanio- 1Departamento de Imunologia, Instituto de Ciências Biomédicas, Universidade de São Paulo, São Paulo, Brazil
- 2Departamento de Parasitologia, Instituto de Ciências Biomédicas, Universidade de São Paulo, São Paulo, Brazil
- 3Departamento de Análises Clínicas e Toxicológicas, Faculdade de Ciências Farmacêuticas, Universidade de São Paulo, São Paulo, Brazil
Malaria is a serious disease and was responsible for 429,000 deaths in 2015. Acute lung injury/acute respiratory distress syndrome (ALI/ARDS) is one of the main clinical complications of severe malaria; it is characterized by a high mortality rate and can even occur after antimalarial treatment when parasitemia is not detected. Rodent models of ALI/ARDS show similar clinical signs as in humans when the rodents are infected with murine Plasmodium. In these models, it was shown that the induction of the enzyme heme oxygenase 1 (HO-1) is protective against severe malaria complications, including cerebral malaria and ALI/ARDS. Increased lung endothelial permeability and upregulation of VEGF and other pro-inflammatory cytokines were found to be associated with malaria-associated ALI/ARDS (MA-ALI/ARDS), and both were reduced after HO-1 induction. Additionally, mice were protected against MA-ALI/ARDS after treatment with carbon monoxide- releasing molecules or with carbon monoxide, which is also released by the HO-1 activity. However, high HO-1 levels in inflammatory cells were associated with the respiratory burst of neutrophils and with an intensification of inflammation during episodes of severe malaria in humans. Here, we review the main aspects of HO-1 in malaria and ALI/ARDS, presenting the dual role of HO-1 and possibilities for therapeutic intervention by modulating this important enzyme.
Malaria: General Overview
Malaria is a serious disease caused by the Plasmodium parasite and transmitted by the bite of the Anopheles mosquito. About 212 million cases of malaria were estimated to occur in 2015, killing approximately 429,000 people in the same year, with the majority of these cases (92%) originating from sub-Saharan Africa and occurring in children under 5 years of age (70%) (WHO, 2016). In Africa and Southeast Asia, of the 3.4 billion people at risk of malaria in 2015, 1.1 billion lived in high-risk areas with more than one case of malaria reported per 1000 inhabitants (WHO, 2016). The species that cause malaria in humans are Plasmodium falciparum, Plasmodium vivax, Plasmodium malariae, Plasmodium ovale, and Plasmodium knowlesi, with the first two being responsible for the greatest number of malaria cases (WHO, 2016). A natural infection by P. cynomolgi, a monkey parasite, was described in a woman in Malaysia (Ta et al., 2014).
The Plasmodium life cycle begins when an infected female Anopheles mosquito bites the intermediate host, injecting sporozoites into the host's blood circulation via the mosquito's saliva. Once in the circulatory system, sporozoites reach the host's liver, where they establish an intracellular asymptomatic infection in hepatocytes (Mota and Rodriguez, 2002). In the next 7–30 days (7–15 days for P. falciparum and P. vivax and 30 days for P. malariae), the parasite differentiates into thousands of merozoites, which are released in the hepatic sinusoid after rupturing the hepatocytes (Prudêncio et al., 2006; Bartoloni and Zammarchi, 2012). In the blood, the merozoites will in turn infect erythrocytes. During the symptomatic phase of malaria, several clinical complications can occur, which together define what is known as severe malaria (Trampuz et al., 2003).
The species P. falciparum is the most virulent and predominates in Africa. On the other hand, the species P. vivax is less virulent and is widely distributed throughout the world (WHO, 2015). However, despite P. vivax being less virulent, cases of severe P. vivax malaria have been reported (Lacerda et al., 2012a; Baird, 2013; Rahimi et al., 2014; Im et al., 2017). In addition, P. vivax, as with P. cynomolgi and P. ovale, can form hypnozoites, the dormant parasite form, in the liver stage, and these hypnozoites can be activated weeks or months after the initial infection, causing a relapse of symptomatic blood stage infection (White and Imwong, 2012).
Severe malaria has been described as a syndrome that may affect multiple organs and is, in many ways, similar to sepsis. This makes it difficult to diagnose at an early stage because it can be confused with other febrile diseases, such as pneumonia and central nervous system infection. The major clinical complications that characterize severe malaria are severe anemia, cerebral malaria, placental malaria, acute renal failure (Kurth et al., 2017) and acute lung injury/ acute respiratory distress syndrome (ALI/ARDS) (Cowman et al., 2016).
The main pathophysiological events that occur during malaria infection are: release of pro-inflammatory cytokines, adhesion of infected erythrocytes to endothelial cells of capillaries and venules, removal of infected erythrocytes from the bloodstream by splenic macrophages and rupture of parasitized erythrocytes with consequent release of factors that activate the inflammatory response, such as glycosylphosphatidylinositol, hemozoin, DNA and precipitated uric acid derived from the parasite (Gupte, 2013; Gallego-Delgado et al., 2014; Gazzinelli et al., 2014; Wassmer and Grau, 2017). These events are associated with each other and are responsible for the major syndromes of malaria. Splenic macrophages and monocytes are mainly responsible for the release of pro-inflammatory mediators. This is because both cell types are exposed to a high number of infected erythrocytes that they phagocytose. Nevertheless, severe anemia, the most common form of severe malaria, cannot be explained solely by the phenomenon of erythrocyte removal. Moreover, a suppressive effect on erythropoiesis by pro-inflammatory cytokines has been proposed (Gazzinelli et al., 2014). In addition to macrophages and monocytes, CD4+ T cells, gamma-delta T cells and NK cells also produce pro-inflammatory cytokines, such as interleukin (IL) 12, IFN-γ (interferon gamma) and TNF (tumor necrosis factor), at an early phase of malaria infection in humans. Among these cell types, CD14+ monocytes were notable producers of TNF and other chemokines. In CD4+ T cells, IL-10 expression was predominant. The involvement of cytokines such as IL-10, IL-6, MIP-1α (macrophage inflammatory protein 1 alpha), MIP-1β (macrophage inflammatory protein 1 beta), and MCP-2 (monocyte chemoattractant protein 2) is associated with a higher likelihood of severe malaria development in humans (Stanisic et al., 2014).
Of the syndromes of severe malaria, cerebral malaria is the best characterized and understood. In humans, P. falciparum invades erythrocytes, causing them to express a surface protein, P. falciparum erythrocyte membrane protein 1 (PfEMP-1), which is responsible for erythrocyte congestion and sequestration in the brain capillaries (Horata et al., 2009; Dorovini-Zis et al., 2011). This protein causes the infected erythrocyte to adhere to the endothelial membrane via the CD36 receptor and EPCR, consequently leading to the severity of cerebral malaria (Turner et al., 2013; Almelli et al., 2014; Bernabeu et al., 2016; Brazier et al., 2017). Erythrocyte sequestration in the brain was also observed in association with axonal and myelin damage, blood-brain barrier (BBB) disruption, coma and cellular immune responses, such as fibrin–platelet thrombi, intravascular accumulation of hemozoin–containing CD45/CD68-positive monocytes, necrosis of the endothelial lining of the occluded vessel and perivascular hemorrhage (Dorovini-Zis et al., 2011; Ponsford et al., 2012). In addition, concerning severe malaria, the adhesion of infected erythrocytes in intervillous spaces is an important factor in the development of placental malaria, which can lead to impairment of fetal development, low birthweight and miscarriage (Costa et al., 2006; Umbers et al., 2011).
There are a few studies on the possible importance of malaria-associated ALI/ARDS (MA-ALI/ARDS), both in vivo (Corbett et al., 1989; Aitken et al., 2014; Sercundes et al., 2016) and in vitro (Muanza et al., 1996; Carvalho et al., 2010). Several receptors, such as ICAM-1 (intercellular adhesion molecule 1) (Carvalho et al., 2010), and CD36 (Barnwell et al., 1989; Oquendo et al., 1989; Baruch et al., 1995, 1996, 1997; Chen et al., 2000), are involved in the adhesion of parasitized red blood cells (PRBCs), leukocytes and platelets to the endothelium (Sherman et al., 2003; Wassmer et al., 2003; Kirchgatter and Del Portillo, 2005). More recently, it has been demonstrated that EPCR is involved in the cytoadhesion of PfEMP1-expressing parasites to lung endothelial cells (Avril et al., 2016), suggesting that this receptor may be involved in the pathogenesis of MA-ALI/ARDS.
The involvement of different immune cells in various organs during the appearance of the first signs of malaria reinforces the hypothesis that multiple host factors, including endothelial activation, cytoadhesion, the inflammatory response and coagulation dysregulation, are involved in the development of severe malaria syndromes, including ALI/ARDS.
Acute Lung Injury/Acute Respiratory Distress Syndrome (ALI/ARDS)
In 1967, the first description of ALI/ARDS indicated that it was a syndrome characterized by severe dyspnea, tachypnea, hypoxia, loss of pulmonary compliance and the presence of diffuse alveolar infiltrates detected by chest X-ray (Ashbaugh et al., 1967). In 1994, criteria were established by the American-European Consensus to define ALI and its most severe form, ARDS (Bernard et al., 1994). However, experts met in Berlin to re-define the criteria for ALI/ARDS in 2012. According to this new definition, the term ALI was removed from use, and ARDS in humans is now defined by three subcategories, depending on the degree of hypoxemia: benign (200 mm Hg < PaO2/FIO2 ≤ 300 mmHg), moderate (100 mmHg < PaO2/FIO2 ≤ 200 mmHg) and severe (PaO2/FIO2 ≤ 100 mmHg) (Ranieri et al., 2012). In addition, other criteria were considered in the definition of ARDS, such as bilateral opacities detected by chest X-ray or by computerized tomography. The first signs of ALI/ARDS are occur either within a week after the stimulus that led to them, after a worsening of existing symptoms or after the new signs onset. Also, ARDS is defined by non-cardiogenic edema and by a positive end expiratory pressure less than 5 cm H2O for benign ARDS or higher than or equal to 10 cm H2O for severe ARDS. Finally, ARDS is defined by a corrected expired volume per minute higher than 10 L/min and/or by a low pulmonary compliance (less than 40 mL/cm of H2O) (Ranieri et al., 2012). Although the Berlin definition did not reach a general agreement, other authors have proposed new modifications to classify ARDS, due to low feasibility of ventilation, arterial blood gas measurements and chest radiographs, especially in developing countries (Riviello et al., 2017).
Aside from this controversy over definitions, ALI/ARDS affects more adults than children, has a high mortality rate and is characterized by dyspnea, tachypnea and hypoxemia that progress rapidly to acute respiratory failure characterized by reduced lung compliance, increased phagocytic activity and increased levels of inflammatory mediators (Monahan, 2013). The most common causes of ALI/ARDS in humans are bacterial and viral pneumonia. There are other common causes, such as sepsis derived from non-pulmonary infections, gastric aspiration and severe trauma. Other less common causes are acute pancreatitis, transfusions, adverse drug reactions and fungal or parasitic infections of the lungs (Matthay et al., 2012).
Hantavirus infection, caused by RNA virus of Bunyaviridae family, is characterized by flu like symptoms in the first 3 days that rapidly progress to respiratory distress, with dyspnea, lung infiltrates, pleural effusion and peribronchovascular/smooth septal thickening that are typical in ALI/ARDS of different causes (Pinto et al., 2014; de Lacerde Barbosa et al., 2017).
In animal models, there is evidence that hyperoxia-associated ALI leads to elevated levels of reactive oxygen species (ROS) that damage endothelial and lung epithelial cells, including type I pneumocytes (via cell death) and cause damage to the basement membrane (Zhang et al., 2003; Thiel et al., 2005). Furthermore, it is believed that endothelial responses are important in protection against hyperoxia-induced death due to the fact that endothelial cells constitute more than 30% of all lung cells (Crapo et al., 1982). Besides hyperoxia, there are other causes of lung endothelial damage, such as influenza virus infection, which was shown to cause platelet adhesion to the lung endothelium and thereby contribute to lung injury (Sugiyama et al., 2015). Bacterial lipopolysaccharide (LPS) is also a cause of lung endothelial cell activation and dysfunction, and its effects are mitigated by hepatocyte growth factor (HGF) (Meng et al., 2015). Recently, a murine model of hantavirus infection that mimic the lung damage in humans, was created, using immunosuppressed Syrian hamsters (Vergote et al., 2017). Histologic findings are similar to other models of ALI/ARDS: hemorrhagic alveolar edema, inflammatory cell infiltration and fibrin deposition (Vergote et al., 2017).
Endothelial damage with the consequent increase in pulmonary vascular permeability is a key factor in the pathogenesis of ALI/ARDS. This has been observed in ALI/ARDS cases associated with sepsis and pneumonia in humans, in which there is a subsequent inability to remove alveolar fluid from the alveolar epithelium (Holter et al., 1986; Constantin et al., 2007; Jabaudon et al., 2016). In murine models, it has been found that pulmonary vascular permeability is present in ALI/ARDS induced by LPS (Meng et al., 2015; Konrad et al., 2016), thrombin, histamine, TNF-alpha and VEGF. These stimuli are responsible for inducing a downstream cascade that culminates in disruption of endothelial junctions, particularly the adherens junctions, by disassembly of the VE-cadherin complex (Cerutti and Ridley, 2017).
During ALI/ARDS, the removal of extracellular fluid from the alveoli occurs due to the activity of the sodium-potassium ATPase pump in alveolar epithelial cells. In addition to the alveolar epithelium, the endothelium is also affected by the formation of spaces between the junctions of endothelial cells (Gonzales et al., 2015). As mentioned above, these spaces are formed as a result of cytoskeletal remodeling of endothelial cells induced by various inflammatory factors, such as vascular endothelial growth factor (VEGF), which is also responsible for disassembly of the VE-cadherin complex (Sukriti et al., 2014; Cerutti and Ridley, 2017). VEGF, a glycoprotein of the platelet-derived growth factor (PDGF) superfamily that is required for vasculogenesis and angiogenesis, is involved in vascular permeability and in the survival of endothelial cells (Gerber et al., 1998; Drake et al., 2000).
Malaria-Associated ALI/ARDS
MA-ALI/ARDS is a major cause of death in adults. It has a high mortality rate (80% to 100%) and can occur before, during or after anti-malarial treatment (Tran et al., 1996; Maguire et al., 2005; Kochar et al., 2009). ALI/ARDS has been diagnosed in patients suffering from malaria caused by all known species of human malaria, including P. malariae (Lozano et al., 1983), P. ovale (Haydoura et al., 2011) and P. knowlesi (Singh and Daneshvar, 2013). However it is more common in infections due to P. falciparum and P. vivax, with an estimated incidence of 5–25 and 1–10% respectively, in infected adults (Mohan et al., 2008; Taylor et al., 2012; Table 1). Among P. falciparum infected children, the incidence of ALI/ARDS varies between 7% (133 cases in 1844 infected children in Kenya) and 16% (867 cases in 5425 children from nine African countries) (Marsh et al., 1995; Dondorp et al., 2010). Pregnant women infected with P. falciparum are in higher risk of developing MA-ALI/ARDS. The incidence of MA-ALI/ARDS in this group is up to 29% (Taylor et al., 2012).
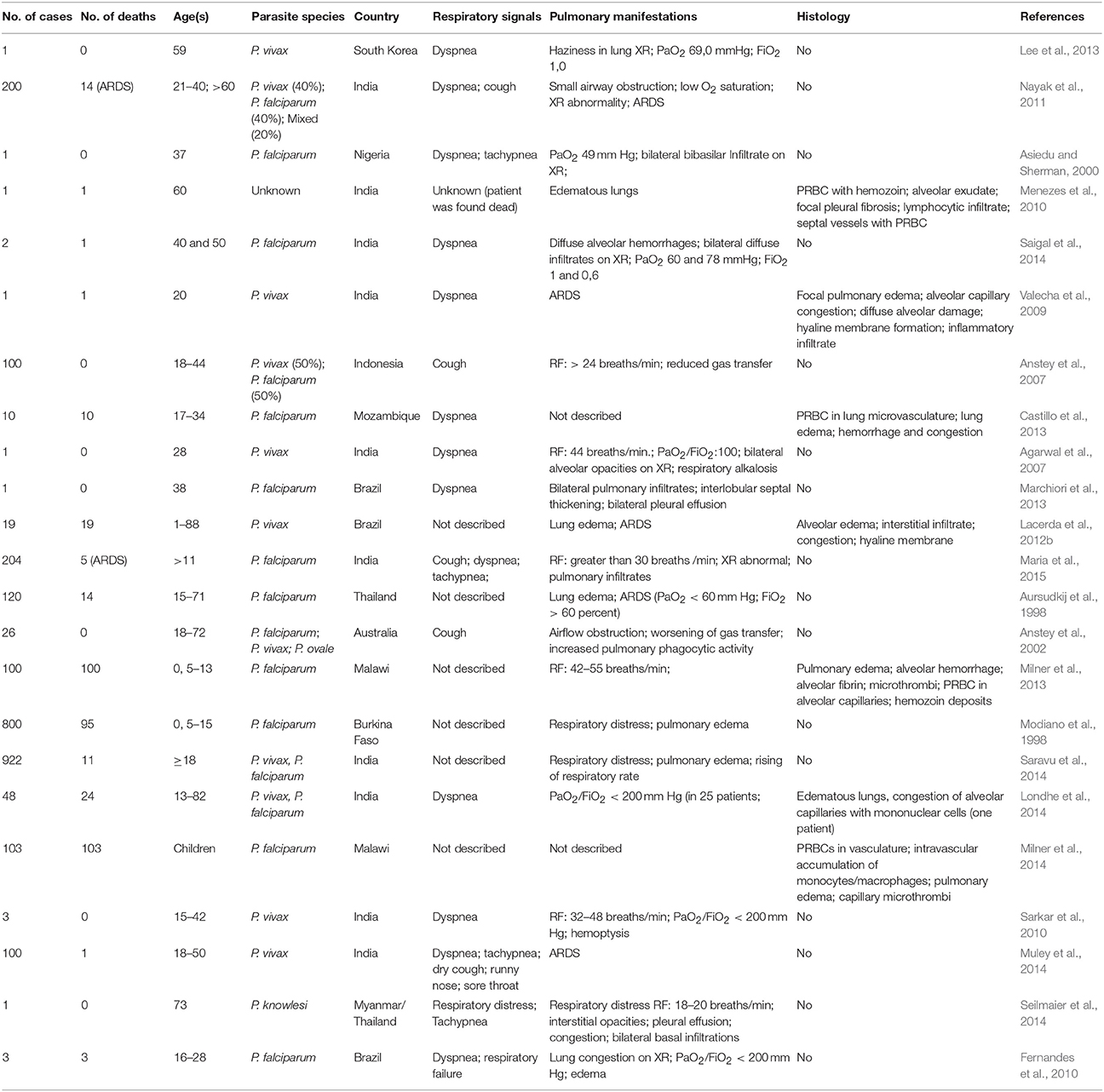
Table 1. Cases of malaria-associated pulmonary complications in humans.
Respiratory symptoms such as cough, respiratory rate increase and a decrease in forced midexpiratory flow can be present even in uncomplicated malaria cases. However, most patients suffering from severe malaria that develop ALI/ARDS are characterized by the presence of pulmonary vascular occlusions and impaired alveolar capillary membrane function (Maguire et al., 2005; Anstey et al., 2007). Additionally, in patients with P. vivax malaria, a reduced pulmonary capillary vascular volume of hemoglobin-containing cells was suggested to occur due to the sequestration of infected erythrocytes within the pulmonary vasculature (Anstey et al., 2007).
It is known that the presence of intravascular fluid in the lungs due to increased alveolar permeability is the key pathophysiological mechanism of MA-ALI/ARDS (Mohan et al., 2008). Multiple factors are possibly involved in this increased permeability, such as increased levels of pro-inflammatory cytokines such as TNF-α, IL-1, IL-6, and IL-8. These factors may also include the endovascular occlusions associated with the accumulation of erythrocytes with reduced deformability, leukocytes and infected erythrocytes as well as the endothelial injury caused by adhesion of infected erythrocytes (Mohan et al., 2008). As previously mentioned, a number of receptors participate in the adhesion of infected erythrocytes, such as CD36, ICAM-1 and EPCR (Carvalho et al., 2010; Avril et al., 2013, 2016; De las Salas et al., 2013). The P. falciparum PfEMP1 protein is known to interact with ICAM-1, an endothelial adhesion molecule that participates in leukocyte transmigration across endothelial cells, in a process that involves the activation of a signaling cascade that leads to disruption of VE-cadherin junctions (Sherman et al., 2003; Cerutti and Ridley, 2017). Additionally, the adhesion of P. falciparum parasitized erythrocytes to CD36 (Sherman et al., 2003) initiates a pro-inflammatory cascade in mononuclear cells through the association of CD36 with Lyn and Fyn, which are members of the Src phosphotyrosine kinase family (Moore et al., 2002). Fyn was shown to be activated in a TLR4 mediated cascade, culminating in the disruption of VE-cadherin junctions and a consequent increase in permeability (Gong et al., 2008). More recently, it was shown that knockdown of CD36 and Fyn reduced lung endothelial barrier dysfunction in a murine model of malaria (Anidi et al., 2013). Expression of VEGF was found to be increased in a murine model and was involved in the increase in vascular permeability observed in MA-ALI/ARDS (Epiphanio et al., 2010). This syndrome may also be associated with other factors, such as heart failure or tachypnea associated with cerebral malaria (Good et al., 2005). Thus, many factors are associated with endothelial permeability and are essential mechanisms of MA-ALI/ARDS. However, the biomarkers that are critical to the development of ALI/ARDS are not yet well defined (Janz and Ware, 2013). Therefore, the study of pro-inflammatory biomarkers, particularly cytokines or proteins related to cellular stress (such as heme oxygenase), is of central importance.
Malaria-Associated ALI/ARDS Models
It has been shown that the pathogenesis of MA-ALI/ARDS is multifactorial and that PRBC endothelium sequestration, inflammation and hemostasis are all involved in the pathogenesis of severe malaria, especially cerebral malaria (van der Heyde et al., 2006), are also relevant in the development of MA-ALI/ARDS.
Murine models are an important connection between patients from an endemic zone and the research laboratory. Emerging hypotheses in human studies can be tested in animal models, which provide valuable information on the pathogenesis of a disease. Additionally, different murine models have been developed to study MA-ALI/ARDS and show aspects similar to human ALI/ARDS cases (Table 2).
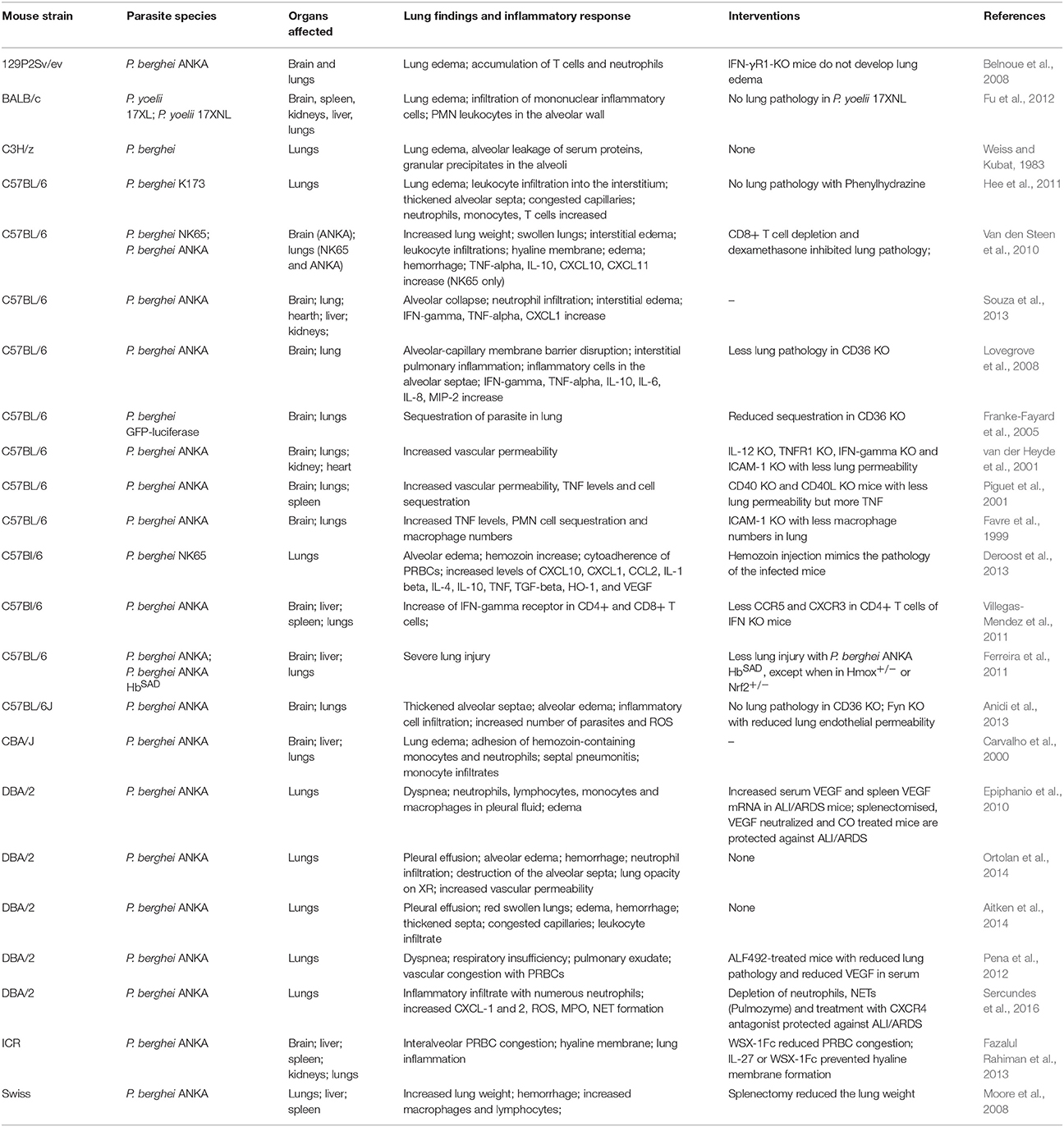
Table 2. Mouse models of ALI/ARDS.
The DBA/2 mouse strain develops ALI/ARDS when infected with the parasite P. berghei ANKA (Epiphanio et al., 2010; Ortolan et al., 2014). Around 50% of the mice from this strain that die between the 7th and 12th days after infection have dyspnea, hypoxemia, reduced respiratory rate and lung opacification identified by X-ray analysis (Epiphanio et al., 2010; Ortolan et al., 2014). Post-mortem studies revealed that these mice had pleural effusions containing neutrophils, lymphocytes, monocytes and macrophages (Aitken et al., 2014; Ortolan et al., 2014; Sercundes et al., 2016). When infected with P. berghei NK65, C57BL/6 mice also develop severe malaria that is characterized by cellular infiltration, edema in lung interstitial tissue, alveolar edema and hyaline membrane formation, which is typical of ALI/ARDS. In this model, approximately 90% of infected mice die from ALI/ARDS (Van den Steen et al., 2010). In these two models, the mice develop MA-ALI/ARDS without developing signs of cerebral malaria, which make them useful for studying this syndrome. Furthermore, VEGF has been identified as being a critical mediator of increased pulmonary vascular permeability, a hallmark of ALI/ARDS (Epiphanio et al., 2010; Pena et al., 2012). In addition to VEGF, cytokines and chemokines, such as TNF, IL-10 (interleukin 10), IL-1β (interleukin 1 beta), IL-6, IL-4, INF-γ, chemokine (C-C motif) ligand 2 (CCL-2), chemokine (C-X-C motif) ligands (CXC) CXCL1, CXCL2, CXCL10, CXCL11, and myeloperoxidase (MPO), were shown to be increased during ALI/ARDS in other studies (Favre et al., 1999; Piguet et al., 2001; Lovegrove et al., 2008; Van den Steen et al., 2010; Deroost et al., 2013; Souza et al., 2013; Sercundes et al., 2016). Recently, the formation of Plasmodium-induced neutrophil extracellular traps (NETs) has been found to contribute to MA-ALI/ARDS pathogenesis (Sercundes et al., 2016). In addition to causing inflammatory factor induction, malaria infection also leads to the release of ROS and free heme from infected erythrocytes during the blood stage (Ferreira et al., 2008). These compounds are harmful to the host, which reacts by increasing the expression of heme oxygenase (HO-1), a heme catabolizing enzyme (Ferreira et al., 2008).
The Heme Oxygenase System
The heme oxygenase system has been intensively studied due to its regulatory properties in both physiological and pathological processes. Heme oxygenase has two isoforms: HO-1, which is inducible and heme oxygenase-2 (HO-2), which is constitutive. The enzymatic activities of both isoforms are identical. HO-2 possibly regulates normal physiological cellular functions, whereas HO-1 is expressed in all cells and is highly inducible by a variety of stimuli. HO-1, encoded by the hmox-1 gene, is considered protective due to its anti-inflammatory, anti-apoptotic and anti-proliferative actions in different cell types, including endothelial cells (Soares et al., 1998). HO-1 is expressed in all cells at low levels, but is rapidly inducible by various stimuli, including heme (Agarwal and Bolisetty, 2013). This enzyme participates in the degradation of free heme, derived from hemoglobin and myoglobin, into equimolar amounts of CO, iron and biliverdin. Biliverdin is subsequently reduced to bilirubin by biliverdin reductase. HO-1 plays a protective role in various organs by modulating tissue responses to injuries, including the lung injury associated with hyperoxia as well as pulmonary hypertension and pulmonary fibrosis (Otterbein et al., 1999; Christou et al., 2000; Tsuburai et al., 2002).
HO-1 provides cellular protection through the degradation of free heme, because this action has pro-oxidant effects (Balla et al., 1991; Tracz et al., 2007). Additionally, the HO-1 reaction products also contribute to the protective response. In response to oxidative stress, cells change their gene expression to activate protective genes. The transcription factor Nrf2 and its target genes (involved in antioxidative cell protection) are important in protecting against ROS and cell damage. (Stocker et al., 1987; Nakagami et al., 1993; Motterlini and Otterbein, 2010).
Several drugs are known to modulate the expression of HO-1. A compound called desoxyrhapontigenin decreases ROS and peroxynitrite production and induces HO-1 via activation of Nrf2. This drug also leads to an improvement in pulmonary inflammation induced by LPS in ICR (imprinting control region) mice (Joo Choi et al., 2014). Besides desoxyrhapontigenin, other compounds such as statins, hemin, curcumin, quercetin, carnosol and cobalt (inducers) and tin and zinc protoporphyrins (inhibitors) are also known modulators of HO-1. (Pamplona et al., 2007; Fei et al., 2012; Zhou et al., 2013; Luo et al., 2014; Chi et al., 2016; Pereira et al., 2016; Yu et al., 2016; Immenschuh et al., 2017). Moreover, carbon monoxide (CO), one of the end products of the HO-1 reaction, shows the same anti-inflammatory, anti-apoptotic, and anti-proliferative effects as HO-1. For this reason, CO-releasing molecules (CO-RMs) (lipid-soluble CORM-2 [Ru(CO)3Cl2]2, and water-soluble CORM-3 [Ru(CO)3Cl2 (H2NCH2CO)2], CORM-401 and ALF499) (Pena et al., 2012; Choi et al., 2015; Hettiarachchi et al., 2017; Inoue et al., 2017; Sun et al., 2017) have been broadly used. Hence, modulating HO-1 expression by pharmacologically targeting this enzyme has promise as a therapeutic application in humans and could be used to inhibit pulmonary inflammation.
The Modulation of HO-1 in ALI/ARDS
It has been shown that the inhibition of HO-1 led to a worsening of ALI/ARDS signs in rats after ischemia-reperfusion (I/R) of the lower limbs (Boutros et al., 2005). On the other hand, cobalt protoporphyrin and hemin, both inducers of HO-1, were shown to be protective against ALI/ARDS (Yin et al., 2011; Pereira et al., 2016; Wang et al., 2017). HO-1 is also involved in endotoxemia, and the induction of HO-1 expression led to a reduction of LPS-induced ALI/ARDS in rats (Otterbein et al., 1995).
Likewise, in a sepsis-induced ALI/ARDS murine model it has been shown that hemin inhibits NLRP3 inflammasome activation through the action of HO-1 (Luo et al., 2014). The NLRP3 inflammasome regulates the maturation of the pro-inflammatory cytokines IL-1β and IL-18 and can be activated by various stimulating factors such as bacteria, viruses, fungi and DAMPS (danger associated molecular patterns) (Schroder and Tschopp, 2010; Anand et al., 2011). Deregulated activation of NLRP3 is present in sepsis-associated ALI/ARDS in mice, and in the absence of NLRP3 activation there is a reduction in pro-inflammatory cytokine and neutrophil levels in bronchoalveolar lavage fluid (Grailer et al., 2014).
Additionally, hemin treatment reduces the levels of oxidative stress markers in lung tissue and reduces the severity of ALI/ARDS in treated mice compared to untreated mice or mice treated with the HO-1 inhibitor zinc protoporphyrin IX (ZnPPIX) (Luo et al., 2014). However, it was recently shown that ZnPPIX has a deleterious effect in a cecal ligation and puncture (CLP) mouse model. In this study, it was shown that the antimalarial drug artesunate induced HO-1 gene expression, and treated mice exhibited less pronounced sepsis mortality, lung injury and neutrophil infiltration (Cao et al., 2016). Also, in a recent study, cobalt protoporphyrin IX was shown induce HO-1 in human cells and in mice infected by human respiratory syncytial virus (hRSV) (Espinoza et al., 2017). The hRSV virus is the main cause of lower respiratory tract illness in children up to five years old worldwide (Rudan et al., 2013). The induction of HO-1 in human respiratory tract epithelial cells led to a reduction of hRSV replication. Additionally, the levels of HO-1 were increased in infected dendritic cells in vitro and in HO-1 inducer-treated mice, which were protected due to a reduction in virus replication and to a decrease of neutrophil infiltration and inflammation in their lungs (Espinoza et al., 2017).
In another study it was observed that HO-1 deficient mice had decreased survival to influenza virus infection, increased levels of lung inflammation and also had impaired production of antibodies following influenza vaccination when compared with wild type mice (Cummins et al., 2012). Also, humans with SNPs in both HO-1 and HO-2, expressing lower levels of them, showed to have lower production of antibodies after influenza vaccination (Cummins et al., 2012). These results reinforce the hypothesis that the observed increased expression of HO-1 in patients may be due to host efforts to reverse the phenotype of ALI/ARDS.
To study the role of HO-1 in ALI/ARDS, chemical inhibitors, such as ZnPPIX, or hmox-1 gene deletion (HO-1-KO) can be used in murine models. However, the use of metalloporphyrins has been shown to have adverse effects, which may interfere with the results of studies on HO-1 (Grundemar and Ny, 1997). Additionally, HO-1-KO tests have the following problems: partial lethality during prenatal development; infertility; smaller size of the HO-1-KO mice relative to wild-type mice; development of microcytic normochromic anemia in HO-1-KO animals; iron deposition in organs, such as the liver and kidneys, which can affect the results of the experiments (Agarwal and Nick, 2000; Gozzelino et al., 2010).
To overcome these problems, it was found that specific inhibition of pulmonary HO-1 through interfering RNA (siRNA) makes it possible to study the effect of post-natal silencing of HO-1 in a lung-targeted manner. To achieve this, Zhang et al. designed specific lentiviral constructs for certain lung cells (Zhang et al., 2013). In animal models, hyperoxia leads to the presence of high amounts of ROS, which damage lung endothelial and epithelial cells, including type I pneumocytes (via cell death), and cause damage to the basement membrane (Zhang et al., 2003; Thiel et al., 2005). When lentiviruses were used to silence HO-1 expression in lung endothelial cells in a model of hyperoxia-induced acute lung injury (HALI), increases in inflammatory cytokines (IL-1β, IL-6 and TNF-α), apoptosis (caspase 3-mediated) and a decrease in autophagy were observed (Zhang et al., 2013). Finally, it was concluded that the HO-1-knockdown in endothelial cells was just as bad as in the whole lung in a murine model of hyperoxia, since there was a reduction in survival compared to the controls in both cases (Zhang et al., 2013). This emphasizes the importance of endothelial cells in controlling the inflammatory response via HO-1 expression.
The Dual Role of HO-1 in Malaria
The role of HO-1 in malaria has long attracted interest among the scientific community and clinicians (Pamplona et al., 2007; Walther et al., 2012; Pereira et al., 2016). There is a number of studies in which HO-1 was observed to be important in malaria infection in humans and in murine models, because its expression was increased during the infection or because its modulation had an effect on the development of the disease (Table 3).
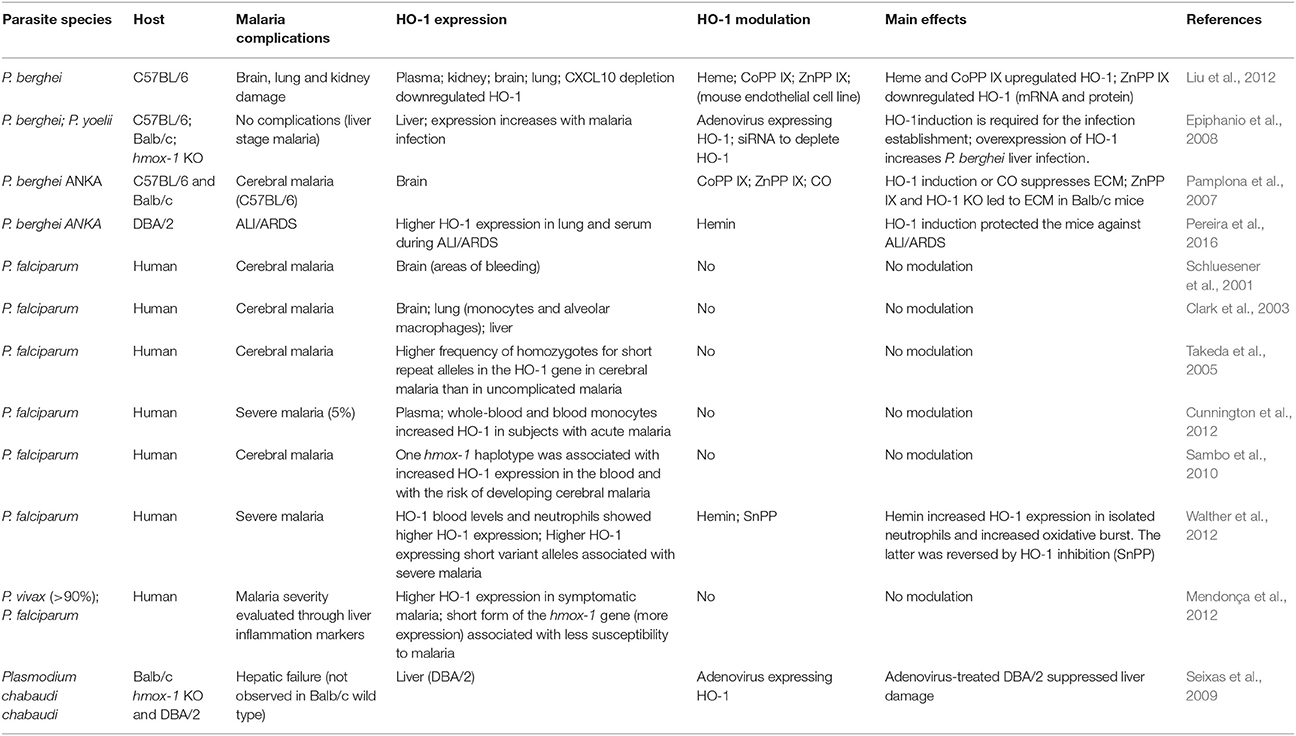
Table 3. Studies on the role of HO-1 in malaria.
As in the cases of hyperoxia, endotoxemia and I/R, malaria infection also leads to the release of ROS and free heme, both of which are harmful to the host's endothelial cells. In addition, heme influences neutrophil activation, leading to a respiratory burst, chemotaxis and formation of NETs (Immenschuh et al., 2017). However, HO-1 in turn catabolizes free heme into iron, biliverdin and CO, which are less toxic to the cells. When exposed to free heme, host cells increase expression of HO-1 (Ferreira et al., 2008).
However, although some of those studies conclude that HO-1 plays a protective role (Pamplona et al., 2007; Seixas et al., 2009), it is known that expression of this inducible enzyme is not always beneficial. At the beginning of the malaria infection, that is, in the asymptomatic phase (hepatic stage of the disease), HO-1 expression is harmful, since it reduces inflammation and consequently provides a favorable environment for the development and multiplication of parasites (Epiphanio et al., 2008).
In addition, a study performed in Gambian children (Walther et al., 2012) showed that high levels of HO-1 were associated with severe malaria. The authors also observed an increase in HMOX1 mRNA expression in inflammatory cells. Surprisingly, they induced HO-1 expression in neutrophils using hemin, which increases the oxidative burst (Walther et al., 2012), suggesting that serious damage to endothelial cells is the mechanism by which HO-1 contributes to ALI/ARDS. On the other hand, the increase in HO-1 induced by chronic hemolysis induces an accumulation of immature granulocytes in the blood, leading to a reduction in the oxidative burst (Cunnington et al., 2011). This decrease in respiratory burst could favor new bacterial infections (Cunnington et al., 2011), predisposing patients with malaria to co-infections, which is also a potential mechanism that could facilitate ALI/ARDS (Lacerda et al., 2012b; Mueller et al., 2012).
Recently, it has been found that Mycobacterium tuberculosis induced the HO-1 overexpression in macrophages from humans and mice, in a mechanism that was dependent on the presence of ROS. HO-1 increased levels were also observed during HIV coinfection, which were directly proportional to the viral load and to the severity of tuberculosis. It was also concluded that HO-1 is useful as a biomarker for both tuberculosis infection and treatment (Rockwood et al., 2017). Additionally, the dependence of HO-1 levels on ROS reinforces the possibility that the HO-1 increase constitutes an effort to revert the exacerbated inflammatory response and minimizes the damage caused by ROS. Thus, despite being increased during the worsening phase of tuberculosis and malaria, the role of HO-1 in these diseases seems to be beneficial. However, the stage of malaria infection seems to be important in defining the role of HO-1. At the beginning of a malarial infection at the liver stage, an increase in the HO-1 levels might be deleterious because it lowers the inflammatory response, which is important to reduce the parasite numbers at this stage. Nevertheless, in posterior stages of infection (blood stage), characterized by the accumulation of free heme as a result of parasitized erythrocyte burst and inflammatory response, HO-1 increase would be more beneficial as it would prevent the toxic effects of heme and reduce an exacerbated inflammation.
It has been observed that MA-ALI/ARDS in humans frequently occurs at late stages of malaria infection and even after the completion of the malaria treatment (Tong et al., 1972; Taylor et al., 2006). Additionally, it has been hypothesized that the accumulation of hemozoin in lung, which stays in tissue even after the parasite clearance, plays an important role in MA-ALI/ARDS. As it has been shown, hemozoin is a potent inducer of the inflammatory response and is correlated with MA-ALI/ARDS (Deroost et al., 2013). Therefore, HO-1 might be a target to prevent MA-ALI/ARDS, avoiding the oxidative burst and deleterious effects of an exacerbated and harmful inflammatory response.
Besides the previous observations in humans, different studies have shown the effects of modulating HO-1 in different infection models (Table 4). These studies have also demonstrated that inducers of HO-1, such as hemin and cobalt protoporphyrin IX (CoPPIX), protected mice infected with malaria or suffering from other diseases, such as polymicrobial sepsis, from ALI/ARDS (Pamplona et al., 2007; Fei et al., 2012; Luo et al., 2014). Treatment with HO-1 inhibitors, such as ZnPPIX or tin-protoporphyrin, led to a worsening of ALI/ARDS signs in cases of sepsis and hyperoxia, but had no effect in experimental cerebral malaria (ECM) (Pamplona et al., 2007; Siner et al., 2007; Ballinger et al., 2012; Fei et al., 2012). In C57BL/6 mice, the induction of HO-1 suppressed the pathogenesis of ECM, preventing the breakdown of the blood brain barrier and reducing neuroinflammation (Pamplona et al., 2007). However, chemically inhibiting HO-1 with ZnPPIX had no effect on pathogenesis in C57BL/6 mice. Nevertheless, it was observed that HO-1-KO Balb/c mice died due to this form of severe malaria, while the wild-type strain did not develop ECM. The same result was observed in Balb/c mice treated with ZnPPIX (Pamplona et al., 2007). Furthermore, it was shown that the induction of HO-1 expression by inoculation of mice with a recombinant adenovirus containing the HO-1 gene protected them against malaria-associated hepatic failure (Seixas et al., 2009). We previously observed that DBA/2 ALI/ARDS-developing mice infected by Plasmodium berghei have increased levels of the HO-1 protein in their lungs and serum, and HO-1 induction improved respiratory parameters, reduced inflammatory cytokines and protected the alveolar capillary barrier in these mice (Pereira et al., 2016). On the other hand, in a study performed in humans, it was found that the variant of the HO-1 promoter (hmox1) with the shortest repeating (GT)n (n < 27), was associated with higher HO-1 expression in peripheral blood leukocytes compared to the variant with the longest repetition (n > 32) (Walther et al., 2012). High HO-1 expression led to higher levels of HO-1, and those elevated levels were associated with severe malaria complications, including ALI/ARDS (Takeda et al., 2005; Sambo et al., 2010; Cunnington et al., 2012; Walther et al., 2012). However, in a study in Brazil, the opposite was observed. Asymptomatic malaria was more frequent in individuals carrying the short form of the HO-1 allele (Mendonça et al., 2012). Importantly, it was shown that the development of MA-ALI/ARDS is suppressed by administration of exogenous CO, and high VEGF levels are associated with the development of this syndrome (Epiphanio et al., 2010). In addition, pulmonary endothelial permeability levels were increased in this murine model, which also exhibited alveolar edema and pleural effusion (Epiphanio et al., 2010; Ortolan et al., 2014). The induction of HO-1 by hemin treatment led to not only a reduction in the mortality due to MA-ALI/ARDS, but also to a decrease in VEGF protein levels in serum and to protection of the endothelial cells against apoptosis (Pereira et al., 2016). Therefore, understanding the mechanisms of endothelial survival factors, such as HO-1 and VEGF, may have important therapeutic implications. Our data have shown that there might be a mechanism of regulation between HO-1 and VEGF. Consequently, it is important to determine the mechanism by which HO-1 has a protective role in MA-ALI/ARDS.
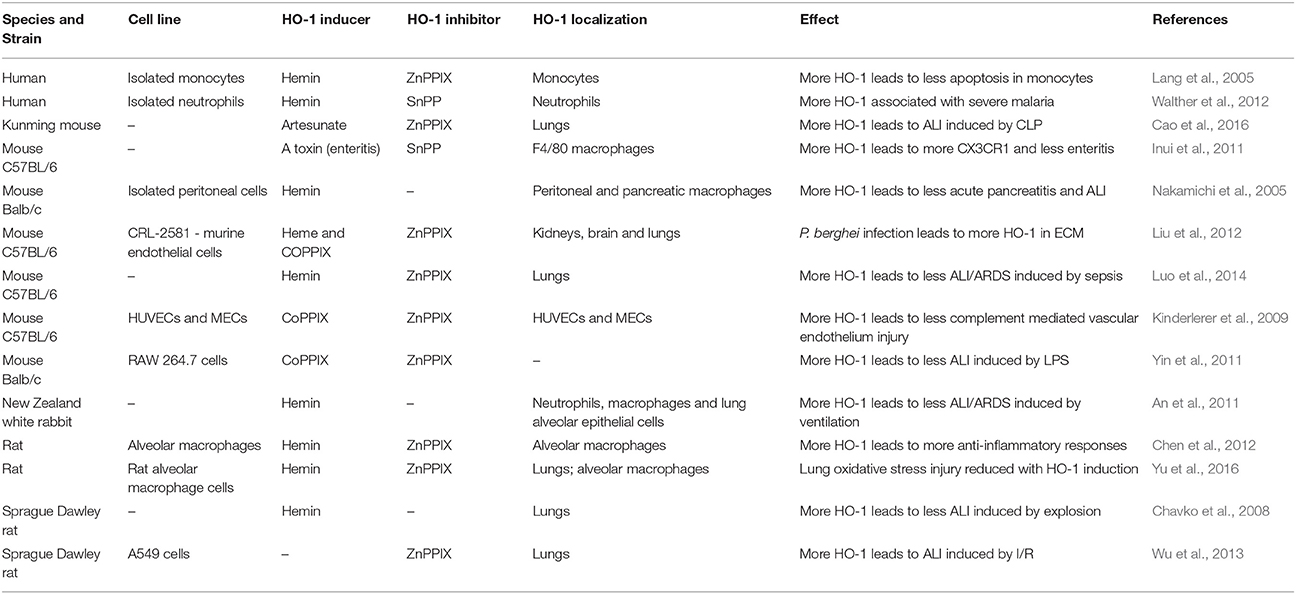
Table 4. Modulation of HO-1 in different disease experimental models.
As shown in the literature reviewed here, HO-1 has an important role in the development of severe malaria (Figure 1). It has also been shown that HO-1 can be modulated in a number of different ways to ameliorate the complications of malaria, particularly ALI/ARDS. This makes HO-1 a target for new drugs such as ALF492, which is efficient in reducing the severity of both ECM and MA-ALI/ARDS (Pena et al., 2012). Although most of the studies on HO-1 show that its induction is beneficial, (Table 4) the roles of this enzyme in diseases such as malaria must be determined in more detail to know which drugs, dosages and time points will be best in preventing ALI/ARDS.
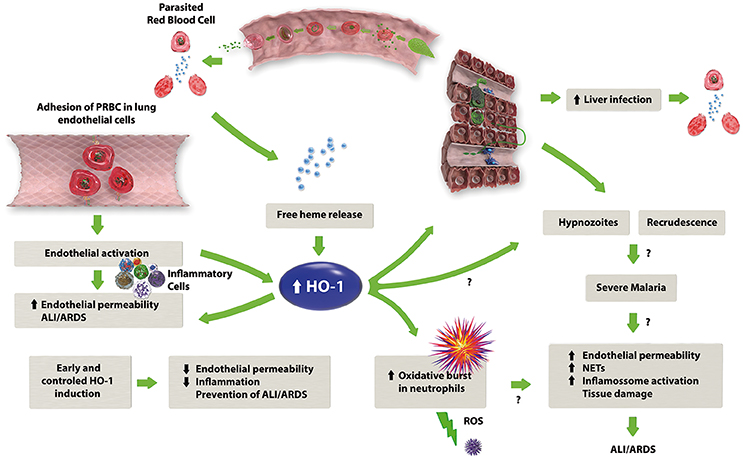
Figure 1. The proposed mechanism of HO-1 in the development of MA-ALI/ARDS. Malaria infection leads to the lysis of erythrocytes with consequent release of free heme, which in combination with the adhesion of infected erythrocytes to the endothelium, causes endothelial activation with subsequent release of pro-inflammatory cytokines and an increase in endothelial permeability. The release of free heme also upregulates HO-1 expression, which may have a dual role: on one hand, HO-1 activity results in the production of anti-inflammatory factors, such as CO and biliverdin, reducing the inflammation caused by malaria. On the other hand, HO-1 could stimulate the oxidative burst in neutrophils, leading to an increase in the inflammatory response.
Final Remarks and Perspectives
The highly complex signaling and cellular arrangements that orchestrate lung injury represent serious challenges in developing new therapies for severe malaria complications, especially MA-ALI/ARDS.
Merely controlling malarial infections with anti-malarial drugs does not regulate pulmonary vascular permeability, since patients can develop ALI/ARDS after anti-malarial treatment is completed. Lung edema results in tissue hypoxia and organ dysfunction, leading to the high mortality observed among ALI/ARDS patients (Mohan et al., 2008).
The induction of HO-1 may be an important target in controlling pulmonary vascular permeability and inflammation (Pereira et al., 2016). However, we must not forget that malaria blood infection already increases HO-1 expression (Pamplona et al., 2007; Epiphanio et al., 2010; Pereira et al., 2016), which is associated with disease severity (Walther et al., 2012). The liver stage may coexist with the blood stage (HO-1 increase) in endemic areas where the infection rate is high and one individual could be infected multiple times in a short time frame (al-Yaman et al., 1997). As HO-1 has an anti-inflammatory effect, it promotes Plasmodium development and multiplication in the hepatocytes (Epiphanio et al., 2008), which increases parasite load and could contribute to the severity of the disease. In addition, it is completely unknown whether HO-1 expression might have some influence on hypnozoites or recrudescence cases, which increases the complexity in understanding the action of HO-1 in this context. Despite this, HO-1 induction was shown to be beneficial in preventing severe malaria syndromes, as cerebral malaria and MA-ALI/ARDS (Pamplona et al., 2007; Pena et al., 2012; Pereira et al., 2016). Additionally, HO-1 was increased during those syndromes not only in mice but also in humans (Takeda et al., 2005; Pamplona et al., 2007; Sambo et al., 2010; Cunnington et al., 2012; Walther et al., 2012; Pereira et al., 2016). Therefore, we hypothesize that high levels of HO-1 are a pausible biomarker of the severity of malaria. However, despite favoring the malaria parasite replication in the liver stage, HO-1 activity has significant role to prevent severe malarial syndromes, including MA-ALI/ARDS.
Understanding the HO-1 mechanisms at play during the different stages of Plasmodium infection is fundamentally important. Using new models, such as the human liver-chimeric FRG KO huHep mouse (Mikolajczak et al., 2015; in which it is possible to study the liver stage, the formation and activation of hypnozoites and the blood stage of infection at the same time) might be useful for discovering the true role of HO-1 in Plasmodium infections and its importance in severe malaria, especially MA-ALI/ARDS.
Author Contributions
MP and CM wrote the manuscript. SE wrote the manuscript and funded this work. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Financial support was provided by 2014/20451-0 (SE) and 2016/07030-0 (CM) grants from the São Paulo Research Foundation (FAPESP). In addition, SE (302566/2015-5) and CM (302951/2016-4) were supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). MP was supported by the 2012/10081-5 FAPESP fellowship.
Abbreviations
ALI/ARDS, acute lung injury/acute respiratory distress syndrome; CCR, C-C chemokine receptor; CLP, cecal ligation and puncture; CO, carbon monoxide; CoPPIX, cobalt protoporphyrin IX; CXCL, chemokine (C-X-C motif) ligand; CXCR, chemokine (C-X-C motif) receptor; DHA, dihydroartemisinin; EBA, Evans blue dye albumin assay; ECM, experimental cerebral malaria; ELISA, enzyme-linked immunosorbent assay; ELISPOT, enzyme-linked immunospot; FACS - fluorescence-activated cell sorting; FiO2, fraction of inspired oxygen; HbSAD, sickle hemoglobin; HO-1, heme oxygenase 1; HUVEC, human umbilical vein endothelial cells; ICAM-1, intercellular adhesion molecule 1; IFA, immunofluorescence assay; IFN, interferon; IHC, immunohistochemistry; IL, interleukin; KO, knockout; LPS, lipopolysaccharide; MA-ALI/ARDS, malaria-associated ALI/ARDS; MEC, murine vascular endothelial cells; MPO, myeloperoxidase; NET - neutrophil extracellular trap; NK, natural killer; PaO2, arterial blood oxygen tension; PCR, polymerase chain reaction; PMN, polymorphonuclear; PRBC, parasitized red blood cell; RF, respiratory frequency; ROS, reactive oxygen species; siRNA, small interfering RNA; SnPP, tin protoporphyrin; TGF, transforming growth factor; TNF, tumor necrosis factor; TNFR1, tumor necrosis factor receptor 1; VEGF, vascular endothelial growth factor; XR, X ray; ZnPPIX, zinc protoporphyrin IX.
References
Agarwal, A., and Bolisetty, S. (2013). Adaptive responses to tissue injury: role of heme oxygenase-1. Trans. Am. Clin. Climatol. Assoc. 124, 111–122.
Agarwal, A., and Nick, H. S. (2000). Renal response to tissue injury: lessons from heme oxygenase-1 gene ablation and expression. J. Am. Soc. Nephrol. 11, 965–973.
Agarwal, R., Nath, A., and Gupta, D. (2007). Noninvasive ventilation in Plasmodium vivax related ALI/ARDS. Intern. Med. 46, 2007–2011. doi: 10.2169/internalmedicine.46.0401
Aitken, E. H., Negri, E. M., Barboza, R., Lima, M. R. I., Álvarez, J. M., Marinho, C. R. F., et al. (2014). Ultrastructure of the lung in a murine model of malaria-associated acute lung injury/acute respiratory distress syndrome. Malar. J. 13:230. doi: 10.1186/1475-2875-13-230
Almelli, T., Ndam, N. T., Ezimegnon, S., Alao, M. J., Ahouansou, C., Sagbo, G., et al. (2014). Cytoadherence phenotype of Plasmodium falciparum-infected erythrocytes is associated with specific pfemp-1 expression in parasites from children with cerebral malaria. Malar. J. 13:333. doi: 10.1186/1475-2875-13-333
al-Yaman, F., Genton, B., Reeder, J. C., Anders, R. F., Smith, T., and Alpers, M. P. (1997). Reduced risk of clinical malaria in children infected with multiple cl0ones of Plasmodium falciparum in a highly endemic area: a prospective community study. Trans. R. Soc. Trop. Med. Hyg. 91, 602–605. doi: 10.1016/S0035-9203(97)90046-8
An, L., Liu, C.-T., Qin, X.-B., Liu, Q.-H., Liu, Y., and Yu, S.-Y. (2011). Protective effects of hemin in an experimental model of ventilator-induced lung injury. Eur. J. Pharmacol. 661, 102–108. doi: 10.1016/j.ejphar.2011.04.032
Anand, P. K., Malireddi, R. K., and Kanneganti, T. D. (2011). Role of the Nlrp3 inflammasome in microbial infection. Front. Microbiol. 2:12. doi: 10.3389/fmicb.2011.00012
Anidi, I. U., Servinsky, L. E., Rentsendorj, O., Stephens, R. S., Scott, A. L., and Pearse, D. B. (2013). CD36 and Fyn kinase mediate malaria-induced lung endothelial barrier dysfunction in mice infected with Plasmodium berghei. PLoS ONE 8:e71010. doi: 10.1371/journal.pone.0071010
Anstey, N. M., Handojo, T., Pain, M. C., Kenangalem, E., Tjitra, E., Price, R. N., et al. (2007). Lung injury in vivax malaria: pathophysiological evidence for pulmonary vascular sequestration and posttreatment alveolar-capillary inflammation. J. Infect. Dis. 195, 589–596. doi: 10.1086/510756
Anstey, N. M., Jacups, S. P., Cain, T., Pearson, T., Ziesing, P. J., Fisher, D. A., et al. (2002). Pulmonary manifestations of uncomplicated falciparum and vivax malaria: cough, small airways obstruction, impaired gas transfer, and increased pulmonary phagocytic activity. J. Infect. Dis. 185, 1326–1334. doi: 10.1086/339885
Ashbaugh, D. G., Bigelow, D. B., Petty, T. L., and Levine, B. E. (1967). Acute respiratory distress in adults. Lancet 2, 319–323. doi: 10.1016/S0140-6736(67)90168-7
Asiedu, D. K., and Sherman, C. B. (2000). Adult respiratory distress syndrome complicating Plasmodium falciparum malaria. Heart Lung 29, 294–297. doi: 10.1067/mhl.2000.106724
Aursudkij, B., Wilairatana, P., Vannaphan, S., Walsh, D. S., Gordeux, V. R., and Looareesuwan, S. (1998). Pulmonary edema in cerebral malaria patients in Thailand. Southeast Asian J. Trop. Med. Public Health 29, 541–545.
Avril, M., Bernabeu, M., Benjamin, M., Brazier, A. J., and Smith, J. D. (2016). Interaction between Endothelial Protein C Receptor and Intercellular Adhesion Molecule 1 to Mediate Binding of Plasmodium falciparum-Infected Erythrocytes to Endothelial Cells. MBio 7:e00615–16. doi: 10.1128/mBio.00615-16
Avril, M., Brazier, A. J., Melcher, M., Sampath, S., and Smith, J. D. (2013). DC8 and DC13 var genes associated with severe malaria bind avidly to diverse endothelial cells. PLoS Pathog. 9:e1003430. doi: 10.1371/journal.ppat.1003430
Baird, J. K. (2013). Evidence and implications of mortality associated with acute Plasmodium vivax malaria. Clin. Microbiol. Rev. 26, 36–57. doi: 10.1128/CMR.00074-12
Balla, G., Vercellotti, G. M., Muller-Eberhard, U., Eaton, J., and Jacob, H. S. (1991). Exposure of endothelial cells to free heme potentiates damage mediated by granulocytes and toxic oxygen species. Lab. Invest. 64, 648–655.
Ballinger, M. N., Newstead, M. W., Zeng, X., Bhan, U., Horowitz, J. C., Moore, B. B., et al. (2012). TLR signaling prevents hyperoxia-induced lung injury by protecting the alveolar epithelium from oxidant-mediated death. J. Immunol. 189, 356–364. doi: 10.4049/jimmunol.1103124
Barnwell, J. W., Asch, A. S., Nachman, R. L., Yamaya, M., Aikawa, M., and Ingravallo, P. (1989). A human 88-kD membrane glycoprotein (CD36) functions in vitro as a receptor for a cytoadherence ligand on Plasmodium falciparum-infected erythrocytes. J. Clin. Invest. 84, 765–772. doi: 10.1172/JCI114234
Bartoloni, A., and Zammarchi, L. (2012). Clinical aspects of uncomplicated and severe malaria. Mediterr. J. Hematol. Infect. Dis. 4:e2012026. doi: 10.4084/mjhid.2012.026
Baruch, D. I., Gormely, J. A., Ma, C., Howard, R. J., and Pasloske, B. L. (1996). Plasmodium falciparum erythrocyte membrane protein 1 is a parasitized erythrocyte receptor for adherence to CD36, thrombospondin, and intercellular adhesion molecule 1. Proc. Natl. Acad. Sci. U.S.A. 93, 3497–3502. doi: 10.1073/pnas.93.8.3497
Baruch, D. I., Ma, X. C., Singh, H. B., Bi, X., Pasloske, B. L., and Howard, R. J. (1997). Identification of a region of PfEMP1 that mediates adherence of Plasmodium falciparum infected erythrocytes to CD36: conserved function with variant sequence. Blood 90, 3766–3775.
Baruch, D. I., Pasloske, B. L., Singh, H. B., Bi, X., Ma, X. C., Feldman, M., et al. (1995). Cloning the P. falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell 82, 77–87. doi: 10.1016/0092-8674(95)90054-3
Belnoue, E., Potter, S. M., Rosa, D. S., Mauduit, M., Grüner, A. C., Kayibanda, M., et al. (2008). Control of pathogenic CD8+ T cell migration to the brain by IFN-gamma during experimental cerebral malaria. Parasite Immunol. 30, 544–553. doi: 10.1111/j.1365-3024.2008.01053.x
Bernabeu, M., Danziger, S. A., Avril, M., Vaz, M., Babar, P. H., Brazier, A. J., et al. (2016). Severe adult malaria is associated with specific PfEMP1 adhesion types and high parasite biomass. Proc. Natl. Acad. Sci. U.S.A. 113, E3270–E3279. doi: 10.1073/pnas.1524294113
Bernard, G. R., Artigas, A., Brigham, K. L., Carlet, J., Falke, K., Hudson, L., et al. (1994). The American-European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. Am. J. Respir. Crit. Care Med. 149, 818–824. doi: 10.1164/ajrccm.149.3.7509706
Boutros, C. N., Zegdi, R., Lila, N., Combillau, M., Fornes, P., Carpentier, A., et al. (2005). Pulmonary expression of inducible heme-oxygenase after ischemia/reperfusion of the lower extremities in rats. J. Surg. Res. 129, 306–312. doi: 10.1016/j.jss.2005.06.031
Brazier, A. J., Avril, M., Bernabeu, M., Benjamin, M., and Smith, J. D. (2017). Pathogenicity determinants of the human malaria parasite Plasmodium falciparum have ancient origins. mSphere 2:e00348–16. doi: 10.1128/mSphere.00348-16
Cao, T.-H., Jin, S.-G., Fei, D.-S., Kang, K., Jiang, L., Lian, Z.-Y., et al. (2016). Artesunate protects against sepsis-induced lung injury via heme oxygenase-1 modulation. Inflammation 39, 651–662. doi: 10.1007/s10753-015-0290-2
Carvalho, B. O., Lopes, S. C., Nogueira, P. A., Orlandi, P. P., Bargieri, D. Y., Blanco, Y. C., et al. (2010). On the cytoadhesion of Plasmodium vivax-infected erythrocytes. J. Infect. Dis. 202, 638–647. doi: 10.1086/654815
Carvalho, L. J., Lenzi, H. L., Pelajo-Machado, M., Oliveira, D. N., Daniel-Ribeiro, C. T., and Ferreira-da-Cruz, M. F. (2000). Plasmodium berghei: cerebral malaria in CBA mice is not clearly related to plasma TNF levels or intensity of histopathological changes. Exp. Parasitol. 95, 1–7. doi: 10.1006/expr.2000.4508
Castillo, P., Menéndez, C., Mayor, A., Carrilho, C., Ismail, M. R., Lorenzoni, C., et al. (2013). Massive plasmodium falciparum visceral sequestration: a cause of maternal death in Africa. Clin. Microbiol. Infect. 19, 1035–1041. doi: 10.1111/1469-0691.12068
Cerutti, C., and Ridley, A. J. (2017). Endothelial cell-cell adhesion and signaling. Exp. Cell Res. 358, 31–38. doi: 10.1016/j.yexcr.2017.06.003
Chavko, M., Prusaczyk, W. K., and McCarron, R. M. (2008). Protection against blast-induced mortality in rats by hemin. J. Trauma 65, 1140–1145; Discussion 1145. doi: 10.1097/TA.0b013e3181870a8c
Chen, H., Xu, W., Liu, D., Li, X., Pan, X., and Pang, Q. (2012). The anti-inflammatory mechanism of heme oxygenase-1 induced by hemin in primary rat alveolar macrophages. Inflammation 35, 1087–1093. doi: 10.1007/s10753-011-9415-4
Chen, Q., Heddini, A., Barragan, A., Fernandez, V., Pearce, S. F., and Wahlgren, M. (2000). The semiconserved head structure of Plasmodium falciparum erythrocyte membrane protein 1 mediates binding to multiple independent host receptors. J. Exp. Med. 192, 1–10. doi: 10.1084/jem.192.1.1
Chi, X., Guo, N., Yao, W., Jin, Y., Gao, W., Cai, J., et al. (2016). Induction of heme oxygenase-1 by hemin protects lung against orthotopic autologous liver transplantation-induced acute lung injury in rats. J. Transl. Med. 14:35. doi: 10.1186/s12967-016-0793-0
Choi, E.-Y., Choe, S.-H., Hyeon, J.-Y., Choi, J.-I., Choi, I. S., and Kim, S.-J. (2015). Carbon monoxide-releasing molecule-3 suppresses Prevotella intermedia lipopolysaccharide-induced production of nitric oxide and interleukin-1β in murine macrophages. Eur. J. Pharmacol. 764, 22–29. doi: 10.1016/j.ejphar.2015.06.039
Christou, H., Morita, T., Hsieh, C. M., Koike, H., Arkonac, B., Perrella, M. A., et al. (2000). Prevention of hypoxia-induced pulmonary hypertension by enhancement of endogenous heme oxygenase-1 in the rat. Circ. Res. 86, 1224–1229. doi: 10.1161/01.RES.86.12.1224
Clark, I. A., Awburn, M. M., Harper, C. G., Liomba, N. G., and Molyneux, M. E. (2003). Induction of HO-1 in tissue macrophages and monocytes in fatal falciparum malaria and sepsis. Malar. J. 2:41. doi: 10.1186/1475-2875-2-41
Constantin, J.-M., Cayot-Constantin, S., Roszyk, L., Futier, E., Sapin, V., Dastugue, B., et al. (2007). Response to recruitment maneuver influences net alveolar fluid clearance in acute respiratory distress syndrome. Anesthesiology 106, 944–951. doi: 10.1097/01.anes.0000265153.17062.64
Corbett, C. E., Duarte, M. I., Lancellotti, C. L., Silva, M. A., and Andrade Júnior, H. F. (1989). Cytoadherence in human falciparum malaria as a cause of respiratory distress. J. Trop. Med. Hyg. 92, 112–120.
Costa, F. T., Avril, M., Nogueira, P. A., and Gysin, J. (2006). Cytoadhesion of Plasmodium falciparum-infected erythrocytes and the infected placenta: a two-way pathway. Brazilian J. Med. Biol. Res. 39, 1525–1536. doi: 10.1590/S0100-879X2006001200003
Cowman, A. F., Healer, J., Marapana, D., and Marsh, K. (2016). Malaria: biology and disease. Cell 167, 610–624. doi: 10.1016/j.cell.2016.07.055
Crapo, J. D., Barry, B. E., Gehr, P., Bachofen, M., and Weibel, E. R. (1982). Cell number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis. 126, 332–337.
Cummins, N. W., Weaver, E. A., May, S. M., Croatt, A. J., Foreman, O., Kennedy, R. B., et al. (2012). Heme oxygenase-1 regulates the immune response to influenza virus infection and vaccination in aged mice. FASEB J. 26, 2911–2918. doi: 10.1096/fj.11-190017
Cunnington, A. J., de Souza, J. B., Walther, M., and Riley, E. M. (2011). Malaria impairs resistance to Salmonella through heme- and heme oxygenase-dependent dysfunctional granulocyte mobilization. Nat. Med. 18, 120–127. doi: 10.1038/nm.2601
Cunnington, A. J., Njie, M., Correa, S., Takem, E. N., Riley, E. M., and Walther, M. (2012). Prolonged neutrophil dysfunction after Plasmodium falciparum malaria is related to hemolysis and heme oxygenase-1 induction. J. Immunol. 189, 5336–5346. doi: 10.4049/jimmunol.1201028
de Lacerde Barbosa, D., Hochhegger, B., Souza, A. S. Jr., Zanetti, G., Escuissato, D. L., de Souze Portes Meirelles, G., et al. (2017). High-resolution computed tomography findings in eight patients with hantavirus pulmonary syndrome. Radiol. Bras. 50, 148–153. doi: 10.1590/0100-3984.2016.0093
De las Salas, B., Segura, C., Pabón, A., Lopes, S. C. P., Costa, F. T., and Blair, S. (2013). Adherence to human lung microvascular endothelial cells (HMVEC-L) of Plasmodium vivax isolates from Colombia. Malar. J. 12, 347. doi: 10.1186/1475-2875-12-347
Deroost, K., Tyberghein, A., Lays, N., Noppen, S., Schwarzer, E., Vanstreels, E., et al. (2013). Hemozoin induces lung inflammation and correlates with malaria-associated acute respiratory distress syndrome. Am. J. Respir. Cell Mol. Biol. 48, 589–600. doi: 10.1165/rcmb.2012-0450OC
Dondorp, A. M., Fanello, C. I., Hendriksen, I. C., Gomes, E., Seni, A., Chhaganlal, K. D., et al. (2010). Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomised trial. Lancet (Lond. Engl.) 376, 1647–1657. doi: 10.1016/S0140-6736(10)61924-1
Dorovini-Zis, K., Schmidt, K., Huynh, H., Fu, W., Whitten, R. O., Milner, D., et al. (2011). The neuropathology of fatal cerebral malaria in malawian children. Am. J. Pathol. 178, 2146–2158. doi: 10.1016/j.ajpath.2011.01.016
Drake, C. J., LaRue, A., Ferrara, N., and Little, C. D. (2000). VEGF regulates cell behavior during vasculogenesis. Dev. Biol. 224, 178–188. doi: 10.1006/dbio.2000.9744
Epiphanio, S., Campos, M. G., Pamplona, A., Carapau, D., Pena, A. C., Ataíde, R., et al. (2010). VEGF promotes malaria-associated acute lung injury in mice. PLoS Pathog. 6:e1000916. doi: 10.1371/journal.ppat.1000916
Epiphanio, S., Mikolajczak, S. A., Gonçalves, L. A., Pamplona, A., Portugal, S., Albuquerque, S., et al. (2008). Heme oxygenase-1 is an anti-inflammatory host factor that promotes murine plasmodium liver infection. Cell Host Microbe 3, 331–338. doi: 10.1016/j.chom.2008.04.003
Espinoza, J. A., León, M. A., Céspedes, P. F., Gómez, R. S., Canedo-Marroquín, G., Riquelme, S. A., et al. (2017). Heme Oxygenase-1 modulates human respiratory syncytial virus replication and lung pathogenesis during infection. J. Immunol. 199, 212–223. doi: 10.4049/jimmunol.1601414
Favre, N., Da Laperousaz, C., Ryffel, B., Weiss, N. A., Imhof, B. A., Rudin, W., et al. (1999). Role of ICAM-1 (CD54) in the development of murine cerebral malaria. Microb. Infect. 1, 961–968. doi: 10.1016/S1286-4579(99)80513-9
Fazalul Rahiman, S. S., Basir, R., Talib, H., Tie, T. H., Chuah, Y. K., Jabbarzare, M., et al. (2013). Interleukin-27 exhibited anti-inflammatory activity during Plasmodium berghei infection in mice. Trop. Biomed. 30, 663–680.
Fei, D., Meng, X., Kang, K., Nan, C., Zhao, M., Pan, S., et al. (2012). Heme oxygenase-1 modulates thrombomodulin and activated protein C levels to attenuate lung injury in cecal ligation and puncture-induced acute lung injury mice. Exp. Lung Res. 38, 173–182. doi: 10.3109/01902148.2012.660559
Fernandes, F. B., Lopes, R. G. C., and Mendes Filho, S. P. D. M. (2010). Malária grave em gestantes. Rev. Bras. Ginecol. e Obs. 32, 579–583. doi: 10.1590/S0100-72032010001200003
Ferreira, A., Balla, J., Jeney, V., Balla, G., and Soares, M. P. (2008). A central role for free heme in the pathogenesis of severe malaria: the missing link? J. Mol. Med. (Berl). 86, 1097–1111. doi: 10.1007/s00109-008-0368-5
Ferreira, A., Marguti, I., Bechmann, I., Jeney, V., Chora, Â., Palha, N. R., et al. (2011). Sickle hemoglobin confers tolerance to plasmodium infection. Cell 145, 398–409. doi: 10.1016/j.cell.2011.03.049
Franke-Fayard, B., Janse, C. J., Cunha-Rodrigues, M., Ramesar, J., Büscher, P., Que, I., et al. (2005). Murine malaria parasite sequestration: CD36 is the major receptor, but cerebral pathology is unlinked to sequestration. Proc. Natl. Acad. Sci. U.S.A. 102, 11468–11673. doi: 10.1073/pnas.0503386102
Fu, Y., Ding, Y., Zhou, T.-L., Ou, Q., and Xu, W. (2012). Comparative Histopathology of Mice Infected With the 17XL and 17XNL Strains of Plasmodium yoelii. J. Parasitol. 98, 310–315. doi: 10.1645/GE-2825.1
Gallego-Delgado, J., Ty, M., Orengo, J. M., van de Hoef, D., and Rodriguez, A. (2014). A surprising role for uric acid: the inflammatory malaria response. Curr. Rheumatol. Rep. 16:401. doi: 10.1007/s11926-013-0401-8
Gazzinelli, R. T., Kalantari, P., Fitzgerald, K. A., and Golenbock, D. T. (2014). Innate sensing of malaria parasites. Nat. Rev. Immunol. 14, 744–757. doi: 10.1038/nri3742
Gerber, H. P., Dixit, V., and Ferrara, N. (1998). Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J. Biol. Chem. 273, 13313–13316. doi: 10.1074/jbc.273.21.13313
Gong, P., Angelini, D. J., Yang, S., Xia, G., Cross, A. S., Mann, D., et al. (2008). TLR4 signaling is coupled to SRC family kinase activation, tyrosine phosphorylation of zonula adherens proteins, and opening of the paracellular pathway in human lung microvascular endothelia. J. Biol. Chem. 283, 13437–13449. doi: 10.1074/jbc.M707986200
Gonzales, J. N., Lucas, R., and Verin, A. D. (2015). The Acute respiratory distress syndrome: mechanisms and perspective therapeutic approaches. Austin J. Vasc. Med. 2:1009.
Good, M. F., Xu, H., Wykes, M., and Engwerda, C. R. (2005). Development and regulation of cell-mediated immune responses to the blood stages of malaria: implications for vaccine research. Annu. Rev. Immunol. 23, 69–99. doi: 10.1146/annurev.immunol.23.021704.115638
Gozzelino, R., Jeney, V., and Soares, M. P. (2010). Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 50, 323–354. doi: 10.1146/annurev.pharmtox.010909.105600
Grailer, J. J., Canning, B. A., Kalbitz, M., Haggadone, M. D., Dhond, R. M., Andjelkovic, A. V., et al. (2014). Critical role for the NLRP3 inflammasome during acute lung injury. J. Immunol. 192, 5974–5983. doi: 10.4049/jimmunol.1400368
Grundemar, L., and Ny, L. (1997). Pitfalls using metalloporphyrins in carbon monoxide research. Trends Pharmacol. Sci. 18, 193–195. doi: 10.1016/S0165-6147(97)90622-2
Gupte, S. (2013). Recent Advances in Pediatrics Special, Vol. 22, Immunology, Infections and Immunization. 1st Edn. eds S. -B. Gupte and M. Gupte (New Delhi: JP Medical Ltd.).
Haydoura, S., Mazboudi, O., Charafeddine, K., Bouakl, I., Baban, T. A., Taher, A. T., et al. (2011). Transfusion-related Plasmodium ovale malaria complicated by acute respiratory distress syndrome (ARDS) in a non-endemic country. Parasitol. Int. 60, 114–116. doi: 10.1016/j.parint.2010.10.005
Hee, L., Dinudom, A., Mitchell, A. J., Grau, G. E., Cook, D. I., Hunt, N. H., et al. (2011). Reduced activity of the epithelial sodium channel in malaria-induced pulmonary oedema in mice. Int. J. Parasitol. 41, 81–88. doi: 10.1016/j.ijpara.2010.07.013
Hettiarachchi, N. T., Boyle, J. P., Dallas, M. L., Al-Owais, M. M., Scragg, J. L., and Peers, C. (2017). Heme oxygenase-1 derived carbon monoxide suppresses Aβ1-42 toxicity in astrocytes. Cell Death Dis. 8:e2884. doi: 10.1038/cddis.2017.276
Holter, J. F., Weiland, J. E., Pacht, E. R., Gadek, J. E., and Davis, W. B. (1986). Protein permeability in the adult respiratory distress syndrome. Loss of size selectivity of the alveolar epithelium. J. Clin. Invest. 78, 1513–1522. doi: 10.1172/JCI112743
Horata, N., Kalambaheti, T., Craig, A., and Khusmith, S. (2009). Sequence variation of PfEMP1-DBLalpha in association with rosette formation in Plasmodium falciparum isolates causing severe and uncomplicated malaria. Malar. J. 8:184. doi: 10.1186/1475-2875-8-184
Im, J. H., Kwon, H. Y., Baek, J., Park, S. W., Durey, A., Lee, K. H., et al. (2017). Severe Plasmodium vivax infection in Korea. Malar. J. 16:51. doi: 10.1186/s12936-017-1684-4
Immenschuh, S., Vijayan, V., Janciauskiene, S., and Gueler, F. (2017). Heme as a Target for Therapeutic Interventions. Front. Pharmacol. 8:146. doi: 10.3389/fphar.2017.00146
Inoue, K., Patterson, E. K., Capretta, A., Lawendy, A. R., Fraser, D. D., and Cepinskas, G. (2017). Carbon monoxide-releasing molecule-401 suppresses polymorphonuclear leukocyte migratory potential by modulating F-actin dynamics. Am. J. Pathol. 187, 1121–1133. doi: 10.1016/j.ajpath.2016.12.025
Inui, M., Ishida, Y., Kimura, A., Kuninaka, Y., Mukaida, N., and Kondo, T. (2011). Protective roles of CX3CR1-mediated signals in toxin A-induced enteritis through the induction of heme oxygenase-1 expression. J. Immunol. 186, 423–431. doi: 10.4049/jimmunol.1000043
Jabaudon, M., Blondonnet, R., Lutz, J., Roszyk, L., Bouvier, D., Guérin, R., et al. (2016). Net alveolar fluid clearance is associated with lung morphology phenotypes in acute respiratory distress syndrome. Anaesth. Crit. Care Pain Med. 35, 81–86 doi: 10.1016/j.accpm.2015.11.006
Janz, D. R., and Ware, L. B. (2013). Biomarkers of ALI/ARDS: pathogenesis, discovery, and relevance to clinical trials. Semin. Respir. Crit. Care Med. 34, 537–548. doi: 10.1055/s-0033-1351124
Joo Choi, R., Cheng, M.-S., and Shik Kim, Y. (2014). Desoxyrhapontigenin up-regulates Nrf2-mediated heme oxygenase-1 expression in macrophages and inflammatory lung injury. Redox Biol. 2, 504–512. doi: 10.1016/j.redox.2014.02.001
Kinderlerer, A. R., Pombo Gregoire, I., Hamdulay, S. S., Ali, F., Steinberg, R., Silva, G., et al. (2009). Heme oxygenase-1 expression enhances vascular endothelial resistance to complement-mediated injury through induction of decay-accelerating factor: a role for increased bilirubin and ferritin. Blood 113, 1598–1607. doi: 10.1182/blood-2008-04-152934
Kirchgatter, K., and Del Portillo, H. A. (2005). Clinical and molecular aspects of severe malaria. An. Acad. Bras. Cienc. 77, 455–475. doi: 10.1590/S0001-37652005000300008
Kochar, D. K., Das, A., Kochar, S. K., Saxena, V., Sirohi, P., Garg, S., et al. (2009). Severe Plasmodium vivax malaria: a report on serial cases from Bikaner in northwestern India. Am. J. Trop. Med. Hyg. 80, 194–198. doi: 10.4269/ajtmh.2009.80.194
Konrad, F. M., Knausberg, U., Höne, R., Ngamsri, K.-C., and Reutershan, J. (2016). Tissue heme oxygenase-1 exerts anti-inflammatory effects on LPS-induced pulmonary inflammation. Mucosal Immunol. 9, 98–111. doi: 10.1038/mi.2015.39
Kurth, F., Develoux, M., Mechain, M., Malvy, D., Clerinx, J., Antinori, S., et al. (2017). Severe malaria in Europe: an 8-year multi-centre observational study. Malar. J. 16:57. doi: 10.1186/s12936-016-1673-z
Lacerda, M. V., Fragoso, S. C., Alecrim, M. G., Alexandre, M. A., Magalhães, B. M., Siqueira, A. M., et al. (2012b). Postmortem characterization of patients with clinical diagnosis of Plasmodium vivax malaria: to what extent does this parasite kill? Clin. Infect. Dis. 55, e67–e74. doi: 10.1093/cid/cis615
Lacerda, M. V., Mourão, M. P., Alexandre, M. A., Siqueira, A. M., Magalhães, B. M., Martinez-Espinosa, F. E., et al. (2012a). Understanding the clinical spectrum of complicated Plasmodium vivax malaria: a systematic review on the contributions of the Brazilian literature. Malar. J. 11:12. doi: 10.1186/1475-2875-11-12
Lang, D., Reuter, S., Buzescu, T., August, C., and Heidenreich, S. (2005). Heme-induced heme oxygenase-1 (HO-1) in human monocytes inhibits apoptosis despite caspase-3 up-regulation. Int. Immunol. 17, 155–165. doi: 10.1093/intimm/dxh196
Lee, H.-J., Baek, J.-H., Chae, M.-H., Joo, H., Lee, J.-S., Chung, M.-H., et al. (2013). A case of vivax malaria complicated by adult respiratory distress syndrome and successful management with extracorporeal membrane oxygenation. Korean J. Parasitol. 51, 551–555. doi: 10.3347/kjp.2013.51.5.551
Liu, M., Amodu, A. S., Pitts, S., Patrickson, J., Hibbert, J. M., Battle, M., et al. (2012). Heme mediated STAT3 activation in severe malaria. PLoS ONE 7:e34280. doi: 10.1371/journal.pone.0034280
Londhe, C., Ganeriwal, A., and deSouza, R. (2014). Study of clinical profile of acute respiratory distress syndrome and acute lung injury in Plasmodium vivax malaria. J. Vector Borne Dis. 51, 339–342.
Lovegrove, F. E., Gharib, S., a, Peña-Castillo, L., Patel, S. N., Ruzinski, J. T., Hughes, T. R., et al. (2008). Parasite burden and CD36-mediated sequestration are determinants of acute lung injury in an experimental malaria model. PLoS Pathog. 4:e1000068. doi: 10.1371/journal.ppat.1000068
Lozano, F., Leal, M., Lissen, E., Munoz, J., Bautista, A., and Regordan, C. (1983). [P. falciparum and P. malariae malaria complicated by pulmonary edema with disseminated intravascular coagulation]. Presse. Med. 12, 3004–3005.
Luo, Y., Jiang, L., Kang, K., Fei, D., Meng, X., Nan, C., et al. (2014). Hemin inhibits NLRP3 inflammasome activation in sepsis-induced acute lung injury, involving heme oxygenase-1. Int. Immunopharmacol. 20, 24–32. doi: 10.1016/j.intimp.2014.02.017
Maguire, G. P., Handojo, T., Pain, M. C., Kenangalem, E., Price, R. N., Tjitra, E., et al. (2005). Lung injury in uncomplicated and severe falciparum malaria: a longitudinal study in papua, Indonesia. J. Infect. Dis. 192, 1966–1974. doi: 10.1086/497697
Marchiori, E., Zanetti, G., Hochhegger, B., Canella, C., and Irion, K. L. (2013). Plasmodium falciparum malaria: another infection of interest to pulmonologists. J. Bras. Pneumol. 39, 750–752. doi: 10.1590/S1806-37132013000600015
Maria, A. K., Singh, R., and Kaur, M. (2015). Pulmonary complications in Falciparum malaria : a retrospective analytical study baseline demographics. Sch. J. App. Med. Sci. 3, 761–767.
Marsh, K., Forster, D., Waruiru, C., Mwangi, I., Winstanley, M., Marsh, V., et al. (1995). Indicators of life-threatening malaria in African children. N. Engl. J. Med. 332, 1399–1404. doi: 10.1056/NEJM199505253322102
Matthay, M. A., Ware, L. B., and Zimmerman, G. A. (2012). The acute respiratory distress syndrome. J. Clin. Invest. 122, 2731–2740. doi: 10.1172/JCI60331
Mendonça, V. R., Luza, N. F., Santos, N. J. G., Borges, V. M., Gonçalves, M. S., Andrade, B. B., et al. (2012). Association between the haptoglobin and heme oxygenase 1 genetic profiles and soluble CD163 in susceptibility to and severity of human malaria. Infect. Immun. 80, 1445–1454. doi: 10.1128/IAI.05933-11
Menezes, R. G., Kanchan, T., Rai, S., Jagadish Rao, P. P., Naik, R., Suresh Kumar Shetty, B., et al. (2010). An autopsy case of sudden unexplained death caused by malaria. J. Forensic Sci. 55, 835–838. doi: 10.1111/j.1556-4029.2010.01328.x
Meng, F., Meliton, A., Moldobaeva, N., Mutlu, G., Kawasaki, Y., Akiyama, T., et al. (2015). Asef mediates HGF protective effects against LPS-induced lung injury and endothelial barrier dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 308, L452–L463. doi: 10.1152/ajplung.00170.2014
Mikolajczak, S. A., Vaughan, A. M., Kangwanrangsan, N., Roobsoong, W., Fishbaugher, M., Yimamnuaychok, N., et al. (2015). Plasmodium vivax liver stage development and hypnozoite persistence in human liver-chimeric mice. Cell Host Microbe 17, 526–535. doi: 10.1016/j.chom.2015.02.011
Milner, D. A., Whitten, R. O., Kamiza, S., Carr, R., Liomba, G., Dzamalala, C., et al. (2014). The systemic pathology of cerebral malaria in African children. Front. Cell. Infect. Microbiol. 4:104. doi: 10.3389/fcimb.2014.00104
Milner, D., Factor, R., Whitten, R., Carr, R. a, Kamiza, S., Pinkus, G., et al. (2013). Pulmonary pathology in pediatric cerebral malaria. Hum. Pathol. 44, 2719–2726. doi: 10.1016/j.humpath.2013.07.018
Modiano, D., Sirima, B. S., Sawadogo, A., Sanou, I., Paré, J., Konat,é, A., et al. (1998). Severe malaria in Burkina Faso: influence of age and transmission level on clinical presentation. Am. J. Trop. Med. Hyg. 59, 539–542. doi: 10.4269/ajtmh.1998.59.539
Mohan, A., Sharma, S. K., and Bollineni, S. (2008). Acute lung injury and acute respiratory distress syndrome in malaria. J. Vector Borne Dis. 45, 179–193.
Monahan, L. J. (2013). Acute respiratory distress syndrome. Curr. Probl. Pediatr. Adolesc. Health Care 43, 278–284. doi: 10.1016/j.cppeds.2013.10.004
Moore, B. R., Jago, J. D., and Batty, K. T. (2008). Plasmodium berghei: parasite clearance after treatment with dihydroartemisinin in an asplenic murine malaria model. Exp. Parasitol. 118, 458–467. doi: 10.1016/j.exppara.2007.10.011
Moore, K. J., El Khoury, J., Medeiros, L. A., Terada, K., Geula, C., Luster, A. D., et al. (2002). A CD36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J. Biol. Chem. 277, 47373–47379. doi: 10.1074/jbc.M208788200
Mota, M. M., and Rodriguez, A. (2002). Invasion of mammalian host cells by Plasmodium sporozoites. Bioessays 24, 149–156. doi: 10.1002/bies.10050
Motterlini, R., and Otterbein, L. E. (2010). The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 9, 728–743. doi: 10.1038/nrd3228
Muanza, K., Gay, F., Behr, C., and Scherf, A. (1996). Primary culture of human lung microvessel endothelial cells: a useful in vitro model for studying Plasmodium falciparum-infected erythrocyte cytoadherence. Res. Immunol. 147, 149–163. doi: 10.1016/0923-2494(96)83167-1
Mueller, A.-K., Behrends, J., Hagens, K., Mahlo, J., Schaible, U. E., and Schneider, B. E. (2012). Natural transmission of Plasmodium berghei exacerbates chronic tuberculosis in an experimental co-infection model. PLoS ONE 7:e48110. doi: 10.1371/journal.pone.0048110
Muley, A., Lakhani, J., Bhirud, S., and Patel, A. (2014). Thrombocytopenia in Plasmodium vivax Malaria: how significant? J. Trop. Med. 2014, 1–4. doi: 10.1155/2014/567469
Nakagami, T., Toyomura, K., Kinoshita, T., and Morisawa, S. (1993). A beneficial role of bile pigments as an endogenous tissue protector: anti-complement effects of biliverdin and conjugated bilirubin. Biochim. Biophys. Acta 1158, 189–193. doi: 10.1016/0304-4165(93)90013-X
Nakamichi, I., Habtezion, A., Zhong, B., Contag, C. H., Butcher, E. C., and Omary, M. B. (2005). Hemin-activated macrophages home to the pancreas and protect from acute pancreatitis via heme oxygenase-1 induction. J. Clin. Invest. 115, 3007–3014. doi: 10.1172/JCI24912
Nayak, K. C., Mohini Kumar, S., Tanwar, R. S., Kulkarni, V., Gupta, A., et al. (2011). A study on pulmonary manifestations in patients with malaria from northwestern India (Bikaner). J. Vector Borne Dis. 48, 219–223.
Oquendo, P., Hundt, E., Lawler, J., and Seed, B. (1989). CD36 directly mediates cytoadherence of Plasmodium falciparum parasitized erythrocytes. Cell 58, 95–101. doi: 10.1016/0092-8674(89)90406-6
Ortolan, L. S., Sercundes, M. K., Barboza, R., Debone, D., Murillo, O., Hagen, S. C. F., et al. (2014). Predictive criteria to study the pathogenesis of malaria-associated ALI/ARDS in mice. Med. Inflamm. 2014:872464. doi: 10.1155/2014/872464
Otterbein, L. E., Mantell, L. L., and Choi, A. M. (1999). Carbon monoxide provides protection against hyperoxic lung injury. Am. J. Physiol. 276, L688–L694. doi: 10.1152/ajplung.1999.276.4.L688
Otterbein, L., Sylvester, S. L., and Choi, A. M. (1995). Hemoglobin provides protection against lethal endotoxemia in rats: the role of heme oxygenase-1. Am. J. Respir. Cell Mol. Biol. 13, 595–601. doi: 10.1165/ajrcmb.13.5.7576696
Pamplona, A., Ferreira, A., Balla, J., Jeney, V., Balla, G., Epiphanio, S., et al. (2007). Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat. Med. 13, 703–710. doi: 10.1038/nm1586
Pena, A. C., Penacho, N., Mancio-Silva, L., Neres, R., Seixas, J. D., Fernandes, A. C., et al. (2012). A novel carbon monoxide-releasing molecule fully protects mice from severe malaria. Antimicrob. Agents Chemother. 56, 1281–1290. doi: 10.1128/AAC.05571-11
Pereira, M. L., Ortolan, L. S., Sercundes, M. K., Debone, D., Murillo, O., Lima, F. A., et al. (2016). Association of Heme Oxygenase 1 with lung protection in malaria-associated ALI/ARDS. Mediat. Inflamm. 2016:4158698. doi: 10.1155/2016/4158698
Piguet, P. F., Kan, C. D., Vesin, C., Rochat, A., Donati, Y., and Barazzone, C. (2001). Role of CD40-CVD40L in mouse severe malaria. Am. J. Pathol. 159, 733–742. doi: 10.1016/S0002-9440(10)61744-0
Pinto, V. L. Jr., Hamidad, A. M., Albuquerque Filho, Dde. O., and dos Santos, V. M. (2014). Twenty years of hantavirus pulmonary syndrome in Brazil: a review of epidemiological and clinical aspects. J. Infect. Dev. Ctries. 8, 137–142. doi: 10.3855/jidc.3254
Ponsford, M. J., Medana, I. M., Prapansilp, P., Hien, T. T., Lee, S. J., Dondorp, A. M., et al. (2012). Sequestration and microvascular congestion are associated with coma in human cerebral malaria. J. Infect. Dis. 205, 663–671. doi: 10.1093/infdis/jir812
Prudêncio, M., Rodriguez, A., and Mota, M. M. (2006). The silent path to thousands of merozoites: the Plasmodium liver stage. Nat. Rev. Microbiol. 4, 849–856. doi: 10.1038/nrmicro1529
Rahimi, B. A., Thakkinstian, A., White, N. J., Sirivichayakul, C., Dondorp, A. M., and Chokejindachai, W. (2014). Severe vivax malaria: a systematic review and meta-analysis of clinical studies since 1900. Malar. J. 13, 481. doi: 10.1186/1475-2875-13-481
Ranieri, V. M., Rubenfeld, G. D., Thompson, B. T., Ferguson, N. D., Caldwell, E., Fan, E., et al. (2012). Acute respiratory distress syndrome: the Berlin Definition. JAMA 307, 2526–2533. doi: 10.1001/jama.2012.5669
Riviello, E. D., Buregeya, E., and Twagirumugabe, T. (2017). Diagnosing acute respiratory distress syndrome in resource limited settings: the Kigali modification of the Berlin definition. Curr. Opin. Crit. Care 23, 18–23. doi: 10.1097/MCC.0000000000000372
Rockwood, N., Costa, D. L., Amaral, E. P., Du Bruyn, E., Kubler, A., Gil-Santana, L., et al. (2017). Mycobacterium tuberculosis induction of heme oxygenase-1 expression is dependent on oxidative stress and reflects treatment outcomes. Front. Immunol. 8:542. doi: 10.3389/fimmu.2017.00542
Rudan, I., O'Brien, K. L., Nair, H., Liu, L., Theodoratou, E., Qazi, S., et al. (2013). Epidemiology and etiology of childhood pneumonia in 2010: estimates of incidence, severe morbidity, mortality, underlying risk factors and causative pathogens for 192 countries. J. Glob. Health 3:10401. doi: 10.7189/jogh.03.010401
Saigal, S., Kapoor, G., Gurjar, M., and Singh, D. K. (2014). Diffuse alveolar hemorrhage due to Plasmodium falciparum: a rare entity–are steroids indicated? J. Vector Borne Dis. 51, 66–68.
Sambo, M. R., Trovoada, M. J., Benchimol, C., Quinhentos, V., Gonçalves, L., Velosa, R., et al. (2010). Transforming growth factor beta 2 and heme oxygenase 1 genes are risk factors for the cerebral malaria syndrome in Angolan children. PLoS ONE 5:e11141. doi: 10.1371/journal.pone.0011141
Saravu, K., Rishikesh, K., Kamath, A., and Shastry, A. B. (2014). Severity in Plasmodium vivax malaria claiming global vigilance and exploration - a tertiary care centre-based cohort study. Malar. J. 13:304. doi: 10.1186/1475-2875-13-304
Sarkar, S., Saha, K., and Das, C. S. (2010). Three cases of ARDS: An emerging complication of Plasmodium vivax malaria. Lung India 27, 154–157. doi: 10.4103/0970-2113.68323
Schluesener, H. J., Kremsner, P. G., and Meyermann, R. (2001). Heme oxygenase-1 in lesions of human cerebral malaria. Acta Neuropathol. 101, 65–68. doi: 10.1007/s004010000250
Schroder, K., and Tschopp, J. (2010). The inflammasomes. Cell 140, 821–832. doi: 10.1016/j.cell.2010.01.040
Seilmaier, M., Hartmann, W., Beissner, M., Fenzl, T., Haller, C., Guggemos, W., et al. (2014). Severe Plasmodium knowlesi infection with multi-organ failure imported to Germany from Thailand/Myanmar. Malar. J. 13, 422. doi: 10.1186/1475-2875-13-422
Seixas, E., Gozzelino, R., Chora, A., Ferreira, A., Silva, G., Larsen, R., et al. (2009). Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc. Natl. Acad. Sci. U.S.A. 106, 15837–15842. doi: 10.1073/pnas.0903419106
Sercundes, M. K., Ortolan, L. S., Debone, D., Soeiro-Pereira, P. V., Gomes, E., Aitken, E. H., et al. (2016). Targeting neutrophils to prevent malaria-associated acute lung injury/acute respiratory distress syndrome in mice. PLoS Pathog. 12:e1006054. doi: 10.1371/journal.ppat.1006054
Sherman, I. W., Eda, S., and Winograd, E. (2003). Cytoadherence and sequestration in Plasmodium falciparum: defining the ties that bind. Microbes Infect. 5, 897–909. doi: 10.1016/S1286-4579(03)00162-X
Siner, J. M., Jiang, G., Cohen, Z. I., Shan, P., Zhang, X., Lee, C. G., et al. (2007). VEGF-induced heme oxygenase-1 confers cytoprotection from lethal hyperoxia in vivo. FASEB J. 21, 1422–1432. doi: 10.1096/fj.06-6661com
Singh, B., and Daneshvar, C. (2013). Human infections and detection of Plasmodium knowlesi. Clin. Microbiol. Rev. 26, 165–184. doi: 10.1128/CMR.00079-12
Soares, M. P., Lin, Y., Anrather, J., Csizmadia, E., Takigami, K., Sato, K., et al. (1998). Expression of heme oxygenase-1 can determine cardiac xenograft survival. Nat. Med. 4, 1073–1077. doi: 10.1038/2063
Souza, M. C., Silva, J. D., Pádua, T. a, Capelozzi, V. L., Rocco, P. R. M., and Henriques, M. D. G. (2013). Early and late acute lung injury and their association with distal organ damage in murine malaria. Respir. Physiol. Neurobiol. 186, 65–72. doi: 10.1016/j.resp.2012.12.008
Stanisic, D. I., Cutts, J., Eriksson, E., Fowkes, F. J., Rosanas-Urgell, A., Siba, P., et al. (2014). γδ T cells and CD14+ monocytes are predominant cellular sources of cytokines and chemokines associated with severe malaria. J. Infect. Dis. 210, 295–305. doi: 10.1093/infdis/jiu083
Stocker, R., Yamamoto, Y., McDonagh, A. F., Glazer, A. N., and Ames, B. N. (1987). Bilirubin is an antioxidant of possible physiological importance. Science 235, 1043–1046. doi: 10.1126/science.3029864
Sugiyama, M. G., Gamage, A., Zyla, R., Armstrong, S. M., Advani, S., Advani, A., et al. (2015). Influenza virus infection induces platelet-endothelial adhesion which contributes to lung injury. J. Virol. 90, 1812–1823. doi: 10.1128/JVI.02599-15
Sukriti, S., Tauseef, M., Yazbeck, P., and Mehta, D. (2014). Mechanisms regulating endothelial permeability. Pulm. Circ. 4, 535–551. doi: 10.1086/677356
Sun, J., Guo, E., Yang, J., Yang, Y., Liu, S., Hu, J., et al. (2017). Carbon monoxide ameliorates hepatic ischemia/reperfusion injury via sirtuin 1-mediated deacetylation of high-mobility group box 1 in rats. Liver Transpl. 23, 510–526. doi: 10.1002/lt.24733
Ta, T. H., Hisam, S., Lanza, M., Jiram, A. I., Ismail, N., and Rubio, J. M. (2014). First case of a naturally acquired human infection with Plasmodium cynomolgi. Malar. J. 13:68. doi: 10.1186/1475-2875-13-68
Takeda, M., Kikuchi, M., Ubalee, R., Na-Bangchang, K., Ruangweerayut, R., Shibahara, S., et al. (2005). Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to cerebral malaria in Myanmar. Jpn. J. Infect. Dis. 58, 268–271.
Taylor, W. R. J., Cañon, V., and White, N. J. (2006). Pulmonary manifestations of malaria: recognition and management. Treat. Respir. Med. 5, 419–428. doi: 10.2165/00151829-200605060-00007
Taylor, W. R. J., Hanson, J., Turner, G. D. H., White, N. J., and Dondorp, A. M. (2012). Respiratory manifestations of malaria. Chest 142, 492–505. doi: 10.1378/chest.11-2655
Thiel, M., Chouker, A., Ohta, A., Jackson, E., Caldwell, C., Smith, P., et al. (2005). Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol. 3:e174. doi: 10.1371/journal.pbio.0030174
Tong, M. J., Ballantine, T. V., and Youel, D. B. (1972). Pulmonary function studies in Plasmodium falciparum malaria. Am. Rev. Respir. Dis. 106, 23–29. doi: 10.1164/arrd.1972.106.1.23
Tracz, M. J., Alam, J., and Nath, K. A. (2007). Physiology and pathophysiology of heme: implications for kidney disease. J. Am. Soc. Nephrol. 18, 414–420. doi: 10.1681/ASN.2006080894
Trampuz, A., Jereb, M., Muzlovic, I., and Prabhu, R. M. (2003). Clinical review: severe malaria. Crit. Care 7, 315–323. doi: 10.1186/cc2183
Tran, T. H., Day, N. P., Nguyen, H. P., Nguyen, T. H., Tran, T. H., Pham, P. L., et al. (1996). A controlled trial of artemether or quinine in Vietnamese adults with severe Falciparum malaria. N. Engl. J. Med. 335, 76–83. doi: 10.1056/NEJM199607113350202
Tsuburai, T., Suzuki, M., Nagashima, Y., Suzuki, S., Inoue, S., Hasiba, T., et al. (2002). Adenovirus-mediated transfer and overexpression of heme oxygenase 1 cDNA in lung prevents bleomycin-induced pulmonary fibrosis via a Fas-Fas ligand-independent pathway. Hum. Gene Ther. 13, 1945–1960. doi: 10.1089/10430340260355356
Turner, L., Lavstsen, T., Berger, S. S., Wang, C. W., Petersen, J. E., Avril, M., et al. (2013). Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature 498, 502–505. doi: 10.1038/nature12216
Umbers, A. J., Aitken, E. H., and Rogerson, S. J. (2011). Malaria in pregnancy: small babies, big problem. Trends Parasitol. 27, 168–175. doi: 10.1016/j.pt.2011.01.007
Valecha, N., Pinto, R. G. W., Turner, G. D. H., Kumar, A., Rodrigues, S., Dubhashi, N. G., et al. (2009). Case report: histopathology of fatal respiratory distress caused by Plasmodium vivax malaria. Am. J. Trop. Med. Hyg. 81, 758–762. doi: 10.4269/ajtmh.2009.09-0348
Van den Steen, P. E., Geurts, N., Deroost, K., Van Aelst, I., Verhenne, S., Heremans, H., et al. (2010). Immunopathology and dexamethasone therapy in a new model for malaria-associated acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 181, 957–968. doi: 10.1164/rccm.200905-0786OC
van der Heyde, H. C., Bauer, P., Sun, G., Chang, W. L., Yin, L., Fuseler, J., et al. (2001). Assessing vascular permeability during experimental cerebral malaria by a radiolabeled monoclonal antibody technique. Infect. Immun. 69, 3460–3465. doi: 10.1128/IAI.69.5.3460-3465.2001
van der Heyde, H. C., Nolan, J., Combes, V., Gramaglia, I., and Grau, G. E. (2006). A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 22, 503–508. doi: 10.1016/j.pt.2006.09.002
Vergote, V., Laenen, L., Vanmechelen, B., Van Ranst, M., Verbeken, E., Hooper, J. W., et al. (2017). A lethal disease model for New World hantaviruses using immunosuppressed Syrian hamsters. PLoS Negl. Trop. Dis. 11:e0006042. doi: 10.1371/journal.pntd.0006042
Villegas-Mendez, A., de Souza, J. B., Murungi, L., Hafalla, J. C. R., Shaw, T. N., Greig, R., et al. (2011). Heterogeneous and tissue-specific regulation of effector T cell responses by IFN-gamma during Plasmodium berghei ANKA infection. J. Immunol. 187, 2885–2897. doi: 10.4049/jimmunol.1100241
Walther, M., De Caul, A., Aka, P., Njie, M., Amambua-Ngwa, A., Walther, B., et al. (2012). HMOX1 gene promoter alleles and high HO-1 levels are associated with severe malaria in Gambian children. PLoS Pathog. 8:e1002579. doi: 10.1371/journal.ppat.1002579
Wang, C., Zhang, Y., Han, L., Guo, L., Zhong, H., and Wang, J. (2017). Hemin ameliorates influenza pneumonia by attenuating lung injury and regulating the immune response. Int. J. Antimicrob. Agents 49, 45–52. doi: 10.1016/j.ijantimicag.2016.09.030
Wassmer, S. C., and Grau, G. E. R. (2017). Severe malaria: what's new on the pathogenesis front? Int. J. Parasitol. 47, 145–152. doi: 10.1016/j.ijpara.2016.08.002
Wassmer, S. C., Combes, V., and Grau, G. E. (2003). Pathophysiology of cerebral malaria: role of host cells in the modulation of cytoadhesion. Ann. N.Y. Acad. Sci. 992, 30–38. doi: 10.1111/j.1749-6632.2003.tb03135.x
Weiss, M. L., and Kubat, K. (1983). Plasmodium berghei: a mouse model for the “sudden death” and “malarial lung” syndromes. Exp. Parasitol. 56, 143–151. doi: 10.1016/0014-4894(83)90105-4
White, N. J., and Imwong, M. (2012). Relapse. Adv. Parasitol. 80, 113–150. doi: 10.1016/B978-0-12-397900-1.00002-5
Wu, S.-Y., Li, M.-H., Ko, F.-C., Wu, G.-C., Huang, K.-L., and Chu, S.-J. (2013). Protective effect of hypercapnic acidosis in ischemia-reperfusion lung injury is attributable to upregulation of heme oxygenase-1. PLoS ONE 8:e74742. doi: 10.1371/journal.pone.0074742
Yin, H., Li, X., Yuan, B., Zhang, B., Hu, S., Gu, H., et al. (2011). Heme oxygenase-1 ameliorates LPS-induced acute lung injury correlated with downregulation of interleukin-33. Int. Immunopharmacol. 11, 2112–2117. doi: 10.1016/j.intimp.2011.09.004
Yu, J., Wang, Y., Li, Z., Dong, S., Wang, D., Gong, L., et al. (2016). Effect of heme oxygenase-1 on Mitofusin-1 protein in LPS-induced ALI/ARDS in rats. Sci. Rep. 6:36530. doi: 10.1038/srep36530
Zhang, X., Shan, P., Sasidhar, M., Chupp, G. L., Flavell, R. A., Choi, A. M. K., et al. (2003). Reactive oxygen species and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase mediate hyperoxia-induced cell death in lung epithelium. Am. J. Respir. Cell Mol. Biol. 28, 305–315. doi: 10.1165/rcmb.2002-0156OC
Zhang, Y., Jiang, G., Sauler, M., and Lee, P. J. (2013). Lung endothelial HO-1 targeting in vivo using lentiviral miRNA regulates apoptosis and autophagy during oxidant injury. FASEB J. 27, 4041–4058. doi: 10.1096/fj.13-231225
Zhou, Z.-H., Kumari, N., Nekhai, S., Clouse, K. A., Wahl, L. M., Yamada, K. M., et al. (2013). Heme oxygenase-1 induction alters chemokine regulation and ameliorates human immunodeficiency virus-type-1 infection in lipopolysaccharide-stimulated macrophages. Biochem. Biophys. Res. Commun. 435, 373–377. doi: 10.1016/j.bbrc.2013.04.095
Keywords: heme oxygenase, malaria, ALI/ARDS, inflammation, endothelium, heme
Citation: Pereira MLM, Marinho CRF and Epiphanio S (2018) Could Heme Oxygenase-1 Be a New Target for Therapeutic Intervention in Malaria-Associated Acute Lung Injury/Acute Respiratory Distress Syndrome? Front. Cell. Infect. Microbiol. 8:161. doi: 10.3389/fcimb.2018.00161
Received: 29 October 2017; Accepted: 26 April 2018;
Published: 16 May 2018.
Edited by:
Vincent Munster, National Institutes of Health (NIH), United StatesReviewed by:
Sergey P. Morzunov, University of Nevada, Reno, United StatesDaniel Ferreira Feijó, Instituto Gonçalo Moniz (IGM), Brazil
Copyright © 2018 Pereira, Marinho and Epiphanio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sabrina Epiphanio, c2FicmluYWVAdXNwLmJy
†Present Address: Marcelo L. M. Pereira, Instituto de Biossistemas e Ciências Integrativas, Faculdade de Ciências, Universidade de Lisboa, Lisbon, Portugal