 Xiaodan Zhong1,2†
Xiaodan Zhong1,2† Ping Cui2,3†
Ping Cui2,3† Junjun Jiang2
Junjun Jiang2 Chuanyi Ning2,4
Chuanyi Ning2,4 Bingyu Liang2
Bingyu Liang2 Jie Zhou2
Jie Zhou2 Li Tian2
Li Tian2 Yu Zhang2
Yu Zhang2 Ting Lei1,2
Ting Lei1,2 Taiping Zuo1,2
Taiping Zuo1,2 Li Ye2,3*
Li Ye2,3* Jiegang Huang2*
Jiegang Huang2* Hui Chen1,2*
Hui Chen1,2*- 1Geriatrics Digestion Department of Internal Medicine, The First Affiliated Hospital of Guangxi Medical University, Nanning, China
- 2Guangxi Key Laboratory of AIDS Prevention and Treatment, School of Public Health, Guangxi Medical University, Nanning, China
- 3Life Science Institute, Guangxi Medical University, Nanning, China
- 4Nursing College, Guangxi Medical University, Nanning, China
Background: New evidence implies that the imbalance of gut microbiota is associated with the progression of alcoholic liver disease (ALD) and that the composition of gut microbiota is altered in ALD patients. However, the predominant bacterium in patients involved in the progress of ALD has not been identified. The purpose of this study is to investigate the predominant bacterium in the early and end-stages of ALD as well as the relationship between the bacterium and the degree of liver injury.
Methods: We enrolled 21 alcoholic fatty liver (AFL) patients, 17 alcoholic liver cirrhosis (ALC) patients and 27 healthy controls, and sequenced the 16S rRNA gene of their fecal microbiota. The gut microbiota composition and its relationship with the indicators of clinical hepatic function were assessed using canonical correspondence analysis (CCA), spearman correlation heatmap and multivariate association with linear (MaAsLin) Models.
Results: The composition and structure of gut microbiota changed greatly in different stages of ALD, and the degree of disorder was aggravated with the progression of ALD, even in the early stage. Moreover, the relative abundance of Streptococcus was highly enriched only in patients with ALC (P <0.001), and positively correlated with AST level (P = 0.029). The abundance of Streptococcus distinguished the liver injury of ALC patients from the controls with an area under the receiver-operating characteristic curve (AUC) of 0.877 (P < 0.001).
Conclusions: These findings indicate that the imbalance of gut microbiota exists at the early and end-stages of ALD, and the degree of disorder is aggravated with the progression of ALD. Streptococcus, as the predominant bacterium, may be a microbiological marker to evaluate the severity of liver injury in ALD patients.
Introduction
Alcoholic liver disease (ALD) is a chronic liver disease caused by long-term alcohol-use disorder, and its pathological changes include steatosis, steatohepatitis, fibrosis, cirrhosis, and hepatocellular carcinoma (Dunn and Shah, 2016).Worldwide, alcohol-related liver diseases are the leading cause of liver disease-related mortality in most countries (Savolainen et al., 1993; Gao and Bataller, 2011). Although it is known that the amount and mode of alcohol consumption are certain risk factors for the onset of ALD (Stickel and Hampe, 2012), the pathogenesis of ALD is not fully understood. Recent studies have provided evidence that ALD may be related to oxidative stress injury from alcohol metabolism, abnormal methionine metabolism, injury of inflammatory mediators, intestinal microbiota imbalance and bacterial translocation (Shao et al., 2018).
The gut microbiota, also named intestinal microbiome or intestinal microbiota, plays an important role in the metabolism and immune regulation of the host (Burkholder and McVeigh, 1942; Chung et al., 2012; McDermott and Huffnagle, 2014; Spasova and Surh, 2014; Morrison and Preston, 2016). The gut microbiota contributes to host physiology through the production of a myriad of metabolites (Krautkramer et al., 2021). As the main product of gut microbiota, short-chain fatty acids (SCFA) have been found to play an important role in lipid turnover and energy homeostasis by reducing adipocyte lipolysis and adipogenesis (Hong et al., 2005). Furthermore, SCFA, in particular propionate and butyrate, have also been shown a strong anti-inflammatory effect through downregulating of lipopolysaccharide (LPS)-induced cytokines expression (Nastasi et al., 2015). Once the gut microbiota is out of balance, the important pathogenic substances produced by the maleficent bacteria enter the liver via the portal vein and then cause liver damage (Szabo, 2015).
Previous studies have shown that alcohol abuse leads to alterations in the structure and composition of gut microbiota and promotes the occurrence and progression of ALD (Sarin et al., 2019). The imbalance of gut microbiota happens both in alcohol-related human disorders and mice models (Yan et al., 2011; Leclercq et al., 2014). Intestinal metabolism of alcohol produces a high concentration of toxic acetaldehyde that alters the intestinal barrier and promotes LPS translocation (Tang et al., 2008; Malaguarnera et al., 2014). Acetaldehyde and LPS then induce Kupffer cells to release ROS, proinflammatory cytokines and chemokines that contribute to hepatocyte damage (Ceni et al., 2014).
It is also found that the degree of dysbiosis in gut microbiota is closely related to the severity of ALD. The liver function test is often used to assess liver function in the clinic, and ALD patients are usually identified by increasing levels of liver enzymes such as aspartate aminotransferase (AST) and gamma-glutamyltranspeptidase (GGT). In particular, the indexes are elevated with the severity of ALD. It’s also reported that the abundance of Akkermansia muciniphila was reduced in patients with alcoholic steatohepatitis compared with healthy controls, which was indirectly correlated with the severity of hepatic disease (Grander et al., 2018). However, the correlation between these predominant bacteria in gut microbiota and abnormal indicators of hepatic function in ALD patients is still unclear, and its significance remains to be elucidated.
In this study, we investigated the change of structure and composition of gut microbiota in patients at the early and end-stages of ALD. Furthermore, the correlation between the indexes of the clinical liver function examination and the bacterial community was analyzed. The aim of this study was also to identify the predominant gut bacterium of ALD patients, which could be related to the progression of the disease and used as a microbiological marker to assess the severity of ALD.
Materials and Methods
Ethics Statement
This study was approved by the Ethics Committee of Guangxi Medical University, Nanning, Guangxi, China. All the study subjects provided written informed consent, and volunteered to participate in the investigation for scientific research.
Patient Recruitment
Due to the higher susceptibility to ALDs in women than in men (Dunn and Shah, 2016), only males (who are more tolerant to alcohol) were chosen as participants in this study. The ALD patients in this study were separated into two groups: alcoholic fatty liver (AFL) and alcoholic liver cirrhosis (ALC), and enrolled from Department of Gastroenterology of the First Affiliated Hospital of Guangxi Medical University if they satisfied the following criteria: 1) long-term alcohol consumption history or drink more than 40 g per day, 2) clinical imageological examination (ultrasonic B or CT) showed steatosis or cirrhosis, 3) no antibiotics or probiotics were used at least two weeks before fecal sample collection. In addition, the patients who met the following criteria were excluded from the study: 1) if the patients were suffering from viral hepatitis, drug hepatitis, autoimmune liver disease or other specific etiology of liver disease, 2) if the patients were suffering from the diseases that confirmed related to microbiome, such as inflammatory bowel disease, gastrointestinal tumors, heart disease, diabetes and so on, 3) if the patients regularly use or recently use specific drugs, which may affect the condition of gut microbiota, such as proton pump inhibitor (PPI), laxatives and so on.
Healthy controls were selected from the Physical Examination Centre of the First Affiliated Hospital of Guangxi Medical University and met the following standards: 1) did not have a long-term alcohol consumption history or drank less than 40 g per day, 2) no abnormalities in the clinical imageological examination (ultrasonic B or CT), 3) no antibiotics or probiotics were used at least two weeks before fecal sample collection, 4) matched with the patients by age and gender. The exclusion criteria mentioned above is also applied to the healthy controls.
Fecal Sample and Basic Data Collection
Fresh stools were collected from the IC patients in the morning after their admission to the hospital, and from the healthy subjects during their physical examination. The fecal samples should be put in stool collection tubes with stool DNA stabilizer (Stratec, Berlin, GER), and stored in -80°C lab freezers immediately. All the study subjects’ general information, including past medical history, medication history, past surgical history, bowel habit and so on, was acquired from our questionnaire. And the results of clinical image examination and the indicators of hepatic function were collected from the participants’ medical report.
DNA Extractions and Amplification of 16S rRNA
Genomic DNA was extracted from fecal samples using the EZNA® DNA Kit (Omega Bio-tek, Norcross, GA, USA) by a tissue lyser (Bao et al., 2017), following the standard protocol. PCR amplification was performed with the 338F (5’-ACTCCTACGGGAGGCAGCAG-3’) and 806R (5’-GGACTACHVGGGTWTCTAAT-3’) primers, which targeted the V3–V4 region of the bacterial 16S rRNA gene. Amplified products were detected by 1% agarose gel electrophoresis and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA).
16S rRNA Sequencing and Microbiota Analysis
Sequencing was conducted by Shanghai Majorbio Bio-pharm Technology (Shanghai, China). The purified amplicons were pooled and paired-end sequenced (2 × 300) on an Illumina MiSeq platform (San Diego, CA, USA) according to the manufacturer’s instructions. The sequencing was removed using the following criteria: 1) sequences shorter than 200 bp, 2) ambiguous bases, 3) sequences with an average mass less than 25. Sequences were assigned to operational taxonomic units (OTUs) with 97% similarity. The sequences were classified using the SILVA database containing bacterial and fungal ribosomal RNA sequences. The raw reads were deposited into the NCBI Sequence Read Archive (SRA) database (BioProject ID: PRJNA690835).
The community diversity of gut microbiota was analyzed after the sequence data subsampled by minimum sequence numbers. In addition, partial least squares discriminant analysis (PLS-DA) and analysis of similarities (ANOSIM) were also performed to compare the bacterial composition among groups. Linear discriminant analysis (LDA), coupled with effect size measurement (LEfSe) analysis, was conducted to elucidate the differences of bacterial taxa between groups (Segata et al., 2011). The relationships between gut microbial community structure and the indicators of hepatic function were analyzed by canonical correspondence analysis (CCA), spearman correlation heatmap and multivariate association with linear (MaAsLin) Models. CCA eliminates redundant variables depending on other measured variables, automatically selecting variables with large effects, and on the variance inflation factor values to gradually remove redundant parameters, and the significance levels are based on 999 Monte Carlo permutations.
Statistical Analysis
All results were presented as mean ± standard deviation (SD) or median (interquartile range), depending on whether they fit the normal distribution. The one-way ANOVA test, nonparametric tests and receiver operating characteristic (ROC) curve were performed using SPSS version 26.0. The Wilcoxon rank-sum test was used for alpha diversity analysis, including Shannon, Chao and Ace indexes. And Kruskal–Wallis H test was used to compare the relative abundance of gut microbiota among groups. Results with P <0.05 between groups were declared statistically significant.
Results
Grouping Situation and Data Output
A total of 27 healthy controls, 21 alcoholic fatty liver (AFL group) and 17 alcoholic liver cirrhosis (ALC group) participants were enrolled for stool sample and clinical data collection (Table 1). To profile the differences in the composition of gut microbiota at different stages of ALD, the V3 region of bacterial 16S rRNA gene from 65 samples were sequenced. After quality trimming and chimaera checking, we obtained 3,308,525 valid sequences in total. Based on the 97% similarity, unique representative sequences were classified into 1486 operational taxonomic units (OTUs) through a clustering operation, from which, 30 phyla, 625 genus and 1065 species were detected.
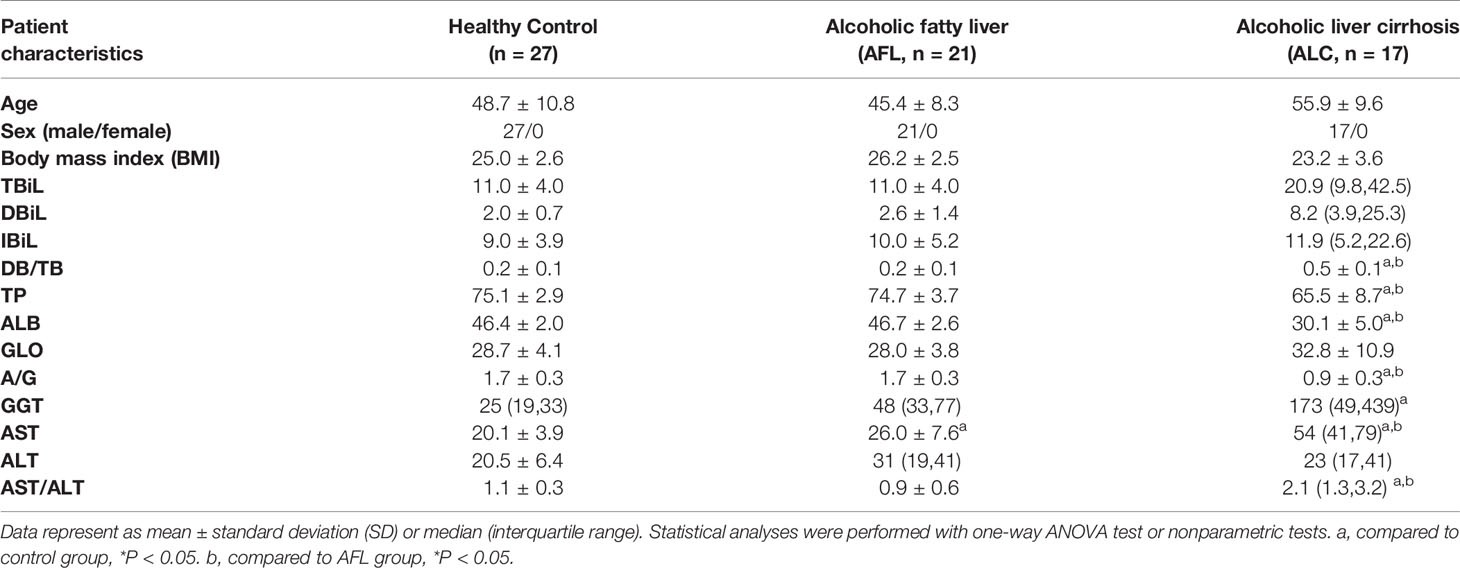
Table 1 Patient characteristics.
Analysis of similarities (ANOSIM) was performed based on Bray -Curtis distances (***P < 0.001). It showed that the difference between the groups was greater than that within groups, which indicated that the grouping method, based on the processes of ALD, was effective and meaningful.
The Community Diversity of Gut Microbiota in ALD Patients Declined
Alpha diversity refers to the diversity of a specific area or ecosystem, of which, Chao and Ace are commonly used to estimate the total number of species of gut microbiota. As shown in Figures 1A, B and Table 2, there were significant differences in the estimators of OTU richness indices including Ace (139.86 ± 46.57 vs. 192.96 ± 62.27, P = 0.004) and Chao (140.69 ± 46.01 vs. 192.22 ± 62.92, P = 0.005) between ALC and control groups. Furthermore, there were also significant differences in Ace and Chao indexes between ALC and AFL groups. The Chao index in ALD patients decreased gradually, indicating that the imbalance of gut microbiota was aggravated with the development of the disease. In addition, the coverage curves of each group eventually flattened out (Figure 1C), showing that the sequencing data of each group approached saturation point and the data at the OTU level could cover most bacteria in the intestinal tract.
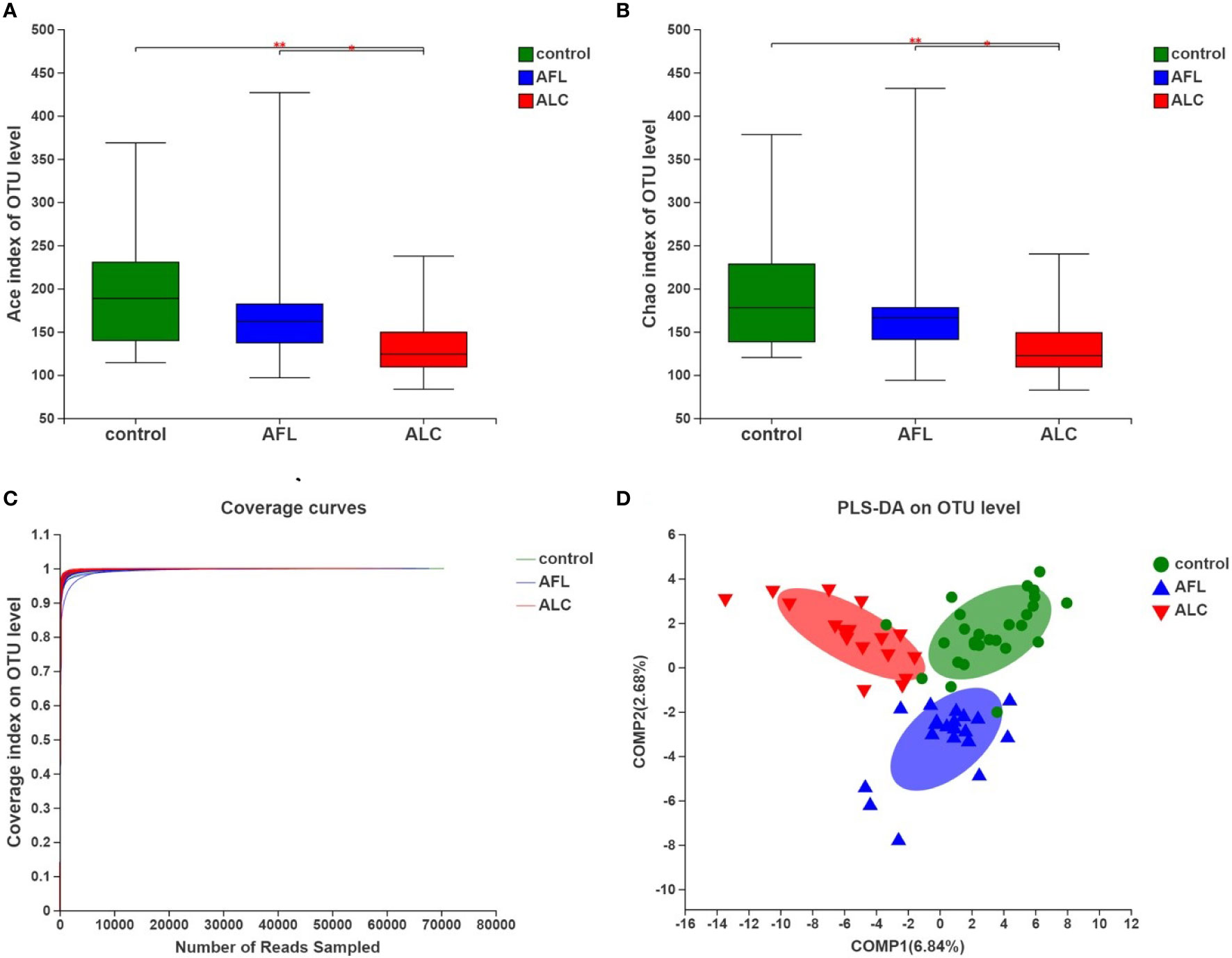
Figure 1 The community diversity of gut microbiota in patients with alcoholic fatty liver (AFL) or alcoholic liver cirrhosis (ALC) compared with healthy controls (control). (A) The Ace diversity indexes, (B) The Chao diversity indexes, (C) Coverage curves on OTU level, and (D) PLS-DA on OTU level. *P < 0.05, **P < 0.01.
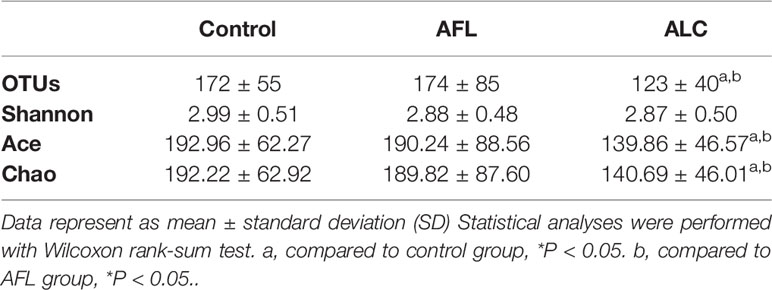
Table 2 Sequencing data summary and community diversity.
To evaluate the similarity of the gut microbiota communities among different samples within the groups, partial least squares discriminant analysis (PLS-DA) was performed at the OTU level. The samples of each group were clustered, and the grouped ellipses of three groups on PLS-DA were separated (Figure 1D). It suggested that there were significant differences in the overall structures of the gut microbiota communities in different periods of ALD.
The Difference Analysis of Composition in Gut Microbiota
Except for the differences of the structures in gut microbiota, the composition of bacterial communities in patients with fatty liver or cirrhosis was indeed different from healthy controls. The most abundant bacterial phylum in the different groups was shown in Figure 2A. We found that Proteobacteria gradually enriched in patients as the disease progressed (***P <0.001), as well as Fusobacteria (*P <0.05). However, the relative abundance of Bacteroidetes showed a protracted downward trend among the three groups.
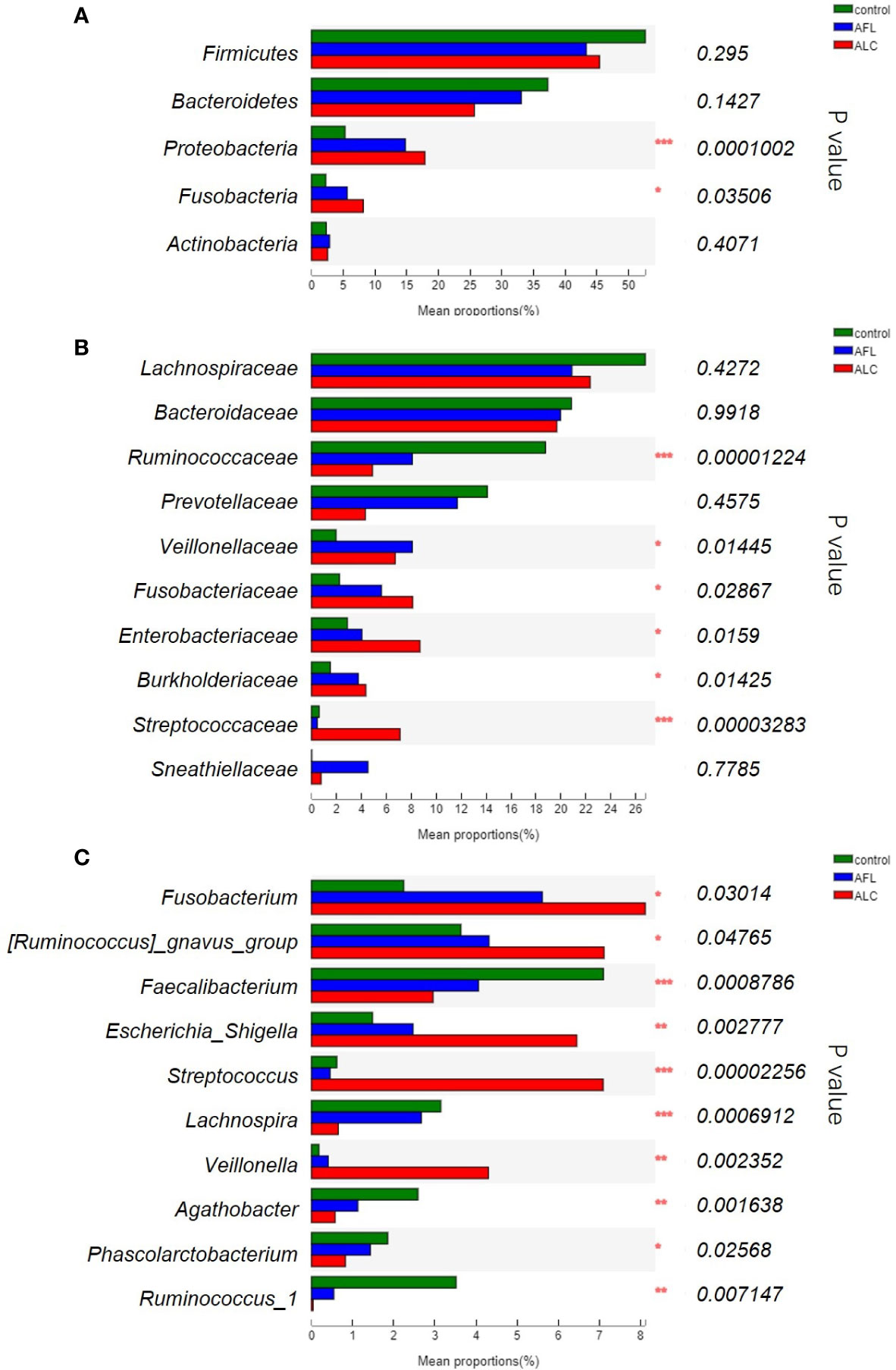
Figure 2 The relative abundance of the gut microbiota between three groups. (A) At the phylum level, (B) at the family level, (C) significant differences between bacteria at the genus level. Statistical analysis was performed by the Kruskal–Wallis H test. *P < 0.05, **P < 0.01, ***P < 0.001.
We further compared the relative abundance of gut microbiota of the three groups at family levels. As shown in Figure 2B, the relative abundance of Ruminococcaceae decreased in ALD patients was lower than that in the healthy controls (***P <0.001), while Fusobacteriaceae, Enterobacteriaceae and Burkholderiaceae (*P <0.05) was increased in the AFL and ALC groups. Streptococcaceae (***P <0.001) were remarkably enriched in the ALC group only.
At the genus level, the top ten predominant bacteria with statistical differences in three groups were shown in Figure 2C. The relative abundance of Faecalibacterium (***P <0.001), Lachnospira (***P <0.001), Agathobacter (**P <0.01) and Ruminococcus_1 (**P <0.01) in the AFL and ALC groups decreased as the disease progressed, while Fusobacterium (*P <0.05) and Escherichia-Shigella (**P <0.01) increased gradually. Similarly, Streptococcus (***P <0.001) and Veillonella (**P <0.01) were significantly enriched in the ALC group only.
Through analyzing the structure and composition of gut microbiota among the three groups, we implemented the linear discriminant analysis (LDA) effect size (LEfSe) method to further validate the specialized communities within the groups. As shown in Figure 3A, a cladogram displayed all the significantly enriched bacterial structure at phylum and genus levels. LDA scores of 3.5 or above were confirmed by LEfSe (Figure 3B), indicating that the listed bacteria were the dominant bacterial community in each group. At the genus level, Faecalibacterium, Sphingomonas and Streptococcus were the most significant enrichment bacteria in the healthy control, AFL and ALC group, respectively.
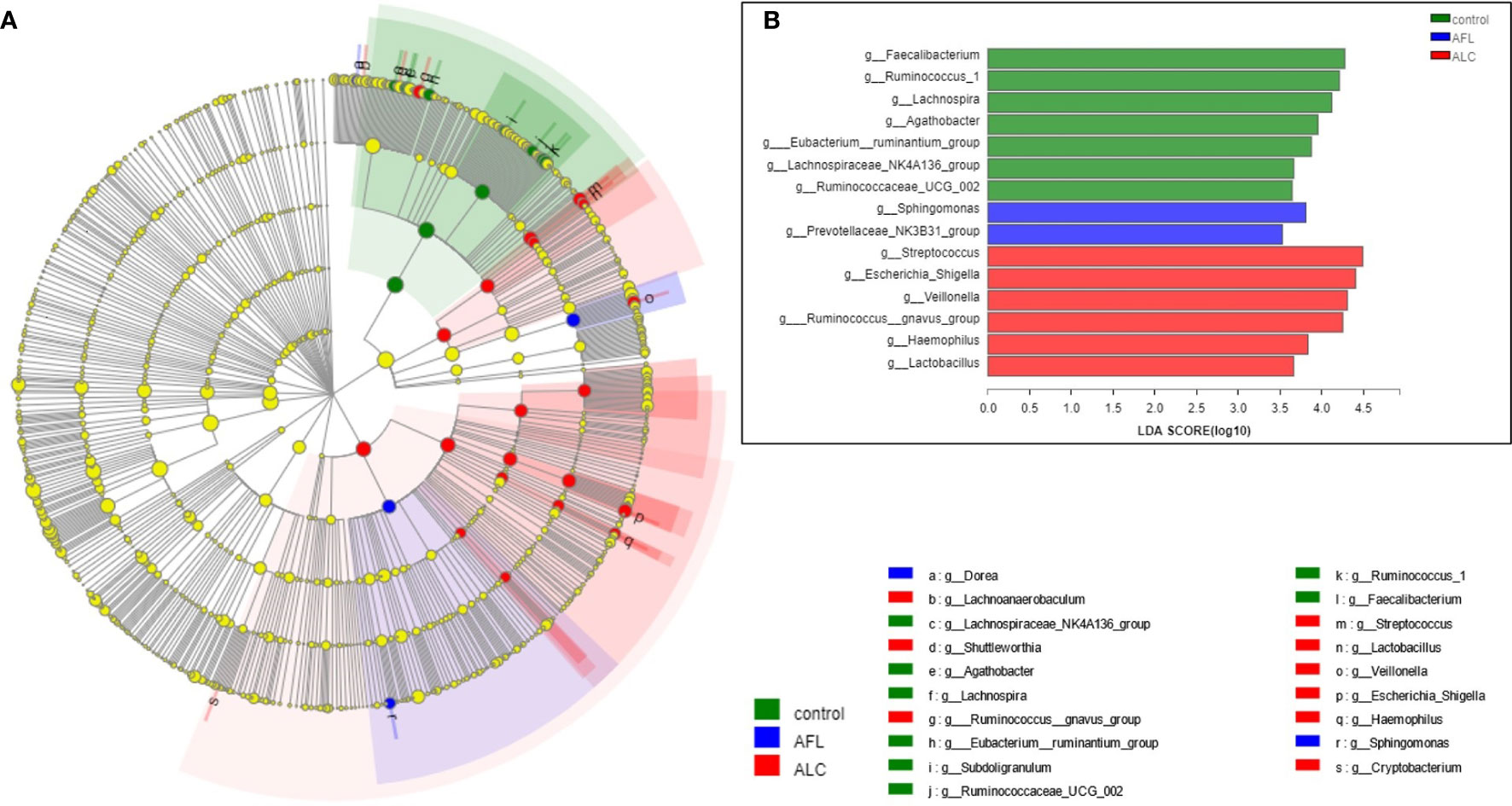
Figure 3 Microbial taxa identified in different groups using LEfSe analysis. (A) Cladogram showed the phylogenetic distribution of bacteria that were significantly enriched between the three groups. Different colors spots represent significantly enriched bacterial in the corresponding group. (B) LDA scores showed significant differences in bacterial within groups at the genus level.
The Correlation Between Relative Abundance of Bacterial and Clinical Hepatic Function Indexes
From clinical indexes of hepatic function in each group (Table 1), we found that ALC patients were associated with abnormal clinical indicators of hepatic function, such as direct bilirubin/total bilirubin (DB/TB), total protein (TP), albumin (ALB), the ratio of albumin to globulin (A/G), AST, GGT and aspartate aminotransferase/alanine aminotransferase (AST/ALT). The hepatic function abnormalities were characterized by damaged hepatic cells and impaired liver function. Meanwhile, AST, ALT and GGT were also elevated in AFL patients, indicating that ALD patients at early stage might have hepatocyte damage without any clinical symptoms.
Usually, the liver function impairment in ALD patients was assessed through clinical hepatic function upon biochemical examination. AST and GGT were the main abnormal indicators of clinical hepatic function in ALD patients. Therefore, we studied whether the indicators of the clinical laboratory tests were related to microbial community structure. After using variance inflation factor (VIF) analysis to filter suitable clinical indexes, we selected CCA, based on a single peak model to verify the relationship between samples, clinical indexes and gut microbiota.
As shown in Figure 4A, the red arrow represents the indexes of the clinical liver function test, and its length means the extent to which this index affects the gut microbiota. Total bilirubin (TBiL), A/G, TP, AST/ALT, DB/TB, GGT and AST were significantly associated with the bacterial community structure. As the indicators reflected the severity of impaired liver function, AST/ALT, DB/TB, GGT, AST were significantly related to ALC patients (red inverted triangles), and the species of intestinal bacteria in genus level, marked in black triangles, were closely linked with the clinical indexes, including Bacteroides, Ruminococcus, Faecalibacterium and Streptococcus.
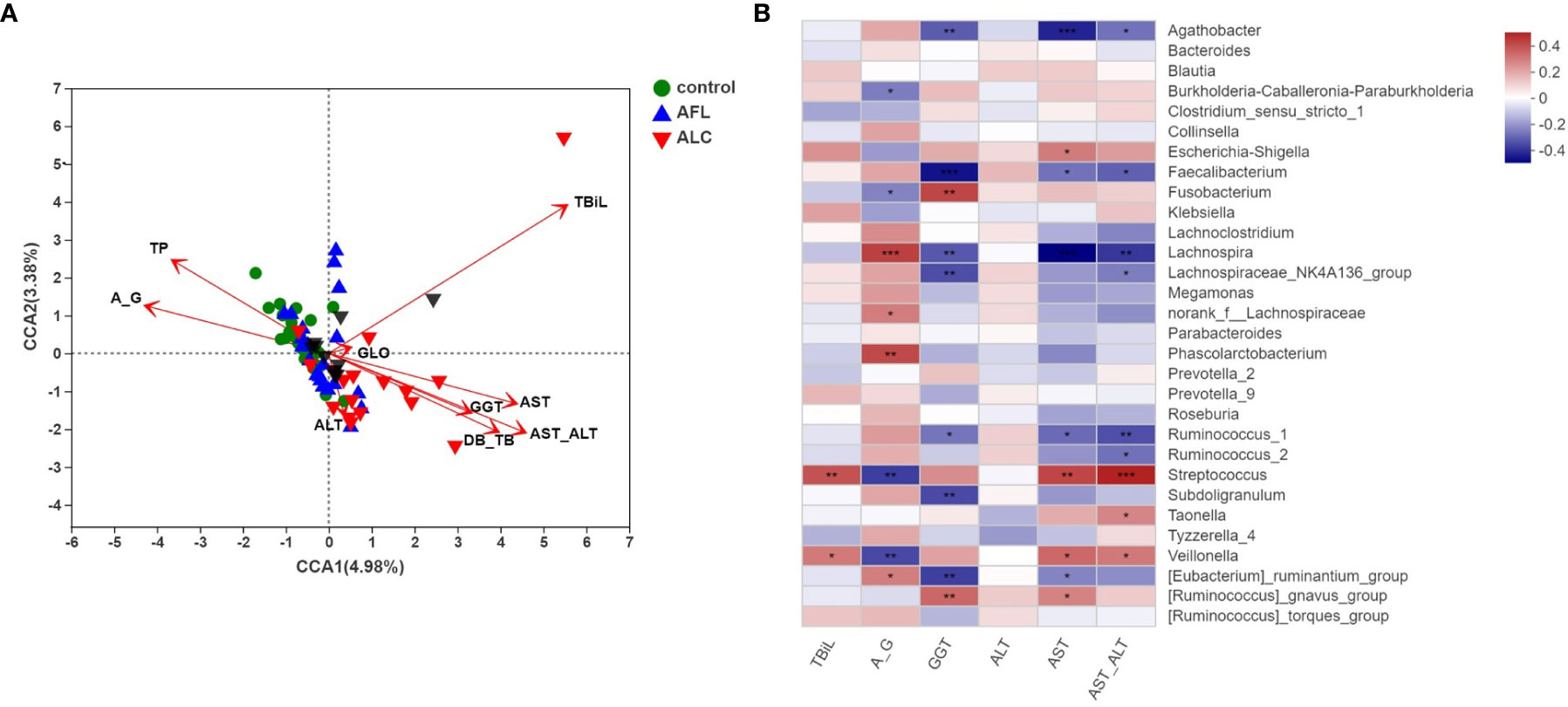
Figure 4 Correlation analysis of the structures of gut microbiota and clinical factors. (A) CCA analysis on genus level showed the correlations between the bacterial community structures and the indicators of clinical laboratory tests. (B) Heatmap analysis of the correlation between the top 30 predominant bacteria at genus level and the clinical factors. Red spots represent a positive correlation, while blue spots represent a negative correlation. *P < 0.05, **P < 0.01, ***P < 0.001.
Then, we utilized a heat map to show the association between microbial species at the genus level and the selected clinical factors (Figure 4B). It’s worth noting that Streptococcus, as a significantly enriched bacteria in the ALC group, was positively correlated with TBiL, AST and AST/ALT, while negatively correlated with A/G. The results reflected that the relative abundance of Streptococcus is closely associated with the indicators of hepatic function.
Streptococcus Correlated with Liver Clinical Factor, which might be an Indicator to Evaluate the Severity of Liver Injury
MaAsLin analysis was used to analyze the relationship between the relative abundance of Streptococcus and individual specific clinical factor, such as AST, AST/ALT and A/G. The relative abundance of Streptococcus was positively correlated with the levels of AST and AST/ALT, while negatively correlated with A/G in the ALD patients (Figures 5A–C; coefficient = 0.0015, p = 0.029; coefficient = 0.0318, p = 0.002; coefficient = –0.0566, p = 0.0007, respectively).
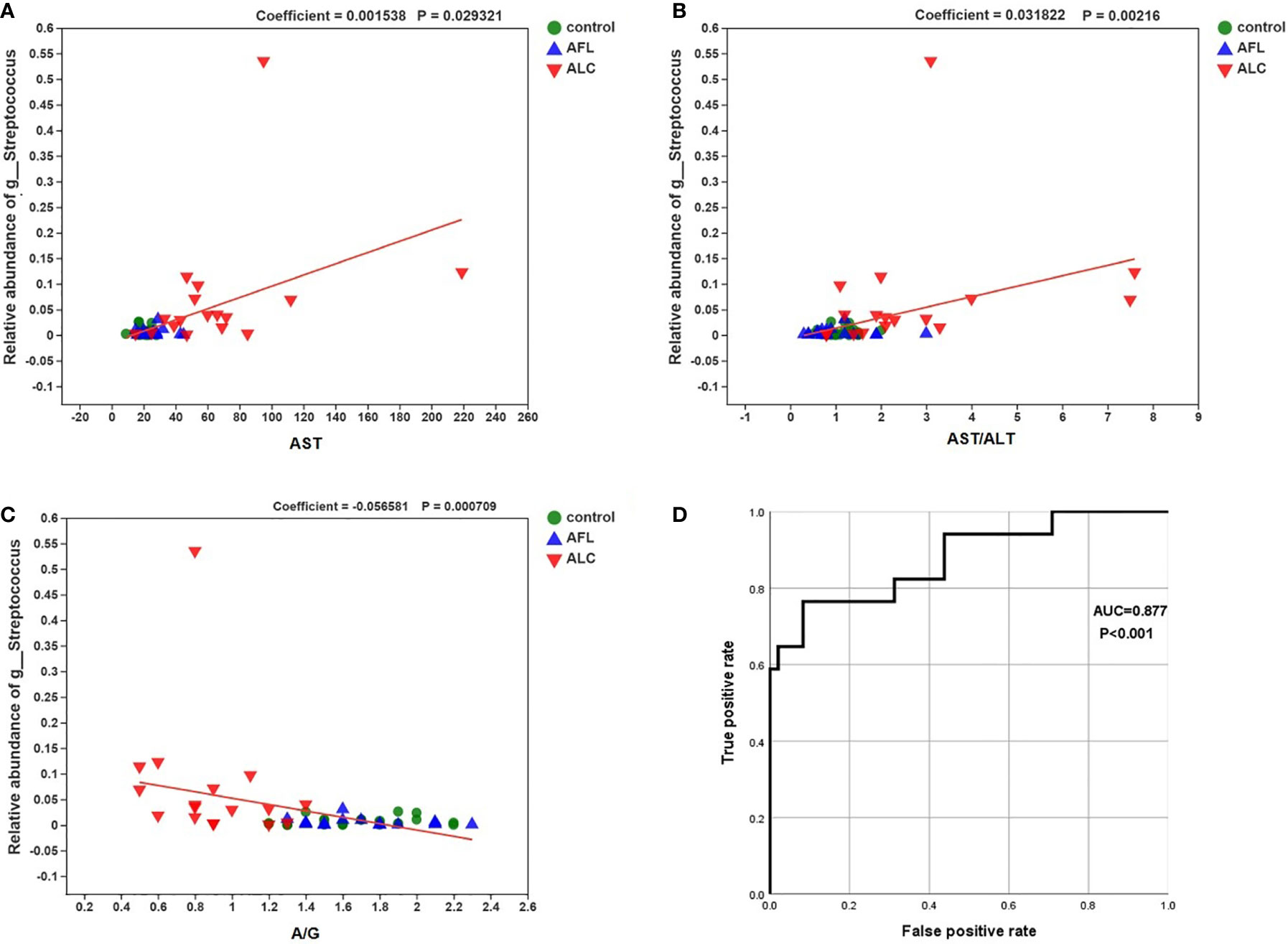
Figure 5 Correlations between the relative abundance of Streptococcus and the level of the specific clinical factor. The correlations were calculated in MaAsLin. (A) AST (coefficient = 0.0015, p = 0.029), (B) AST/ALT (coefficient = 0.0318, p = 0.002) and (C) A/G (–0.0566, p = 0.0007). (D) the ROC curve about the relative abundance of Streptococcus and ALC.
Finally, we performed a receiver operating characteristic curve (ROC) and calculated the area under the curve (AUC) to evaluate the diagnostic efficiency of the relative abundance of Streptococcus on the liver function of ALC patients. The results showed that AUC was close to 1 (Figure 5D; AUC = 0.877, P < 0.001), suggesting that it is probable to assess the severity of liver injury in ALC patients by the relative abundance of Streptococcus.
Discussion
Recently, numerous studies have suggested that the progression of alcoholic liver disease is associated with dysbacteriosis (Sarin et al., 2019), although the relationship between the altered gut microbiota and the severity or progression of ALD is still unknown. We compared the structure and composition of gut microbiota in the early and end-stages of ALD patients and found that the community diversity and composition of gut microbiota in ALD patients were changed with disease progressed. Moreover, we discovered the linear correlation between the predominant bacterium and the main indicators of hepatic function in ALC patients. Through the correlation analysis, we inferred a new potential key species, which was integrally involved in the progression of ALD.
Generally speaking, ALD patients have elevated serum aminotransferase levels. However, in the early stage of ALD, patients’ serum aminotransferase levels can be normal. ALT, AST and TBil are sensitive indexes to evaluate the degree of liver injury among many liver function indexes. It is well known that chronic alcohol consumption can lead to elevated serum GGT (Conigrave et al., 2003; Hietala et al., 2005). GGT is a sensitive marker of alcohol consumption and liver dysfunction, which could return to normal after 2–3 weeks of abstaining from drinking (Niemela, 2016). We found that AST, ALT and GGT were slightly elevated in AFL patients, while almost all the indicators were abnormal in ALC patients, except GGT, suggesting that AFL patients possibly have hepatocyte damage caused by alcohol consumption, and that ALC patients have sustained impaired liver function even after abstinence from alcohol.
Growing evidence suggests that one of the manifestations of dysbiosis in gut microbiota is the changed community diversity. Similar to previous studies (Addolorato et al., 2020; Zheng et al., 2020), we also found that the alpha diversity and beta diversity of the gut microbiota in ALD patients were changed, even in its early stages, which showed that the richness of species decreased and the structure was altered due to alcohol consumption. Previous studies have reported that alcohol-induced changes in gut microbiota composition and metabolic function may contribute to alcohol-induced oxidative stress and the subsequent development of ALD (Ceni et al., 2014), meaning that alcohol abuse may be a main factor leading to the imbalance of gut microbiota.
Moreover, as the severity of ALD changes with time, so does the composition of gut microbiota and the relative abundance of bacteria. We found that Enterobacteriaceae and Streptococcaceae families were significantly increased in fecal samples from ALC patients, while Ruminococcaceae families were significantly reduced. Bajaj et al. pointed out that the change of the abundance of “good” vs. “bad” bacteria in liver cirrhosis patients was reflected by the cirrhosis dysbiosis ratio, which is determined by the abundance of Lachnospiraceae and Ruminococcaceae divided by the abundance of Enterobacteriaceae and Bacteroidaceae (Bajaj et al., 2014). As mentioned above, the changed abundance of the bacterium at the family level in three groups reduced the cirrhosis dysbiosis ratio, meaning the lower the ratio, the more serious the imbalance of gut microbiota in ALC patients. Furthermore, the specific bacterial families (Ruminococcaceae, Streptococcaceae, Fusobacteraceae) are strongly associated with cognition and inflammation in liver cirrhosis with hepatic encephalopathy (Bajaj et al., 2012).
Unlike some frequently reported bacterium, Streptococcus was observed to be the predominant gut bacterium of ALD patients. Previous studies have shown that Streptococcus was characteristically higher in liver cirrhosis and alcoholics (Chen et al., 2011; Tsuruya et al., 2016). And coincidentally, duodenal dysbiosis showed a dominance shift toward specific potential pathogenic bacteria genera (Streptococcus) in alcohol use disorders (Maccioni et al., 2020). In our study, we demonstrated that the relative abundance of Streptococcus was positively correlated with AST, which, as a major abnormal indicator of alcoholic liver injury, is typically more than twice that of ALT (Diehl, 2002). It revealed that the increased abundance of Streptococcus was correlated with the severity of hepatocyte damage in ALC. Similarly, Streptococcus bovis bacteremia has been associated with gastrointestinal diseases, especially colon cancer (Alvarez et al., 2015). According to multifaceted evidence that Streptococcus was closely related to liver cirrhosis, we propose a bold hypothesis that the abundance of Streptococcus could be used as an indicator to forecast the severity of cirrhosis. The ROC curve and AUC effectively proved the feasibility of this hypothesis. The greater the AUC means the stronger accuracy of the new bioindicator. Coincidentally, a core gut microbiota signature can identify cirrhosis across geographically separated cohorts, independent of other influencing factors on the gut microbiota (Oh et al., 2020). Therefore, a specificity of bacteria in gut microbiota, as a non-invasive diagnostic test for cirrhosis, has a sound theoretical basis, and the early diagnosis through the detection of changed bacterium is necessary, because of difficult diagnosis at the early stage of ALD.
Altogether, we demonstrated that the degree of imbalance of gut microbiota is more severe as the progression of ALD worsens, and the relative abundance of Streptococcus is related to the important indicators of clinical hepatic function. We can look forward that Streptococcus may become a microbial marker to evaluate the severity of liver injury in liver cirrhosis patients. As a potential pathogenic bacteria genera, Streptococcus was positively related to tryptophan, whose metabolites have been proved to be related to fat metabolism and proinflammatory reaction (Krishnan et al., 2018; Oh et al., 2020). Therefore, we hypothesis that the enriched Streptococcus possibly could cause hepatocyte damage by the proinflammatory effect of its metabolites.
Data Availability Statement
The data sets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: BioSample database, BioProject ID: PRJNA690835 (https://www.ncbi.nlm.nih.gov/sra/PRJNA690835).
Ethics Statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
HC, JH, and LY designed the study. XZ and PC contributed to data analysis and wrote the paper. JZ, LT, YZ, TL, and TZ participated in data acquisition. JJ, CN, and BL participated in interpreting the results. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China (NSFC, 81460305, 82002134, 81660334, 82060366), Youth Science Foundation of Guangxi Medical University (GXMUYSF201826), Guangxi Natural Science Foundation (2018GXNSFAA050099, 2017GXNSFAA198190), Guangxi Bagui Scholar (to JJ), Guangxi Medical University Training Program for Distinguished Young Scholars (to JJ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Addolorato G., Ponziani F. R., Dionisi T., Mosoni C., Vassallo G. A., Sestito L., et al. (2020). Gut microbiota compositional and functional fingerprint in patients with alcohol use disorder and alcohol-associated liver disease. Liver Int. 40, 878–888. doi: 10.1111/liv.14383
Alvarez A., Garcia C. J., Jia Y., Boman D., Zuckerman M. J. (2015). Streptococcus bovis Bacteremia: Association with Gastrointestinal and Liver Disease in a Predominantly Hispanic Population. South Med. J. 108, 425–429. doi: 10.14423/SMJ.0000000000000310
Bajaj J. S., Ridlon J. M., Hylemon P. B., Thacker L. R., Heuman D. M., Smith S., et al. (2012). Linkage of gut microbiome with cognition in hepatic encephalopathy. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G168–G175. doi: 10.1152/ajpgi.00190.2011
Bajaj J. S., Heuman D. M., Hylemon P. B., Sanyal A. J., White M. B., Monteith P., et al. (2014). Altered profile of human gut microbiome is associated with cirrhosis and its complications. J. Hepatol. 60, 940–947. doi: 10.1016/j.jhep.2013.12.019
Bao Y. J., Xu Z., Li Y., Yao Z., Sun J., Song H. (2017). High-throughput metagenomic analysis of petroleum-contaminated soil microbiome reveals the versatility in xenobiotic aromatics metabolism. J. Environ. Sci. (China) 56, 25–35. doi: 10.1016/j.jes.2016.08.022
Burkholder P. R., McVeigh I. (1942). Synthesis of Vitamins by Intestinal Bacteria. Proc. Natl. Acad. Sci. U.S.A. 28, 285–289. doi: 10.1073/pnas.28.7.285
Ceni E., Mello T., Galli A. (2014). Pathogenesis of alcoholic liver disease: role of oxidative metabolism. World J. Gastroenterol. 20, 17756–17772. doi: 10.3748/wjg.v20.i47.17756
Chen Y., Yang F., Lu H., Wang B., Chen Y., Lei D., et al. (2011). Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 54, 562–572. doi: 10.1002/hep.24423
Chung H., Pamp S. J., Hill J. A., Surana N. K., Edelman S. M., Troy E. B., et al. (2012). Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149, 1578–1593. doi: 10.1016/j.cell.2012.04.037
Conigrave K. M., Davies P., Haber P., Whitfield J. B. (2003). Traditional markers of excessive alcohol use. Addiction 98 ( Suppl 2), 31–43. doi: 10.1046/j.1359-6357.2003.00581.x
Diehl A. M. (2002). Liver disease in alcohol abusers: clinical perspective. Alcohol 27, 7–11. doi: 10.1016/s0741-8329(02)00204-5
Dunn W., Shah V. H. (2016). Pathogenesis of Alcoholic Liver Disease. Clin. Liver Dis. 20, 445–456. doi: 10.1016/j.cld.2016.02.004
Gao B., Bataller R. (2011). Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 141, 1572–1585. doi: 10.1053/j.gastro.2011.09.002
Grander C., Adolph T. E., Wieser V., Lowe P., Wrzosek L., Gyongyosi B., et al. (2018). Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut 67, 891–901. doi: 10.1136/gutjnl-2016-313432
Hietala J., Puukka K., Koivisto H., Anttila P., Niemela O. (2005). Serum gamma-glutamyl transferase in alcoholics, moderate drinkers and abstainers: effect on gt reference intervals at population level. Alcohol Alcohol 40, 511–514. doi: 10.1093/alcalc/agh201
Hong Y. H., Nishimura Y., Hishikawa D., Tsuzuki H., Miyahara H., Gotoh C., et al. (2005). Acetate and propionate short chain fatty acids stimulate adipogenesis via GPCR43. Endocrinology 146, 5092–5099. doi: 10.1210/en.2005-0545
Krautkramer K. A., Fan J., Backhed F. (2021). Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Microbiol. 19, 77–94. doi: 10.1038/s41579-020-0438-4
Krishnan S., Ding Y., Saedi N., Choi M., Sridharan G. V., Sherr D. H., et al. (2018). Gut Microbiota-Derived Tryptophan Metabolites Modulate Inflammatory Response in Hepatocytes and Macrophages. Cell Rep. 23, 1099–1111. doi: 10.1016/j.celrep.2018.03.109
Leclercq S., Matamoros S., Cani P. D., Neyrinck A. M., Jamar F., Starkel P., et al. (2014). Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc. Natl. Acad. Sci. U.S.A. 111, E4485–E4493. doi: 10.1073/pnas.1415174111
Maccioni L., Gao B., Leclercq S., Pirlot B., Horsmans Y., De Timary P., et al. (2020). Intestinal permeability, microbial translocation, changes in duodenal and fecal microbiota, and their associations with alcoholic liver disease progression in humans. Gut Microbes 12:1782157. doi: 10.1080/19490976.2020.1782157
Malaguarnera G., Giordano M., Nunnari G., Bertino G., Malaguarnera M. (2014). Gut microbiota in alcoholic liver disease: pathogenetic role and therapeutic perspectives. World J. Gastroenterol. 20, 16639–16648. doi: 10.3748/wjg.v20.i44.16639
McDermott A. J., Huffnagle G. B. (2014). The microbiome and regulation of mucosal immunity. Immunology 142, 24–31. doi: 10.1111/imm.12231
Morrison D. J., Preston T. (2016). Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 7, 189–200. doi: 10.1080/19490976.2015.1134082
Nastasi C., Candela M., Bonefeld C. M., Geisler C., Hansen M., Krejsgaard T., et al. (2015). The effect of short-chain fatty acids on human monocyte-derived dendritic cells. Sci. Rep. 5:16148. doi: 10.1038/srep16148
Niemela O. (2016). Biomarker-Based Approaches for Assessing Alcohol Use Disorders. Int. J. Environ. Res. Public Health 13:166. doi: 10.3390/ijerph13020166
Oh T. G., Kim S. M., Caussy C., Fu T., Guo J., Bassirian S., et al. (2020). A Universal Gut-Microbiome-Derived Signature Predicts Cirrhosis. Cell Metab. 32, 878–888 e6. doi: 10.1016/j.cmet.2020.06.005
Sarin S. K., Pande A., Schnabl B. (2019). Microbiome as a therapeutic target in alcohol-related liver disease. J. Hepatol. 70, 260–272. doi: 10.1016/j.jhep.2018.10.019
Savolainen V. T., Liesto K., Mannikko A., Penttila A., Karhunen P. J. (1993). Alcohol consumption and alcoholic liver disease: evidence of a threshold level of effects of ethanol. Alcohol Clin. Exp. Res. 17, 1112–1117. doi: 10.1111/j.1530-0277.1993.tb05673.x
Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. doi: 10.1186/gb-2011-12-6-r60
Shao T., Zhao C., Li F., Gu Z., Liu L., Zhang L., et al. (2018). Intestinal HIF-1alpha deletion exacerbates alcoholic liver disease by inducing intestinal dysbiosis and barrier dysfunction. J. Hepatol. 69, 886–895. doi: 10.1016/j.jhep.2018.05.021
Spasova D. S., Surh C. D. (2014). Blowing on embers: commensal microbiota and our immune system. Front. Immunol. 5:318. doi: 10.3389/fimmu.2014.00318
Stickel F., Hampe J. (2012). Genetic determinants of alcoholic liver disease. Gut 61, 150–159. doi: 10.1136/gutjnl-2011-301239
Szabo G. (2015). Gut-liver axis in alcoholic liver disease. Gastroenterology 148, 30–36. doi: 10.1053/j.gastro.2014.10.042
Tang Y., Banan A., Forsyth C. B., Fields J. Z., Lau C. K., Zhang L. J., et al. (2008). Effect of alcohol on miR-212 expression in intestinal epithelial cells and its potential role in alcoholic liver disease. Alcohol Clin. Exp. Res. 32, 355–364. doi: 10.1111/j.1530-0277.2007.00584.x
Tsuruya A., Kuwahara A., Saito Y., Yamaguchi H., Tsubo T., Suga S., et al. (2016). Ecophysiological consequences of alcoholism on human gut microbiota: implications for ethanol-related pathogenesis of colon cancer. Sci. Rep. 6:27923. doi: 10.1038/srep27923
Yan A. W., Fouts D. E., Brandl J., Starkel P., Torralba M., Schott E., et al. (2011). Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 53, 96–105. doi: 10.1002/hep.24018
Keywords: alcoholic liver disease, gut microbiota, hepatic function, Streptococcus, aspartate aminotransferase
Citation: Zhong X, Cui P, Jiang J, Ning C, Liang B, Zhou J, Tian L, Zhang Y, Lei T, Zuo T, Ye L, Huang J and Chen H (2021) Streptococcus, the Predominant Bacterium to Predict the Severity of Liver Injury in Alcoholic Liver Disease. Front. Cell. Infect. Microbiol. 11:649060. doi: 10.3389/fcimb.2021.649060
Received: 03 January 2021; Accepted: 01 March 2021;
Published: 17 March 2021.
Edited by:
Rodolfo García-Contreras, National Autonomous University of Mexico, MexicoReviewed by:
Esther Nistal, University of Leon, SpainRafael Franco-Cendejas, National Institute of Rehabilitation Luis Guillermo Ibarra Ibarra, Mexico
Copyright © 2021 Zhong, Cui, Jiang, Ning, Liang, Zhou, Tian, Zhang, Lei, Zuo, Ye, Huang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Chen, Y2hlbmh1aXlmeUBneG11LmVkdS5jbg==; Jiegang Huang, amllZ2FuZ2h1YW5nQGd4bXUuZWR1LmNu; Li Ye, eWVsaUBneG11LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship