 Soon-Hwan Oh1
Soon-Hwan Oh1 Klaus Schliep2
Klaus Schliep2 Allyson Isenhower3Rubi Rodriguez-Bobadilla3
Allyson Isenhower3Rubi Rodriguez-Bobadilla3 Vien M. Vuong4
Vien M. Vuong4 Christopher J. Fields5
Christopher J. Fields5 Alvaro G. Hernandez5
Alvaro G. Hernandez5 Lois L. Hoyer1*
Lois L. Hoyer1*- 1Department of Pathobiology, College of Veterinary Medicine, University of Illinois at Urbana-Champaign, Urbana, IL, United States
- 2Institute of Environmental Biotechnology, Graz University of Technology, Graz, Austria
- 3Department of Biology, Millikin University, Decatur, IL, United States
- 4Department of Computer Science, University of Illinois at Urbana-Champaign, Urbana, IL, United States
- 5Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign, Urbana, IL, United States
The Candida albicans agglutinin-like sequence (ALS) family is studied because of its contribution to cell adhesion, fungal colonization, and polymicrobial biofilm formation. The goal of this work was to derive an accurate census and sequence for ALS genes in pathogenic yeasts and other closely related species, while probing the boundaries of the ALS family within the Order Saccharomycetales. Bioinformatic methods were combined with laboratory experimentation to characterize 47 novel ALS loci from 8 fungal species. AlphaFold predictions suggested the presence of a conserved N-terminal adhesive domain (NT-Als) structure in all Als proteins reported to date, as well as in S. cerevisiae alpha-agglutinin (Sag1). Lodderomyces elongisporus, Meyerozyma guilliermondii, and Scheffersomyces stipitis were notable because each species had genes with C. albicans ALS features, as well as at least one that encoded a Sag1-like protein. Detection of recombination events between the ALS family and gene families encoding other cell-surface proteins such as Iff/Hyr and Flo suggest widespread domain swapping with the potential to create cell-surface diversity among yeast species. Results from the analysis also revealed subtelomeric ALS genes, ALS pseudogenes, and the potential for yeast species to secrete their own soluble adhesion inhibitors. Information presented here supports the inclusion of SAG1 in the ALS family and yields many experimental hypotheses to pursue to further reveal the nature of the ALS family.
Introduction
Most knowledge about agglutinin-like sequence (Als) protein structure and function comes from the study of Candida albicans (reviewed in Hoyer and Cota, 2016). C. albicans Als proteins are adhesins that promote interactions between fungi and host cells, establishing colonization that provides the opportunity to cause disease. Als proteins also function in C. albicans adhesion to abiotic surfaces such as indwelling medical devices, and between fungi and other microbes such as bacteria, seeding polymicrobial biofilm development (Peters et al., 2012).
Using Als protein features known at the time, Hoyer and Cota (2016) suggested the NT/T/TR/CT model for proteins in the Als family with each protein possessing an NT-Als adhesive domain (NT; Lin et al., 2014), followed by a Thr-rich region (T), tandemly repeated sequences (TR), and a C-terminal (CT) domain rich in Ser and Thr. Other key features include a secretory signal peptide and GPI anchor addition sequence that direct the mature protein to β-1,6-glucan crosslinkage in the fungal cell wall (Lu et al., 1994). Display of the NT-Als adhesive domain on the cell surface is key for its ability to interact with other cells and materials (Lin et al., 2014).
The crystallographic structure of the NT-Als adhesive domain revealed protein folding driven by formation of 4 disulfide bonds (8 Cys); an invariant Lys is located at the end of the NT-Als binding pocket (Salgado et al., 2011). The positively charged Lys establishes a salt bridge with the C-terminal carboxylic acid of an incoming peptide ligand. Als binding affinity is at the micromolar level; avidity of Als protein interactions is increased by a peptide sequence with high β-aggregation potential (called the amyloid-forming region; AFR; Lipke et al., 2018).
The first ALS gene was named for the similarity of its predicted protein to Saccharomyces cerevisiae alpha-agglutinin (Sag1; Hoyer et al., 1995). At that time, considerably less DNA sequence information was available in public databases. Sag1 is an adhesin that promotes cell-cell contact between haploid yeasts during the mating process (Wojciechowicz et al., 1993). Sag1 binds the carboxyl-terminal peptide of a-agglutinin with nanomolar affinity (Cappellaro et al., 1994; Zhao et al., 2001). In contrast to Als proteins, the sequence of the N-terminal region of Sag1 has 6 Cys and an Arg residue at the position of the invariant Lys. The structure of Sag1 was not solved experimentally although molecular modeling and biochemical analyses contributed considerable insight into protein function and potential structure (Chen et al., 1995; Lipke et al., 1995; Grigorescu et al., 2000). Despite inherent differences between Als proteins and Sag1, various authors recognized the common features between these proteins and proposed that Sag1 is part of the Als family (Hoyer and Cota, 2016; Lipke, 2018).
The rich foundation of experimental evidence from C. albicans was used to define ALS genes and their encoded proteins in other fungal species including Candida dubliniensis (Jackson et al., 2009), Candida parapsilosis (Oh et al., 2019), Candida orthopsilosis (Lombardi et al., 2019), Candida metapsilosis (Oh et al., 2019) and Candida tropicalis (Oh et al., 2021). The burgeoning number of publicly available draft genome sequences provides the opportunity to locate other ALS sequences using a simple BLAST search. However, lack of proper gene assembly is a frequent stumbling block due to the tendency for each species to have multiple ALS loci and for ALS genes to encode long stretches of highly conserved, tandemly repeated sequences. Computer assembly of short-read sequences often breaks down in ALS coding regions because the length of repeated sequences is similar to the length of sequence reads (Lombardi et al., 2019). Emergence of long-read DNA sequencing technology provided datasets with more-accurate ALS assemblies, however, PCR amplification and Sanger sequencing are still frequently required to complete ALS loci (Oh et al., 2019; Oh et al., 2021).
The initial focus of this work was to derive an accurate census and sequence for the ALS genes in the human pathogenic Saccharomycetales species. Closely related species, including some with biotechnological importance, were also studied because their genome sequences are incorporated into available analysis tools (Maguire et al., 2013). Information presented here expands the list of known ALS loci, adding 47 carefully validated genes from 8 fungal species. Four long-read-based genome assemblies were generated. Predicted proteins were evaluated in the context of the model C. albicans Als and S. cerevisiae Sag1 characteristics. The availability of new, accurate structural prediction tools (Jumper et al., 2021) provided the opportunity to assess similarities within this group of proteins. Recent release of many new genome sequences for budding yeasts in the subphylum Saccharomycotina (Shen et al., 2018) allowed new insight into whether C. albicans Als proteins and S. cerevisiae Sag1 arose from a common ancestor. The resulting information redefines the ALS family, its diversity, and taxonomic boundaries. Information presented here also provides the basis for new hypotheses regarding Als protein function.
Materials and Methods
Fungal Strain and Public Genome Sequence Resources
To ensure use of authenticated materials, Lodderomyces elongisporus strain NRRL YB-4239 and Spathaspora passalidarum strain NRRL Y-27907 were obtained from the Agricultural Research Service Culture Collection (Peoria, IL; https://nrrl.ncaur.usda.gov). Meyerozyma guilliermondii strain ATCC 6260 and Scheffersomyces stipitis strain CBS 6054 (ATCC 58785) were purchased from the American Type Culture Collection (https://www.atcc.org). Table 1 lists genome sequences used for this study.
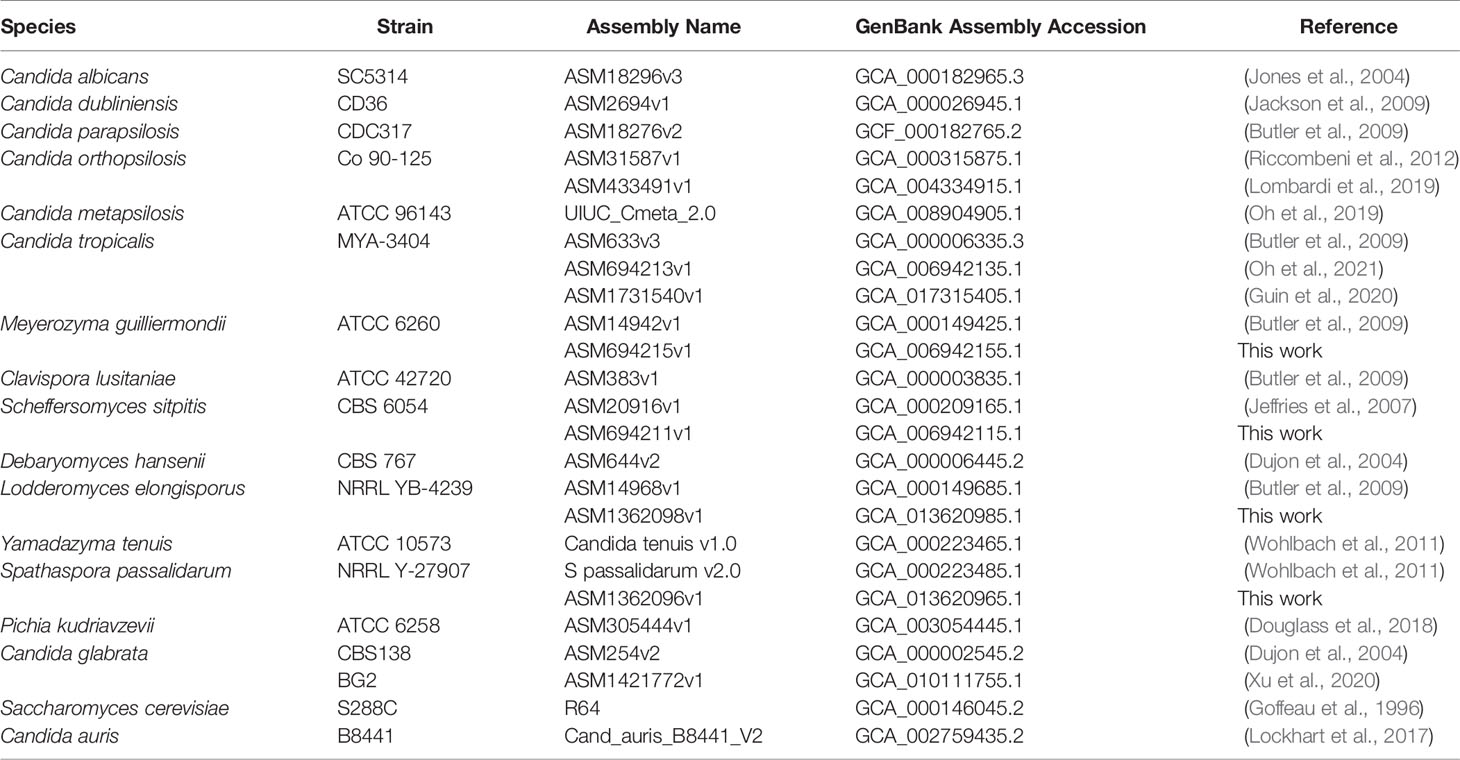
Table 1 Principal genome sequences used for ALS gene detection.
Genome Sequencing and Assembly
New genome assemblies were constructed for L. elongisporus NRRL YB-4239 (ASM1362098v1), M. guilliermondii ATCC 6260 (ASM6942125v1), S. stipitis CBS 6054 (ASM694211v1), and S. passalidarum NRRL Y-27907 (ASM362096v1) using a combination of Illumina MiSeq and Oxford Nanopore MinION data. Details of the method and genome assembly are in Supplementary Files S1–S4, respectively. Similar methods were described previously (Oh et al., 2019; Oh et al., 2021) and were reproduced here for the reader’s convenience.
Fungal cells were stored at -80°C and streaked to YPD agar plates (per liter: 10 g yeast extract, 20 g Bacto peptone, 20 g dextrose, 20 g Bacto agar). A single colony was inoculated into YPD liquid medium and the culture grown 16 h at 30°C and 200 r/min shaking. Genomic DNA was isolated according to Sherman et al. (1986). Briefly, cells were spheroplasted with zymolyase and lysed with sodium dodecyl sulfate. The lysate was extracted with phenol, DNA precipitated with isopropanol, and the final preparation treated with Proteinase K. DNA shearing was minimized by use of wide-bore pipet tips and gentle mixing. Agarose gel electrophoresis was used to verify the presence of high-molecular-weight DNA prior to subsequent library preparation.
Libraries were constructed and sequenced at the Roy J. Carver Biotechnology Center, University of Illinois at Urbana-Champaign. Individually barcoded shotgun libraries were made using the Hyper Library construction kit (Roche) and pooled in equimolar concentration. The pooled libraries were quantitated by qPCR and sequenced on one MiSeq flowcell for 251 cycles from each end of the fragment using a MiSeq 500-cycle sequencing kit (version 2). PhiX DNA was used as a spike-in control for MiSeq sequencing runs. FASTQ files were generated and demultiplexed with bcl2fastq Conversion Software (Illumina, version 2.17.1.14). MiSeq reads were quality trimmed using Trimmomatic v0.36 (Bolger et al., 2014) to remove adaptors from the 3’ end of the reads prior to assembly, retaining all reads greater than or equal to 50 nt in length.
Libraries for Oxford Nanopore long-read sequencing used 1 μg of genomic DNA that was sheared in a gTube (Covaris, Woburn, MA, United States) for 1 min at 6,000 r/min in a MiniSpin plus microcentrifuge (Eppendorf, Hauppauge, NY, United States). The sheared DNA was converted to a shotgun library with the LSK-108 kit (Oxford Nanopore) with the Expansion barcoding kit (EXP-NBD103). The libraries were pooled in equimolar concentration and the pool was sequenced on two SpotON MK I (R9.5) flowcells for 48 h using a MinION MK 1B sequencer. Basecalling was done with software MinKNOW version 1.7.7 and demultiplexing was performed with the Albacore software version 1.2.4. Sixty bp were trimmed from each end of the reads to remove barcodes using a custom script.
Detailed assembly steps are outlined in Supplemental Files S1–S4 for the four genomes. In brief, trimming was performed using a simple Perl script to remove 60 nt from the ends of the reads, retaining reads longer than or equal to 1000 nt for the final assembly. Canu v1.5 (Koren et al., 2017) was used for assembly with the following parameters: “canu -p asm -d useGrid = false -nanopore-raw genomeSize=<GENOME_SIZE> <TRIMMED_FASTQ>”, using the relevant quality-trimmed reads from Porechop and the following estimated genome sizes as input: S. stipitis = 15.4 Mb, L. elongisporus = 15.5 Mb, M. guilliermondii = 10.6 Mb, S. passalidarum = 13.2 Mb.
Trimmed Oxford Nanopore reads were then aligned against their associated assembly using bwa v0.7.5 (Li, 2018) by first indexing the assembly using “bwa index <ASSEMBLY>”, then aligning reads using the parameters “bwa mem -x ont2d <ASSEMBLY_INDEX> <TRIMMED_READS>”. The alignment was then used to polish the assembly using nanopolish v0.7.1 (Senol Cali et al., 2018). Quality-trimmed MiSeq data were used to further polish the assembly using Pilon v1.22 for error correction (Walker et al., 2014). Assembly names and GenBank accession numbers for the genome sequences for L. elongisporus NRRL YB-4239, M. guilliermondii ATCC 6260, S. passalidarum NRRL Y-27907, and S. stipitis CBS 6054 are located in Table 1.
Identification of ALS Genes and Predicted Als Protein Features
Methods for identifying ALS genes and deducing predicted protein features were nearly identical to those reported in previous publications (Oh et al., 2019; Oh et al., 2021). Details were reproduced here for the reader’s convenience. BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to identify potential ALS genes and Als proteins in the genome sequences (Table 1). BLAST searches were also conducted using the Candida Genome Database (www.candidagenome.org; Skrzypek et al., 2017). Query sequences included all C. albicans ALS genes as reported by Oh et al. (2019). As new ALS/Als sequences were identified, they were also used as BLAST queries until search reports failed to reveal new sequences.
ALS gene sequences were verified or corrected by Sanger sequencing of PCR-amplified products. Primers were designed using the Primer Quest Tool (https://www.idtdna.com) and primers purchased from Integrated DNA Technologies (Coralville, IA). PCR used Q5 High Fidelity DNA Polymerase (New England Biolabs) according to the manufacturer’s instructions. Extension time was adjusted to promote amplification of the desired product size. PCR products were purified using the MultiScreen HTS 96-well Filtration System (Millipore), then Sanger sequenced at the Roy J. Carver Biotechnology Center (University of Illinois at Urbana-Champaign). A list of primer sequences is located in Supplementary Table S1.
SignalP-5.0 Server (http://www.cbs.dtu.dk/services/SignalP; Nielsen, 2017) was used to locate putative secretory signal peptides. The Big-PI Fungal Predictor (https://mendel.imp.ac.at/gpi/fungi_server.html; Eisenhaber et al., 2004) identified potential GPI anchor addition sites. Dotmatcher (http://emboss.bioinformatics.nl/cgi-bin/emboss/dotmatcher) was used to detect repeated sequences. The ExPASy Server was used to assess amino acid sequence composition (https://web.expasy.org/protparam; Gasteiger et al., 2005). European Bioinofrmatics Institute (EMBL-EBI) tools were used for translating nucleotide sequences, sequence alignment, and other general processes (https://www.ebi.ac.uk/services; Cook et al., 2017). β-aggregation potential in amino acid sequences was assessed using Tango (http://tango.crg.es; Fernandez-Escamilla et al., 2004; Rousseau et al., 2006). All nucleotide sequences were translated with the alternative yeast nuclear code (translation table 12) except those from C. glabrata and S. cerevisiae which used the standard code (translation table 1). There was disagreement about the translation table for L. elongisporus: the NCBI database used translation table 1 even though the species was recognized as part of the CUG clade that translates CUG as Ser instead of Leu (translation table 12; Santos et al., 2011).
Phylogenetics Analysis
The kingdom Fungi phylogeny of Li et al. (2021), based on 290 genes from 1644 species, was pruned using the R package ape (Paradis and Schliep, 2019). C. metapsilosis was not included in the phylogeny. Because of its close relationship to C. parapsilosis and C. orthopsilosis (Tavanti et al., 2005), C. metapsilosis was attached at the root of these sister species.
Sequences for phylogenetic analysis were trimmed to include only the NT-Als domain, which has adhesive function (Lin et al., 2014). Secretory signal peptide sequences were included in the analysis. Tango (http://tango.crg.es; Rousseau et al., 2006; Fernandez-Escamilla et al., 2004) was used to define the amyloid-forming region (AFR) that marks the C-terminal end of NT-Als (Lin et al., 2014). Sequences that did not encode an AFR were trimmed to a length similar to others in the dataset. Amino acid sequences used for phylogenetic analysis were compiled into Supplementary File S5. Sequences were aligned using MAFFT v7.453 (Katoh and Standley, 2013). A phylogeny was inferred using the best fit model WAG+G+I and subsequent bootstrap analysis was performed using RAxML-NG (Kozlov et al., 2019).
Structural Predictions
Protein structural predictions were derived using AlphaFold (Jumper et al., 2021), accessed using AlphaFold Colab (https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb). ColabFold (Mirdita et al., 2021; https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb) was used for more-rapid generation of structural prototypes. ColabFold utilized MMSeqs2 (UniRef+Environmental), as well as a custom MSA file constructed using Hhblits Toolkit server (Zimmerman et al., 2018; Gabler et al., 2020).
Clustal Omega (Madeira et al., 2019) was used to align amino acid sequences from Supplementary File S5 with those from 4LE8, the Protein Data Bank (https://www.rcsb.org) entry for the crystallographic structure of C. albicans NT-Als3 (chain A, 299 amino acids). This molecule did not include the secretory signal peptide at the N-terminal end nor the amyloid-forming region at the C-terminal end. Sequences in Supplementary File S5 were trimmed according to the Clustal Omega alignment and the trimmed sequences were used for AlphaFold analysis. Sequences for other fungal cell-surface proteins were included as controls in the structural analysis: C. albicans Hyr1 (NCBI Reference Sequence: XP_722183.2, amino acids 21 to 309) and S. cerevisiae Flo1 (NCBI Reference Sequence: NP_009424.1; amino acids 25 to 273). Structural visualization and alignments used the PyMOL Molecular Graphics System, Version 2.5.2 (Schrödinger, LLC).
Results
Detection and Assembly of ALS Sequences in Eight Fungal Species
Examination of new and existing genome sequence data (Table 1) provided the information needed to locate, assemble and/or validate the sequence for 47 ALS loci (5 from Lodderomyces elongisporus NRRL YB-4239; 3 from Candida auris B8441; 3 from Scheffersomyces stipitis CBS 6054; 4 from Meyerozyma guilliermondii ATCC 6260; 29 from Spathaspora passalidarum NRRL Y-27907; and 1 each from Clavispora lusitaniae ATCC 42720, Yamadazyma tenuis ATCC 10573, and Debaryomyces hansenii CBS 767). These sequences are presented here in the context of previously described ALS loci including 8 from Candida albicans SC5314 (Hoyer et al., 2008), 7 from Candida dubliniensis CD36 (Jackson et al., 2009), 13 from Candida tropicalis MYA-3404 (Oh et al., 2021), 5 from Candida parapsilosis CDC 317 (Oh et al., 2019), 3 from Candida orthopsilosis Co 90-125 (Lombardi et al., 2019), and 4 from Candida metapsilosis ATCC 96143 (Oh et al., 2019). Supplementary Table S2 details all gene names, GenBank accession numbers, and predicted protein characteristics. Gene names were taken from locus tags associated with the annotated reference genome for each species. For genes that were not annotated in the reference genome, “0.5” was added to the name, indicating that the ORF was located between two previously annotated genes. Figure 1 displays the phylogenetic relationship between the species described here.
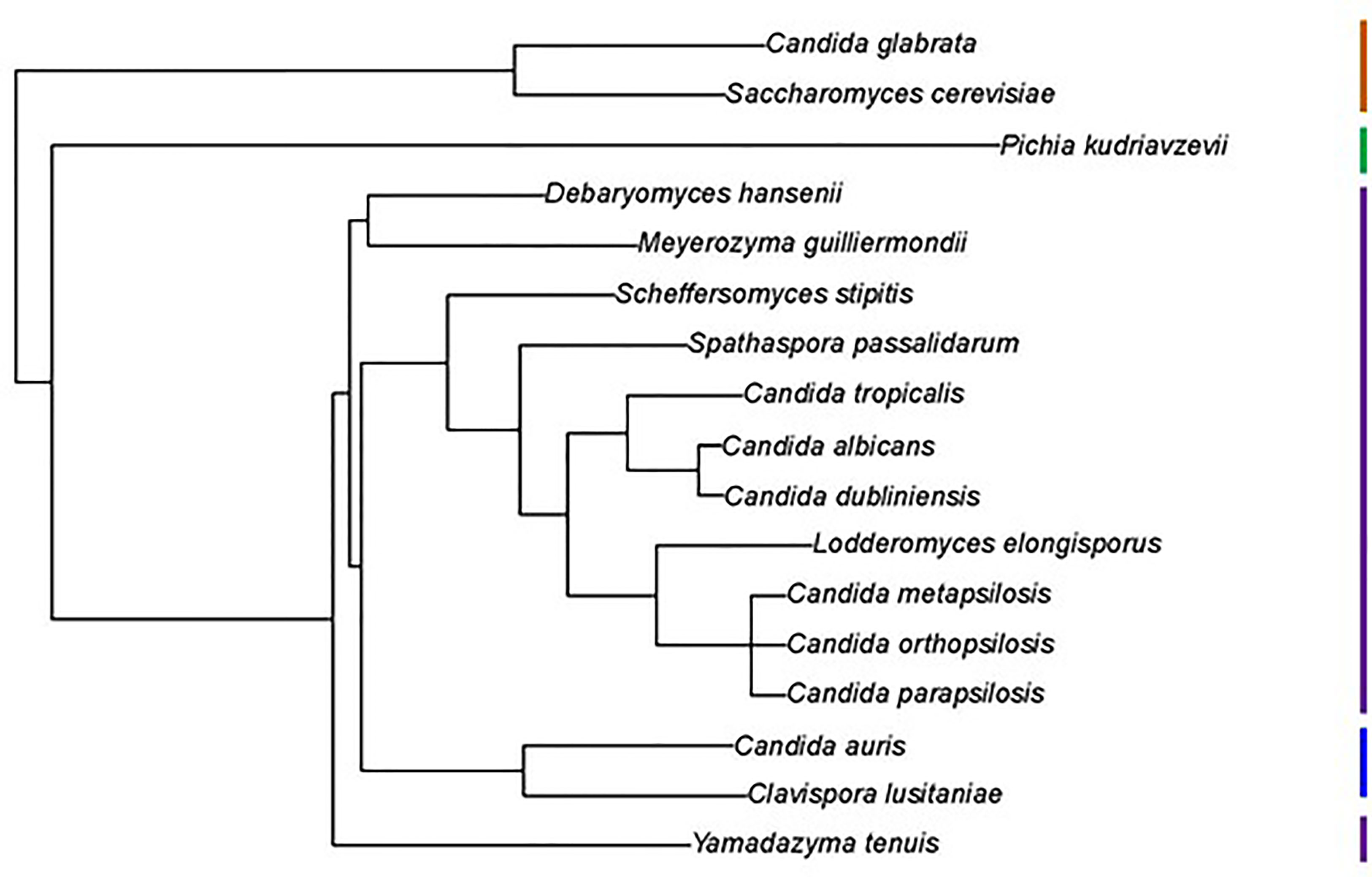
Figure 1 Phylogenetic tree showing relationships between fungal species used in this study. The tree was pruned from the genome-scale phylogeny of the kingdom Fungi developed by Li et al. (2021). The phylogeny was based on 290 concatenated sequences in 1644 species. C. metapsilosis was not part of the original analysis and was added to the tree based on its close relationship with C. parapsilosis and C. orthopsilosis (Tavanti et al., 2005). All species listed were Phylum Ascomycota, Subphylum Saccharomycotina, Class Saccharomycetes, Order Saccharomycetales. Vertical bars on the right of the image indicate Family designations according to the NCBI Taxonomy Database (https://www.ncbi.nlm.nih.gov/taxonomy). Brown = Family Saccharomycetaceae, Green = Family Pichiaceae, Purple = Family Debaryomycetaceae, and Blue = Family Metschnikowiaceae.
L. elongisporus
L. elongisporus is an infrequent human pathogen and, as accurately portrayed in Figure 1, closely related to C. parapsilosis (Lockhart et al., 2008). Two L. elongisporus genome sequences were available through the National Center for Biotechnology Information (NCBI; https://www.ncbi.nlm.nih.gov) database; both were for strain NRRL YB-4239 (Table 1). Assembly ASM14968v1 was comprised of 145 contigs arranged into 28 scaffolds (i.e. composed of contigs and gaps that are indicated by insertion of “NNN”) while ASM1362098v1 had 53 contigs. ALS gene names were derived from the original annotation of the reference genome (ASM14968v1). Although a L. elongisporus karyotype was not located in the literature, the number of contigs/scaffolds was far higher than an expected number of chromosomes, indicating that the genome assemblies required refinement.
BLAST analysis using ALS query sequences consistently revealed 6 L. elongisporus loci (Supplementary File S6). LeALS734 was located in the middle of a > 3 Mb supercontig in each genome assembly. LeALS2536, LeALS2716 and LeALS2721 were located in the center of another supercontig in ASM14968v1 with the latter two genes located near each other and transcribed in the same direction (Figure 2). The remaining genes (LELG_04272, LeALS5708) were each on their own supercontig.
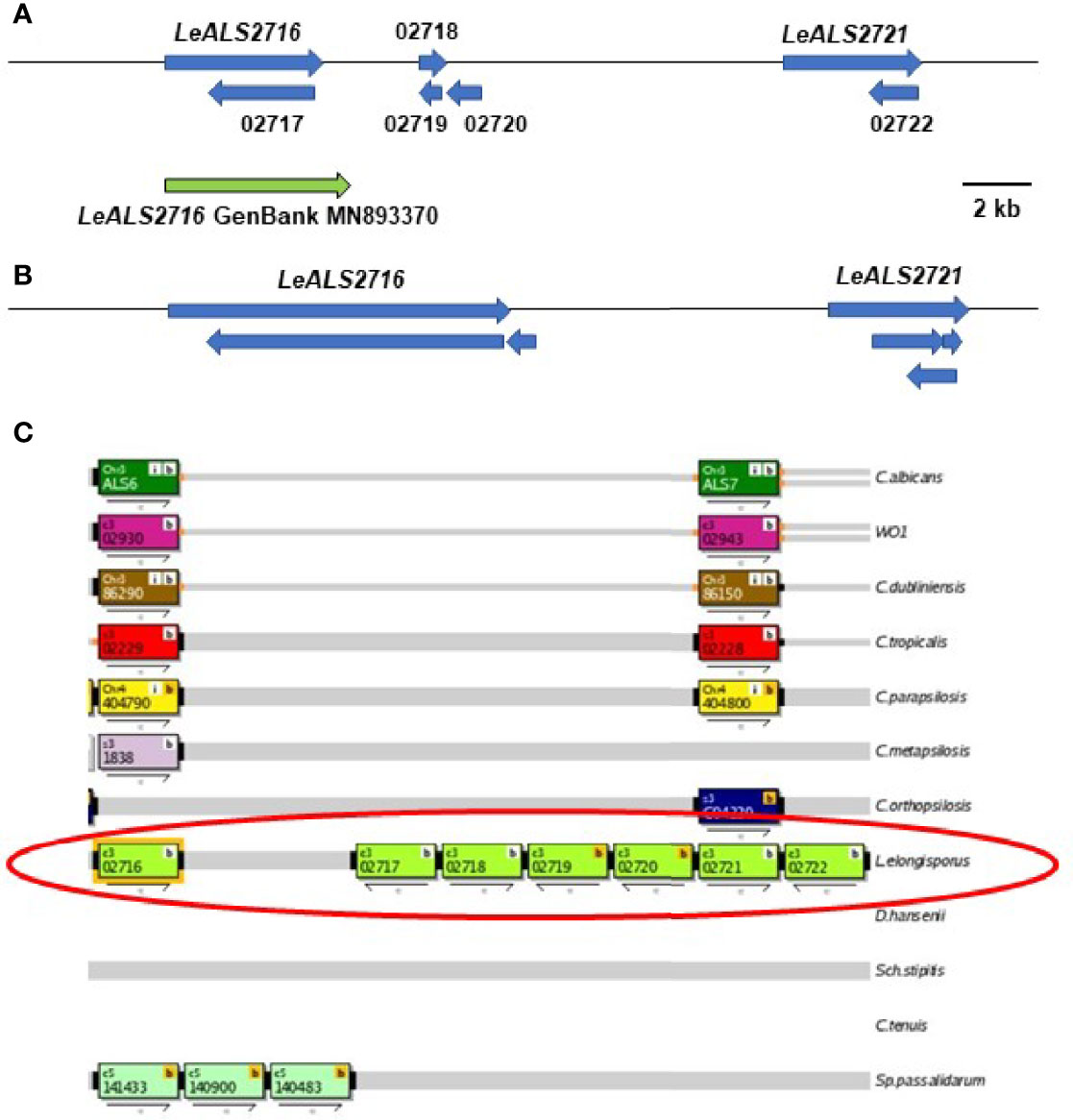
Figure 2 Schematic of the genome region that included LeALS2716 and LeALS2721 from (A) assembly ASM14968v1 and (B) assembly ASM1362098v1. (C) shows the region as represented in the Candida Genome Order Browser (CGOB; https://cgob.ucd.ie; Maguire et al., 2013) based on ASM14968v1 data. L. elongisporus information is circled in red; ORF numbers are shown in each rectangle and the direction of transcription indicated by the arrow below. ORFs 02718, 02719, and 02720 featured the IFF/HYR repeated sequences that were also found in LeALS2716 suggesting that ORF 02716 was longer than initially annotated. The large number of repeated sequences complicated genome sequence assembly in this region. The predicted size of LeALS2716 in ASM1362098v1 was greater than the final PCR-amplified/Sanger-sequenced fragment that was deposited into GenBank (accession number MN893370; green arrow).
Proteins predicted from five loci (LeALS734, LeALS2536, LeALS2716, LeALS2721, and LeALS5708) had features expected for an Als protein including a secretory signal peptide, NT-Als domain, Ser/Thr-rich sequences, and GPI anchor addition site (Supplementary Table S2). Although the protein translated from the sixth locus (LELG_04272) had tandemly repeated sequences like those in Als proteins, the ORF did not predict an NT-Als domain (Supplementary File S6). BLAST of sequences N-terminal to the LELG_04272 tandem repeats best matched M. guilliermondii PGUG_02520, a 660-amino acid protein of unknown function. The hybrid LELG_04272 was likely the result of recombination between an ALS open reading frame (ORF) and another gene. Such recombination was observed previously between ALS and IFF/HYR genes in C. metapsilosis CmALS2265 which encoded an Als N-terminal domain on the stalk of an Iff/Hyr protein (Oh et al., 2019). Because LELG_04272 lacked an NT-Als adhesive domain, it was not given an Als family name.
Similar to C. metapsilosis, recombination between ALS and IFF/HYR genes was evident in L. elongisporus (Supplementary File S6). LeALS734 and LeALS2716 each had the 5’ end of an ALS gene and the 3’ end of an IFF/HYR gene. Consensus tandem repeat sequences for each locus closely matched that for CmALS2265 (Oh et al., 2019). The remaining LeALS genes did not include a central domain of tandemly repeated sequences.
The predicted amino acid sequence of the NT-Als domain was highly conserved between LeAls2536, LeAls2721, and LeAls5708 (70-80% identity; Supplementary File S6). LeAls734 and LeAls2716 were approximately 40% identical to each other and to the other L. elogisporus Als proteins. Since the crystallographic structure of C. albicans NT-Als3 is known (Lin et al., 2014), the CaAls3 sequence was included for comparison to newly characterized proteins. The LeAls proteins were 33-45% identical to the CaAls3 NT-Als sequence.
The incomplete L. elongisporus genome assemblies provided multiple opportunities for repairing ORFs. LELG_04272 lacked a GPI anchor addition site in its original annotation in ASM14968v1 (Supplementary File S6). Primer design, PCR amplification, and Sanger DNA sequencing indicated that the downstream LELG_04271 was actually the 3’ end of the LELG_04272 coding region. The full sequence was deposited into GenBank (accession number OK172378). The region encoding LeALS2716 and LeALS2721 was also assembled poorly in the available genome sequences (Figure 2). Insertion of many NNN (designating unknown sequence) in this region of assembly ASM14968v1 made it difficult to locate the gene boundaries. The size of the verified LeALS2716 was larger than predicted in assembly ASM14968v1, but smaller than suggested in assembly ASM1362098v1. The difference was possibly attributable to variation in tandem repeat copy number between alleles in the diploid L. elongisporus. A similar scenario was encountered for completing the sequence of LeALS734: several previously designated ORFs were combined into the final LeALS734, yet the sequence deposited into GenBank was shorter than anticipated from the genome assemblies that guided PCR primer design (details not shown).
M. guilliermondii
In addition to its role as a human pathogen, M. guilliermondii has applications in bioprocessing (e.g. production of riboflavin and xylitol) and bioremediation (reviewed in Papon et al., 2013). Eight M. guilliermondii genome assemblies were reported in the NCBI database. Two sequences were available for strain ATCC 6260, which was the focus of this work. ASM14942v1 consisted of 71 contigs arranged on 9 scaffolds. ASM694215v1 had 9 contigs.
Two of the 4 ALS loci in strain ATCC 6260 (MgALS2302, MgALS3259) were on the same chromosome, approximately 1.7 Mb away from each other at opposite telomeres, transcribed in opposite directions (Supplementary File S7). The 3’ end of the 9681-bp MgALS2302 was located approximately 10 kb from one telomere while the 3’ end of the 6963-bp MgAls3259 was about 7 kb from the opposite telomere. The other two MgALS loci (MgALS673 and MgALS3330) were each on other large contigs, likely representing M. guilliermondii chromosomes.
The coding region for MgALS3259 predicted two potential start sites, one 60 bp upstream from the other. The second predicted Met start site provided a clear secretory signal peptide and was included in GenBank accession MH753514. PGUG_03259 (accession number XM_001485480) included the upstream start site instead. Another difference between sequences deduced here and those from the ASM14942v1 assembly included the length of MgALS2302. The gene was considerably longer in the current work compared to previous GenBank deposits (9681 bp vs. 3840 bp). The new gene name reflected its inclusion of multiple ORFs (PGUG_02299, PGUG_02300, PGUG_02301, and PGUG_02302) and start of the gene in PGUG_02302.
Three of the 4 MgALS loci had an extensive central region of tandemly repeated sequences that were very similar to consensus repeats from previously characterized Candida genes (Supplementary File S7). The most-common repeat unit size was 35 amino acids with a range of 34-36 amino acids. The predicted MgAls2302 and MgAls3259 NT-Als protein sequences were the most similar to each other. They shared 57% identity while other NT-Als comparisons were only about 30% identical.
C. auris
Availability of genome sequences for over 100 isolates of C. auris in the NCBI database reflected its emergence as an important pathogen (Du et al., 2020). Strain B8441 (18 contigs arranged into 15 scaffolds; assembly Cand_auris_B8441_V2) was used for this work (Table 1). The three ALS loci in strain B8441 were on different scaffolds with CauALS2582 in a subtelomeric location (Supplementary File S8). Only one predicted C. auris Als protein (CauAls4112) had a central domain of tandemly repeated (34-39 amino acids) sequences. C. auris NT-Als sequences were 38-50% identical to each other and 25-33% identical to C. albicans NT-Als3. BLAST searching of other C. auris genomes showed that many were missing the CauALS2582 locus. These observations matched the report of Muñoz et al. (2021) that described loss of subtelomeric adhesins such as CauALS2582 in clade II C. auris isolates.
S. stipitis
S. stipitis is studied because of its ability to ferment xylose, and thus its potential to contribute to biofuel production from hemicellulose (Jeffries et al., 2007). Because S. stipitis is within the CUG clade of the Family Debaryomycetaceae, the genome of this beetle-associated commensal yeast is often included in comparisons that feature human-pathogenic species (Wohlbach et al., 2011; Maguire et al., 2013). The NCBI database included three S. stipitis genome sequences: two for strain CBS 6054 (ASM20916v1 = 9 contigs arranged on 8 scaffolds; ASM694211v1 = 30 contigs) and one for strain NRRL Y-7124. Efforts here focused on strain CBS 6054 (Table 1).
Strain CBS 6054 had three ALS loci (Supplementary Table S2). Two (SsALS2386 and SsALS2786) were on chromosome 2, approximately 700 kb apart and transcribed in opposite directions (Supplementary File S9). The third (SsALS4579) was on chromosome 4. NT domains of the predicted chromosome 2 proteins were 58% identical. However, each was only 27-29% identical to SsAls4579. Both SsAls2386 and SsAls2786 had a central domain of tandemly repeated sequences that resembled those from previously studied Candida species.
BLAST searches and text searches of the NCBI database revealed other repeat-containing loci that were annotated as ALS genes in assembly ASM20916v1. For example, PICST_4391 (called “ALS1”) was a fragment of SsALS2786 described above. PICST_4539 (called “ALS1.2”) was a fragment of SsALS2386 and PICST_31759 (called “ALS6”) corresponded to SsALS4579. PICST_31095, despite being named “ALS2”, belonged to the IFF/HYR family. Another, called “ALS4” (PICST_30411), was comprised only of repeated sequences. It was flanked by sequences that contained many stop codons with a low likelihood of encoding a larger ORF. In assembly ASM20916v1, PICST_30411 was adjacent to an IFF/HYR gene in a subteromeric region on chromosome 2.
S. passalidarum
The S. passalidarum genome was also sequenced to study xylose metabolism in fungal species (Wohlbach et al., 2011). Because initial BLAST searching revealed an unusually high number of ALS loci in S. passalidarum, additional analysis was pursued. Two genome assemblies for strain NRRL Y-27907 were located in the NCBI database (Table 1). One was created by the DOE Joint Genome Institute (Spathaspora passalidarum v2.0; 26 contigs arranged onto 8 scaffolds) and the other was part of the current work (ASM1362096v1; 10 contigs).
BLAST searching revealed 29 physical loci with sequence similarity to ALS genes (Supplementary Table S2). Seven of the 8 contigs from assembly Spathaspora passalidarum v2.0 had at least 1 ALS locus, and as many as 7 ALS loci (Table 2). Four locations featured ALS genes adjacent to each other: SpALS64434 and SpALS64435 on scaffold 1, SpALS50348 and SpALS50349 on scaffold 3, SpALS153035 and SpALS153035.5 on scaffold 4, and SpALS68952 and SpALS68952.5 on scaffold 8. Two ALS genes were subtelomeric: SpALS131476 on scaffold 1 and SpALS137089 on scaffold 3.
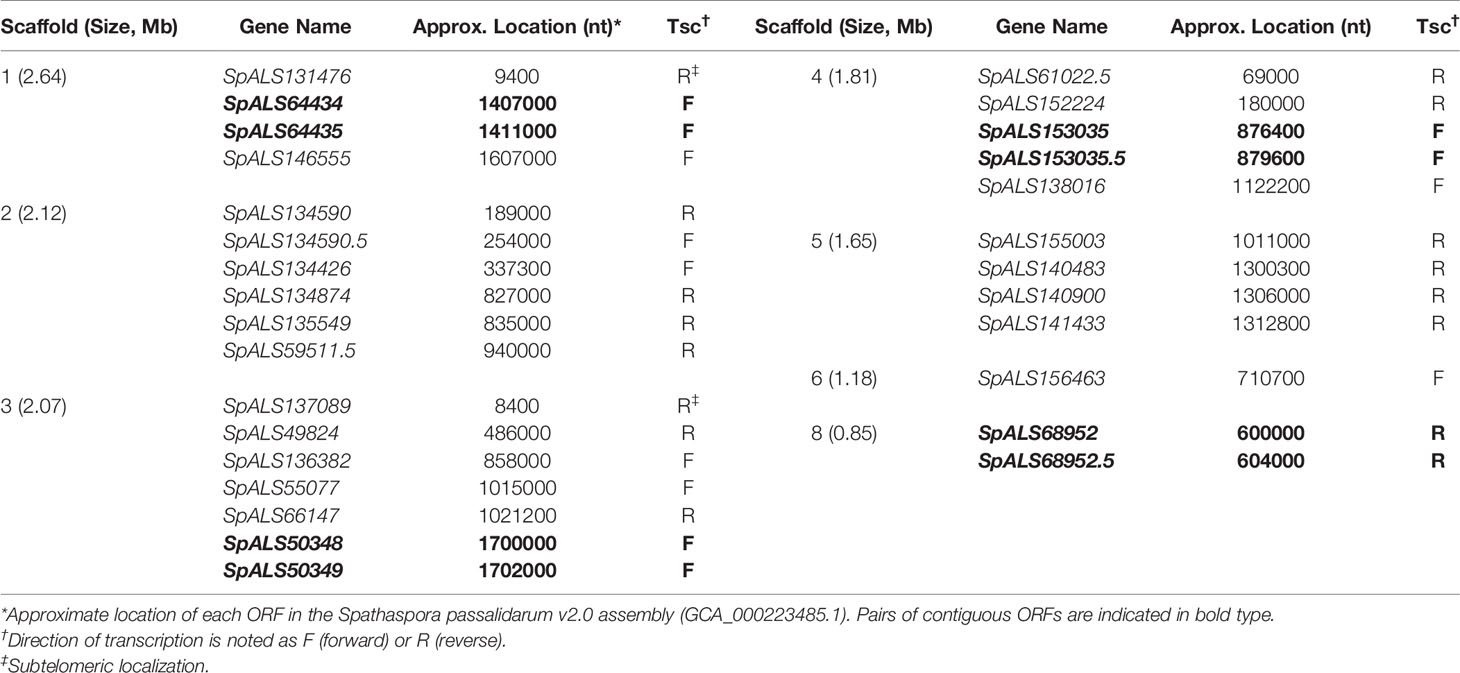
Table 2 Physical location of ALS loci in the S. passalidarum NRRL Y-27907 genome.
Examination of the encoded protein sequences showed that many did not have repeated sequences in the center (Supplementary File S10). When present, repeated sequences were unusually long (Supplementary Table S2), highly variable in the length of the repeat unit, and did not necessarily have tandemly arrayed repeat copies. A consensus of the 80-amino-acid and 89-amino-acid versions of the S. passalidarum repeated sequence resembled the T (Thr-rich) domain of C. albicans Als proteins that is located between the end of NT-Als and the start of the tandem repeat region (reviewed in Hoyer and Cota, 2016). Some manuscripts depict this region separately from “NT-Als” for which the crystal structure is known (e.g. Oh et al., 2019). Other manuscripts, like this one, include the T domain in the “NT-Als +” region for each protein.
S. passalidarum had several partial ALS genes (Supplementary File S10). In previous analyses, partial ALS genes indicated poor genome assembly that could be corrected by PCR amplification and Sanger sequencing (Oh et al., 2021). Here, however, PCR amplification and Sanger sequencing validated the accuracy of the partial ALS genes including the contiguous loci SpALS50348 and SpALS50349 that each only encoded an NT-Als domain with a secretory signal peptide. A clear stop codon between SpALS68952.5 and SpALS68952 created pseudogenes from a perhaps previously long, functional gene. Some predicted proteins lacked features such as a secretory signal peptide that would direct the protein toward cell-surface localization. Signal-peptide-encoding sequences were noted upstream of SpALS66147 and SpALS134590.5 but were separated from the ORF by at least one clear stop codon. Other predicted proteins (SpAls49824, SpAls61022.5, SpAls140483, and SpAls140900; Supplementary File S10) showed only a weak signal for GPI anchor addition, suggesting that the mature proteins may not be localized to the cell surface. Among the proteins predicted to have both secretory signal peptide and strong GPI anchor addition signal, many were short, raising the question of how far the adhesive NT-Als domain could be projected away from the S. passalidarum cell surface to contact binding partners.
Comparison between the NT-Als domains for the predicted S. passalidarum proteins showed considerable sequence divergence from C. albicans NT-Als3 and from each other (Supplementary File S10). Many comparisons were in the range of 22-45% identity although greater sequence conservation was observed for some pairs (60-80% range). Contiguous loci (37-62% identity) were not necessarily more conserved than other comparisons. The most-conserved pair was SpAls55077 and SpAls134590 (89%) potentially indicating a gene duplication.
Y. tenuis
Interest in the Y. tenuis genome also arose from its xylose fermentation capabilities (Wohlbach et al., 2011). A single complete ALS locus was detected in the only Y. tenuis genome assembly available in the NCBI database (strain ATCC 10573; Candida tenuis v1.0; Table 1). The predicted YtAls93631 protein was > 3000 amino acids and included a central domain of tandemly repeated sequences (32-45 amino acids; Supplementary File S11). The most common repeat unit was 35 amino acids. The Y. tenuis genome sequence was quite fragmented, consisting of 74 contigs arranged onto 25 scaffolds. A second potential ALS locus was detected at the end of a scaffold sequence (CANTEscaffold_00010; 12.1 kb total length) suggesting that other Y. tenuis ALS loci may be revealed as the genome assembly matures.
C. lusitaniae
C. lusitaniae is a less-frequent cause of human candidiasis (reviewed in François et al., 2001). Of the species considered here, C. lusitaniae was the most closely related to C. auris (Figure 1). More than 30 C. lusitaniae genome assemblies were available in the NCBI database. Observations here focused on strain ATCC 42720 (ASM383v1) which was comprised of 88 contigs arranged onto 9 scaffolds. One long ALS gene (> 8 kb) was detected in this sequence (Supplementary Table S2). BLAST of other available genomes also suggested a single ALS locus, although incomplete genome assemblies with many contigs may obscure the presence of other loci. Two different types of repeated DNA sequences were found in the center of ClALS3274 (Supplementary File S11). BLAST searching of the non-redundant nucleotide database suggested that the repeats predicting a 33 amino acid sequence were unique. The other repeated sequences were primarily 67 amino acids in length, although some repeat units expanded to 102 amino acids. Sequences within these longer repeats were reminiscent of motifs found in the Als consensus repeat from C. parapilsosis/C.orthopsilosis/C. metapsilosis, as well as the 80-amino-acid consensus repeat from S. passalidarum Als proteins (Supplementary File S10).
D. hansenii
Rather than being studied as a pathogen, D. hansenii is discussed commonly in the context of food production (summarized in Gori et al., 2011). D. hansenii growth requires surface adhesion (Mortensen et al., 2005), a property that might involve Als proteins. The NCBI database displayed 12 D. hansenii genome sequences, representing 11 different strains. This work focused on CBS 767 (ASM644v2), which was designated as the reference genome and had 15 contigs arranged on 8 scaffolds. A single ALS locus was detected, located on the chromosome G contig (Supplementary Table S2). The predicted protein, DhAls2178, did not contain tandemly repeated sequences (Supplementary File S11).
C. glabrata
C. glabrata is well-recognized as a human pathogen, particularly for its ability to rapidly evolve resistance to antifungal drugs (Vale-Silva and Sanglard, 2015). Eighteen C. glabrata genomes were present in the NCBI database. BLAST of the strain CBS 138 sequence using ALS and Als queries revealed CAGL0G04125g, which was annotated to indicate similarity to S. cerevisiae Sag1 (Supplementary File S11). Like Sag1, the C. glabrata predicted protein did not encode a central domain of tandemly repeated sequences.
NT-Als Structural Predictions
The C. albicans NT-Als3 structure was solved crystallographically (Lin et al., 2014; Protein Data Bank accession number 4LE8), providing experimental information that can be used to produce structural predictions for other NT-Als proteins. A crystallographic structure has not been reported for S. cerevisiae Sag1. The recent development of the highly accurate protein structural prediction program AlphaFold (Jumper et al., 2021) and widespread access to its use (e.g. Mirdita et al., 2021) provided the opportunity to assess predicted structural relatedness between the diverse set of NT-Als sequences and their similarity to Sag1.
Figure 3 shows the crystallographic structure of C. albicans NT-Als3 from 4LE8 (Figure 3A) and the AlphaFold-predicted structure for the same amino acid sequence (Figure 3B). The root-mean-square-deviation (RMSD) value for alignment of the two structures was 0.53, indicating the ability of AlphaFold to reproduce the crystallographic structure of NT-Als3 from its amino acid sequence. AlphaFold was then used to predict the structures of other NT-Als proteins, particularly those with limited sequence identity to C. albicans NT-Als3 (Figures 3C–G). Strikingly, a similar structure was predicted for each protein, including Sag1. Analysis of each NT-Als sequence in Supplementary File S5 produced the same general protein shape (data not shown) suggesting that all, including Sag1, were unified as a family. To demonstrate diversity in structural predictions, the amino acid sequences for the N-terminal domain of the cell-surface proteins C. albicans Hyr1 (amino acids 21 to 309) and S. cerevisiae Flo1 (amino acids 25 to 273) were entered into AlphaFold. The resulting structural predictions were different from the Als family and from each other (Figures 3H, I).
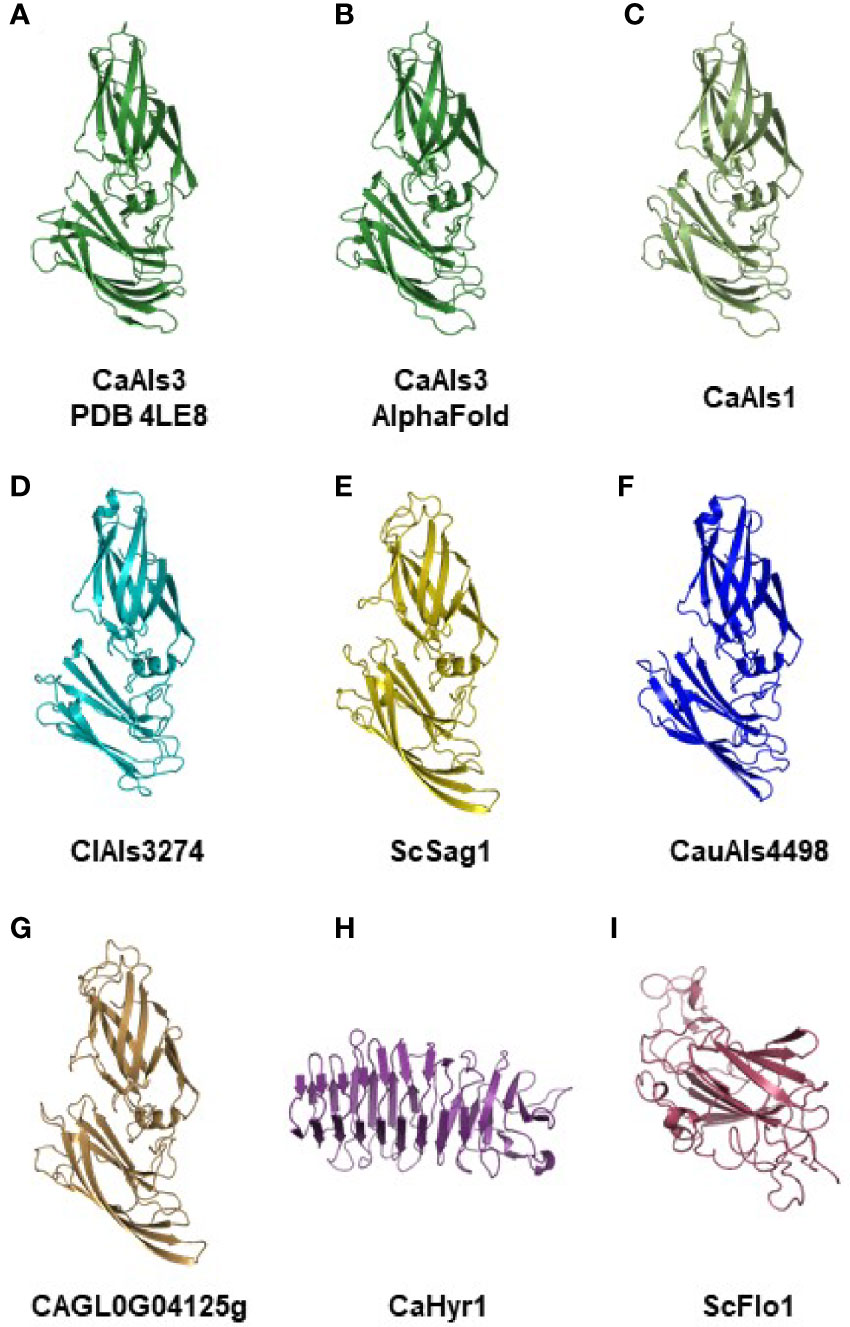
Figure 3 Experimental and AlphaFold-predicted protein structures. (A) Crystallographic structure of C. albicans NT-Als3 (Protein Data Bank accession 4LE8) visualized using PyMOL. (B) C. albicans NT-Als3 structure predicted by AlphaFold from the 4LE8 amino acid sequence. AlphaFold structural predictions for the corresponding region of C. albicans NT-Als1 (C; 83% identical to NT-Als3), ClAls3274 (D; 33% identical to NT-Als3), ScSag1 (E; 25% identity), CauAls4498 (F; 31% identity), and CAGL0G04125g (G; 21% identity). An AlphaFold structural prediction was also completed for the N-terminal functional domain of C. albicans Hyr1 (H) and S. cerevisiae Flo1 (I) to demonstrate structural diversity among cell-surface proteins that contain a central domain of repeated sequences. The AlphaFold prediction for C. albicans NT-Als3 (B) recapitulated the known experimental structure (A; RMSD = 0.53 as calculated using PyMOL align). Predictions for molecules (B–G) produced the same general structure suggesting that all should be included in the Als protein family. Supplementary File S12 shows the structures of C. albicans NT-Als3 (B) and ScSag1 (E) aligned with the disulfide bonds highlighted.
Adhesive Activity Predictions From NT-Als Protein Features
In order to provide biologically meaningful adhesive activity on the fungal cell surface, Als proteins need a secretory signal peptide and GPI anchor addition site to achieve an effective localization (Lu et al., 1994). The crystallographic structure of C. albicans NT-Als3 (Lin et al., 2014) demonstrated the involvement of 8 Cys residues in folding of the mature protein and the function of an invariant Lys at the end of the binding cavity that sinks the negative charge of incoming peptide ligands. Other work demonstrated the importance of the amyloid-forming region (AFR) for increasing the avidity of aggregative interactions (Ho et al., 2019). Supplementary Table S2 evaluated each of these features for the previously characterized and newly introduced Als proteins. The data were summarized in Figure 4 from which various hypotheses emerged.

Figure 4 Visualization of NT-Als sequence features from Supplementary Table S2. Each spot represents an Als protein; color coding matches Supplementary Table S2. C. albicans NT-Als3 has 8 Cys that create four disulfide bonds (Lin et al., 2014) while S. cerevisiae Sag1 has only 6 and is missing the C57-C133 disulfide bond that is present in NT-Als3 (Salgado et al., 2011). Most NT-Als proteins had 8 Cys like NT-Als3 (center column), some had 6 Cys like ScSag1 (left column), and others had varying numbers of Cys (range of 4 to 14; right column). Presence of the amyloid-forming region (AFR) in C. albicans Als proteins promotes protein aggregation (Ho et al., 2019). While many Als proteins had the expected AFR (strength and location; top row), others had a predicted weak AFR (second row) or none (bottom row). Some S. passalidarum Als proteins had a strong AFR 20-30 amino acids C-terminal to the expected location known in C. albicans (third row). It was unknown whether this alternative location contributed to aggregative potential. An invariant Lys in C. albicans NT-Als establishes a salt bridge with the C-terminal carboxylic acid of an incoming peptide ligand (Salgado et al., 2011). Arg (R) in this location may serve a similar function. Some predicted proteins did not have a positively charged amino acid in this position (X). A red asterisk indicates proteins featured in the structural predictions (Figure 3).
The cluster of dots in the top center of Figure 4 contained proteins with 8 Cys and an invariant Lys. Each protein represented in the top center cluster of dots also had an AFR with high predicted β-aggregation potential and a location immediately C-terminal to the NT-Als domain as in C. albicans Als proteins (Salgado et al., 2011; Lipke et al., 2018). The top center cluster of dots included 7 of the 8 C. albicans Als proteins, 6 of 7 from C. dubliniensis, and 9 of 13 from C. tropicalis suggesting robust potential for functional adhesive interactions by these species. Also present in the top center cluster of dots were 2 of the S. stipitis Als proteins, 2 from S. passalidarum, and 1 each from C. parapsilosis, C. metapsilosis, L. elongisporus, and Y. tenuis.
Viewing potential Als function according to these criteria provided predictions about the adhesive activity in other species. For example, C. parapsilosis, C. orthopsilosis, and C. metapsilosis Als proteins almost universally had a weaker or absent AFR leading to the hypothesis that these species are functionally less adherent than C. albicans, C. dubliniensis, and C. tropicalis. Data in Figure 4 suggested a similar hypothesis for S. passalidarum: despite its overabundance of ALS-like loci, S. passalidarum encoded only two proteins (SpAls138016 and SpAls156463; Supplementary File S10) that possessed the functional features present in NT-Als3 and therefore may not be a very adherent species. However, nine SpAls proteins had AFR sequences 20-30 amino acids C-terminal to the location where they are found in C. albicans Als3 (Supplementary Table S2). Experimentation is required to evaluate whether sequences with high β-aggregation potential in this alternative location contribute to S. passalidarum aggregative interactions. Using the criteria in Figure 4, C. lusitaniae was an outlier. Its sole Als protein, ClAls3274, had only 4 Cys residues and no AFR. Despite these key differences, the predicted structure for the NT-ClAls3274 domain was similar to those from other species (Figure 3D). The contribution of ClAls3274 remains to be tested, as does the overall adhesion profile for the species.
Proteins with features that more closely resemble Sag1 than C. albicans Als3 segregated to the left column of the diagram. Substitution of Arg (R printed in the dot) in place of the invariant Lys was a common theme. Three of the four M. guilliermondii Als proteins were in the left column. Perhaps these proteins have more-specific, higher-affinity adhesive interactions that more-closely resemble those of Sag1, rather than NT-Als3. The same prediction could be made for two of the C. auris proteins, as well as the sole protein from D. hansenii.
Information presented in Figure 4 and Supplementary Table S2 showed that some species had a complement of Als proteins that possessed features of both S. cerevisiae Sag1 and C. albicans Als3. For example, S. stipitis SsAls4579 had Sag1-like features (lower left, Figure 4) while SsAls2386 and SsAls2786 looked more like C. albicans Als3 (top center, Figure 4). The line diagrams in Supplementary File S9 reinforced this idea, illustrating the extensive regions of tandemly repeated sequences in the latter two proteins (C. albicans Als-like) and lack of a repeated domain in SsAls4579 (Sag1-like). M. guilliermondii was a similar mixing bowl of Als proteins: MgAls673 was the most Sag1-like of the group. Each of the other MgAls proteins had a long stalk of tandemly repeated units with NT domains that perhaps were more like Sag1 than Als3 (Supplementary File S7). L. elongisporus NT-Als proteins also appeared in more than one column in Figure 4 with one (LeAls734) sharing greater similarity with Sag1. The recombinant nature of some of the LeAls proteins presented a more-complex picture than for S. stipitis or M. guilliermondii (Supplementary File S6).
Drawing a Tree to Visualize NT-Als Relatedness
In addition to comparing NT-Als proteins based on the features described above, more-global comparisons were made. Clustal Omega alignment of the NT-Als sequences (Supplementary File S5) showed identity values ranging from approximately 20% to 90% (data not shown). A maximum likelihood tree was drawn to visualize the results (Figure 5).
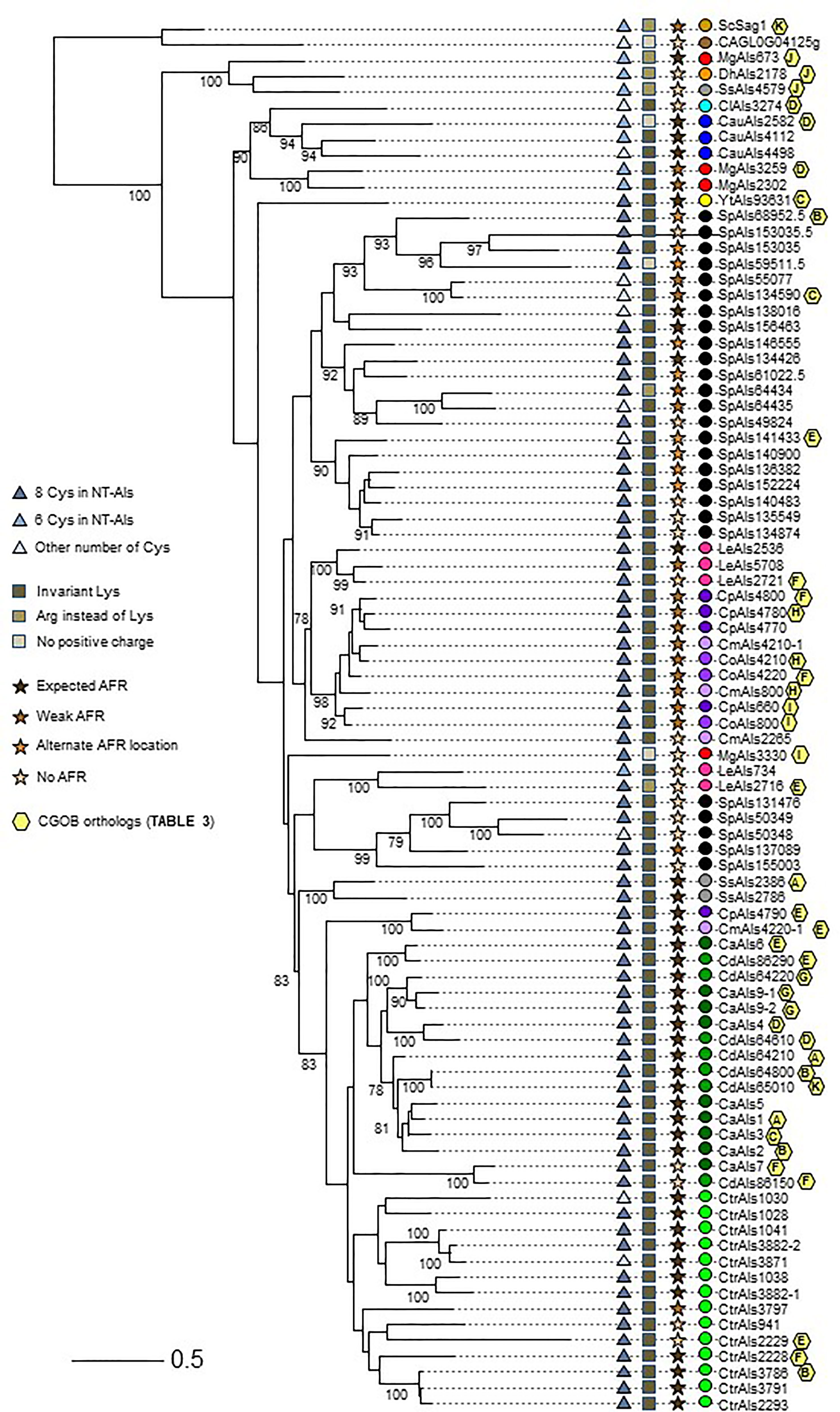
Figure 5 Relationships between NT-Als sequences depicted in a tree format. Information from Figure 4 (number of Cys in NT-Als, presence of the invariant Lys, nature of the AFR) is included using symbols at the left of the tree. Yellow hexagons on the right of the branches depict ortholog group information from Table 3. Colored dots indicating species follow the color scheme from Supplementary Table S2 and Figure 4. The scale bar represents substitutions per site.
The topology of the tree reflected the overall diversity in NT-Als sequence identity. For example, NT-Als sequences from the closely related species C. parapsilosis/C. orthopsilosis/C. metapsilosis appeared on short branches in tight groups. In constrast, many branches showing the relationship between the highly diverse S. passalidarum NT-Als sequences were quite long, most notably for SpAls153035.5, which was the least related to other proteins in that species.
Als sequences from the same species tended to cluster together which made the overall tree organization similar to the species tree in Figure 1. The close relationship between C. albicans and C. dubliniensis (Jackson et al., 2009), as well as that between C. parapsilosis/C. orthopsilosis/C. metapsilosis (Tavanti et al., 2005), were reflected by the proteins from these groups being localized to distinct regions of the tree. C. tropicalis sequences were similarly clustered as were most of the S. passalidarum sequences. C. auris and C. lusitaniae sequences were grouped closely, similar to their position on the species tree (Figure 1). Within the groupings of closely related species, orthologs were obvious (e.g. CaAls6/CdAls86290, CaAls7/CdAls86150, CpAls660/CoAls800 and CoAls4210/CmAls4210-1). Examples of paralogs, likely the result of gene duplication, included CpAls4770/CpAls4780/CpAls4800, CtrAls3791/CtrAls2293/CtrAls3786, and SpAls64434/SpAls64435.
Similar to predictions made from the Figure 4 diagram, NT-Als sequences from some species were spread across the tree. The four M. guilliermondii Als sequences were found in three different locations: MgAls673 was grouped more closely to sequences like ScSag1; MgAls2302 and MgAls3259 were clustered together near proteins from C. auris; and MgAls3330 was on its own branch elsewhere on the tree. Low bootstrap support for the latter location suggested that MgAls3330 could easily be located differently on the tree. The three sequences from S. stipitis were also dispersed with one near ScSag1 and the two others closer to the C. albicans/C. dubliniensis proteins. The five L. elongisporus Als proteins were split into two groups: the first including LeAls2536/LeAls5708/LeAls2721 (likely paralogs) and the second including LeAls734/LeAls2716 (recombinants between Als and Iff/Hyr). The Sag1-like features for SsAls4579 and MgAls673 were reflected on the tree (Figure 5), while LeAls734 and LeAls2716 (both Als recombinants with Iff/Hyr) clustered together in a different location.
The tree in Figure 5 was drawn from a portion of each protein sequence. Another way to evaluate phylogenetic relationships is via conservation of physical location (synteny) as depicted using the Candida Gene Order Browser (CGOB; Maguire et al., 2013). CGOB includes all species in Figure 5 except C. glabrata (CAGL0G04125g). A screenshot from one region of the CGOB was included in Figure 2C. It showed genes from several different fungal species that were orthologs of C. albicans ALS6 and ALS7. Orthologs reported by CGOB are shown in Table 3. Yellow hexagons with alphabetical labels were used to display the CGOB relationships on the tree in Figure 5.
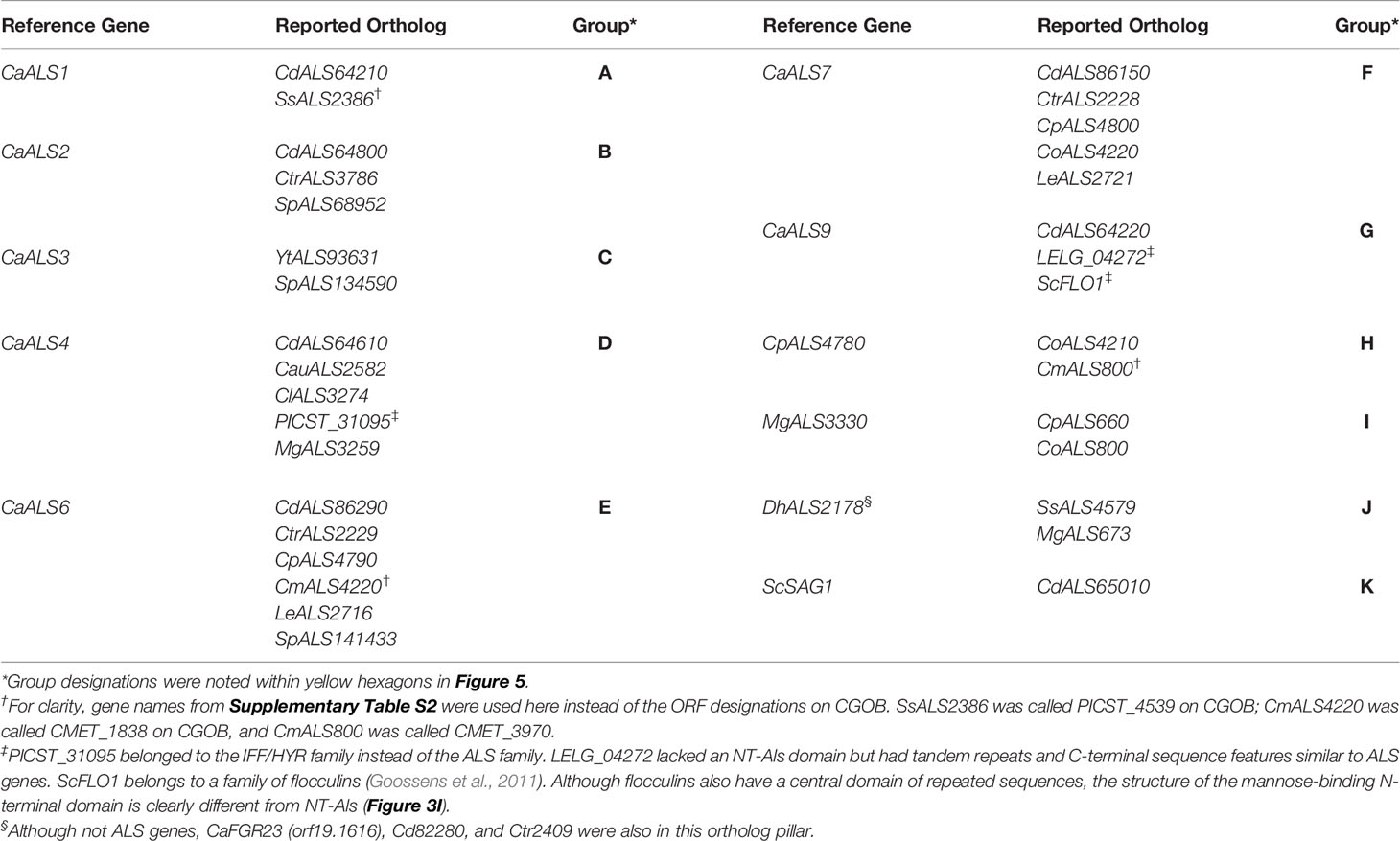
Table 3 Orthologous ALS loci reported on Candida Gene Order Browser (CGOB).
CGOB ortholog designations were most reliable in closely related species such as C. albicans and C. dubliniensis: each C. albicans locus had a C. dubliniensis ortholog except CaALS3 and CaALS5 which are missing from the C. dubliniensis genome (Jackson et al., 2009). These ortholog groups were frequently augmented with genes from other species such as Group A which included SsALS2386. Groups E and F were the largest, suggesting ortholog relationships between genes in 7 and 6 species, respectively.
CGOB also proposed ortholog groups that did not include C. albicans or C. dubliniensis sequences. One example was Group J that included the SAG1-like MgALS673 and SsALS4579, as well as DhAls2178, the lone ALS sequence from the primarily haploid D. hansenii. Examination of the predicted protein sequence showed lack of a tandem repeat domain as well as other Sag1-like features (Supplementary File S11 and Figure 4).
Some CGOB ortholog designations were perhaps problematic. For example, PICST_31095 (Group D) belonged to the IFF/HYR family instead of ALS and ScFLO1(Group G) encoded a flocculin with a mannose-binding N-terminal domain that was structurally diverse from the Als family (Figure 3). Perhaps the most intriguing ortholog designation was Group K which included ScSAG1 and CdALS65010. CdALS65010 was nearly 100% identical to CdALS64800, which was included in ortholog Group B. These loci are near each other on C. dubliniensis chromosome 6 and the appear to be paralogs (i.e. recently duplicated; Jackson et al., 2009). As species become more divergent, assessing ortholog designations becomes more challenging. However, the suggestion of orthology between ScSAG1 and a C. dubliniensis ALS gene supported the general idea that the genes were part of the same family and arose from a common ancestor.
Probing the Taxonomic Boundaries of the ALS Family
The rapidly increasing number of genome sequences present in public databases provided an opportunity to further explore the breadth and potential origin of the ALS family. The NCBI database can be queried using taxonomic identification (taxid) numbers to detect potential ALS loci more broadly across the phylogenetic tree. Table 4 lists the taxids for designations of the major Families within the Order Saccharomycetales. Inputs from NCBI (lifemap-ncbi-univ-lyon1.fr), Shen et al. (2018), and Li et al. (2021) were included although the taxonomic designations were not identical among the sources. The portion of the kingdom Fungi tree (Li et al., 2021) that represented the Subphylum Saccharomycotina was reproduced in Figure 6.
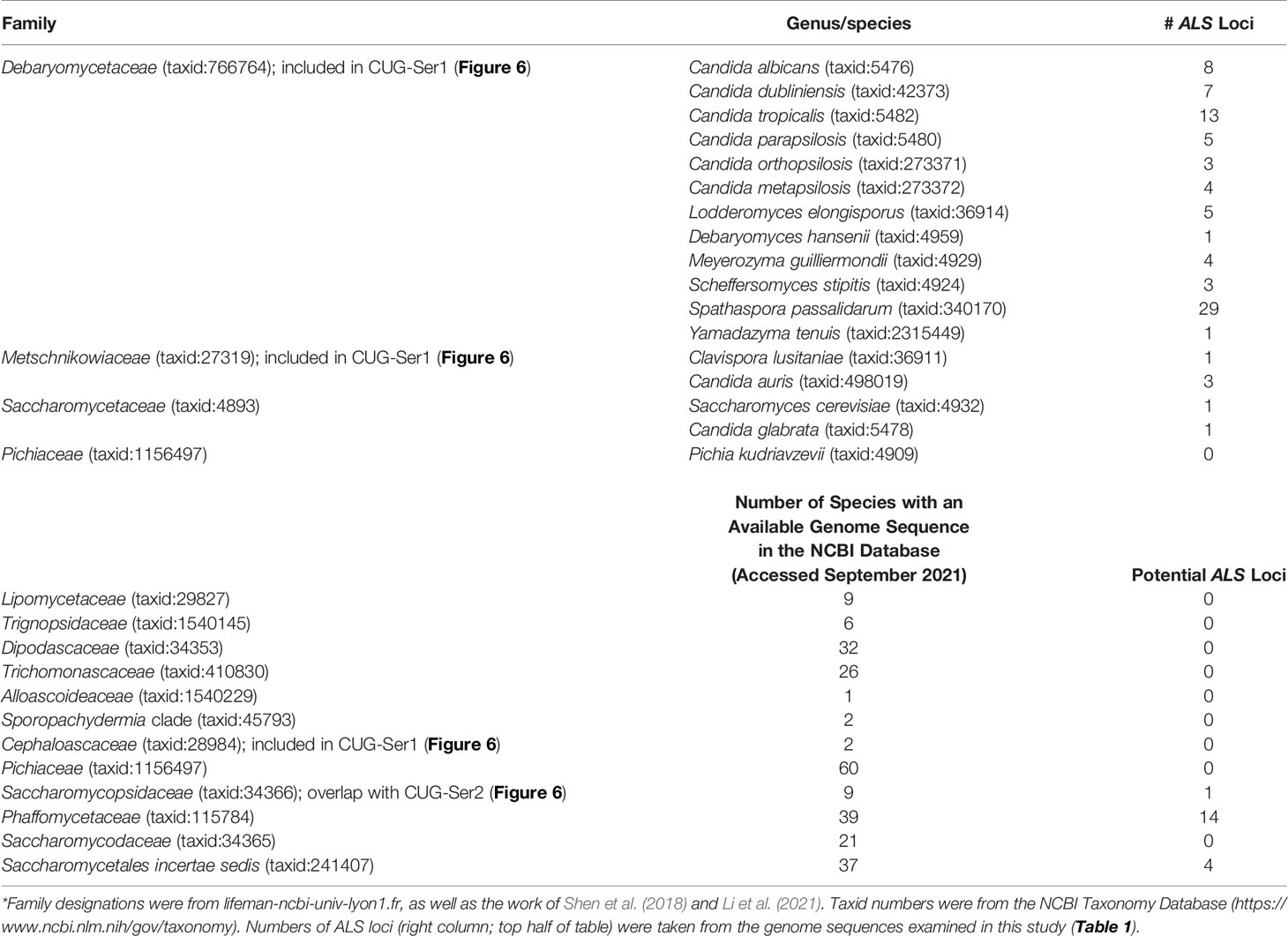
Table 4 ALS gene representation within the Order Saccharomycetales*.
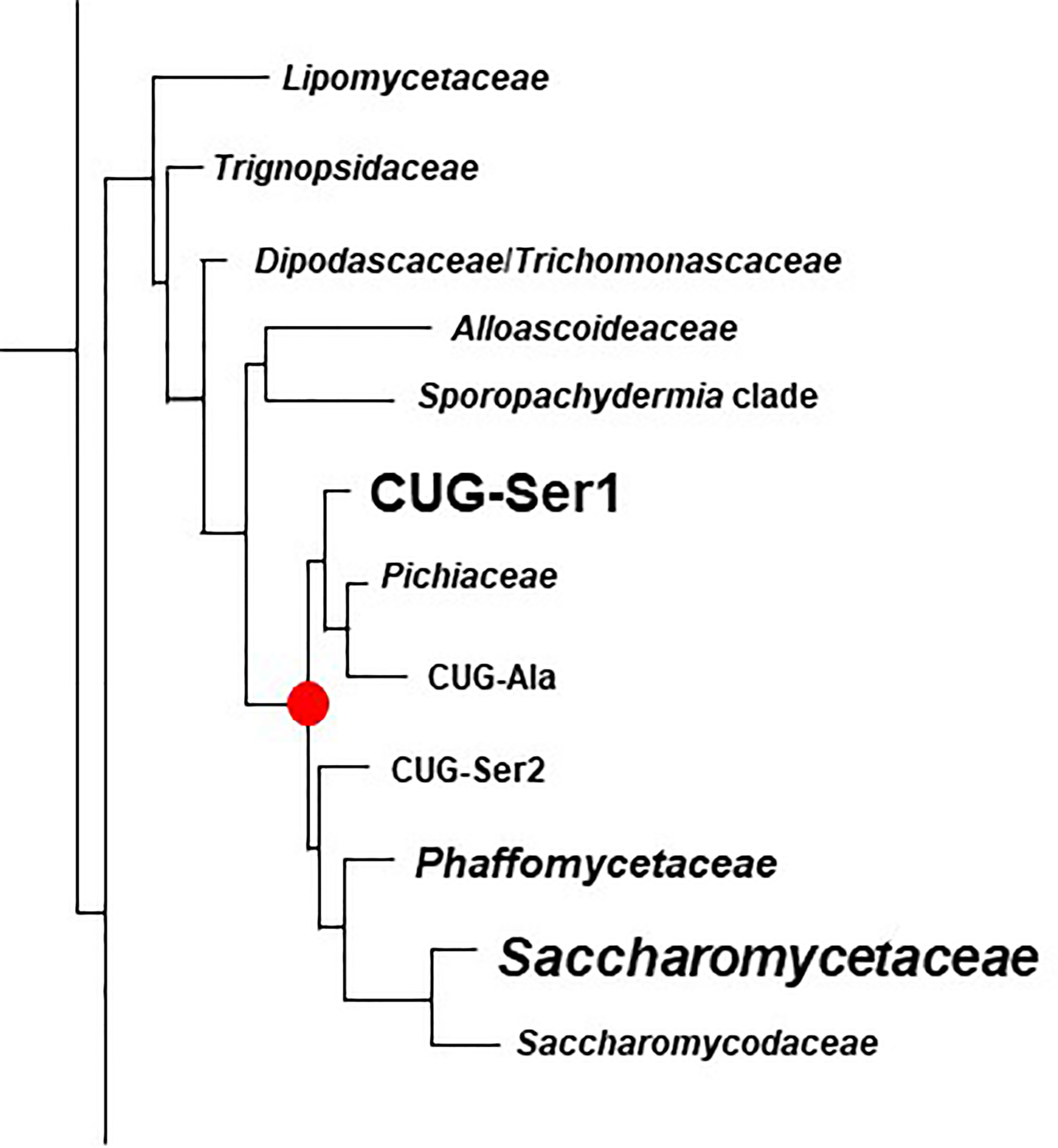
Figure 6 Phylogenetic tree of Families in the Order Saccharomycetales. The tree was traced from the data of Li et al. (2021) to highlight the region of interest for the current study. Family names were those used by Li et al. (2021). Table 4 placed these names into the context of Family designations used on the NCBI Taxonomy Database (Schoch et al., 2020). The red dot denotes the common ancestor of the CUG-Ser1 clade and Saccharomycetaceae, the two groups in which ALS genes were most common (indicated by larger font). ALS genes were also detected in the Phaffomycetaceae.
Figure 6 indicated the position of a common ancestor (red dot) for the ALS family that would include both S. cerevisiae SAG1 (in the Saccharomycetaceae) and genes more closely resembling C. albicans ALS loci (in CUG-Ser1). BLAST searching using taxids did not reveal ALS loci in available genome sequences from the Lipomycetaceae, Trignopsidaceae, Dipodascaceae, Trichomonascaceae, Alloascoidaceae, or the Sporopachydermia clade (Table 4). CUG-Ser1 was comprised of the Debaryomycetaceae, Metschnikowiaceae, and Cephaloascaceae (Shen et al., 2018). BLAST searching using taxids suggested that there were numerous additional ALS genes among the 88 Debaryomycetaceae species genomes in NCBI (accessed September 2021). Among the 62 Metschnikowiaceae species, ALS sequences were found primarily in species closely related to C. auris and C. lusitaniae including Candida haemulonii, Candida duobushaemulonis, and Candida pseudohaemulonii, all of which have been observed as pathogens (Jurardo-Martin et al., 2020). C. intermedia, noted for its ability to ferment xylose, as well as a rare cause of catheter-related fungemia (Ruan et al., 2010; Geijer et al., 2020) also had ALS sequences. No ALS sequences were detected in the Cephaloascaceae, although only 2 genomes were available (Table 4).
Pichia kudriavzevii (formerly Candida krusei; Douglass et al., 2018) was included in Table 4 and in Figure 1 since the species is among those associated with clinical disease (Gomez-Gaviria and Mora-Montes, 2020). However, ALS genes were not detected in Pichia kudriavzevii, nor in the genomes of 60 other species from the Family Pichiaceae that were available in the NCBI database (Table 4). No ALS sequences were detected in the species included in the CUG-Ala clade (Figure 6). The lone BLAST hit in the Saccharomycopsidaceae (Table 4; overlap with CUG-Ser2 in Figure 6) was based more on the presence of Ser/Thr-rich repeated sequences than on an NT-Als adhesive domain. Additional work would be required to validate the sequence and determine if an NT-Als domain was present.
Potential ALS genes in the Phaffomycetaceae (Table 4) were located in two species each in the genera Wickerhamomyces and Cyberlindnera; each locus encoded a potential NT-Als domain. Many additional ALS sequences were found among the 104 Saccharomycetaceae genomes available in NCBI. This family included S. cerevisiae, for which SAG1 was the only BLAST hit. For other species such as Torulospora delbrueckii, a species of interest for wine making and baking (Fernandes et al., 2021), more than one ALS sequence was detected in the genome. One (TDEL_0E04030) predicted a protein that more closely resembled S. cerevisiae Sag1, while another (TDEL_0G04810) encoded an NT-Als domain (validated using AlphaFold; data not shown) attached to a complex C-terminal Ser/Thr-rich sequence. This predicted C-terminal domain was notable because it included 5 copies of the 45-amino acid tandem repeat found in S. cerevisiae Flo1 (Soares, 2010), suggesting recombination between the ALS and FLO families. A predicted protein in Lachancea thermotolerans (KLTH0C00440p) suggested an even-more-convincing recombinant that had an NT-Als domain attached to > 30 copies of the Flo1 tandem repeat. These sequences require verification to ensure the recombinants were part of the genome, rather than an artifact of computational genome assembly. ALS sequences were also detected in Kazachstania species extending the representation of ALS sequences throughout the Saccharomycetaceae as defined by Shen et al. (2018). Many of these sequences appeared partial, suggesting the need for PCR amplification and repair to reveal the true nature of their encoded proteins. No ALS sequences were detected among the 21 Saccharomycodaceae species queried.
The taxonomic category Saccharomycetales incerta sedis had four ALS sequences, all in the species Diutina rugosa (formerly Candida rugosa), which is noted as an infrequent human pathogen (Ming et al., 2019). Evaluation of the best hit (DIURU_002000) showed a predicted secretory signal peptide and GPI anchor addition site. Within the NT-Als region, DIURU_002000 was 37% identical to C. albicans NT-Als3, had 8 Cys in the expected locations, Arg instead of the invariant Lys, and a strong predicted AFR. AlphaFold produced the same general structure for the DIURU_002000 NT-Als protein as for those shown in Figure 3 (data not shown).
Collectively, these observations pinpointed the location of the ALS family among currently available genome sequences and suggested potential taxonomic boundaries that will continue to be tested as more genome sequences are added to this region of the phylogenetic tree.
Discussion
Availability of genome resources for pathogenic yeasts, as well as other closely related species, provided a comparative view of the ALS family across a broader expanse of the phylogenetic tree than previously investigated. Despite advances in long-read DNA sequence technologies, many ALS loci still required PCR amplification and Sanger sequencing to derive an accurate gene count and sequence. The availability of the unprecedented protein structural prediction accuracy of AlphaFold (Jumper et al., 2021) provided the opportunity to demonstrate conservation of NT-Als domain shape for the proteins listed in Supplementary Table S2. Recent addition of more than 200 new budding yeast genome sequences into the NCBI database (Shen et al., 2018) further bridged the gap between S. cerevisiae and C. albicans and provided a clearer picture of the ALS family breadth. In total, the information leads to the conclusions that S. cerevisiae Sag1 is an Als protein and the Als family is far more diverse than previously described.
Presence of the NT-Als domain is universal among the proteins in Supplementary Table S2, and clearly is a defining characteristic for inclusion in the Als family. The AFR is recognized as part of the NT-Als domain (Lin et al., 2014); this sequence was absent, weaker, or perhaps in another location in many of the newly characterized Als proteins described here. Immediately following the AFR in C. albicans Als proteins is a short Thr-rich region that some publications refer to as the T domain (reviewed in Hoyer and Cota, 2016). The T domain in C. albicans Als proteins ends at the start of the tandem repeat region. Designating a T domain in many of the proteins described here would require arbitrary judgement calls so the region was not included in any of the line diagrams (Supplementary Files S6-S11). NT-Als domains were drawn to include all amino acids up to the start of the repeated sequences or Ser/Thr-rich regions.
In some proteins (e.g. SpAls55077, SpAls134590, ClAls3274), sequences similar to the C. albicans T domain are expanded into repeated sequences of variable length in the center of the protein. While all C. albicans Als proteins have a region of highly conserved, 36-amino-acid tandemly repeated sequences, this part of the Als protein is quite variable among those from other species. Variations include tandem copies of novel lengths and sequences, repeated sequences that are not necessarily tandem copies, and complete lack of repeated sequences. The latter presentation is characteristic of S. cerevisiae Sag1 and other proteins more closely related to ScSag1 on the Figure 5 tree. Sequences most C-terminal in the Als family are also diverse, many featuring an enriched Ser/Thr composition. Ser/Thr richness promotes O-glycosylation that can form an extended structure on which the NT-Als domain is displayed on the cell surface (Jentoft, 1990). Some of the Als proteins have other amino acids (e.g. Glu, Gly, Pro) that are also enriched in the C-terminal portion of the protein or in novel, short repeated sequences.
Inclusion in the Als family is focused on the presence of an NT-Als domain that has the same general shape as the proteins described here (Figure 3). This Als family definition also recognizes instances of recombination between ALS genes and others encoding cell-surface proteins. A previous report (Oh et al., 2019) described CmALS2265, the product of recombination between ALS genes and the IFF/HYR family (Bailey et al., 1996; Bates et al., 2007; Chaffin, 2008; Boisramé et al., 2011; Riccombeni et al., 2012). In the present study, additional recombinants between ALS and IFF/HYR genes were noted in L. epongisporus (LeALS734 and LeALS2716). The tantalizing possibility of ALS/FLO recombinants in T. delbrueckii and L. thermotolerans suggests even-greater interfamily recombination as a mechanism for generating diversity on the fungal cell surface. Recombination, as the mechanism to generate novel adhesins, has been discussed previously (Verstrepen et al., 2004; Verstrepen et al., 2005) although these descriptions involved recombination between genes in the same family. In contrast, interfamily recombination places the functional NT domain from one family onto the glycosylated stalk from another. Predicted structures for the NT domains of Als, Iff/Hyr, and Flo proteins were included in Figure 3 to highlight their obvious differences. Potential for hybrid function expands if the glycosylated stalk is more than just a pedestal for displaying the NT domain. These observations provide ample experimental hypotheses to pursue to better understand cell-surface diversity among the yeast species.
Another novel observation from the current work is the subtelomeric localization of ALS genes in multiple species including M. guilliermondii (MgALS2302, MgALS3259), C. auris (CauALS2582), and S. passalidarum (SpALS131476, SpALS137089). C. albicans subtelomeric regions are described as hotspots for genomic rearrangement, although adhesin-encoding genes are not located there (Dunn and Anderson, 2019). Subtelomeric location of adhesin genes is well-documented in other species such as C. glabrata (Xu et al., 2020). However, analysis of long-read-based genome sequences from three C. glabrata isolates suggested that changes in subtelomeric adhesin-encoding genes is due to break-induced replication, a relatively rare event that affects only the gene closest to the telomere (Xu et al., 2021). Muñoz et al. (2021) noted C. auris subtelomeric regions rich in genes encoding Iff/Hyr proteins, candidate cell wall proteins unique to that species, and CauAls2582. Analysis of genome sequences from 12 C. auris isolates representing diverse clades showed that Clade II isolates lose large subtelomeric regions and the cell wall genes associated with them (Muñoz et al., 2021). The consequences of ALS gene location in the subtelomeric region remain to be further explored in species such as M. guilliermondii and S. passalidarum.
By including Sag1 in the Als family, the definition of Als function also becomes more diverse. Sag1 binds a very specific ligand (the C-terminal peptide of a-agglutinin; Cappellaro et al., 1994) with high affinity. In comparison, C. albicans Als proteins can bind many ligands with lower affinity and rely on AFR-mediated protein aggregation to increase avidity (reviewed in Hoyer and Cota, 2016; Ho et al., 2019). Similar to those in C. albicans, several of the more-recently described Als proteins in other species function in adhesion to host cells or protein-coated surfaces (Bertini et al., 2016; Neale et al., 2018; Zoppo et al., 2018; Lombardi et al., 2019; Zoppo et al., 2020a; Zoppo et al., 2020b). However, considerable effort is still required to understand the specifics of these interactions. Information in Figures 3, 5 can be used to select proteins from along the newly defined Als family spectrum to better understand how structure contributes to functional variation. Experiments involving purified proteins and biophysical techniques could be pursued, or perhaps individual genes or chimeric constructs could be expressed in model systems such as the recently described ALS-negative C. orthopsilosis strain (Zoppo et al., 2019). Experimentally demonstrated structural solutions could also be pursued, and in place of the difficult-to-crystallize ScSag1, proteins close to ScSag1 on the Figure 5 tree may serve as informative surrogates (e.g. Group J and CAGL0G04125G, which may be mating glycoproteins from the haploid species M. guilliermondii, D. hansenii, and S. stipitis).
The ALS alleles listed in Supplementary Table S2 are quite long: many are at least 5 kb and one is over 15 kb. In C. albicans, where multiple strains and ALS alleles have been examined, these long ORFs are maintained without interruption. One notable example is C. albicans ALS7 that experiences frequent expansions and contractions of multiple different repeated sequences and is transcribed at an extremely low level that may not produce sufficient protein for biologically meaningful adhesive interactions (Zhang et al., 2003; Green et al., 2005). In contrast, the current study revealed the first example of ALS pseudogenes. They are found in S. passalidarum that has amplified the ALS family to 29 loci, well beyond the census in other characterized species (Supplementary File S10). In addition to the ALS pseudogenes, many of the other S. passalidarum ALS loci encode proteins that are predicted to mislocalize compared to the C. albicans model: lack of a secretory signal peptide suggests protein that accumulates inside the cell rather than on the surface. The presence of loci that encode proteins with secretory potential, but lacking the ability to cross-link into the cell wall (i.e. no GPI anchor addition site), suggests the potential for S. passalidarum to export its own adhesion inhibitors. For example, a secreted, high-affinity binding protein could inhibit S. passalidarum adhesion to host cells, or perhaps release individual cells from host interactions or from polymicrobial structures such as biofilms. Wohlbach et al. (2011) pointed out that the wood-boring-beetle-associated S. passalidarum is also a commensal, similar to the typical relationship between humans and many of the fungi described here. However, only two of the SpAls proteins include all NT-Als features (8 Cys, invariant Lys, AFR) that are important for function of C. albicans Als3 (Supplementary Table S2; Supplementary File S10; Figure 4). The long branches associated with the S. passalidarum Als sequences in Figure 5 could result from sequence substitutions due to lack of selective pressure or could indicate functional diversification that serves S. passalidarum in its ecological niche. The role of the Als proteins in S. passalidarum is another topic with many hypotheses for future exploration.
Additional hypotheses to test involve allelic variability and gene expression. Information reported here is limited to one allele from each species. Previous work in C. albicans revealed tremendous allelic variation, often involving copy numbers of repeated sequences, but sometimes affecting the NT-Als coding region (reviewed in Hoyer et al., 2008). Considerable variation is frequently observed between alleles within the same strain of a diploid species (Zhao et al., 2003; Zhao et al., 2007). It is reasonable to hypothesize that allelic variation exists among the genes and species described here. It is also reasonable to expect examples of differential gene expression with growth stage and morphology, gene expression variations between strains of the same species, and gene expression patterns that differ between in vitro and in vivo growth conditions (reviewed in Hoyer et al., 2008; Coleman et al., 2012; Galán-Ladero et al., 2019; Lombardi et al., 2019). There are many exciting avenues for investigation as the nature of the ALS family continues to be discovered.
Data Availability Statement
The genome assembly for Lodderomyces elongisporus strain NRRL YB-4239, generated using Illumina MiSeq and Oxford Nanopore MinION data, is available in GenBank under BioProject accession number PRJNA432415, BioSample accession number SAMN08448784, and Genome Assembly accession number GCA_013620985.1 (ASM1362098v1). The genome assembly for Meyerozyma guilliermondii strain ATCC 6260, generated using Illumina MiSeq and Oxford Nanopore MinION data, is available in GenBank under BioProject accession number PRJNA432394, BioSample accession number SAMN08448312, and Genome Assembly accession number GCA_006942155.1 (ASM694215v1). The genome assembly for Scheffersomyces stipitis strain CBS 6054, generated using Illumina MiSeq and Oxford Nanopore MinION data, is available in GenBank under BioProject accession number PRJNA432364, BioSample accession number SAMN08446935, and Genome Assembly accession number GCA_006942115.1 (ASM694211v1). The genome assembly for Spathaspora passalidarum strain NRRL Y-27907, generated using Illumina MiSeq and Oxford Nanopore MinION data, is available in GenBank under BioProject accession number PRJNA432249, BioSample accession number SAMN08439027, and Genome Assembly accession number GCA_013620965.1 (ASM1362096v1).
Author Contributions
LH conceptualized the study, acquired funding, and administered the project. KS, VV, CF, and AH conducted formal analysis. S-HO, AI, RR-B, CF, AH, and LH performed the investigation. CF, AH, and LH developed the study methodology. S-HO, KS, CF, AH, and LH wrote the original draft. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by R15 DE026401 from the National Institute of Dental and Craniofacial Research, National Institutes of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
This article is dedicated to the memory of Professor W. LaJean Chaffin, an outstanding scientist, mentor, and friend. Professor Chaffin shared her enthusiasm for medical mycology with many young researchers and helped them become established investigators. AI and RR-B were part of an Undergraduate Program in Fungal Genomics, which is a collaboration between the University of Illinois at Urbana-Champaign and Millikin University. Other students who contributed to this effort included Kaia Ball, Madeline Batek, Anton Bershanskiy, Stephen DeMartini, Erica Forbes, Zeidy Garcia, Jillian Jones, Jessie Kirk, Mariah McNamer, Deniz Namik, Quinn Nguyen, and Brooke Smith. The authors thank Drs. Travis Wilcoxen and Laura Zimmerman, Millikin University Department of Biology, for coordinating student participation and supervising research credit for students in the program.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2021.794529/full#supplementary-material
References
Bailey, D. A., Feldmann, P. J., Bovey, M., Gow, N. A., Brown, A. J. (1996). The Candida albicans HYR1 Gene, Which is Activated in Response to Hyphal Development, Belongs to a Gene Family Encoding Yeast Cell Wall Proteins. J. Bacteriol. 178, 5353–5360. doi: 10.1128/jb.178.18.5353-5360.1996
Bates, S., de la Rosa, J. M., MacCallum, D. M., Brown, A. J. P., Gow, N. A. R., Odds, F. C. (2007). Candida albicans Iff11, a Secreted Protein Required for Cell Wall Structure and Virulence. Infect. Immun. 75, 2922–2928. doi: 10.1128/IAI.00102-07
Bertini, A., Zoppo, M., Lombardi, L., Rizzato, C., De Carolis, E., Vella, A., et al. (2016). Targeted Gene Disruption in Candida parapsilosis Demonstrates a Role for CPAR2_404800 in Adhesion to a Biotic Surface and in a Murine Model of Ascending Urinary Tract Infection. Virulence 7, 85–97. doi: 10.1080/21505594.2015.1112491
Boisramé, A., Cornu, A., Da Costa, G., Richard, M. L. (2011). Unexpected Role for a Serine/Threonine-Rich Domain in the Candida albicans Iff Protein Family. Eukaryot Cell 10, 1317–1330. doi: 10.1128/EC.05044-11
Bolger, A. M., Lohse, M., Usadel, B.. (2014). Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Butler, G., Rasmussen, M. D., Lin, M. F., Santos, M. A., Sakthikumar, S., Munro, C. A., et al. (2009). Evolution of Pathogenicity and Sexual Reproduction in Eight Candida Genomes. Nature 459, 657–662. doi: 10.1038/nature08064
Cappellaro, C., Baldermann, C., Rachel, R., Tanner, W. (1994). Mating Type-Specific Cell-Cell Recognition of Saccharomyces cerevisiae: Cell Wall Attachment and Active Sites of a- and α-Agglutinin. EMBO J. 20, 4737–4744. doi: 10.1002/j.1460-2075.1994.tb06799.x
Chaffin, W. L. (2008). Candida albicans Cell Wall Proteins. Microbiol. Mol. Biol. Rev. 72, 495–544. doi: 10.1128/MMBR.00032-07
Chen, M. H., Shen, Z. M., Bobin, S., Kahn, P. C., Lipke, P. N. (1995). Structure of Saccharomyces cerevisiae Alpha-Agglutinin: Evidence for a Yeast Cell Wall Protein With Multiple Immunoglobulin-Like Domains With Atypical Disulfides. J. Biol. Chem. 270, 26168–26177. doi: 10.1074/jbc.270.44.26168
Coleman, D. A., Oh, S.-H., Manfra-Maretta, S. L., Hoyer, L. L. (2012). A Monoclonal Antibody Specific for Candida albicans Als4 Demonstrates Overlapping Localization of Als Family Proteins on the Fungal Cell Surface and Highlights Differences Between Als Localization In Vitro and In Vivo. FEMS Immunol. Med. Microbiol. 64, 321–333. doi: 10.1111/j.1574-695X.2011.00914.x
Cook, C. E., Bergman, M. T., Cochrane, G., Apweiler, R., Birney, E. (2017). The European Bioinformatics Institute in 2017: Data Coordination and Integration. Nucleic Acids Res. 46, D21–D29. doi: 10.1093/nar/gkx1154
Douglass, A. P., Offei, B., Braun-Galleani, S., Coughlan, A. Y., Martos, A. A. R., Ortiz-Merino, R. A., et al. (2018). Population Genomics Shows No Distinction Between Pathogenic Candida krusei and Environmental Pichia kudriavzevii: One Species, Four Names. PLoS Pathog. 14, e1007138. doi: 10.1371/journal.ppat.1007138
Du, H., Bing, J., Hu, T., Ennis, C. E., Nobile, C. J., Huang, G. (2020). Candida auris: Epidemiology, Biology, Antifungal Resistance, and Virulence. PLoS Pathog. 16, e1008921. doi: 10.1371/journal.ppat.1008921
Dujon, B., Sherman, D., Fischer, G., Durrens, P., Casaregola, S., Lafontaine, I., et al. (2004). Genome Evolution in Yeasts. Nature 430, 35–44. doi: 10.1038/nature02579
Dunn, M. J., Anderson, M. Z. (2019). To Repeat or Not to Repeat: Repetitive Sequences Regulate Genome Stability in Candida albicans. Genes 10:866. doi: 10.3390/genes10110866
Eisenhaber, B., Schneider, G., Wildpaner, M., Eisenhaber, F. (2004). A Sensitive Predictor for Potential GPI Lipid Modification Sites in Fungal Protein Sequences and its Application to Genome-Wide Studies for Aspergillus nidulans, Candida albicans, Neurospora crassa, Saccharomyces cerevisiae, and Schizosaccharmyces pombe. J. Mol. Biol. 337, 243–253. doi: 10.1016/j.jmb.2004.01.025
Fernandes, T., Silva-Sousa, F., Pereira, F., Rito, T., Soares, P., Franco-Duarte, R., et al. (2021). Biotechnological Importance of Torulaspora delbrueckii: From the Obscurity to the Spotlight. J. Fungi 7:712. doi: 10.3390/jof090712
Fernandez-Escamilla, A.-M., Rousseau, F., Schymkowitz, J., Serrano, L. (2004). Prediction of Sequence-Dependent and Mutational Effects on the Aggregation of Peptides and Proteins. Nat. Biotechnol. 22, 1302–1306. doi: 10.1038/nbt1012
François, F., Noël, T., Pépin, R., Brulfert, A., Chastin, C., Favel, A., et al. (2001). Alternative Identification Test Relying Upon Sexual Reproductive Abilities of Candida lusitaniae Strains Isolated From Hospitalized Patients. J. Clin. Microbiol. 39, 3906–3914. doi: 10.1128/JCM.39.11.3906-3914.2001
Gabler, F., Nam, S. Z., Till, S., Mirdita, M., Steinegger, M., Söding, J., et al. (2020). Protein Sequence Analysis Using the MPI Bioinformatics Toolkit. Curr. Protoc. Bioinf. 72, e108. doi: 10.1002/cpbi.108
Galán-Ladero, M. A., Blanco-Blanco, M. T., Fernández-Calderón, M. C., Lucio, L., Gutiérrez-Martín, Y., Blanco, M. T., et al. (2019). Candida tropicalis Biofium Formation and Expression Levels of the CTRG ALS-Like Genes in Sessile Cells. Yeast 36, 107–115. doi: 10.1002/yea.3370
Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M. R., Appel, R. D., et al. (2005). Protein Identification and Analysis Tools on the ExPASy Server, In: Walker, J. M. Ed. The Proteomics Protocols Handbook (Totowa: Humana Press), 571–607. doi: 10.1385/1-59259-890-0:571
Geijer, C., Faria-Oliveira, F., Moreno, A. D., Stenberg, S., Mazurkewich, S., Olsson, L. (2020). Genomic and Transcriptomic Analysis of Candida intermedia Reveals the Genetic Determinants for its Xylose-Converting Capacity. Biotechnol. Biofuels 13, 48. doi: 10.1186/s13068-020-1663-9
Goffeau, A., Barrell, B. G., Bussey, H., Davis, R. W., Dujon, B., Feldmann, H., et al. (1996). Life With 6000 Genes. Science 274, 563–567. doi: 10.1126/science.274.5287.546
Gomez-Gaviria, M., Mora-Montes, H. M. (2020). Current Aspects in the Biology, Pathogeny, and Treatment of Candida krusei, a Neglected Fungal Pathogen. Infect. Drug Resist. 13, 1673–1689. doi: 10.2147/IDR.S247944
Goossens, K. V. Y., Stassen, C., Stals, I., Donohue, D. S., Devreese, B., De Greve, H., et al. (2011). The N-Terminal Domain of the Flo1 Flocculation Protein From Saccharomyces cerevisiae Binds Specifically to Mannose Carbohydrates. Eukarot Cell 10, 110–117. doi: 10.1128/EC.00185-10
Gori, K., Knudsen, P. B., Nielsen, K. F., Arneborg, N., Jespersen, L. (2011). Alcohol-Based Quorum Sensing Plays a Role in Adhesion and Sliding Motility of the Yeast Debaryomyces hansenii. FEMS Yeast. Res. 11, 643–652. doi: 10.1111/j.1567-1364.2011.00755.x
Green, C. B., Zhao, X., Yeater, K. M., Hoyer, L. L. (2005). Construction and Real-Time RT-PCR Validation of Candida albicans PALS-GFP Reporter Strains and Their Use in Flow Cytometry Analysis of ALS Gene Expression in Budding and Filamentous Cells. Microbiology 151, 1051–1060. doi: 10.1099/mic.0.27696-0
Grigorescu, A., Chen, M.-H., Zhao, H., Khan, P. C., Lipke, P. N. (2000). A CD2-Based Model of Yeast Alpha-Agglutinin Elucidates Solution Properties and Binding Characteristics. IUBMB Life 50, 105–113. doi: 10.1080/713803692
Guin, K., Chen, Y., Mishra, R., Muzaki, S. R. B., Thimmappa, B. C., O’Brien, C. E., et al. (2020). Spatial Inter-Centromeric Interactions Facilitated the Emergence of Evolutionary New Centromeres. Elife 9, e58556. doi: 10.7554/eLife.58556
Ho, V., Herman-Bausier, P., Shaw, C., Conrad, K. A., Garcia-Sherman, M. C., Draghi, J., et al. (2019). An Amyloid Core Sequence in the Major Candida albicans Adhesin Als1p Mediates Cell-Cell Adhesion. mBio 10, e01766–e01719. doi: 10.1128/mBio.01766-19
Hoyer, L. L., Cota, E. (2016). Candida albicans Agglutinin-Like Sequence (Als) Family Vignettes: A Review of Als Protein Structure and Function. Front. Microbiol. 7, 280. doi: 10.3389/fmicb.2016.00280
Hoyer, L. L., Green, C. B., Oh, S.-H., Hoyer, L. L. (2008). Discovering the Secrets of the Candida albicans Agglutinin-Like Sequence (ALS) Gene Family – a Sticky Pursuit. Med. Mycol. 46, 1–15. doi: 10.1080/13693780701435317
Hoyer, L. L., Scherer, S., Shatzman, A. R., Livi, G. P. (1995). Candida albicans ALS1: Domains Related to a Saccharomyces cerevisiae Sexual Agglutinin Separated by a Repeating Motif. Mol. Microbiol. 15, 39–54. doi: 10.1111/j.1365-2958.1995. tb02219.x.
Jackson, A. P., Gamble, J. A., Yeomans, T., Moran, G. P., Saunders, D., Harris, D., et al. (2009). Comparative Genomics of the Fungal Pathogens Candida dubliniensis and Candida albicans. Genome Res. 19, 2231–2244. doi: 10.1101/gr.097501.109
Jeffries, T. W., Grigoriev, I. V., Grimwood, J., Laplaza, J. M., Aerts, A., Salamov, A., et al. (2007). Genome Sequence of the Lignocellulose-Bioconverting and Xylose-Fermenting Yeast Pichia stipitis. Nat. Biotechnol. 25, 319–326. doi: 10.1038/nbt1290
Jentoft, N. (1990). Why are Proteins O-Glycosylated? Trends Biochem. Sci. 15, 291–294. doi: 10.1016/0968-0004(90)90014-3
Jones, T., Federspiel, N. A., Chibana, H., Dungan, J., Kalman, S., Magee, B. B., et al. (2004). The Diploid Genome Sequence of Candida albicans. Proc. Natl. Acad. Sci. U.S.A. 101, 7329–7334. doi: 10.1073/pnas.0401648101
Jumper, J., Evans, R., Pritzel, Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly Accurate Protein Structure Prediction With AlphaFold. Nature 596, 583–589. doi: 10.1038/s41586-021-03819-2
Jurardo-Martin, I., Marcos-Arias, C., Tamayo, E., Guridi, A., de Groot, P. W. J., Quindós, G., et al. (2020). Candida duobushaemulonii: An Old But Unreported Pathogen. J. Fungi 6, 374. doi: 10.3390/jof6040374
Katoh, K., Standley, D. M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., Phillippy, A. M. (2017). Canu: Scalable and Accurate Long-Read Assembly via Adaptive k-mer Weighting and Repeat Separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B., Stamatakis, A. (2019). RAxML-NG: A Fast, Scalable and User-Friendly Tool for Maximum Likelihood Phylogenetic Inference. Bioinformatics 35, 4453–4455. doi: 10.1093/bioinformatics/btz305
Li, H. (2018). Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 34, 3094–3100. doi: 10.1093/bioinformatics/bty191
Li, Y., Steenwyk, J. L., Chang, Y., Wang, Y., James, T. Y., Stajich, J. E., et al. (2021). A Genome-Scale Phylogeny of the Kingdom Fungi. Curr. Biol. 26, 1653–1665. doi: 10.1016/j.cub.2021.01.074
Lin, J., Oh, S.-H., Jones, R., Garnett, J. A., Salgado, P. S., Rushnakova, S., et al. (2014). The Peptide-Binding Cavity is Essential for Als3-Mediated Adhesion of Candida albicans to Human Cells. J. Biol. Chem. 289, 18401–18412. doi: 10.1074/jbc.M114.547877
Lipke, P. N. (2018). What We Do Not Know About Fungal Cell Adhesion Molecules. J. Fungi 4:59. doi: 10.3390/jof4020059
Lipke, P. N., Chen, M. H., de Nobel, H., Kurjan, J., Kahn, P. C. (1995). Homology Modeling of an Immunoglobulin-Like Domain in the Saccharomyces cerevisiae Adhesion Protein Alpha-Agglutinin. Protein Sci. 4, 2168–2178. doi: 10.1002/pro.5560041023
Lipke, P. N., Klotz, S. A., Dufrene, Y. F., Jackson, D. N., Garcia-Sherman, M. C. (2018). Amyloid-Like β-Aggregates as Force-Sensitive Switches in Fungal Biofilms and Infections. Microbiol. Mol. Biol. Rev. 82, e00035–e00017. doi: 10.1128/MMBR.00035-17
Lockhart, S. R., Etienne, K. A., Vallabhaneni, S., Chowdhary, A., Govender, N. P., Colombo, A. L., et al. (2017). Simultaneous Emergence of Multidrug-Resistant Candida auris on 3 Continents Confirmed by Whole-Genome Sequencing and Epidemiological Analyses. Clin. Infect. Dis. 64, 134–140. doi: 10.1093/cid/ciw691
Lockhart, S. R., Messer, S. A., Pfaller, M. A., Diekema, D. J. (2008). Lodderomyces elongisporus Masquerading as Candida parapsilosis as a Cause of Bloodsream Infections. J. Clin. Microbiol. 46, 374–376. doi: 10.1128/JCM.01790-07
Lombardi, L., Zoppo, M., Rizzato, C., Bottai, D., Hernandez, A. G., Hoyer, L. L., et al. (2019). Characterization of the Candida orthopsilosis Agglutinin-Like Sequence (ALS) Genes. PLoS One 14, e0215912. doi: 10.1371/journal.pone.0215912
Lu, C. F., Kurjan, J., Lipke, P. N. (1994). A Pathway for Cell Wall Anchorage of Saccharomyces cerevisiae Alpha-Agglutinin. Mol. Cell Biol. 14, 4825–4833. doi: 10.1128/mcb.14.7.4825-4833
Madeira, M., Park, Y. M., Lee, J., Buso, M., Gur, T., Madhusoodanan, N., et al. (2019). The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019. Nucl. Acids Res. 47, W636–W641. doi: 10.1093/nar/gkz268
Maguire, S. L., ÓhÉigeartaigh, S. S., Byrne, K. P., Schröder, M. S., O’Gaora, P., Wolfe, K. H., et al (2013). Comparative Genome Analysis and Gene Finding in Candida Species Using CGOB. Mol. Biol. Evol. 30, 1281–1291. doi: 10.1093/molbev/mst042
Ming, C., Huang, J., Wang, Y., Lv, Q., Zhou, B., Liu, T., et al. (2019). Revision of the Medically Relevant Species of the Yeast Genus Diutina. Med. Mycol. 57, 226–233. doi: 10.1093/mmy/myy001
Mirdita, M., Ovchinnikov, S., Steinegger, M. (2021). ColabFold – Making Protein Folding Accessible to All. bioRxiv. preprint. doi: 10.1101/2021.08.15.456425
Mortensen, H. D., Gori, K., Jespersen, L., Arneborg, N. (2005). Debaryomyces hansenii Strains With Different Cell Sizes and Surface Physicochemical Properties Adhere Differently to a Solid Agarose Surface. FEMS Microbiol. Lett. 249, 165–170. doi: 10.1016/j.femsle.2005.06.009
Muñoz, J. F., Welsh, R. M., Shea, T., Batra, D., Gade, L., Howard, D., et al. (2021). Clade-Specific Chromosomal Rearrangements and Loss of Subtelomeric Adhesins in Candida auris. Genetics 218, iyab029. doi: 10.1093/genetics/iyab029
Neale, M. N., Glass, K. A., Longley, S. J., Kim, D. J., Laforce-Nesbitt, S. S., Wortzel, J. D., et al. (2018). Role of the Inducible Adhesin CpAls7 in Binding of Candida parapsilosis to the Extracellular Matrix Under Fluid Shear. Infect. Immun. 86, e00892–e00817. doi: 10.1128/IAI.00892-17
Nielsen, H. (2017). “Predicting Secretory Proteins With SignalP,” in Protein Function Prediction, Methods in Molecular Biology, vol. 1611. Ed. Kihara, D. (New York, NY: Humana Press), 59–73. doi: 10.1007/978-1-4939-7015-5_6
Oh, S.-H., Isenhower, A., Rodriguez-Bobadilla, R., Smith, B., Jones, J., Hubka, V., et al. (2021). Pursuing Advances in DNA Sequencing Technology to Solve a Complex Genomic Jigsaw Puzzle: The Agglutinin-Like Sequence (ALS) Genes of Candida tropicalis. Front. Microbiol. 11, 594531. doi: 10.3389/fmicb.2020.594531
Oh, S.-H., Smith, B., Miller, A. N., Staker, B., Fields, C., Hernandez, A., et al. (2019). Agglutinin-Like Sequence (ALS) Genes in the Candida parapsilosis Species Complex: Blurring the Boundaries Between Gene Families That Encode Cell-Wall Proteins. Front. Microbiol. 10, 781. doi: 10.3389/fmicb.2019.00781
Papon, N., Savini, V., Lanoue, A., Simkin, A. J., Crèche, J., Giglioli-Guivarc’h, N., et al. (2013). Candida guilliermondii: Biotechnological Applications, Perspectives for Biological Control, Emerging Clinical Importance and Recent Advances in Genetics. Curr. Genet. 59, 73–90. doi: 10.1007/s00294-013-0391-0
Paradis, E., Schliep, K. (2019). ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R. Bioinformatics 35, 526–528. doi: 10.1093/bioinformatics/bty633
Peters, B. M., Ovchinnikova, E. S., Krom, B. P., Schlecht, L. M., Zhou, H., Hoyer, L. L., et al. (2012). Staphylococcus aureus Adherence to Candida albicans Hyphae is Mediated by the Hyphal Adhesin Als3p. Microbiology 158, 2975–2986. doi: 10.1099/mic.0.062109-0
Riccombeni, A., Vidanes, G., Proux.Wéra, E., Wolfe, K. H., Butler, G. (2012). Sequence and Analysis of the Genome of the Pathogenic Yeast Candida orthopsilosis. PLoS One 7, e35750. doi: 10.1371/journal.pone.0035750
Rousseau, F., Schymkowitz, J., Serrano, L. (2006). Protein Aggregation and Amyloidosis: Confusion of the Kinds? Curr. Opin. Struct. Biol. 16, 118–126. doi: 10.1016/j.sbi.2006.01.011
Ruan, S. Y., Chein, J. Y., Hou, Y. C., Hsueh, P. R. (2010). Catheter-Related Fungemia Caused by Candida intermedia. Int. J. Infect. Dis. 14, 147–149. doi: 10.1016/j.ijid.2009.03.015
Salgado, P. S., Yan, R., Taylor, J. D., Burchell, L., Jones, R., Hoyer, L. L., et al. (2011). Structural Basis for the Broad Specificity to Host-Cell Ligands by the Pathogenic Fungus Candida albicans. Proc. Natl. Acad. Sci. U.S.A. 108, 15775–15779. doi: 10.1073/pnas.1103496108
Santos, M. A. S., Gomes, A. C., Santos, M. C., Carreto, L. C., Moura, G. R. (2011). The Genetic Code of the Fungal CTG Clade. C R. Biologies 334, 607–611. doi: 10.1016/j.cvri.2011.05.008
Schoch, C. L., Ciufo, S., Domrachev, M., Hotton, C. L., Kannan, S., Khovanskaya, R., et al. (2020). NCBI Taxonomy: A Comprehensive Update on Curation, Resources and Tools. Database 2020, baaa062. doi: 10.1093/database/baaa062
Senol Cali, D., Kim, J. S., Ghose, S., Alkan, C., Mutlu, O. (2018). Nanopore Sequencing Technology and Tools for Genome Assembly: Computational Analysis of the Current State, Bottlenecks and Future Directions. Brief. Bioinform. 20, 1542–1559. doi: 10.1093/bib/bby017
Shen, X.-X., Opulente, D. A., Kominek, J., Zhou, X., Steenwyk, J. L., Buh, K. V., et al. (2018). Tempo and Mode of Genome Evolution in the Budding Yeast Subphylum. Cell 175, 1533–1545. doi: 10.1016/j.cell.2018.10.023
Sherman, F., Fink, G. R., Hicks, J. B. (1986). Laboratory Course Manual for Methods in Yeast Genetics (Cold Spring Harbor, NY: Cold Spring Harbor Press).
Skrzypek, M. S., Binkley, J., Binkley, G., Miyasato, S. R., Simison, M., Sherlock, G. (2017). The Candida Genome Database (CGD): Incorporation of Assembly 22, Systematic Identifiers and Visualization of High Throughput Sequencing Data. Nucleic Acids Res. 45, D592–D596. doi: 10.1093/nar/gkw924
Soares, E. V. (2010). Flocculation in Saccharomyces cerevisiae: A Review. J. Appl. Microbiol. 110, 1–18. doi: 10.1111/j.1365-2672.2010.04897.x
Tavanti, A., Davidson, A. D., Gow, N. A., Maiden, M. C., Odds, F. C. (2005). Candida orthopsilosis and Candida metapsilosis spp. nov. Replace Candida parapsilosis Groups II and III. J. Clin. Microbiol. 43, 284–292. doi: 10.1128/JCM. 43.1.284-292.2005.
Vale-Silva, L. A., Sanglard, D. (2015). Tipping the Balance Both Ways: Drug Resistance and Virulence in Candida glabrata. FEMS Yeast. Res. 15:fov025. doi: 10.1093/femsyr/fov025
Verstrepen, K. J., Jansen, A., Lewitter, F., Fink, G. R. (2005). Intragenic Tandem Repeats Generate Functional Variability. Nat. Genet. 37, 986–990. doi: 10.1038/ng1618
Verstrepen, K. J., Reynolds, T. B., Fink, G. R. (2004). Origins of Variation in the Fungal Cell Surface. Nat. Rev. 2, 533–540. doi: 10.1038/nrmicro927
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS One 9, e112963. doi: 10.1371/journal. pone.0112963.
Wohlbach, D. J., Kuo, A., Sato, T. K., Potts, K. M., Salamov, A. A., Labutti, K. M., et al. (2011). Comparative Genomics of Xylose-Fermenting Fungi for Enhanced Biofuel Production. Proc. Natl. Acad. Sci. U.S.A. 108, 13212–13217. doi: 10.1073/pnas.1103039108
Wojciechowicz, D., Lu, C. F., Kurjan, J., Lipke, P. N. (1993). Cell Surface Anchorage and Ligand-Binding Domains of the Saccharomyces cerevisiae Cell Adhesion Protein Alpha-Agglutinin, a Member of the Immunoglobulin Superfamily. Mol. Cell. Biol. 13, 2554–2563. doi: 10.1128/mcb.13.4.2554-2563.1993
Xu, Z., Green, B., Benoit, N., Schatz, M., Wheelan, S., Cormack, B. (2020). De Novo Genome Assembly of Candida glabrata Reveals Cell Wall Protein Complement and Structure of Dispersed Tandem Repeat Arrays. Mol. Microbiol. 113, 1209–1224. doi: 10.1111/mmi.14488
Xu, Z., Green, B., Benoit, N., Sobel, J. D., Schatz, M. C., Wheelan, S., et al. (2021). Cell Wall Protein Variation, Break-Induced Replication, and Subtelomere Dynamics in Candida glabrata. Mol. Microbiol. 116, 260–276. doi: 10.1111/mmi.14707
Zhang, N., Harrex, A. L., Holland, B. R., Fenton, L. E., Cannon, R. D., Schmid, J. (2003). Sixty Alleles of the ALS7 Open Reading Frame in Candida albicans: ALS7 is a Hypermutable Contingency Locus. Genome Res. 13, 2005–2017. doi: 10.1101/gr.1024903
Zhao, S., Oh, S.-H., Jajko, R., Diekema, D. J., Pfaller, M. A., Pujol, C., et al. (2007). Analysis of ALS5 and ALS6 Allelic Variability in a Geographically Diverse Collection of Candida albicans Isolates. Fungal Genet. Biol. 44, 1298–1309. doi: 10.1016/j.fgb.2007.05.004
Zhao, H., Shen, Z.-M., Kahn, P. C., Lipke, P. N. (2001). Interaction of α-Agglutinin and a-Agglutinin, Saccharomyces cerevisiae Sexual Cell Adhesion Molecules. J. Bacteriol. 183, 2874–2880. doi: 10.1128/JB.183.9.2874-2880.2001
Zimmerman, L., Stephens, A., Nam, S. Z., Rau, D., Kübler, J., Lozajic, M., et al. (2018). A Completely Reimplemented MPI Bioinformatics Toolkit With a New HHpred Server at its Core. J. Mol. Biol. 430, 2237–2243. doi: 10.1016/j.jmb.2017.12.007
Zoppo, M., Di Luca, M., Franco, M., Rizzato, C., Lupetti, A., Stringaro, A., et al. (2020a). CpALS4770 and CpALS4780 Contribution to the Virulence of Candida parapsilosis. Microbiol. Res. 231, 126351. doi: 10.1016/j.micres.2019.126351
Zoppo, M., Fiorentini, F., Rizzato, C., Di Luca, M., Lupetti, A., Bottai, D., et al. (2020b). Role of CpALS4780 and CpALS0660 in Candida parapsilosis Virulence: Evidence From a Murine Model of Vaginal Candidiasis. J. Fungi 6:86. doi: 10.3390/jof6020086
Zoppo, M., Lombardi, L., Rizzato, C., Lupetti, A., Bottai, D., Papp, C., et al. (2018). CORT0C04210 is Required for Candida orthopsilosis Adhesion to Human Buccal Cells. Fungal Genet. Biol. 120, 19–29. doi: 10.1016/j.fgb.2018.09.001
Keywords: ALS genes, adhesion, comparative genomics, protein structure, fungi, repeated sequences
Citation: Oh S-H, Schliep K, Isenhower A, Rodriguez-Bobadilla R, Vuong VM, Fields CJ, Hernandez AG and Hoyer LL (2021) Using Genomics to Shape the Definition of the Agglutinin-Like Sequence (ALS) Family in the Saccharomycetales. Front. Cell. Infect. Microbiol. 11:794529. doi: 10.3389/fcimb.2021.794529
Received: 13 October 2021; Accepted: 09 November 2021;
Published: 14 December 2021.
Edited by:
Priya Uppuluri, University of California, Los Angeles, United StatesReviewed by:
Govind Vediyappan, Kansas State University, United StatesCi Fu, University of Toronto, Canada
Copyright © 2021 Oh, Schliep, Isenhower, Rodriguez-Bobadilla, Vuong, Fields, Hernandez and Hoyer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lois L. Hoyer, bGhveWVyQGlsbGlub2lzLmVkdQ==