 Cody Shaw
Cody Shaw Mallory C. Ringham2,3
Mallory C. Ringham2,3- 1Department of Chemistry, Stony Brook University, Stony Brook, NY, United States
- 2Department of Electrical and Computer Engineering, Stony Brook University, Stony Brook, NY, United States
- 3Ebb Carbon, Inc., South San Francisco, CA, United States
- 4Independent Researcher, Seattle, WA, United States
- 5Pacific Marine Environmental Laboratory, National Oceanic and Atmospheric Administration, Seattle, WA, United States
- 6Cooperative Institute for Climate, Ocean, and Ecosystem Studies, University of Washington, Seattle, WA, United States
- 7Google Inc., Seattle, WA, United States
- 8Department of Earth & Planetary Sciences, Yale University, New Haven, CT, United States
- 9Yale Center for Natural Carbon Capture, Yale University, New Haven, CT, United States
Mg(OH)2 holds potential as an alkalinity source for Ocean Alkalinity Enhancement (OAE). In this study, Mg(OH)2 was produced via precipitation from the alkalinity exchange of NaOH addition into brine. Mineralogy characterization (XRD, EDS, SEM) and dissolution kinetics (total alkalinity release rates) were used to investigate the adoption of seawater-precipitated Mg(OH)2 for OAE and compared to industrial sources. XRD revealed industrial sources contained a higher degree of crystallinity of 0.83–0.85 compared to 0.16–0.33 for seawater-precipitated paste. Mg(OH)2 at a higher degree of crystallinity (>80%) had significantly slower dissolution rates than Mg(OH)2 with a lower degree of crystallinity (<20%). A strong inverse relation between degree of crystallinity and dissolution rate was found for both seawater-precipitated and industrial sourced Mg(OH)2. Despite similar elemental composition to industrial sources, seawater-precipitated Mg(OH)2 exhibited lower crystallinity resulting in faster and more complete dissolution, suggesting a potential advantage over other alkalinity sources. Its seemingly tunable dissolution kinetics due to crystallinity provides an opportunity to optimize the material for OAE and carbon dioxide removal efficiency.
1 Introduction
There is scientific consensus that avoiding the most extreme effects of climate change will not only require decarbonization, but also the removal of carbon dioxide (CO2) from the atmosphere. Most scenarios that prevent more than 2 °C of warming require around 10–20 GtCO2/yr. of carbon dioxide removal (CDR) by 2,100 (IPCC, 2022; Rogelj et al., 2018). The scale of the problem means that an adoption of multiple methods will likely be required. Ocean alkalinity enhancement (OAE) refers to a promising set of CDR methods that leverage the natural role of the oceans to remove CO2 from the air (Renforth and Henderson, 2017; Committee on A Research Strategy for Ocean-based Carbon Dioxide Removal and Sequestration, Ocean Studies Board, Division on Earth and Life Studies, & National Academies of Sciences, Engineering, and Medicine, 2022). Alkalinity is a measure of the capacity of seawater to neutralize acids, in particular carbonic acid, which forms when CO2 reacts with water (Wolf-Gladrow et al., 2007). OAE methods increase the alkalinity of seawater, pulling additional CO2 from the atmosphere into the ocean, where it is stably stored as bicarbonate ions (HCO3−). As the ocean already contains a large inventory of bicarbonate (37,200 GtC, Keppler et al., 2020), the additional bicarbonate is not expected to appreciably change the ecological environment and is estimated to have a durability of 10,000 years (Renforth and Henderson, 2017) to 100,000 years (Cai et al., 2008). Multiple alkalinity sources have been proposed, including CaO (Kheshgi, 1995), Mg(OH)2 (Davies et al., 2018; Yang et al., 2023), crushed mafic and ultramafic rocks (Hangx and Spiers, 2009; Köhler et al., 2010; Meysman and Montserrat, 2017; Montserrat et al., 2017; Rigopoulos et al., 2018; and Schuiling and de Boer, 2011), limestone-derived Ca(OH)2 via electrochemistry (Rau, 2009), and aqueous NaOH generated electrochemically from aqueous brine streams (Eisaman et al., 2023; House et al., 2007; Schiffer et al. 2024).
Electrochemically generated aqueous NaOH is a promising approach because it avoids uncertainties in the dissolution rates of mined minerals (Hangx and Spiers, 2009; Montserrat et al., 2017) and also avoids environmental concerns regarding trace impurities released from geologically sourced alkalinity (Bach et al., 2019). However, aqueous alkalinity is gravimetrically and volumetrically less dense (0.5–1 mol(alk)/kg) than solid sources of alkalinity (20–25 mol(alk)/kg), so the costs and emissions of alkalinity transport tend to restrict NaOH alkalinity dispersal to coastal locations rather than the offshore ship-based dispersal that is possible with solid forms of alkalinity. The rate of alkalinity dispersal from any single location will be limited by permitted bounds of key metrics such as maximum allowable pH (He and Tyka, 2023). As an example, the addition of alkalinity in the surface ocean locally and temporarily increases seawater pH, with the pH increase being maximal at the point of introduction within the mixing zone and then decreasing as the added alkalinity mixes with untreated ocean water. OAE practitioners also aim to keep the carbonate chemistry in a range that avoids runaway precipitation of calcium carbonate (CaCO3; Moras et al., 2022; Fuhr et al., 2021; Suitner et al., 2024), which would tend to reverse the goal of OAE by removing net alkalinity (Zeebe and Wolf-Gladrow, 2001). The maximum rate of alkalinity addition at a given location will then be determined by the dissolution kinetics of mineral sources and the rate at which alkalinity in the mixing zone is diluted by the surrounding ocean. With this limitation, the maximum rate of OAE is expected to scale with the area over which the alkalinity can be added, and therefore also scale with the transportability of the alkalinity source.
Here an approach called “alkalinity exchange” is proposed that will allow OAE employing aqueous alkalinity to continue to grow in scale even once it starts to run up against practical limitations imposed by the need to avoid extreme chemical changes. The general concept of alkalinity exchange is to take advantage of the presence of divalent cations, like Mg2+ and Ca2+ in seawater and waste brines such as reverse osmosis concentrate (ROC), to precipitate alkalinity-containing solids, such as Mg(OH)2, upon addition of aqueous NaOH. This effectively exchanges one form of alkalinity (dilute aqueous NaOH) for another form (concentrated solid Mg(OH)2) that is more gravimetrically and volumetrically dense: from a molecular point of view, the hydroxide bound in the solid Mg(OH)2 is the same hydroxide that was added as NaOH. The Mg(OH)2 can then be economically dispersed further out at sea via ship, helping to overcome limitations on the rate at which alkalinity can be dispersed near the coast (He and Tyka, 2023). Mg(OH)2 also has a lower pKA than NaOH, lowering the risk of extreme perturbations to seawater pH compared to strategies that release NaOH directly. Alkalinity exchange is especially attractive because in some approaches to the electrochemical generation of alkalinity, the incoming brine must be pretreated to reduce the concentration of divalent cations such as Mg2+ and Ca2+ to prevent unwanted scaling in the electrochemical system (De Lannoy et al., 2018; Eisaman et al., 2012; Eisaman et al., 2018; Eisaman et al., 2023; Schiffer et al. 2024). In this case, alkalinity exchange can be used to partially pretreat the incoming brine by reducing the Mg2+ ion concentration to levels that are acceptable for input into the electrochemical system. While the Ca2+ ion concentration will still need to be reduced, the removal of Mg2+ during alkalinity exchange significantly reduces the cost of pretreatment. This process is conceptually illustrated in Figure 1A.
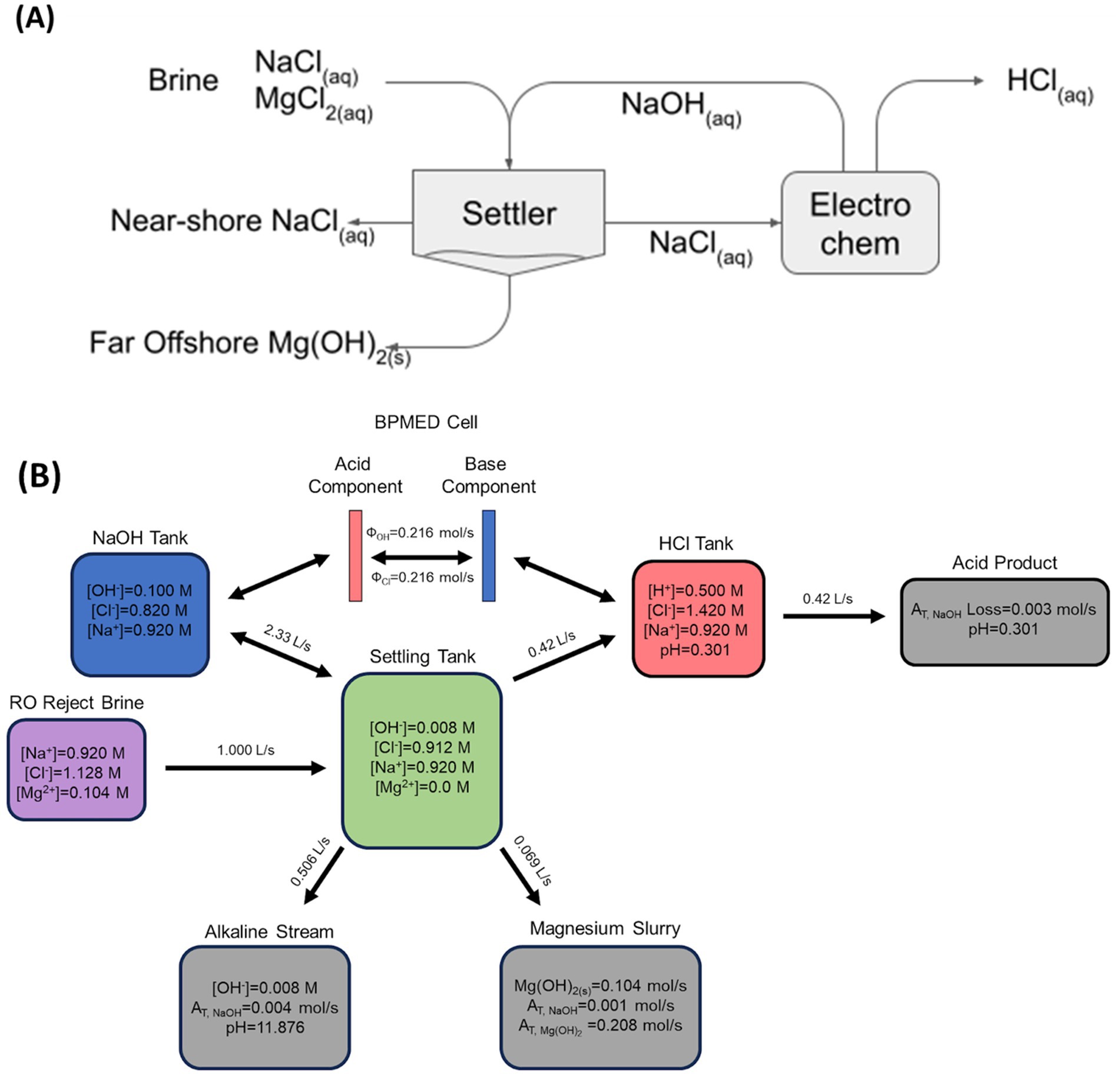
Figure 1. Conceptual (A) and mass-balanced (B) flow in an alkalinity exchange process, where generated electrochemical alkalinity is transferred to Mg(OH)2 which is used as a carrier.
In Figure 1B, a simulated balanced mass flow of a bipolar membrane electrodialysis (BPMED) alkalinity exchange reactor is shown (Eisaman et al., 2012). The initial brine was assumed to have twice the ionic concentration of seawater. Additional Cl− ions were introduced to adjust for charge differences with Mg2+. The desired Mg(OH)2 slurry output was 3 mol AT/kg based on water content. The BPMED system uses energy to continuously exchange and separate Cl− and OH− creating basic and acidic streams which enter the NaOH and HCl Tanks. The generated NaOH is then pumped into the “Settling Tank” where it meets fresh RO reject brine (which is rich in Mg2+). Precipitation of Mg(OH)2 occurs, which removes essentially all Mg2+ in solution together with a stochiometric amount of OH−. A pH below 14 is maintained to prevent Ca(OH)2 precipitation which would reduce the yield of Mg(OH)2. CaCO3 precipitation is limited by the availability of CO32− in the solution. The values displayed in the “Settling Tank” represent the ion concentrations after precipitation. The Mg(OH)2 or “Magnesium Slurry” is continuously removed from the tank and forms the desired solid alkalinity carrier. Excess liquid also leaves the settling tank in order to balance the overall volume flows. On the other side of the BPMED stack, the acidic effluent is circulated into the “HCl Tank.” The HCl byproduct is then removed for beneficial use in other processes such as enhanced mineral weathering (Li and Hitch, 2015; Zhang et al., 2024), algal cultivation (Hibbeln et al., 2024), and upgrading of concrete aggregates (Jin et al., 2025). To maintain a constant pH in the HCl tank, “clarified” brine from the “Settling Tank” is pumped to mix with the acidic solution to generate the acidic waste stream. Instead of using the clarified brine, as shown here, it is also possible to use a different source of water for this purpose.
Understanding the Mg(OH)2 precipitation process and the kinetics of its subsequent dissolution in seawater when used as an alkalinity source for OAE is of critical importance for an OAE approach based on alkalinity exchange. Precipitation of Mg(OH)2 from brines using NaOH has been explored as a means of mineral extraction (Vassallo et al., 2021) and pretreatment (Ayoub et al., 2014; El-Manharawy and Hafez, 2003), but not in the context of its subsequent dissolution in seawater. Re-dissolution kinetics are critical for OAE approaches because sinking particulates that dissolve slowly release the alkalinity deeper in the water column, or potentially not at all, thereby delaying or decreasing the impact of the mineral addition on surface-ocean air-sea exchange of CO2. On the other extreme, highly soluble chemicals like CaO can dissolve rapidly and lead to large unwanted perturbations in surface water chemistry. The dissolution speed of magnesium hydroxide particles in water depends on the particle size distribution, the hydration state, and the surface dissolution rate. Dissolution is highly pH and temperature dependent and dissolution surface rates at seawater pH of 8.1 have been reported by a number of studies: Pokrovsky and Schott (2004) reports a surface dissolution rate of ground Brucite, 100–200 μm diameter particles, using a flow reactor as R = 1 × 10−8 mol/m2/s at pH 8.1 at 25 °C. Bharadwaj et al. (2013) reported dissolution of lab grade Mg(OH)2, 6 μm diameter particles, in a stirred, pH-controlled reactor, once converted to surface rate, as R= 𝑘𝑟r0p/M𝑟=4.07 ×10−6 mol/m2/s (r0 is initial particle radius, p is density, M𝑟 is molar mass) at pH 8.6 at 22 °C. Vermilyea (1969) finds rates from 1.5 × 10−5 to 3 × 10−7 mol/m2/s with 20 mm diameter particles. Kudoh et al. (2006) found the rate to be R = 5.0 × 10−9 mol/m2/s at pH 8 and 25 °C. These rates (Figure 2) span nearly 3 orders of magnitude suggesting that factors other than size, temperature, and pH may be at play, such as the degree of crystallinity and/or the presence of crystal defects. In this paper we examine the re-dissolution of precipitated Mg(OH)2, and the dependence of its material properties.
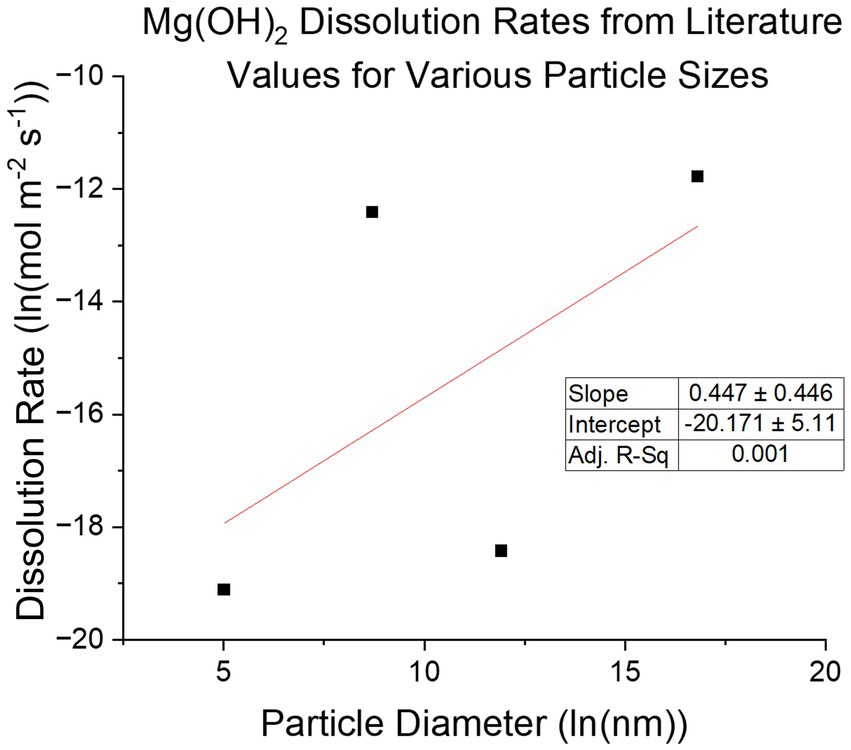
Figure 2. Compilation of various literature reported dissolution rates of Mg(OH)2 with respective particle sizes reported. There is no strong correlation between dissolution rate and particle size, suggesting other properties of the material are influential.
In this study, the role of morphological changes to Mg(OH)2 is investigated by altering the time that the material is left in its saturated supernatant after precipitation. By storing it in this type of environment, changes in chemical composition and crystallinity could occur that impact the kinetics of dissolution. This tests the sensitivity of Mg(OH)2 precipitation to this delay time as well as the ability to control the subsequent dissolution characteristics of the product to meet the needs of OAE. We also separately test the impacts of stirring during Mg(OH)2 precipitation and its subsequent dissolution. This research aims to inform potential future efforts to effectively manufacture and store Mg(OH)2 as a transportable alkalinity source for use in OAE.
2 Methods
2.1 Industrial Mg(OH)2 sources
Mg(OH)2 from several manufacturers was used for comparison with seawater-precipitated Mg(OH)2, described below. Lab grade commercial Mg(OH)2 was sourced from Sigma-Aldrich (“Industrial A”: Magnesium Hydroxide, BioUltra, >99.0%, Source#: BCCH8744) and Thermo Scientific (“Industrial B”: Magnesium Hydroxide, 95% pure, Lot#: A0451713). These materials are a fine, white powder. “Mined Powder” was sourced from Garrison Minerals AlkapHixⓇ, an industrial manufacturer of Mg(OH)2 materials. It is a white powder but noticeably more coarse than the Industrial sources stated as 97–100% Mg(OH)2 and reported specific gravity of 1.42 g/cm3. “Mined Slurry” was also sourced from Garrison Minerals AlkapHixⓇ, which is Mg(OH)2 in a semi-liquid state containing 50–60% Mg(OH)2 and 40–50% H2O with a pH of ~10 and reported specific gravity of 1.42 g/cm3. Its appearance was pinkish-white and viscous. Common industrial production of Mg(OH)2 is precipitation by addition of CaO into filtered seawater or MgCl2 salt into NaOH (Seeger et al., 2011; Song et al., 2013). Other processes include hydration of MgO via exothermic reaction in H2O (Wright, 2022; Fontana et al., 2023). Specific production methods for each source in this study are not known.
2.2 Seawater and brine production
This study leveraged seawater collected from experimental tanks used by Ringham et al. (2024), in which natural seawater was pumped from Flax Pond, part of a salt marsh tidal wetlands on Long Island Sound in Stony Brook, NY, United States. Natural seawater was UV-treated and filtered before experiments with a 0.45 μm filter capsule to remove large debris and to reduce any biological presence. Temperature and salinity of this seawater were measured with a Sea-Bird 38 Digital Oceanographic Thermometer and a Sea-Bird 45 MicroTSG Thermosalinograph. pH was measured using both a SAMI-pH and a Thermo-Scientific Orion Star A211 probe. AT (total alkalinity) was determined with an open-cell potentiometric titrator. DIC was measured via a coulometer and a single-operator multiparameter analyzer at NOAA Pacific Marine Environmental Laboratory (NOAA/PMEL). Temperature, salinity, pH, and AT were measured continuously while DIC was discreetly measured. Seawater salinity was increased by simple ambient evaporation until a target salinity was reached to create a brine used for precipitation of Mg(OH)2. Brine was chosen as the source for Mg rather than “normal” seawater to mimic the concentrations found within reject brine from desalination plants. Utilizing brine from these sources provides more Mg at lesser volumes, while pre-treating reject brine by removing the Mg ions which is required before brine usage in electrochemical processes. Salinity was determined using a handheld salinity refractometer. The average initial properties of seawater used in these experiments are provided in Table 1. Seawater properties and data for all experiments completed in this study can be found in published dataset (Shaw, 2025).
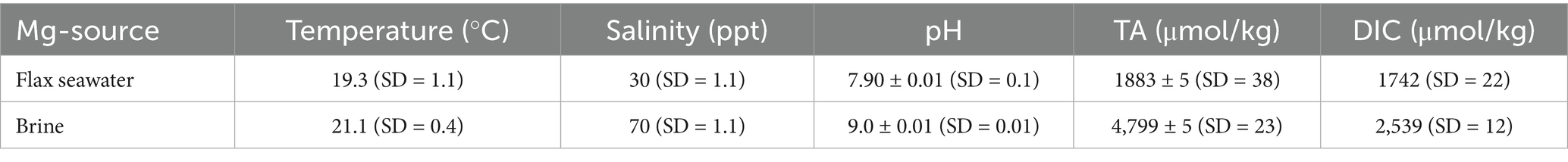
Table 1. Average properties of collected seawater from Flax Pond Laboratory and brine.
2.3 Precipitation of Mg(OH)2
Mg(OH)2 for “stirred” conditions was precipitated by adding 1.57 mL of prepared brine into 0.75 mL 0.5 M NaOH (Sigma-Aldrich; Yang et al., 2023). For “non-stirred” conditions 11.45 mL brine was mixed into 5.5 mL 0.5 M NaOH (Karl, 1968). The precipitated Mg(OH)2 and supernatant were not filter separated to ensure no material was lost to prevent any alkalinity yield error.
To investigate the influence of delay time, the brine and NaOH mixture was kept in a series of 50 mL closed falcon tubes undisturbed for 0, 2, 16, 24, 48, 96, and 336 h (depending on the delay time of the specific sample) and wrapped in film to further reduce gas exchange. Delay time was defined as the time at which the Mg(OH)2 (and supernatant) remained closed between initial precipitation and addition to seawater for dissolution. Over time, the precipitated Mg(OH)2 settled to the bottom of the tube with a distinct, separate supernatant (Figures 3A–C). Calculations were made to estimate the amount of CO2 within the headspace of the falcon tubes (See in Supplementary material). Results showed the CO2 concentration in the headspace being 20-220x smaller than the expected OH− after precipitation. Therefore, the effects of CO2 absorption into the supernatant are non-significant.
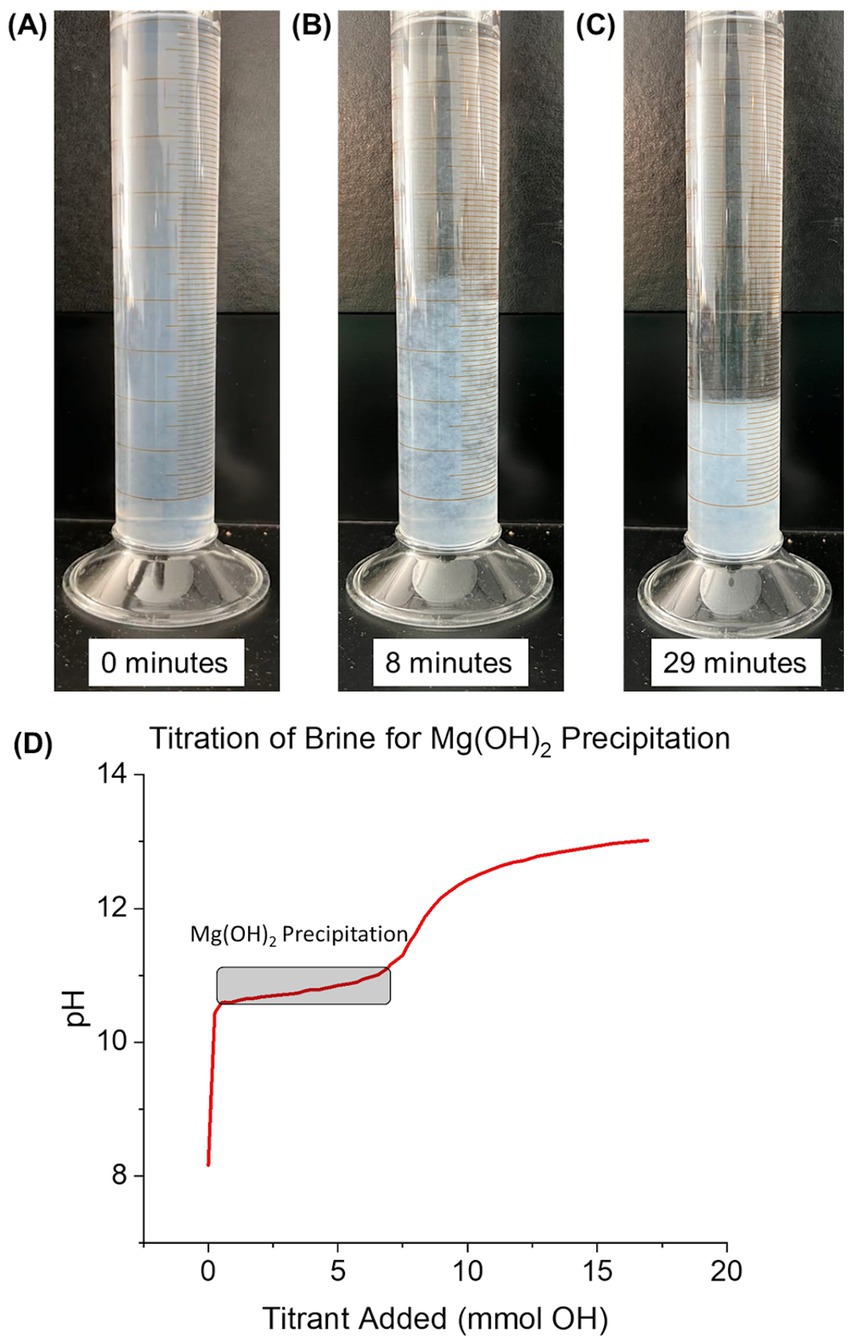
Figure 3. Visible case of seawater-precipitated Mg(OH)2 settling over time while in supernatant. Progress of settling after precipitation is (A) 0 min, (B) 8 min, and (C) 29 min. (D) Titration curve of 100 mL of seawater for the precipitation of Mg(OH)2. Titrant was 0.5 M NaOH and added until pH reached 13. Mg(OH)2 precipitation began at a pH of ~10.5, consuming a total of 5.5 mmol of OH−.
For continuously mixed Mg(OH)2 the entire precipitation and delay time sequence was performed in a 20 mL flat bottom glass bottle with a miniature stir bar added before sealing. The mixture was placed on a stir plate and was mixed for the duration of the delay time before continuing with the alkalinity dissolution experiment.
For tests using industrial Mg(OH)2, 11 mg of Mg(OH)2 was added to the beakers before continuing the alkalinity dissolution experiment. This amount was chosen as it holds a comparable total alkalinity to the seawater-precipitated. To compare industrial Mg(OH)2 to various delay times of seawater-precipitated Mg(OH)2, a solution of 1.57 mL distilled water with an adjusted salinity (using NaCl and handheld salinity refractometer) of 75 was mixed with 20 μL of 0.5 M NaOH. This solution used minimum NaOH to produce a pH of 10.3 to ensure no industrial Mg(OH)2 dissolved before dissolution. Then 11 mg of industrial Mg(OH)2 was added before being stored for the desired delay time.
2.4 Measuring Mg(OH)2 dissolution (stirred-conditions—no dead zones)
The experimental procedure for Mg(OH)2 dissolution contained conditions listed in Table 2. 1 L of UV-treated seawater was placed in each of 4 borosilicate beakers (3 replicates and 1 control). Each beaker was equipped with a standard glass electrode to measure pH and temperature (Thermo Scientific Orion ROSS Ultra pH/ATC Triode combination electrode, 8157BNUMD). Samples were drawn from seawater stock to measure total alkalinity for the initial conditions. Initial salinity was measured. Ideal conditions (known as “stirred-conditions” in this study) consisted of the use of a stir bar and ~600 RPM that allowed for homogenizing the solution, which prevented dead zones.
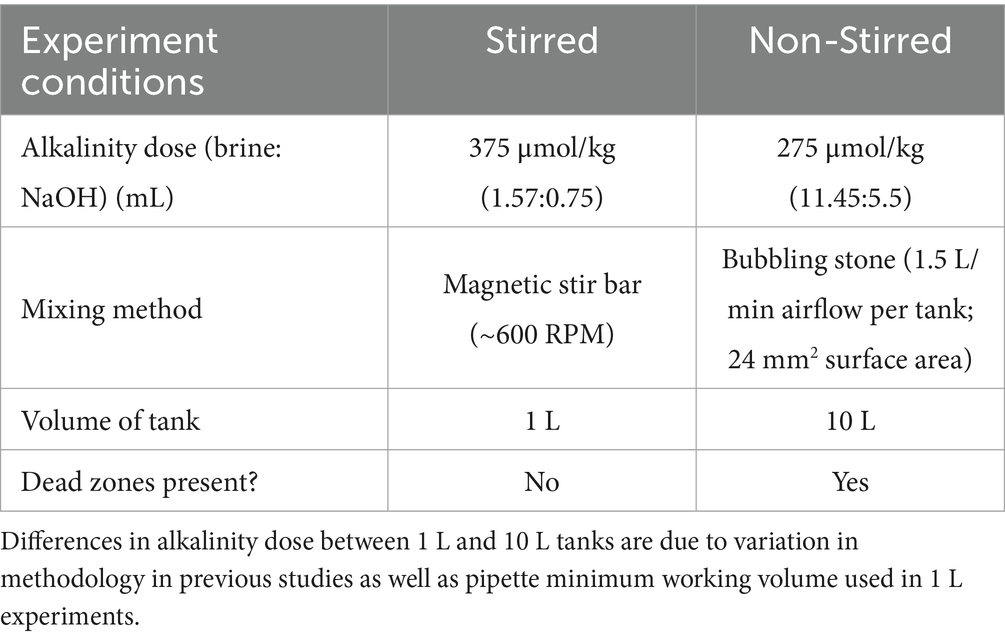
Table 2. Experimental tank set-up for “Stirred” and “Non-Stirred” conditions.
Measurements were collected for baseline conditions before alkalinity additions. One beaker was selected as the control, to which no alkalinity was added. The seawater was allowed to mix for ~30 s to allow for baseline data collection and equilibration, then the contents of the 50 mL falcon tubes from the precipitation step (precipitated Mg(OH)2 and supernatant) were stirred to homogenize and added to the remaining 3 replicate beakers. The tubes were rinsed with distilled water to ensure all alkalinity was transferred into the beaker keeping total DI water use below 2 mL to ensure minimal impacts to salinity and seawater properties. Experiments proceeded for 35 min to observe the initial and bulk dissolution kinetics of the Mg(OH)2. Seawater samples were then collected from each beaker and AT was measured to determine the efficiency of alkalinity release during dissolution. To account for evaporation and salinity changes, the beakers were covered to decrease surface area of water to the atmosphere and AT calculations were normalized to salinity at the end of the experiment (Friis et al., 2003).
2.5 Measuring Mg(OH)2 dissolution (non-stirred-conditions—dead zones)
Cylindrical tanks (10 L) were filled with seawater stored at room temperature (20 ± 2 °C) for the duration of the experiment and subjected to conditions listed in Table 2. Tanks were wrapped in aluminum foil to reduce light exposure and covered with lids to limit evaporation and possible introduction of foreign particles. The filled tanks were aerated with an aquarium air bubble strip for 24 h to equilibrate with the atmosphere before any alkalinity additions or measurements began. Air flow was 1.5 L/min per tank and air stone surface area was 20 × 1.2 mm. This provided an air velocity of 1.04 m/s with bubbles 0.1–0.2 mm in diameter rising at 0.01 m/s. The surface area of the bottom of the tank was 491 mm2 with the air stone comprising ~4.9% of total area and therefore with the remaining 95.1% of tank bottom being classified as mixing deadzones. The precipitate and supernatant in the Mg(OH)2 falcon tubes were shaken to create a homogenous mixture, then quickly poured into the tank and rinsed with <1 mL DI H2O. This mixture was initially stirred into tanks to ensure better dissolution. Because bubbling was the only active mixing for this experiment, dead zones were present along the bottom of the tanks and around the air bubble strip; therefore this condition is described as non-stirred mixing. Non-stirred mixing provides a closer representation of real-world applications of OAE as ocean mixing is not homogenous.
Control tanks with no alkalinity additions and tanks with only 5.5 mL 0.5 M NaOH (aq) were measured. Both cases were used as baseline data to correct for variation in seawater and NaOH solution concentration differences. Measurements of pH and alkalinity were taken before additions to serve as the initial conditions of each tank, followed by 2 measurements after alkalinity addition on the same day (within 2 min after addition and approximately 2 h after, respectively). Afterward, daily measurements were collected for 4 days.
2.6 Total alkalinity determination
AT and pH samples were collected using a 30 mL disposable plastic syringe with a 0.45 μm glass fiber filter (GF/F) to remove any undissolved particles that would interfere with sample analysis. A series of 3 replicates were analyzed for each sample to collect an average AT and pH. An open-cell potentiometric titrator (Dickson et al., 2007) was used to track the titration of 15 mL seawater samples with a dilute HCl solution following a modified Gran titration procedure (Ringham et al., 2024; Song et al., 2013; Wang et al., 2023). This system used a Thermo Scientific Orion ROSS Ultra pH/ATC Triode combination electrode (8157BNUMD) calibrated using three buffer solutions (pHNBS 4.01, 7, and 10.01), a dilute HCl solution (~0.1 M in 0.7 M NaCl), and Certified Reference Material seawater standards sourced from the Dickson laboratory at Scripps Institution of Oceanography. The precision of AT measurements was ±5–10 μmol/kg (Ringham et al., 2024). Temperature and pH was measured via a Thermo-Scientific Orion Star A211 probe that was calibrated each day before tank measurements using a three-pH buffer system (4.01, 7.00, and 10.01).
2.7 Precipitate analysis
Mg(OH)2 was precipitated with 5.5 mL of 0.5 M NaOH and 11.45 mL of brine to produce enough solid material for precipitation analysis using the same NaOH to brine ratio as was used in dissolution experiments. For each delay time, 3 samples were created to determine average properties. The precipitate was separated from the supernatant using a 0.45 GF/F filter, then thoroughly rinsed with DI water 3 times to wash away any solid NaCl to improve solids characterization (Song et al., 2013; Ziegenheim et al., 2020). Washing the Mg(OH)2 with DI water posed no risk to the material properties due to the small volume of water used for rinsing and the low solubility of Mg(OH)2 (0.0069 g/100 g H2O) in water compared to NaCl (36.0 g/100 g H2O; Hodgman, 1952). The supernatant was then discarded. The Mg(OH)2 paste was transferred and closed in an airtight container along with the wet filter paper to prevent drying during storage for wet material characterization. For dried analysis, the paste was transferred to a watch glass, where it was placed in an oven at 90 °C until completely dried (~1–2 h). The solid was crushed using a mortar and pestle into a uniform Mg(OH)2 powder then stored in a sealed container until analysis.
Characterization was performed at the Center for Functional Nanomaterials (CFN) at Brookhaven National Laboratory. Surface images of samples were obtained using a Hitachi 4800 Scanning Electron Microscopy (SEM) at 5 kV by placing a portion of the dried powder onto carbon conductive tape adhered to the instrument’s sample plate, ensuring all powder was pressed onto tape to prevent loose material within the instrument. The same procedure was used for the JEOL JSM-7600F equipped with Oxford Instruments’ attachment for elemental dispersive X-ray spectroscopy (EDS). Elemental composition scans were done at 2 different sites within each sample to ensure a representative analysis. External standard calibration was done by creating 5 mixtures of Mg(OH)2 and CaCO3 powders to create a series of known wt% of Mg (0, 8.17, 21.04, 33.18, and 41.68 wt%) comparing to the measured Mg wt% of each sample. The calibration equation was then used to adjust measured values of all unknown samples.
Phase composition was measured from Rigaku SmartLab X-ray Diffraction (XRD) with Cu K⍺ (1.5406 Å) radiation. Analysis software (SmartLab Studio II) was used containing database of standard reference diffractograms of each phase for identification of samples. Dried powder was placed onto a silicon zero-diffraction plate until a 1 mm diameter circle was formed at minimum. The powder was left loose on the plate which was then carefully moved into the SmartLab XRD. 1D scans were conducted from 10 to 80° 2θ at 4°/min with a step size of 0.0400° and count time of 0.65 s per step. 2 scans were completed on each sample to collect an average diffractogram. Typical instrument broadening ranged from 0.025–0.05°. For Mg(OH)2 paste characterization, a holding cell was created by building a 3 mm tall 3 cm x 2 cm wall of silicone on top of a quartz crystal slide. The paste, collected after filtration and washing but before drying, was placed into the cell, making sure the Mg(OH)2 was level with the silicone walls. Then the same XRD procedure was used. To account for instrument broadening, an external standard (pure Si) was analyzed using the same procedure as done for our samples. A calibration curve of 2θ vs. FWHM of the standard was created which was then applied to the data from samples to adjust the measured FWHM. The adjusted values represent peaks where broadening is only from the crystal structure and not instrument contributions.
Various physical characteristics of the seawater-precipitated Mg(OH)2 were calculated for both powder and paste forms of Mg(OH)2 using XRD. The degree of crystallinity was calculated based on the ratio of the area under each major XRD peak divided by the total area under all XRD peaks. Using Williamson-Hall Plot Analysis (Williamson and Hall, 1953), the broadening of the major peaks of Mg(OH)2 was used along with the scattering angle to calculate the average crystallite size. The d-spacing of the samples were also calculated from XRD data (Bragg, 1913).
Fourier-Transform Infrared Spectroscopy (FTIR) spectra of seawater-precipitated Mg(OH)2 was collected using a Nicolet 6700 FT-IR spectrometer. Scans were from 650 to 4,000 cm-1 on an ATR base with diamond crystal (Data found in Supplementary material). Dynamic Light Scattering (DLS) was attempted however Mg(OH)2 samples were incompatible with a more detailed explanation in the Supplementary material.
3 Results
3.1 Seawater-precipitated Mg(OH)2
A titration curve for seawater-precipitated Mg(OH)2, measured by titrating seawater with 0.5 M NaOH, is shown in Figure 3D. 100 mL of UV-treated seawater from Flax Pond was titrated with 0.5 M NaOH to observe the precipitation reactions at elevated pH. The pH value rose sharply during initial NaOH additions until leveling off around 10.5 after 1.2 mmol NaOH had been added. The shallow-sloped plateau in pH versus NaOH addition indicates the precipitation of Mg(OH)2; most of the additional OH− ions were precipitated as Mg(OH)2 rather than increasing the solution pH during this phase (Equation 1).
The starting point of Mg(OH)2 precipitation was similar to previous studies including Battaglia et al. (2022), Phillips et al. (1977), Yousefi et al. (2017), and Pilarska et al. (2017). The formation of Mg(OH)2 was observed as the change in solution turbidity from clear to cloudy white. The pH remained relatively stable as NaOH was added until all free Mg2+ was removed from the solution via the precipitation of solid Mg(OH)2. The removal of Mg2+ required an addition of 5.5 mmol NaOH to the seawater sample. Once all Mg2+ was removed from the solution by Mg(OH)2 precipitation, the pH began to rise again until another plateau was reached after a total of 9.25 mmol NaOH had been added. The pH change began to slow at a pH of ~12.1, indicating Ca(OH)2 formation and therefore the removal of Ca2+ from solution (Equation 2; Carrotte, 2004):
Using this approach, all free Mg2+ in seawater can be selectively removed without removing Ca2+ in the form of Ca(OH)2. This sequential separation of ions allows for the creation of Mg(OH)2 with a purity of >98% while requiring minimal refining methods (the precipitate was only washed with DI water to remove NaCl). The purity of the Mg(OH)2 makes it a viable alkalinity source for OAE as all initial alkalinity used for precipitation was successfully transferred to Mg(OH)2 and not other species with different dissolution kinetics. The high efficiency of alkalinity production can save on costs as well as skipping further purification methods to prevent introducing less soluble and more basic hydroxides into the ocean (such as Ca(OH)2).
3.2 Industrial Mg(OH)2 characterization
XRD diffraction patterns of Industrial A (Mg(OH)2, Sigma-Aldrich), Industrial B (Mg(OH)2, ThermoFisher), Mined Powder (Mg(OH)2, Garrison Minerals), Mined Slurry (Mg(OH)2, Garrison Minerals), and seawater-precipitated Mg(OH)2 (0 Hour Delay Time) were collected to compare our seawater-precipitated material to industrial-produced Mg(OH)2 (Figure 4A). The industrial sources contained no impurities above the background noise and XRD showed sharp peaks indicating a high degree of crystallinity that agrees with previous patterns of brucite (Lv et al., 2024; Turek and Gnot, 1995). This is compared to the seawater-precipitated Mg(OH)2 which contained broader XRD peaks, but in the same position as the industrial sources. The broader peaks indicate that seawater-precipitated Mg(OH)2 is more amorphous than that obtained from industrial sources.
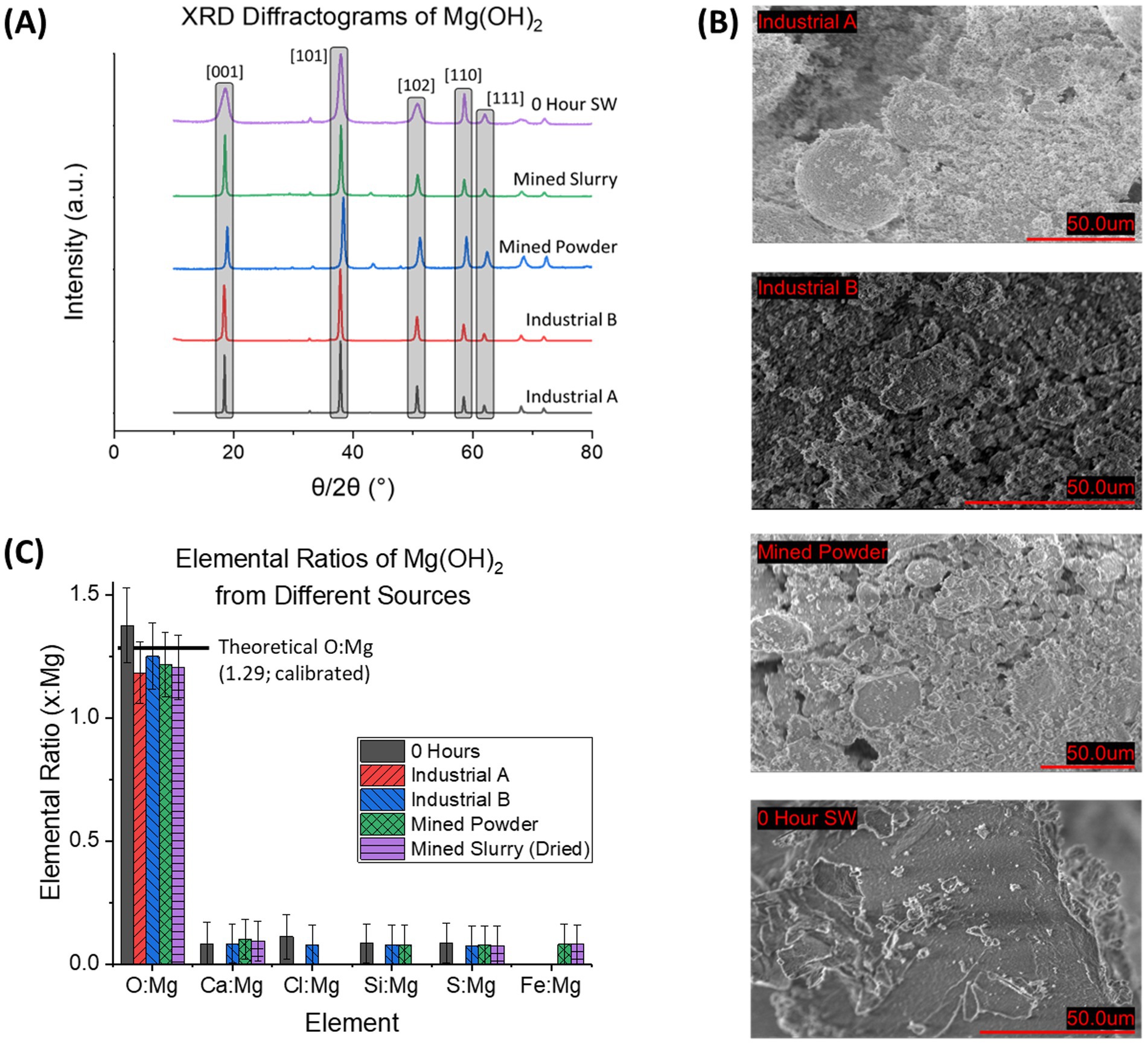
Figure 4. Characterization of the Industrial Sources of Mg(OH)2 with seawater-precipitated Mg(OH)2. (A) XRD diffractogram of Industrial Sources of Mg(OH)2 alongside 0 Hour delay time seawater-precipitated Mg(OH)2. Patterns reveal that Industrial sources are more crystalline than seawater-precipitated based on sharp, narrow peaks. (B) SEM images of each Industrial source and 0 Hour seawater-precipitate. In comparison to the seawater based sample, all others exhibited more granular materials, seen by the distinct shapes in the images. This is compared to the seawater-precipitate sample, which is more or less a solid piece of material that is only broken due to mechanical milling. (C) Elemental analysis from EDS of the 4 samples is shown by reporting elemental ratios. Theoretical O: Mg is based on stoichiometric calculations. All samples contained similar ratios for all elements. Ca: Mg is the largest impurity with Mined Powder containing 0.10 ± 0.08, Mined Slurry with 0.09 ± 0.08, Industrial B with 0.08 ± 0.08, Industrial A having 0, and 0 Hour seawater-precipitated with 0.08 ± 0.08.
SEM images (Figure 4B) further revealed the difference between the source of Mg(OH)2, with industrial brucite containing granular formations in the mean diameter of 6.43 ± 1.6 μm2 (Industrial A), 0.594 ± 0.18 μm2 (Industrial B), 1.47 ± 0.61 μm2 (Mined Powder). The seawater-precipitated Mg(OH)2 contained no distinct shapes. The seawater-precipitated Mg(OH)2 is a thick white paste after filtration. Once dried, it is a hard, brittle solid that is milled before characterization. The industrial sources of brucite were purchased as a fine white powder so no additional milling was necessary. The mined slurry was a viscous pinkish material that was stated to contain 45–55% H2O with the remainder being Mg(OH)2.
Elemental composition information was determined by EDS for each of the sources of Mg(OH)2 (Figure 4C). The relationship of the concentration of selected elements (O, Ca, Cl, Si, S, and Fe) compared to the detected amount of Mg was used instead of absolute wt% to avoid the uncertainty associated with wt% in EDS analysis. Both Al and C were omitted due to interference from sample holder and carbon tape. The theoretical ratio of O: Mg in pure Mg(OH)2 was calculated to be 1.29 after adjusted for EDS calibration (1.32 before adjustment). The average O: Mg value for all industrial sources was 1.21 ± 0.03 compared to 1.38 ± 0.15 for 0 h seawater-precipitated. The mined powder and slurry had a Fe: Mg of 0.08 ± 0.08 while all other sources had 0. Ca: Mg was found to be 0 for Industrial A, 0.08 ± 0.08 Industrial B, 0.10 ± 0.08 Mined Powder, 0.09 ± 0.08 Mined Slurry, and 0.08 ± 0.08 in 0 h seawater-precipitated.
Within the industrial sources, the mined powder and slurry exhibited insignificant but measurable levels of iron, which can be a result of the mining process itself when using certain magnesium-rich minerals such as serpentine (Teir et al., 2007). Multiple of the sources contained trace amounts of Ca which can be associated with CaCO3 which is often found with Mg(OH)2 due to lower precipitation pH threshold in solution (Koutsoukos and Kontoyannis, 1984).
3.3 Seawater-precipitated delay time characterization
Experiments were performed to determine if the time that the Mg(OH)2 was held in suspension before being redissolved in a larger volume of seawater (delay time) affected the dissolution kinetics of the Mg(OH)2. Figures 5A,B shows the normalized XRD diffractograms of seawater-precipitated Mg(OH)2 for different delay times for powder (A) and paste (B) forms. For both forms, the major diffraction angle peaks associated with Mg(OH)2 are labeled: [001] = 18.5°, [101] = 37.5°, [102] = 50.6°, [110] = 58.4°, and [111] = 61.8° (where the numbers in the square brackets denote the specific crystal lattice orientation (Miller indices) found with the material). NaCl appeared in various samples, however this was due to inadequate washing after filtration and not related to the purity of the Mg(OH)2 itself. NaCl presence had no impact on the characterization or dissolution kinetics due to the solubility differences (NaCl is immediately redissolved) once in seawater and only trace amounts are present (Haynes, 2014). The data show a sharpening of the diffraction peaks the longer the material is allowed to sit in suspension. This is consistent with a model where the initial sudden increase in pH first leads to the creation of relatively amorphous or nanocrystalline material with poorly formed crystals. Over time, these crystals reform and grow more regular, with the diffraction pattern gradually resembling that of industrial Mg(OH)2 samples.
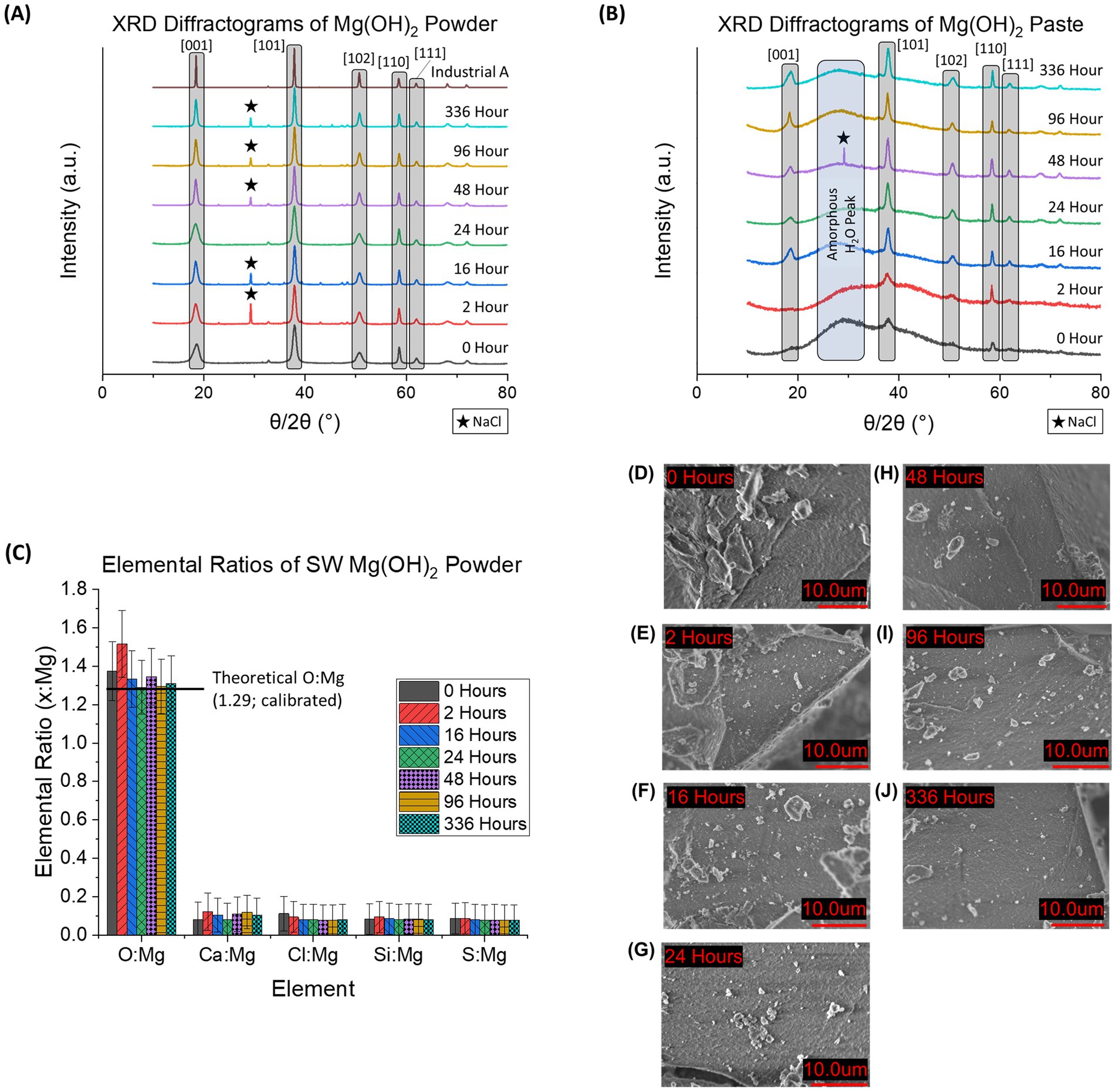
Figure 5. (A) XRD diffractogram of seawater-precipitated Mg(OH)2 characterized as a dried fine powder and (B) as a wet paste. The five major peaks associated with Mg(OH)2 are highlighted in gray. Presence of NaCl impurities is denoted by a star (★). In 4B, the amorphous region associated with water content within the Mg(OH)2 is marked by a blue column. (C) EDS data provides elemental ratios of 6 major elements (oxygen (O), magnesium (Mg), calcium (Ca), chloride (Cl), silicon (Si), and sulfur (S)) of powdered, seawater-precipitated Mg(OH)2 at each delay time used in this study. Theoretical O: Mg is based on stoichiometric calculations and were found in good agreement with the measurements. All samples contained Ca, which ranged from 0.08 ± 0.08 to 0.12 ± 0.09 Ca: Mg indicating possible presence of CaCO3. (D–J) SEM images of powdered seawater-precipitated Mg(OH)2 are shown at each delay time. Samples were dried and then crushed using mortar and pestle until a homogeneous powder was obtained. No distinct grains were observed for any of the delay times.
When observing the difference between powder and paste forms, the paste always appeared as a more amorphous material with broader peaks. This is due to the presence of significant amounts of H2O within the paste during analysis. Powder does not contain the same levels of H2O due to being dried before analysis. The inclusion of H2O in the paste prevents long-range ordered structures and keeps the arrangement more liquid-like (Hu et al., 2017). This lack of order results in more broad peaks in XRD diffractograms.
SEM images of the powdered, seawater-precipitated Mg(OH)2 are shown at each delay time used in this study (Figures 5D–J). No distinct grains or crystals were observed at this scale (10 μm), consistent with the lack of well-formed crystals. However, even at 336 h of delay time, no visible crystals could be seen. No visible impurities were observed in any of the samples, such as CaCO3 crystals, which were described by Fernandez-Diaz et al. (1996).
The elemental ratios of the powdered seawater-precipitated Mg(OH)2 samples were determined (Figure 5C). The 5 most common elements detected (O, Ca, Cl, Si, S, and Fe) were compared to the detected amount of Mg. Al and C were omitted due to interference from sample holder and carbon tape. The theoretical ratio of O: Mg in pure Mg(OH)2 was calculated to be 1.29 after EDS calibration (1.32 before adjustment). The average O; Mg for all delay times of seawater-precipitated samples was 1.35 ± 0.08. All samples had a detected Ca: Mg ratio that ranged from 0.08 to 0.12. Presence of Ca indicates possible CaCO3 formation during the precipitation process contributing to impurities. No trend of elemental composition as a function of delay time was discernible.
3.4 Dissolution kinetics of Mg(OH)2
The dissolution rate was calculated with units of [OH]/min using the measured alkalinity change of the seawater before and after the addition of Mg(OH)2, and the continuous tracking of pH during dissolution. The results shown occurred at a time that ranged from 3 to 21 min after addition of alkalinity, which is based on the maximum pH reached. The observed trend revealed that shorter delay times correspond to faster dissolution rates. Rates significantly decreased during the first few hours of delay time (Figure 6A) and then began to plateau at delay times beyond 96 h. Visually, the Mg(OH)2 settled as delay time progressed, decreasing in volume within the solution until a semi-solid pit at the bottom of the test tube was formed (a similar phenomenon was observed in Figures 3A–C).
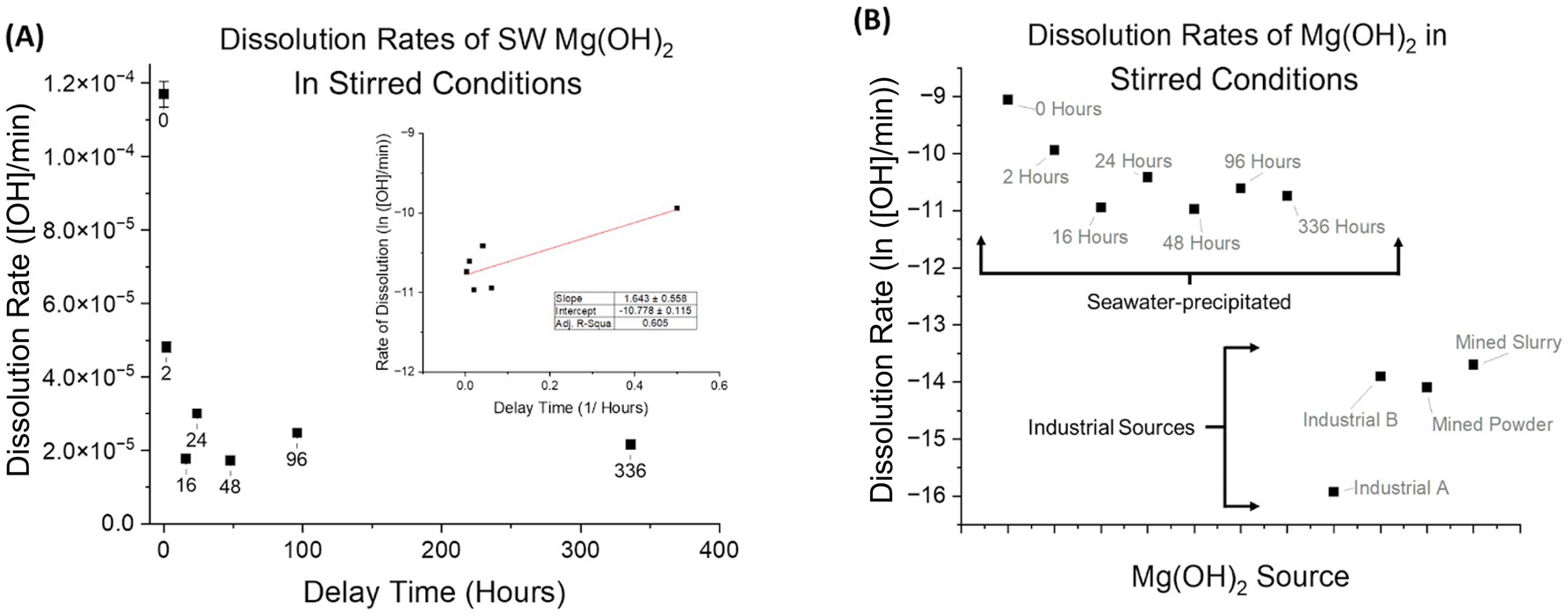
Figure 6. (A) Dissolution rates ([OH]/min) of seawater-precipitated Mg(OH)2 are plotted against their respective delay times in stirred-conditions. Stirred-conditions for complete dissolution include mechanical stirring by stir bar, sufficient mixing to ensure homogenization and no dead zones in solution. Each experiment had continuous pH measuring and discrete AT analysis. Shorter delay times produced the fastest dissolution rates up to 24 h. After 24 h of delay time, the dissolution rate plateaued. (B) Dissolution rates (ln([OH]/min)) of Mg(OH)2 in stirred-conditions are shown. Industrial A performed at the slowest rate whereas Industrial B, Mined Powder, and Mined Slurry had rates an order of magnitude higher. Compared to those of seawater-precipitated, all industrial sources have slower rates by at least 2 orders of magnitude.
3.5 XRD calculated properties
The observed trend of degree of crystallinity with delay time is approximately the inverse of the trend for dissolution rates, with the crystallinity increasing initially, but then plateauing as the delay time increases (Figure 7A). For the powdered form, a delay time of 0 h produced material that was 82 ± 4.1% crystalline, increasing to 89 ± 4.4% for 336 h. The paste form of Mg(OH)2 exhibited lower crystallinity overall, compared to the powdered form, with a more significant fractional dependence of crystallinity on delay time, increasing from 16 ± 0.82% to 33 ± 1.6% for 0 and 336 h, respectively. Industrial sources of Mg(OH)2 were measured to have an average degree of crystallinity of 84 ± 4.2%.
![Three graphs display crystallinity data for SW Mg(OH)₂ over time. (A) Shows degree of crystallinity (%) versus delay time (hours) with industrial sources as a reference. (B) Illustrates crystallite size (nm) against delay time, comparing powder and paste forms to industrial standards. (C) Represents d-spacing difference (nm) of crystal planes [001] and [101] over time, highlighting differences between powder and paste. Each graph includes data points, error bars, and legends indicating powder and paste forms.](https://www.frontiersin.org/files/Articles/1616362/fclim-07-1616362-HTML/image_m/fclim-07-1616362-g007.jpg)
Figure 7. (A) Seawater-precipitated Mg(OH)2 degree of crystallinity (calculated from XRD data) was compared to delay times for both powder and paste form of the precipitated material. A trend of increasing crystallinity is seen with increasing delay time for both forms. Industrial sources, which were only measured in powder form, were also calculated for comparison. There is a range of 8.8% in the degree of crystallinity in powder form within the delay times and 16.5% for paste. (B) Crystallite size for each form of seawater-precipitated Mg(OH)2 is shown. The powdered form exhibits a sharp initial increase in size for the first 48 h, and then plateaus. Meanwhile, the paste form sees no significant shift in size as delay time increases. This difference could be attributed to the evaporation of water from heating, which reduces amorphousness and promotes further crystallization. The average crystallite size for industrial sources was 36.3 ± 17.6 nm. Compared to the 336 Hour of powder, the industrial average was 23.0 nm larger, while industrial average was 27.7 nm larger for 336 Hours paste form. (C) d-spacing (interatomic spacing) was calculated for each delay time of seawater-precipitated Mg(OH)2 of powder and paste form. Powder consisted of a higher d-spacing difference on average compared to paste, with a separation of 0.054 nm. The d-spacing difference of Industrial A was 2.42 nm. This was 0.02 nm smaller than the average for powdered form SW Mg(OH)2 and was 0.04 nm larger than the paste form.
Powders with longer delay times were observed to have larger crystallite sizes (Figure 7B), ranging from 4.6 ± 0.25 nm (0 Hours) to 13.3 ± 0.25 nm (336 Hours). Little change was observed in crystallite size of paste within the range of delay times studied: 3.2 ± 0.08 nm (0 Hours) to 6.5 ± 0.20 nm (48 Hours) to 4.3 ± 0.21 nm (336 h). The crystallite size of sample “Industrial A” was found to be 61 ± 0.36 nm, which was 4.6 times greater than the largest size observed for any seawater-precipitated samples (powder value of 13.3 nm at 336 h). The Williamson-Hall method was used to provide quantitative calculation of crystallite size that can account for peak broadening due to micro-strains which the Scherrer equation lacks (Yousefi et al., 2017). Uncertainty for crystallite size was propagated by accounting for instrumental broadening in XRD analysis and uncertainty in both measured FWHM and 2θ of diffractograms. These calculated uncertainties are not visible resulting in no error bars in Figure 7B.
In crystallography, d-spacing is the interplanar spacing between atomic layers in a material. The d-spacing is unique for each crystal and therefore used to identify material phase composition (Hammond, 2002). The two largest peaks of Mg(OH)2, [001] and [101], were selected (Figure 7C). For the powdered form, all samples besides the 0 Hours had a d-spacing difference greater than that of Industrial A which was used as reference. For paste, all except 96 Hours were below the reference spacing. D-spacing uncertainty was calculated based on the errors provided for 2θ peak locations.
3.6 Precipitate mixing influence on alkalinity release
Dissolution rates of Mg(OH)2 at stirred-tank conditions (Table 2) were compared with varying procedures for mixing of the precipitate (Figure 8). The “unstirred” system was the standard procedure used during the study, compared with samples that were “stirred-before-addition” and “continuously-stirred.” For all 3 systems, the same trend was observed, with lower delay times having faster dissolution and then quickly reaching a plateau as the delay time increases.
![Graph showing dissolution rates of SW Mg(OH)₂ over time with different mixing methods. The y-axis represents the dissolution rate ([OH⁻]/min), and the x-axis represents delay time (hours). Data points include squares (unstirred), circles (stirred before addition), and triangles (continuously stirred). Notable points are labeled with corresponding hours.](https://www.frontiersin.org/files/Articles/1616362/fclim-07-1616362-HTML/image_m/fclim-07-1616362-g008.jpg)
Figure 8. Dissolution rates of seawater-precipitated Mg(OH)2 in stirred-tank conditions with different precipitate mixing. “Unstirred” is the original and standard form of precipitation throughout this study. “Stirred-before-addition” refers to experiments where the precipitate and supernatant were mixed right before addition to the tank. “Continuously-stirred” refers to experiments where the precipitate and supernatant were always kept homogenized until addition. Following a similar trend in earlier experiments, shorter delay time leads to the fastest dissolution rates, across all three types of precipitate stirring used.
3.7 Alkalinity release in stirred-tank conditions
Alkalinity recovery for all seawater-precipitated samples was >0.95 after 35 min due to the high mixing within the tank without the presence of dead zones (Figure 9). For the industrial samples (experiments continued for 4 days due to slow dissolution), a large range was observed between Industrial A (0.12 ± 0.02) and Industrial B (0.61 ± 0.05), showcasing how much industrial Mg(OH)2 varies for OAE applications. A control using aqueous NaOH of equivalent alkalinity achieved a recovery fraction of 0.995 ± 0.03.
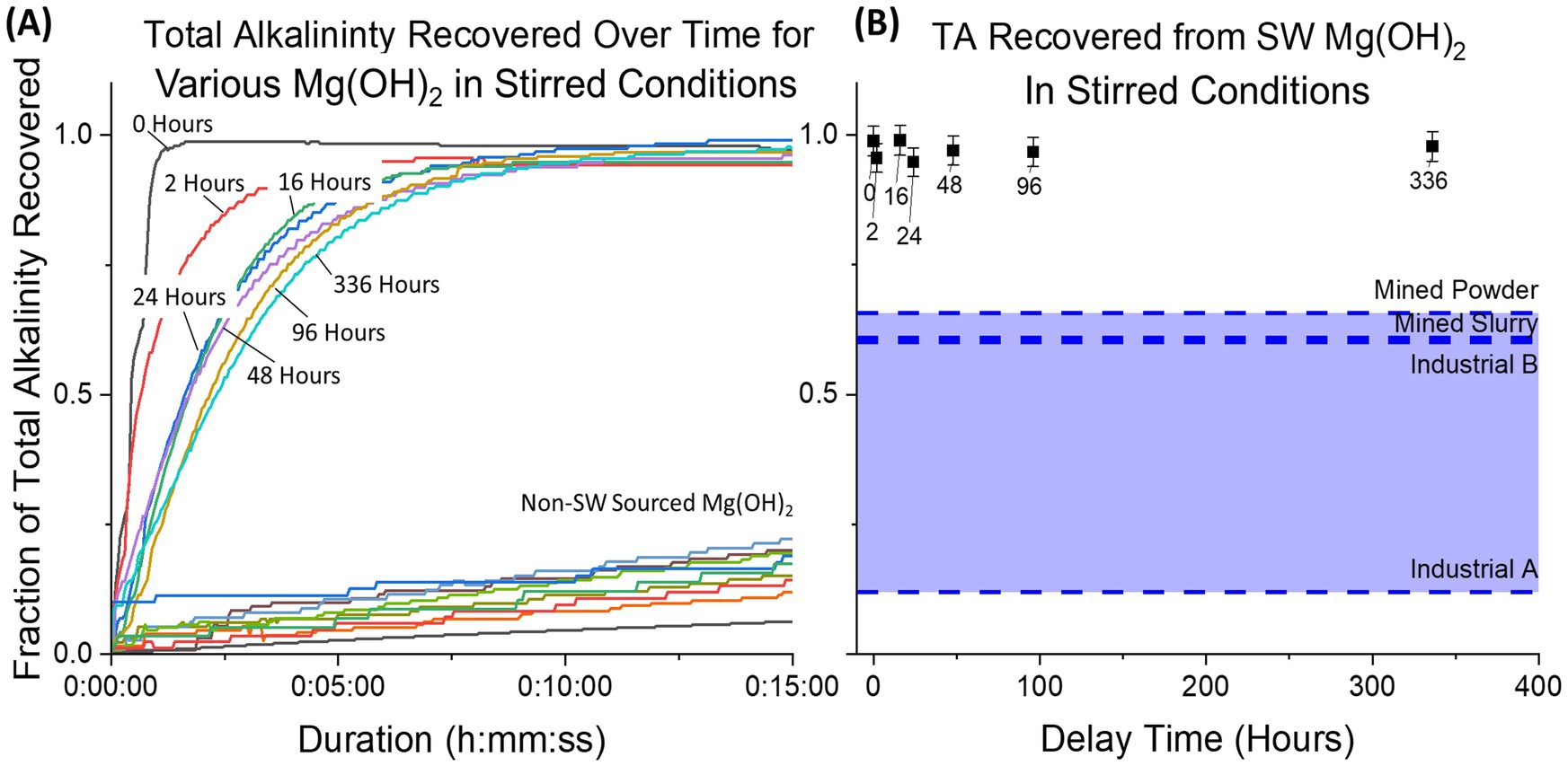
Figure 9. The fraction of total alkalinity recovered from Mg(OH)2 is plotted for continuous measurement (initial 15 min) (A) and final measurement (B). All datapoints for seawater-precipitated Mg(OH)2 (labeled for each delay time) exhibit high recovery fractions, while all industrial sources have much lower recovery. Likewise, final recovery for all seawater-precipitated samples achieved 0.95 or higher compared to industrial samples which ranged from 0.12 to 0.65.
3.8 Alkalinity release in non-stirred-tank conditions
Dissolution rates and AT recovery were determined for seawater-precipitated Mg(OH)2 in non-stirred-conditions, where only air bubbling was used to circulate seawater in the tank (Figures 10A–C). A similar trend for dissolution rates was observed here compared to stirred-conditions, however the rates themselves were 3 orders of magnitude slower than under stirred-conditions. Experiments without substantial pH data were excluded from these calculations. It should be noted that immediate dissolution rates cannot be derived from this data due to the limited frequency at which AT analysis was performed.
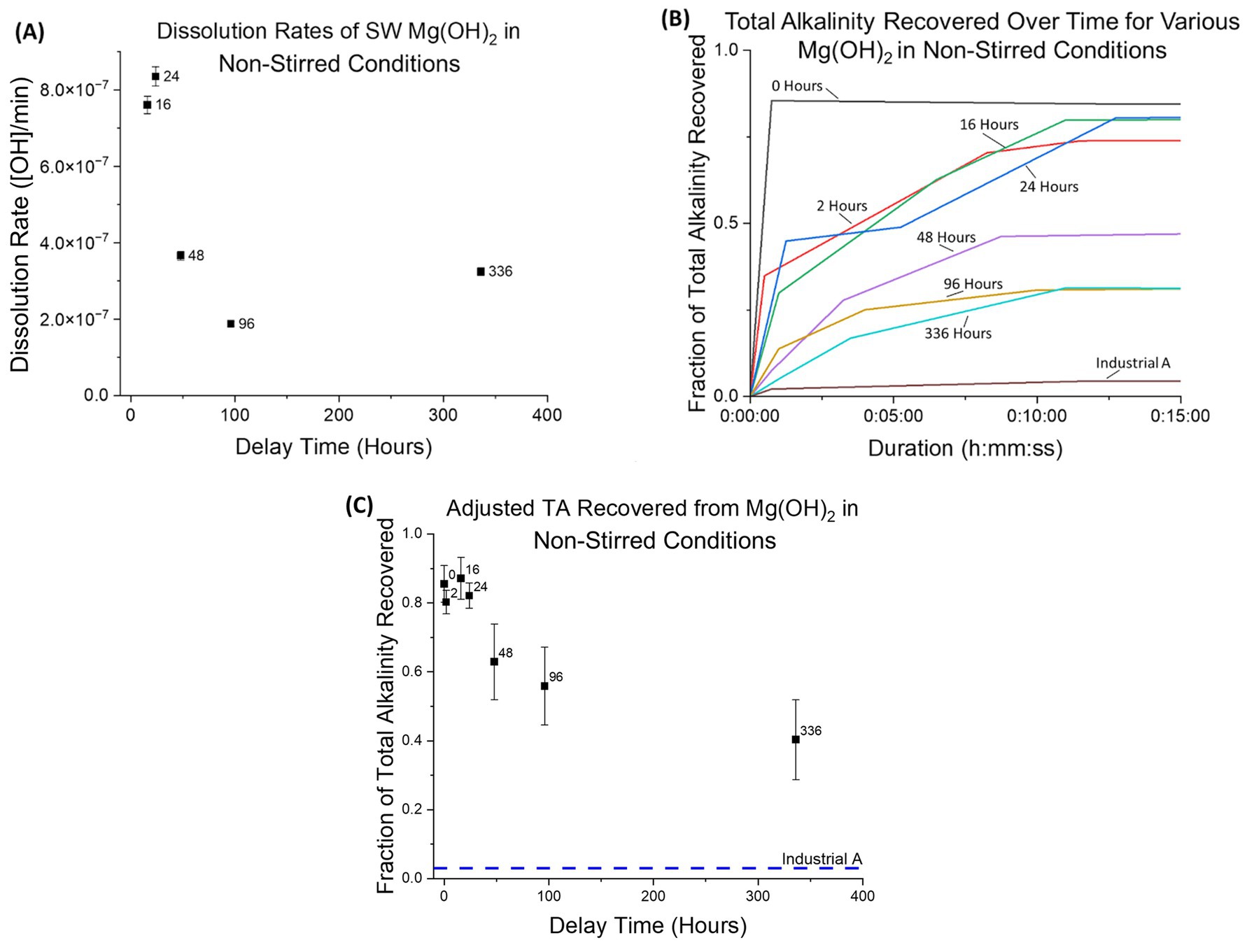
Figure 10. (A) Dissolution rates of seawater-precipitated Mg(OH)2 in non-stirred condition. This includes no mechanical stirring, dead zones present, and bubbling. Similar to stirred-conditions, the dissolution rate decreases as delay time increases, with a strong decrease in the initial 48 h. (B) Fraction of total alkalinity recovered plotted against time for non-stirred-conditions using continuous observation in the initial 15 min. All seawater-precipitated Mg(OH)2 rates are distinctly separated from the industrial source rate. (C) Total alkalinity recovered for seawater-precipitated Mg(OH)2 in non-stirred-conditions. While stirred-conditions allowed for recovery of 0.95 or higher for all delay times, seawater-precipitated Mg(OH)2 in non-stirred-conditions performed significantly lower, ranging from 0.85 ± 0.05 with 0 Hours down to 0.40 ± 0.12 with 336 Hours. Here, with the presence of dead zones and no mechanical mixing, the morphological differences in crystallinity can impact the recovery.
Unlike the AT recovery with stirred-conditions, a decreasing trend was observed for non-stirred-conditions as delay time increased. Within the first 24 h, the fraction remained between 0.80 ± 0.03 and 0.87 ± 0.06. The fraction of recovery then decreased to 0.40 ± 0.12 for a delay of 336 h, compared to 0.98 ± 0.09 for stirred-conditions (at a delay of 336 h). Sample “Industrial A” also exhibited a reduction in the fraction of recovery under change of conditions, from 0.12 ± 0.02 (stirred) to 0.03 ± 0.03 (non-stirred).
4 Discussion
4.1 Mg(OH)2 source influence
Our observations showed that the morphology of seawater-precipitated Mg(OH)2 varied drastically from industrial sources of brucite (Figure 4). With a higher crystallinity, the industrial brucite was expected to have a less favorable Gibbs Free energy of dissolution and lower solubility (Gurunath et al., 2013). Immediate precipitation, such as the one used to create the seawater-precipitated samples, produced a lower crystalline material. By introducing NaOH immediately into the seawater solution, a supersaturation mixture is quickly made which triggers primary precipitation. The primary precipitation leads to nucleation being favored and creates an abundance of small crystal nuclei instead of crystal growth. This is compared to the industrial sources whose synthesis is most likely based on (1) a purer Mg2+ source solution and (2) a different base. Turek et al. suggests this by stating precipitation methods can influence the properties of particles, including addition speed and type of base (Thanh et al., 2014; Wu et al., 2022). Abundant and rapid nucleation resulted in the seawater-precipitated Mg(OH)2 having an amorphous structure that was less ordered and more easily dissolved (Novakovic et al., 2018; Chu and Yalkowsky, 2009). When comparing the elemental ratios of the materials, both the seawater-precipitated and industrial sources contained the same major elements and were relatively pure Mg(OH)2.
Different industrial sources of Mg(OH)2 appeared to be similar based on chemical and phase composition as well as crystallinity. As predicted, we observed that all tested industrial sources of Mg(OH)2 dissolve at similar rates to each other but at distinct and slower rates from seawater-precipitated Mg(OH)2 (Figure 6B). The faster dissolution of seawater-precipitated Mg(OH)2 is consistent with its lower degree of crystallinity.
4.2 Delay time impact on crystallinity
The dissolution kinetics of the seawater-precipitated Mg(OH)2 were determined for different amounts of delay time mentioned earlier in Methods 2.3, which kept the precipitated Mg(OH)2 in high pH and supersaturated supernatant between precipitation and re-dissolution. It was found that there was a significant decrease in dissolution rate with increasing delay time, with the largest decreases occurring over the first 2 h of delay (Figure 6A). The exponential decay of the dissolution rate with delay time suggests that there was an order and energy change within the structure of Mg(OH)2 due to ripening. This agrees with findings that found ripening to be a lead influence in the growth of primary crystals over time. At the same time the crystal lattice became more structured while it remained in saturated solution without elemental composition changes. This suggests that because the Mg(OH)2 remains in the supernatant, it allows for more particles to develop due to an excess of OH− and for the growth of existing particles through the improvement of crystal structure (Dong et al., 2018). These two changes can both be observed with the XRD diffractograms.
A clear shift in XRD diffractograms was observed with the profile of the peaks that reveals a shift in crystallinity within delay times which can be attributed to Ostwald ripening. This recrystallization occurs when smaller crystals dissolve into solution and then are deposited onto larger crystals that grow in size (Kahlweit, 1975). For both the powder and paste forms of precipitated Mg(OH)2, the peaks at each wavelength became sharper at longer delay times (Figures 5A,B). This is an indication that the material was experiencing an improvement in the crystal lattice by remaining in the supernatant. This optimization led to a higher crystallinity (Lv et al., 2024). The peaks highlighted in both forms corresponded to the major diffraction peaks of Mg(OH)2, qualitatively confirming that the material produced is of high purity. The [101] peak was the most intense among delay times, which suggested that surface polarity is higher in the Mg(OH)2 which resulted in the very gel like material due to agglomeration.
Degree of crystallinity, crystallite size, and d-spacing were calculated for both powder and paste forms of the seawater-precipitated Mg(OH)2 (Figures 7A–C). Both forms exhibited the same trend in degree of crystallinity: an initial increase, followed by a plateau after the 24–48 h mark. The powdered form resulted in similar crystallinity as the industrial forms of Mg(OH)2, suggesting that the powder will perform similarly when dissolved. The sharp decrease in the crystallinity of the paste compared to the powdered form can be attributed to the water content in the hydrated material. Mg2+ has a high affinity to water molecules, causing it to create stable complexes and led to these hydrated states. During precipitate filtration, the material is almost gel like due to these Mg-H2O states (Xue et al., 2009). The paste form cannot have a long-range ordered lattice structure due to the presence of H2O making it more amorphous (Shirasaki, 1961). Crystallite size for both forms revealed a similar pattern with delay time allowing for an increase in size. However, crystallite size for all of the seawater-precipitated samples was far below that of the industrial samples visible in SEM images. This is due to the same precipitation conditions and method used. Seawater-precipitated Mg(OH)2 exhibited smaller crystallite sizes with no distinct granules or shapes regardless of delay time. A smaller crystallite size would result in larger surface area and therefore promote a faster dissolution rate. The d-spacing difference between the 2 most prominent crystal planes of Mg(OH)2 ([001] and [101]) was calculated to elucidate the morphological changes in the material as delay time progressed. The d-spacing of the powder form was found to be routinely higher than “Industrial A” which was used as a standard, and the precipitated Mg(OH)2 paste had a larger d-spacing than the standard. Differences among the powder and paste form of seawater Mg(OH)2 are assumed to be due to the water content of the Mg(OH)2 seen by having a broad peak around 30°. Before drying in an oven, the paste form exhibits more amorphous-like properties seen with XRD findings. The broader peaks for amorphous materials are due to a lack of long-range lattice order and a wide distribution of atomic spacings (in this case because there is H2O present) which emits a diffuse scattering of X-rays. A less significant contribution to the crystallinity difference is that the heat applied during drying could provide additional energy for continued crystallization.
The paste form of seawater Mg(OH)2 was investigated further, since it was the closest to what was used in the dissolution experiments. When the degree of crystallinity was examined, a clear trend was seen of increasing degree as delay time increased (Figure 11). This agreed with our hypothesis that an energy barrier within the structure is the leading cause for the difference in dissolution rates. Therefore, the degree of crystallinity and dissolution rate were compared. A clear trend was observed after linearization of the dissolution rates. As the degree of crystallinity of the paste increased, the dissolution rate decreased. With an adjusted R-square value of 0.90, there is a good fit and a significant relationship between the variables. Crystallinity was influenced by the precipitation method having an excess of OH− which caused rapid nucleation that favored large quantity of small crystals. Along with delay time where the crystals experienced growth and lattice improvement due to longer time in the supernatant. These factors resulted in differences in crystallinity that saw a change in dissolution rates.
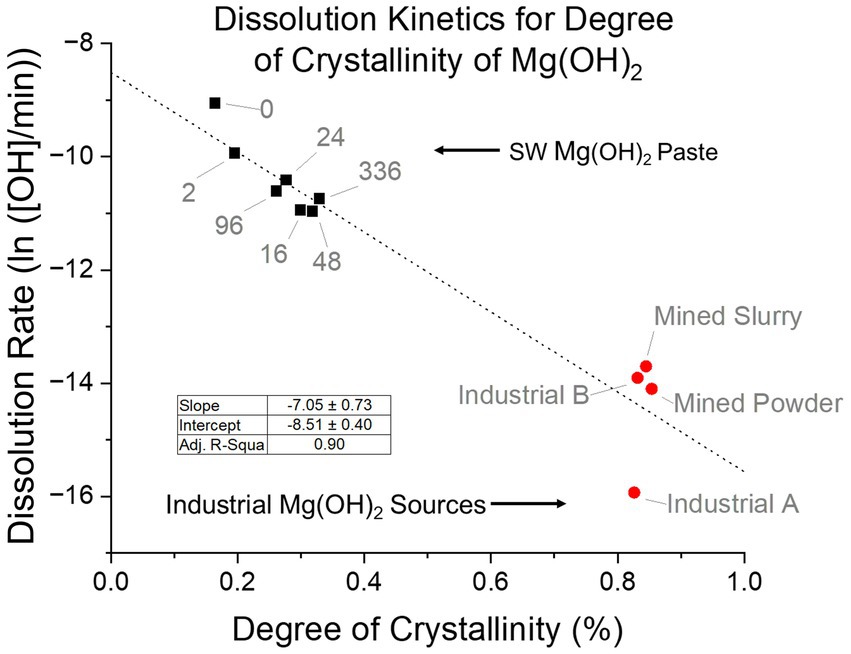
Figure 11. Relationship between the degree of crystallinity and the dissolution rate of seawater-precipitated Mg(OH)2 paste and industrial sources. Linear regression shows a slope of −7.05 degree of crystallinity/ln(rate) with a R-square value of 0.90. If industrial sources are removed, the slope becomes −10.22 degree of crystallinity/ln(rate) with a R-square value of 0.84.
Delay times were a major influence in crystallinity and can be used as a method to control the rate of dissolution. This holds significance during OAE applications because the rate at which alkalinity dissolves determine the pH disruption to the water at the release site. Excessive pH can be harmful to the local ecology and cause runaway precipitation of CaCO3. Such precipitation effects have been observed in experiments in which alkalinity-induced supersaturation of CaCO3 resulted in enough precipitation that more alkalinity was lost from solution than was initially added (Moras et al., 2022; Hartmann et al., 2022; Ringham et al., 2024; Suitner et al., 2024). This poses challenges for the carbon removal efficiency of OAE and may have implications for ecosystem health due to the increase in seawater turbidity. Understanding the dissolution of Mg(OH)2 that was previously precipitated from solution is also directly relevant for several proposed methods which seek to extract magnesium hydroxide from forsterite (Scott et al. 2021; Charnay et al., 2023), rather than from seawater. In this approach, olivine is dissolved in electrochemically generated hydrochloric acid. After neutralization with the simultaneously produced base and removal of the silica and unwanted heavy metal contaminants, clean Mg(OH)2 is obtained. This material can be used as the alkalinity source for OAE. Most previous OAE Mg(OH)2 studies focused on commercially available sources such as calcined magnesite (Kitidis et al., 2024), dissolved olivine material (Hartmann et al., 2022; Yang et al., 2023; Moras et al., 2022, Montserrat et al., 2017), or mined brucite (Yang et al., 2023). With the understanding that the synthesis method having a significant influence on the precipitation and properties of Mg(OH)2, the novelty of this work is highlighted.
There was a significant difference in alkalinity released over time from seawater-precipitated Mg(OH)2 and industrial sources, with the former dissolving considerably faster. We attribute this difference to the higher crystallinity and larger crystallite sizes seen in the industrial sources led by larger energy barriers for dissolution and smaller surface areas. Even when industrial samples were allowed to remain in solution for extended periods of time (5–7 days), complete dissolution was not achieved. This is compared to the seawater samples that are seen to reach complete (98% or higher) dissolution within ~15 min of introduction. Within the seawater samples, there was a decrease in the redissolution rates as the delay times between adding NaOH/brine and testing redissolution rates increased. This pattern was the result of the increasing crystallinity of the internal structure of the Mg(OH)2 as the amount of time provided for re-ordering the crystal structure increased. Collectively, these results show that variability in the speed, quantity, and predictability of AT delivery for various Mg(OH)2 sources must be considered for Mg(OH)2 based OAE approaches.
4.3 Sample preparation impact
Sample preparation methods (unstirred, stirring-before-addition, and continuously-stirred) were designed to ensure that physical agglomeration of seawater Mg(OH)2 was not the cause of the correlation between dissolution rates and delay times (Figure 8). It was found that all sample preparation methods conducted resulted in no significant variation in dissolution rate for the same delay times. This suggests that the change in rate was due to the chemical and morphological differences among the delay time samples and not the macroscopic physical characteristics. This experiment confirmed that the methodology used to produce ideal dissolution rates was not limited to the mechanical stirring of the beakers during Mg(OH)2 precipitation and settling.
4.4 Alkalinity release in non-stirred-tanks
Another set of tank tests (labeled “non-stirred”) did not employ mechanical stirring and instead relied on bubbling from an aquarium bubble stone (Table 2). These conditions observed a similar trend, where shorter delay times resulted in faster rates. However, the absolute magnitude of the rates were several orders slower than that found in the stirred-conditions (Figure 10).
The fraction of total alkalinity recovered for non-stirred dissolution conditions produced a different pattern than the equivalent for stirred-conditions. There is a decrease in recovered alkalinity as delay increases unlike stirred cases where all seawater-precipitated Mg(OH)2 solutions were essentially 100%. Additionally, the fractions determined were all below 90% and reached as low as 42% for seawater samples in non-stirred-conditions. Under these conditions, there was the lack of mechanical stirring in tanks which resulted in a presence of dead zones where portions of seawater sample sink and are never re-introduced to solution. This lack of homogenous mixing combined with the difference in crystallinity for delay times is what resulted in the differences between stirred and non-stirred-conditions. The non-stirred-conditions enhances the difference of crystallinity among delay times. Under bubble-agitated dissolution conditions the slower dissolution speeds correlated with higher crystallinity of the starting material. Due to the larger energy needed to dissolve the more crystalline samples when dead zones are added and samples are no longer continuously suspended, full dissolution was not realized. This is shown even more drastically with the use of Industrial A sample where an alkalinity recovery fraction of only 0.04 was achieved due to the majority of material simply sinking into the dead zones and not dissolving. This is crucial to understand for an OAE application as the ocean is not an ideal mixing environment. Steps will need to be taken to avoid materials getting buried in sediments or sinking below the surface mixing layer with active air-sea exchange.
5 Conclusion
In this study, seawater-precipitated Mg(OH)2 was characterized, and its properties were compared to those of available industrial sources. The two groups of materials were found to vary greatly in their morphology and physical properties including particle size, degree of crystallinity, and crystallite size. Industrial sources had a degree of crystallinity of 0.83 to 0.85 while seawater-precipitated ranged from 0.16 to 0.33 for paste or 0.81 to 0.90 in powdered form. The elemental ratios were found to be the same for all samples, producing high purity (>96%) Mg(OH)2 with only trace impurities such as Ca or Fe. There is a strong relationship between the dissolution rate and the degree of crystallinity that is present in the material. The slower dissolution rates found with higher crystallinity can be attributed to increased energy required to overcome the ordered crystal structure. These results suggest that a longer delay between adding NaOH/brine and redissolution allows for ripening to occur which results in a larger and more stable crystal with higher crystallinity that is less prone to redissolution. Other factors such as sample preparation may also be influential to the degree of crystallinity, such as using a powdered or paste form, and we find that the higher water content within Mg(OH)2 pastes is linked to faster redissolution. Additionally, future experiments such as those focused on precipitation storage in the complete absence of CO2 could provide further understanding of the recrystallization of Mg(OH)2. The study also compared the effect that stirred and non-stirred mixing conditions have on the efficiency of Mg(OH)2 redissolution. When conditions are ideal and there is sufficient mechanical mixing to prevent dead zones, the alkalinity recovery remains above 95% for all seawater-precipitated samples. In non-stirred-conditions that included dead zones and no mechanical mixing (only bubbling), longer delays before analysis again resulted in higher crystallinity and slower redissolution, but this time also resulted in lower recovery rates of ~40% compared to >80% for shorter delays. This same effect of mixing was seen with the industrial sources as well.
For Mg(OH)2-based OAE applications, this study highlights the importance of alkalinity source preparation and deployment location mixing. To achieve the fastest and highest alkalinity recovery after dissolution, Mg(OH)2 material should be kept as a hydrated paste and dispersed as quickly as possible to retain the initial amorphous crystalline properties. However, drawbacks of such measures would include the higher costs and logistical barriers of transporting the hydrated water weight and of rapidly dispersing the product. These results suggest that the alkalinity recovery of seawater-precipitated Mg(OH)2 can be controlled by modulating the length of time allowed crystals to precipitate from seawater after adding the concentrated base. This approach therefore also holds potential in sites where adequate mixing is available, yet slower dissolution is needed. An area of future interest that was not under the main subject of this study would be the scalability of Mg(OH)2 production for OAE practices. Preliminary estimates based on schematics provided in Figure 1, would suggest requiring approximately 88,400 L of alkalinity to raise a 1 km2 ocean area with a depth of 10 m to raise the alkalinity 375 μmol/kg (the target used in this study). Evaluation to understand the size of tanks needed for material production would be a key part in the further implementation of Mg(OH)2 and other solid alkalinity sources for ocean-scale deployment. In summary, we suggest that Mg(OH)2 should be considered for future OAE deployments because it can be generated from seawater (or reject brine treatment) and converted into an alkalinity-dense material that redissolves with rapid and potentially controllable kinetics.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: Zenodo Repository, https://10.5281/zenodo.15121979.
Author contributions
CS: Conceptualization, Methodology, Investigation, Writing – review & editing, Writing – original draft. MR: Methodology, Conceptualization, Writing – review & editing. BC: Conceptualization, Writing – review & editing. MT: Methodology, Conceptualization, Writing – review & editing. ME: Conceptualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. We acknowledge funding from The Grantham Foundation for the Protection of the Environment under the SEA MATE (Safe Elevation of Alkalinity for the Mitigation of Acidification Through Electrochemistry) grant. In addition, this research used the XRD, SEM, and EDS facility of the Center for Functional Nanomaterials (CFN), which is a US Department of Energy Office of Science User Facility, at Brookhaven National Laboratory under Contract No. DE-SC0012704. For the initial stages of this research BRC was funded through the Cooperative Institute for Climate, Ocean, and Ecosystem Studies (CICOES) under NOAA Cooperative Agreement NA20OAR4320271 and supported by NOAA‘s PMEL (CICOES contribution number 2025–1442).
Acknowledgments
We would like to thank Stephen Abrams and Thomas Wilson at Stony Brook University Flax Pond Marine Lab for technical assistance in experiment setup. Unfortunately, our final author, Matthew D. Eisaman, passed away in February 2025. This work would not have been possible without his crucial contributions and unwavering support as an advisor, mentor, and friend.
Conflict of interest
MR is Lead Oceanographer and Head of MRV at Ebb Carbon, Inc. ME is Co-Founder and Chief Scientific Advisor at Ebb Carbon, Inc. MT is employed by the company Google Inc. MR was employed by Ebb Carbon, Inc., South San Francisco, CA, Unites States.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor MF declared a past co-authorship with the author BC.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fclim.2025.1616362/full#supplementary-material
References
Ayoub, G. M., Zayyat, R. M., and Al-Hindi, M. (2014). Precipitation softening: a pretreatment process for seawater desalination. Environ. Sci. Pollut. Res. 21, 2876–2887. doi: 10.1007/s11356-013-2237-1
Bach, L. T., Gill, S. J., Rickaby, R. E. M., Gore, S., and Renforth, P. (2019). CO2 removal with Enhanced Weathering and Ocean Alkalinity Enhancement: potential risks and co-benefits for marine pelagic ecosystems. Front. Clim. 1. doi: 10.3389/fclim.2019.00007
Battaglia, G., Romano, S., Raponi, A., Marchisio, D., Ciofalo, M., and Tamburini, A. (2022). Analysis of particles size distributions in Mg(OH)2 precipitation from highly concentrated MgCl2 solutions. Powder Technol. 398:117106. doi: 10.1016/j.powtec.2021.117106
Bharadwaj, H. K., Lee, J.-Y., Li, X., Liu, Z., and Keener, T. C. (2013). Dissolution kinetics of magnesium hydroxide for CO2 separation from coal-fired power plants. Journal of Hazardous Materials 250–251, 292–297. doi: 10.1016/j.jhazmat.2013.02.009
Bragg, W. H. (1913). The reflection of X-rays by crystals. Proceedings of the Royal Society of London. Series a, containing papers of a mathematical and physical character 88, 428–438.
Cai, W.-J., Guo, X., Chen, C.-T. A., Dai, M., Zhang, L., and Zhai, W. (2008). A comparative overview of weathering intensity and HCO3− flux in the world’s major rivers with emphasis on the Changjiang, Huanghe, Zhujiang (pearl) and Mississippi Rivers. Cont. Shelf Res. 28, 1538–1549. doi: 10.1016/j.csr.2007.10.014
Carrotte, P. (2004). Endodontics: part 9 calcium hydroxide, root resorption, endo-perio lesions. Br. Dent. J. 197, 735–743. doi: 10.1038/sj.bdj.4811897
Charnay, B., Chen, Y., Agarwal, R., Misleh, J., Wright, G., Sauve, E., et al. (2023). Membrane-free electrochemical production of acid and base solutions capable of processing ultramafic rocks. ChemRxiv. doi: 10.26434/chemrxiv-2023-5tndz
Chu, K., and Yalkowsky, S. (2009). Predicting aqueous solubility: the role of crystallinity. Curr. Drug Metab. 10, 1184–1191. doi: 10.2174/138920009790820110
Committee on A Research Strategy for Ocean-based Carbon Dioxide Removal and Sequestration, Ocean Studies Board, Division on Earth and Life Studies, & National Academies of Sciences, Engineering, and Medicine. (2022). A research strategy for ocean-based carbon dioxide removal and sequestration 26278. Washington, D.C.: National Academies Press.
Davies, P. A., Yuan, Q., and De Richter, R. (2018). Desalination as a negative emissions technology. Environ. Sci.: Water Res. Technol. 4, 839–850. doi: 10.1039/C7EW00502D
De Lannoy, C.-F., Eisaman, M. D., Jose, A., Karnitz, S. D., DeVaul, R. W., Hannun, K., et al. (2018). Indirect ocean capture of atmospheric CO2: part I. Prototype of a negative emissions technology. Int. J. Greenhouse Gas Control 70, 243–253. doi: 10.1016/j.ijggc.2017.10.007
Dickson, A. G., Sabine, C. L., Christian, J. R., and Bargeron, C. P. (2007). Guide to best practices for ocean CO2 measurements. Sidney, BC: North Pacific Marine Science Organization.
Dong, H., Wang, A., Smart, G., Johnson, D., and Koenig, G. M. (2018). In-situ analysis of nucleation and growth of transition metal oxalate battery precursor particles via time evolution of solution composition and particle size distribution. Colloids and Surfaces A: Physicochemical and Engineering Aspects 558, 8–15. doi: 10.1016/j.colsurfa.2018.08.047
Eisaman, M. D., Geilert, S., Renforth, P., Bastianini, L., Campbell, J., Dale, A. W., et al. (2023). Assessing the Technical Aspects of Ocean-Alkalinity-Enhancement Approaches. State of the Planet 2-oae2023, 1–29. doi: 10.5194/sp-2-oae2023-3-2023
Eisaman, M. D., Parajuly, K., Tuganov, A., Eldershaw, C., Chang, N., and Littau, K. A. (2012). CO2 extraction from seawater using bipolar membrane electrodialysis. Energy Environ. Sci. 5:7346. doi: 10.1039/c2ee03393c
Eisaman, M. D., Rivest, J. L. B., Karnitz, S. D., De Lannoy, C.-F., Jose, A., DeVaul, R. W., et al. (2018). Indirect ocean capture of atmospheric CO2: part II. Understanding the cost of negative emissions. Int. J. Greenhouse Gas Control 70, 254–261. doi: 10.1016/j.ijggc.2018.02.020
El-Manharawy, S., and Hafez, A. (2003). Study of seawater alkalization as a promising RO pretreatment method. Desalination 153, 109–120. doi: 10.1016/S0011-9164(02)01110-4
Fernandez-Diaz, L., Putnis, A., Prieto, M., and Putnis, C. V. (1996). The role of magnesium in the crystallization of calcite and aragonite in a porous medium. J. Sediment. Res. 66, 482–491.
Fontana, D., Forte, F., Pietrantonio, M., Pucciarmati, S., and Marcoaldi, C. (2023). Magnesium recovery from seawater desalination brines: a technical review. Environ. Dev. Sustain. 25, 13733–13754. doi: 10.1007/s10668-022-02663-2
Friis, K., Körtzinger, A., and Wallace, D. W. R. (2003). The salinity normalization of marine inorganic carbon chemistry data. Geophys. Res. Lett. 30:36-1-36-4. doi: 10.1029/2002GL015898
Fuhr, M., Geilert, S., Schmidt, M., and Wallmann, K. (2021). “Kinetics of olivine weathering in seawater: an experimental study” in Goldschmidt2021 abstracts (Virtual: European Association of Geochemistry).
Gurunath, S., Pradeep Kumar, S., Basavaraj, N. K., and Patil, P. A. (2013). Amorphous solid dispersion method for improving oral bioavailability of poorly water-soluble drugs. J. Pharm. Res. 6, 476–480. doi: 10.1016/j.jopr.2013.04.008
Hammond, C. (2002). The basics of crystallography and diffraction (2nd edn). Meas. Sci. Technol. 13:232. doi: 10.1088/0957-0233/13/2/708
Hangx, S. J. T., and Spiers, C. J. (2009). Coastal spreading of olivine to control atmospheric CO2 concentrations: a critical analysis of viability. Int. J. Greenhouse Gas Control 3, 757–767. doi: 10.1016/j.ijggc.2009.07.001
Hartmann, J., Suitner, N., Lim, C., Schneider, J., Marín-Samper, L., Arístegui, J., et al. (2022). Stability of alkalinity in Ocean Alkalinity Enhancement (OAE) approaches – consequences for durability of CO2 storage. doi: 10.5194/bg-2022-126
He, J., and Tyka, M. D. (2023). Limits and CO2 equilibration of near-coast alkalinity enhancement. Biogeosciences 20, 27–43. doi: 10.5194/bg-20-27-2023
Hibbeln, C. F., Marsh, P., Myers, C. R., Valdez, P. J., Edmundson, S. J., and Subban, C. V. (2024). Maximizing marine carbon removal by coupling electrochemical and biological methods. Environ. Sci. Technol. Lett. 11, 438–444. doi: 10.1021/acs.estlett.4c00107
Hodgman, C. D. (1952). Handbook of chemistry and physics. 34th Edn. Cleveland: Chemical Rubber Publishing Co.
House, K. Z., House, C. H., Schrag, D. P., and Aziz, M. J. (2007). Electrochemical acceleration of chemical weathering as an energetically feasible approach to mitigating anthropogenic climate change. Environ. Sci. Technol. 41, 8464–8470. doi: 10.1021/es0701816
Hu, Z.-Q., Wang, A.-M., and Zhang, H.-F. (2017). “Amorphous materials” in Modern inorganic synthetic chemistry (Amsterdam, The Netherlands and Cambridge, MA, USA: Elsevier), 641–667.
IPCC (2022). “Summary for policymakers” in Climate change 2021: The physical science basis, contribution of working group I to the sixth assessment report of the intergovernmental panel on climate change. eds. V. Masson-Delmotte, P. Zhai, A. Pirani, S. L. Connors, and C. Péan (Cambridge, United Kingdom, New York, NY, USA: Cambridge University Press), 3–32.
Jin, S., Tyka, M. D., Rodriguez Martinez, C., Davis, C. E., Van Arsdale, C., and Papania-Davis, A. R. (2025). Negative emission enabled by combining ocean alkalinity enhancement and waste concrete upcycling. ACS Sustain. Chem. Eng. 13, 1532–1543. doi: 10.1021/acssuschemeng.4c07507
Keppler, L., Landschützer, P., Gruber, N., Lauvset, S. K., and Stemmler, I. (2020). Seasonal carbon dynamics in the near-global ocean. Global Biogeochemical Cycles 34:e2020GB006571. doi: 10.1029/2020GB006571
Kheshgi, H. S. (1995). Sequestering atmospheric carbon dioxide by increasing ocean alkalinity. Energy 20, 915–922.
Kitidis, V., Rackley, S. A., Burt, W. J., Rau, G. H., Fawcett, S., Taylor, M., et al. (2024). Magnesium hydroxide addition reduces aqueous carbon dioxide in wastewater discharged to the ocean. Communications Earth & Environment 5:354. doi: 10.1038/s43247-024-01506-4
Köhler, P., Hartmann, J., and Wolf-Gladrow, D. A. (2010). Geoengineering potential of artificially enhanced silicate weathering of olivine. Proc. Natl. Acad. Sci. 107, 20228–20233. doi: 10.1073/pnas.1000545107
Koutsoukos, P. G., and Kontoyannis, C. G. (1984). Precipitation of calcium carbonate in aqueous solutions. J. Chemical Society, Faraday Trans. 1: Physical Chem. Condensed Phases 80:1181.
Kudoh, Y., Kameda, J., and Kogure, T. (2006). Dissolution of brucite on the (001) surface at neutral pH: in situ atomic force microscopy observations. Clay Clay Miner. 54, 598–604. doi: 10.1346/CCMN.2006.0540506
Li, J., and Hitch, M. (2015). Carbon dioxide sorption isotherm study on pristine and acid-treated olivine and its application in the vacuum swing adsorption process. Minerals 5, 259–275. doi: 10.3390/min5020259
Lv, Y., Bai, L., Ma, Y., and Zhao, L. (2024). Investigation of crystallization growth characteristics of Mg(OH)2 crystals under unconstrained conditions. Materials 17:1956. doi: 10.3390/ma17091956
Meysman, F. J. R., and Montserrat, F. (2017). Negative CO2 emissions via enhanced silicate weathering in coastal environments. Biol. Lett. 13:20160905. doi: 10.1098/rsbl.2016.0905
Montserrat, F., Renforth, P., Hartmann, J., Leermakers, M., Knops, P., and Meysman, F. J. R. (2017). Olivine dissolution in seawater: implications for CO2 sequestration through enhanced weathering in coastal environments. Environ. Sci. Technol. 51, 3960–3972. doi: 10.1021/acs.est.6b05942
Moras, C. A., Bach, L. T., Cyronak, T., Joannes-Boyau, R., and Schulz, K. G. (2022). Ocean alkalinity enhancement – avoiding runaway CaCO 3 precipitation during quick and hydrated lime dissolution. Biogeosciences 19, 3537–3557. doi: 10.5194/bg-19-3537-2022
Novakovic, D., Isomäki, A., Pleunis, B., Fraser-Miller, S. J., Peltonen, L., Laaksonen, T., et al. (2018). Understanding dissolution and crystallization with imaging: a surface point of view. Mol. Pharm. 15, 5361–5373. doi: 10.1021/acs.molpharmaceut.8b00840
Phillips, V. A., Kolbe, J. L., and Opperhauser, H. (1977). Effect of pH on the growth of Mg(OH)2 crystals in an aqueous environment at 60°C. J. Cryst. Growth 41, 228–234.
Pilarska, A. A., Klapiszewski, Ł., and Jesionowski, T. (2017). Recent development in the synthesis, modification and application of Mg(OH)2 and MgO: a review. Powder Technol. 319, 373–407. doi: 10.1016/j.powtec.2017.07.009
Pokrovsky, O. S., and Schott, J. (2004). Experimental study of brucite dissolution and precipitation in aqueous solutions: Surface speciation and chemical affinity control. Geochimica et Cosmochimica Acta 68, 31–45. doi: 10.1016/S0016-7037(03)00238-2
Rau, G. H. (2009). Electrochemical CO2 capture and storage with hydrogen generation. Energy Procedia 1, 823–828. doi: 10.1016/j.egypro.2009.01.109
Renforth, P., and Henderson, G. (2017). Assessing Ocean alkalinity for carbon sequestration. Rev. Geophys. 55, 636–674. doi: 10.1002/2016RG000533
Rigopoulos, I., Harrison, A. L., Delimitis, A., Ioannou, I., Efstathiou, A. M., Kyratsi, T., et al. (2018). Carbon sequestration via enhanced weathering of peridotites and basalts in seawater. Appl. Geochem. 91, 197–207. doi: 10.1016/j.apgeochem.2017.11.001
Ringham, M. C., Hirtle, N., Shaw, C., Lu, X., Herndon, J., Carter, B. R., et al. (2024). An assessment of ocean alkalinity enhancement using aqueous hydroxides: kinetics, efficiency, and precipitation thresholds. Biogeosciences 21, 3551–3570. doi: 10.5194/bg-21-3551-2024
Rogelj, J., Popp, A., Calvin, K. V., Luderer, G., Emmerling, J., Gernaat, D., et al. (2018). Scenarios towards limiting global mean temperature increase below 1.5 °C. Nat. Clim. Chang. 8, 325–332. doi: 10.1038/s41558-018-0091-3
Schiffer, Z. J., Lucas, É., Watkins, N. B., Ardo, S., Xiang, C., and Atwater, H. A. (2024). Acid and Base Generation via an Electrochemical Hydrogen-Looping Cell Tailored for Carbon Removal Applications. Device 2:100506. doi: 10.1016/j.device.2024.100506
Schuiling, R. D., and de Boer, P. L. (2011). Rolling stones; fast weathering of olivine in shallow seas for cost-effective CO2 capture and mitigation of global warming and ocean acidification. Earth Syst. Dynam. Discuss. 2, 551–568. doi: 10.5194/esdd-2-551-2011
Scott, A., Oze, C., Shah, V., Yang, N., Shanks, B., Cheeseman, C., et al. (2021). Transformation of abundant magnesium silicate minerals for enhanced CO2 sequestration. Communications Earth & Environment 2:25. doi: 10.1038/s43247-021-00099-6
Seeger, M., Otto, W., Flick, W., Bickelhaupt, F., and Akkerman, O. S. (2011). “Magnesium compounds” in Ullmann’s Encyclopedia of industrial chemistry. ed. E. Wiley-VCH (Weinheim, Germany: Wiley).
Shaw, C. (2025). Data set for "using magnesium hydroxide for ocean alkalinity enhancement: elucidating the role of formation conditions on material properties and dissolution kinetics" [data set]. Zenodo. doi: 10.5281/zenodo.15121979
Shirasaki, T. (1961). ABSTRACTS from the Denki-Kagaku Vol. 29. J. Electrochem. Society Japan, 29, E202–E206.
Song, X., Tong, K., Sun, S., Sun, Z., and Yu, J. (2013). Preparation and crystallization kinetics of micron-sized Mg(OH)2 in a mixed suspension mixed product removal crystallizer. Front. Chem. Sci. Eng. 7, 130–138. doi: 10.1007/s11705-013-1332-7
Suitner, N., Faucher, G., Lim, C., Schneider, J., Moras, C. A., Riebesell, U., et al. (2024). Ocean Alkalinity Enhancement Approaches and the Predictability of Runaway Precipitation Processes: Results of an Experimental Study to Determine Critical Alkalinity Ranges for Safe and Sustainable Application Scenarios. Biogeosciences 21, 4587–4604. doi: 10.5194/bg-21-4587-2024
Teir, S., Revitzer, H., Eloneva, S., Fogelholm, C.-J., and Zevenhoven, R. (2007). Dissolution of natural serpentinite in mineral and organic acids. Int. J. Miner. Process. 83, 36–46. doi: 10.1016/j.minpro.2007.04.001
Thanh, N. T. K., Maclean, N., and Mahiddine, S. (2014). Mechanisms of nucleation and growth of nanoparticles in solution. Chem. Rev. 114, 7610–7630. doi: 10.1021/cr400544s
Turek, M., and Gnot, W. (1995). Precipitation of magnesium hydroxide from brine. Ind. Eng. Chem. Res. 34, 244–250.
Vassallo, F., La Corte, D., Cancilla, N., Tamburini, A., Bevacqua, M., Cipollina, A., et al. (2021). A pilot-plant for the selective recovery of magnesium and calcium from waste brines. Desalination 517:115231. doi: 10.1016/j.desal.2021.115231
Vermilyea, D. A. (1969). The Dissolution of MgO and Mg(OH)2 in Aqueous Solutions. Journal of The Electrochemical Society 116:1179. doi: 10.1149/1.2412273
Wang, H., Pilcher, D. J., Kearney, K. A., Cross, J. N., Shugart, O. M., Eisaman, M. D., et al. (2023). Simulated impact of ocean alkalinity enhancement on atmospheric CO2 removal in the Bering Sea. Earths Future 11:e2022EF002816. doi: 10.1029/2022EF002816
Williamson, G. K., and Hall, W. H. (1953). X-ray line broadening from filed aluminium and wolfram. Acta Metallurgica 1, 22–31. doi: 10.1016/0001-6160(53)90006-6
Wolf-Gladrow, D. A., Zeebe, R. E., Klaas, C., Körtzinger, A., and Dickson, A. G. (2007). Total alkalinity: The explicit conservative expression and its application to biogeochemical processes. Marine Chemistry 106, 287–300. doi: 10.1016/j.marchem.2007.01.006
Wright, H. (2022). The Industrial Side of Magnesium Hydroxide (Part 1). Garrison Minerals. Available online at: https://www.garrisonminerals.com/post/the-industrial-side-part-1
Wu, K.-J., Tse, E. C. M., Shang, C., and Guo, Z. (2022). Nucleation and growth in solution synthesis of nanostructures – from fundamentals to advanced applications. Prog. Mater. Sci. 123:100821. doi: 10.1016/j.pmatsci.2021.100821
Xue, D., Yan, X., and Wang, L. (2009). Production of specific Mg(OH)2 granules by modifying crystallization conditions. Powder Technology 191, 98–106. doi: 10.1016/j.powtec.2008.09.012
Yang, B., Leonard, J., and Langdon, C. (2023). Seawater alkalinity enhancement with magnesium hydroxide and its implication for carbon dioxide removal. Mar. Chem. 253:104251. doi: 10.1016/j.marchem.2023.104251
Yousefi, S., Ghasemi, B., Tajally, M., and Asghari, A. (2017). Optical properties of MgO and Mg(OH)2 nanostructures synthesized by a chemical precipitation method using impure brine. J. Alloys Compd. 711, 521–529. doi: 10.1016/j.jallcom.2017.04.036
Zeebe, R. E., and Wolf-Gladrow, D. A. (2001). CO₂ in seawater: Equilibrium, kinetics, isotopes. Amsterdam; New York: Elsevier.
Zhang, Q., Awolayo, A. N., Phelps, P. R., Vadsariya, S., Laureijs, C. T., Eisaman, M. D., et al. (2024). Enhanced cation release via acid pretreatment for gigaton-scale geologic CO2 sequestration in basalt. Int. J. Greenhouse Gas Control 139:104266. doi: 10.1016/j.ijggc.2024.104266
Keywords: magnesium hydroxide, ocean alkalinity enhancement, marine carbon dioxide removal, seawater precipitation, crystallinity, materials science, dissolution kinetics
Citation: Shaw C, Ringham MC, Carter BR, Tyka MD and Eisaman MD (2025) Using magnesium hydroxide for ocean alkalinity enhancement: elucidating the role of formation conditions on material properties and dissolution kinetics. Front. Clim. 7:1616362. doi: 10.3389/fclim.2025.1616362
Edited by:
Michael Fong, Independent Researcher, Gaithersburg, MD, United StatesReviewed by:
Mohammed Hashim, University of Southern California, United StatesPhilip Davies, University of Birmingham, United Kingdom
Copyright © 2025 Shaw, Ringham, Carter, Tyka and Eisaman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cody Shaw, Y29keS5zaGF3QHN0b255YnJvb2suZWR1
†Deceased