 Raffaele Altara1,2,3*
Raffaele Altara1,2,3* Fouad A. Zouein4*
Fouad A. Zouein4* Rita Dias Brandão5
Rita Dias Brandão5 Saeed N. Bajestani3,6
Saeed N. Bajestani3,6 Alessandro Cataliotti1,2
Alessandro Cataliotti1,2 George W. Booz7
George W. Booz7- 1Institute for Experimental Medical Research, Oslo University Hospital and University of Oslo, Oslo, Norway
- 2KG Jebsen Center for Cardiac Research, Oslo, Norway
- 3Department of Pathology, School of Medicine, University of Mississippi Medical Center, Jackson, MS, United States
- 4Faculty of Medicine, Department of Pharmacology and Toxicology, American University of Beirut, Beirut, Lebanon
- 5Department of Clinical Genetics, Maastricht University Medical Centre, Maastricht, Netherlands
- 6Department of Ophthalmology, School of Medicine, University of Mississippi Medical Center, Jackson, MS, United States
- 7Department of Pharmacology and Toxicology, School of Medicine, University of Mississippi Medical Center, Jackson, MS, United States
Standard therapies for heart failure with preserved ejection fraction (HFpEF) have been unsuccessful, demonstrating that the contribution of the underlying diastolic dysfunction pathophysiology differs from that of systolic dysfunction in heart failure and currently is far from being understood. Complicating the investigation of HFpEF is the contribution of several comorbidities. Here, we selected three established rat models of diastolic dysfunction defined by three major risk factors associated with HFpEF and researched their commonalities and differences. The top differentially expressed genes in the left ventricle of Dahl salt sensitive (Dahl/SS), spontaneous hypertensive heart failure (SHHF), and diabetes 1 induced HFpEF models were derived from published data in Gene Expression Omnibus and used for a comprehensive interpretation of the underlying pathophysiological context of each model. The diversity of the underlying transcriptomic of the heart of each model is clearly observed by the different panel of top regulated genes: the diabetic model has 20 genes in common with the Dahl/SS and 15 with the SHHF models. Advanced analytics performed in Ingenuity Pathway Analysis (IPA®) revealed that Dahl/SS heart tissue transcripts triggered by upstream regulators lead to dilated cardiomyopathy, hypertrophy of heart, arrhythmia, and failure of heart. In the heart of SHHF, a total of 26 genes were closely linked to cardiovascular disease including cardiotoxicity, pericarditis, ST-elevated myocardial infarction, and dilated cardiomyopathy. IPA Upstream Regulator analyses revealed that protection of cardiomyocytes is hampered by inhibition of the ERBB2 plasma membrane-bound receptor tyrosine kinases. Cardioprotective markers such as natriuretic peptide A (NPPA), heat shock 27 kDa protein 1 (HSPB1), and angiogenin (ANG) were upregulated in the diabetes 1 induced model; however, the model showed a different underlying mechanism with a majority of the regulated genes involved in metabolic disorders. In conclusion, our findings suggest that multiple mechanisms may contribute to diastolic dysfunction and HFpEF, and thus drug therapies may need to be guided more by phenotypic characteristics of the cardiac remodeling events than by the underlying molecular processes.
Introduction
Comparatively isolated diastolic dysfunction or stiffness of the left ventricle is a chronic pathological condition that evolves into heart failure with preserved ejection fraction (HFpEF). An increasing body of evidence reports that HFpEF is frequent in women and the elderly (1–3). In fact, HFpEF accounts for about half the cases of heart failure (HF) and is the leading cause of hospital admission in patients over 65 years of age. Although individuals with HFpEF exhibit similar mortality rates as those that have heart failure with reduced ejection fraction (HFrEF) or impaired cardiac contractility, there are currently no proven effective medicines for this condition (1, 4–6) Thus, HFpEF is a major unmet medical need and there is an urgency for new therapeutic approaches and strategies that target mechanisms specific for HFpEF. Notably, HFpEF is associated with the co-presence of several comorbidities (7). Particularly among younger persons, HFpEF is associated with the interrelated cardiovascular risk factors of obesity and type II diabetes (8). Given the obesity epidemic that is attributed to the Western diet and lifestyle, and an aging population, it is not surprising then that HFpEF is predicted to be the primary cause of HF within a decade.
A clinical cardiac phenotype like HFpEF is difficult to study due to its complex nature that involves genetic, molecular, and environmental factors. Ultimately, increased cardiac stiffness in HFpEF is thought to arise from loss of nitric oxide signaling due to microvascular inflammation, increased cardiac fibrosis, and a concentric pattern of cardiac hypertrophy that also compromises ventricular chamber size. Several animal models are characterized by diastolic dysfunction and have been used to study HFpEF. However, it is unclear what these different models have in common, or their differences, as far as disease etiology and cardiac remodeling, which could reveal common or fundamental signaling pathways representing potential therapeutic targets. Given the rapid growth of experimental genetic data in repositories, such as Gene Expression Omnibus (GEO), data mining is a powerful approach that can create countless openings for holistic investigations using assorted studies.
Our in silico investigation aimed towards identifying novel pathological pathways in the heart that are relevant for the development of HFpEF. In our study, we make use of public datasets, to perform top-notch biostatistics and bioinformatics that aid in deciphering genetic events that drive the detrimental cardiac remodeling in three rodent models with diastolic dysfunction, the Dahl salt sensitive rat (Dahl/SS), the spontaneously hypertensive heart failure (SHHF) rat, and the type I diabetic rat.
Methods
GEO Search
GEO search for microarray data from heart in three animal models with underlying diastolic disfunction: the Dahl Salt Sensitive (GSE66617), the Spontaneous Hypertensive Heart Failure (GSE2876), and the Streptozotocin/Diabetes1 (GSE6880). The total number of samples of each dataset was n = 28, 10 and 6 for Dahl SS, SHHF, and diabetic, respectively.
Rat Models Selected
Dahl Salt Sensitive (SS) rats were fed either a 0.3% NaCl (normal salt, NS, control group) or with 8% NaCl (high salt, HS, heart failure group) diet until evidence of left ventricular dysfunction (9). The authors extracted total RNA from left ventricular samples at the stage of heart failure using mirVana (Applied Biosystems) according to the manufacturer’s instructions (9). Three samples from each group were pooled. cDNA and Amino Allyl aRNA was synthesized by Amino Allyl MessageAmp II aRNA Amplification Kit (Ambion). CyeDye Coupling and fragmentation were performed according to the manufacturer’s protocol (TORAY Industries, Inc, Tokyo, Japan). Samples were then hybridized for 16 h at 37°C on a Rat Oligo chip 20 K (TORAY Industries, Inc, Tokyo, Japan). Scanning was performed on a 3D-Gene Scanner (Toray Industries Inc., Tokyo, Japan).
Spontaneously Hypertensive and Heart Failure-prone (SHHF) rats were obtained by backcrossing a Koletsky obese rat to a Spontaneously Hypertensive Rat (SHR/N) (10). Controls were 4 months old lean rats and heart failure rats were 10 months old obese rats (10). Total RNA was extracted with TRIzol (GIBCO) according to the manufacturer’s protocol and reverse transcribed with Superscript enzyme (Invitrogen) and labelling the cDNA with 33P. Purification of cDNA was performed using NucleoSpin Extract (Macherey-Nagel). The kit Array-Advantage GF (Ambion) was used to hybridize the samples into pan-genomic macroarray nylon membranes (RZPD, Berlin), which were exposed for 24 h and scanned with a Scan Typhoon 9400 (resolution at 50 µm per pixel).
Diabetic rats were induced with streptozotocin injection at 8 weeks, which led to a model of insulin-deficient type 1 diabetes (11). Rats were sacrificed at 12 weeks (so diabetic for 4 weeks) (11). RNA was extracted with TRIzol (GIBCO) and cleaned with RNeasy total RNA mini kit (Qiagen). Superscript II (Life Technologies) was used for first strand cDNA synthesis. Subsequent steps necessary to hybridize the samples on Affymetrix RAE 230A microarrays were performed according to the Affymetrix protocol. Arrays were scanned on an Hewlett-Packard Gene Array scanner.
Microarray Data Analysis
GEO2R has been used as a base for the analysis of the Dahl/SS dataset, whereas the Limma package was used to analyze the Diabetic and SHHF model. Distribution of value data for the Samples used was calculated (data not shown) and median-centered values indicated that the data were normalized and cross-comparable. Fold-changes per group were calculated using average Disease/Control. Statistical significance of differences in gene expression was assessed using the nonparametric Wilcoxon signed-rank test and the log2-transformed data.
The differentially expressed genes (DEGs) with fold-change (FC) >2 and < −2 and adjusted p-value < 0.05 have been selected for Dahl/SS and SHHF, whereas FC >1.5 and FC < −1.5 was chosen for Diabetic 1 due to the high level of confidence [false discovery rate (FDR) = 0, (Figure S1)] at p-values < 0.05.
The Benjamini and Hochberg false discovery rate method was selected by default because it is the most commonly used adjustment for microarray data and provides a good balance between discovery of statistically significant genes and limitation of false positives.
Gene Set Enrichment Analysis (GSEA) was performed for each of the three datasets using QIAGEN’s Ingenuity Pathway Core Analysis (IPA, QIAGEN Redwood City, www.qiagen.com/ingenuity). The Ingenuity Knowledge Base was used as reference set and both direct and indirect experimentally confirmed relationships from humans and rodents. Z-scores of >2 or < −2 were considered significant. Details about the statistics used can be found at the Ingenuity website: http://www.ingenuity.com/products/ipa#/?tab=resources.
Genes have been mapped in IPA for further analysis (see Table 1).
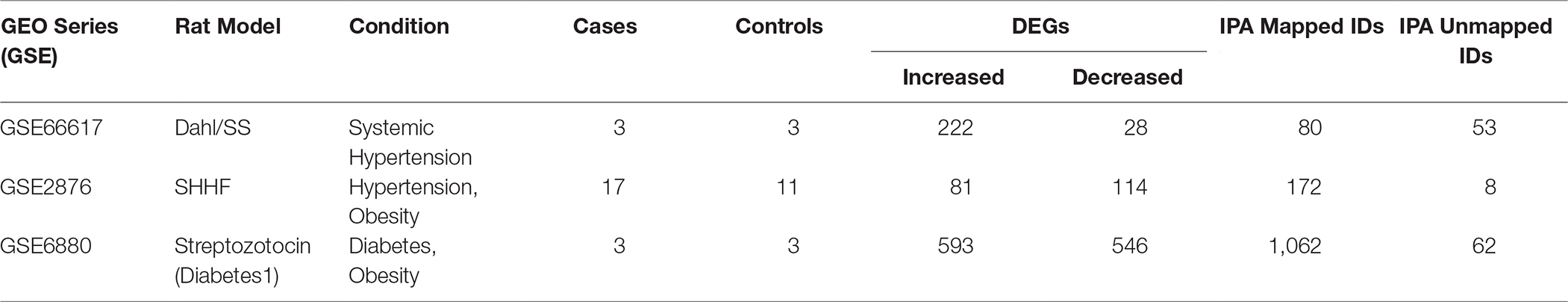
Table 1. DEGs analysed in IPA
Results
Animal Models of HFpEF
Our GEO search for published microarray data on hearts from animal models of diastolic dysfunction was narrowed down to the Dahl/SS, streptozotocin-induced diabetic, and SHHF rat models. Table 1 shows the overall number of DEGs found for each animal model. The top 250 DEGs (genes with the smallest p-value) calculated with GEO2R indicated that the diabetic model has 20 genes in common with the Dahl/SS and 15 with the SHHF models (Table 2). Given the high cumulative number of significant calls for the diabetic rat model (Figure S1) we included up to 1,139 genes with a local FDR (false discovery rate) of 0.
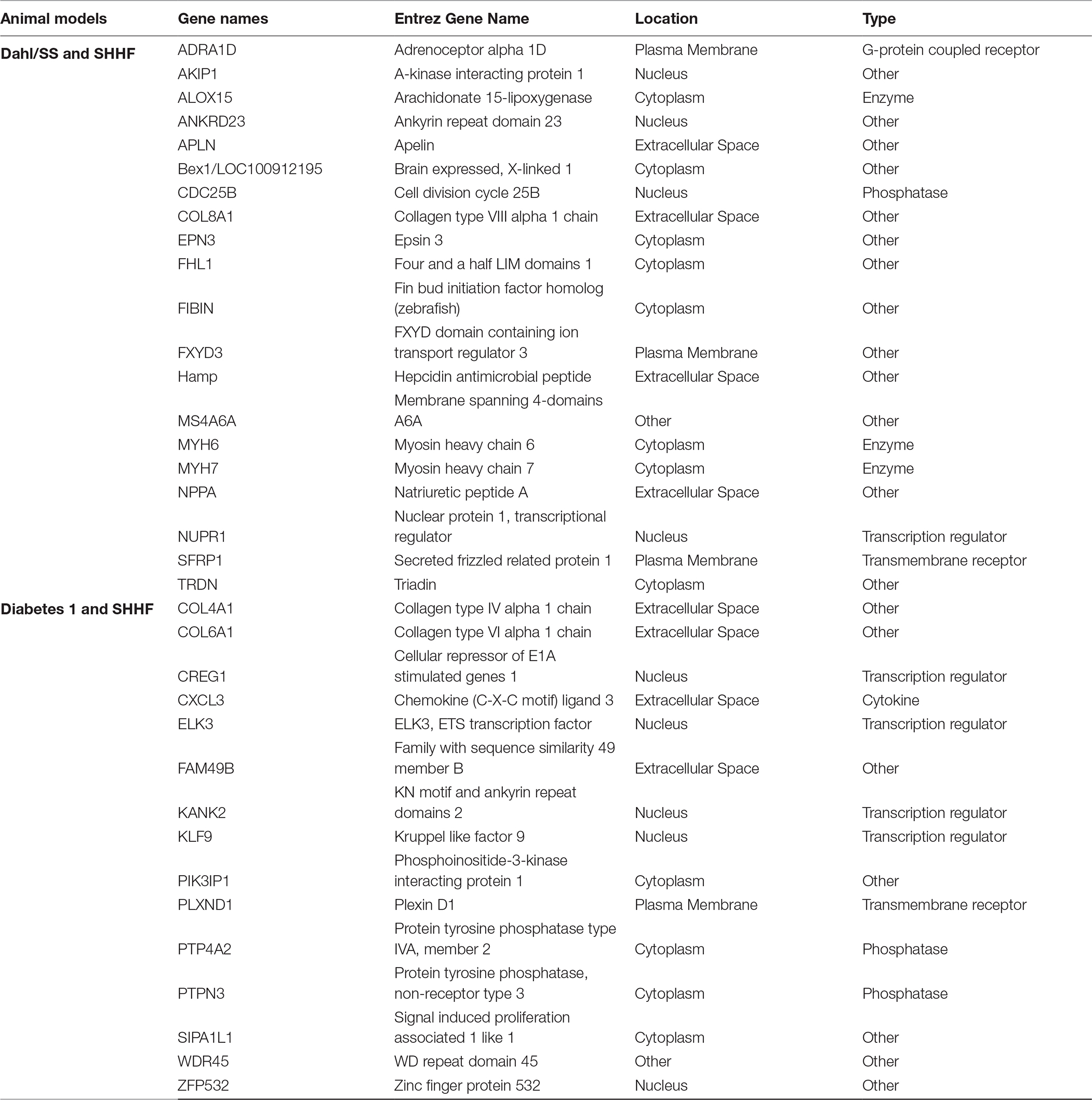
Table 2. Common DEGs among the HRpEF animal models
Cardiac Genes Assessed by GSEA
IPA generated annotations were derived for the DEGs in each of the datasets. Among the three animal models we selected, only the Dahl/SS and SHHF models showed that over-represented genes (n = 25 and n = 26, respectively) determined by DEG analysis are strongly associated with cardiovascular diseases. The cardiovascular disease phenotypes of these two models appear to have different gene datasets as none of the DEGs identified are in common (Table 3). SHHF with 17 genes is also associated with metabolic disease, whereas the diabetic rat is solely associated with metabolic disease with 175 genes (Table 4). These two datasets appear to have in common phosphoinositide-3-kinase interacting protein 1 (PIK3IP1), a negative regulator of PI3K and physiological, but not pathological cardiac hypertrophy (12).
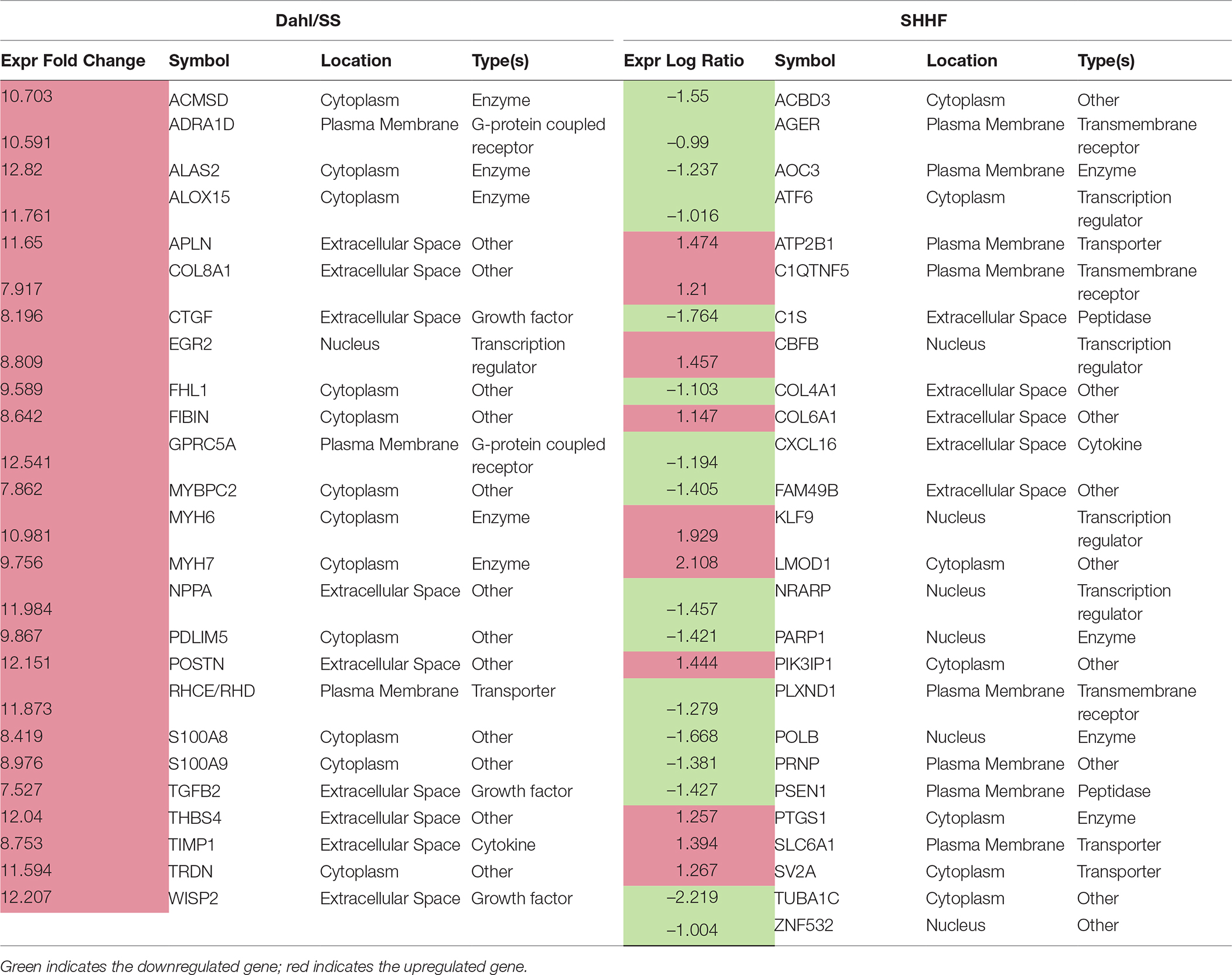
Table 3. DEGs linked to cardiovascular disease
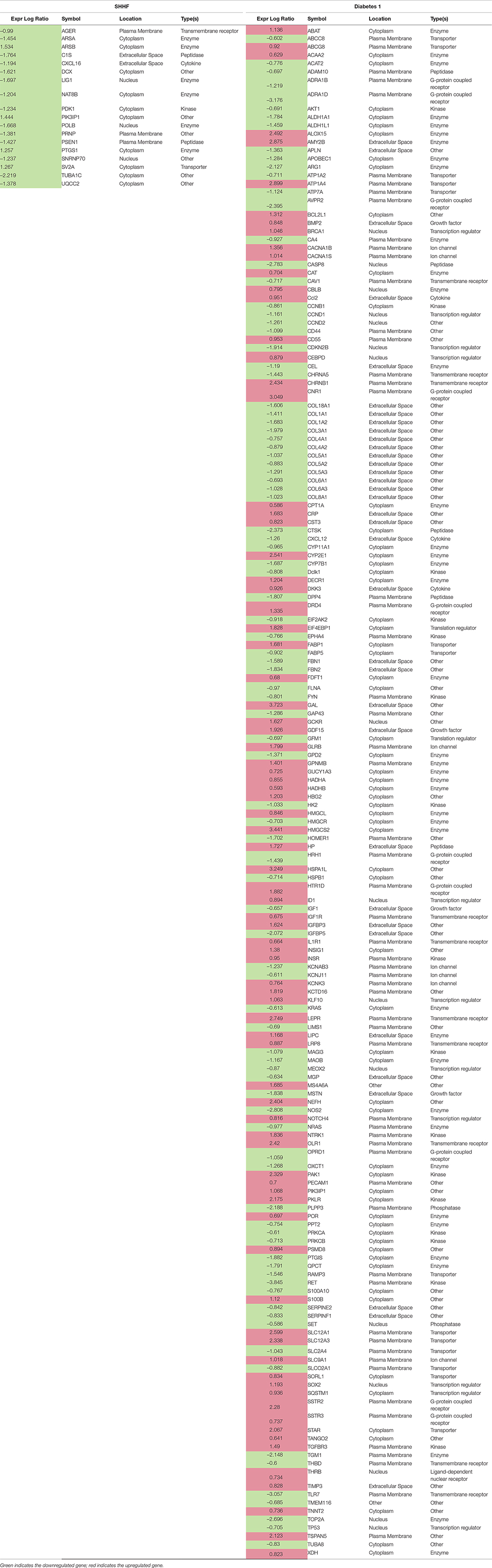
Table 4. DEGs associated with metabolic disease
Pathway Analysis and Pathophysiological Mechanisms
The contribution of every DEGs to the development of diastolic dysfunction was calculated in the context of molecular network and biological progresses using the algorithms described in the Materials and Methods section.
Dahl/SS
The biological activities occurring in the heart tissue were identified using the IPA Upstream Regulator analytic on the gene expression changes we observed in the dataset. The analysis inferred the likely activation states of 5 transcription regulators given by our DEG dataset. Hence, based on the comparison of the change of direction of the DEGs (i.e., expression in the Dahl/SS model to control) and what is known from the literature we found that the transcription regulators TGFB1, RETNLB, TNF, INHBA, and IL17RA (z-score 2.4, 2.2, 2.1, 2.0, 2.0, respectively) were predicted to be activated (Figure 1). A further examination of the underlying biological activities prompted by our dataset from the hearts of Dahl/SS rats revealed biological activities such as dilated cardiomyopathy, hypertrophy of heart, arrhythmia, and failure of heart (Figure 2).
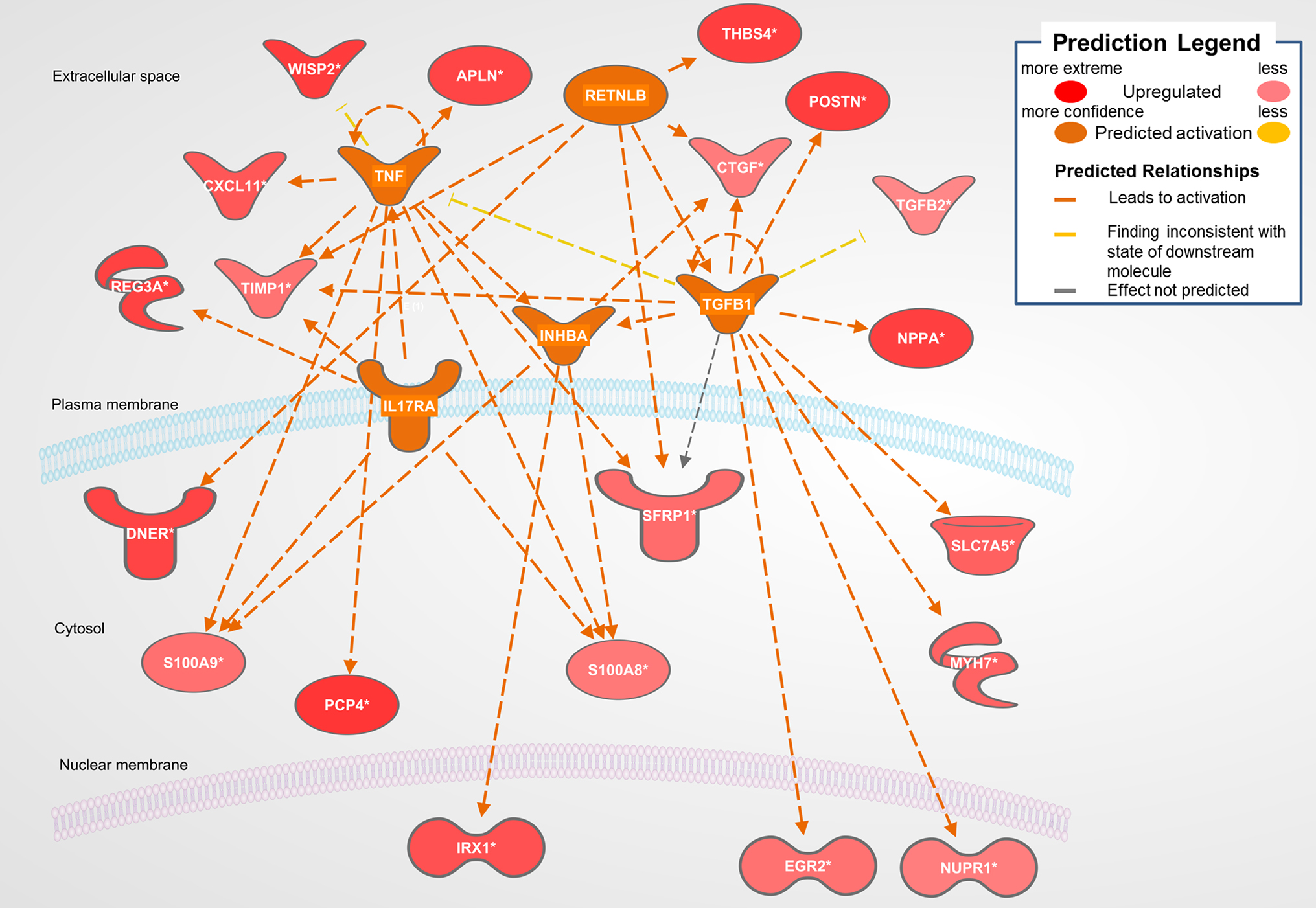
Figure 1. Top predicted activation state in Dahl SS. Asterisks (*) indicate that multiple identifiers in the dataset file map to a single gene in the Global Molecular Network. Figure has been generated using Ingenuity Pathways Analysis (IPA; Ingenuity Systems, Inc., Cambridge, MA, USA).
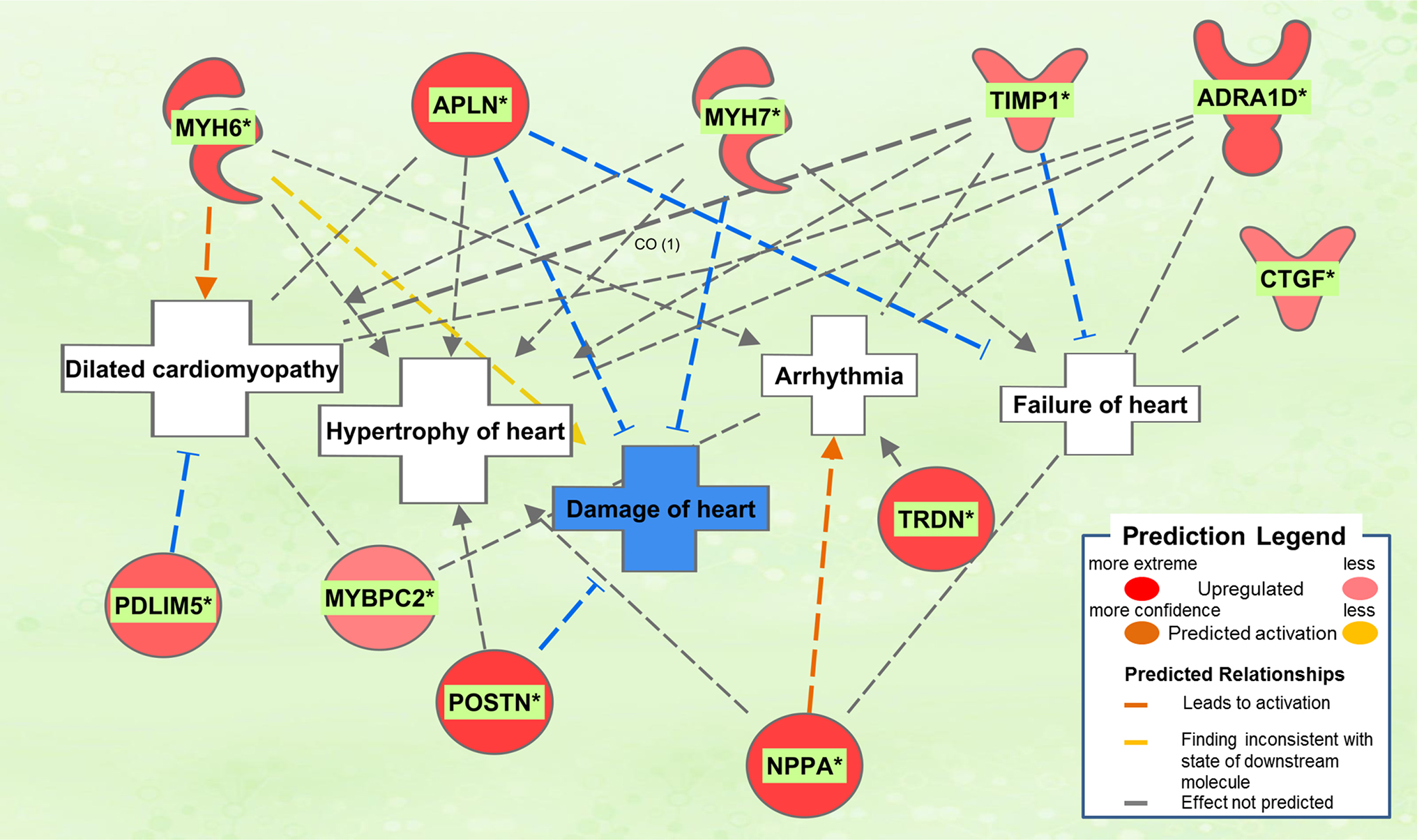
Figure 2. Top pathological functions in Dahl/SS. Asterisks (*) indicate that multiple identifiers in the dataset file map to a single gene in the Global Molecular Network. Figure has been generated using Ingenuity Pathways Analysis (IPA; Ingenuity Systems, Inc., Cambridge, MA, USA).
SHHF
A total of 186 DEGs were observed. Of the 78 significantly upregulated genes, 43 fell into the rather nondescript category of “other”, followed by 10 in the second largest group transcription regulator. Of the 108 downregulated genes, 61 were characterized as “other”, 17 as enzyme, and 6 as transcription regulator. Differently from the Dahl/SS, the SHHF model did not show significant inferred activation of upstream transcription regulators, apart from a moderate activation (z-score 0.3) of NUPR1. In this model, the ERBB2 upstream regulator was predicted to be inhibited (z-score −2.2). As shown in Figure 3, ERBB2 is a plasma membrane-bound receptor tyrosine kinases that is involved in the protection of cardiac myocytes, which is illustrated by the cardiotoxicity of ERBB2-targeted cancer therapies (13). Out of the 250 DEGs determined in the SHHF heart tissue, 26 genes were linked to cardiovascular disease including cardiotoxicity, pericarditis, ST-elevated myocardial infarction, and dilated cardiomyopathy. A list of these genes is provided in Table 3.
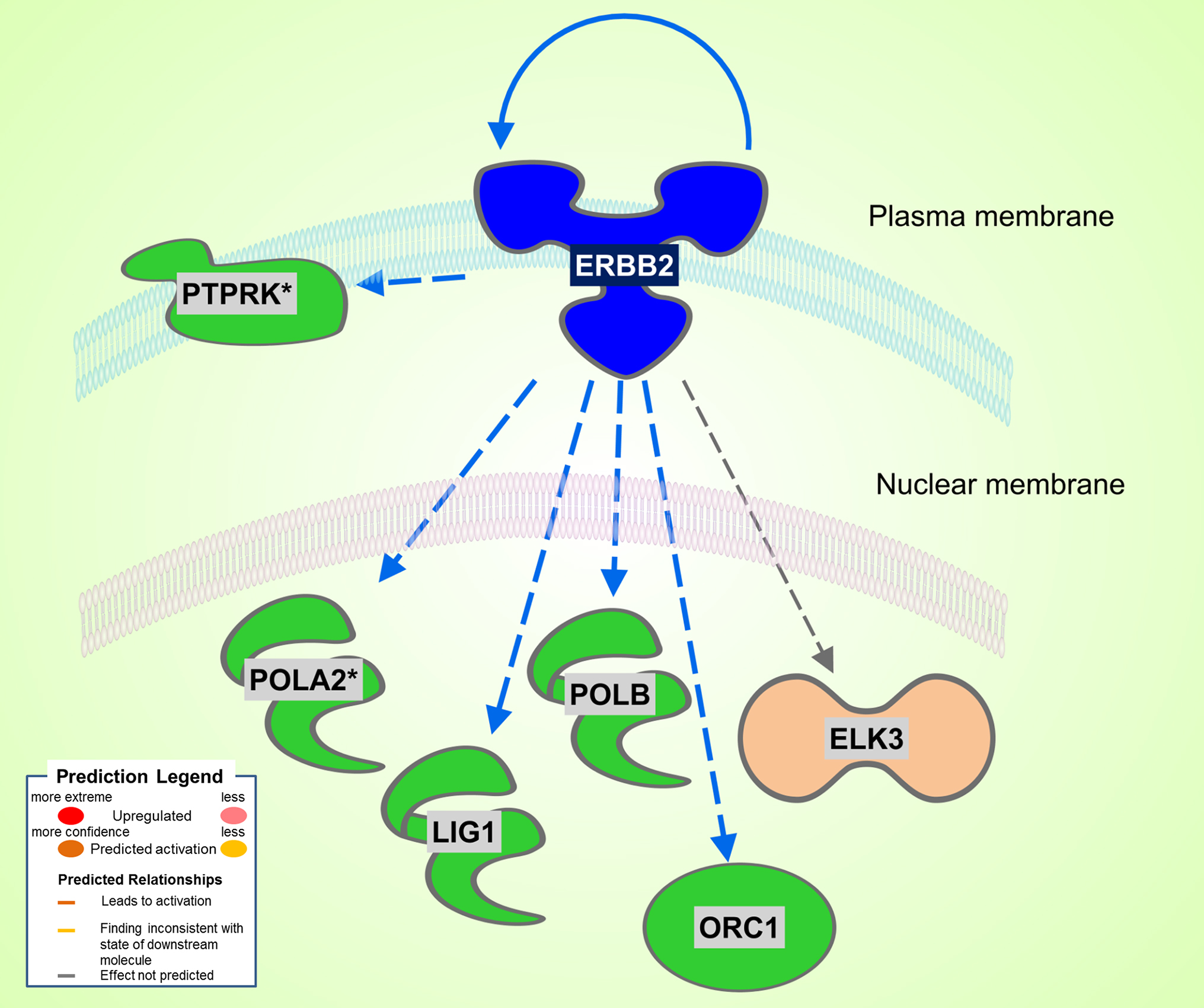
Figure 3. Significant inhibition of ERBB2 kinase in SHHF. Asterisks (*) indicate that multiple identifiers in the dataset file map to a single gene in the Global Molecular Network. Figure has been generated using Ingenuity Pathways Analysis (IPA; Ingenuity Systems, Inc., Cambridge, MA, USA).
Diabetic 1
The diabetic rat model resulted in the highest number of DEGs (see Table 1) consistent with a FDR = 0. A total of 18 (9 activated and 9 inhibited) significant upstream transcriptional regulators that can explain the observed gene expression profile were identified. A list of these genes is displayed in Table 5. Not surprisingly, given the metabolic underpinnings of the model, most (110) of the 558 genes that were upregulated code for enzymes. Other groupings include 30 kinases, 20 G-protein-coupled receptors, 18 ion channels, and 6 growth factors. Upregulated as well is NPPA encoding for atrial natriuretic peptide (ANP), a biomarker for cardiac hypertrophy and heart failure (Figure 4). Two genes that code for recently proposed novel biomarkers for HFpEF were also upregulated: HSPB1, which codes for cardioprotective heat shock protein 27 (hsp27) (14, 15) and ANG which codes for angiogenin, a protein important for vascularization (16).
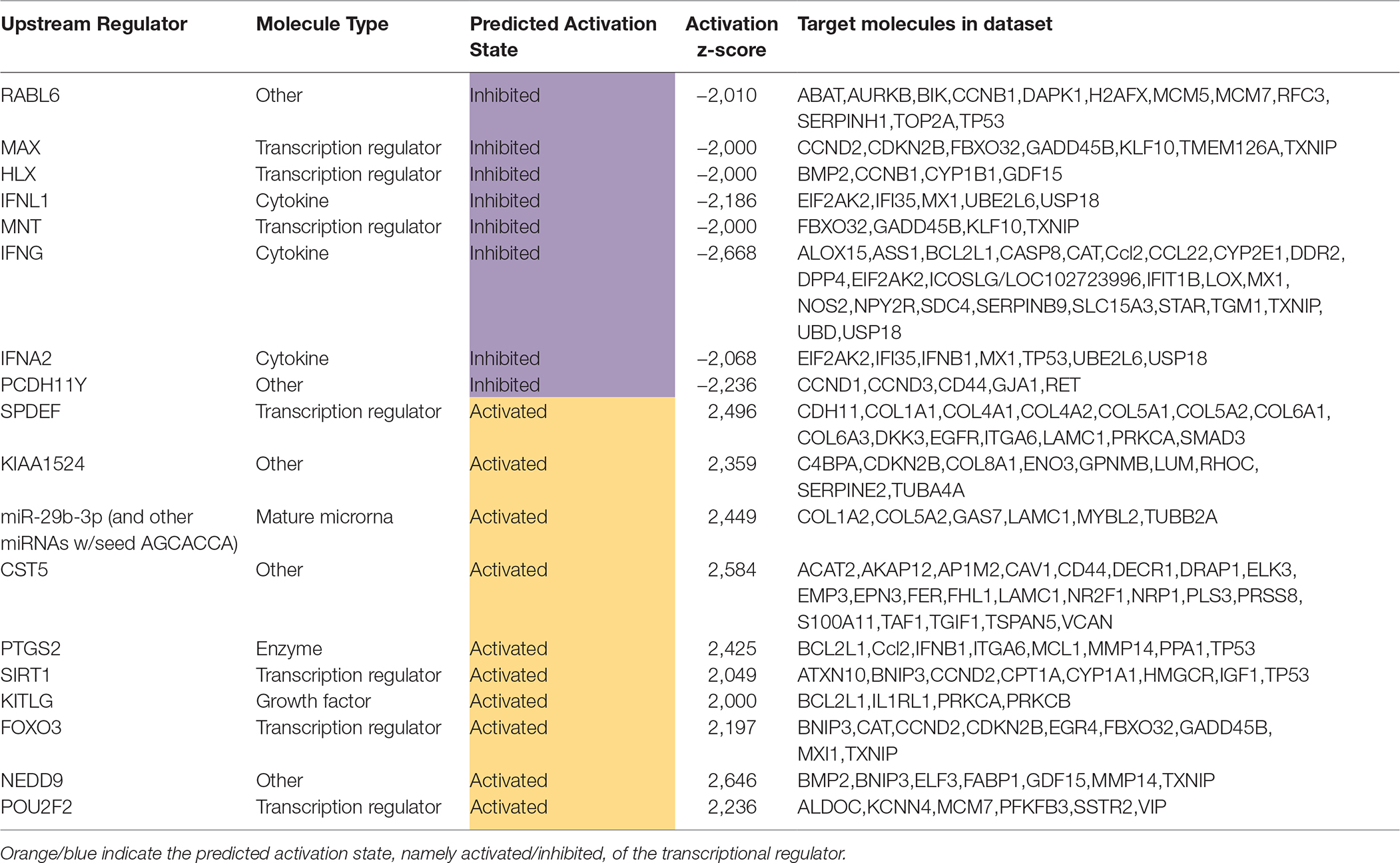
Table 5. Significant upstream transcriptional regulators in the diabetic rat model
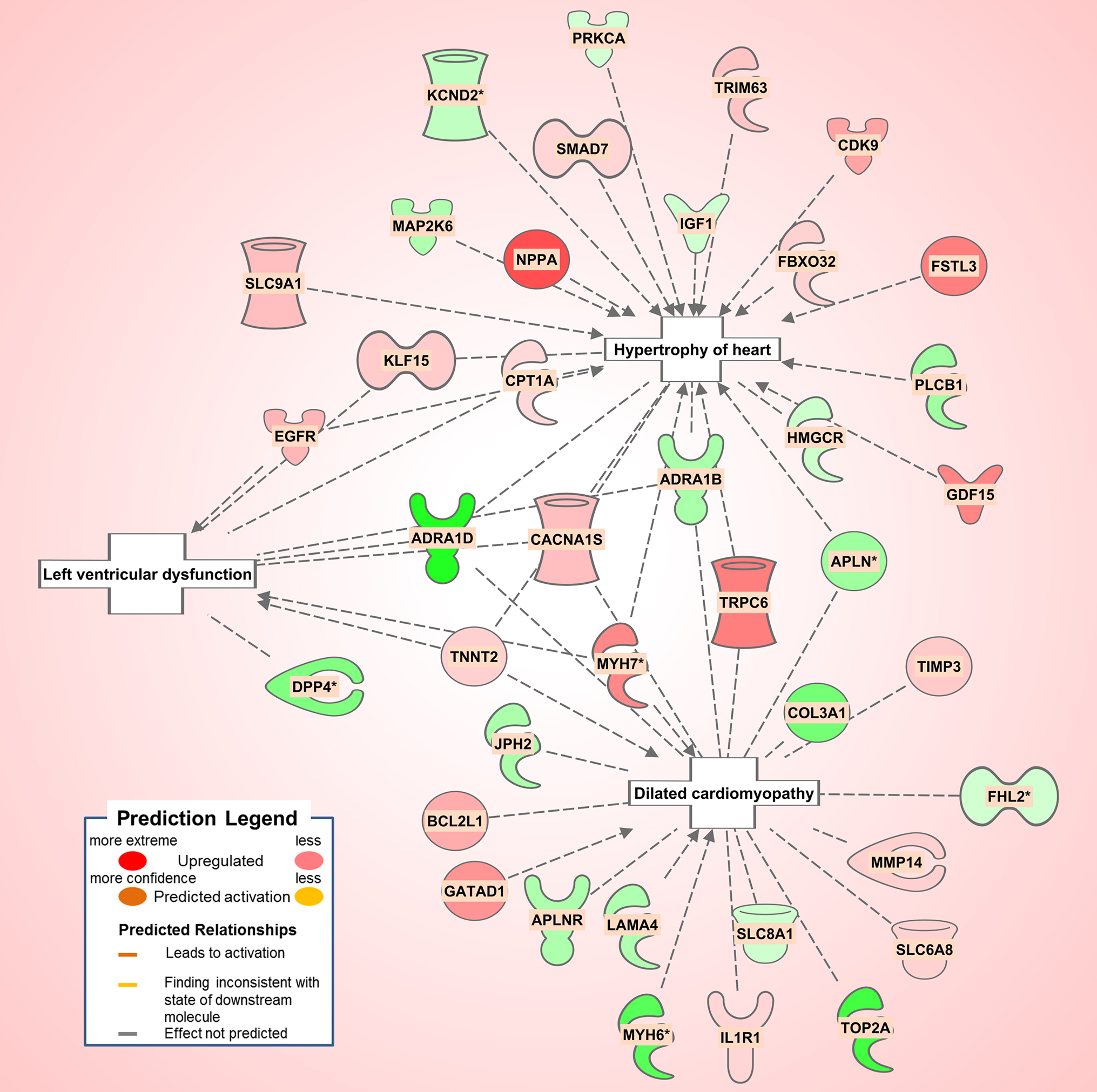
Figure 4. Pathological processes in the diabetic rat model. Asterisks (*) indicate that multiple identifiers in the dataset file mapto a single gene in the Global Molecular Network. Figure has been generated using Ingenuity Pathway Analysis (IPA; Ingenuity Systems, Inc., Cambridge, MA, USA).
Discussion
In our study, we selected three established rat models of diastolic dysfunction that are defined by three major risk factors associated with HFpEF. Remarkably, we found that the cardiac transcriptomics of the models demonstrated little commonality, either in differentially regulated genes or specific biological functions. Overall, our findings highlight the case that divergent molecular processes may underpin a shared phenotype.
Diastolic dysfunction is primarily characterized by stiffening of the left ventricular (LV) tissue, which contributes to impaired relaxation and filling of the ventricle. The specific mechanisms leading to myocardial stiffening are undetermined, but broadly LV stiffness has active and passive components. Passive stiffness describes the inherent stretchiness of the myocardium and is governed by the pliability of both the extracellular matrix and the contractile units. The latter is set by the giant sarcomeric filament, titin, which also determines the elastic recoil of the myocardium, thereby contributing in a major way to LV filling via diastolic suction (17). Evidence indicates that abnormal high-energy phosphate metabolism, specifically increased free cytosolic ADP and enhanced AMP catabolism, may contribute to passive myocardial diastolic stiffness as well (18, 19).
Active stiffness, or reduced relaxation, of the LV tissue depends on Ca2+ homeostasis (20). There are two interrelated aspects of intracellular Ca2+ handling that determine diastolic function; the rate of cytosolic Ca2+ decline following its release from the sarcoplasmic reticulum (SR), and the resting or end-diastolic Ca2+ concentration (21). These parameters are set by the rate of Ca2+ removal from the cell via the sodium calcium exchanger (NCX) and reuptake of Ca2+ into stores by SERCA2, in addition to Ca2+ leak from the SR. We did not observe any changes in the gene expression of calcium handling proteins (SERCA2, NCX, PLN, or CASQ2) in any of the three models of diastolic dysfunction. Of course, our approach does not address the possibility that in any of the models of diastolic dysfunction the activities of the Ca2+ handling protein are negatively affected by posttranslational modification or change in their environment.
Figure 1 displays the targets of TGFB1 that are differentially expressed in the Dahl/SS dataset. TGFB1 plays a central role in hypertension-induced cardiac hypertrophy and fibrosis (22, 23), and consistent with that fact, genes involved in fibrosis/extracellular matrix (ECM) accumulation (TGFB1, POSTN, CTGF, TIMP1) (24) and hypertension-related pathological hypertrophy (NPPA and MYH7) (25–28) are upregulated with hypertension. Interestingly, the Dahl/SS model of heart failure is associated with upregulation of the Wnt signaling inhibitor secreted frizzled-related protein-1 (SFRP1), which protects against ventricular dilation and hypertrophy (29), as well as increased expression of the transporter of large neutral amino acids (SLC7A5). Downstream targets of TGFB1 upregulated in Dahl/SS rats include EGR2 and NUPR1. Based on computational analysis of the transcriptomes at five time points in male murine hearts subjected to transverse aorta banding, the transcription factor EGR2 was recently identified as a new candidate key regulator of cardiovascular-associated genes (30). Nupr1 is a nuclear basic helix-loop-helix protein and transcriptional coregulator that is induced in response to stress in the heart (31). Recent evidence indicates that Nupr1 activation contributes to cardiac fibrosis, while protecting against autophagy and apoptosis that lead to heart failure (32). Elevated circulating levels of both interleukin (IL)-6 and TNF are observed in HFpEF (33), and in the Dahl/SS model TNF shares several downstream targets with TGFB1, including TIMP1, INHBA, and SFRP1 (Figure 1). The role of INHBA or RETNLB in cardiac remodeling is unreported. The predicted downstream target of RETNLB, THBS4, has been shown to attenuate pressure-overload cardiac fibrosis (34). TNF is also linked to S100A9 and S100A8 expression (both significantly increased in our DEG list), as is IL17RA (Interleukin 17 receptor A). IL17RA has been linked to myocardial collagen metabolism in hypertension-induced diastolic dysfunction and heart failure (35, 36). The “damage-associated molecular patterns” (DAMPs), proteins S100A8/A9 contribute to cardiac fibrosis by activating pro-inflammatory NF-κB signaling in cardiac fibroblasts via the receptor for advanced glycation end products (RAGE) and inducing the expression of multiple chemokines and cytokines (37). NF-κB is predicted to be activated in the Dahl/SS model (z-score of 1.92), as evidenced by the marked upregulation of CTGF, CXCL11, SLC7A5, TIMP1, and CDC25B. TNF is also linked to upregulation of the chemokine CXCL11 and the hormone Apelin, which appear to have conflicting actions on cardiac inflammation (38) and hypertrophy (39), respectively. Interestingly, TNF is linked to upregulation of Purkinje cell protein-4 (PCP4), a putative regulator of calmodulin and Ca2+/calmodulin-dependent kinase II (CaMKII) signaling, within the His-Purkinje network and contributor to cardiac arrhythmias (40). The role if any played by DNER, REG3A, and IRX1 to diastolic dysfunction and cardiac hypertrophy is not reported, although genetic variants of the IRX1 gene were recently reported to contribute to the pathogenesis of congenital heart disease (41). Finally, expression levels of CXCL11 were increased in hearts of Dahl/SS rats fed a high salt diet, which is consistent with the concept that inflammation plays a key role in the pathogenesis of HFpEF. The closely related CXCL9, CXCL10, and CXCL11 are chemoattractants for monocytes and lymphocytes. Accumulating evidence has implicated these chemokines in several cardiovascular diseases, including atherosclerosis, hypertension, cardiac hypertrophy, and heart failure, as well as in transplant coronary artery disease and heart transplant rejection (42). Of note, circulating levels of these chemokines were found to provide additional diagnostic utility beyond NT-pro BNP levels for detecting LV diastolic dysfunction in hypertensive patients (43). In addition, we observed that serum levels of CXCL10 are increased in patients with symptomatic heart failure (44). Although CXCL9, -10, and -11 all bind to CXCR3, there is some evidence that these agonists activate opposing responses due to biased signaling that is a fixture of G protein-coupled receptors (44). Whereas CXCL9/CXCL10/CXCR3 interactions drive effector Th1 polarization, CXCL11/CXCR3 binding seems to induce an immunotolerizing state characterized by polarization into regulatory Tr1 lymphocytes that produce anti-inflammatory IL-10.
For the SHHF dataset, none of the DEGs stood out mechanistically in the context of heart failure. Expression of the gene for frizzled class receptor 5 (FZD5), which plays a role in endothelial cells in angiogenesis (45), was increased, as was the gene for the repressor of the protective, anti-oxidant transcription factor Nrf2, KEAP1 (46).
Unexpectedly, the genes for 9 different collagen types were downregulated in diabetic 1 hearts, although a number of genes involved in ECM remodeling and fibrosis were upregulated, including MMP14 (24), as well as SDC4 and LOX, which together have been linked to myocardial stiffness and diastolic dysfunction (47). PDGFD, which codes for platelet-derived growth factor D, a potent pro-fibrotic cytokine (48), was upregulated. Two signaling components of the pro-fibrotic cytokine TGF-β were upregulated, SMAD3 (49) and TGFBR3 (TGF-β receptor 3), although TGFBR1 (TGF-β receptor 1) was downregulated and the inhibitory SMAD7 was upregulated. However, CAV1 was downregulated. CAV1 encodes for caveolin 1, which may inhibit both TGF-β-dependent and –independent cardiac fibrosis (50). Other relevant pro-fibrotic genes that were upregulated, include: the chemokines CCL2 and CXCL3, linked to fibrosis and inflammation (51–53); pro-inflammatory cytokine Il-3 (54) and the innate immune cytokine IFNB1; and the K+ channel KCNN4, which is linked to angiotensin II-induced cardiac fibroblast proliferation and collagen production (55). The gene for the tyrosine kinase receptor EGFR was also upregulated. EGFR is implicated in cardiac fibrosis and hypertrophy (56–58), as well as heart protection (59). In contrast, the gene for TIMP3, which has anti-fibrotic actions (60), was upregulated. Several pro-fibrotic genes were downregulated, including: VCAN (Versican) (61); LUM (Lumican), an ECM-localized proteoglycan that binds collagen and is important for fibrosis, is associated with inflammation, and is increased in experimental and clinical heart failure (62); SERPINE2, which is increased in pressure overload- and angiotensin II-induced cardiac remodeling downstream of ERK1/2 signaling and increases collagen deposition (63); and the integrin ITGA6, associated with collagen deposition (64).
Notable genes linked to metabolism were increased in diabetic hearts, including FABP1 and GDF15. The later encodes a heart-derived hormone, which regulates body growth (65). PFKFB3 encodes a pro-glycolytic enzyme that is upregulated in cardiac progenitor cells by diabetes (66). The gene for the leptin receptor (LEPR), which is implicated to both cardiac hypertrophy and cardioprotection in obesity (67), was upregulated. However, a number of genes associated with cardiac hypertrophy and heart failure due to pressure-overload or angiotensin II infusion were downregulated. CCND2 (cyclin D2), which was implicated in cardiac hypertrophy, was downregulated (68), but so was the cyclin-dependent kinase inhibitor CDKN2B. TP53, a major player in the development of systolic heart failure (69, 70), was downregulated, as was PRKCA. Activation of PKCα is implicated in cardiac hypertrophy and heart failure (71–73).
Oxidative stress is a feature of the diabetic heart (74) and several genes associated with inflammation and oxidative stress were upregulated. CAT, encoding the key antioxidant enzyme catalase was upregulated. On the other hand, TXNIP, which inhibits the anti-oxidative function of thioredoxin, was also upregulated. TXNIP is linked to increased oxidative stress, along with fibrosis and arrhythmias in diabetes (75), as well as diabetic cardiomyopathy (76). The thioredoxin system 2 is a major buffer against H2O2 emission from mitochondria (77), and the present evidence showing that TXNIP is strongly upregulated in the diabetic heart may account for the overall oxidative environment found in the diabetic heart. TSPAN5, encoding a cell surface protein was upregulated. Evidence was recently reported that endothelial Tspan5- and Tspan17-ADAM10 complexes contribute to inflammation by maintaining VE-cadherin expression and promoting T lymphocyte transmigration (78). Another injury-related gene that was upregulated was BNIP3, which is associated with mitochondrial autophagy and apoptosis and is increased in the heart by pressure overload and various stresses (79, 80).
Notably, a number of protective genes were upregulated in the type 1 diabetic heart. The gene for the Smad corepressor, transforming growth interacting factor (TGIF) was increased, as was BMP2. BMP2 may antagonize BMP4-induced cardiomyocyte hypertrophy and apoptosis (81) and TGF-β-induced fibrosis (82). Three genes linked to inhibition of cardiac hypertrophy were upregulate, viz., DKK3 (Dickkopf-3) (83), FBXO32 (encoding E3 ligase, Fbxo32) (84), and GADD45B, which blocks MKK7-induced JNK activation (85). The anti-apoptotic gene BCL2L1 (86) was upregulated, as well as MCL1, which is important in mitochondrial function and autophagy (87). VIP (vasoactive intestinal peptide), cardioprotective in heart failure (88), and SMAD5, which has anti-apoptotic actions in cardiac myocytes (89) were upregulated. On the other hand, AKAP12, which may protect against IR injury (90), was downregulated, as were both MYBL2 and TUBB2A, which were recently shown to be upregulated in the heart after acute MI (91, 92).
Interestingly, we found that there were no genes similarly affected among all three models of diastolic dysfunction, nor between the Dahl SS and SHHF models. However, the type 1 diabetes model shared 20 regulated genes with the Dahl SS model and 15 with the SHHF model. Human diseases traditionally have been differentiated and categorized on the basis of which organ system they primarily affect. An alternate view is now emerging, which emphasizes that different diseases typically have common underlying mechanisms and intermediate pathophenotypes or endo(pheno)types. According to this construct, expression of a specific disease reflects the interplay between relevant endophenotypes and the local organ-based environment. Important examples of such endophenotypes are inflammation, fibrosis, and metabolic dysfunction, which play essential roles in many developing diseases. In this study, we identified the endophenotype networks of three models of diastolic dysfunction and explored their relation to cardiovascular diseases in particular. We identified the subnetworks of the top regulated microarray genes that are playing a role in inflammation, fibrosis, and metabolism. Although our three models of diastolic dysfunction exhibit little overlap in regulated genes, each is still significantly enriched with disease-associated genes. Moreover, they are enriched also with differentially expressed genes linked to cardiovascular disease (risk).
Our findings illustrate as well the observation that HFpEF is a syndrome with multiple extracardiac comorbidities, such as aging, hypertension, and obesity, rather than a disease of clearly defined etiology (93). Pharmacological targeting of HFpEF may need to be informed more by phenotypic characteristics of the cardiac remodeling events that occur than by the underlying molecular processes alone. These phenotypic events may include concentric hypertrophy, myocardial stiffness, fibrosis, endothelial dysfunction, capillary rarefaction, and abnormal ventricular-arterial coupling, as well as changes in the shape and stiffness of the left atrium.
There are some limitations and caveats to our approach. Changes in gene expression may not necessarily be reflective of changes in protein levels, given the contribution of additional levels of post-transcriptional regulation, including microRNA mediated regulation. Alterations in protein stability and function may occur as well. In addition, in any disease context, changes in gene expression have a temporal and dynamic component, with alterations in expression of any particular gene and its protein levels at a given time point being impacted upon by changes in other network or system components. Nonetheless, global changes in gene expression are reflective of basic underlying pathological processes and offer insight into stress-related responsiveness to the disease process. In addition, there are several other models of HFpEF, such as abdominal aortic constriction and partial nephrectomy. Certainly, it would be of interest to extend our analysis in the future to include these other models. It should be mentioned as well that streptozotocin affects other organ systems, including vascular function; however, HFpEF in many patients is increasingly recognized as a systemic disorder involving vascular inflammation and dysfunction (94).
In conclusion, our in silico analysis of differential gene expression in three common rat models of diastolic dysfunction highlights the diversity in causality and molecular basis for the impaired cardiac function. A better understanding of the phenotype changes that accompany diastolic dysfunction, such as concentric hypertrophy, and their impact on genetic changes, is required to gain better insight into the disease process. An effective pharmacological approach for diastolic dysfunction and HFpEF may arise from a global strategy based on a better understanding of altered cardiac ultrastructure.
Author Contributions
RA conceived the study, analyzed the data, designed the figures and tables, and helped write the manuscript. FZ and RB assisted in the analysis of the data and writing of the manuscript. SB and AC helped write the manuscript. GB assisted in the analysis and interpretation of the results, and contributed to the writing and editing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer VO and handling Editor declared their shared affiliation.
Acknowledgement
The authors acknowledge the computational assistance of Dr. Michael R. Garrett of the UMMC Molecular and Genomics Core Facility.
Funding
This work was supported by grants from the American University of Beirut (grant number 100410) to FZ. RA is supported by the South-Eastern Norway Regional Health Authority (HSØ-RHF), project #2016089, and Olav Raagholt og Gerd Meidel Raagholts, ≪Validation of CXCR3 ligands as biomarkers for HFpEF≫.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2018.00011/full#supplementary-material
References
1. Upadhya B, Taffet GE, Cheng CP, Kitzman DW. Heart failure with preserved ejection fraction in the elderly: scope of the problem. J Mol Cell Cardiol (2015) 83:73–87. doi: 10.1016/j.yjmcc.2015.02.025
2. Taylor AL. Heart failure in women. Curr Heart Fail Rep (2015) 12(2):187–95. doi: 10.1007/s11897-015-0252-x
3. Borlaug BA. The pathophysiology of heart failure with preserved ejection fraction. Nat Rev Cardiol (2014) 11(9):507–15. doi: 10.1038/nrcardio.2014.83
4. Tschöpe C, Lam CS. Diastolic heart failure: What we still don't know. Looking for new concepts, diagnostic approaches, and the role of comorbidities. Herz (2012) 37(8):875–9. doi: 10.1007/s00059-012-3719-5
5. Miljkovik LV, Spiroska V. Heart failure with preserved ejection fraction - concept, pathophysiology, diagnosis and challenges for treatment. Open Access Maced J Med Sci (2015) 3(3):521–7. doi: 10.3889/oamjms.2015.087
6. von Lueder TG, Krum H. New medical therapies for heart failure. Nat Rev Cardiol (2015) 12(12):730–40. doi: 10.1038/nrcardio.2015.137
7. Lekavich CL, Barksdale DJ, Neelon V, Wu JR. Heart failure preserved ejection fraction (HFpEF): an integrated and strategic review. Heart Fail Rev (2015) 20(6):643–53. doi: 10.1007/s10741-015-9506-7
8. Altara R, Giordano M, Nordén ES, Cataliotti A, Kurdi M, Bajestani SN, et al. Targeting obesity and diabetes to treat heart failure with preserved ejection fraction. Front Endocrinol (2017) 8:160. doi: 10.3389/fendo.2017.00160
9. Ono T, Kamimura N, Matsuhashi T, Nagai T, Nishiyama T, Endo J, et al. The histone 3 lysine 9 methyltransferase inhibitor chaetocin improves prognosis in a rat model of high salt diet-induced heart failure. Sci Rep (2017) 7:39752. doi: 10.1038/srep39752
10. Roncalli J, Smih F, Desmoulin F, Dumonteil N, Harmancey R, Hennig S, et al. NMR and cDNA array analysis prior to heart failure reveals an increase of unsaturated lipids, a glutamine/glutamate ratio decrease and a specific transcriptome adaptation in obese rat heart. J Mol Cell Cardiol (2007) 42(3):526–39. doi: 10.1016/j.yjmcc.2006.11.007
11. van Lunteren E, Moyer M. Oxidoreductase, morphogenesis, extracellular matrix, and calcium ion-binding gene expression in streptozotocin-induced diabetic rat heart. Am J Physiol Endocrinol Metab (2007) 293(3):E759–68. doi: 10.1152/ajpendo.00191.2007
12. Song HK, Kim J, Lee JS, Nho KJ, Jeong HC, Kim J, et al. Pik3ip1 modulates cardiac hypertrophy by inhibiting PI3K pathway. PLoS One (2015) 10(3):e0122251. doi: 10.1371/journal.pone.0122251
13. Dokmanovic M, King KE, Mohan N, Endo Y, Wu WJ. Cardiotoxicity of ErbB2-targeted therapies and its impact on drug development, a spotlight on trastuzumab. Expert Opin Drug Metab Toxicol (2017) 13(7):755–66. doi: 10.1080/17425255.2017.1337746
14. Lin S, Wang Y, Zhang X, Kong Q, Li C, Li Y, et al. HSP27 Alleviates Cardiac Aging in Mice via a Mechanism Involving Antioxidation and Mitophagy Activation. Oxid Med Cell Longev (2016) 2016:1–13. doi: 10.1155/2016/2586706
15. Liu S, Iskandar R, Chen W, Zhang J, Wang Y, Chen X, et al. Soluble glycoprotein 130 and heat shock protein 27 as novel candidate biomarkers of chronic heart failure with preserved ejection fraction. Heart Lung Circ (2016) 25(10):1000–6. doi: 10.1016/j.hlc.2016.02.011
16. Jiang H, Zhang L, Yu Y, Liu M, Jin X, Zhang P, et al. A pilot study of angiogenin in heart failure with preserved ejection fraction: a novel potential biomarker for diagnosis and prognosis? J Cell Mol Med (2014) 18(11):2189–97. doi: 10.1111/jcmm.12344
17. Lewinter MM, Granzier HL. Cardiac titin and heart disease. J Cardiovasc Pharmacol (2014) 63(3):207–12. doi: 10.1097/FJC.0000000000000007
18. Paolocci N, Tavazzi B, Biondi R, Gluzband YA, Amorini AM, Tocchetti CG, et al. Metalloproteinase inhibitor counters high-energy phosphate depletion and AMP deaminase activity enhancing ventricular diastolic compliance in subacute heart failure. J Pharmacol Exp Ther (2006) 317(2):506–13. doi: 10.1124/jpet.105.099168
19. Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res (2009) 81(3):412–9. doi: 10.1093/cvr/cvn301
20. King NM, Methawasin M, Nedrud J, Harrell N, Chung CS, Helmes M, et al. Mouse intact cardiac myocyte mechanics: cross-bridge and titin-based stress in unactivated cells. J Gen Physiol (2011) 137(1):81–91. doi: 10.1085/jgp.201010499
21. Louch WE, Stokke MK, Sjaastad I, Christensen G, Sejersted OM. No rest for the weary: diastolic calcium homeostasis in the normal and failing myocardium. Physiology (2012) 27(5):308–23. doi: 10.1152/physiol.00021.2012
22. Schultz JJ, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, et al. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest (2002) 109(6):787–96. doi: 10.1172/JCI0214190
23. Zhang H, Wu J, Dong H, Khan SA, Chu ML, Tsuda T. Fibulin-2 deficiency attenuates angiotensin II-induced cardiac hypertrophy by reducing transforming growth factor-β signalling. Clin Sci (2014) 126(4):275–88. doi: 10.1042/CS20120636
24. Tsoutsman T, Wang X, Garchow K, Riser B, Twigg S, Semsarian C. CCN2 plays a key role in extracellular matrix gene expression in severe hypertrophic cardiomyopathy and heart failure. J Mol Cell Cardiol (2013) 62:164–78. doi: 10.1016/j.yjmcc.2013.05.019
25. Ding X, Xu X, Yan Y, Song X, Liu S, Wang G, et al. Effects of renal sympathetic denervation and angiotensin-converting enzyme inhibitor on left ventricular hypertrophy. Comparison in spontaneously hypertensive rats. Herz (2015) 40(4):695–701. doi: 10.1007/s00059-014-4110-5
26. Rehman A, Leibowitz A, Yamamoto N, Rautureau Y, Paradis P, Schiffrin EL. Angiotensin type 2 receptor agonist compound 21 reduces vascular injury and myocardial fibrosis in stroke-prone spontaneously hypertensive rats. Hypertension (2012) 59(2):291–9. doi: 10.1161/HYPERTENSIONAHA.111.180158
27. Montgomery RL, Hullinger TG, Semus HM, Dickinson BA, Seto AG, Lynch JM, et al. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation (2011) 124(14):1537–47. doi: 10.1161/CIRCULATIONAHA.111.030932
28. Yap LB, Mukerjee D, Timms PM, Ashrafian H, Coghlan JG. Natriuretic peptides, respiratory disease, and the right heart. Chest (2004) 126(4):1330–6. doi: 10.1378/chest.126.4.1330
29. Sklepkiewicz P, Shiomi T, Kaur R, Sun J, Kwon S, Mercer B, et al. Loss of secreted frizzled-related protein-1 leads to deterioration of cardiac function in mice and plays a role in human cardiomyopathy. Circ Heart Fail (2015) 8(2):362–72. doi: 10.1161/CIRCHEARTFAILURE.114.001274
30. Chang YM, Ling L, Chang YT, Chang YW, Li WH, Shih AC, et al. Three TF co-expression modules regulate pressure-overload cardiac hypertrophy in male mice. Sci Rep (2017) 7(1):7560. doi: 10.1038/s41598-017-07981-4
31. Goruppi S, Patten RD, Force T, Kyriakis JM. Helix-loop-helix protein p8, a transcriptional regulator required for cardiomyocyte hypertrophy and cardiac fibroblast matrix metalloprotease induction. Mol Cell Biol (2007) 27(3):993–1006. doi: 10.1128/MCB.00996-06
32. Georgescu SP, Aronovitz MJ, Iovanna JL, Patten RD, Kyriakis JM, Goruppi S. Decreased metalloprotease 9 induction, cardiac fibrosis, and higher autophagy after pressure overload in mice lacking the transcriptional regulator p8. Am J Physiol Cell Physiol (2011) 301(5):C1046–56. doi: 10.1152/ajpcell.00211.2011
33. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol (2013) 62(4):263–71. doi: 10.1016/j.jacc.2013.02.092
34. Frolova EG, Sopko N, Blech L, Popovic ZB, Li J, Vasanji A, et al. Thrombospondin-4 regulates fibrosis and remodeling of the myocardium in response to pressure overload. Faseb J (2012) 26(6):2363–73. doi: 10.1096/fj.11-190728
35. Sandip C, Tan L, Huang J, Li Q, Ni L, Cianflone K, et al. Common variants in IL-17A/IL-17RA axis contribute to predisposition to and progression of congestive heart failure. Medicine (2016) 95(27):e4105. doi: 10.1097/MD.0000000000004105
36. Liu W, Wang X, Feng W, Li S, Tian W, Xu T, et al. Lentivirus mediated IL-17R blockade improves diastolic cardiac function in spontaneously hypertensive rats. Exp Mol Pathol (2011) 91(1):362–7. doi: 10.1016/j.yexmp.2011.04.003
37. Wu Y, Li Y, Zhang C, A X, Wang Y, Cui W, et al. S100a8/a9 released by CD11b+Gr1+ neutrophils activates cardiac fibroblasts to initiate angiotensin II-Induced cardiac inflammation and injury. Hypertension (2014) 63(6):1241–50. doi: 10.1161/HYPERTENSIONAHA.113.02843
38. Altara R, Mallat Z, Booz GW, Zouein FA. The CXCL10/CXCR3 axis and cardiac inflammation: implications for immunotherapy to treat infectious and noninfectious diseases of the heart. J Immunol Res (2016) 2016:4396368:1–12. doi: 10.1155/2016/4396368
39. Lu L, Wu D, Li L, Chen L. Apelin/APJ system: a bifunctional target for cardiac hypertrophy. Int J Cardiol (2017) 230:164–70. doi: 10.1016/j.ijcard.2016.11.215
40. Kim EE, Shekhar A, Lu J, Lin X, Liu FY, Zhang J, et al. PCP4 regulates Purkinje cell excitability and cardiac rhythmicity. J Clin Invest (2014) 124(11):5027–36. doi: 10.1172/JCI77495
41. Guo C, Wang Q, Wang Y, Yang L, Luo H, Cao XF, et al. Exome sequencing reveals novel IRXI mutation in congenital heart disease. Mol Med Rep (2017) 15(5):3193–7. doi: 10.3892/mmr.2017.6410
42. Altara R, Manca M, Brandão RD, Zeidan A, Booz GW, Zouein FA. Emerging importance of chemokine receptor CXCR3 and its ligands in cardiovascular diseases. Clin Sci (2016) 130(7):463–78. doi: 10.1042/CS20150666
43. Altara R, Gu YM, Struijker-Boudier HA, Thijs L, Staessen JA, Blankesteijn WM. Left ventricular dysfunction and CXCR3 ligands in hypertension: from animal experiments to a population-based pilot study. PLoS One (2015) 10(10):e0141394. doi: 10.1371/journal.pone.0141394
44. Altara R, Manca M, Hessel MH, Gu Y, van Vark LC, Akkerhuis KM, et al. CXCL10 is a circulating inflammatory marker in patients with advanced heart failure: a pilot study. J Cardiovasc Transl Res (2016) 9(4):302–14. doi: 10.1007/s12265-016-9703-3
45. Arderiu G, Espinosa S, Peña E, Aledo R, Badimon L. Monocyte-secreted Wnt5a interacts with FZD5 in microvascular endothelial cells and induces angiogenesis through tissue factor signaling. J Mol Cell Biol (2014) 6(5):380–93. doi: 10.1093/jmcb/mju036
46. Barančík M, Grešová L, Barteková M, Dovinová I. Nrf2 as a key player of redox regulation in cardiovascular diseases. Physiol Res (2016) 65(Suppl 1):S1–10.
47. Herum KM, Lunde IG, Skrbic B, Louch WE, Hasic A, Boye S, et al. Syndecan-4 is a key determinant of collagen cross-linking and passive myocardial stiffness in the pressure-overloaded heart. Cardiovasc Res (2015) 106(2):217–26. doi: 10.1093/cvr/cvv002
48. Pontén A, Folestad EB, Pietras K, Eriksson U. Platelet-derived growth factor D induces cardiac fibrosis and proliferation of vascular smooth muscle cells in heart-specific transgenic mice. Circ Res (2005) 97(10):1036–45. doi: 10.1161/01.RES.0000190590.31545.d4
49. Su SA, Yang D, Wu Y, Xie Y, Zhu W, Cai Z, et al. EphrinB2 regulates cardiac fibrosis through modulating the interaction of stat3 and TGF-β/Smad3 signaling. Circ Res (2017) 121(6):617–27. doi: 10.1161/CIRCRESAHA.117.311045
50. Shihata WA, Putra MRA, Chin-Dusting JPF. Is there a potential therapeutic role for Caveolin-1 in fibrosis? Front Pharmacol (2017) 8:567. doi: 10.3389/fphar.2017.00567
51. Dobaczewski M, Frangogiannis NG. Chemokines and cardiac fibrosis. Front Biosci (2009) 1:391–405. doi: 10.2741/s33
52. Clancy RM, Markham AJ, Jackson T, Rasmussen SE, Blumenberg M, Buyon JP. Cardiac fibroblast transcriptome analyses support a role for interferogenic, profibrotic, and inflammatory genes in anti-SSA/Ro-associated congenital heart block. Am.J Physiol Heart Circ Physiol (2017) 313(3):H631–40. doi: 10.1152/ajpheart.00256.2017
53. Guilherme L, Kalil J. Rheumatic heart disease: molecules involved in valve tissue inflammation leading to the autoimmune process and anti-S. pyogenes vaccine. Front Immunol (2013) 4:352. doi: 10.3389/fimmu.2013.00352
54. Quast C, Alter C, Ding Z, Borg N, Schrader J. Adenosine formed by CD73 on T cells inhibits cardiac inflammation and fibrosis and preserves contractile function in transverse aortic constriction-induced heart failure. Circ Heart Fail (2017) 10(4):e003346. doi: 10.1161/CIRCHEARTFAILURE.116.003346
55. Wang LP, Fan SJ, Li SM, Wang XJ, Gao JL, Yang XH. Protective role of ACE2-Ang-(1-7)-Mas in myocardial fibrosis by downregulating KCa3.1 channel via ERK1/2 pathway. Pflugers Arch (2016) 468(11-12):2041–51. doi: 10.1007/s00424-016-1875-9
56. Chan HW, Smith NJ, Hannan RD, Thomas WG. Tackling the EGFR in pathological tissue remodelling. Pulm Pharmacol Ther (2006) 19(1):74–8. doi: 10.1016/j.pupt.2005.04.005
57. Peng K, Tian X, Qian Y, Skibba M, Zou C, Liu Z, et al. Novel EGFR inhibitors attenuate cardiac hypertrophy induced by angiotensin II. J Cell Mol Med (2016) 20(3):482–94. doi: 10.1111/jcmm.12763
58. Liao Y, Zhao H, Ogai A, Kato H, Asakura M, Kim J, et al. Atorvastatin slows the progression of cardiac remodeling in mice with pressure overload and inhibits epidermal growth factor receptor activation. Hypertens Res (2008) 31(2):335–44. doi: 10.1291/hypres.31.335
59. Reichelt ME, O'Brien S, Thomas WG, Headrick JP. Transactivation of the epidermal growth factor receptor in responses to myocardial stress and cardioprotection. Int J Biochem Cell Biol (2017) 83:97–110. doi: 10.1016/j.biocel.2016.12.014
60. Fan D, Takawale A, Basu R, Patel V, Lee J, Kandalam V, et al. Differential role of TIMP2 and TIMP3 in cardiac hypertrophy, fibrosis, and diastolic dysfunction. Cardiovasc Res (2014) 103(2):268–80. doi: 10.1093/cvr/cvu072
61. Gabrielsen A, Lawler PR, Yongzhong W, Steinbrüchel D, Blagoja D, Paulsson-Berne G, et al. Gene expression signals involved in ischemic injury, extracellular matrix composition and fibrosis defined by global mRNA profiling of the human left ventricular myocardium. J Mol Cell Cardiol (2007) 42(4):870–83. doi: 10.1016/j.yjmcc.2006.12.016
62. Engebretsen KV, Lunde IG, Strand ME, Waehre A, Sjaastad I, Marstein HS, et al. Lumican is increased in experimental and clinical heart failure, and its production by cardiac fibroblasts is induced by mechanical and proinflammatory stimuli. Febs J (2013) 280(10):2382–98. doi: 10.1111/febs.12235
63. Li X, Zhao D, Guo Z, Li T, Qili M, Xu B, et al. Overexpression of SerpinE2/protease nexin-1 contribute to pathological cardiac fibrosis via increasing Collagen deposition. Sci Rep (2016) 6:37635. doi: 10.1038/srep37635
64. Lodder EM, Scicluna BP, Beekman L, Arends D, Moerland PD, Tanck MW, et al. Integrative genomic approach identifies multiple genes involved in cardiac collagen deposition. Circ Cardiovasc Genet (2014) 7(6):790–8. doi: 10.1161/CIRCGENETICS.114.000537
65. Wang T, Liu J, Mcdonald C, Lupino K, Zhai X, Wilkins BJ, et al. GDF15 is a heart-derived hormone that regulates body growth. EMBO Mol Med (2017) 9(8):1150–64. doi: 10.15252/emmm.201707604
66. Salabei JK, Lorkiewicz PK, Mehra P, Gibb AA, Haberzettl P, Hong KU, et al. Type 2 diabetes dysregulates glucose metabolism in cardiac progenitor cells. J Biol Chem (2016) 291(26):13634–48. doi: 10.1074/jbc.M116.722496
67. Leifheit-Nestler M, Wagner NM, Gogiraju R, Didié M, Konstantinides S, Hasenfuss G, et al. Importance of leptin signaling and signal transducer and activator of transcription-3 activation in mediating the cardiac hypertrophy associated with obesity. J Transl Med (2013) 11:170. doi: 10.1186/1479-5876-11-170
68. Zhong W, Mao S, Tobis S, Angelis E, Jordan MC, Roos KP, et al. Hypertrophic growth in cardiac myocytes is mediated by Myc through a Cyclin D2-dependent pathway. Embo J (2006) 25(16):3869–79. doi: 10.1038/sj.emboj.7601252
69. Fujita T, Ishikawa Y. Apoptosis in heart failure. -the role of the β-adrenergic receptor-mediated signaling pathway and p53-mediated signaling pathway in the apoptosis of cardiomyocytes. Circ J (2011) 75(8):1811–8.
70. Matsumoto S, Sakata Y, Suna S, Nakatani D, Usami M, Hara M, et al. Circulating p53-responsive microRNAs are predictive indicators of heart failure after acute myocardial infarction. Circ Res (2013) 113(3):322–6. doi: 10.1161/CIRCRESAHA.113.301209
71. Hamdani N, Bishu KG, von Frieling-Salewsky M, Redfield MM, Linke WA. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc Res (2013) 97(3):464–71. doi: 10.1093/cvr/cvs353
72. Choi JH, Ke Q, Bae S, Lee JY, Kim YJ, Kim UK, et al. Doxercalciferol, a pro-hormone of vitamin D, prevents the development of cardiac hypertrophy in rats. J Card Fail (2011) 17(12):1051–8. doi: 10.1016/j.cardfail.2011.08.006
73. Liu Q, Molkentin JD. Protein kinase Cα as a heart failure therapeutic target. J Mol Cell Cardiol (2011) 51(4):474–8. doi: 10.1016/j.yjmcc.2010.10.004
74. Varma U, Koutsifeli P, Benson VL, Mellor KM, Delbridge LMD. Molecular mechanisms of cardiac pathology in diabetes - experimental insights. Biochim Biophys Acta (2017). doi: 10.1016/j.bbadis.2017.10.035
75. Saito S, Teshima Y, Fukui A, Kondo H, Nishio S, Nakagawa M, et al. Glucose fluctuations increase the incidence of atrial fibrillation in diabetic rats. Cardiovasc Res (2014) 104(1):5–14. doi: 10.1093/cvr/cvu176
76. Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C, et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS One (2014) 9(8):e104771. doi: 10.1371/journal.pone.0104771
77. Stanley BA, Sivakumaran V, Shi S, Mcdonald I, Lloyd D, Watson WH, et al. Thioredoxin reductase-2 is essential for keeping low levels of H(2)O(2) emission from isolated heart mitochondria. J Biol Chem (2011) 286(38):33669–77. doi: 10.1074/jbc.M111.284612
78. Reyat JS, Chimen M, Noy PJ, Szyroka J, Rainger GE, Tomlinson MG. ADAM10-interacting tetraspanins Tspan5 and Tspan17 regulate VE-Cadherin expression and promote T Lymphocyte transmigration. J Immunol (2017) 199(2):666–76. doi: 10.4049/jimmunol.1600713
79. Chaanine AH, Sreekumaran Nair K, Bergen RH, Klaus K, Guenzel AJ, Hajjar RJ, et al. Mitochondrial integrity and function in the progression of early pressure overload-induced left ventricular remodeling. J Am Heart Assoc (2017) 6(6):e005869. doi: 10.1161/JAHA.117.005869
80. Chaanine AH, Kohlbrenner E, Gamb SI, Guenzel AJ, Klaus K, Fayyaz AU, et al. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am J Physiol Heart Circ Physiol (2016) 311(6):H1540–59. doi: 10.1152/ajpheart.00549.2016
81. Lu J, Sun B, Huo R, Wang YC, Yang D, Xing Y, et al. Bone morphogenetic protein-2 antagonizes bone morphogenetic protein-4 induced cardiomyocyte hypertrophy and apoptosis. J Cell Physiol (2014) 229(10):1503–10. doi: 10.1002/jcp.24592
82. Wang S, Sun A, Li L, Zhao G, Jia J, Wang K, et al. Up-regulation of BMP-2 antagonizes TGF-β1/ROCK-enhanced cardiac fibrotic signalling through activation of Smurf1/Smad6 complex. J Cell Mol Med (2012) 16(10):2301–10. doi: 10.1111/j.1582-4934.2012.01538.x
83. Zhang Y, Liu Y, Zhu XH, Zhang XD, Jiang DS, Bian ZY, et al. Dickkopf-3 attenuates pressure overload-induced cardiac remodelling. Cardiovasc Res (2014) 102(1):35–45. doi: 10.1093/cvr/cvu004
84. Tsui H, Zi M, Wang S, Chowdhury SK, Prehar S, Liang Q, et al. Smad3 couples pak1 with the antihypertrophic pathway through the E3 ubiquitin ligase, Fbxo32. Hypertension (2015) 66(6):1176–83. doi: 10.1161/HYPERTENSIONAHA.115.06068
85. Wang J, Wang H, Chen J, Wang X, Sun K, Wang Y, et al. GADD45B inhibits MKK7-induced cardiac hypertrophy and the polymorphisms of GADD45B is associated with inter-ventricular septum hypertrophy. Biochem Biophys Res Commun (2008) 372(4):623–8. doi: 10.1016/j.bbrc.2008.05.122
86. Ikeda S, Hamada M, Hiwada K. Contribution of non-cardiomyocyte apoptosis to cardiac remodelling that occurs in the transition from compensated hypertrophy to heart failure in spontaneously hypertensive rats. Clin Sci (1999) 97(2):239–46. doi: 10.1042/cs0970239
87. Thomas RL, Gustafsson AB. MCL1 is critical for mitochondrial function and autophagy in the heart. Autophagy (2013) 9(11):1902–3. doi: 10.4161/auto.26168
88. Szema AM, Dang S, Li JC. Emerging novel therapies for heart failure. Clin Med Insights Cardiol (2015) 9(Suppl 2):57–64. doi: 10.4137/CMC.S29735
89. Sun Y, Zhou J, Liao X, Lu Y, Deng C, Huang P, et al. Disruption of Smad5 gene induces mitochondria-dependent apoptosis in cardiomyocytes. Exp Cell Res (2005) 306(1):85–93. doi: 10.1016/j.yexcr.2005.02.012
90. Yang X. Identification of risk genes associated with myocardial infarction based on the recursive feature elimination algorithm and support vector machine classifier. Mol Med Rep (2018) 17(1):1555–60. doi: 10.3892/mmr.2017.8044
91. Cheng M, An S, Li J. Identifying key genes associated with acute myocardial infarction. Medicine (2017) 96(42):e7741. doi: 10.1097/MD.0000000000007741
92. Zhang T, Zhao LL, Cao X, Qi LC, Wei GQ, Liu JY, et al. Bioinformatics analysis of time series gene expression in left ventricle (LV) with acute myocardial infarction (AMI). Gene (2014) 543(2):259–67. doi: 10.1016/j.gene.2014.04.002
93. Zouein FA, de Castro Brás LE, da Costa DV, Lindsey ML, Kurdi M, Booz GW. Heart failure with preserved ejection fraction: emerging drug strategies. J Cardiovasc Pharmacol (2013) 62(1):13–21. doi: 10.1097/FJC.0b013e31829a4e61
Keywords: metabolic disease, endothelial and microvascular dysfunction, inflammation, heart failure, hypertension
Citation: Altara R, Zouein FA, Brandão RD, Bajestani SN, Cataliotti A and Booz GW (2018). In Silico Analysis of Differential Gene Expression in Three Common Rat Models of Diastolic Dysfunction. Front. Cardiovasc. Med. 5:11. doi: 10.3389/fcvm.2018.00011
Received: 16 December 2017; Accepted: 05 February 2018;
Published: 21 February 2018
Edited by:
Yeong-Renn Chen, Northeast Ohio Medical University, United StatesReviewed by:
Vahagn Ohanyan, Northeast Ohio Medical University, United StatesNazareno Paolocci, Johns Hopkins University, United States
Copyright © 2018 Altara, Zouein, Brandão, Bajestani, Cataliotti and Booz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raffaele Altara, cmFmZmFlbGUuYWx0YXJhQG1lZGlzaW4udWlvLm5v; Fouad A. Zouein, ZnoxNUBhdWIuZWR1Lmxi