 Christian Steinberg*
Christian Steinberg* Charles Nadeau-Routhier
Charles Nadeau-Routhier Philippe AndréFrançois PhilipponJean-François SarrazinIsabelle NaultGilles O'HaraLouis BlierFranck MolinBenoit PlourdeKarine RoyEric LaroseMarie ArsenaultJean Champagne
Philippe AndréFrançois PhilipponJean-François SarrazinIsabelle NaultGilles O'HaraLouis BlierFranck MolinBenoit PlourdeKarine RoyEric LaroseMarie ArsenaultJean Champagne- Division of Cardiology, Multidisciplinary Cardiovascular Department, Institut Universitaire de Cardiologie et Pneumologie de Québec (IUCPQ-UL), Université Laval, Québec City, QC, Canada
Background: Apical hypertrophic cardiomyopathy (aHCM) is thought to have a more benign clinical course compared to septal HCM (sHCM), but most data have been derived from Asian cohorts. Comparative data on clinical outcome in Caucasian aHCM cohorts are scarce, and the results are conflicting. The aim of this study was to estimate the prevalence and outcome of aHCM in French-Canadians of Caucasian descent.
Methods and results: We conducted a retrospective, single-center cohort study. The primary endpoint was a composite of documented sustained ventricular arrhythmia (VA), appropriate ICD therapy, arrhythmogenic syncope, cardiac arrest, or all-cause mortality. A total of 301 HCM patients (65% males) were enrolled including 80/301 (27%) with aHCM and 221/301 (73%) with sHCM. Maximal wall thickness was similar in both groups. Left ventricular apical aneurysm was significantly more common in aHCM (10 vs. 0.5%; p < 0.001). The proportion of patients with myocardial fibrosis ≥ 15% of the left ventricular mass was similar between aHCM and sHCM (21 vs. 24%; p = 0.68). Secondary prevention ICDs were more often implanted in aHCM patients (16 vs. 7%; p = 0.02). The primary endpoint occurred in 26% of aHCM and 10.4% of sHCM patients (p = 0.001) and was driven by an increased incidence of sustained VA (10 vs. 2.3%; p = 0.01). Multivariate analysis identified apical aneurysm and a phenotype of aHCM as independent predictors of the primary endpoint and the occurrence of sustained ventricular tachycardia. Unexplained syncope and a family history of sudden cardiac death were additional predictors for sustained VA. Apical HCM was associated with an increased risk of ventricular arrhythmia even when excluding patients with apical aneurysm.
Conclusions: The phenotype of apical HCM is much more common in French-Canadians (27%) of Caucasian descent compared to other Caucasian HCM populations. Apical HCM in French-Canadians is associated with an increased risk for ventricular arrhythmia.
Introduction
Hypertrophic cardiomyopathy (HCM) is the most common hereditary cardiomyopathy with an estimated prevalence of 1:500 and is a leading cause of sudden unexplained death, in particular in young competitive athletes (1, 2). Depending on the predominant localization of segmental myocardial hypertrophy, different HCM phenotypes can be distinguished including septal HCM (sHCM) and apical HCM (aHCM) (3, 4). Apical HCM is characterized by focal hypertrophy of the apical segments resulting in a characteristic ace-of-spade-like end-systolic configuration of the left ventricle that is associated with deep, symmetric precordial T-wave inversions on the surface ECG (5). Additional findings may include midventricular obstruction/gradients and/or left ventricular apical aneurysm (6). The prevalence of aHCM varies among different ethnic groups and is usually more common in East-Asian and Afro-Caribbean populations compared to Caucasians (6–9). Apical HCM is considered to have a more benign course compared to sHCM, but most data are derived from Asian cohorts (10–12). There are conflicting data with regard to the outcome of aHCM in Caucasian North American populations (10, 13).
The French-Canadian population of the province of Quebec is predominantly of European Caucasian descent and represents a unique North American population from a genetic point of view. This feature is largely explained by the particular immigration history of Quebec together with regional colonization patterns resulting in founder mutations for various hereditary diseases (14).
The prevalence and outcome of aHCM in the Caucasian French-Canadian population is unknown, so far. The aim of this study was to estimate the prevalence of aHCM in the Caucasian French-Canadian population of Quebec and to assess the incidence and predictors of ventricular arrhythmia in this particular cohort.
Methods
Study Population and Definition of HCM
We conducted a retrospective cohort study at a single tertiary University center. The study was approved by the local ethics and review board. Eligible patients had to be ≥ 18 years old and were identified from the electronic medical records as well as the institutional cardiac imaging and pacemaker/defibrillator databases including the period from 2000 to 2017. To study our abovementioned hypothesis, we included only French-Canadians of Caucasian descent (self-reported ethnicity) in this study. Hypertrophic cardiomyopathy was diagnosed according to current guidelines and defined as otherwise unexplained left ventricular wall thickness ≥ 15 mm in one or more segments as measured by either transthoracic echocardiography or cardiac magnetic resonance imaging (CMR) (15–17). With regard to septal HCM, best efforts were made to include only patients with reverse curve septal hypertrophy. In the case of discordant imaging results with regard to HCM phenotype, maximal wall thickness, and presence/absence of apical aneurysm, we hierarchized CMR findings over echo results, which has also been proposed by recent literature (18). In the case of borderline segmental wall thickening (13–14 mm) HCM was diagnosed in the presence of at least one of the following findings: (1) positive genetic test for a known sarcomeric HCM variant, (2) a positive family history of HCM, (3) a ratio of end-diastolic septal/lateral wall thickness of ≥ 1.5, or (4) typical ECG findings of aHCM (16, 17). Apical HCM was defined as segmental hypertrophy of a ≥ 1 of the left ventricular apical segments with or without minor involvement of the inferoseptal or midseptal wall segments (13–14 mm). Dynamic obstruction of the left ventricular outflow tract, midventricular obstruction, and left ventricular apical aneurysm were defined according to current guidelines (4, 19). For the purpose of this study, we excluded mixed or diffuse forms of HCM, which represented <10% of the entire study cohort.
Patients were excluded in the presence of long-standing (≥ 5 years) uncontrolled arterial hypertension (systolic blood pressure ≥ 160 mmHg), moderate or severe aortic stenosis, subvalvular membranes of the left ventricular outflow tract, prosthetic aortic valve, evidence or strong suspicion of underlying HCM phenocopies including infiltrative cardiomyopathies (ex cardiac amyloidosis, Fabry disease, etc.), or left ventricular non-compaction.
Cardiac Magnetic Resonance Imaging
Cardiac magnetic resonance imaging was performed using a 1.5-T system (Philips Achieva 1.5 Tesla, operating release 2.6 level 3, Philips Healthcare, Netherlands). All CMR exams were reviewed by a single experienced level-3 certified cardiologist who was blinded to the clinical data and myocardial phenotype of the patients. Myocardial fibrosis was quantified using validated software (CVI42, Circle, Canada) as previously described by our group (20). We set limits for myocardial fibrosis at ≥ 6 SD from the normal myocardium in accordance with published criteria for the assessment of myocardial fibrosis in hypertrophic cardiomyopathy (21, 22).
Implantable Cardioverter Defibrillator (ICD) Implantation
The indications for ICD implantation were independently reevaluated by two independent electrophysiologists and assigned to primary or secondary prevention at the moment of the most recent follow-up visit according to current guidelines including traditional risk factors and new risk prediction models (16, 17). ICD programming including antitachycardia pacing (ATP) was standardized for all primary and secondary prevention ICDs with focus on delayed arrhythmia detection at higher ventricular rates based on available evidence and current guidelines (23–27). Supplementary Table 1 displays the ICD programming for primary prevention. Secondary prevention ICDs with documented ventricular fibrillation or polymorphic VT as index arrhythmia are programmed like primary prevention ICDs as recommended by current guidelines (26, 27). In the case of monomorphic VT with known rate, the slowest therapy zone is programmed 10 bpm below the documented tachycardia rate as recommended (26, 27).
Arrhythmic Outcomes and All-Cause Mortality
The primary endpoint was a composite of documented sustained ventricular arrhythmia (VT) (monomorphic VT, polymorphic VT, or ventricular fibrillation) as index arrhythmia or during follow-up, any appropriate ICD therapy (ATP or shock), likely arrhythmogenic syncope, resuscitated cardiac arrest from ventricular arrhythmia, or all-cause mortality. Likely arrhythmogenic syncope was defined as sudden, otherwise unexplained loss of consciousness in the absence of dynamic obstruction of the left ventricular outflow tract, bradycardia, and absence of a neurovegetative prodrome/postdrome (28).
ECG Analysis
Resting standard 12-lead ECGs (paper speed 25 mm/s, 10 mm/mV) showing intrinsic ventricular depolarization were eligible for analysis. We excluded ECGs with paced QRS complexes. Eligible ECGs were analyzed for the presence of symmetric precordial T-wave inversion, signs of early repolarization, and ECG criteria suggesting left ventricular apical aneurysm (convex ST elevation of ≥ 1 mm in ≥ 2 contiguous leads through V1–V4 associated with loss of giant T-wave negativity) as published before (29–32). Early repolarization pattern was defined according to published recommendations requiring a J-point elevation of ≥ 1 mm in ≥ 2 contiguous inferior and/or lateral leads while excluding leads V1–V3 (33). Ventricular paced rhythms and QRS duration ≥ 110 ms were excluded for the analysis of the ER pattern. Symmetric T-wave inversion of precordial leads required at least 2 leads with a T-wave amplitude ≥ 1 mm. T-wave inversion with amplitudes ≥ 10 mm was classified as giant T-waves according to published criteria (5, 34).
Genetic Testing
Genetic data were collected from the medical records and analyzed if available. For the purpose of this study, we only included genetic testing using contemporary next-generation sequencing (NGS). DNA sequencing targeted all coding exons, all exon–intron boundaries, and some potential mutation sites located outside the coding regions of the target genes. Deletion/duplication analysis was performed for the majority of samples. The following 19 target genes for HCM and HCM phenocopies were selected: ACTC1, ALPK3, CSRP3, FLNC, GAA, GLA, LAMP2, MYBPC3, MYH7, MYL2, MYL3, PRKAG2, RAF1, SOS1, TNNI3, TNNT2, TPM1, and TTR. Variant classification was conducted according to current guidelines (35).
Statistical Analysis
Categorical variables were analyzed using Fisher's exact test or χ2 test where appropriate. All continuous variables were tested for normality and are displayed as mean ± standard deviation (SD) or median with interquartile range (IQR) according to data distribution. Continuous variables were analyzed using Student's t-test or Mann–Whitney U-test where appropriate. Being survival-free from composite endpoints was compared using Kaplan–Meier curves and log-rank tests. Univariate and multivariate analyses for clinical predictors of the composite endpoint as well as sustained monomorphic ventricular tachycardia were conducted using logistic regression and a Cox proportional hazards model. Statistically significant univariate predictors with p ≤ 0.05 were selected for multivariate analysis. Statistical significance was considered for p < 0.05. All statistical analyses were conducted using Stata 14.1 Software (StataCorp, College Station, Texas, USA).
Results
Study Population
A total of 301 individuals with HCM were included in the study. Mean age was 55 ± 17 years, 65% were males, and all patients were Caucasians. Apical HCM was diagnosed in 27% (80/301) and sHCM in 73% (221/301) individuals. Baseline characteristics of the study population are displayed in Table 1. There were no differences between both groups with regard to age, sex, and medical comorbidities. Familial HCM accounted for 20% of all cases, and a family history of sudden cardiac death was present in 21% of the study population showing no difference between aHCM and sHCM. Overall, 36% of all study subjects had undergone ICD implantation with similar proportions of primary prevention ICDs in both groups (74% primary prevention ICDs). However, there were significantly more secondary prevention ICD carriers in the group of aHCM (16% vs 7%; p = 0.02). The subgroup of sHCM included 60 individuals who had undergone septal reduction therapy for previous, severe left ventricular outflow tract obstruction (Table 1).
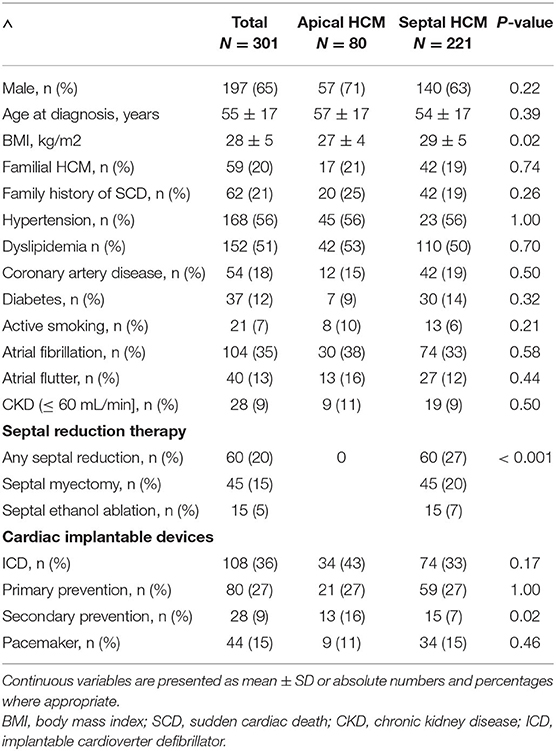
Table 1. Baseline characteristics.
Findings of Cardiac Imaging
All individuals had at least one transthoracic echocardiogram, and in those with serial echocardiograms, the most recent exam in our database was used for analysis. The majority of patients with sHCM presented a reverse curve septal hypertrophy. In addition, 162/301 (54%) of all study subjects had at least one cardiac magnetic resonance (CMR) imaging during the study period. The proportion of patients with CMR was similar in both groups (aHCM 54 vs. sHCM 52%; p = 0.80). The results of cardiac imaging are outlined in Tables 2, 3. With regard to the maximal segmental wall thickness, there was no significant different between aHCM and sHCM. Indexed LV mass, biventricular volumes, and systolic function showed also no difference between both groups. The overall proportion of patients with detectable myocardial fibrosis was high (86%); however, scar volume and extension were similar in both groups. The median percentage of left ventricular fibrosis was 7% (IQR 10). The proportion of individuals with more advanced myocardial scarring (i.e., ≥ 15% of left ventricular mass) was slightly higher in patients with aHCM compared to sHCM, but the difference was not statistically significant. A left ventricular apical aneurysm was present in 10% of individuals with aHCM compared to only 0.5% of patients with sHCM (p < 0.001).
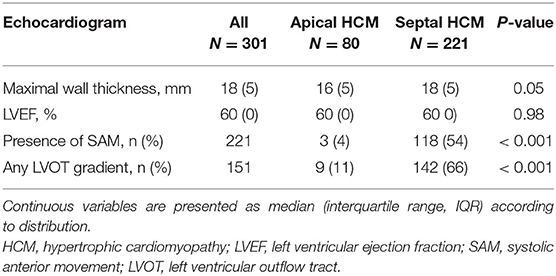
Table 2. Echocardiographic characteristics.
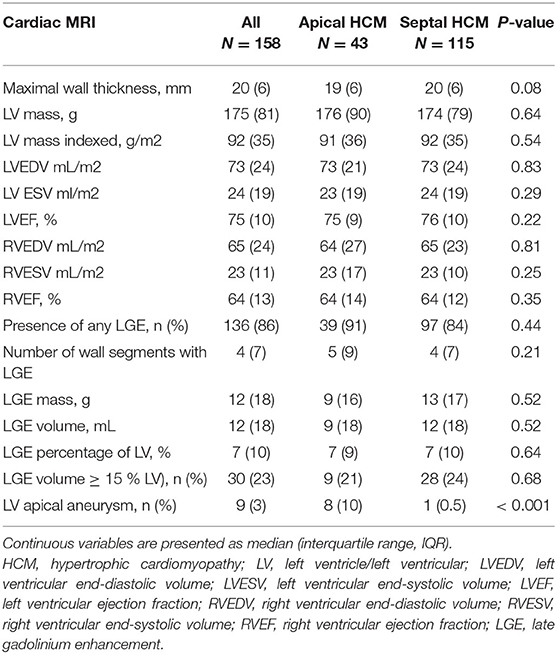
Table 3. Cardiac magnetic resonance data.
Arrhythmic Outcomes
The median follow-up duration 47 months (IQR 87 months) for patients with aHCM and 77 months (IQR 111 months) for patients with sHCM. The follow-up duration was significantly longer for patients with sHCM (p = 0.02). Overall survival free from predefined endpoints is displayed in Figure 1A and was significantly decreased in patients with aHCM (log rank: p < 0.001) compared to sHCM. The median time to a first arrhythmic event of the composite endpoint was 48 months (IQR 109 months) for patients with aHCM and 56 months (IQR 80 months) for patients with sHCM (p = 0.83). A detailed distribution of arrhythmic events, all-cause mortality, and ICD therapies is illustrated in Figure 2. Arrhythmic events over time were significantly more common in individuals with aHCM. The combined primary endpoint occurred in 21/80 (26%) patients with aHCM compared to 23/221 (10.4%) with sHCM (p = 0.001). This was primarily driven by a significantly higher rate of sustained monomorphic VT occurring four times more often in aHCM compared to sHCM (10% vs. 2.3%; p = 0.01) (Figures 1B, 2). The 5-year incidence rate for the primary endpoint was 28.9% in aHCM and 11.9% in sHCM. The corresponding 5-year incidence of sustained monomorphic VT was 10.9% for patients with aHCM and 3.6% for patients with sHCM. Cardiac arrest as index event occurred in 7.5% of aHCM and 2.7% of sHCM patients (p = 0.07). Although the rate of index cardiac arrest was 2.8-fold higher in aHCM compared to sHCM, this difference did not reach statistical significance, which may be related to the overall small number of resuscitated cardiac arrests. Cardiac arrest as index event occurred at a similar median age in aHCM and sHCM (57 [14] vs. 56 [20] years; p = 0.89). Apical HCM was associated with significantly more arrhythmic events even when excluding cardiac arrest as index event (p = 0.01). With regard to the indication for initial ICD implantation, sustained monomorphic VT occurred in 19.4% of secondary prevention carriers compared to 8.8% of primary prevention carriers (p = 0.01). Patients with index cardiac arrest had a significantly increased risk of sustained monomorphic VT during follow-up (p = 0.04); however, 75% of all sustained VT events occurred in HCM individuals without initial cardiac arrest. Appropriate ICD therapies were similar in both groups, and the cumulative incidence of shocks and ICD was low. The overall rate of inappropriate ICD therapies (inappropriate shock or ATP) was low. A total of 9 inappropriate ICD shocks (3% per group, p = 1.0) and a total of 5 inappropriate ATP therapies (1% in aHCM and 2% in sHCM; p = 1.0) were recorded over the entire study period. Eighteen individuals died during the study period (6% overall mortality). The all-cause mortality rate in aHCM was more than twice as high compared to sHCM (1.5/100 patient-years vs. 0.6/100 patient-years), but this difference did not reach statistical significance. The vast majority out of hospital and without detailed information about the medical circumstances and a final ICD interrogation was not available for any of the deceased ICD carriers.
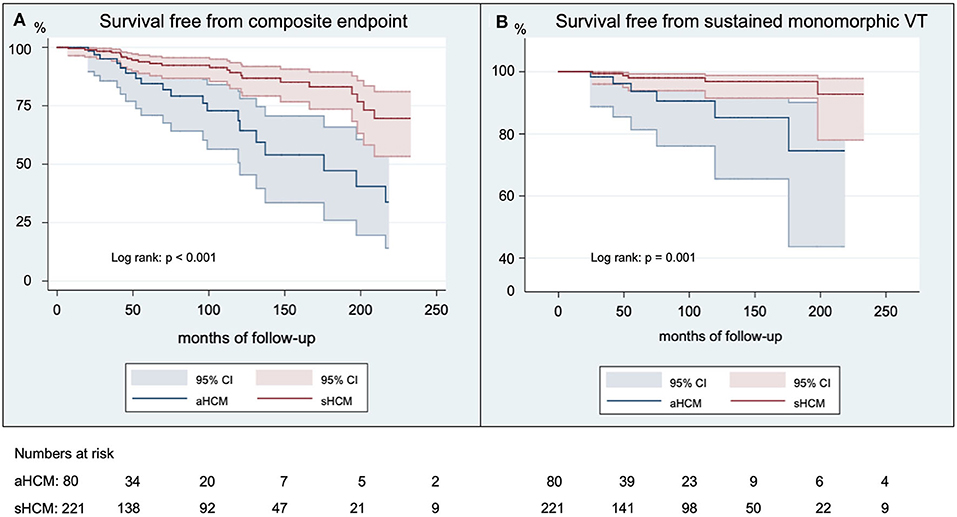
Figure 1. Survival free from arrhythmic outcomes. Shown are Kaplan-Meier curves for the survival free from the composite endpoint (A) and free from sustained monomorphic VT (B) in French Canadians with aHCM compared to sHCM. Kaplan-Meier curves are truncated at 250 months of follow-up.
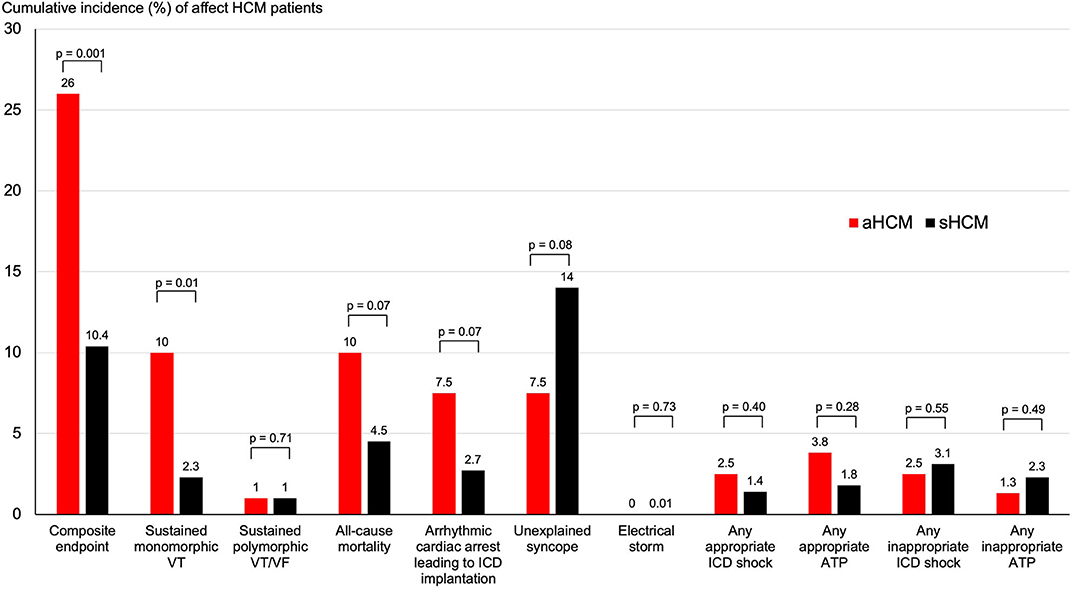
Figure 2. Cumulative incidence of arrhythmic outcomes and endpoints in apical and septal HCM. Shown are the cumulative incidence of mortality and arrhythmic endpoints over the follow-up period for patients with apical and septal HCM.
Those circumstances prevented any adjudication of the mortality causes. Cumulative all-cause mortality over time showed no difference between aHCM and sHCM (10% vs. 5%; p = 0.10), but the overall event rate was low. Interestingly, 20% (12/60) of sHCM patients post septal reduction therapy experienced a primary endpoint. Septal reduction therapy was not associated with an increased or decreased risk of an arrhythmic event or all-cause mortality on univariate analysis (HR 1.15 95% CI 0.56–2.35; p = 0.70).
ECG Data
All eligible resting ECGs were analyzed to look for additional electrocardiographic markers that may correlate with the underlying HCM phenotype and the risk of arrhythmic events. ECG results are displayed in Table 4. PR intervals and QRS durations were slightly longer in individuals with sHCM. Conduction abnormalities were common and observed in 111/301 individuals (37%) including permanent ventricular pacing in 12% and complete left or right bundle branch block in 77/301 (26%) of all study subjects. Left bundle branch block was significantly more common in sHCM (19% vs. 1%; p < 0.001) and was typically related to previous septal reduction therapy (43 vs. 11%; p < 0.001). As expected, symmetric precordial T-wave inversion was significantly more common in aHCM compared to sHCM. The amplitude of T-wave inversion was also significantly more pronounced in aHCM. None of the HCM patients with confirmed left ventricular apical aneurysm displayed typical ECG findings, suggestive of aneurysm formation. Since a recent study has suggested an association between arrhythmic events and the co-presence of an early repolarization (ER) pattern, all eligible ECGs were in addition analyzed with regard to J-point abnormalities. An (ER) pattern was observed in 29/139 (21%) of all eligible ECGs with similar prevalence in both groups. The median J-point elevation was more pronounced in aHCM compared to sHCM (2 [2] vs. 1.5 [0.75] mm; p = 0.03). However, the presence of an ER pattern was not associated with an increased probability of ventricular arrhythmia in our study population (OR 1.19, 95% CI 0.46–3.07; p = 0.72).
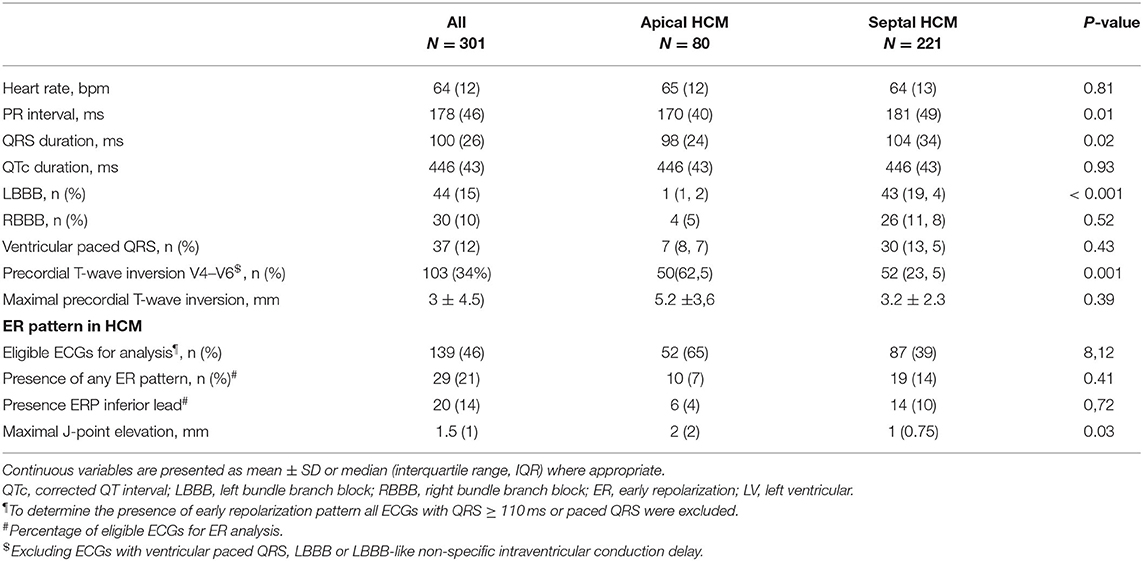
Table 4. ECG data.
Interestingly, none of the individuals with confirmed left ventricular apical aneurysm had typical ER findings.
Genetics
Although our study cohort includes patients from 2,000 on, routine genetic testing at our institution is only available since more recent. At the time of manuscript preparation, genetic data were available for 117 individuals (39%) with similar proportions of genetic testing in aHCM and sHCM (45% vs. 36%; p = 0.23) (Table 5). Genetic test results are displayed in Table 5. The overall yield of genetic testing was low, and a pathogenic or likely pathogenic variant was found in only 26 individuals (22%% of all tested individuals). Variants of MYBPC3 were most common accounting for 33% of all pathogenic/likely pathogenic mutations. The yield of genetic testing was significantly higher in sHCM compared to aHCM, and a pathogenic/likely pathogenic variant was found 5 times more often in sHCM (6% vs. 30%; p = 0.02). A positive family history of HCM did not influence the yield of genetic testing in this small genetic sample size (25% aHCM vs. 25% sHCM; p = 1.00), neither did the presence of a family history of SCD (6% aHCM vs. 6% sHCM; p = 1.00). The likelihood of a positive genetic test result remained unaffected by the absence or occurrence of arrhythmic events (p = 0.59).
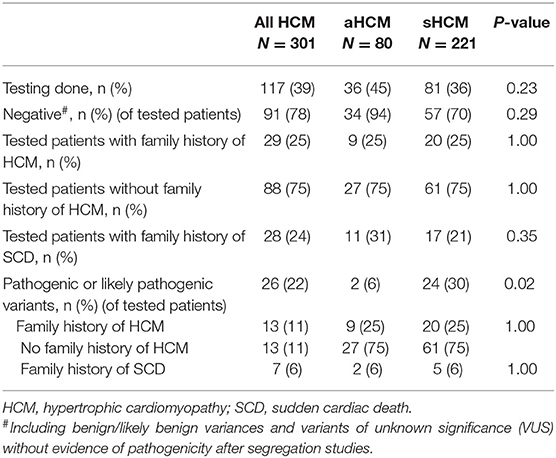
Table 5. Targeted genetic testing using next-generation sequencing.
Predictors of Arrhythmic Events
To identify risk factors of arrhythmic events, we conducted univariate and multivariate regression analysis using a Cox proportional hazard model. In addition to conventional risk factors (maximal wall thickness ≥ 30 mm, unexplained syncope, family history of SCD), we also included more recent risk factors (LGE, atrial fibrillation) and the presence and absence of left ventricular apical aneurysm. Given the low number of patients with segmental wall thickness ≥ 30 mm, we included grading of the maximal wall thickness as documented by CMR. The proportion of missing data about non-sustained ventricular tachycardia and/or exerciser-related drop of blood pressure prevented their inclusion for analysis. The results of the univariate analysis are displayed in Table 6. Apical HCM (HR 2.42, 95% CI 1.38–4.22; p = 0.002) and the presence of LV apical aneurysm (HR 3.70; 95% CI 1.12–12.27; p = 0.03) were predictors of the composite endpoint and the occurrence of sustained monomorphic VT. Even after exclusion of patients with left ventricular apical aneurysm, individuals with aHCM had significantly more arrhythmic events compared to sHCM (p = 0.004). A percentage of ≥ 15% of LGE was borderline predictive for the occurrence of a composite endpoint (HR 3.65 95% CI 0.98–13.62; p = 0.05). Among the conventional risk factors, unexplained syncope and a family history of unexplained SCD predicted sustained monomorphic VT at univariate analysis. Univariate predictors with a p ≤ 0.05 were entered into the multivariate analysis. Because of a strong collinearity between aHCM and the presence of a left ventricular apical aneurysm (p < 0.001), we conducted separate multiple regressions for those two factors. All other parameters did not show collinearity. Results of the multivariate analysis are displayed in Table 7. Apical HCM (HR 4.58, 95% CI 1.10–10.16; p = 0.04) and a percentage of left ventricular LGE ≥ 15% (HR 3.91, 95% CI 1.04–14.74; p = 0.04) were independent predictors of the composite endpoint in our study cohort. With regard to the occurrence of sustained monomorphic VT, aHCM (HR 5.17, 95% CI 1.65–16.15; p = 0.01), LV apical aneurysm (HR 4.89, 95% CI 1.20–19.81; p = 0.03), and a family history of unexplained SCD (HR 4.79, 95% CI 1.51–15.19; p = 0.01) were identified as independent predictors.
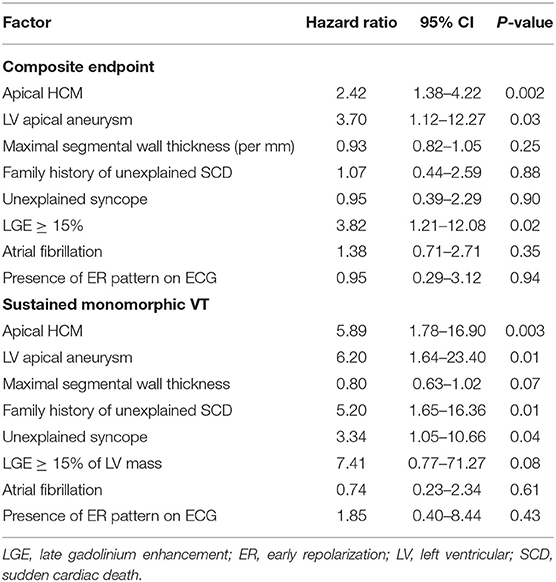
Table 6. Univariate analysis of predictors of arrhythmic outcomes.
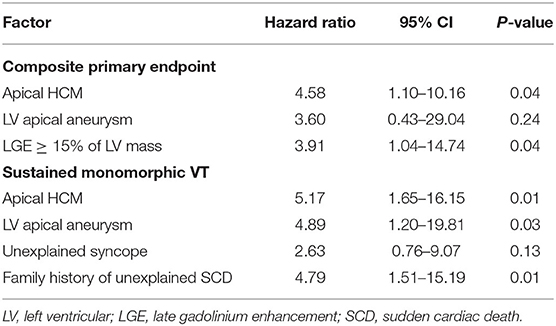
Table 7. Multivariate analysis of predictors of arrhythmic outcomes.
Discussion
Apical HCM is a distinct HCM phenotype with typical electrocardiographic and imaging findings that has been initially described in Japan where it accounts for 30–41% of all HCM cases (5, 6, 36). Over many years, aHCM was thought to affect predominantly Southeast Asian populations and to have a more benign clinical course compared to classic asymmetric sHCM (10, 36, 37). The findings of our study shed further light on the role of aHCM in Caucasian populations challenging the traditional concept of aHCM as a more benign phenotype.
First, we found that aHCM in the Quebec population was significantly more common than previously reported in other Caucasian populations accounting for 27% of all HCM cases followed at our center. Previous studies have reported prevalences of aHCM in the range of 3–11% among various Caucasian populations including the U.S. and the United Kingdom (7–9). This striking difference may be partially explained by differences in diagnostic criteria, infrequent use of cardiac magnetic resonance imaging in the past, or non-standardized classification of mixed/overlapping HCM phenotypes. For the present study, we excluded mixed HCM phenotypes and strictly applied the definitions of aHCM and sHCM as recommended by current guidelines (16, 17, 19). Additional reasons may be related to unique, yet unknown, genetic features of the French-Canadian population of Quebec (38, 39).
The most important observation of the present study is the significantly higher rate of ventricular arrhythmia in patients with aHCM compared to sHCM. The cumulative event rate in aHCM was 30%, which is among the highest rates reported so far for this particular HCM phenotype. The increased event rate in aHCM was largely related to a relatively high proportion of sustained monomorphic ventricular tachycardia (15%), which has not been reported before. Although aHCM was not associated with increased all-cause mortality compared to sHCM in our study, it caused increased morbidity due to the higher rate of recurrent ventricular arrhythmia.
Our results challenge the traditional concept of aHCM as a more “benign” form of HCM that has been previously reported in Asian and Non-Asian populations (10–12, 29). At least in the French-Canadian Caucasian population of Quebec, aHCM displays a more aggressive phenotype that is characterized by an increased risk of ventricular arrhythmia.
Previous studies reporting adverse cardiac events in patients with aHCM have mostly focused on all-cause mortality or included cohorts with different ethnic backgrounds (13, 40, 41). The study of Klarich et al. reported high all-cause mortality of 29% over a mean follow-up of 78 months that was mostly driven by non-cardiac deaths and the overall incidence of sudden cardiac death was low (13). The same study also identified female sex and atrial fibrillation as predictors of all-cause mortality, which was not reproduced by our study. Our study population was otherwise comparable to previous contemporary HCM cohorts with regard to age, medical comorbidities, and findings on cardiac imaging (8, 12, 13). The proportion of familial HCM and a positive family history of SCD was comparable to previous studies and was similar between aHCM and sHCM in our study (8). The high proportion of familial aHCM (20%) in our population is significantly higher than the 6% reported in a large Japanese cohort. This suggests different inheritance patterns and leaves room for speculation about the possibility of different pathophysiological and genetic mechanisms (42).
The increased risk of ventricular arrhythmia in our aHCM population also highlights the challenge of adequate risk stratification in aHCM. Preventive ICD implantation is currently recommended based on the presence of traditional risk factors or the more recent risk score developed by O'Mahony et al. (17, 28, 43). However, current risk prediction models were typically derived from patient populations with sHCM and may not be accurate for risk stratification in apical HCM.
The degree of segmental hypertrophy in aHCM and sHCM was similar in our study and did not show any differences with regard to the left ventricular mass index contrasting the results Kim et al. who found larger LV mass indices in sHCM (12). Those differences could be related to ethnic characteristics as the study of Kim et al. only included Korean individuals (12).
In addition to traditional risk factors, the presence of left ventricular myocardial fibrosis has been established as a prognostic factor for adverse cardiovascular outcomes in HCM with increased risk at scarring of ≥ 15 % of the left ventricular mass (22). In contrast to Asian HCM cohorts (12), the overall degree of left ventricular fibrosis in our study was similar between aHCM and sHCM. Interestingly, the proportion of aHCM patients with ≥ 15% of left ventricular fibrosis in our cohort was 10 times higher compared to the Korean aHCM cohort (12).
An important structural finding in our study cohort was a 10% prevalence of left ventricular apical aneurysm in the group of aHCM, which is among the highest reported in Caucasian cohorts with aHCM. Higher prevalences of apical aneurysm up to 23% have been described in Asian, but not Caucasian populations (12, 44). A crucial element for the assessment of left ventricular apical aneurysm is cardiac MRI, which has been shown to be more sensitive and accurate for the diagnosis of apical aneurysm compared to contrast-enhanced or standard echocardiography (45, 46). The lack of systematic CMR assessment of HCM patients in the past may have influenced the low prevalence overall and in aHCM in older studies. Even in our study, there are still 45% of study subjects who did not undergo CMR imaging—often because of previous implantation of a non-MRI compatible pacemaker or ICD. Therefore, the true proportion of LV apical aneurysm in our study cohort may actually be even higher. Apical aneurysm of the left ventricle has been identified as a predictor of worse prognosis and increased risk of ventricular arrhythmia in HCM (46, 47). However, most previous studies did not differentiate between aHCM and sHCM (46, 48). In our study, the presence of LV apical aneurysm was an independent predictor of ventricular arrhythmia, which is consistent with previous reports (46, 47). The presence of LV apical aneurysm is currently not part of recommended criteria for the indication of a primary prevention ICD (17, 43, 49). Our data and the reports of others, however, have shown that apical aneurysm is strongly associated with ventricular arrhythmia highlighting the need for a novel risk prediction model targeting specifically the subgroup of patients with aHCM.
Limitations
Our study has several limitations. First, this is a retrospective single-center registry with limitations inherent to any retrospective study design.
Referral bias is a typical problem of registry data from tertiary centers and may have also influenced the present study. Because of that and a modest sample size, we cannot exclude that the true prevalence of aHCM may be different and that the overall cardiac risk in the general HCM population may be lower. It is possible that a subset of aHCM patients with very mild phenotypes and low cardiac risk never comes to medical attention. Second, the overall event rate in this study was relatively low, which may be related to the modest sample size, but is consistent with other large-scale contemporary studies and also reflects contemporary treatment strategies that have significantly improved the clinical course of HCM (50).
Inadvertent inclusion of hypertensive cardiomyopathies with isolated sigmoid hypertrophy of the basal septum is a potential concern for outcome analysis in all studies of sHCM cohorts and could potentially lower arrhythmic event rates. Despite very specific and detailed inclusion criteria, we cannot exclude that a small proportion of patients with sHCM actually may have hypertensive cardiomyopathy. True sHCM is typically characterized by reverse curve septal hypertrophy representing the majority of sHCM cases in our study. This phenotype should be distinguished from isolated sigmoid hypertrophy of the basal septum, which represents a more benign form and is often no true primary HCM (51).
A detailed mortality comparison between aHCM and sHCM was not possible given the low rate of all-cause mortality and the fact that deaths could not been adjudicated. Third, our detailed cardiac MRI analysis did not find significant differences between aHCM and sHCM in our French-Canadian population. Only 50% of our study population had a cardiac MRI performed, so it is possible that we may have missed differences in scar burden between the two study groups. Missing MRI studies were predominantly related to the presence of non-MRI compatible pacemaker or ICD systems and limited access to MRI until the early 2000's. Despite that, the proportion of patients with cardiac MRI was still higher than in other contemporary cohorts of aHCM patients (8, 13).
Only 39% of individuals underwent genetic testing, which mostly reflects missing genetic infrastructure at our center in the past. Although the genetic yield in aHCM is expected to be lower compared to sHCM, the results of our study were even lower compared to data from the literature (6). This may be partially explained by the overall low rates of genetic testing, but we cannot completely exclude the presence of an unidentified genetic substrate in the French-Canadian population.
Conclusions
The phenotype of apical HCM is much more common in French-Canadians (27%) of Caucasian descent compared to other Caucasian HCM populations. Apical HCM in French-Canadians is associated with an increased risk for ventricular arrhythmia.
Data Availability Statement
The authors acknowledge that the data presented in this study must be deposited and made publicly available in an acceptable repository, prior to publication. Frontiers cannot accept a article that does not adhere to our open data policies.
Ethics Statement
The studies involving human participants were reviewed and approved by ERB of the IUCPQ-UL. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author Contributions
CS designed and planned the study. Data collection was performed by CS, CN-R, and PA. Data analysis and statistical analysis was performed by CS, CN-R, PA, and J-FS. Cardiac MRI interpretation was performed by EL. Interpretation of echocardiographies was performed by MA. CS wrote the manuscript that was subsequently revised and edited by all co-authors. All authors have read and approved the final version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the HCM patients and their families who made this work possible.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2020.548564/full#supplementary-material
Abbreviations
AF, Atrial Fibrillation; CMR, Cardiac magnetic resonance; ER, Early repolarization; aHCM, Apical hypertrophic cardiomyopathy; HCM, Hypertrophic cardiomyopathy; sHCM, Septal hypertrophic cardiomyopathy; LV, Left ventricle; LVEF, Left ventricular ejection fraction; ICD, Implantable cardioverter defibrillator; VF, Ventricular fibrillation; VT, Ventricular tachycardia; SCD, Sudden cardiac death.
References
1. Maron BJ, Haas TS, Ahluwalia A, Murphy CJ, Garberich RF. Demographics and epidemiology of sudden deaths in young competitive athletes: from the United States national registry. Am J Med. (2016) 129:1170–7. doi: 10.1016/j.amjmed.2016.02.031
2. Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. (2017) 121:749–70. doi: 10.1161/CIRCRESAHA.117.311059
3. Maron BJ, Gottdiener JS, Epstein SE. Patterns and significance of distribution of left ventricular hypertrophy in hypertrophic cardiomyopathy. A wide angle, two dimensional echocardiographic study of 125 patients. Am J Cardiol. (1981) 48:418–28. doi: 10.1016/0002-9149(81)90068-0
4. Parato VM, Antoncecchi V, Sozzi F, Marazia S, Zito A, Maiello M, et al. Echocardiographic diagnosis of the different phenotypes of hypertrophic cardiomyopathy. Cardiovasc Ultrasound. (2016) 14:30. doi: 10.1186/s12947-016-0072-5
5. Yamaguchi H, Ishimura T, Nishiyama S, Nagasaki F, Nakanishi S, Takatsu F, et al. Hypertrophic nonobstructive cardiomyopathy with giant negative T waves (apical hypertrophy): ventriculographic and echocardiographic features in 30 patients. Am J Cardiol. (1979) 44:401–12. doi: 10.1016/0002-9149(79)90388-6
6. Jan MF, Todaro MC, Oreto L, Tajik AJ. Apical hypertrophic cardiomyopathy: present status. Int J Cardiol. (2016) 222:745–59. doi: 10.1016/j.ijcard.2016.07.154
7. Kitaoka H, Doi Y, Casey SA, Hitomi N, Furuno T, Maron BJ. Comparison of prevalence of apical hypertrophic cardiomyopathy in Japan and the United States. Am J Cardiol. (2003) 92:1183–6. doi: 10.1016/j.amjcard.2003.07.027
8. Towe EC, Bos JM, Ommen SR, Gersh BJ, Ackerman MJ. Genotype-phenotype correlations in apical variant hypertrophic cardiomyopathy. Congenit Heart Dis. (2015) 10:E139–45. doi: 10.1111/chd.12242
9. Sheikh N, Papadakis M, Panoulas VF, Prakash K, Millar L, Adami P, et al. Comparison of hypertrophic cardiomyopathy in Afro-Caribbean vs. white patients in the UK. Heart. (2016) 102:1797–804. doi: 10.1136/heartjnl-2016-309843
10. Eriksson MJ, Sonnenberg B, Woo A, Rakowski P, Parker TG, Wigle ED, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol. (2002) 39:638–45. doi: 10.1016/S0735-1097(01)01778-8
11. Kim SH, Kim SO, Han S, Hwang KW, Lee CW, Nam GB, et al. Long-term comparison of apical vs. asymmetric hypertrophic cardiomyopathy. Int Heart J. (2013) 54:207–11. doi: 10.1536/ihj.54.207
12. Kim EK, Lee SC, Hwang JW, Chang SA, Park SJ, On YK, et al. Differences in apical and non-apical types of hypertrophic cardiomyopathy: a prospective analysis of clinical, echocardiographic, and cardiac magnetic resonance findings and outcome from 350 patients. Eur Heart J Cardiovasc Imag. (2016) 17:678–86. doi: 10.1093/ehjci/jev192
13. Klarich KW, Attenhofer Jost CH, Binder J, Connolly HM, Scott CG, Freeman WK, et al. Risk of death in long-term follow-up of patients with apical hypertrophic cardiomyopathy. Am J Cardiol. (2013) 111:1784–91. doi: 10.1016/j.amjcard.2013.02.040
14. Gauvin H, Moreau C, Lefebvre JF, Laprise C, Vezina H, Labuda D, et al. Genome-wide patterns of identity-by-descent sharing in the French Canadian founder population. Eur J Hum Genet. (2014) 22:814–21. doi: 10.1038/ejhg.2013.227
15. American College of Cardiology Foundation Task Force on Expert Consensus D Hundley WG Bluemke DA Finn JP Flamm SD Fogel MA . ACCF/ACR/AHA/NASCI/SCMR 2010 expert consensus document on cardiovascular magnetic resonance: a report of the American college of cardiology foundation task force on expert consensus documents. J Am Coll Cardiol. (2010) 55:2614–62. doi: 10.1016/j.jacc.2009.11.011
16. Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American college of cardiology foundation/American heart association task force on practice guidelines. Developed in collaboration with the American association for thoracic surgery, American society of echocardiography, American society of nuclear cardiology, heart failure society of America, heart rhythm society, society for cardiovascular angiography and interventions, and society of thoracic surgeons. J Am Coll Cardiol. (2011) 58:e212–60. doi: 10.1016/j.jacc.2011.06.011
17. Authors/Task Force M, Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. (2014) 35:2733–79. doi: 10.1093/eurheartj/ehu284
18. Hindieh W, Weissler-Snir A, Hammer H, Adler A, Rakowski H, Chan RH. Discrepant measurements of maximal left ventricular wall thickness between cardiac magnetic resonance imaging and echocardiography in patients with hypertrophic cardiomyopathy. Circ Cardiovasc Imag. (2017) 10:e006309. doi: 10.1161/CIRCIMAGING.117.006309
19. Nagueh SF, Bierig SM, Budoff MJ, Desai M, Dilsizian V, Eidem B, et al. American society of echocardiography clinical recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: endorsed by the American society of nuclear cardiology, society for cardiovascular magnetic resonance, and society of cardiovascular computed tomography. J Am Soc Echocardiogr. (2011) 24:473–98. doi: 10.1016/j.echo.2011.03.006
20. Larose E, Rodes-Cabau J, Pibarot P, Rinfret S, Proulx G, Nguyen CM, et al. Predicting late myocardial recovery and outcomes in the early hours of ST-segment elevation myocardial infarction traditional measures compared with microvascular obstruction, salvaged myocardium, and necrosis characteristics by cardiovascular magnetic resonance. J Am Coll Cardiol. (2010) 55:2459–69. doi: 10.1016/j.jacc.2010.02.033
21. Harrigan CJ, Peters DC, Gibson CM, Maron BJ, Manning WJ, Maron MS, et al. Hypertrophic cardiomyopathy: quantification of late gadolinium enhancement with contrast-enhanced cardiovascular MR imaging. Radiology. (2011) 258:128–33. doi: 10.1148/radiol.10090526
22. Chan RH, Maron BJ, Olivotto I, Pencina MJ, Assenza GE, Haas T, et al. Prognostic value of quantitative contrast-enhanced cardiovascular magnetic resonance for the evaluation of sudden death risk in patients with hypertrophic cardiomyopathy. Circulation. (2014) 130:484–95. doi: 10.1161/CIRCULATIONAHA.113.007094
23. Moss AJ, Schuger C, Beck CA, Brown MW, Cannom DS, Daubert JP, et al. Reduction in inappropriate therapy and mortality through ICD programming. N Engl J Med. (2012) 367:2275–83. doi: 10.1056/NEJMoa1211107
24. Gasparini M, Proclemer A, Klersy C, Kloppe A, Lunati M, Ferrer JB, et al. Effect of long-detection interval vs standard-detection interval for implantable cardioverter-defibrillators on antitachycardia pacing and shock delivery: the ADVANCE III randomized clinical trial. JAMA. (2013) 309:1903–11. doi: 10.1001/jama.2013.4598
25. Saeed M, Hanna I, Robotis D, Styperek R, Polosajian L, Khan A, et al. Programming implantable cardioverter-defibrillators in patients with primary prevention indication to prolong time to first shock: results from the PROVIDE study. J Cardiovasc Electrophysiol. (2014) 25:52–9. doi: 10.1111/jce.12273
26. Wilkoff BL, Fauchier L, Stiles MK, Morillo CA, Al-Khatib SM, Almendral J, et al. 2015 HRS/EHRA/APHRS/SOLAECE expert consensus statement on optimal implantable cardioverter-defibrillator programming and testing. Heart Rhythm. (2016) 13:e50–86. doi: 10.1016/j.hrthm.2015.11.018
27. Philippon F, Sterns LD, Nery PB, Parkash R, Birnie D, Rinne C, et al. Management of implantable cardioverter defibrillator recipients: care beyond guidelines. Can J Cardiol. (2017) 33:977–90. doi: 10.1016/j.cjca.2017.05.012
28. O'Mahony C, Jichi F, Pavlou M, Monserrat L, Anastasakis A, Rapezzi C, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J. (2014) 35:2010–20. doi: 10.1093/eurheartj/eht439
29. Webb JG, Sasson Z, Rakowski H, Liu P, Wigle ED. Apical hypertrophic cardiomyopathy: clinical follow-up and diagnostic correlates. J Am Coll Cardiol. (1990) 15:83–90. doi: 10.1016/0735-1097(90)90180-W
30. Moon JC, Fisher NG, McKenna WJ, Pennell DJ. Detection of apical hypertrophic cardiomyopathy by cardiovascular magnetic resonance in patients with non-diagnostic echocardiography. Heart. (2004) 90:645–9. doi: 10.1136/hrt.2003.014969
31. Ichida M, Nishimura Y, Kario K. Clinical significance of left ventricular apical aneurysms in hypertrophic cardiomyopathy patients: the role of diagnostic electrocardiography. J Cardiol. (2014) 64:265–72. doi: 10.1016/j.jjcc.2014.02.011
32. Pennacchini E, Musumeci MB, Conte MR, Stollberger C, Formisano F, Bongioanni S, et al. Electrocardiographic evolution in patients with hypertrophic cardiomyopathy who develop a left ventricular apical aneurysm. J Electrocardiol. (2015) 48:818–25. doi: 10.1016/j.jelectrocard.2015.06.004
33. Macfarlane PW, Antzelevitch C, Haissaguerre M, Huikuri HV, Potse M, Rosso R, et al. The early repolarization pattern: a consensus paper. J Am Coll Cardiol. (2015) 66:470–7. doi: 10.1016/j.jacc.2015.05.033
34. Flett AS, Maestrini V, Milliken D, Fontana M, Treibel TA, Harb R, et al. Diagnosis of apical hypertrophic cardiomyopathy: T-wave inversion and relative but not absolute apical left ventricular hypertrophy. Int J Cardiol. (2015) 183:143–8. doi: 10.1016/j.ijcard.2015.01.054
35. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
36. Sakamoto T, Tei C, Murayama M, Ichiyasu H, Hada Y. Giant T wave inversion as a manifestation of asymmetrical apical hypertrophy (AAH) of the left ventricle. Echocardiographic and ultrasono-cardiotomographic study. Jpn Heart J. (1976) 17:611–29. doi: 10.1536/ihj.17.611
37. Moro E, D'Angelo G, Nicolosi GL, Mimo R, Zanuttini D. Long-term evaluation of patients with apical hypertrophic cardiomyopathy. Correlation between quantitative echocardiographic assessment of apical hypertrophy and clinical-electrocardiographic findings. Eur Heart J. (1995) 16:210–7. doi: 10.1093/oxfordjournals.eurheartj.a060887
38. Laberge AM, Michaud J, Richter A, Lemyre E, Lambert M, Brais B, et al. Population history and its impact on medical genetics in Quebec. Clin Genet. (2005) 68:287–301. doi: 10.1111/j.1399-0004.2005.00497.x
39. Low-Kam C, Rhainds D, Lo KS, Provost S, Mongrain I, Dubois A, et al. Whole-genome sequencing in French Canadians from Quebec. Hum Genet. (2016) 135:1213–21. doi: 10.1007/s00439-016-1702-6
40. Kim KH, Kim HK, Hwang IC, Lee SP, Park EA, Lee W, et al. Myocardial scarring on cardiovascular magnetic resonance in asymptomatic or minimally symptomatic patients with “pure” apical hypertrophic cardiomyopathy. J Cardiovasc Magn Reson. (2012) 14:52. doi: 10.1186/1532-429X-14-52
41. Hanneman K, Crean AM, Williams L, Moshonov H, James S, Jimenez-Juan L, et al. Cardiac magnetic resonance imaging findings predict major adverse events in apical hypertrophic cardiomyopathy. J Thorac Imaging. (2014) 29:331–9. doi: 10.1097/RTI.0000000000000115
42. Kubo T, Kitaoka H, Okawa M, Hirota T, Hoshikawa E, Hayato K, et al. Clinical profiles of hypertrophic cardiomyopathy with apical phenotype–comparison of pure-apical form and distal-dominant form. Circ J. (2009) 73:2330–6. doi: 10.1253/circj.CJ-09-0438
43. Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American college of cardiology foundation/American heart association task force on practice guidelines. Circulation. (2011) 124:2761–96. doi: 10.1161/CIR.0b013e318223e2bd
44. Matsubara K, Nakamura T, Kuribayashi T, Azuma A, Nakagawa M. Sustained cavity obliteration and apical aneurysm formation in apical hypertrophic cardiomyopathy. J Am Coll Cardiol. (2003) 42:288–95. doi: 10.1016/S0735-1097(03)00576-X
45. Rowin EJ, Maron MS. The role of cardiac MRI in the diagnosis and risk stratification of hypertrophic cardiomyopathy. Arrhythm Electrophysiol Rev. (2016) 5:197–202. doi: 10.15420/aer.2016:13:3
46. Rowin EJ, Maron BJ, Haas TS, Garberich RF, Wang W, Link MS, et al. Hypertrophic cardiomyopathy with left ventricular apical aneurysm: implications for risk stratification and management. J Am Coll Cardiol. (2017) 69:761–73. doi: 10.1016/j.jacc.2016.11.063
47. Igarashi M, Nogami A, Kurosaki K, Hanaki Y, Komatsu Y, Fukamizu S, et al. Radiofrequency catheter ablation of ventricular tachycardia in patients with hypertrophic cardiomyopathy and apical aneurysm. JACC Clin Electrophysiol. (2018) 4:339–50. doi: 10.1016/j.jacep.2017.12.020
48. Maron MS, Finley JJ, Bos JM, Hauser TH, Manning WJ, Haas TS, et al. Prevalence, clinical significance, and natural history of left ventricular apical aneurysms in hypertrophic cardiomyopathy. Circulation. (2008) 118:1541–9. doi: 10.1161/CIRCULATIONAHA.108.781401
49. Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: executive summary: a report of the American college of cardiology/American heart association task force on clinical practice guidelines and the heart rhythm society. Heart Rhythm. (2018) 15:e190–e252. doi: 10.1161/CIR.0000000000000548
50. Maron BJ, Rowin EJ, Casey SA, Link MS, Lesser JR, Chan RH, et al. Hypertrophic cardiomyopathy in adulthood associated with low cardiovascular mortality with contemporary management strategies. J Am Coll Cardiol. (2015) 65:1915–28. doi: 10.1016/j.jacc.2015.02.061
Keywords: hypertrophic cardiomyopathy, apical hypertrophic cardiomyopathy, ventricular arrhythmia, septal hypertrophic cardiomyopathy, French-Canadian
Citation: Steinberg C, Nadeau-Routhier C, André P, Philippon F, Sarrazin J-F, Nault I, O'Hara G, Blier L, Molin F, Plourde B, Roy K, Larose E, Arsenault M and Champagne J (2020) Ventricular Arrhythmia in Septal and Apical Hypertrophic Cardiomyopathy: The French-Canadian Experience. Front. Cardiovasc. Med. 7:548564. doi: 10.3389/fcvm.2020.548564
Received: 03 April 2020; Accepted: 25 August 2020;
Published: 22 October 2020.
Edited by:
Valeria Novelli, Agostino Gemelli University Polyclinic, Catholic University of the Sacred Heart, ItalyReviewed by:
Steven Clive Greenway, University of Calgary, CanadaGeorges Nemer, American University of Beirut, Lebanon
Copyright © 2020 Steinberg, Nadeau-Routhier, André, Philippon, Sarrazin, Nault, O'Hara, Blier, Molin, Plourde, Roy, Larose, Arsenault and Champagne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Steinberg, Y2hyaXN0aWFuLnN0ZWluYmVyZ0Bjcml1Y3BxLnVsYXZhbC5jYQ==