Abstract
Plasminogen activator inhibitor 1 (PAI-1) is a member of the serine protease inhibitor (serpin) superfamily. PAI-1 is the principal inhibitor of the plasminogen activators, tissue plasminogen activator (tPA), and urokinase-type plasminogen activator (uPA). Turbulence in the levels of PAI-1 tilts the balance of the hemostatic system resulting in bleeding or thrombotic complications. Not surprisingly, there is strong evidence that documents the role of PAI-1 in cardiovascular disease. The more recent uncovering of the coalition between the hemostatic and inflammatory pathways has exposed a distinct role for PAI-1. The storm of proinflammatory cytokines liberated during inflammation, including IL-6 and TNF-α, directly influence PAI-1 synthesis and increase circulating levels of this serpin. Consequently, elevated levels of PAI-1 are commonplace during infection and are frequently associated with a hypofibrinolytic state and thrombotic complications. Elevated PAI-1 levels are also a feature of metabolic syndrome, which is defined by a cluster of abnormalities including obesity, type 2 diabetes, hypertension, and elevated triglyceride. Metabolic syndrome is in itself defined as a proinflammatory state associated with elevated levels of cytokines. In addition, insulin has a direct impact on PAI-1 synthesis bridging these pathways. This review describes the key physiological functions of PAI-1 and how these become perturbed during disease processes. We focus on the direct relationship between PAI-1 and inflammation and the repercussion in terms of an ensuing hypofibrinolytic state and thromboembolic complications. Collectively, these observations strengthen the utility of PAI-1 as a viable drug target for the treatment of various diseases.
Introduction
Plasminogen activator inhibitor-1 (PAI-1) is a fast-acting serpin that regulates the fibrinolytic system through inhibition of tissue plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA). PAI-1 quenches the enzymatic activity of these proteases to constrain fibrin degradation and stabilize the hemostatic plug. Like other serpins, PAI-1 forms a 1:1 enzyme-inhibitor complex with its target proteases, rendering them enzymatically inactive and resulting in rapid clearance from the circulation via the hepatic system. However, PAI-1 is an unusual serpin in that it can lose activity by spontaneous insertion of the reactive center loop into the body of the molecule, forming “latent” PAI-1 (1). The active form of PAI-1 is very unstable and has a short half-life of 1 h (2), whereas conversion to its thermodynamically stable latent form allows a prolonged half-life of 2–4 h (3). In healthy individuals' plasma, PAI-1 circulates in excess over its target protease tPA but at relatively low concentrations compared with other serpins, and is highly variable in normal individuals (1–40 ng/ml). PAI-1 is relatively unstable with a half-life of around 1–2 h in the circulation (4), however, binding to the extracellular matrix protein, vitronectin, stabilizes the active form of PAI-1 (5), and augments its half-life (6).
Circulating PAI-1 levels are under genetic control that is directly related to an insertion/deletion (5G/4G) polymorphism at position −675 of the promoter (7). The 4G allele gives rise to elevated plasma PAI-1 levels (8–11). PAI-1 displays circadian rhythm with a peak in early morning that coincides with the time of onset of myocardial infarction (MI) (12). The 4G/5G polymorphism differs according to ethnic group which has a direct impact on PAI-1 circulating levels (13). Interestingly, the levels of PAI-1 vary according to gender and show a positive correlation with increasing age (14).
Platelets contain the major pool of circulating PAI-1, which when activated following vessel injury, release this cargo thereby protecting the developing thrombus from premature fibrinolysis. Not surprisingly, the platelet precursor cell, megakaryocytes, are a major site of PAI-1 synthesis (15) and platelets themselves are now known to retain some PAI-1 mRNA which can produce functional protein (16). However, PAI-1 is also synthesized by other cells including endothelial (17), adipocytes (18–20), hepatocytes (21), and cardiomyocytes (22) (Figure 1A). Given the crucial role of PAI-1 in hemostasis, a deficiency in this serpin gives rise to a moderate bleeding diathesis (23). Conversely, increased levels of PAI-1 are associated with thrombotic complications. In addition to its hemostatic role, PAI-1 functions in several physiological processes such as inflammation, wound healing, and tumor progression. A strong relationship between PAI-1 and obesity, diabetes, and metabolic syndrome (MetS) was recognized many years ago with this serpin now being considered central to these pathophysiological processes (24). This review will focus on the impact and relationship of this unusual serpin in dictating and orchestrating the development of thromboinflammation and cardiovascular complications as a result of its participation in the pathogenesis of associated diseases.
Figure 1
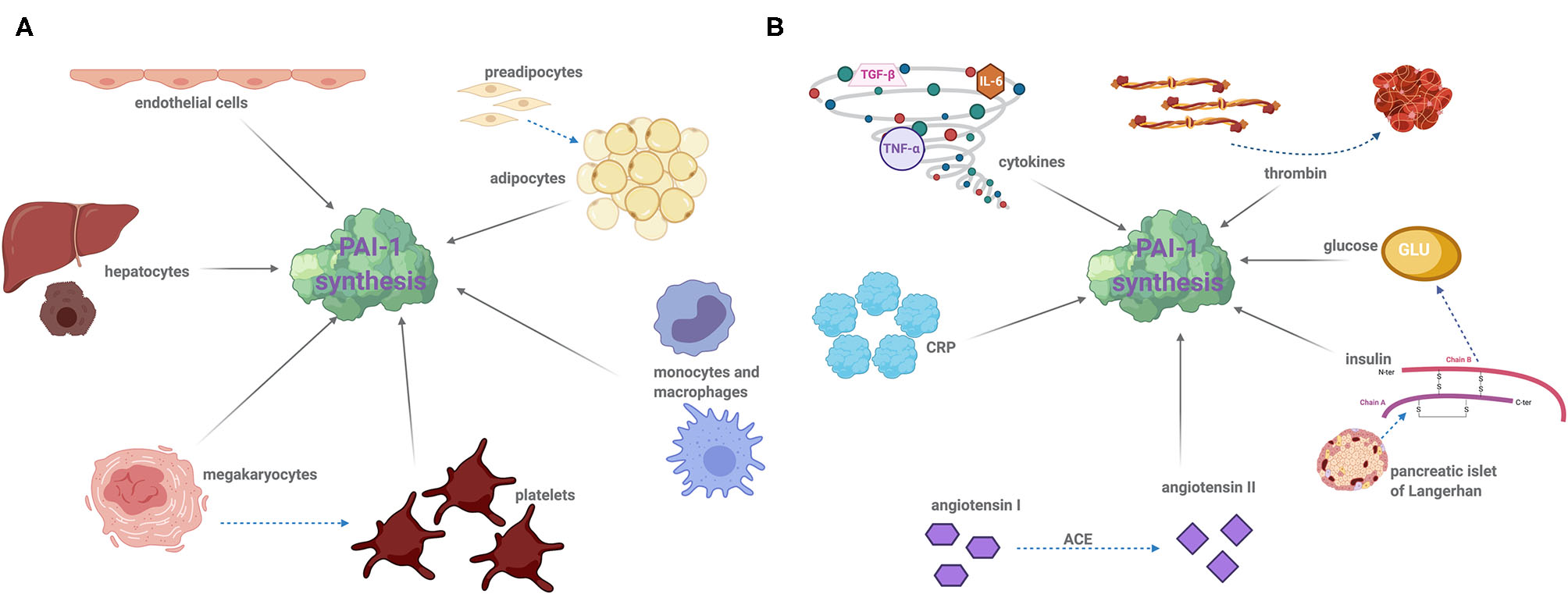
Cellular sources of PAI-1 of modulators of synthesis: (A) PAI-1 is produced by a variety of cell types including the following: endothelial cells, adipocytes, hepatocytes, leukocytes (monocytes and macrophages), megakaryocytes, and platelets. Dotted arrows demonstrated cell differentiation and solid arrows indicate PAI-1 synthesis. (B) A number of factors induce PAI-1 synthesis and secretion. Proinflammatory cytokines, namely interleukin-6 (IL-6), tissue necrosis factor alpha (TNF-α), and transforming growth factor beta (TGF-β) significantly augment PAI-1 synthesis. Thrombin, insulin, glucose, angiotensin II, and C-reactive protein (CRP) can also stimulate PAI-1 expression. Drugs that target insulin and the angiotensin converting enzyme (ACE), such as metformin and captopril, are known to decrease plasma PAI-1 concentration. Dotted arrows represent the reaction of an enzyme or protein synthesis, and solid arrows indicate PAI-1 synthesis.
PAI-1 and Thrombosis
The fate of a forming thrombus is determined by platelet deposition and the balance of coagulation and fibrinolytic factors. An increase in circulating levels of PAI-1 or augmented local release of this inhibitor due to platelet activation shifts the balance to a hypofibrinolytic state. PAI-1 has been recognized as a pivotal protein in the progression of vascular events and is linked to MI (25–27), stroke (28), deep vein thrombosis (DVT) (29), and microvascular thrombosis (30). Elevated levels of plasma PAI-1 precede the occurrence of MI (26), and survivors exhibit consistently high levels (25). Acute increases in plasma PAI-1 levels within 24 h in patients with acute ST-elevated myocardial infarction are associated with heart failure and death and are a strong independent predictor of mortality at 30 days (31). The renin-angiotensin II system (RAS) is strongly activated following acute MI, and angiotensin II triggers PAI-1 synthesis (32) (Figure 1B). The RAS has been linked to the circadian variation in PAI-1, suggesting that the use of angiotensin-converting enzyme (ACE) inhibitors to inhibit the RAS system may blunt PAI-1 levels thereby reducing the risk of “early morning” MI (33).
Elevated levels of PAI-1 have been detected in atherosclerotic plaques in humans (34–36), which are significantly inflated in type 2 diabetes mellitus (T2DM) subjects (37). Dysregulation of the fibrinolytic system appears to play a significant role in atherosclerotic plaque development by perturbing the wound-healing response and neointimal formation (38). This can in part be attributed to reduced vascular smooth muscle cell migration via inhibition of binding of vitronectin to integrin αvβ3 (39). Moreover, PAI-1 stabilizes the fibrin matrix within the developing plaque by attenuating plasmin formation. Increased serum levels of PAI-1 have been noted in patients with atherosclerotic disease, including coronary artery disease (40) and stroke (41). A recent large meta-analysis has indicated that PAI-1 is implicated in the pathogenesis of atherosclerotic disease (42). The two-pronged approach of PAI-1 inducing a hypofibrinolytic state in atherosclerotic diseases and impacting on lesion formation and progression indicates the perilous complications of this serpin in the pathogenesis of these diseases. Animal models of ablation (43, 44) of the PAI-1 gene and pharmacological inhibition of PAI-1 (45) in murine models have proved insightful in our understanding of these processes. Nevertheless, it is challenging to extrapolate all of these findings to humans, which combined with the lack of a licensed inhibitor, leaves a gap in our knowledge and understanding of this serpin in the intricate mechanisms of this complex disease.
Perioperative DVT has been linked to elevated levels of circulating PAI-1 (29). A higher incidence of DVT (46) and venous thrombosis (47) has been noted in Asian Indian patients harboring the 4G polymorphism, leading to a suggestion that it be included in all laboratory testing panels for thrombophilia. Similar studies in white Caucasian populations have described association of the 4G polymorphism with idiopathic DVT and inherited thrombophilia (48). More recently, preoperative plasma PAI-1 has been revealed as an independent risk factor for the onset of DVT in patients undergoing total hip arthroplasty (49). Elevated levels of plasma PAI-1 largely account for delayed clot lysis times in healthy individuals and are associated with first incidence of venous thrombosis (50). These lines of evidence highlight the importance of this serpin in predisposing individuals to a hypofibrinolytic state, which is directly linked to an increased frequency of venous thrombosis. Yet, the exact mechanisms underpinning this pathophysiological process and cellular source of PAI-1 remain to be elucidated.
Infection and Inflammation
PAI-1 is a positive acute phase protein that is dramatically elevated in the proinflammatory state, such as acute tissue injury, sepsis, and inflammation. The role of PAI-1 in this context is primarily considered a protective mechanism to limit dissemination of pathogens and promote tissue repair. Augmented levels of PAI-1 in non-typeable Haemophilus influenzae infection are associated with bacterial clearance and shortening of the disease duration (51). Pharmacological inhibition of PAI-1 in a Pseudomonas aeruginosa pneumonia mouse model attenuates neutrophil migration, thereby dampening the innate immune response (52). PAI-1 modulation of neutrophil migration has also been demonstrated in Escherichia coli infection (53). However, aberrant activation of this defense mechanism produces a hypofibrinolytic state which promotes thrombotic complications.
Sepsis occurs due to overreaction of the host defense mechanism, most commonly in response to bacterial infection, but can also be caused by viral and fungal pathogens. Sepsis leads to enhanced exposure of the coagulation protein, tissue factor, inciting fibrin deposition and microthrombi throughout the vasculature (54). Multiorgan failure is a frequent complication in sepsis patients, and the development of disseminated intravascular coagulation (DIC) is a major contributor (54) and is associated with aberrant thrombin generation. Endothelial dysfunction induces release of proinflammatory cytokines (55, 56) which combined with augmented levels of thrombin provoke PAI-1 synthesis (Figure 1B). Endothelial cells produce enhanced levels of PAI-1 in response to C-reactive protein (CRP) (57, 58) (Figure 1B), which is a proinflammatory marker in critically ill patients such as sepsis patients (59–61). A recent meta-analysis has reported PAI-1 as a predictor of disease severity in sepsis and overall mortality (62), but the prognostic value of this biomarker in disease progression requires further attention.
A hypofibrinolytic state has been observed in multiple viral infections and is associated with elevated PAI-1 levels (63–67). Most recently, elevated PAI-1 and tPA antigen levels have been described in patients infected with severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) which causes coronavirus-19 disease (COVID-19) (64, 68). However, the net effect of the increased PAI-1 and tPA levels may differ between patients, with variations in this axis being attributed to both a hypo- (64) and hyperfibrinolytic (68) phenotype. In the severe acute respiratory syndrome coronavirus (SARS-CoV) epidemic in 2002 and 2003, the hypofibrinolytic state was attributed to overexpression of PAI-1 which inhibited plasminogen activator activity causing persistence of fibrin deposition (63, 69).
Several proinflammatory cytokines significantly augment PAI-1 synthesis (70–72). Interleukin-6 (IL-6) is an acute-phase inflammatory protein that has been reported to significantly increase PAI-1 and tPA antigen (73). Cytokine release syndrome (CRS) is an acute systemic inflammatory response that can be triggered by various infections and can be observed in sepsis and acute respiratory distress syndrome (ARDS). Endothelial IL-6 trans-signaling promotes IL-6, IL-8, and monocyte chemoattractant protein-1 (MCP-1) and PAI-1 synthesis (55). Inhibition of this trans-signaling circuit by the IL-6R antagonist, tocilizumab, has recently been shown to reduce PAI-1 expression in a small study of COVID-19 patients (55), and is now a recommended treatment for ICU patients after improved outcomes in patients on the Remap-Cap trial (74).
Tumor necrosis factor-alpha (TNF-α), acting via NFκB, is a strong stimulator of PAI-1 expression (56) (Figure 1B). TNF-α is an important regulator of PAI-1 expression in adipose tissue, and neutralizing TNF-α significantly reduces both plasma and adipose tissue levels (75, 76). Several studies support the hypothesis that TNF-α may be responsible for expression of TGF-β (75, 77), another major stimulant of PAI-1 biosynthesis (77–80). Elevated levels of PAI-1 in turn block conversion of latent transforming growth factor beta (TGF-β) contributing to a self-regulation mechanism (80). The affiliation between proinflammatory cytokines and heightened PAI-1 synthesis provides a definitive link between this serpin and the inflammatory response.
Metabolic Syndrome
Metabolic syndrome (MetS) is a multifaceted disorder that encompasses several conditions that considerably elevate the risk of CVD and T2DM. Definitions vary, but in general, diagnosis of MetS requires individuals to meet at least three of the following criteria (81, 82);
Abdominal obesity
Dyslipidemia—elevated triglycerides and apolipoprotein B and low levels of high-density lipoprotein (HDL)
Hypertension
Hyperglycemia
Insulin resistance
Whether the clustering of these conditions elevates an individual's risk over that of a single disorder is a matter of ongoing debate (83, 84). Nonetheless, given the prevalence of MetS worldwide and the fact that this cluster of risk factors predicts CVD in multiple settings, it is clear that we require a stronger understanding of the pathophysiology to develop predictive tools and improve therapeutic options.
Abdominal obesity is an essential criterion in the development of MetS (85, 86). Adipose tissue is an endocrine organ composed of multiple cell types, that secrete adipokines of diverse biological function, such as adiponectin (87–90), leptin (91), and various cytokines, including IL-6 and TNF-α. PAI-1 is a known adipokine (8, 92–94). Adipokine secretion is dependent on the location of the fat store in the body and the composition of cells comprised within the adipose tissue. A correlation between MetS and PAI-1 levels was established in the late 1980s (95). Elevated levels of PAI-1 in individuals with MetS has been demonstrated using criteria defined by both the World Health Organization (WHO) (96) and the National Cholesterol Education Program Expert Panel on Detection, Evaluation and Treatment of High Blood Cholesterol in Adults (NCEP-ATPIII) (97). Elevated levels of PAI-1 in humans predict incidence of MetS in two prospective studies (98, 99). It is well-established that PAI-1 can predict the risk of future CVD (100) and onset of T2DM (101). Together, these data have led to the interpretation that PAI-1 is a true component of MetS (102) and could be an important clinical criterion for development of future CVD (103).
Obesity
Obesity is a global epidemic (104) that is inextricably linked with increased risk of CVD, including arterial, venous, and microvascular thrombosis (105, 106); DVT (107, 108), coronary thrombosis (105, 106), pulmonary embolism (PE) (108, 109) and stroke (110). Obesity-related thrombosis is linked to decreased fibrinolytic activity (111–119) which can be largely attributed to escalating levels of plasma PAI-1 antigen and activity (112, 120). Increased PAI-1 synthesis by adipocytes in response to protractedly elevated levels of TNF-α, insulin, and TGF-β is primarily responsible (112, 114, 120–122) (Figure 1B). Intriguingly, elevated tPA levels have been reported in studies on MetS (96, 97) and obesity (114, 123), which could be the result of impairment of the endothelium; however, the dominant phenotype is hypofibrinolysis.
Plasma PAI-1 significantly correlates with a variety of adiposity measures, including body mass index (BMI), waist-to-hip ratio, total fat mass, and visceral and subcutaneous adipose tissue (124–126). The insulin resistance atherosclerosis study (IRAS) was the first to report that PAI-1 antigen and activity positively correlate with BMI (r = 0.314/0.425, respectively) (127). Adipocytes from obese humans harbor twice as much PAI-1 mRNA resulting in a ≥six-fold increase in secretion of PAI-1 and plasma PAI-1 activity compared with lean individuals (8). Weight loss in obese subjects reduces plasma PAI-1 (122, 125, 128), indicative that circulating levels are directly related to the degree of adipose tissue. In line with this, pharmacological inhibition of plasma PAI-1 in animal models results in weight loss, as well as a reduction in adipose tissue and adipocyte volume (19, 129–131). The number and size of lipid-containing vesicles in adipocytes are also decreased, as well as plasma glucose and triglyceride levels and insulin resistance (129, 132, 133). These data indicate that adipocyte-derived PAI-1 functions in an autocrine role, with one study indicating that PAI-1 inhibition limits differentiation of preadipocytes into mature adipocytes (129, 134).
Interestingly, PAI-1 synthesis is not uniform, with adipocytes from visceral fat depots harboring significantly more PAI-1 mRNA than subcutaneous or femoral fat depots (92, 135). Indeed, visceral fat has been suggested as a determinant of PAI-1 activity in overweight and obese women (136). To fully exploit PAI-1 as a biomarker of MetS, and future CVD, it may be necessary to correlate this serpin with additional factors such as visceral fat levels, rather than more general measurements of BMI.
Type 2 Diabetes
T2DM is intrinsically linked to obesity (92, 137–139), and elevated levels of PAI-1 are strongly correlated with insulin resistance (137, 140), impaired glucose tolerance (137, 140), and T2DM (141, 142). Many studies have reported strong associations between PAI-1 and development of T2DM (143–150). Furthermore, lifestyle and pharmacological interventions to manage diabetes have been shown to decrease circulating plasma PAI-1 levels (151–153). Indeed, PAI-1 activity is significantly reduced upon treatment with the antidiabetic drug, metformin, with a corresponding improvement in glycemic control and reduction in insulin resistance (152).
The IRAS study revealed that PAI-1 was a reliable predictor for developing T2DM, despite adjustments for adiposity, body fat distribution, and insulin sensitivity in patients (p = 0.002) (101). Interestingly, this study also found that PAI-1 activity was increased, but not correlated with insulin concentration, in plasma from non-obese children with T2DM parents (101). Various murine models have determined that PAI-1 plays a pivotal role in development of insulin resistance (132, 133). A mouse model of diet-induced obesity was used to study the relationship between PAI-1, obesity, and insulin in PAI-1 deficient PAI-1−/− and wild type (WT) mice (132). Obesity and insulin resistance that developed in WT mice in response to a high-fat diet was prevented in PAI-1−/− mice (132). Furthermore, PAI-1−/− mice showed increased resting metabolic rates and total energy expenditure, compared with WT. Treatment of WT mice with an angiotensin type I receptor antagonist reduced PAI-1 levels, attenuated diet-induced obesity, hyperglycemia, and hyperinsulinemia (132). Genetically obese (ob/ob) mice deficient in PAI-1 weigh significantly less than those with normal PAI-1 levels; as a result, these mice demonstrate a significant improvement in hyperglycemia and hyperinsulinemia (133). Intraperitoneal glucose administration markedly augments serum insulin levels in WT ob/ob mice; however, the increase in PAI-1−/− mice was dramatically reduced (133). In situ hybridization studies revealed that TNF-α expression was significantly reduced in PAI-1−/− ob/ob mice compared with WT ob/ob mice (133). Together with the well-documented role of TNF-α in stimulating PAI-1 expression (75), this study exposes a complex reciprocal relationship between PAI-1 and TNF-α which merits further study.
Insulin directly stimulates PAI-1 synthesis and secretion from adipocytes (154) (Figure 1), a process which is upregulated in hyperinsulinemia and hyperglycemia (154, 155). Glucose also upregulates PAI-1 expression in vascular smooth muscle cells, endothelial cells, and adipose tissue (156–159). Clinical studies consistently demonstrate a strong correlation between plasma PAI-1 and insulin resistance (154, 160); however, cause or consequence is less clear, that is whether elevated PAI-1 is a result of insulin resistance or if it occurs independently. The role of TNF-α and TGF-β in stimulating PAI-1 expression in adipose tissue (76, 154) suggests that the increase in plasma PAI-1 and insulin resistance may be bi-directional.
There is evidence to support that chronic inflammation and insulin resistance are linked (101, 143). It is hypothesized that this may be due to increased expression of proinflammatory cytokines, namely IL-6 and TNF-α, from adipose tissue (161, 162) which in turn can stimulate acute phase proteins, including PAI-1 (13, 163). Furthermore, several studies (143, 164, 165) have reported that tPA antigen and activity are associated with developing T2DM, and tPA and PAI-1 antigens are strongly correlated in plasma (166). Despite the concordant increase in tPA and PAI-1, a hypofibrinolytic state prevails in T2DM individuals.
Hypertension
There is accumulating evidence implicating PAI-1 in the development of hypertension (167), and plasma PAI-1 is associated with several risk factors for hypertension, including obesity (168, 169), insulin resistance (140, 169), and inflammation (70) as discussed above. Genetic ablation of PAI-1 protects against hypertension and perivascular fibrosis induced by nitric oxide synthase (NOS) inhibition (170, 171). NOS plays a key role in regulating vascular tone and remodeling of the vessel wall (172–174), and inhibition of NOS induces progressive hypertension and vascular fibrosis (175–177). Furthermore, inhibition of PAI-1 with a novel small molecule inhibitor (PAI-039) protects a mouse model against angiotensin II-induced aortic remodeling and cardiac fibrosis (178).
Human studies indicate a direct correlation between plasma PAI-1 and hypertension and its associated conditions (179–185), such as arterial stiffness (186) and atherosclerosis (187). Interestingly, the 4G allele for PAI-1 is associated with increased systolic, diastolic, and mean arterial blood pressure (188), indicative of a direct link between plasma PAI-1 and blood pressure. A study examining two longitudinal cohorts of American Indians revealed that baseline PAI-1 is predictive of hypertension independent of other variables (189). Participants with the highest concentration of PAI-1 (>58 ng/ml) had a 63% increased risk of hypertension compared with those in the lowest group (<33 ng/ml) (189). A similar prospective study, the Framingham Offspring Study, confirmed that a higher concentration of plasma PAI-1 was associated with an increased risk of hypertension (odds ratio = 1.28) (190). Despite plasma PAI-1 correctly predicting the risk of hypertension in human studies, it did not provide a significant advantage over conventional risk factors, such as fasting glucose, alcohol consumption, BMI, cigarette smoking, or C-reactive protein (189). Given the close relationship of PAI-1 with the RAS system and the documented increase in the levels of PAI-1 in hypertension, the mechanisms underpinning this relationship warrant further investigation.
Potential Therapeutic Options and Discussion
The driving force of PAI-1 in thrombosis, inflammation, and metabolic syndrome is evident (Figure 2). In addition, this serpin functions in a variety of pathophysiological processes, outwith the subject matter of this review, including wound healing (191), cardiac fibrosis (192), cancer (193), and senescence (194). These numerous roles underscore the potential of PAI-1 as an attractive therapeutic target; nevertheless, to date, no PAI-1 inhibitors have been approved for clinical use.
Figure 2
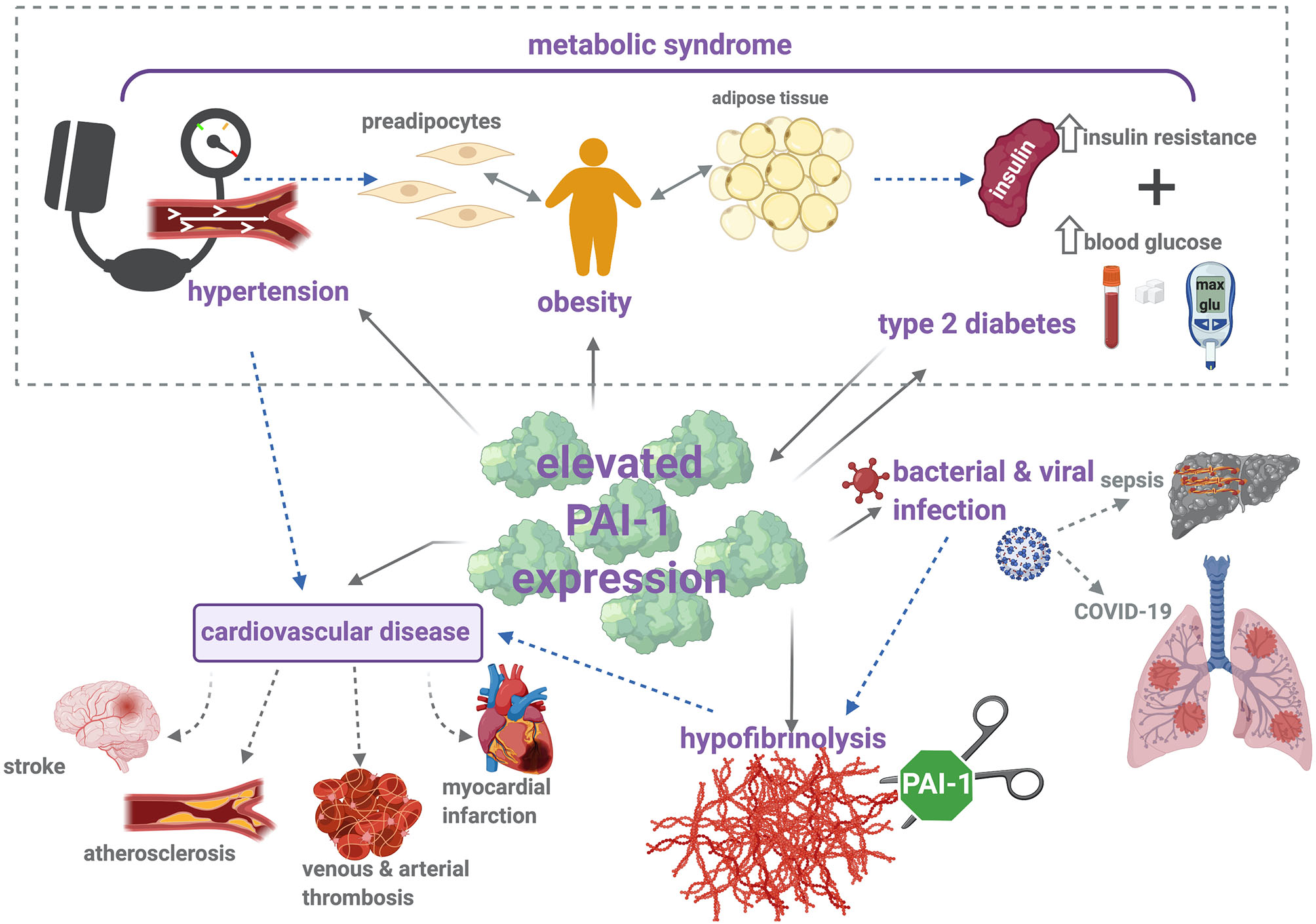
PAI-1 modulates thrombosis and inflammation via multiple pathophysiological mechanisms. Metabolic syndrome is characterized by increased insulin resistance, obesity, and hypertension, which contribute to elevated risk of cardiovascular disease (CVD). Increased levels of PAI-1 antigen and activity are positively associated with hypertension, obesity, type 2 diabetes (T2DM), and CVD. Briefly, elevated levels of PAI-1 antigen and activity occur in obesity, which is a known risk factor for CVD and T2DM. Strong correlations between increased PAI-1 and development of T2DM have been identified, including increased insulin resistance and impaired glucose tolerance. Conversely, insulin and glucose can stimulate PAI-1 secretion from adipose tissue. Elevated levels of PAI-1 attenuate plasmin formation and downregulate fibrin degradation. This hypofibrinolytic state provokes thromboembolic complications, including stroke, atherosclerosis, myocardial infarction, and venous and arterial thrombosis. Hypofibrinolysis and elevated PAI-1 levels have been associated with bacterial and viral infections, including sepsis and COVID-19. Sepsis is characterized by the development of disseminated intravascular coagulation (DIC), a major contributor to resulting organ failure. In COVID-19 patients, disruption between coagulation and fibrinolysis leads to fibrin deposits in the lung parenchyma and thrombosis. Solid arrows represent (patho)physiological processes that arise as a result of increased PAI-1 levels, and dotted arrows illustrate the links between each of these individual pathologies.
Small molecules, peptides, monoclonal antibodies, and antibody fragments have all been used to modulate PAI-1 activity by interfering at different stages of the PAI-1/plasminogen activator interaction [(195–199), reviewed in detail by (200)]. A number of clinically approved drugs indirectly reduce plasma PAI-1; these include insulin sensitizing agents for management of T2DM, such as metformin, and ACE inhibitors (used to treat hypertension) (201). However, these drugs have been studied in experimental models (202–205), and as yet, there is limited information available from human studies.
Drugs targeting PAI-1 in the experimental phase have produced promising results (206–208). A potent neutralizing diabody to PAI-1 and activated thrombin activatable fibrinolysis inhibitor (TAFIa) rapidly enhances clot breakdown (207). Simultaneous inhibition of PAI-1 and TAFIa may improve current thrombolytic therapy; e.g., co-administration with tPA thereby permitting a lower dose and thus enhancing its safety profile (207, 209). Tiplaxtinin, a PAI-1 antagonist, prevents venous thrombosis, angiotensin II-induced atherosclerosis, and obesity in a ferric chloride-induced vascular injury model in rats (206). More recently, a nanobody to PAI-1 has been developed that selectively stabilizes the active form of PAI-1, which may be used as a diagnostic or analytical tool (195, 208).
Other drugs that elicit pharmacological inhibition of PAI-1 have reached phases 1 and 2 clinical trials. A small molecule inhibitor, TM5614, is currently being trialed in a single-center, randomized controlled trial for high-risk patients hospitalized with severe COVID-19 and requiring oxygen (210). Another PAI-1 inhibitor, ACT001, is currently in phase 1 clinical trials for treatment of glioblastoma, the most aggressive primary malignant brain tumor in adults (211, 212).
This review predominantly focuses on the role of PAI-1 in the thrombosis-inflammation axes and associated diseases (Figure 2). It is evident that a proinflammatory state, whether it arises from infection, vascular thrombotic complications, a condition such as MetS, or its associated cluster of diseases spark a dramatic elevation in plasma PAI-1. The underlying motive for this can be attributed to the many cytokines and proteins that can elicit PAI-1 synthesis, thereby inextricably bridging these conditions with this complex serpin. The cellular source of PAI-1 may vary according to the disease processes, e.g., of platelet or endothelial origin during infection or venous thrombosis but of adipose tissue origin in obesity. Many of the conditions described herein predominantly affect the aging population, and it is noted that there is also a clear link between circulating PAI-1 levels and age (213). These observations further confound the relationship between elevated PAI-1 and thromboinflammation, leading to cardiovascular complications. This review serves as an aide-mémoire on the consequences of PAI-1 elevation and highlights the utility of this serpin as a potential therapeutic target in the treatment of various pathological conditions which are associated with a hypofibrinolytic state and development of thromboembolic diseases.
Statements
Author contributions
GM and CW researched the data, wrote, and revised the manuscript. NM conceived the article, wrote the article, and edited the article. All authors contributed to the article and approved the submitted version.
Funding
NM and CW are funded by grants from the British Heart Foundation PG/15/82/31721 and PG/20/17/35050 and Friends of Anchor RS 2019 003 and Aberdeen Development Trust. GM, CW, and NM were also funded by NHS Grampian Endowment Fund COV19-004.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
MottonenJStrandASymerskyJSweetRMDanleyDEGeogheganKFet al. Structural basis of latency in plasminogen activator inhibitor-1. Nature. (1992) 355:270–3. 10.1038/355270a0
2.
LawrenceDAOlsonSTPalaniappanSGinsburgD. Engineering plasminogen activator inhibitor 1 mutants with increased functional stability. Biochemistry. (1994) 33:3643–8. 10.1021/bi00178a022
3.
VaughanDEDeclerckPJVan HoutteEDe MolMCollenD. Studies of recombinant plasminogen activator inhibitor-1 in rabbits. Pharmacokinetics and evidence for reactivation of latent plasminogen activator inhibitor-1 in vivo. Circ Res. (1990) 67:1281–6. 10.1161/01.RES.67.5.1281
4.
LawrenceDAPalaniappanSStefanssonSOlsonSTFrancis-ChmuraAMShoreJDet al. Characterization of the binding of different conformational forms of plasminogen activator inhibitor-1 to vitronectin. Implications for the regulation of pericellular proteolysis. J Biol Chem. (1997) 272:7676–80. 10.1074/jbc.272.12.7676
5.
DeclerckPJDe MolMAlessiMCBaudnerSPaquesEPPreissnerKTet al. Purification and characterization of a plasminogen activator inhibitor 1 binding protein from human plasma. Identification as a multimeric form of S protein (vitronectin). J Biol Chem. (1988) 263:15454–61. 10.1016/S0021-9258(19)37610-0
6.
ZhouAHuntingtonJAPannuNSCarrellRWReadRJ. How vitronectin binds PAI-1 to modulate fibrinolysis and cell migration. Nat Struct Biol. (2003) 10:541–4. 10.1038/nsb943
7.
KohlerHPGrantPJ. Plasminogen-activator inhibitor type 1 and coronary artery disease. N Engl J Med. (2000) 342:1792–801. 10.1056/NEJM200006153422406
8.
ErikssonPReynisdottirSLonnqvistFStemmeVHamstenAArnerP. Adipose tissue secretion of plasminogen activator inhibitor-1 in non-obese and obese individuals. Diabetologia. (1998) 41:65–71. 10.1007/s001250050868
9.
HoekstraTGeleijnseJMSchoutenEGKluftC. Diurnal variation in PAI-1 activity predominantly confined to the 4G-allele of the PAI-1 gene. Thromb Haemost. (2002) 88:794–8. 10.1055/s-0037-1613304
10.
van der BomJGBotsMLHaverkateFKluftCGrobbeeDE. The 4G5G polymorphism in the gene for PAI-1 and the circadian oscillation of plasma PAI-1. Blood. (2003) 101:1841–4. 10.1182/blood-2002-07-2181
11.
KathiresanSGabrielSBYangQLochnerALLarsonMGLevyDet al. Comprehensive survey of common genetic variation at the plasminogen activator inhibitor-1 locus and relations to circulating plasminogen activator inhibitor-1 levels. Circulation. (2005) 112:1728–35. 10.1161/CIRCULATIONAHA.105.547836
12.
AndreottiFDaviesGJHackettDRKhanMIDe BartACAberVRet al. Major circadian fluctuations in fibrinolytic factors and possible relevance to time of onset of myocardial infarction, sudden cardiac death and stroke. Am J Cardiol. (1988) 62:635–7. 10.1016/0002-9149(88)90669-8
13.
FestaAD'AgostinoRJrRichSSJennyNSTracyRPet al. Promoter (4G/5G) plasminogen activator inhibitor-1 genotype and plasminogen activator inhibitor-1 levels in blacks, Hispanics, and non-Hispanic whites: the Insulin Resistance Atherosclerosis Study. Circulation. (2003) 107:2422–7. 10.1161/01.CIR.0000066908.82782.3A
14.
KrishnamurtiCTangDBBarrCFAlvingBM. Plasminogen activator and plasminogen activator inhibitor activities in a reference population. Am J Clin Pathol. (1988) 89:747–52. 10.1093/ajcp/89.6.747
15.
KonkleBASchickPKHeXLiuRJMazurEM. Plasminogen activator inhibitor-1 mRNA is expressed in platelets and megakaryocytes and the megakaryoblastic cell line CHRF-288. Arterioscler Thromb. (1993) 13:669–74. 10.1161/01.ATV.13.5.669
16.
BrogrenHKarlssonLAnderssonMWangLErlingeDJernS. Platelets synthesize large amounts of active plasminogen activator inhibitor 1. Blood. (2004) 104:3943–8. 10.1182/blood-2004-04-1439
17.
van MourikJALawrenceDALoskutoffDJ. Purification of an inhibitor of plasminogen activator (antiactivator) synthesized by endothelial cells. J Biol Chem. (1984) 259:14914–21. 10.1016/S0021-9258(17)42691-3
18.
MorangePEAlessiMCVerdierMCasanovaDMagalonGJuhan-VagueI. PAI-1 produced ex vivo by human adipose tissue is relevant to PAI-1 blood level. Arterioscler Thromb Vasc Biol. (1999) 19:1361–5. 10.1161/01.ATV.19.5.1361
19.
LoskutoffDJSamadF. The adipocyte and hemostatic balance in obesity: studies of PAI-1. Arterioscler Thromb Vasc Biol. (1998) 18:1–6. 10.1161/01.ATV.18.1.1
20.
CrandallDLQuinetEMMorganGABuslerDEMcHendry-RindeBKralJG. Synthesis and secretion of plasminogen activator inhibitor-1 by human preadipocytes. J Clin Endocrinol Metab. (1999) 84:3222–7. 10.1210/jcem.84.9.5987
21.
CwikelBJBarouski-MillerPAColemanPLGelehrterTD. Dexamethasone induction of an inhibitor of plasminogen activator in HTC hepatoma cells. J Biol Chem. (1984) 259:6847–51. 10.1016/S0021-9258(17)39805-8
22.
ChunTYPrattJH. Aldosterone increases plasminogen activator inhibitor-1 synthesis in rat cardiomyocytes. Mol Cell Endocrinol. (2005) 239:55–61. 10.1016/j.mce.2005.03.016
23.
FayWPParkerACCondreyLRShapiroAD. Human plasminogen activator inhibitor-1 (PAI-1) deficiency: characterization of a large kindred with a null mutation in the PAI-1 gene. Blood. (1997) 90:204–8. 10.1182/blood.V90.1.204.204_204_208
24.
AlessiMCJuhan-VagueI. PAI-1 and the metabolic syndrome: links, causes, and consequences. Arterioscler Thromb Vasc Biol. (2006) 26:2200–7. 10.1161/01.ATV.0000242905.41404.68
25.
HamstenAWimanBde FaireUBlombackM. Increased plasma levels of a rapid inhibitor of tissue plasminogen activator in young survivors of myocardial infarction. N Engl J Med. (1985) 313:1557–63. 10.1056/NEJM198512193132501
26.
Juhan-VagueIPykeSDAlessiMCJespersenJHaverkateFThompsonSG. Fibrinolytic factors and the risk of myocardial infarction or sudden death in patients with angina pectoris. ECAT Study Group. European concerted action on thrombosis and disabilities. Circulation. (1996) 94:2057–63. 10.1161/01.CIR.94.9.2057
27.
HamstenAde FaireUWalldiusGDahlenGSzamosiALandouCet al. Plasminogen activator inhibitor in plasma: risk factor for recurrent myocardial infarction. Lancet. (1987) 2:3–9. 10.1016/S0140-6736(87)93050-9
28.
NagaiNSuzukiYVan HoefBLijnenHRCollenD. Effects of plasminogen activator inhibitor-1 on ischemic brain injury in permanent and thrombotic middle cerebral artery occlusion models in mice. J Thromb Haemost. (2005) 3:1379–84. 10.1111/j.1538-7836.2005.01466.x
29.
WimanB. Plasminogen activator inhibitor 1 (PAI-1) in plasma: its role in thrombotic disease. Thromb Haemost. (1995) 74:71–6. 10.1055/s-0038-1642655
30.
YamamotoKSaitoH. A pathological role of increased expression of plasminogen activator inhibitor-1 in human or animal disorders. Int J Hematol. (1998) 68:371–85.
31.
ColletJPMontalescotGVicautEAnkriAWalyloFLestyCet al. Acute release of plasminogen activator inhibitor-1 in ST-segment elevation myocardial infarction predicts mortality. Circulation. (2003) 108:391–4. 10.1161/01.CIR.0000083471.33820.3C
32.
VaughanDELazosSATongK. Angiotensin II regulates the expression of plasminogen activator inhibitor-1 in cultured endothelial cells. A potential link between the renin-angiotensin system and thrombosis. J Clin Invest. (1995) 95:995–1001. 10.1172/JCI117809
33.
BrownNJAgirbasliMAWilliamsGHLitchfieldWRVaughanDE. Effect of activation and inhibition of the renin-angiotensin system on plasma PAI-1. Hypertension. (1998) 32:965–71. 10.1161/01.HYP.32.6.965
34.
SchneidermanJSawdeyMSKeetonMRBordinGMBernsteinEFDilleyRBet al. Increased type 1 plasminogen activator inhibitor gene expression in atherosclerotic human arteries. Proc Natl Acad Sci USA. (1992) 89:6998–7002. 10.1073/pnas.89.15.6998
35.
LupuFBergonzelliGEHeimDACousinEGentonCYBachmannFet al. Localization and production of plasminogen activator inhibitor-1 in human healthy and atherosclerotic arteries. Arterioscler Thromb. (1993) 13:1090–100. 10.1161/01.ATV.13.7.1090
36.
RaghunathPNTomaszewskiJEBradySTCaronRJOkadaSSBarnathanES. Plasminogen activator system in human coronary atherosclerosis. Arterioscler Thromb Vasc Biol. (1995) 15:1432–43. 10.1161/01.ATV.15.9.1432
37.
SobelBEWoodcock-MitchellJSchneiderDJHoltREMarutsukaKGoldH. Increased plasminogen activator inhibitor type 1 in coronary artery atherectomy specimens from type 2 diabetic compared with nondiabetic patients: a potential factor predisposing to thrombosis and its persistence. Circulation. (1998) 97:2213–21. 10.1161/01.CIR.97.22.2213
38.
CarmelietPMoonsLLijnenRJanssensSLupuFCollenDet al. Inhibitory role of plasminogen activator inhibitor-1 in arterial wound healing and neointima formation: a gene targeting and gene transfer study in mice. Circulation. (1997) 96:3180–91. 10.1161/01.CIR.96.9.3180
39.
StefanssonSLawrenceDAArgravesWS. Plasminogen activator inhibitor-1 and vitronectin promote the cellular clearance of thrombin by low density lipoprotein receptor-related proteins 1 and 2. J Biol Chem. (1996) 271:8215–20. 10.1074/jbc.271.14.8215
40.
DrinaneMCShermanJAHallAESimonsMMulligan-KehoeMJ. Plasminogen and plasmin activity in patients with coronary artery disease. J Thromb Haemost. (2006) 4:1288–95. 10.1111/j.1538-7836.2006.01979.x
41.
de Paula SabinoARibeiroDDDominguetiCPDos SantosMSGadelhaTDusseLMet al. Plasminogen activator inhibitor-1 4G/5G promoter polymorphism and PAI-1 plasma levels in young patients with ischemic stroke. Mol Biol Rep. (2011) 38:5355–60. 10.1007/s11033-011-0687-4
42.
LiuYChengJGuoXMoJGaoBZhouHet al. The roles of PAI-1 gene polymorphisms in atherosclerotic diseases: a systematic review and meta-analysis involving 149,908 subjects. Gene. (2018) 673:167–73. 10.1016/j.gene.2018.06.040
43.
EitzmanDTWestrickRJXuZTysonJGinsburgD. Plasminogen activator inhibitor-1 deficiency protects against atherosclerosis progression in the mouse carotid artery. Blood. (2000) 96:4212–5. 10.1182/blood.V96.13.4212.h8004212_4212_4215
44.
SjolandHEitzmanDTGordonDWestrickRNabelEGGinsburgD. Atherosclerosis progression in LDL receptor-deficient and apolipoprotein E-deficient mice is independent of genetic alterations in plasminogen activator inhibitor-1. Arterioscler Thromb Vasc Biol. (2000) 20:846–52. 10.1161/01.ATV.20.3.846
45.
KhoukazHBJiYBraetDJVadaliMAbdelhamidAAEmalCDet al. Drug targeting of plasminogen activator inhibitor-1 inhibits metabolic dysfunction and atherosclerosis in a murine model of metabolic syndrome. Arterioscler Thromb Vasc Biol. (2020) 40:1479–90. 10.1161/ATVBAHA.119.313775
46.
AkhterMSBiswasARanjanRMeenaAYadavBKSharmaAet al. Plasminogen activator inhibitor-1 (PAI-1) gene 4G/5G promoter polymorphism is seen in higher frequency in the Indian patients with deep vein thrombosis. Clin Appl Thromb Hemost. (2010) 16:184–8. 10.1177/1076029609333673
47.
PrabhudesaiAShettySGhoshKKulkarniB. Investigation of Plasminogen Activator Inhibitor-1 (PAI-1) 4G/5G promoter polymorphism in Indian venous thrombosis patients: a case-control study. Eur J Haematol. (2017) 99:249–54. 10.1111/ejh.12912
48.
SartoriMTDanesinCSaggioratoGTormeneDSimioniPSpieziaLet al. The PAI-1 gene 4G/5G polymorphism and deep vein thrombosis in patients with inherited thrombophilia. Clin Appl Thromb Hemost. (2003) 9:299–307. 10.1177/107602960300900405
49.
TangJZhuWMeiXZhangZ. Plasminogen activator inhibitor-1: a risk factor for deep vein thrombosis after total hip arthroplasty. J Orthop Surg Res. (2018) 13:8. 10.1186/s13018-018-0716-2
50.
MeltzerMELismanTde GrootPGMeijersJCle CessieSDoggenCJet al. Venous thrombosis risk associated with plasma hypofibrinolysis is explained by elevated plasma levels of TAFI and PAI-1. Blood. (2010) 116:113–21. 10.1182/blood-2010-02-267740
51.
LimJHWooCHLiJD. Critical role of type 1 plasminogen activator inhibitor (PAI-1) in early host defense against nontypeable Haemophilus influenzae (NTHi) infection. Biochem Biophys Res Commun. (2011) 414:67–72. 10.1016/j.bbrc.2011.09.023
52.
GoolaertsALafargueMSongYMiyazawaBArjomandiMCarlesMet al. PAI-1 is an essential component of the pulmonary host response during Pseudomonas aeruginosa pneumonia in mice. Thorax. (2011) 66:788–96. 10.1136/thx.2010.155788
53.
RoelofsJJTeskeGJBontaPIde VriesCJMeijersJCWeeningJJet al. Plasminogen activator inhibitor-1 regulates neutrophil influx during acute pyelonephritis. Kidney Int. (2009) 75:52–9. 10.1038/ki.2008.454
54.
SemeraroNAmmolloCTSemeraroFColucciM. Sepsis-associated disseminated intravascular coagulation and thromboembolic disease. Mediterr J Hematol Infect Dis. (2010) 2:e2010024. 10.4084/mjhid.2010.024
55.
KangSTanakaTInoueHOnoCHashimotoSKioiYet al. IL-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc Natl Acad Sci USA. (2020) 117:22351–6. 10.1073/pnas.2010229117
56.
SwiatkowskaMSzemrajJCierniewskiCS. Induction of PAI-1 expression by tumor necrosis factor alpha in endothelial cells is mediated by its responsive element located in the 4G/5G site. FEBS J. (2005) 272:5821–31. 10.1111/j.1742-4658.2005.04979.x
57.
ChenCNanBLinPYaoQ. C-reactive protein increases plasminogen activator inhibitor-1 expression in human endothelial cells. Thromb Res. (2008) 122:125–33. 10.1016/j.thromres.2007.09.006
58.
DevarajSXuDYJialalI. C-reactive protein increases plasminogen activator inhibitor-1 expression and activity in human aortic endothelial cells: implications for the metabolic syndrome and atherothrombosis. Circulation. (2003) 107:398–404. 10.1161/01.CIR.0000052617.91920.FD
59.
LoboSMLoboFRBotaDPLopes-FerreiraFSolimanHMMelotCet al. C-reactive protein levels correlate with mortality and organ failure in critically ill patients. Chest. (2003) 123:2043–9. 10.1378/chest.123.6.2043
60.
PovoaPCoelhoLAlmeidaEFernandesAMealhaRMoreiraPet al. C-reactive protein as a marker of infection in critically ill patients. Clin Microbiol Infect. (2005) 11:101–8. 10.1111/j.1469-0691.2004.01044.x
61.
BaidoshviliANijmeijerRLagrandWKHackCENiessenHW. Localisation of C reactive protein in infarcted tissue sites of multiple organs during sepsis. J Clin Pathol. (2002) 55:152–3. 10.1136/jcp.55.2.152
62.
TipoeTLWuWKKChungLGongMDongMLiuTet al. Plasminogen activator inhibitor 1 for predicting sepsis severity and mortality outcomes: a systematic review and meta-analysis. Front Immunol. (2018) 9:1218. 10.3389/fimmu.2018.01218
63.
GralinskiLEBankheadAIIIJengSMenacheryVDProllSBelisleSEet al. Mechanisms of severe acute respiratory syndrome coronavirus-induced acute lung injury. mBio. (2013) 4:e00271–13. 10.1128/mBio.00271-13
64.
NougierCBenoitRSimonMDesmurs-ClavelHMarcotteGArgaudLet al. Hypofibrinolytic state and high thrombin generation may play a major role in SARS-COV2 associated thrombosis. J Thromb Haemost. (2020) 18:2215–9. 10.1111/jth.15016
65.
WoodroffeSBKuanS. Human cytomegalovirus infection induces mRNA expression and secretion of plasminogen inhibitor type-1 in endothelial cells. J Med Virol. (1998) 55:268–71. 10.1002/(SICI)1096-9071(199808)55:4<268::AID-JMV3>3.0.CO;2-Z
66.
KoppelKBrattGSchulmanSBylundHSandstromE. Hypofibrinolytic state in HIV-1-infected patients treated with protease inhibitor-containing highly active antiretroviral therapy. J Acquir Immune Defic Syndr. (2002) 29:441–9. 10.1097/00126334-200204150-00003
67.
SosothikulDSeksarnPPongsewalakSThisyakornULusherJ. Activation of endothelial cells, coagulation and fibrinolysis in children with Dengue virus infection. Thromb Haemost. (2007) 97:627–34. 10.1160/TH06-02-0094
68.
ZuoYWarnockMHarbaughAet al. Plasma tissue plasminogen activator and plasminogen activator inhibitor-1 in hospitalized COVID-19 patients. Sci Rep. (2021) 11:1580. 10.1038/s41598-020-80010-z
69.
BertozziPAstedtBZenziusLLynchKLeMaireFZapolWet al. Depressed bronchoalveolar urokinase activity in patients with adult respiratory distress syndrome. N Engl J Med. (1990) 322:890–7. 10.1056/NEJM199003293221304
70.
KruithofEK. Regulation of plasminogen activator inhibitor type 1 gene expression by inflammatory mediators and statins. Thromb Haemost. (2008) 100:969–75. 10.1160/TH08-04-0269
71.
KruithofEKMestriesJCGasconMPYthierA. The coagulation and fibrinolytic responses of baboons after in vivo thrombin generation–effect of interleukin 6. Thromb Haemost. (1997) 77:905–10. 10.1055/s-0038-1656076
72.
van der PollTLeviMBullerHRvan DeventerSJde BoerJPHackCEet al. Fibrinolytic response to tumor necrosis factor in healthy subjects. J Exp Med. (1991) 174:729–32. 10.1084/jem.174.3.729
73.
MestriesJCKruithofEKGasconMPHerodinFAgayDYthierA. In vivo modulation of coagulation and fibrinolysis by recombinant glycosylated human interleukin-6 in baboons. Eur Cytokine Netw. (1994) 5:275–81. 10.1016/0268-9499(94)90299-2
74.
WiseJ. Covid-19: Critically ill patients treated with arthritis drug tocilizumab show improved outcomes, researchers report. BMJ. (2020) 371:m4530. 10.1136/bmj.m4530
75.
SamadFUysalKTWiesbrockSMPandeyMHotamisligilGSLoskutoffDJ. Tumor necrosis factor alpha is a key component in the obesity-linked elevation of plasminogen activator inhibitor 1. Proc Natl Acad Sci USA. (1999) 96:6902–7. 10.1073/pnas.96.12.6902
76.
SamadFYamamotoKLoskutoffDJ. Distribution and regulation of plasminogen activator inhibitor-1 in murine adipose tissue in vivo. Induction by tumor necrosis factor-alpha and lipopolysaccharide. J Clin Invest. (1996) 97:37–46. 10.1172/JCI118404
77.
BirgelMGottschling-ZellerHRohrigKHaunerH. Role of cytokines in the regulation of plasminogen activator inhibitor-1 expression and secretion in newly differentiated subcutaneous human adipocytes. Arterioscler Thromb Vasc Biol. (2000) 20:1682–7. 10.1161/01.ATV.20.6.1682
78.
Gottschling-ZellerHBirgelMRohrigKHaunerH. Effect of tumor necrosis factor alpha and transforming growth factor beta 1 on plasminogen activator inhibitor-1 secretion from subcutaneous and omental human fat cells in suspension culture. Metabolism. (2000) 49:666–71. 10.1016/S0026-0495(00)80046-3
79.
AlessiMCBastelicaDMorangePBerthetBLeducIVerdierMet al. Plasminogen activator inhibitor 1, transforming growth factor-beta1, and BMI are closely associated in human adipose tissue during morbid obesity. Diabetes. (2000) 49:1374–80. 10.2337/diabetes.49.8.1374
80.
SatoYTsuboiRLyonsRMosesHRifkinDB. Characterization of the activation of latent TGF-beta by co-cultures of endothelial cells and pericytes or smooth muscle cells: a self-regulating system. J Cell Biol. (1990) 111:757–63. 10.1083/jcb.111.2.757
81.
EckelRHAlbertiKGGrundySMZimmetPZ. The metabolic syndrome. Lancet. (2010) 375:181–3. 10.1016/S0140-6736(09)61794-3
82.
SaklayenMG. The global epidemic of the metabolic syndrome. Curr Hypertens Rep. (2018) 20:12. 10.1007/s11906-018-0812-z
83.
SatishMSaxenaSKAgrawalDK. Adipokine dysregulation and insulin resistance with atherosclerotic vascular disease: metabolic syndrome or independent sequelae?J Cardiovasc Transl Res. (2019) 12:415–24. 10.1007/s12265-019-09879-0
84.
KassiEPervanidouPKaltsasGChrousosG. Metabolic syndrome: definitions and controversies. BMC Med. (2011) 9:48. 10.1186/1741-7015-9-48
85.
AlbertiKGZimmetPShawJGroupIDFETFC. The metabolic syndrome–a new worldwide definition. Lancet. (2005) 366:1059–62. 10.1016/S0140-6736(05)67402-8
86.
ZimmetPMaglianoDMatsuzawaYAlbertiGShawJ. The metabolic syndrome: a global public health problem and a new definition. J Atheroscler Thromb. (2005) 12:295–300. 10.5551/jat.12.295
87.
SchererPEWilliamsSFoglianoMBaldiniGLodishHF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem. (1995) 270:26746–9. 10.1074/jbc.270.45.26746
88.
MaedaKOkuboKShimomuraIFunahashiTMatsuzawaYMatsubaraK. cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (AdiPose Most abundant Gene transcript 1). Biochem Biophys Res Commun. (1996) 221:286–9. 10.1006/bbrc.1996.0587
89.
HuELiangPSpiegelmanBM. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem. (1996) 271:10697–703. 10.1074/jbc.271.18.10697
90.
DiezJJIglesiasP. The role of the novel adipocyte-derived hormone adiponectin in human disease. Eur J Endocrinol. (2003) 148:293–300. 10.1530/eje.0.1480293
91.
ZhangYProencaRMaffeiMBaroneMLeopoldLFriedmanJM. Positional cloning of the mouse obese gene and its human homologue. Nature. (1994) 372:425–32. 10.1038/372425a0
92.
AlessiMCPeirettiFMorangePHenryMNalboneGJuhan-VagueI. Production of plasminogen activator inhibitor 1 by human adipose tissue: possible link between visceral fat accumulation and vascular disease. Diabetes. (1997) 46:860–7. 10.2337/diabetes.46.5.860
93.
CigoliniMTonoliMBorgatoLFrigottoLManzatoFZeminianSet al. Expression of plasminogen activator inhibitor-1 in human adipose tissue: a role for TNF-alpha?Atherosclerosis. (1999) 143:81–90. 10.1016/S0021-9150(98)00281-0
94.
KajiH. Adipose tissue-derived plasminogen activator inhibitor-1 function and regulation. Compr Physiol. (2016) 6:1873–96. 10.1002/cphy.c160004
95.
VaguePJuhan-VagueIAillaudMFBadierCViardRAlessiMCet al. Correlation between blood fibrinolytic activity, plasminogen activator inhibitor level, plasma insulin level, and relative body weight in normal and obese subjects. Metabolism. (1986) 35:250–3. 10.1016/0026-0495(86)90209-X
96.
AgewallSBokemarkLWikstrandJLindahlAFagerbergB. Insulin sensitivity and hemostatic factors in clinically healthy 58-year-old men. Thromb Haemost. (2000) 84:571–5. 10.1055/s-0037-1614069
97.
AnandSSYiQGersteinHLonnEJacobsRVuksanVet al. Relationship of metabolic syndrome and fibrinolytic dysfunction to cardiovascular disease. Circulation. (2003) 108:420–5. 10.1161/01.CIR.0000080884.27358.49
98.
AlessiMCNicaudVScroyenILangeCSautNFumeronFet al. Association of vitronectin and plasminogen activator inhibitor-1 levels with the risk of metabolic syndrome and type 2 diabetes mellitus. Results from the D.E.S.I.R. prospective cohort. Thromb Haemost. (2011) 106:416–22. 10.1160/TH11-03-0179
99.
IngelssonEPencinaMJToflerGHBenjaminEJLanierKJJacquesPFet al. Multimarker approach to evaluate the incidence of the metabolic syndrome and longitudinal changes in metabolic risk factors: the Framingham Offspring Study. Circulation. (2007) 116:984–92. 10.1161/CIRCULATIONAHA.107.708537
100.
ThogersenAMJanssonJHBomanKNilssonTKWeinehallLHuhtasaariFet al. High plasminogen activator inhibitor and tissue plasminogen activator levels in plasma precede a first acute myocardial infarction in both men and women: evidence for the fibrinolytic system as an independent primary risk factor. Circulation. (1998) 98:2241–7. 10.1161/01.CIR.98.21.2241
101.
FestaAD'AgostinoRJrTracyRPHaffnerSMInsulin Resistance Atherosclerosis S. Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: the insulin resistance atherosclerosis study. Diabetes. (2002) 51:1131–7. 10.2337/diabetes.51.4.1131
102.
Juhan-VagueIThompsonSGJespersenJ. Involvement of the hemostatic system in the insulin resistance syndrome. A study of 1500 patients with angina pectoris. The ECAT Angina Pectoris Study Group. Arterioscler Thromb. (1993) 13:1865–73. 10.1161/01.ATV.13.12.1865
103.
MertensIVerrijkenAMichielsJJVan der PlankenMRuigeJBVan GaalLF. Among inflammation and coagulation markers, PAI-1 is a true component of the metabolic syndrome. Int J Obes (Lond). (2006) 30:1308–14. 10.1038/sj.ijo.0803189
104.
JaacksLMVandevijvereSPanAMcGowanCJWallaceCImamuraFet al. The obesity transition: stages of the global epidemic. Lancet Diabetes Endocrinol. (2019) 7:231–40. 10.1016/S2213-8587(19)30026-9
105.
WolkRBergerPLennonRJBrilakisESSomersVK. Body mass index: a risk factor for unstable angina and myocardial infarction in patients with angiographically confirmed coronary artery disease. Circulation. (2003) 108:2206–11. 10.1161/01.CIR.0000095270.85646.E8
106.
HubertHBFeinleibMMcNamaraPMCastelliWP. Obesity as an independent risk factor for cardiovascular disease: a 26-year follow-up of participants in the Framingham Heart Study. Circulation. (1983) 67:968–77. 10.1161/01.CIR.67.5.968
107.
YangGDe StaerckeCHooperWC. The effects of obesity on venous thromboembolism: a review. Open J Prev Med. (2012) 2:499–509. 10.4236/ojpm.2012.24069
108.
SteinPDBeemathAOlsonRE. Obesity as a risk factor in venous thromboembolism. Am J Med. (2005) 118:978–80. 10.1016/j.amjmed.2005.03.012
109.
SteinPDMattaFGoldmanJ. Obesity and pulmonary embolism: the mounting evidence of risk and the mortality paradox. Thromb Res. (2011) 128:518–23. 10.1016/j.thromres.2011.10.019
110.
SukSHSaccoRLBoden-AlbalaBCheunJFPittmanJGElkindMSet al. Abdominal obesity and risk of ischemic stroke: the Northern Manhattan Stroke Study. Stroke. (2003) 34:1586–92. 10.1161/01.STR.0000075294.98582.2F
111.
BlokhinIOLentzSR. Mechanisms of thrombosis in obesity. Curr Opin Hematol. (2013) 20:437–44. 10.1097/MOH.0b013e3283634443
112.
LentzSR. Thrombosis in the setting of obesity or inflammatory bowel disease. Hematology Am Soc Hematol Educ Program. (2016) 2016:180–7. 10.1182/asheducation-2016.1.180
113.
Cesarman-MausGHajjarKA. Molecular mechanisms of fibrinolysis. Br J Haematol. (2005) 129:307–21. 10.1111/j.1365-2141.2005.05444.x
114.
Janand-DelenneBChagnaudCRaccahDAlessiMCJuhan-VagueIVagueP. Visceral fat as a main determinant of plasminogen activator inhibitor 1 level in women. Int J Obes Relat Metab Disord. (1998) 22:312–7. 10.1038/sj.ijo.0800585
115.
LandinKStigendalLErikssonEKrotkiewskiMRisbergBTengbornLet al. Abdominal obesity is associated with an impaired fibrinolytic activity and elevated plasminogen activator inhibitor-1. Metabolism. (1990) 39:1044–8. 10.1016/0026-0495(90)90164-8
116.
GiltayEJElbersJMGoorenLJEmeisJJKooistraTAsschemanHet al. Visceral fat accumulation is an important determinant of PAI-1 levels in young, nonobese men and women: modulation by cross-sex hormone administration. Arterioscler Thromb Vasc Biol. (1998) 18:1716–22. 10.1161/01.ATV.18.11.1716
117.
McGillJBSchneiderDJArfkenCLLucoreCLSobelBE. Factors responsible for impaired fibrinolysis in obese subjects and NIDDM patients. Diabetes. (1994) 43:104–9. 10.2337/diab.43.1.104
118.
Potter van LoonBJKluftCRadderJKBlankensteinMAMeindersAE. The cardiovascular risk factor plasminogen activator inhibitor type 1 is related to insulin resistance. Metabolism. (1993) 42:945–9. 10.1016/0026-0495(93)90005-9
119.
Juhan-VagueIVaguePAlessiMCBadierCValadierJAillaudMFet al. Relationships between plasma insulin triglyceride, body mass index, and plasminogen activator inhibitor 1. Diabete Metab. (1987) 13 (3 Pt 2):331–6.
120.
SkurkTHaunerH. Obesity and impaired fibrinolysis: role of adipose production of plasminogen activator inhibitor-1. Int J Obes Relat Metab Disord. (2004) 28:1357–64. 10.1038/sj.ijo.0802778
121.
ShimomuraIFunahashiTTakahashiMMaedaKKotaniKNakamuraTet al. Enhanced expression of PAI-1 in visceral fat: possible contributor to vascular disease in obesity. Nat Med. (1996) 2:800–3. 10.1038/nm0796-800
122.
MavriAAlessiMCBastelicaDGeel-GeorgelinOFinaFSentocnikJTet al. Subcutaneous abdominal, but not femoral fat expression of plasminogen activator inhibitor-1 (PAI-1) is related to plasma PAI-1 levels and insulin resistance and decreases after weight loss. Diabetologia. (2001) 44:2025–31. 10.1007/s001250100007
123.
LadenvallPNilssonSJoodKRosengrenABlomstrandCJernC. Genetic variation at the human tissue-type plasminogen activator (tPA) locus: haplotypes and analysis of association to plasma levels of tPA. Eur J Hum Genet. (2003) 11:603–10. 10.1038/sj.ejhg.5201011
124.
PeverillRETeedeHJMalanEKotsopoulosDSmolichJJMcGrathBP. Relationship of waist and hip circumference with coagulation and fibrinolysis in postmenopausal women. Clin Sci (Lond). (2007) 113:383–91. 10.1042/CS20070084
125.
MavriAStegnarMKrebsMSentocnikJTGeigerMBinderBR. Impact of adipose tissue on plasma plasminogen activator inhibitor-1 in dieting obese women. Arterioscler Thromb Vasc Biol. (1999) 19:1582–7. 10.1161/01.ATV.19.6.1582
126.
SylvanARutegardJNJanungerKGSjolundBNilssonTK. Normal plasminogen activator inhibitor levels at long-term follow-up after jejuno-ileal bypass surgery in morbidly obese individuals. Metabolism. (1992) 41:1370–2. 10.1016/0026-0495(92)90110-V
127.
SkurkTLeeYMNicuta-RolfsTOHaastertBWirthAHaunerH. Effect of the angiotensin II receptor blocker candesartan on fibrinolysis in patients with mild hypertension. Diabetes Obes Metab. (2004) 6:56–62. 10.1111/j.1463-1326.2004.00316.x
128.
EstellesADalmauJFalcoCBerbelOCastelloREspanaFet al. Plasma PAI-1 levels in obese children–effect of weight loss and influence of PAI-1 promoter 4G/5G genotype. Thromb Haemost. (2001) 86:647–52. 10.1055/s-0037-1616100
129.
CrandallDLQuinetEMEl AyachiSHrehaALLeikCESavioDAet al. Modulation of adipose tissue development by pharmacological inhibition of PAI-1. Arterioscler Thromb Vasc Biol. (2006) 26:2209–15. 10.1161/01.ATV.0000235605.51400.9d
130.
SawdeyMSLoskutoffDJ. Regulation of murine type 1 plasminogen activator inhibitor gene expression in vivo. Tissue specificity and induction by lipopolysaccharide, tumor necrosis factor-alpha, and transforming growth factor-beta. J Clin Invest. (1991) 88:1346–53. 10.1172/JCI115440
131.
WangLChenLLiuZLiuYLuoMChenNet al. PAI-1 exacerbates white adipose tissue dysfunction and metabolic dysregulation in high fat diet-induced obesity. Front Pharmacol. (2018) 9:1087. 10.3389/fphar.2018.01087
132.
MaLJMaoSLTaylorKLKanjanabuchTGuanYZhangYet al. Prevention of obesity and insulin resistance in mice lacking plasminogen activator inhibitor 1. Diabetes. (2004) 53:336–46. 10.2337/diabetes.53.2.336
133.
SchaferKFujisawaKKonstantinidesSLoskutoffDJ. Disruption of the plasminogen activator inhibitor 1 gene reduces the adiposity and improves the metabolic profile of genetically obese and diabetic ob/ob mice. FASEB J. (2001) 15:1840–2. 10.1096/fj.00-0750fje
134.
CorreiaMLHaynesWG. A role for plasminogen activator inhibitor-1 in obesity: from pie to PAI?Arterioscler Thromb Vasc Biol. (2006) 26:2183–5. 10.1161/01.ATV.0000244018.24120.70
135.
HalleuxCMDeclerckPJTranSLDetryRBrichardSM. Hormonal control of plasminogen activator inhibitor-1 gene expression and production in human adipose tissue: stimulation by glucocorticoids and inhibition by catecholamines. J Clin Endocrinol Metab. (1999) 84:4097–105. 10.1210/jcem.84.11.6127
136.
MertensIVan der PlankenMCorthoutsBWautersMPeifferFDe LeeuwIet al. Visceral fat is a determinant of PAI-1 activity in diabetic and non-diabetic overweight and obese women. Horm Metab Res. (2001) 33:602–7. 10.1055/s-2001-17907
137.
FestaAD'AgostinoRJrMykkanenLTracyRPZaccaroDJet al. Relative contribution of insulin and its precursors to fibrinogen and PAI-1 in a large population with different states of glucose tolerance. The Insulin Resistance Atherosclerosis Study (IRAS). Arterioscler Thromb Vasc Biol. (1999) 19:562–8. 10.1161/01.ATV.19.3.562
138.
ErshowAG. Environmental influences on development of type 2 diabetes and obesity: challenges in personalizing prevention and management. J Diabetes Sci Technol. (2009) 3:727–34. 10.1177/193229680900300418
139.
Al-GoblanASAl-AlfiMAKhanMZ. Mechanism linking diabetes mellitus and obesity. Diabetes Metab Syndr Obes. (2014) 7:587–91. 10.2147/DMSO.S67400
140.
MeigsJBMittlemanMANathanDMToflerGHSingerDEMurphy-SheehyPMet al. Hyperinsulinemia, hyperglycemia, and impaired hemostasis: the Framingham Offspring Study. JAMA. (2000) 283:221–8. 10.1001/jama.283.2.221
141.
Al-HamodiZIsmailISSaif-AliRAhmedKAMuniandyS. Association of plasminogen activator inhibitor-1 and tissue plasminogen activator with type 2 diabetes and metabolic syndrome in Malaysian subjects. Cardiovasc Diabetol. (2011) 10:23. 10.1186/1475-2840-10-23
142.
NagiDKMohamed AliVJainSKWaljiSYudkinJS. Plasminogen activator inhibitor (PAI-1) activity is elevated in Asian and Caucasian subjects with non-insulin-dependent (type 2) diabetes but not in those with impaired glucose tolerance (IGT) or non-diabetic Asians. Diabet Med. (1996) 13:59–64. 10.1002/(SICI)1096-9136(199601)13:1<59::AID-DIA2>3.0.CO;2-Z
143.
EliassonMCJanssonJHLindahlBStegmayrB. High levels of tissue plasminogen activator (tPA) antigen precede the development of type 2 diabetes in a longitudinal population study. The Northern Sweden MONICA study. Cardiovasc Diabetol. (2003) 2:19. 10.1186/1475-2840-2-19
144.
KanayaAMHarrisTGoodpasterBHTylavskyFCummingsSRHealthA. Adipocytokines attenuate the association between visceral adiposity and diabetes in older adults. Diabetes Care. (2004) 27:1375–80. 10.2337/diacare.27.6.1375
145.
KanayaAMWassel FyrCVittinghoffEHarrisTBParkSWGoodpasterBHet al. Adipocytokines and incident diabetes mellitus in older adults: the independent effect of plasminogen activator inhibitor 1. Arch Intern Med. (2006) 166:350–6. 10.1001/archinte.166.3.350
146.
SoaresALRosarioPWBorgesMASousaMOFernandesAPCarvalhoM. PAI-1 and D-dimer in type 2 diabetic women with asymptomatic macrovascular disease assessed by carotid Doppler. Clin Appl Thromb Hemost. (2010) 16:204–8. 10.1177/1076029609334626
147.
VerkleijCJBruijnREMeestersEWGerdesVEMeijersJCMarxPF. The hemostatic system in patients with type 2 diabetes with and without cardiovascular disease. Clin Appl Thromb Hemost. (2011) 17:E57–63. 10.1177/1076029610384112
148.
KitagawaNYanoYGabazzaECBrunoNEArakiRMatsumotoKet al. Different metabolic correlations of thrombin-activatable fibrinolysis inhibitor and plasminogen activator inhibitor-1 in non-obese type 2 diabetic patients. Diabetes Res Clin Pract. (2006) 73:150–7. 10.1016/j.diabres.2005.12.008
149.
HughesKChooMKuperanPOngCNAwTC. Cardiovascular risk factors in non-insulin-dependent diabetics compared to non-diabetic controls: a population-based survey among Asians in Singapore. Atherosclerosis. (1998) 136:25–31. 10.1016/S0021-9150(97)00180-9
150.
DeclerckPJAlessiMCVerstrekenMKruithofEKJuhan-VagueICollenD. Measurement of plasminogen activator inhibitor 1 in biologic fluids with a murine monoclonal antibody-based enzyme-linked immunosorbent assay. Blood. (1988) 71:220–5. 10.1182/blood.V71.1.220.220
151.
LeeNHuiDWuAChanPCameronPJoyntGMet al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N Engl J Med. (2003) 348:1986–94. 10.1056/NEJMoa030685
152.
NagiDKYudkinJS. Effects of metformin on insulin resistance, risk factors for cardiovascular disease, and plasminogen activator inhibitor in NIDDM subjects. A study of two ethnic groups. Diabetes Care. (1993) 16:621–9. 10.2337/diacare.16.4.621
153.
FonsecaVAReynoldsTHemphillDRandolphCWallJValiquetTRet al. Effect of troglitazone on fibrinolysis and activated coagulation in patients with non-insulin-dependent diabetes mellitus. J Diabetes Complications. (1998) 12:181–6. 10.1016/S1056-8727(97)00109-8
154.
SamadFPandeyMBellPALoskutoffDJ. Insulin continues to induce plasminogen activator inhibitor 1 gene expression in insulin-resistant mice and adipocytes. Mol Med. (2000) 6:680–92. 10.1007/BF03402048
155.
BastardJPPieroniL. Plasma plasminogen activator inhibitor 1, insulin resistance and android obesity. Biomed Pharmacother. (1999) 53:455–61. 10.1016/S0753-3322(00)88103-2
156.
PandolfiAIacovielloLCapaniFVitacolonnaEDonatiMBConsoliA. Glucose and insulin independently reduce the fibrinolytic potential of human vascular smooth muscle cells in culture. Diabetologia. (1996) 39:1425–31. 10.1007/s001250050594
157.
SuzukiMAkimotoKHattoriY. Glucose upregulates plasminogen activator inhibitor-1 gene expression in vascular smooth muscle cells. Life Sci. (2002) 72:59–66. 10.1016/S0024-3205(02)02182-3
158.
GabrielyIYangXMCasesJAMaXHRossettiLBarzilaiN. Hyperglycemia induces PAI-1 gene expression in adipose tissue by activation of the hexosamine biosynthetic pathway. Atherosclerosis. (2002) 160:115–22. 10.1016/S0021-9150(01)00574-3
159.
MaielloMBoeriDPodestaFCaglieroEVichiMOdettiPet al. Increased expression of tissue plasminogen activator and its inhibitor and reduced fibrinolytic potential of human endothelial cells cultured in elevated glucose. Diabetes. (1992) 41:1009–15. 10.2337/diabetes.41.8.1009
160.
Juhan-VagueIAlessiMC. PAI-1, obesity, insulin resistance and risk of cardiovascular events. Thromb Haemost. (1997) 78:656–60. 10.1055/s-0038-1657607
161.
RajiMAAl SnihSRayLAPatelKVMarkidesKS. Cognitive status and incident disability in older Mexican Americans: findings from the Hispanic established population for the epidemiological study of the elderly. Ethn Dis. (2004) 14:26–31.
162.
LutseyPLCushmanMSteffenLMGreenDBarrRGHerringtonDet al. Plasma hemostatic factors and endothelial markers in four racial/ethnic groups: the MESA study. J Thromb Haemost. (2006) 4:2629–35. 10.1111/j.1538-7836.2006.02237.x
163.
OldroydJBanerjeeMHealdACruickshankK. Diabetes and ethnic minorities. Postgrad Med J. (2005) 81:486–90. 10.1136/pgmj.2004.029124
164.
Hernestal-BomanJNorbergMJanssonJHEliassonMErikssonJWLindahlBet al. Signs of dysregulated fibrinolysis precede the development of type 2 diabetes mellitus in a population-based study. Cardiovasc Diabetol. (2012) 11:152. 10.1186/1475-2840-11-152
165.
WannametheeSGSattarNRumleyAWhincupPHLennonLLoweGD. Tissue plasminogen activator, von Willebrand factor, and risk of type 2 diabetes in older men. Diabetes Care. (2008) 31:995–1000. 10.2337/dc07-1569
166.
LoweGDRumleyAWhincupPHDaneshJ. Hemostatic and rheological variables and risk of cardiovascular disease. Semin Vasc Med. (2002) 2:429–39. 10.1055/s-2002-36771
167.
ManciaGFagardRNarkiewiczKRedonJZanchettiABohmMet al. ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J. (2013) 34:2159–219. 10.1093/eurheartj/eht151
168.
EksteenPPietersMde LangeZKrugerHS. The association of clot lysis time with total obesity is partly independent from the association of PAI-1 with central obesity in African adults. Thromb Res. (2015) 136:415–21. 10.1016/j.thromres.2015.05.033
169.
LalicKJoticARajkovicNSinghSStosicLPopovicLet al. Altered daytime fluctuation pattern of plasminogen activator inhibitor 1 in type 2 diabetes patients with coronary artery disease: a strong association with persistently elevated plasma insulin, increased insulin resistance, and abdominal obesity. Int J Endocrinol. (2015) 2015:390185. 10.1155/2015/390185
170.
KaikitaKFogoABMaLSchoenhardJABrownNJVaughanDE. Plasminogen activator inhibitor-1 deficiency prevents hypertension and vascular fibrosis in response to long-term nitric oxide synthase inhibition. Circulation. (2001) 104:839–44. 10.1161/hc3301.092803
171.
KaikitaKSchoenhardJAPainterCARipleyRTBrownNJFogoABet al. Potential roles of plasminogen activator system in coronary vascular remodeling induced by long-term nitric oxide synthase inhibition. J Mol Cell Cardiol. (2002) 34:617–27. 10.1006/jmcc.2002.2001
172.
MoncadaSHiggsA. The L-arginine-nitric oxide pathway. N Engl J Med. (1993) 329:2002–12. 10.1056/NEJM199312303292706
173.
PalmerRMAshtonDSMoncadaS. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature. (1988) 333:664–6. 10.1038/333664a0
174.
PalmerRMFerrigeAGMoncadaS. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. (1987) 327:524–6. 10.1038/327524a0
175.
BaylisCMitrukaBDengA. Chronic blockade of nitric oxide synthesis in the rat produces systemic hypertension and glomerular damage. J Clin Invest. (1992) 90:278–81. 10.1172/JCI115849
176.
ZatzRBaylisC. Chronic nitric oxide inhibition model six years on. Hypertension. (1998) 32:958–64. 10.1161/01.HYP.32.6.958
177.
RibeiroMOAntunesEde NucciGLovisoloSMZatzR. Chronic inhibition of nitric oxide synthesis. A new model of arterial hypertension. Hypertension. (1992) 20:298–303. 10.1161/01.HYP.20.3.298
178.
WeisbergADAlbornozFGriffinJPCrandallDLElokdahHFogoABet al. Pharmacological inhibition and genetic deficiency of plasminogen activator inhibitor-1 attenuates angiotensin II/salt-induced aortic remodeling. Arterioscler Thromb Vasc Biol. (2005) 25:365–71. 10.1161/01.ATV.0000152356.85791.52
179.
GavriilakiEGkaliagkousiENikolaidouBTriantafyllouGChatzopoulouFDoumaS. Increased thrombotic and impaired fibrinolytic response to acute exercise in patients with essential hypertension: the effect of treatment with an angiotensin II receptor blocker. J Hum Hypertens. (2014) 28:606–9. 10.1038/jhh.2014.18
180.
PoliKAToflerGHLarsonMGEvansJCSutherlandPALipinskaIet al. Association of blood pressure with fibrinolytic potential in the Framingham offspring population. Circulation. (2000) 101:264–9. 10.1161/01.CIR.101.3.264
181.
EliassonMJanssonJHNilssonPAsplundK. Increased levels of tissue plasminogen activator antigen in essential hypertension. A population-based study in Sweden. J Hypertens. (1997) 15:349–56. 10.1097/00004872-199715040-00005
182.
MakrisTStavroulakisGPapadopoulosDPaizisIKrespiPTsoukalaCet al. White coat hypertension and haemostatic/fibrinolytic balance disorders. Eur Cytokine Netw. (2006) 17:137–41.
183.
CobanEOzdoganM. The plasma levels of plasminogen activator inhibitor-1 in subjects with white coat hypertension. Int J Clin Pract. (2004) 58:541–4. 10.1111/j.1368-5031.2004.00119.x
184.
TiryakiOBuyukhatipogluHUsalanC. Plasma plasminogen activator inhibitor 1 (PAI-1) and P-selectin levels in urgent hypertension: effect of single dose captopril and nifedipine on fibrinolytic activity. Clin Exp Hypertens. (2010) 32:347–51. 10.3109/10641961003628478
185.
Armas-HernandezMJHernandez-HernandezRArmas-PadillaMCSosa-CanacheBCammarataRPachecoBet al. Fibrinolytic system in normotensive subjects and hypertensive patients. Am J Ther. (2007) 14:177–82. 10.1097/01.pap.0000249923.06373.95
186.
LiebWLarsonMGBenjaminEJYinXToflerGHSelhubJet al. Multimarker approach to evaluate correlates of vascular stiffness: the Framingham Heart Study. Circulation. (2009) 119:37–43. 10.1161/CIRCULATIONAHA.108.816108
187.
ZahranMNasrFMMetwalyAAEl-SheikhNKhalilNSHarbaT. The role of hemostatic factors in atherosclerosis in patients with chronic renal disease. Electron Physician. (2015) 7:1270–6. 10.14661/1270
188.
BjorckHMErikssonPAlehagenUDe BassoRLjungbergLUPerssonKet al. Gender-specific association of the plasminogen activator inhibitor-1 4G/5G polymorphism with central arterial blood pressure. Am J Hypertens. (2011) 24:802–8. 10.1038/ajh.2011.63
189.
PengHYehFde SimoneGBestLGLeeETHowardBVet al. Relationship between plasma plasminogen activator inhibitor-1 and hypertension in American Indians: findings from the Strong Heart Study. J Hypertens. (2017) 35:1787–93. 10.1097/HJH.0000000000001375
190.
ToflerGHMassaroJO'DonnellCJWilsonPWFVasanRSSutherlandPAet al. Plasminogen activator inhibitor and the risk of cardiovascular disease: The Framingham Heart Study. Thromb Res. (2016) 140:30–5. 10.1016/j.thromres.2016.02.002
191.
SimoneTMLongmateWMLawBKHigginsPJ. Targeted inhibition of PAI-1 activity impairs epithelial migration and wound closure following cutaneous injury. Adv Wound Care (New Rochelle). (2015) 4:321–8. 10.1089/wound.2014.0611
192.
FlevarisPVaughanD. The role of plasminogen activator inhibitor type-1 in fibrosis. Semin Thromb Hemost. (2017) 43:169–77. 10.1055/s-0036-1586228
193.
PlacencioVRDeClerckYA. Plasminogen activator inhibitor-1 in cancer: rationale and insight for future therapeutic testing. Cancer Res. (2015) 75:2969–74. 10.1158/0008-5472.CAN-15-0876
194.
VaughanDRaiRKhanSSErenMG. Plasminogen activator inhibitor-1 is a marker and a mediator of senescence. Arterioscler Thromb Vasc Biol. (2017) 37:1446–52. 10.1161/ATVBAHA.117.309451
195.
SillenMWeeksSDZhouXKomissarovAAFlorovaGIdellSet al. Molecular mechanism of two nanobodies that inhibit PAI-1 activity reveals a modulation at distinct stages of the PAI-1/plasminogen activator interaction. J Thromb Haemost. (2020) 18:681–92. 10.1111/jth.14716
196.
VousdenKALundqvistTPopovicBNaimanBCarruthersAMNewtonPet al. Discovery and characterisation of an antibody that selectively modulates the inhibitory activity of plasminogen activator inhibitor-1. Sci Rep. (2019) 9:1605. 10.2210/pdb6i8s/pdb
197.
BijnensAPGilsAStassenJMKomissarovAAKnockaertIBrouwersEet al. The distal hinge of the reactive site loop and its proximity: a target to modulate plasminogen activator inhibitor-1 activity. J Biol Chem. (2001) 276:44912–8. 10.1074/jbc.M103077200
198.
KomissarovAADeclerckPJShoreJD. Mechanisms of conversion of plasminogen activator inhibitor 1 from a suicide inhibitor to a substrate by monoclonal antibodies. J Biol Chem. (2002) 277:43858–65. 10.1074/jbc.M204110200
199.
VerhammeIKvassmanJODayDDebrockSVleugelsNDeclerckPJet al. Accelerated conversion of human plasminogen activator inhibitor-1 to its latent form by antibody binding. J Biol Chem. (1999) 274:17511–7. 10.1074/jbc.274.25.17511
200.
SillenMDeclerckPJ. Targeting PAI-1 in cardiovascular disease: structural insights into pai-1 functionality and inhibition. Front Cardiovasc Med. (2020) 7:622473. 10.3389/fcvm.2020.622473
201.
VaughanDE. PAI-1 antagonists: the promise and the peril. Trans Am Clin Climatol Assoc. (2011) 122:312–25.
202.
HadiganCMeigsJBRabeJD'AgostinoRBWilsonPWLipinskaIet al. Increased PAI-1 and tPA antigen levels are reduced with metformin therapy in HIV-infected patients with fat redistribution and insulin resistance. J Clin Endocrinol Metab. (2001) 86:939–43. 10.1210/jcem.86.2.7410
203.
ErsoyCKiyiciSBudakFOralBGucluMDuranCet al. The effect of metformin treatment on VEGF and PAI-1 levels in obese type 2 diabetic patients. Diabetes Res Clin Pract. (2008) 81:56–60. 10.1016/j.diabres.2008.02.006
204.
PretoriusMMurpheyLJMcFarlaneJAVaughanDEBrownNJ. Angiotensin-converting enzyme inhibition alters the fibrinolytic response to cardiopulmonary bypass. Circulation. (2003) 108:3079–83. 10.1161/01.CIR.0000105765.54573.60
205.
FogariRZoppiAMugelliniAMaffioliPLazzariPDerosaG. Role of angiotensin II in plasma PAI-1 changes induced by imidapril or candesartan in hypertensive patients with metabolic syndrome. Hypertens Res. (2011) 34:1321–6. 10.1038/hr.2011.137
206.
ElokdahHAbou-GharbiaMHennanJKMcFarlaneGMugfordCPKrishnamurthyGet al. Tiplaxtinin, a novel, orally efficacious inhibitor of plasminogen activator inhibitor-1: design, synthesis, and preclinical characterization. J Med Chem. (2004) 47:3491–4. 10.1021/jm049766q
207.
DevelterJBoothNADeclerckPJGilsA. Bispecific targeting of thrombin activatable fibrinolysis inhibitor and plasminogen activator inhibitor-1 by a heterodimer diabody. J Thromb Haemost. (2008) 6:1884–91. 10.1111/j.1538-7836.2008.03137.x
208.
SillenMWeeksSDStrelkovSVDeclerckPJ. Structural insights into the mechanism of a nanobody that stabilizes PAI-1 and modulates its activity. Int J Mol Sci. (2020) 21:5859. 10.3390/ijms21165859
209.
WyseureTRubioMDenormeFMartinez de LizarrondoSPeetersMGilsAet al. Innovative thrombolytic strategy using a heterodimer diabody against TAFI and PAI-1 in mouse models of thrombosis and stroke. Blood. (2015) 125:1325–32. 10.1182/blood-2014-07-588319
210.
Study To antagOnize Plasminogen Activator Inhibitor-1 in Severe COVID-19: Trial identifier NCT04634799. Available online at: https://clinicaltrials.gov/ct2/show/NCT04634799?term=PAI-1+inhibitor&draw=2&rank=1
211.
ThakkarJPDolecekTAHorbinskiCOstromQTLightnerDDBarnholtz-SloanJSet al. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol Biomarkers Prev. (2014) 23:1985–96. 10.1158/1055-9965.EPI-14-0275
212.
XiXLiuNWangQChuYYinZDingYet al. ACT001, a novel PAI-1 inhibitor, exerts synergistic effects in combination with cisplatin by inhibiting PI3K/AKT pathway in glioma. Cell Death Dis. (2019) 10:757. 10.1038/s41419-019-1986-2
213.
YamamotoKTakeshitaKKojimaTTakamatsuJSaitoH. Aging and plasminogen activator inhibitor-1 (PAI-1) regulation: implication in the pathogenesis of thrombotic disorders in the elderly. Cardiovasc Res. (2005) 66:276–85. 10.1016/j.cardiores.2004.11.013
Summary
Keywords
PAI-1, fibrinolysis, metabolic syndrome, obese, diabetes, thrombosis, inflammation
Citation
Morrow GB, Whyte CS and Mutch NJ (2021) A Serpin With a Finger in Many PAIs: PAI-1's Central Function in Thromboinflammation and Cardiovascular Disease. Front. Cardiovasc. Med. 8:653655. doi: 10.3389/fcvm.2021.653655
Received
14 January 2021
Accepted
23 February 2021
Published
16 April 2021
Volume
8 - 2021
Edited by
Marie-Christine Bouton, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Reviewed by
Margarethe Geiger, Medical University of Vienna, Austria; Benoît Ho-Tin-Noé, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Updates
Copyright
© 2021 Morrow, Whyte and Mutch.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola J. Mutch n.j.mutch@abdn.ac.uk
This article was submitted to Atherosclerosis and Vascular Medicine, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.