Abstract
Obesity has a strong impact on the pathogenesis of cardiovascular disease, which raises enthusiasm to understand how excess adiposity causes vascular injury. Adipose tissue is an essential regulator of cardiovascular system through its endocrine and paracrine bioactive products. Obesity induces endothelial dysfunction, which often precedes and leads to the development of cardiovascular diseases. Connecting adipose tissue-endothelial cell interplay to endothelial dysfunction may help us to better understand obesity-induced cardiovascular disease. This Mini Review discussed (1) the general interactions and obesity-induced endothelial dysfunction, (2) potential targets, and (3) the outstanding questions for future research.
Introduction
The increased incidence of obesity contributes to the prevalence of various metabolic diseases. About 1.9 billion people are predicted to be obese or overweight, worldwide (1). Obesity is an established risk factor for cardiovascular disease (CVD) (2); obese individuals are predisposed to a range of cardiometabolic abnormalities (3). Thus, great attention has been drawn to the topic of how excess adiposity leads to vascular dysfunction. Mechanistically, adipose tissue (AT) affects the cardiovascular system through the secretion of bioactive products (e.g., adipocytokines and microvesicles), inorganic molecules, and reactive oxygen species (ROS). AT becomes dysfunctional in obesity and generates a pro-inflammatory, hyperlipidemic, and insulin-resistant environment, which ultimately leads to the development of metabolic complications (e.g., diabetes) and cardiovascular complications (e.g., atherosclerosis) (4). Arteries residing in visceral adipose display impaired vascular responses to endothelium-dependent agonists (e.g., intraluminal flow), characteristic of endothelial dysfunction (ED), which occurs long before CVD has developed. Recognizing the roles of AT in obesity-induced ED/CVD, this mini review will discuss the AT-EC interactions with their potential as therapeutic targets of obesity-induced ED and the remaining research questions that merit further investigation.
The Role of AT in Obesity
AT is the body's largest endocrine organ and secretes hormones, cytokines, and proteins in endocrine and/or paracrine manners that affect cell and tissue function throughout the body (5). AT is also essential in maintaining lipid and glucose homeostasis, which becomes dysfunctional in obesity and excessive deposition of fat occurs. Enlarged adipocytes in obese individuals promoted macrophage-mediated inflammation and adipokine-induced insulin resistance (6). The pathological function of AT is determined by their cellular composition, secretome (secretion profiles), and location in the human body (7). Further, low storage and removal of adipose triglycerides promote dyslipidemia, while high storage and low removal prompt obesity (8). In addition, detection of brown and/or beige AT in adult humans (9, 10) and the realization of adipocyte heterogeneity and plasticity of white AT spurred great interest in targeting AT for possible therapeutic advantages (11).
Obesity-Induced Endothelial Dysfunction (ED)
Impairment of flow-induced vasodilation, arterial dilation prompted by blood flow, is one hallmark of obesity-induced ED. It is demonstrated by the impairment of endothelial nitric oxide synthase (eNOS) and the loss of nitric oxide (NO), a major vasodilator and anti-inflammatory agent (12). Obesity also promotes damage of the endothelial glycocalyx, which responds to mechanical force from blood vessels and regulates NO production. Flow-mediated vasodilation in mouse and human mesenteric arteries was hindered by loss of the endothelial flow-sensitivity of Kir (inwardly rectifying K+) channel due to obesity-induced glycocalyx thinning (13). Defective physiological properties of endothelial cells (EC) will switch the vascular endothelium to a pro-inflammatory, prothrombotic and proatherogenic phenotype, leading to leukocyte adhesion, activation of platelets, and pro-oxidation of mitogens, along with impaired endothelial NO production, decreased synthesis of endothelium-derived hyperpolarizing factors (EDHF), and increased vasoconstriction factors, such as angiotensin II (Ang II) and prostaglandin (PGH2) (14). Through activation of adhesion molecules, leukocyte proliferation, and transmigration, ED reportedly launches CVD progression in obesity (15). Moreover, secretion of angiotensinogen of the renin-angiotensin system (RAS) by dysfunctional adipocytes leads to its overexpression in RAS, enhancing ROS production and increasing the atherogenic and thromboembolic potentials of EC (16). The risks associated with cardiovascular complications can be mitigated through inhibition of inflammatory mechanisms and controlling obesity (16).
Modes of AT-EC Interaction in Obesity
Human AT can be broadly divided into subcutaneous AT (SAT) and visceral AT (VAT). VAT in the heart can be classified as epicardial AT (EAT) and pericardial AT (PAT). Among all AT depots, the perivascular AT (PVAT) is recognized as a vital regulator of vascular biology because of its anatomical proximity to the vessels (17). These ATs regulate cardiovascular system through the secretion of bioactive products, such as adipokines, microvesicles, and gaseous messengers. The secretome is under tight control by homeostatic mechanisms, which can become dysregulated in obesity. There are two modes of AT-EC interaction: the endocrine mode, which is an indirect crosstalk through the circulation, and the paracrine mode, which is a direct interplay. Obesity-initiated systemic or local inflammation and insulin resistance shift the AT secretome from an anti-inflammatory and anti-atherogenic state toward a pro-inflammatory and pro-atherogenic state.
Endocrine Mode
Associated with connective tissue and blood vessel proliferation, inflammation has been regarded as the first stage of vascular dysfunction. AT-derived tumor necrotic factor-α (TNF-α) is one product of inflammation (18). In obesity, microvasculature from VAT is an important source of low-grade inflammation and oxidative stress. Both contribute to vascular changes and favor increased atherosclerosis under clinical conditions. Mechanistically, excessive macronutrients accumulated on the AT promote the secretion and release of inflammatory mediators, including interleukin-6 (IL-6), interleukin-1β (IL-1β), TNF-α, leptin, and stimulation of monocyte chemoattractant protein-1 (MCP-1), which subsequently produce less adiponectin, thereby initiating a proinflammatory state (16) and driving vascular destabilization and leakage (19).
Among AT-secreted factors, MicroRNAs (miRNAs) are short, single-stranded, non-coding RNA molecules that play important roles in a variety of cellular processes, such as differentiation, proliferation, apoptosis, and stress response; their alteration contributes to the development of many pathologies, including obesity (20). Specifically, AT-derived miRNA (21) mediates obesity-induced ED by affecting gene expression of eNOS, SIRT1, cellular producers of ROS, autophagy machinery, and ER stress (22).
Paracrine Mode
PVAT and EAT elicit direct impacts on the adjacent vascular wall or myocardium, respectively, through the paracrine release of bioactive mediators. These mediators travel to neighboring vessels, thereby regulating the biology of entire vascular beds in a “vasocrine” manner (23). In obesity, PVAT-secreted high-concentrations of adipokines (e.g., TNF-α and IL-6) access the vascular lumen and suppress the PI3-K pathway of insulin signaling, which unlocks the vasoconstrictor effects of endothelin 1, leading to a reduction in insulin-mediated muscle nutritive blood flow, contributing to insulin resistance (23). PVAT also releases miRNA, including miR-221-3p, which is highly enriched in obese PVAT and recently reported to induce ED by vascular remodeling (24). EAT has close proximity to the adventitia of the coronary arteries and shares the same microcirculation as the underlying myocardium (25, 26). When adversely remodeled and dysfunctional in obesity, EAT secretes proinflammatory cytokines, contributing directly to the pathogenesis of coronary artery disease (27).
Targeting AT-EC Axis to Treat Obesity-Induced ED/CVD
AT has an essential role in obesity-induced CVD (28) Recent findings suggest that targeting either AT, EC (single-target), or both (dual-target) with a mechanism-based approach would improve obesity-induced ED/CVD (Table 1).
Table 1
| Potential targets/interventions | Effects and mechanisms | |
|---|---|---|
| Single-target | EC (29) | |
| eNOS | Slow-release eNOS substrate arginine (30) or blocking arginase (31, 32) to improve eNOS function and/or NO bioavailability in rodents and patients. | |
| FTO inhibition | Overcame glucose intolerance and insulin resistance and hypertension in mouse models of obesity (33). | |
| NOX inhibition | Inhibition of specific subunits ameliorated ROS-induced ED in rat model of obesity (34, 35). | |
| CD40L inhibition | Improved ROS-induced inflammation and ED in mouse models of obesity (36). | |
| NETs | Blocking formation or increased degradation in EC prevented ED in mouse model of obesity (37, 38). | |
| TRPV4 channels | Activity rescue improved ED (39), involving Ca2+-mediated vasoregulation (40), in mouse models of obesity. | |
| Soluble (pro)renin receptor inhibition | Soluble (pro)renin receptor induced ED and hypertension by activating AT1R leading to RAS hyperactivity in mouse models of obesity (41). | |
| AT (4) | ||
| GRK2 inhibition | Reduced AT-macrophage infiltration and improved ED in mice (42). | |
| Anti-inflammation | Reduced AT-pro-inflammatory cytokine production by adipokine and leptin (43). | |
| Dual-target | GLP1 agonist DPP4 inhibitors | Improved cardiovascular outcomes in patients of type 2 diabetes mellitus (44, 45) related to improved AT function (46). |
| SGLT2 inhibitor | Cardioprotective (47) and beneficial for heart failure in patients (48). | |
| Anti-inflammation | Reduced AT-pro-inflammatory cytokine production and restored endothelial function by metformin (49), resveratrol (50), and methotrexate (51). | |
| Lifestyle intervention | Exercise improved EC function (52) or reversed ED (53). Calorie restriction improved vascular insulin sensitivity and reduced inflammation (54). | |
Recent advances in treating obesity-induced ED.
Single-Target Interventions
Targeting peripheral vascular EC to improve ED has been long exploited (29). Recent studies showed that heterozygous eNOS deficiency was associated with ED in diet-induced obesity (55). Patients with abnormal arginine metabolism and bioavailability due to obesity displayed lower cardiometabolic risk after treatment with a slow-release eNOS substrate, arginine (30). Vascular function could be rescued in obese vessels by targeting EC-Ca2+ toolkit, although this was not tested in obese subjects (56). A recent study showed that deletion of fat mass and obesity-associated protein (FTO) in EC rescued metabolic and vascular function in obesity (33), independent of its known function in regulation of obesity (57). Vascular arginase reduces NO bioavailability, which hastens microvascular remodeling in obesity (31). EC-derived arginase mediates obesity-induced vascular dysfunction and arterial stiffening (32), implicating arginase as a potential target of obesity-induced ED.
Oxidative stress is a key pathogenic factor of microvascular complications in metabolic disease. Renal Nox1, Nox2, and Nox4 contribute differentially to vascular oxidative stress-associated ED in obesity, suggesting a need to identify specific Nox subunits as a target (34), which may result in effective prevention of obesity-related CVD (35). Oxidative stress-associated inflammation is another frequently tested target. CD40 ligand (CD40L) signaling regulates ED via immune cell recruitment and platelet activation in mouse models of hypertension, the mechanism of which extended to mouse models of obesity, implicating CD40L as a therapeutic target for lipid dysmetabolism (36). Neutrophil extracellular traps (NETs) have an inflammatory web-like chromatin structure (37). Inhibition or degradation of NETs prevented ED in mouse model of obesity (38) Further, adipose macrophage infiltration enhanced vascular ED in obese subjects (58). Blocking infiltration of macrophages and T lymphocytes in PVAT prevented obesity-induced ED in mice with G protein-coupled receptor kinase 2 (GRK2) deletion in myeloid cells (42), suggesting GRK2 as a potential therapeutic target. Deletion of lipoxin receptor in leukocytes led to unsolved inflammation in mice, which augmented ED with diabetic cardiomyopathy in obesity (59). Similar therapeutic potentials have been found in other inflammatory mediators, such as adipokine and leptin (43).
Obesity is a strong predictor of hypertension, although it remains unknown how obesity increases blood pressure (60). ED is a hallmark of obesity-induced hypertension. Insulin resistance and increased systolic blood pressure led to ED in obesity; however, targeting these factors presented with different benefits depending on sex (61) and ethnic group (62). RAS hyperactivity was often thought to result from Ang II-dependent stimulation of the Ang II type 1 receptor (AT1R). Recently, the soluble (pro)renin receptor was found to induce ED and hypertension by activating AT1R in high-fat diet (HFD) feeding mice (41). Another recent study reported that endothelial transient receptor potential vanilloid 4 channels (TRPV4) was impaired in a mouse model of diet-induced obesity and obese human resistance vessels, resulting in increased blood pressure (39). This contrasted with findings from another recent study in the same mouse model, but with a longer duration on HFD feeding, which implicated Ca2+-spark vasoregulation as the underlying mechanism (40). Prolonged HFD feeding in mice appeared to improve the vascular response to leptin, which overrode ED induction (63) In any case, strategies to preserve or protect a functional target on EC would bear promise to improve ED and hypertension in obesity.
Dual-Target Interventions
In terms of safety, cost, and effectiveness, most clinical approaches targeting AT have not been successful in the treatment of AT-induced CVD (28). However, some commonly used anti-hyperglycemic medications and lifestyle intervention could elicit a dual action: improving AT function and conferring an appreciable cardiovascular benefit.
Glucagon-like peptide 1 (GLP1) is an incretin, responsible for insulin secretion, glucagon inhibition, and decreased gastrointestinal motility in the post-prandial setting. GLP1 is inactivated by dipeptidyl peptidase 4 (DPP4), an AT-expressing enzyme. GLP1 agonists (e.g., liraglutide) and DPP4 inhibitors (e.g., sitagliptin) are now being used in the management of type 2 diabetes mellitus with improved cardiovascular outcomes in clinical trials (44, 45), implicating AT involvement (46). Sodium–glucose transporter 2 (SGLT2) (e.g., empagliflozin) is responsible for renal glucose reabsorption, the inhibition of which also exerted direct AT effects with a cardioprotective profile (47). As reported in a meta-analysis, SGLT2 inhibitors generated consistent beneficial outcomes for heart failure and kidney disease, with certain heterogeneity in cardiovascular deaths (48). Whether endothelial function and/or inflammatory response are improved and whether they are associated with the favorable outcomes in these trials remain to be determined.
Since positive cardiovascular outcomes have been observed in patients with established CVD by the use of anti-inflammatory agents [e.g., canakinumab (64)], interfering with AT inflammation could generate favorable outcomes. Indeed, in rodent models of obesity, several pharmaceuticals [e.g., metformin (49), resveratrol (50), and methotrexate (51)] are reported to reduce pro-inflammatory cytokine expression in AT and promote adiponectin expression, thereby rescuing eNOS phosphorylation and endothelial function.
Lifestyle intervention (e.g., exercise and diet) is one of the best approaches, especially when medicines are not available or existing medicines have failed. ED directly impairs basic vascular function (e.g., blood flow alteration), which made it a great target for pharmacological and/or exercise intervention with insulin-based therapies (57). Obese individuals who performed an acute high-intensity interval exercise presented with improved plasma pentraxin 3 and endothelial function (52). Even a short-term weight loss could reverse obesity-induced microvascular ED (53). Calorie restriction improved vascular insulin sensitivity, which was associated with downregulation of pro-inflammatory cytokine production in aged AT (54).
Given the functional AT-EC interaction, targeting both AT and EC (“dual-action therapies”) would be a better approach for obesity (65–67), which might lead to the cardiovascular benefits observed in different large-scale clinical trials (68).
Discussion: Unresolved Questions and Future Directions
AT-induced ED is central to the development of CVD, the major cause of morbidity and mortality (69). In obesity, AT induces ED by releasing bioactive products locally and systematically (Figure 1). ED is the very first step in CVD pathogenesis; understanding the molecular mechanism could help us to identify therapeutic targets. Progress has been made in this regard, but important questions remain unanswered.
Figure 1
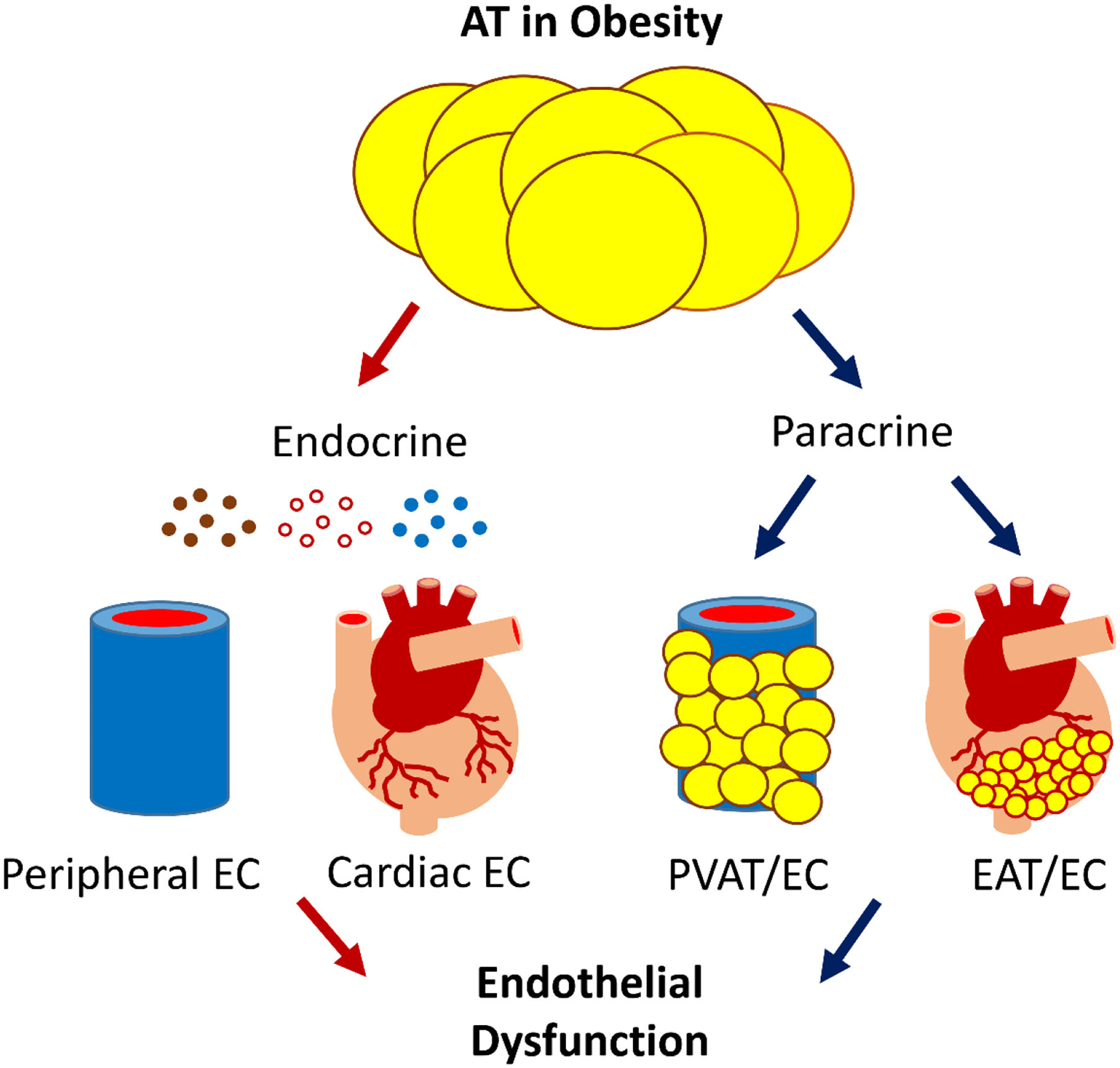
The scheme of AT-EC interplay in obesity-induced ED. AT interacts with the cardiovascular system via endocrine and paracrine secretion of bioactive products, e.g., adipokines, gaseous messengers, and microvesicles that carry bioactive molecules such as miRNA. Dysfunctional AT-EC interactions may induce ED in obesity, leading to CVD.
AT has striking biological variability due to its location and metabolic state, affecting the individual's overall cardiometabolic risk (70–73). For example, excess visceral AT has been linked to diabetogenic/atherogenic metabolic abnormalities more so than subcutaneous AT (73), partly because the former has more glucocorticoid receptors, which accelerated fat deposition when the hypothalamic-pituitary-adrenal axis was activated, leading to insulin resistance in the liver and in the skeletal muscle. To uncover how AT promotes CVD in obesity, we should consider both AT- expansion and its heterogeneous nature as an endocrine organ (74–78).
Large-scale epidemiological studies have questioned the exact nature of adiposity-adverse outcomes association, implicating an “obesity paradox,” in which individuals with overweight and even obesity present survival benefit compared with their normal-weight counterparts in general population and those with chronic diseases (79, 80) or critical illness (e.g., heart failure) (74–78). Although under debate (81), the survival benefits may be attributable to higher energy reserves, inflammatory preconditioning, endotoxin neutralization, adrenal steroid synthesis, activation of RAS, secretion of cardioprotective factors, and prevention of muscle wasting (82). Given the methodological flaws in these studies, further randomized and controlled clinical trials and prospective studies are required to validate the concept. Future research should focus on the pathophysiologic role of AT in critical illness. In this regard, the role and mechanism of endothelial (dys)function in the obesity paradox (83) remains to be elucidated.
Recent studies demonstrated that vascular-derived and heart-derived signals, such as pro-inflammatory and oxidative stimuli released from diseased vessels and/or myocardium, modified AT biology, e.g., by providing adipocyte precursors and driving angiogenesis in response to excess calories (84). Thus, AT can be regulated by feedback signals from the vascular EC, suggestive of a bidirectional interaction. Similar to EC control of CVD development (e.g., atherogenesis) by interacting with VSMC (85), emerging studies support that EC controls whole-body metabolisms through interactions with metabolic tissues (84), including AT in obesity-associated insulin resistance. The molecular mechanisms for bidirectional regulation of AT and EC merit continued investigation to better translate findings into clinical benefits.
The vasculature is present in all major organs, sustaining homeostasis and function throughout the body. The vascular EC display extensive functional heterogeneity depending on the vessel and tissue in which they reside. It facilitates the unique physiological function of each organ, such as nutrient transport, endocrine signaling, waste disposal, and disease protection. The mechanisms sustaining EC heterogeneity remain unknown (86). EC functional diversity was initially investigated by exploring EC specialization on a global scale [e.g., expression profile of multiple cultured EC with DNA microarrays (87)], followed by attempts to decipher functional and transcriptomic features of organ-specific EC in small populations or seldom-expressed genes in the lung (88), liver (89), heart (90), and other tissues (91). The emerging single-cell RNA sequencing technologies facilitate finding genes and pathways that dictate the organ-specific function of EC (92). A transcriptome study identified distinct gene expression profiles in cardiac EC (when compared with renal, cerebral, or pulmonary EC), e.g., higher expression of CD36 signaling cascade (90), which is a key regulator of fatty acid uptake and involved in atherogenesis (93). A recent study reported a relationship of circulating EC with obesity and cardiometabolic risk factors (94). Future research could identify signatures of the EC in depot-specific AT to determine their pathological roles in vascular complications.
The current COVID-19 pandemic presents an urgent health crisis (95). Numerous studies reported that severe obesity is associated with increased morbidity and mortality from COVID-19 (96–103), suggesting obesity as a risk factor for severe COVID-19 disease (104, 105). This is not surprising given that obesity is generally associated with increased incidence and severity of respiratory viral infection (106, 107). However, the underlying mechanism has yet to be elucidated. Recently, COVID-19 has been suggested as a multiorgan endothelial disease for its association with vasculitis and ED (108–111), which may be gender- and age-dependent (112). Although both the hypothetical role and therapeutic targetability of the vasculature in COVID-19 remain to be validated (113), an urgent need in this pandemic is to identify factors that mediate the physiological interactions between obesity and vasculature that contribute to CVD. Epithelial cell-derived IL-33 (114) was a key player in driving all stages of COVID-19 disease (115). EC also express IL-33 (116), the expression of which was enhanced in AT-EC by severe obesity (117). It would be timely to test whether EC-derived IL-33 mediates COVID-19-associated vascular complications (118). The hope is to bring about clinical breakthroughs for the treatment of COVID-19 in patients with obesity.
Obesity is a major risk factor for common medical conditions beyond CVD, such as type 2 diabetes (119), dyslipidemias (120), fatty liver (121), Alzheimer's disease (122, 123), and some cancers (124). These conditions occur due to obesity-induced insulin resistance and AT-derived endocrine factors (5). Given the essential roles of EC in the development of these disorders individually [diabetes (14), dyslipidemias, fatty liver (125, 126), Alzheimer's disease (127, 128), and cancers (129)], one would wonder whether targeting the AT-EC axis would be a novel avenue to improve these common conditions. Answers to these questions could be clinically significant in preventing or treating obesity-related complications.
Statements
Author contributions
JX proposed the conception. ML, MQ, KK, and JX wrote the article. All authors contributed to the article and approved the submitted version.
Funding
The research in the authors' lab was supported by a National Institutes of Health Grant (R01HL-130845), a Beginning Grant-in-Aid Award (14BGIA20030027), and a National Scientist Development Grant (10SDG2600164) from the American Heart Association, a Junior Faculty Award (1-12-JF-58) from the American Diabetes Association, and Research Awards (HR11-200, HR14-062, and HR17-046) from the Oklahoma Center for the Advancement of Science and Technology, and the Seed Grants from the Presbyterian Health Foundation and the Harold Hamm Diabetes Center (all to JX).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
SaltielAROlefskyJM. Inflammatory mechanisms linking obesity and metabolic disease. J Clin Invest. (2017) 127:1–4. 10.1172/JCI92035
2.
RimmEBStampferMJGiovannucciEAscherioASpiegelmanDColditzGAWillettWC. Body size and fat distribution as predictors of coronary heart disease among middle-aged and older US men. Am J Epidemiol. (1995) 141:1117–27. 10.1093/oxfordjournals.aje.a117385
3.
HartzAJRupleyDCJrKalkhoffRDRimmAA. Relationship of obesity to diabetes: influence of obesity level and body fat distribution. Prev Med. (1983) 12:351–7. 10.1016/0091-7435(83)90244-X
4.
ChaitAden HartighLJ. Adipose tissue distribution, inflammation and its metabolic consequences, including diabetes and cardiovascular disease. Front Cardiovasc Med. (2020) 7:22. 10.3389/fcvm.2020.00022
5.
GestaSTsengYHKahnCR. Developmental origin of fat: tracking obesity to its source. Cell. (2007) 131:242–56. 10.1016/j.cell.2007.10.004
6.
GreenbergASObinMS. Obesity and the role of adipose tissue in inflammation and metabolism. Am J Clin Nutr. (2006) 83:461S−5S. 10.1093/ajcn/83.2.461S
7.
GiraltMVillarroyaF. White, brown, beige/brite: different adipose cells for different functions?Endocrinology. (2013) 154:2992–3000. 10.1210/en.2013-1403
8.
LanginD. In and out: adipose tissue lipid turnover in obesity and dyslipidemia. Cell Metab. (2011) 14:569–70. 10.1016/j.cmet.2011.10.003
9.
SidossisLKajimuraS. Brown and beige fat in humans: thermogenic adipocytes that control energy and glucose homeostasis. J Clin Invest. (2015) 125:478–86. 10.1172/JCI78362
10.
NedergaardJCannonB. The browning of white adipose tissue: some burning issues. Cell Metab. (2014) 20:396–407. 10.1016/j.cmet.2014.07.005
11.
RamseyerVDGrannemanJG. Adrenergic regulation of cellular plasticity in brown, beige/brite and white adipose tissues. Adipocyte. (2016) 5:119–29. 10.1080/21623945.2016.1145846
12.
SteinbergHOChakerHLeamingRJohnsonABrechtelGBaronAD. Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J Clin Invest. (1996) 97:2601–10. 10.1172/JCI118709
13.
FancherISLe MasterEAhnSJAdamosCLeeJCBerdyshevEet al. Impairment of flow-sensitive inwardly rectifying k(+) channels via disruption of glycocalyx mediates obesity-Induced endothelial dysfunction. Arterioscler Thromb Vasc Biol. (2020) 40:e240–55. 10.1161/ATVBAHA.120.314935
14.
XuJZouMH. Molecular insights and therapeutic targets for diabetic endothelial dysfunction. Circulation. (2009) 120:1266–86. 10.1161/CIRCULATIONAHA.108.835223
15.
BartonM. Obesity and aging: determinants of endothelial cell dysfunction and atherosclerosis. Pflugers Arch. (2010) 460:825–37. 10.1007/s00424-010-0860-y
16.
KwaifaIKBahariHYongYKNoorSM. Endothelial dysfunction in obesity-Induced inflammation: molecular mechanisms and clinical implications. Biomolecules. (2020) 10:291. 10.3390/biom10020291
17.
SaxtonSNClarkBJWithersSBEringaECHeagertyAM. Mechanistic links between obesity, diabetes, and blood pressure: role of perivascular adipose tissue. Physiol Rev. (2019) 99:1701–63. 10.1152/physrev.00034.2018
18.
VirdisAColucciRBernardiniNBlandizziCTaddeiSMasiS. Microvascular endothelial dysfunction in human obesity: role of TNF-alpha. J Clin Endocrinol Metab. (2019) 104:341–8. 10.1210/jc.2018-00512
19.
JunaidASchoemanJYangWStamWMashaghiAvan ZonneveldAJHankemeierT. Metabolic response of blood vessels to TNFalpha. Elife. (2020) 9:e54754. 10.7554/eLife.54754
20.
HeynGSCorreaLHMagalhaesKG. The impact of adipose tissue-derived miRNAs in metabolic syndrome, obesity, and cancer. Front Endocrinol. (2020) 11:563816. 10.3389/fendo.2020.563816
21.
ZhouYTanC. miRNAs in adipocyte-derived extracellular vesicles: multiple roles in development of obesity-associated disease. Front Mol Biosci. (2020) 7:171. 10.3389/fmolb.2020.00171
22.
Ait-AissaKNguyenQMGabaniMKassanAKumarSChoiSKet al. MicroRNAs and obesity-induced endothelial dysfunction: key paradigms in molecular therapy. Cardiovasc Diabetol. (2020) 19:136. 10.1186/s12933-020-01107-3
23.
YudkinJSEringaEStehouwerCD. “Vasocrine” signalling from perivascular fat: a mechanism linking insulin resistance to vascular disease. Lancet. (2005) 365:1817–20. 10.1016/S0140-6736(05)66585-3
24.
LiXBallantyneLLYuYFunkCD. Perivascular adipose tissue-derived extracellular vesicle miR-221-3p mediates vascular remodeling. FASEB J. (2019) 33:12704–22. 10.1096/fj.201901548R
25.
SacksHSFainJN. Human epicardial adipose tissue: a review. Am Heart J. (2007) 153:907–17. 10.1016/j.ahj.2007.03.019
26.
MuzurovicEMVujosevicSMikhailidisDP. Can we decrease epicardial and pericardial fat in patients with diabetes?J Cardiovasc Pharmacol Ther. (2021). 10.1177/10742484211006997
27.
GuglielmoMLinADeyDBaggianoAFusiniLMuscogiuriGPontoneG. Epicardial fat and coronary artery disease: role of cardiac imaging. Atherosclerosis. (2021) 321:30–8. 10.1016/j.atherosclerosis.2021.02.008
28.
OikonomouEKAntoniadesC. The role of adipose tissue in cardiovascular health and disease. Nat Rev Cardiol. (2019) 16:83–99. 10.1038/s41569-018-0097-6
29.
DeanfieldJEHalcoxJPRabelinkTJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. (2007) 115:1285–95. 10.1161/CIRCULATIONAHA.106.652859
30.
MariottiF. Arginine supplementation and cardiometabolic risk. Curr Opin Clin Nutr Metab Care. (2020) 23:29–34. 10.1097/MCO.0000000000000612
31.
MasiSColucciRDurantiENannipieriMAnselminoMIppolitoCet al. Aging modulates the influence of arginase on endothelial dysfunction in obesity. Arterioscler Thromb Vasc Biol. (2018) 38:2474–83. 10.1161/ATVBAHA.118.311074
32.
Obesity-induced vascular dysfunction and arterial stiffening requires endothelial cell arginase 1. Cardiovasc Res. (2018) 114:64. 10.1093/cvr/cvx217
33.
KrugerNBiwerLAGoodMERuddimanCAWolpeAGDeLalioLJet al. Loss of endothelial FTO antagonizes obesity-Induced metabolic and vascular dysfunction. Circ Res. (2020) 126:232–42. 10.1161/CIRCRESAHA.119.315531
34.
MunozMLopez-OlivaMERodriguezCMartinezMPSaenz-MedinaJSanchezAet al. Differential contribution of Nox1, Nox2 and Nox4 to kidney vascular oxidative stress and endothelial dysfunction in obesity. Redox Biol. (2020) 28:101330. 10.1016/j.redox.2019.101330
35.
VirdisAMasiSColucciRChiriacoMUlianaMPuxedduIet al. Microvascular endothelial dysfunction in patients with obesity. Curr Hypertens Rep. (2019) 21:32. 10.1007/s11906-019-0930-2
36.
StevenSDibMHausdingMKashaniFOelzeMKroller-SchonSet al. CD40L controls obesity-associated vascular inflammation, oxidative stress, and endothelial dysfunction in high fat diet-treated and db/db mice. Cardiovasc Res. (2018) 114:312–23. 10.1093/cvr/cvx197
37.
PapayannopoulosV. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2018) 18:134–47. 10.1038/nri.2017.105
38.
WangHWangQVenugopalJWangJKleimanKGuoCEitzmanDT. Obesity-induced endothelial dysfunction is prevented by neutrophil extracellular trap inhibition. Sci Rep. (2018) 8:4881. 10.1038/s41598-018-23256-y
39.
OttoliniMHongKCopeELDanevaZDeLalioLJSokolowskiJDet al. Local peroxynitrite impairs endothelial transient receptor potential vanilloid 4 channels and elevates blood pressure in obesity. Circulation. (2020) 141:1318–333. 10.1161/CIRCULATIONAHA.119.043385
40.
GreensteinASKadirSCsatoVSugdenSABaylieRAEisnerDANelsonMT. Disruption of pressure-Induced ca(2+) spark vasoregulation of resistance arteries, rather than endothelial dysfunction, underlies obesity-Related hypertension. Hypertension. (2020) 75:539–48. 10.1161/HYPERTENSIONAHA.119.13540
41.
FuZWangFLiuXHuJSuJLuXet al. Soluble (Pro)Renin receptor induces endothelial dysfunction and hypertension in mice with diet-Induced obesity via activation of angiotensinII type 1 receptor. Clin Sci. (2021) 135:793–810. 10.1042/CS20201047
42.
Gonzalez-AmorMVila-BedmarRRodrigues-DiezRMoreno-CarrilesRArconesACCruces-SandeMet al. Myeloid GRK2 regulates obesity-Induced endothelial dysfunction by modulating inflammatory responses in perivascular adipose tissue. Antioxidants. (2020) 9:953. 10.3390/antiox9100953
43.
SinghMBenenciaF. Inflammatory processes in obesity: focus on endothelial dysfunction and the role of adipokines as inflammatory mediators. Int Rev Immunol. (2019) 38:157–71. 10.1080/08830185.2019.1638921
44.
MarsoSPDanielsGHBrown-FrandsenKKristensenPMannJFNauckMAet al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2016) 375:311–22. 10.1056/NEJMoa1603827
45.
GreenJBBethelMAArmstrongPWBuseJBEngelSSGargJet al. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2015) 373:232–42. 10.1056/NEJMoa1501352
46.
LamersDFamullaSWronkowitzNHartwigSLehrSOuwensDMet al. Dipeptidyl peptidase 4 is a novel adipokine potentially linking obesity to the metabolic syndrome. Diabetes. (2011) 60:1917–25. 10.2337/db10-1707
47.
ZinmanBWannerCLachinJMFitchettDBluhmkiEHantelSet al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. (2015) 373:2117–28. 10.1056/NEJMoa1504720
48.
McGuireDKShihWJCosentinoFCharbonnelBCherneyDZIDagogo-JackSet al. Association of SGLT2 inhibitors with cardiovascular and kidney outcomes in patients with type 2 diabetes: a Meta-analysis. JAMA Cardiol. (2020) 6:148–58. 10.1001/jamacardio.2020.4511
49.
SunYLiJXiaoNWangMKouJQiLet al. Pharmacological activation of AMPK ameliorates perivascular adipose/endothelial dysfunction in a manner interdependent on aMPK and SIRT1. Pharmacol Res. (2014) 89:19–28. 10.1016/j.phrs.2014.07.006
50.
XiaNForstermannULiH. Effects of resveratrol on eNOS in the endothelium and the perivascular adipose tissue. Ann N Y Acad Sci. (2017) 1403:132–41. 10.1111/nyas.13397
51.
MaYLiLShaoYBaiXBaiTHuangX. Methotrexate improves perivascular adipose tissue/endothelial dysfunction via activation of AMPK/eNOS pathway. Mol Med Rep. (2017) 15:2353–9. 10.3892/mmr.2017.6225
52.
SlusherALFicoBGDodgeKMGartenRSFerrandiPJRodriguezAAet al. Impact of acute high-intensity interval exercise on plasma pentraxin 3 and endothelial function in obesity individuals-a pilot study. Eur J Appl Physiol. (2021) 121:1567–77. 10.1007/s00421-021-04632-5
53.
CsipoTFulopGALipeczATarantiniSKissTBalasubramanianPet al. Short-term weight loss reverses obesity-induced microvascular endothelial dysfunction. Geroscience. (2018) 40:337–46. 10.1007/s11357-018-0028-9
54.
AmorSMartin-CarroBRubioCCarrascosaJMHuWHuangYet al. Study of insulin vascular sensitivity in aortic rings and endothelial cells from aged rats subjected to caloric restriction: role of perivascular adipose tissue. Exp Gerontol. (2018) 109:126–36. 10.1016/j.exger.2017.10.017
55.
AliMIChenXDidionSP. Heterozygous ENOS deficiency is associated with oxidative stress and endothelial dysfunction in diet-induced obesity. Physiol Rep. (2015) 3:e12630. 10.14814/phy2.12630
56.
MocciaFNegriSFarisPBerra-RomaniR. Targeting the endothelial ca2+ toolkit to rescue endothelial dysfunction in obesity associated-Hypertension. Curr Med Chem. (2020) 27:240–57. 10.2174/0929867326666190905142135
57.
LuseMAHeistonEMMalinSKIsaksonBE. Cellular and functional effects of insulin based therapies and exercise on endothelium. Curr Pharm Des. (2020) 26:3760–7. 10.2174/1381612826666200721002735
58.
ApovianCMBigorniaSMottMMeyersMRUlloorJGaguaMet al. Adipose macrophage infiltration is associated with insulin resistance and vascular endothelial dysfunction in obese subjects. Arterioscler Thromb Vasc Biol. (2008) 28:1654–9. 10.1161/ATVBAHA.108.170316
59.
TourkiBKainVShaikhSRLeroyXSerhanCNHaladeGV. Deficit of resolution receptor magnifies inflammatory leukocyte directed cardiorenal and endothelial dysfunction with signs of cardiomyopathy of obesity. FASEB J. (2020) 34:10560–73. 10.1096/fj.202000495RR
60.
FultonDJRSteppDW. Origins of hypertension in obesity: plain vanilla(oid) or multiple flavors?Circulation. (2020) 141:1334–7. 10.1161/CIRCULATIONAHA.120.046027
61.
TaylorLERamirezLAMusallJBSullivanJC. Tipping the scales: are females more at risk for obesity- and high-fat diet-induced hypertension and vascular dysfunction?Br J Pharmacol. (2019) 176:4226–42. 10.1111/bph.14783
62.
LteifAAHanKMatherKJ. Obesity, insulin resistance, and the metabolic syndrome: determinants of endothelial dysfunction in whites and blacks. Circulation. (2005) 112:32–8. 10.1161/CIRCULATIONAHA.104.520130
63.
RochaVDSClaudioERGda SilvaVLCordeiroJPDomingosLFda CunhaMRHet al. High-fat diet-Induced obesity model does not promote endothelial dysfunction via increasing leptin/Akt/eNOS signaling. Front Physiol. (2019) 10:268. 10.3389/fphys.2019.00268
64.
RidkerPMEverettBMThurenTMacFadyenJGChangWHBallantyneCet al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. 10.1056/NEJMoa1707914
65.
DraganoNRVFernoJDieguezCLopezMMilbankE. Reprint of: recent updates on obesity treatments: available drugs and future directions. Neuroscience. (2020) 447:191–215. 10.1016/j.neuroscience.2020.08.009
66.
DraganoNRVFernoJDieguezCLopezMMilbankE. Recent updates on obesity treatments: available drugs and future directions. Neuroscience. (2020) 437:215–39. 10.1016/j.neuroscience.2020.04.034
67.
SchinzariFTesauroMCardilloC. Endothelial and perivascular adipose tissue abnormalities in obesity-related vascular dysfunction: novel targets for treatment. J Cardiovasc Pharmacol. (2017) 69:360–8. 10.1097/FJC.0000000000000469
68.
WildingJPHJacobS. Cardiovascular outcome trials in obesity: a review. Obes Rev. (2021) 22:e13112. 10.1111/obr.13112
69.
BenjaminEJBlahaMJChiuveSECushmanMDasSRDeoRet al. Heart disease and stroke statistics-2017 update: a report from the American heart association. Circulation. (2017) 135:e146–603. 10.1161/CIR.0000000000000491
70.
BrittonKAMassaroJMMurabitoJMKregerBEHoffmannUFoxCS. Body fat distribution, incident cardiovascular disease, cancer, and all-cause mortality. J Am Coll Cardiol. (2013) 62:921–5. 10.1016/j.jacc.2013.06.027
71.
SilvaKRCortesILiechockiSCarneiroJRSouzaAABorojevicRet al. Characterization of stromal vascular fraction and adipose stem cells from subcutaneous, preperitoneal and visceral morbidly obese human adipose tissue depots. PLoS ONE. (2017) 12:e0174115. 10.1371/journal.pone.0174115
72.
MarinouKHodsonLVasanSKFieldingBABanerjeeRBrismarKet al. Structural and functional properties of deep abdominal subcutaneous adipose tissue explain its association with insulin resistance and cardiovascular risk in men. Diabetes Care. (2014) 37:821–9. 10.2337/dc13-1353
73.
DespresJP. Body fat distribution and risk of cardiovascular disease: an update. Circulation. (2012) 126:1301–13. 10.1161/CIRCULATIONAHA.111.067264
74.
BowmanKAtkinsJLDelgadoJKosKKuchelGABleAet al. Central adiposity and the overweight risk paradox in aging: follow-up of 130,473 UK biobank participants. Am J Clin Nutr. (2017) 106:130–5. 10.3945/ajcn.116.147157
75.
FlierJSCookKSUsherPSpiegelmanBM. Severely impaired adipsin expression in genetic and acquired obesity. Science. (1987) 237:405–8. 10.1126/science.3299706
76.
AkoumianakisIAntoniadesC. The interplay between adipose tissue and the cardiovascular system: is fat always bad?Cardiovasc Res. (2017) 113:999–1008. 10.1093/cvr/cvx111
77.
LitwinSE. Good fat, bad fat: the increasingly complex interplay of adipose tissue and the cardiovascular system. J Am Coll Cardiol. (2013) 62:136–7. 10.1016/j.jacc.2013.04.028
78.
PicardFDeshaiesY. PPARgamma and novel nuclear factors in the interplay between adipose tissue and the cardiovascular system: view from the chair. Int J Obes. (2005) 29(Suppl. 1):S24–5. 10.1038/sj.ijo.0802909
79.
AuneDSenAPrasadMNoratTJanszkyITonstadSet al. BMI and all cause mortality: systematic review and non-linear dose-response meta-analysis of 230 cohort studies with 3.74 million deaths among 30.3 million participants. BMJ. (2016) 353:i2156. 10.1136/bmj.i2156
80.
Powell-WileyTMNgwaJKebedeSLuDSchultePJBhattDLet al. Impact of body mass index on heart failure by race/ethnicity from the get with the guidelines-heart failure (GWTG-HF) registry. JACC Heart Fail. (2018) 6:233–2. 10.1016/j.jchf.2017.11.011
81.
AkinINienaberCA. “Obesity paradox” in coronary artery disease. World J Cardiol. (2015) 7:603–8. 10.4330/wjc.v7.i10.603
82.
ElagiziACarboneSLavieCJMehraMRVenturaHO. Implications of obesity across the heart failure continuum. Prog Cardiovasc Dis. (2020) 63:561–9. 10.1016/j.pcad.2020.09.005
83.
LavieCJMilaniRV. Weight reduction and improvements in endothelial function: combating the “obesity paradox” in coronary heart disease. Chest. (2011) 140:1395–6. 10.1378/chest.11-1889
84.
LiMQianMXuJ. Vascular endothelial regulation of obesity-associated insulin resistance. Front Cardiovasc Med. (2017) 4:51. 10.3389/fcvm.2017.00051
85.
LiMQianMKylerKXuJ. Endothelial-Vascular smooth muscle cells interactions in atherosclerosis. Front Cardiovasc Med. (2018) 5:151. 10.3389/fcvm.2018.00151
86.
PaikDTTianLWilliamsIMRheeSZhangHLiuCet al. Single-cell RNA-seq unveils unique transcriptomic signatures of organ-specific endothelial cells. Circulation. (2020) 142:1848–62. 10.1161/CIRCULATIONAHA.119.041433
87.
ChiJTChangHYHaraldsenGJahnsenFLTroyanskayaOGChangDSet al. Endothelial cell diversity revealed by global expression profiling. Proc Natl Acad Sci USA. (2003) 100:10623–8. 10.1073/pnas.1434429100
88.
DingBSNolanDJGuoPBabazadehAOCaoZRosenwaksZet al. Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell. (2011) 147:539–53. 10.1016/j.cell.2011.10.003
89.
DingBSNolanDJButlerJMJamesDBabazadehAORosenwaksZet al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature. (2010) 468:310–5. 10.1038/nature09493
90.
LotherABergemannSDengLMoserMBodeCHeinL. Cardiac endothelial cell transcriptome. Arterioscler Thromb Vasc Biol. (2018) 38:566–74. 10.1161/ATVBAHA.117.310549
91.
NolanDJGinsbergMIsraelyEPalikuqiBPoulosMGJamesDet al. Molecular signatures of tissue-specific microvascular endothelial cell heterogeneity in organ maintenance and regeneration. Dev Cell. (2013) 26:204–19. 10.1016/j.devcel.2013.06.017
92.
PaikDTChoSTianLChangHYWuJC. Single-cell RNA sequencing in cardiovascular development, disease and medicine. Nat Rev Cardiol. (2020) 17:457–73. 10.1038/s41569-020-0359-y
93.
FebbraioMSilversteinRL. CD36: implications in cardiovascular disease. Int J Biochem Cell Biol. (2007) 39:2012–30. 10.1016/j.biocel.2007.03.012
94.
SolteroEGSoloveyANHebbelRPPalzerEFRyderJRShaibiGQet al. Relationship of circulating endothelial cells with obesity and cardiometabolic risk factors in children and adolescents. J Am Heart Assoc. (2021) 10:e018092. 10.1161/JAHA.120.018092
95.
LockhartSMO'RahillyS. When two pandemics meet: why is obesity associated with increased COVID-19 mortality?Med (N Y). (2020) 1:33–42. 10.1016/j.medj.2020.06.005
96.
KassDA. COVID-19 and severe obesity: a big problem?Ann Intern Med. (2020) 173:840–1. 10.7326/M20-5677
97.
KalligerosMShehadehFMylonaEKBenitezGBeckwithCGChanPAMylonakisE. Association of obesity with disease severity among patients with coronavirus disease 2019. Obesity. (2020) 28:1200–4. 10.1002/oby.22859
98.
RichardsonSHirschJSNarasimhanMCrawfordJMMcGinnTDavidsonKWet al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with cOVID-19 in the New York city area. JAMA. (2020) 323:2052–9. 10.1001/jama.2020.6775
99.
BornsteinSRDalanRHopkinsDMingroneGBoehmBO. Endocrine and metabolic link to coronavirus infection. Nat Rev Endocrinol. (2020) 16:297–8. 10.1038/s41574-020-0353-9
100.
SimonnetAChetbounMPoissyJRaverdyVNouletteJDuhamelAet al. High prevalence of obesity in severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) requiring invasive mechanical ventilation. Obesity. (2020). 28:1195–9. 10.1002/oby.22831
101.
WangAZhaoWXuZGuJ. Timely blood glucose management for the outbreak of 2019 novel coronavirus disease (COVID-19) is urgently needed. Diabetes Res Clin Pract. (2020) 162:108118. 10.1016/j.diabres.2020.108118
102.
FadiniGPMorieriMLLongatoEAvogaroA. Prevalence and impact of diabetes among people infected with SARS-CoV-2. J Endocrinol Invest. (2020) 43:867–869. 10.1007/s40618-020-01236-2
103.
ThenCBakAMorissetADaderBDucoussoMMaciaJLDruckerM. The N-terminus of the cauliflower mosaic virus aphid transmission protein p2 is involved in transmission body formation and microtubule interaction. Virus Res. (2021) 297:198356. 10.1016/j.virusres.2021.198356
104.
DruckerDJ. Diabetes, obesity, metabolism, and sARS-CoV-2 infection: the end of the beginning. Cell Metab. (2021) 33:479–98. 10.1016/j.cmet.2021.01.016
105.
DalamagaMChristodoulatosGSKarampelaIVallianouNApovianCM. Understanding the co-epidemic of obesity and COVID-19: current evidence, comparison with previous epidemics, mechanisms, and preventive and therapeutic perspectives. Curr Obes Rep. (2021) 1–30. 10.1007/s13679-021-00436-y
106.
LouieJKAcostaMWinterKJeanCGavaliSSchechterRet al. Factors associated with death or hospitalization due to pandemic 2009 influenza a(H1N1) infection in California. JAMA. (2009) 302:1896–902. 10.1001/jama.2009.1583
107.
NeidichSDGreenWDRebelesJKarlssonEASchultz-CherrySNoahTLet al. Increased risk of influenza among vaccinated adults who are obese. Int J Obes. (2017) 41:1324–30. 10.1038/ijo.2017.131
108.
SteinRAYoungLM. From ACE2 to COVID-19: a multiorgan endothelial disease. Int J Infect Dis. (2020) 100:425–30. 10.1016/j.ijid.2020.08.083
109.
LibbyPLuscherT. COVID-19 is, in the end, an endothelial disease. Eur Heart J. (2020) 41:3038–44. 10.1093/eurheartj/ehaa623
110.
Bermejo-MartinJFAlmansaRTorresAGonzalez-RiveraMKelvinDJ. COVID-19 as a cardiovascular disease: the potential role of chronic endothelial dysfunction. Cardiovasc Res. (2020) 116:e132–3. 10.1093/cvr/cvaa140
111.
SarduCGambardellaJMorelliMBWangXMarfellaRSantulliG. Hypertension, thrombosis, kidney failure, and diabetes: is COVID-19 an endothelial disease? A comprehensive evaluation of clinical and basic evidence. J Clin Med. (2020) 9:1417. 10.20944/preprints202004.0204.v1
112.
FroldiGDorigoP. Endothelial dysfunction in coronavirus disease 2019 (COVID-19): gender and age influences. Med Hypotheses. (2020) 144:110015. 10.1016/j.mehy.2020.110015
113.
TeuwenLAGeldhofVPasutACarmelietP. COVID-19: the vasculature unleashed. Nat Rev Immunol. (2020) 20:389–91. 10.1038/s41577-020-0343-0
114.
CayrolCGirardJP. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. (2014) 31:31–7. 10.1016/j.coi.2014.09.004
115.
ZizzoGCohenPL. Imperfect storm: is interleukin-33 the achilles heel of cOVID-19?Lancet Rheumatol. (2020) 2:e779–90. 10.1016/S2665-9913(20)30340-4
116.
de OliveiraMFATalvaniARocha-VieiraE. IL-33 in obesity: where do we go from here?Inflamm Res. (2019) 68:185–94. 10.1007/s00011-019-01214-2
117.
ZeydaMWernlyBDemyanetsSKaunCHammerleMHantuschBet al. Severe obesity increases adipose tissue expression of interleukin-33 and its receptor ST2, both predominantly detectable in endothelial cells of human adipose tissue. Int J Obes. (2013) 37:658–65. 10.1038/ijo.2012.118
118.
SouthKMcCullochLMcCollBWElkindMSAllanSMSmithCJ. Preceding infection and risk of stroke: an old concept revived by the COVID-19 pandemic. Int J Stroke. (2020) 15:722–32. 10.1177/1747493020943815
119.
PicheMETchernofADespresJP. Obesity phenotypes, diabetes, and cardiovascular diseases. Circ Res. (2020) 126:1477–500. 10.1161/CIRCRESAHA.120.316101
120.
VekicJZeljkovicAStefanovicAJelic-IvanovicZSpasojevic-KalimanovskaV. Obesity and dyslipidemia. Metabolism. (2019) 92:71–81. 10.1016/j.metabol.2018.11.005
121.
LiLLiuDWYanHYWangZYZhaoSHWangB. Obesity is an independent risk factor for non-alcoholic fatty liver disease: evidence from a meta-analysis of 21 cohort studies. Obes Rev. (2016) 17:510–9. 10.1111/obr.12407
122.
VerdileGKeaneKNCruzatVFMedicSSabaleMRowlesJet al. Inflammation and oxidative stress: the molecular connectivity between insulin resistance, obesity, and alzheimer's disease. Mediators Inflamm. (2015) 2015:105828. 10.1155/2015/105828
123.
KellarDCraftS. Brain insulin resistance in Alzheimer's disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol. (2020) 19:758–766. 10.1016/S1474-4422(20)30231-3
124.
WilsonKMChoE. Obesity and kidney cancer. Recent Results Cancer Res. (2016) 208:81–93. 10.1007/978-3-319-42542-9_5
125.
PasarinMAbraldesJGLiguoriEKokBLa MuraV. Intrahepatic vascular changes in non-alcoholic fatty liver disease: potential role of insulin-resistance and endothelial dysfunction. World J Gastroenterol. (2017) 23:6777–87. 10.3748/wjg.v23.i37.6777
126.
FedericoADallioMMasaroneMPersicoMLoguercioC. The epidemiology of non-alcoholic fatty liver disease and its connection with cardiovascular disease: role of endothelial dysfunction. Eur Rev Med Pharmacol Sci. (2016) 20:4731–41. Available online at: https://pubmed.ncbi.nlm.nih.gov/27906428/
127.
SalminaABInzhutovaAIMalinovskayaNAPetrovaMM. Endothelial dysfunction and repair in alzheimer-type neurodegeneration: neuronal and glial control. J Alzheimers Dis. (2010) 22:17–36. 10.3233/JAD-2010-091690
128.
FukuiYHishikawaNShangJSatoKNakanoYMoriharaRet al. Peripheral arterial endothelial dysfunction of neurodegenerative diseases. J Neurol Sci. (2016) 366:94–99. 10.1016/j.jns.2016.04.042
129.
GeorgeDJKaelinWGJr. The von Hippel-Lindau protein, vascular endothelial growth factor, and kidney cancer. N Engl J Med. (2003) 349:419–21. 10.1056/NEJMp030061
Summary
Keywords
adipose tissue, endothelial dysfuction, obesity, cell interaction, cardiovascular disease
Citation
Li M, Qian M, Kyler K and Xu J (2021) Adipose Tissue-Endothelial Cell Interactions in Obesity-Induced Endothelial Dysfunction. Front. Cardiovasc. Med. 8:681581. doi: 10.3389/fcvm.2021.681581
Received
18 March 2021
Accepted
01 June 2021
Published
01 July 2021
Volume
8 - 2021
Edited by
Manfredi Tesauro, University of Rome Tor Vergata, Italy
Reviewed by
Kimie Tanaka, Juntendo University, Japan; Zhihua Wang, Chinese Academy of Medical Sciences and Peking Union Medical College, China
Updates
Copyright
© 2021 Li, Qian, Kyler and Xu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Xu jian-xu@ouhsc.edu
This article was submitted to Cardiovascular Metabolism, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.