Abstract
Atherosclerosis is a fundamental disease of the cardiovascular system that leads to high morbidity and mortality worldwide. The endothelium is the first protective barrier in atherosclerosis. Endothelial cells have the potential to be transformed into mesenchymal cells, in a process termed endothelial to mesenchymal transition (EndMT). On the one hand, EndMT is known to contribute to atherosclerosis by inducing a number of phenotypes ranging from endothelial cell dysfunction to plaque formation. On the other hand, risk factors for atherosclerosis can lead to EndMT. A substantial body of evidence has suggested that EndMT induces the development of atherosclerosis; therefore, a deeper understanding of the molecular mechanisms underlying EndMT in atherosclerosis might provide insights to reverse this condition.
Introduction
Atherosclerosis is a common disease of the cardiovascular system characterized by plaque formation in the artery wall (1). Although some hypothesis based on “inflammation” (2), “lipid” (3) and “immunology” (4) have been proposed to explain the development of atherosclerosis, the pathogenesis of this condition is still not fully understood. Recently, the concept of endothelial to mesenchymal transition (EndMT) has also been put forward to explain the pathophysiological process of atherosclerosis from the perspective of cell trans differentiation (5). EndMT refers to a process in which endothelial cells can be transformed into mesenchymal cells. In this process, endothelial cells acquire the characteristics of mesenchymal cells and feature with loss of cell–cell contact and cell polarity under the condition of biochemical and biomechanical stimulus (6) (Figure 1). The obvious changes involved in EndMT at the molecular level include decreased expression of endothelial cell markers [such as platelet endothelial cell adhesion molecule-1 (CD31), CD34, von Willebrand Factor (vWF), tyrosine kinase with immunoglobulin-like and EGF-like domains 1 (TIE1), TEK receptor tyrosine kinase (TIE2), and vascular endothelial cadherin (CDH5)] and the increased expression of mesenchymal cell markers [such as alpha-smooth muscle actin (α-SMA), ferroptosis suppressor protein 1 (FSP1), Calponin, and smooth muscle 22 alpha (SM22α) (7, 8)]. During EndMT, endothelial cells undergo morphological changes from a cuboidal to a spindle shape. EndMT is a requirement for the formation of endocardial cushions during heart development; cushions resemble the early endothelium (9). EndMT has not only been described in organogenesis, but it has also been implicated in many diseases including pulmonary arterial hypertension (PAH) (10), fibrosis (11), cancer (12), pathological neovascularization (13), and atherosclerosis (14).
Figure 1
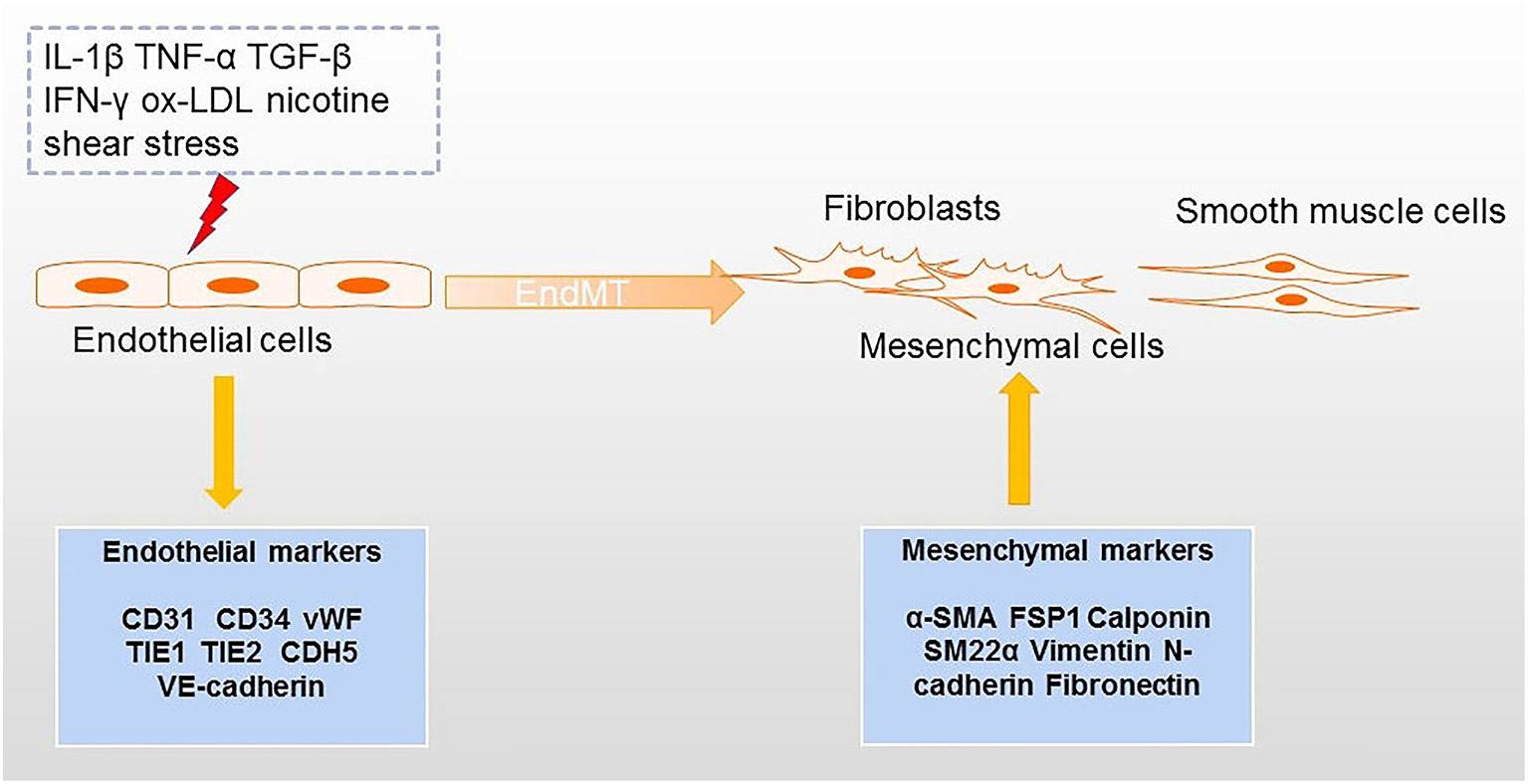
Processes inducing EndMT in atherosclerosis. Factors that can lead to EndMT include several inflammatory cytokines, ox-LDL, nicotine, and shear stress. The transition from endothelial cells to mesenchymal cells (e.g., fibroblasts and smooth muscle cells) is accompanied by downregulation of endothelial markers and upregulation of mesenchymal markers.
In this Review, we focus on the relationship between EndMT and progression of atherosclerosis to help to communicate relevant knowledge for atherosclerosis prevention. We discuss the effects of EndMT and the mechanisms underlying this process in the context of atherosclerosis.
Triggering EndMT Factors in Atherosclerosis
The endothelium plays an important role in withstanding risk factors against atherosclerosis. Various conditions including sustained inflammation, fluid shear stress, ox-LDL, and smoking can facilitate EndMT (15, 16). Sustained inflammation is a typical pathologic feature of atherosclerosis (17). Inflammatory cytokines including interleukin-1beta (IL-1β), tumor necrosis factor alpha (TNF-α), transforming growth factor-beta (TGF-β), and interferon-gamma (IFN-γ) induce endothelial dysfunction and the acquisition of mesenchymal properties, therefore contributing to atherosclerosis (18–20). Long-term exposure to inflammatory cytokines can induce EndMT by altering the morphology of endothelial cells and EndMT-associated markers (21). In addition to the alteration of expression of endothelial/mesenchymal markers, inflammatory cytokines can also activate the TGF-β pathway and the non-TGF-β pathways (19).
Disturbed blood flow is another pathological characteristic of atherosclerosis. Zhou et al. showed that pulsatile shear stress (PS) exerts an atheroprotective role through the maintenance of endothelial cell homeostasis. In contrast, oscillatory shear stress (OS) causes endothelial cell dysfunction (22). Prior literature has also shown that EndMT can be induced by shear stress (23, 24). In addition, data from next-generation sequencing suggested that PS and OS lead to opposite effects in regulating EndMT gene regulation (25). Notably, expression of the twist family basic helix-loop-helix transcription factor (TWIST) expression was observed under conditions of low shear stress in regions of adult arteries (26). (27) demonstrated that low shear stress can result in the depletion of teneleven-translocation 2 (TET2). Overexpression of TET2 alleviated EndMT in atherosclerosis. Other risk factors associated with atherosclerosis can also lead to EndMT. Oxidized low density lipoprotein (ox-LDL) upregulates EndMT transcriptional factor Snail in human aortic endothelial cells (HAECs), in a on ox-LDLreceptor-dependent manner (LOX-1) (28). Moreover, ox-LDL induces accumulation of reactive oxygen species (ROS) accumulation in cells and synergistically promotes radiation-induced EndMT (29, 30). The effect of ox-LDL on EndMT can be inhibited by vaccarin (31) and naringin (32). Smoking is another known essential risk factor of atherosclerosis. Nicotine treated ApoE−/− mice and human aortic endothelial cells (HAECs) presented with mesenchymal phenotypes, and blocking the nicotine receptor alleviated nicotine-induced EndMT and lesions in mice (33).
EndMT is Related To Endothelial Cell Plasticity
EndMT is a hallmark of endothelial plasticity. A detailed Review by William C. Aird previously discussed the phenotypic heterogeneity of the endothelium. In the embryonic period, endothelial cell differentiation plays an important role in organ development (34). Endothelial cells differentiate to meet the needs of organ development. For example, multiple lineage hematopoietic progenitors are derived from hemogenic endothelial cells via process endothelial-to-hematopoietic transition (EHT) (35). EndMT is essential for heart development. Heart endothelial cells give rise to cardiac fibroblasts and smooth muscle cells, contributing to the formation of the endocardial cushion formation (36). In the adult, disease can occur when homeostasis of endothelial cells is disrupted and these cells remain in an EndMT state.
In atherosclerosis, endothelial cells are exposed to various biochemical and physical stimuli derived from the circulating blood. Low-density lipoprotein (LDL), cholesterol, and wall shear stress may induce EndMT and disrupt endothelial cell homeostasis, leading to endothelial dysfunction and thereby contributing to the development of atherosclerosis. In addition, it has been well documented that inflammatory factors such as IL-1β, TNF-α, TGF-β, and endotoxins can induce endothelial dysfunction via EndMT. In inflammation conditions, activation of endothelial cells occurs, leading to phenotypic and molecular changes (16, 37–39). Endothelial cells are heterogeneous and show a high degree of plasticity in both normal and atheromatous conditions. (40) defined eight endothelial cell clusters in the heart and aorta of patients with diabetic atherosclerosis at single cell level, three of which expressed mesenchymal markers, indicating EndMT markers. Further analysis suggested that the proportion of EndMT-derived fibroblast like cells was higher in atherosclerosis group compared to the normal group, with alterations in extracellular-matrix organization, adhesion, and apoptosis.
EndMT Involves Atherosclerotic Plaque Formation and Instability
The presence of endothelial cell-derived mesenchymal like cells in plaques provides robust evidence of the involvement of EndMT in atherosclerosis. Previous studies demonstrated that atherosclerotic plaques contain mesenchymal cells (including fibroblasts and smooth muscle cells), which regulate inflammation, extra-cellular matrix and collagen production, and plaque structural integrity and play a key functional role in atherosclerosis (41, 42). To investigate the origins of atherosclerosis-associated fibroblasts, (43) examined and confirmed the presence of EndMT-derived fibroblast-like cells present in atherosclerotic lesions through lineage-tracking, suggesting a role of EndMT atherosclerosis development. Matrix metalloproteinases (MMPs) are associated with unstable atherosclerotic lesions (44, 45), and EndMT-derived fibroblast-like cells express higher levels of MMP1, MMP9, and MMP10 compared with normal fibroblasts. In addition, TGF-β signaling, oxidative stress, and hypoxia facilitate endothelial cells conversion to mesenchymal cells and are all hallmarks of atherosclerosis. During EndMT process, active endothelial cells express adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), which enhance monocyte, leukocyte, and macrophage recruitment and infiltration (46). Interestingly, (47) found evidence for a crosstalk between macrophages and EndMT: macrophages in atherosclerotic lesions in vivo upregulate the expression of mesothelial markers, promoting EndMT, and, conversely, EndMT cells impact the function, such as the capacity of lipid uptake, and phenotypes of macrophages. In addition to in vivo and in vitro studies, single cell sequencing technology has been helpful to further our understanding of the landscape and pathophysiology of human atherosclerotic plaques. Recently, a study identified 14 cell populations including endothelial cells, smooth muscle cells, mast cells, B cells, myeloid cells, and T cells and identified multiple cellular activation states in plaques (48). One of the identified subclasses of endothelial cells expressed the smooth muscle cell markers such as alpha-actin 2 (ACTA2), notch receptor 3 (NOTCH3), and myosin heavy chain 11 (MYH11), suggesting that this subtype was undergoing EndMT and providing additional evidence on plasticity of endothelial cell plasticity. Altogether, these findings confirm that EndMT is closely associated with plaque initiation and development.
Mechanisms Regulating EndMT in Atherosclerosis
A substantial body of research pinpoints to several features of atherosclerosis that can lead to EndMT via several signaling pathways (49, 50). The TGF-β signaling pathway is a canonical pathway modulating EndMT, which has been shown to crosstalk with other pathways including the fibroblast growth factor (FGF), Notch, and bone morphogenetic protein (BMP) pathways (Figure 2). In addition, non-coding RNAs also paly an essential role in EndMT. We next discuss the mechanisms underlying EndMT in the context of atherosclerosis.
Figure 2
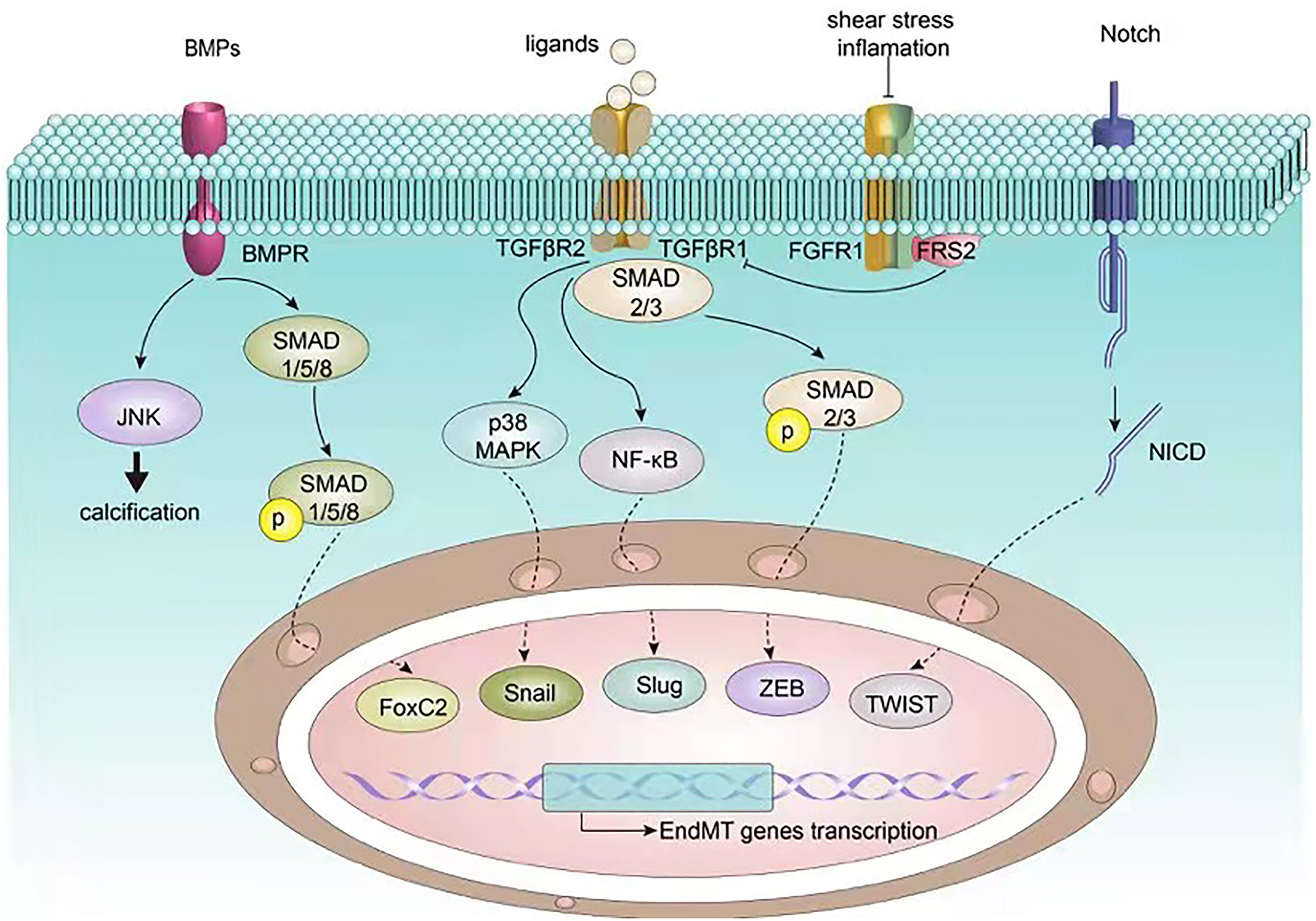
Signaling pathways involved in EndMT in atherosclerosis. EndMT related transcription factors such as Snail, Slug, ZEB, TWIST, and FoxC2 are governed by upstream signalings including BMP, TGF-β, FGF, and Notch, which in turn regulate endothelial and mesenchymal gene expression, ultimately inducing EndMT.
TGF-β Signaling Pathway
The role of TGF-β signaling pathway on EndMT has been extensively studied. In brief, the TGF-β family ligands bind to type I receptors and type II receptors, phosphorylating and thereby activating the transducer small mother against decapentaplegic (SMAD). Nuclear import of active SMAD transmits regulates gene transcription (51). In addition, TGF-β receptors can also activate other signaling pathways. Specifically, TGF-β signaling can be divided into SMAD-dependent and non-SMAD-dependent pathway (52). Growing evidence demonstrated the role of TGF-β signaling pathway in regulating cell proliferation, differentiation, adhesion, migration, and apoptosis not only in both embryonic development and the pathology of human disease (53). (14) showed that risk factors of atherosclerosis such as oscillatory shear stress and inflammation-induced loss of fibroblast growth factor receptor 1 (FGFR1) expression can activate TGF-β signaling and contribute to EndMT. FGFR1 depletion induces EndMT by upregulation of smooth muscle markers and mesenchymal markers which are also targets of TGF-β (54). Although the crucial role of TGF-β in EndMT has been well established, the different roles of the various isoforms of TGF-β are less known. To this aim, Sabbineni et al. (55) compared the effect of three isoforms of TGF-β (TGF-β1–3) on EndMT and found that TGFβ2 was the one most associated with EndMT. In addition, TGFβ2 is required for epithelial-to-mesenchymal cell transformation during endocardial cushion (56). Experiments treating human microvascular endothelial cells (HMECs) with different TGFβ isoforms indicated that only TGF-β2 substantially increased Smad2/3, p38 mitogen-activated protein kinase (MAPK), and mesenchymal transcription factors Snail and forkhead box protein C2 (Foxc2). Furthermore, TGF-β2 and IL-1β co-stimulate EndMT to activate nuclear factor-kappaB (NF-κB) (19).
Given the critical role of TGF-β in regulating EndMT, TGF-β inhibition may be an approach to reverse EndMT. Knockout of endothelium-specific TGF-β receptor relieved EndMT and kidney fibrosis (57). However, TGF-β exhibits potent regulatory functions, and inhibition of this pathway might also result in unwanted side effects.
Altogether, this evidence suggests that TGF-β may also be a promising therapeutic target for atherosclerosis, and further studies to confirm this hypothesis and assess safety are needed.
BMP Signaling Pathway
Bone morphogenetic proteins (BMPs) belong to TGF-β signaling pathway superfamily. The crosstalk of TGF-β/BMP signaling is well understood (58). More than 20 BMPs with different functions have been identified so far (59). The BMP ligand-receptor interaction induces SMAD1/5/8 phosphorylation to mediate downstream signaling (60). BMPs bind to two different receptors to mediate signal transduction through SMAD-dependent and SMAD-independent pathways (61). (62) showed down regulation of BMP type II receptor BMPR2 in pro-inflammatory-induced EndMT, with enhanced BMP-9-induced osteogenic differentiation, which leads to a decrease in c-Jun N-terminal kinase (JNK) signaling, thus contributing to calcification. Similarly, another study suggested a protective role for BMPR2 in endothelial cell homeostasis, in particular by balancing BMP/TGF-β signaling to protect cells from increased responses toward TGF-β (63). In addition to BMPR2, BMP6 also has the ability to induce osteogenic differentiation and mineralization consistent with EndMT. Activation of reactive oxygen species (ROS) is required for BMP6 to regulate osteogenic genes, osteogenic differentiation, and calcification (64). Furthermore, (29) found that brain and muscle ARNT-like protein-1 (BMAL1) suppressed ROS-induced EndMT through BMP signaling, therefore inhibiting atherosclerosis plaque progression.
Notch Signaling Pathway
Notch signaling pathway includes 4 receptors (Notch1–4) and 5 ligands (Delta-like-1, 3, 4, and Jagged1–2) (65). Notch receptor-ligand binding generates Notch intracellular domain (NICD) which is translocated to the nucleus where NICD binds to the transcription factor RBPJK/CSL to regulate target gene expression and thus cell fate specification (66). Within the cardiovascular system, Notch signaling is implicated in both development, such as cardiac valve formation, and pathological process, such as response to vascular injury (67, 68). It was observed that Notch receptors and ligands are expressed in the vasculature (69). Notch activated endothelial cells show the characteristics of EndMT including downregulation of endothelial markers and upregulation of mesenchymal markers (70). In addition, Notch activation induces Slug overexpression in endothelial cells which is associated with a loss of the endothelial phenotype (71). Notch signaling contributes to EndMT independently of or synergistically with TGF-β. TGF-β1 induces upregulation of several Notch components including Jagged-1, the receptor Notch-1, N1ICD, recombination signal binding protein J kappa (RBPJK), as well as target genes hairy enhancer of split-1 (Hes-1) and Hes-5(72). Activation of Notch signaling pathway in vitro induced EndMT by increasing the expression of vascular endothelial (VE)-cadherin and overexpression of α-SMA, whereas inhibition of Notch signaling pathway with gamma-secretase inhibitors (GSI) attenuated the development of atherosclerotic lesions (73).
Non-coding RNAs
Non-coding RNAs including microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs) are involved in the regulation the process of EndMT. (74) have provided a comprehensive description of the non-coding RNAs known to be involved in EndMT regulation. In this Review, we focus on non-coding RNAs influencing EndMT and atherosclerosis progression (Table 1). MiRNAs are a class of non-coding RNAs with a length of 20–40 nucleotides, which suppress target mRNAs by binding to their 3'-UTR (75). miR-449a was highly expressed in ApoE−/− diabetic mice and modulated EndMT by increasing the expression of mesenchymal cell markers and reducing E-cadherin which interacts with adiponectin receptor 2 (AdipoR2) in lipid rafts. ApoE-/- diabetic mice treated with an antagonist of miR-449a showed reduction of atherosclerotic lesions (76). miR-374b induces EndMT by targeting mitogen-activated protein kinase 7 (MAPK7), which is decreased in the atheroprone hyperplastic regions and is known to be inhibit EndMT. In the TGF-β treated endothelial cells, the increased level of miR-374b was counteracted by an inhibitor of ALK5 (SB431542). In addition, silencing miR-374b targets by means of short hairpin RNAs (shRNAs) specifically decreased MAPK7 signaling members, and increased expression of endothelial markers VE-cadherin and endothelial nitric oxide synthase (eNOS), and of the mesenchymal markers SM22α and Calponin (77). In a recent study, high expression of miR-122 was observed in both ApoE−/− mice and in vitro EndMT models, and miR-122 inhibition in ApoE-/- mice reduced the progression of plaque formation. Similarly, inhibiting miR-122 could reverse the EndMT phenotype induced by H2O2, whereas, silencing of the miR-122 target neuronal PAS domain protein 3 (NPAS3) gene abolished EndMT reversal. Therefore, this study suggests that miR-122 promotes plaque formation via NPAS3 mediated EndMT and could be a new therapeutic target in atherosclerosis (78).
Table 1
| Non-coding RNAs | Targets | Regulation | Models | Year | Reference |
|---|---|---|---|---|---|
| miR-449a | AdipoR2 | upregulation | ApoE−/− diabetic mice Human carotid atherosclerotic plaque HUVECs |
2019 | (76) |
| miR-374b | MAPK7 | upregulation | Human coronary arteries Pigs HUVECs |
2019 | (77) |
| miR-122 | NPAS3 | upregulation | ApoE−/− mice HAECs HUVECs |
2021 | (78) |
| H19 | TET1/TGFβR2/TSP1 | upregulation | H19 KO mice Primary mouse pulmonary endothelial cells HAECs HUVECs |
2010 | (83) |
| MALAT1 | Wnt/β-catein | upregulation | ApoE−/− mice HUVECs |
2019 | (85) |
| LIC00657 | miR-30c-5p/Wnt7b/β-catenin | upregulation | Atherosclerosis patients serum HUVECs |
2020 | (86) |
| LncRNA ZFAS1 | miR-150-5P/Notch3 | upregulation | ApoE−/− mice HUVECs | 2021 | (89) |
Non-coding RNAs regulating EndMT in atherosclerosis.
List of non-coding RNAs regulating EndMT associated with atherosclerosis including miRNA and lncRNAs. AdipoR2, Adiponectin receptor 2; MAPK7, Mitogen-activated protein kinase 7; NPAS3, Neuronal PAS Domain Protein 3; TET1, Tet Methylcytosine Dioxygenase 1; TGFβR2, TGF-βreceptor 2; TSP1, Thrombospondin 1; MALAT1, Metastasis-associated lung adenocarcinoma transcript 1; HUVECs, Human umbilical vein endothelial cells; HAECs, Human Aortic Endothelial Cells.
LncRNAs are non-coding RNAs with a length of more than 200 nucleotides that have recently emerged as important regulators in development and disease (79). So far, although more than 5000 lncRNAs have been identified (80), few of them have been implicated in EndMT. LncRNA H19 is increased in aortic tissues and is associated to the extent of cardiovascular disease in a model of atherosclerosis (81, 82). H19 upregulates TGF-β receptor 2 (TGFβR2) and thrombospondin 1 (TSP1) via let-7/TET1, and therefore has the potential to model EndMT markers including Slug, SM22-α, Vimentin, and fibronectin1 (FN1) (83). LncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) has been shown to play a critical role in cardiovascular disease (84). Li et al. (85) observed an increasing expression of MALAT1 in atherosclerotic mice and human umbilical vein endothelial cells (HUVECs) treated with ox-LDL which also showed CD31 and vWF downregulation and α-SMA and vimentin overexpression. MALAT1 promotes β-catenin translocation to nuclear translocation and enhances EndMT induced by ox-LDL in a MALAT1/Wnt/β-catenin dependent manner. Interestingly, LINC00657 was found to be overexpressed in the serum of atherosclerosis patients and in HUVECs treated with ox-LDL. LINC00657 has a similar effect on inducing EndMT. LINC00657 promotes EndMT by acting as a sponge for miRNA-30c-5p and by positively regulating Wnt7b/β-catenin activation (86). LncRNA ZFAS1 is another contributor of atherosclerosis (87, 88). Results from in vivo and in vitro atherosclerosis models suggested that ZFAS1 triggered EndMT via inhibition of miR-150-5p, hence increasing the expression of Notch3, an active regulator of EndMT (89). Currently, the function of lncRNAs and circRNAs in regulating EndMT in atherosclerosis is largely unknown. CircRNAs associated with EndMT have been reported in many diseases including neuro inflammatory disorders (90), bladder carcinoma (91), pulmonary disease (92), and ischaemic stroke (93). Additional research to clarify the role of lncRNAs and circRNAs in disease is needed.
Preventing EndMT as a Potential Approach to Treat Atherosclerosis
Given the role of EndMT in modulating atherosclerosis, disruption of the EndMT might be a therapeutic option for treating atherosclerosis. Indeed, some compounds and clinical drugs may have protective effect on atherosclerosis by inhibiting EndMT. RGFP966 is an inhibitor of histone deacetylase 3 (HDAC3), an important regulator of cardiovascular diseases which was found to be upregulated in atherosclerotic lesions (94), and can reduce atherosclerotic lesions by inhibiting EndMT in the aortic root (95). Icariin, a compound derived from Epimedium, inhibited ox-LDL-induced EndMT via H19/miR-148b-3p/ELF5 (E74-like factor 5). Icariin induced H19 overexpression and led to an attenuation of the EndMT process, exerting a protective effect in atherosclerosis (96). Recently, simvastatin, a clinical lipid-lowering drug, was shown to inhibit EndMT (25). A study by (25) has shown that simvastatin can inhibit EndMT via upregulation of KLF4/miR-483 axis in HUVECs. Additionally, simvastatin attenuated 1-Palmitoyl-2-(5-oxovaleroyl)-sn-glycero-3-phosphocholine (POVC) inducing EndMT, by suppressing oxidative stress and TGF-β/SMAD signaling, suggesting that simvastatin could potentially be used in treating atherosclerosis (97). Altogether, these findings suggest the therapeutic potential of EndMT disruption in atherosclerosis. EndMT is a complex process resulting from the action of many factors, ranging from signaling pathways to non-coding RNAs. However, effective drugs to reverse EndMT are still lacking. Moving forward, single-cell/high-throughput sequencing technology might provide helpful insights to uncover EndMT associated targets for the treatment of atherosclerosis.
Highlighting the Progression of EndMT in Atherosclerosis
The studies by Chen et al. (14) and Evrard et al. (43) provide strong evidence for the involvement of EndMT in atherosclerosis. EndMT drives the initiation of atherosclerosis by accelerating plaque growth and instability of plaque. Previous research has shown that EndMT is regulated in a number of signaling pathways, in particular the canonical TGF-β signaling, and several atherosclerosis risk factors, e.g., inflammation, shear stress, ox-LDL, and smoking. Conversely, FGF signaling plays a protective role on EndMT. Remarkably, several non-coding RNAs were reported to modulating EndMT, offering therapeutic application to treat atherosclerosis (74). Epigenetic mechanisms involved in EndMT regulation are poorly understood. Histone deacetylases (HDACs) play a role in EndMT, in particular HDAC3 and HDAC9, and promoted atherosclerosis progression (98). Overall, the molecular mechanisms underlying EndMT remain largely unknown, and additional research is needed to discover new targets that can be explored in reverse atherosclerosis.
Conclusions
EndMT is associated with the formation of atherosclerotic lesions. Therefore, reversing or inhibiting EndMT might help to prevent the development or progression of atherosclerosis. In vivo, suppression of EndMT shows promising effects in alleviating atherosclerosis. However, animal and cell models of atherosclerosis present many limitations, and the study and detection of EndMT in humans offer great challenges. More research is needed to understand the role of EndMT in atherosclerosis to ultimately offer new insights for the treatment of atherosclerosis.
Funding
This study was supported by the Guangdong Provincial Key Laboratory of Precision Medicine and Clinical Translation Research of Hakka Population under Grant 2018B030322003; the Science and Technology Program of Meizhou under Grant 2019B0202001.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Author contributions
QH, HW, and ZZ contributed to the conception of the study, performed the literature search, and wrote the manuscript. QH, YG, and ZZ edited the manuscript. ZY and YG assisted in the literature search and critically revised the article for important intellectual content. All authors have read and approved the final manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
Libby P Buring JE Badimon L Hansson GK Deanfield J Bittencourt MS et al . Atherosclerosis. Nat Rev Dis Primers. (2019) 5:56. 10.1038/s41572-019-0106-z
2.
Zhu Y Xian X Wang Z Bi Y Chen Q Han X et al . Research progress on the relationship between atherosclerosis and inflammation. Biomolecules. (2018) 8:80. 10.3390/biom8030080
3.
Ahotupa M . Oxidized lipoprotein lipids and atherosclerosis. Free Radic Res. (2017) 51:439–47. 10.1080/10715762.2017.1319944
4.
Gistera A Hansson GK . The immunology of atherosclerosis. Nat Rev Nephrol. (2017) 13:368–80. 10.1038/nrneph.2017.51
5.
Wesseling M Sakkers TR de Jager SCA Pasterkamp G Goumans MJ . The morphological and molecular mechanisms of epithelial/endothelial-to-mesenchymal transition and its involvement in atherosclerosis. Vascul Pharmacol. (2018) 106:1–8. 10.1016/j.vph.2018.02.006
6.
Moonen JR Krenning G Brinker MG Koerts JA van Luyn MJ Harmsen MC . Endothelial progenitor cells give rise to pro-angiogenic smooth muscle-like progeny. Cardiovasc Res. (2010) 86:506–15. 10.1093/cvr/cvq012
7.
Krenning G Moonen JR van Luyn MJ Harmsen MC . Vascular smooth muscle cells for use in vascular tissue engineering obtained by endothelial-to-mesenchymal transdifferentiation (EnMT) on collagen matrices. Biomaterials. (2008) 29:3703–11. 10.1016/j.biomaterials.2008.05.034
8.
Li Y Lui KO Zhou B . Reassessing endothelial-to-mesenchymal transition in cardiovascular diseases. Nat Rev Cardiol. (2018) 15:445–56. 10.1038/s41569-018-0023-y
9.
Markwald RR Fitzharris TP Manasek FJ . Structural development of endocardial cushions. Am J Anat. (1977) 148:85–119. 10.1002/aja.1001480108
10.
Ranchoux B Antigny F Rucker-Martin C Hautefort A Pechoux C Bogaard HJ et al . Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation. (2015) 131:1006–18. 10.1161/CIRCULATIONAHA.114.008750
11.
Li Z Chen B Dong W Kong M Fan Z Yu L et al . MKL1 promotes endothelial-to-mesenchymal transition and liver fibrosis by activating TWIST1 transcription. Cell Death Dis. (2019) 10:899. 10.1038/s41419-019-2101-4
12.
Jiao K Zhen J Wu M Teng M Yang K Zhou Q et al . 27-Hydroxycholesterol-induced EndMT acts via STAT3 signaling to promote breast cancer cell migration by altering the tumor microenvironment. Cancer Biol Med. (2020) 17:88–100. 10.20892/j.issn.2095-3941.2019.0262
13.
Ubil E Duan J Pillai IC Rosa-Garrido M Wu Y Bargiacchi F et al . Mesenchymal-endothelial transition contributes to cardiac neovascularization. Nature. (2014) 514:585–90. 10.1038/nature13839
14.
Chen PY Qin L Baeyens N Li G Afolabi T Budatha M et al . Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest. (2015) 125:4514–28. 10.1172/JCI82719
15.
Chen PY Schwartz MA Simons M . Endothelial-to-mesenchymal transition, vascular inflammation, and atherosclerosis. Front Cardiovasc Med. (2020) 7:53. 10.3389/fcvm.2020.00053
16.
Cho JG Lee A Chang W Lee MS Kim J . Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front Immunol. (2018) 9:294. 10.3389/fimmu.2018.00294
17.
Geovanini GR Libby P . Atherosclerosis and inflammation: overview and updates. Clin Sci. (2018) 132:1243–52. 10.1042/CS20180306
18.
Chrobak I Lenna S Stawski L Trojanowska M . Interferon-gamma promotes vascular remodeling in human microvascular endothelial cells by upregulating endothelin (ET)-1 and transforming growth factor (TGF) beta2. J Cell Physiol. (2013) 228:1774–83. 10.1002/jcp.24337
19.
Maleszewska M Moonen JR Huijkman N van de Sluis B Krenning G Harmsen MC . IL-1beta and TGFbeta2 synergistically induce endothelial to mesenchymal transition in an NFkappaB-dependent manner. Immunobiology. (2013) 218:443–54. 10.1016/j.imbio.2012.05.026
20.
Yoshimatsu Y Wakabayashi I Kimuro S Takahashi N Takahashi K Kobayashi M et al . TNF-alpha enhances TGF-beta-induced endothelial-to-mesenchymal transition via TGF-beta signal augmentation. Cancer Sci. (2020) 111:2385–99. 10.1111/cas.14455
21.
Chen PY Qin L Barnes C Charisse K Yi T Zhang X et al . FGF regulates TGF-beta signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep. (2012) 2:1684–96. 10.1016/j.celrep.2012.10.021
22.
Zhou J Li YS Chien S . Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler Thromb Vasc Biol. (2014) 34:2191–8. 10.1161/ATVBAHA.114.303422
23.
Moonen JRAJ Lee ES Schmidt M Maleszewska M Koerts JA Brouwer LA et al . Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc Res. (2015) 108:377–86. 10.1093/cvr/cvv175
24.
Mahmoud MM Serbanovic-Canic J Feng S Souilhol C Xing R Hsiao S et al . Shear stress induces endothelial-to-mesenchymal transition via the transcription factor Snail. Sci Rep. (2017) 7:3870. 10.1038/s41598-017-03532-z
25.
Lai B Li Z He M Wang Y Chen L Zhang J et al . Atheroprone flow enhances the endothelial-to-mesenchymal transition. Am J Physiol Heart Circ Physiol. (2018) 315:H1293–303. 10.1152/ajpheart.00213.2018
26.
Mahmoud MM Kim HR Xing R Hsiao S Mammoto A Chen J et al . TWIST1 integrates endothelial responses to flow in vascular dysfunction and atherosclerosis. Circ Res. (2016) 119:450–62. 10.1161/CIRCRESAHA.116.308870
27.
Li A Tan L Zhang S Tao J Wang Z Wei D . Low shear stress-induced endothelial mesenchymal transformation via the down-regulation of TET2. Biochem Biophys Res Commun. (2021) 545:20–6. 10.1016/j.bbrc.2021.01.062
28.
Su Q Sun Y Ye Z Yang H Li L . Oxidized low density lipoprotein induces endothelial-to-mesenchymal transition by stabilizing snail in human aortic endothelial cells. Biomed Pharmacother. (2018) 106:1720–6. 10.1016/j.biopha.2018.07.122
29.
Zhu M Tang H Tang X Ma X Guo D Chen F . BMAL1 suppresses ROS-induced endothelial-to-mesenchymal transition and atherosclerosis plaque progression via BMP signaling. Am J Transl Res. (2018) 10:3150–61.
30.
Kim M Choi SH Jin YB Lee HJ Ji YH Kim J et al . The effect of oxidized low-density lipoprotein (ox-LDL) on radiation-induced endothelial-to-mesenchymal transition. Int J Radiat Biol. (2013) 89:356–63. 10.3109/09553002.2013.763193
31.
Gong L Liu Y Tan F Li S Wang X Xu M et al . Vaccarin prevents ox-LDL-induced HUVEEndMT C, inflammation and apoptosis by suppressing ROS/p38 MAPK signaling. Am J Transl Res. (2019) 11:2140–54.
32.
Zhao H Liu M Liu H Suo R Lu C . Naringin protects endothelial cells from apoptosis and inflammation by regulating the Hippo-YAP Pathway. Biosci Rep. (2020) 40:BSR20193431. 10.1042/BSR20193431
33.
Qin W Zhang L Li Z Xiao D Zhang Y Zhang H et al . Endothelial to mesenchymal transition contributes to nicotine-induced atherosclerosis. Theranostics. (2020) 10:5276–89. 10.7150/thno.42470
34.
Aird WC . Phenotypic heterogeneity of the endothelium: I. structure, function, and mechanisms. Circ Res. (2007) 100:158–73. 10.1161/01.RES.0000255691.76142.4a
35.
Gritz E Hirschi KK . Specification and function of hemogenic endothelium during embryogenesis. Cell Mol Life Sci. (2016) 73:1547–67. 10.1007/s00018-016-2134-0
36.
Eisenberg MRLM . Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res. (1995) 77:1–6. 10.1161/01.RES.77.1.1
37.
Rieder F Kessler SP West GA Bhilocha S de la Motte C Sadler TM et al . Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J Pathol. (2011) 179:2660–73. 10.1016/j.ajpath.2011.07.042
38.
Perez L Munoz-Durango N Riedel CA Echeverria C Kalergis AM Cabello-Verrugio C et al . Endothelial-to-mesenchymal transition: cytokine-mediated pathways that determine endothelial fibrosis under inflammatory conditions. Cytokine Growth Factor Rev. (2017) 33:41–54. 10.1016/j.cytogfr.2016.09.002
39.
Good RB Gilbane AJ Trinder SL Denton CP Coghlan G Abraham DJ et al . Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary arterial hypertension. Am J Pathol. (2015) 185:1850–8. 10.1016/j.ajpath.2015.03.019
40.
Zhao G Lu H Liu Y Zhao Y Zhu T Garcia-Barrio MT et al . Single-cell transcriptomics reveals endothelial plasticity during diabetic atherogenesis. Front Cell Dev Biol. (2021) 9:689469. 10.3389/fcell.2021.689469
41.
Brokopp CE Schoenauer R Richards P Bauer S Lohmann C Emmert MY et al . Fibroblast activation protein is induced by inflammation and degrades type I collagen in thin-cap fibroatheromata. Eur Heart J. (2011) 32:2713–22. 10.1093/eurheartj/ehq519
42.
Martínez-González J Berrozpe M Varela O Badimon L . Heterogeneity of smooth muscle cells in advanced human atherosclerotic plaques: intimal smooth muscle cells expressing a fibroblast surface protein are highly activated by platelet-released products. Eur J Clin Invest. (2001) 31:939–49. 10.1046/j.1365-2362.2001.00920.x
43.
Evrard SM Lecce L Michelis KC Nomura-Kitabayashi A Pandey G Purushothaman KR et al . Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. (2016) 7:11853. 10.1038/ncomms11853
44.
Schneider F Sukhova GK Aikawa M Canner J Gerdes N Tang SM et al . Matrix-metalloproteinase-14 deficiency in bone-marrow-derived cells promotes collagen accumulation in mouse atherosclerotic plaques. Circulation. (2008) 117:931–9. 10.1161/CIRCULATIONAHA.107.707448
45.
Furman C Copin C Kandoussi M Davidson R Moreau M McTaggiart F et al . Rosuvastatin reduces MMP-7 secretion by human monocyte-derived macrophages: potential relevance to atherosclerotic plaque stability. Atherosclerosis. (2004) 174:93–8. 10.1016/j.atherosclerosis.2004.01.009
46.
Davignon J Ganz P . Role of endothelial dysfunction in atherosclerosis. Circulation. (2004) 109:III27–32. 10.1161/01.CIR.0000131515.03336.f8
47.
Helmke A Casper J Nordlohne J David S Haller H Zeisberg EM et al . Endothelial-to-mesenchymal transition shapes the atherosclerotic plaque and modulates macrophage function. FASEB J. (2019) 33:2278–89. 10.1096/fj.201801238R
48.
Depuydt MAC Prange KHM Slenders L Ord T Elbersen D Boltjes A et al . Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ Res. (2020) 127:1437–55. 10.1161/CIRCRESAHA.120.316770
49.
Li A Peng W Xia X Li R Wang Y Wei D . Endothelial-to-mesenchymal transition: a potential mechanism for atherosclerosis plaque progression and destabilization. DNA Cell Biol. (2017) 36:883–91. 10.1089/dna.2017.3779
50.
Souilhol C Harmsen MC Evans PC Krenning G . Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc Res. (2018) 114:565–77. 10.1093/cvr/cvx253
51.
Hata A Chen YG . TGF-beta Signaling from receptors to smads. Cold Spring Harb Perspect Biol. (2016) 8:a022061. 10.1101/cshperspect.a022061
52.
Massague J . TGFbeta signalling in context. Nat Rev Mol Cell Biol. (2012) 13:616–30. 10.1038/nrm3434
53.
Jia S Meng A . TGFbeta family signaling and development. Development. (2021) 148:dev188490. 10.1242/dev.188490
54.
Chen PY Qin L Tellides G Simons M . Fibroblast growth factor receptor 1 is a key inhibitor of TGFβ signaling in the endothelium. Sci Signal. (2014) 7:ra90. 10.1126/scisignal.2005504
55.
Sabbineni H Verma A Somanath PR . Isoform-specific effects of transforming growth factor beta on endothelial-to-mesenchymal transition. J Cell Physiol. (2018) 233:8418–28. 10.1002/jcp.26801
56.
Camenisch TD Molin DG Person A Runyan RB Gittenberger-de Groot AC McDonald JA et al . Temporal and distinct TGFbeta ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev Biol. (2002) 248:170–81. 10.1006/dbio.2002.0731
57.
Xavier S Vasko R Matsumoto K Zullo JA Chen R Maizel J et al . Curtailing endothelial TGF-beta signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in CKD. J Am Soc Nephrol. (2015) 26:817–29. 10.1681/ASN.2013101137
58.
Hiepen C Mendez PL Knaus P . It takes two to tango: endothelial TGFbeta/BMP signaling crosstalk with mechanobiology. Cells. (2020) 9:1965. 10.3390/cells9091965
59.
Sampath TK Reddi AH . Discovery of bone morphogenetic proteins - a historical perspective. Bone. (2020) 140:115548. 10.1016/j.bone.2020.115548
60.
Katagiri T Watabe T . Bone morphogenetic proteins. Cold Spring Harb Perspect Biol. (2016) 8:a021899. 10.1101/cshperspect.a021899
61.
Miyazono K Maeda S Imamura T . BMP receptor signaling: transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine Growth Factor Rev. (2005) 16:251–63. 10.1016/j.cytogfr.2005.01.009
62.
Sanchez-Duffhues G Garcia de Vinuesa A van de Pol V Geerts ME de Vries MR Janson SG et al . Inflammation induces endothelial-to-mesenchymal transition and promotes vascular calcification through downregulation of BMPR2. J Pathol. (2019) 247:333–46. 10.1002/path.5193
63.
Hiepen JJC Hildebrandt S Kampfrath B Goktas M Murgai A Cuellar Camacho JL et al . BMPR2 acts as a gatekeeper to protect endothelial cells from increased TGFβ responses and altered cell mechanics. PLoS Biol. (2019) 17:e3000557. 10.1371/journal.pbio.3000557
64.
Yung LM Sanchez-Duffhues G Ten Dijke P Yu PB . Bone morphogenetic protein 6 and oxidized low-density lipoprotein synergistically recruit osteogenic differentiation in endothelial cells. Cardiovasc Res. (2015) 108:278–87. 10.1093/cvr/cvv221
65.
Bray FMS . Notch pathway: making sense of suppressor of hairless. Curr Biol. (2001) 11:R217–21. 10.1016/S0960-9822(01)00109-9
66.
Kopan R Ilagan MX . The canonical notch signaling pathway: unfolding the activation mechanism. Cell. (2009) 137:216–33. 10.1016/j.cell.2009.03.045
67.
Luna-Zurita L Prados B Grego-Bessa J Luxan G del Monte G Benguria A et al . Integration of a Notch-dependent mesenchymal gene program and Bmp2-driven cell invasiveness regulates murine cardiac valve formation. J Clin Invest. (2010) 120:3493–507. 10.1172/JCI42666
68.
Lindner BCV Prudovsky I Small D Maciag T Liaw L . Members of the jagged/notch gene families are expressed in injured arteries and regulate cell phenotype via alterations in cell matrix and cell-cell interaction. Am J Pathol. (2001) 159:875–83. 10.1016/S0002-9440(10)61763-4
69.
Iso T Hamamori Y Kedes L . Notch signaling in vascular development. Arterioscler Thromb Vasc Biol. (2003) 23:543–53. 10.1161/01.ATV.0000060892.81529.8F
70.
Noseda M McLean G Niessen K Chang L Pollet I Montpetit R et al . Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ Res. (2004) 94:910–7. 10.1161/01.RES.0000124300.76171.C9
71.
Niessen K Fu Y Chang L Hoodless PA McFadden D Karsan A . Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J Cell Biol. (2008) 182:315–25. 10.1083/jcb.200710067
72.
Tian D Zeng X Wang W Wang Z Zhang Y Wang Y . Protective effect of rapamycin on endothelial-to-mesenchymal transition in HUVECs through the Notch signaling pathway. Vascul Pharmacol. (2019) 113:20–6. 10.1016/j.vph.2018.10.004
73.
Lin QQ Zhao J Zheng CG Chun J . Roles of notch signaling pathway and endothelial-mesenchymal transition in vascular endothelial dysfunction and atherosclerosis. Eur Rev Med Pharmacol Sci. (2018) 22:6485–91. 10.26355/eurrev_201810_16062
74.
Hulshoff MS Del Monte-Nieto G Kovacic J Krenning G . Non-coding RNA in endothelial-to-mesenchymal transition. Cardiovasc Res. (2019) 115:1716–31. 10.1093/cvr/cvz211
75.
Lee FR Ambros V . The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. (1993) 75:843–54. 10.1016/0092-8674(93)90529-Y
76.
Jiang L Hao C Li Z Zhang P Wang S Yang S et al . miR-449a induces EndMT, promotes the development of atherosclerosis by targeting the interaction between AdipoR2 and E-cadherin in lipid rafts. Biomed Pharmacother. (2019) 109:2293–304. 10.1016/j.biopha.2018.11.114
77.
Vanchin B Offringa E Friedrich J Brinker MG Kiers B Pereira AC et al . MicroRNA-374b induces endothelial-to-mesenchymal transition and early lesion formation through the inhibition of MAPK7 signaling. J Pathol. (2019) 247:456–70. 10.1002/path.5204
78.
Wu X Du X Yang Y Liu X Liu X Zhang N et al . Inhibition of miR-122 reduced atherosclerotic lesion formation by regulating NPAS3-mediated endothelial to mesenchymal transition. Life Sci. (2021) 265:118816. 10.1016/j.lfs.2020.118816
79.
Qian X Zhao J Yeung PY Zhang QC Kwok CK . Revealing lncRNA structures and interactions by sequencing-based approaches. Trends Biochem Sci. (2019) 44:33–52. 10.1016/j.tibs.2018.09.012
80.
Iyer MK Niknafs YS Malik R Singhal U Sahu A Hosono Y et al . The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. (2015) 47:199–208. 10.1038/ng.3192
81.
Yang Y Wei F Yang L Kuang C Zhang H Deng J et al . Silencing of long non-coding RNA H19 downregulates CTCF to protect against atherosclerosis by upregulating PKD1 expression in ApoE knockout mice. Aging. (2019) 11:10016–30. 10.18632/aging.102388
82.
Gao W Zhu M Wang H Zhao S Zhao D Yang Y et al . Association of polymorphisms in long non-coding RNA H19 with coronary artery disease risk in a Chinese population. Mutat Res. (2015) 772:15–22. 10.1016/j.mrfmmm.2014.12.009
83.
Cao T Jiang Y Li D Sun X Zhang Y Qin L et al . H19/TET1 axis promotes TGF-beta signaling linked to endothelial-to-mesenchymal transition. FASEB J. (2020) 34:8625–40. 10.1096/fj.202000073RRRRR
84.
Zhang X Hamblin MH Yin KJ . The long noncoding RNA Malat1: its physiological and pathophysiological functions. RNA Biol. (2017) 14:1705–14. 10.1080/15476286.2017.1358347
85.
Li H Zhao Q Chang L Wei C Bei H Yin Y et al . LncRNA MALAT1 modulates ox-LDL induced EndMT through the Wnt/beta-catenin signaling pathway. Lipids Health Dis. (2019) 18:62. 10.1186/s12944-019-1006-7
86.
Wu H Liu T Hou H . Knockdown of LINC00657 inhibits ox-LDL-induced endothelial cell injury by regulating miR-30c-5p/Wnt7b/beta-catenin. Mol Cell Biochem. (2020) 472:145–55. 10.1007/s11010-020-03793-9
87.
Carrizzo A Damato A Vecchione C . Long non-coding RNA-ZFAS1: a novel possible biomarker to monitor and hamper the atherosclerotic process?Int J Cardiol. (2020) 319:129–30. 10.1016/j.ijcard.2020.05.009
88.
Wang CH Shi HH Chen LH Li XL Cao GL Hu XF . Identification of key lncRNAs associated with atherosclerosis progression based on public datasets. Front Genet. (2019) 10:123. 10.3389/fgene.2019.00123
89.
Yin Q He M Huang L Zhang X Zhan J Hu J . lncRNA ZFAS1 promotes ox-LDL induced EndMT through miR-150-5p/Notch3 signaling axis. Microvasc Res. (2021) 134:104118. 10.1016/j.mvr.2020.104118
90.
Yang L Han B Zhang Y Bai Y Chao J Hu G et al . Engagement of circular RNA HECW2 in the nonautophagic role of ATG5 implicated in the endothelial-mesenchymal transition. Autophagy. (2018) 14:404–18. 10.1080/15548627.2017.1414755
91.
Zhong Z Huang M Lv M He Y Duan C Zhang L et al . Circular RNA MYLK as a competing endogenous RNA promotes bladder cancer progression through modulating VEGFA/VEGFR2 signaling pathway. Cancer Lett. (2017) 403:305–17. 10.1016/j.canlet.2017.06.027
92.
Fang S Guo H Cheng Y Zhou Z Zhang W Han B et al . circHECTD1 promotes the silica-induced pulmonary endothelial-mesenchymal transition via HECTD1. Cell Death Dis. (2018) 9:396. 10.1038/s41419-018-0432-1
93.
Bai Y Zhang Y Han B Yang L Chen X Huang R et al . Circular RNA DLGAP4 ameliorates ischemic stroke outcomes by targeting mir-143 to regulate endothelial-mesenchymal transition associated with blood-brain barrier integrity. J Neurosci. (2018) 38:32–50. 10.1523/JNEUROSCI.1348-17.2017
94.
Hoeksema MA Gijbels MJ Van den Bossche J van der Velden S Sijm A Neele AE et al . Targeting macrophage histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med. (2014) 6:1124–32. 10.15252/emmm.201404170
95.
Chen L Shang C Wang B Wang G Jin Z Yao F et al . HDAC3 inhibitor suppresses endothelial-to-mesenchymal transition via modulating inflammatory response in atherosclerosis. Biochem Pharmacol. (2021) 192:114716. 10.1016/j.bcp.2021.114716
96.
Liu S Xu DS Li M Zhang Y Li Q Li TT et al . Icariin attenuates endothelial-mesenchymal transition via H19/miR-148b-3p/ELF5 in ox-LDL-stimulated HUVECs. Mol Ther Nucleic Acids. (2021) 23:464–75. 10.1016/j.omtn.2020.11.021
97.
Li Y Zhang YX Ning DS Chen J Li SX Mo ZW et al . Simvastatin inhibits POVPC-mediated induction of endothelial to mesenchymal cell transition. J Lipid Res. (2021) 62:100066. 10.1016/j.jlr.2021.100066
98.
Lecce L Xu Y V'Gangula B Chandel N Pothula V Caudrillier A et al . Histone deacetylase 9 promotes endothelial-mesenchymal transition and an unfavorable atherosclerotic plaque phenotype. J Clin Invest. (2021) 131:e131178. 10.1172/JCI131178
Summary
Keywords
atherosclerosis, endothelial to mesenchymal transition, plasticity of endothelial cells, regulating mechanisms, EndMT-associated marker
Citation
Huang Q, Gan Y, Yu Z, Wu H and Zhong Z (2021) Endothelial to Mesenchymal Transition: An Insight in Atherosclerosis. Front. Cardiovasc. Med. 8:734550. doi: 10.3389/fcvm.2021.734550
Received
01 July 2021
Accepted
13 August 2021
Published
17 September 2021
Volume
8 - 2021
Edited by
Lina A. Shehadeh, University of Miami, United States
Reviewed by
Zhihua Wang, Chinese Academy of Medical Sciences and Peking Union Medical College, China; Prachi Umbarkar, University of Alabama at Birmingham, United States
Updates
Copyright
© 2021 Huang, Gan, Yu, Wu and Zhong.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heming Wu wuheming1986@126.comZhixiong Zhong zhongzhixiong01@126.com
†These authors have contributed equally to this work
This article was submitted to Cardiovascular Metabolism, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.