Abstract
Some studies have shown that sodium-glucose cotransporter (SGLT) 2 inhibitors can definitively attenuate the occurrence of cardiovascular diseases such as heart failure (HF), dilated cardiomyopathy (DCM), and myocardial infarction. With the development of research, SGLT2 inhibitors can also reduce the risk of arrhythmias. So in this review, how SGLT2 inhibitors play a role in reducing the risk of arrhythmia from the perspective of electrical remodeling and structural remodeling are explored and then the possible mechanisms are discussed. Specifically, we focus on the role of SGLT2 inhibitors in Na+ and Ca2 + homeostasis and the transients of Na+ and Ca2 +, which could affect electrical remodeling and then lead to arrythmia. We also discuss the protective role of SGLT2 inhibitors in structural remodeling from the perspective of fibrosis, inflammation, oxidative stress, and apoptosis. Ultimately, it is clear that SGLT2 inhibitors have significant benefits on cardiovascular diseases such as HF, myocardial hypertrophy and myocardial infarction. It can be expected that SGLT2 inhibitors can reduce the risk of arrhythmia.
Introduction
With the notion that they lower blood glucose levels, chemists were able to successfully isolate several sodium-glucose cotransporter (SGLT) inhibitors in the 19th century (1). It was understood that, independent of insulin, SGLT inhibitors modify specific phases of kidney metabolism to promote glycosuria, consequently lowering blood glucose (2). SGLT proteins exist in two forms, SGLT1 and SGLT2. Further research revealed that SGLT2 proteins are found mostly in the S1 segment of the proximal convoluted tubule (PCT) and account for approximately 90% of glucose reabsorption (3). SGLT1 proteins are located in the S2/S3 segment of PCT and facilitate reabsorption of the remaining glucose (4). Therefore, blocking SGLT proteins to reduce glucose reabsorption may be a novel therapeutic strategy. Being among the newest hypoglycemics on the market, SGLT2 inhibitors have been extensively studied and demonstrated to significantly reduce the likelihood of suffering cardiovascular events (5). Empagliflozin (EMPA), for example, is a highly specific SGLT2 inhibitor, that has been shown to significantly reduce cardiovascular mortality (6). The EMPA-REG OUTCOME® trial (ClinicalTrials.gov number, NCT01131676), the first trial to report the effects of empagliflozin on cardiovascular outcomes, indicated that empagliflozin can significantly reduce cardiovascular and all-cause mortality by 38 and 32%, respectively (7). Scientific investigation has shown that other SGLT2 inhibitors, such as canagliflozin (1) and dapagliflozin, also have protective effects on the cardiovascular health of diabetes mellitus (DM) type 2 sufferers (2). Canagliflozin can significantly reduce the incidence rate, which is similar to the composite of death from cardiovascular causes, non-fatal myocardial infarction or non-fatal stroke (1). A few years after the EMPA-REG OUTCOME trial, the DECLARE–TIMI 58 trial showed that dapagliflozin has a beneficial effect on non-diabetic patients with heart failure (HF) (3). It has been established that SGLT2 inhibitors reduce the incidence of cardiovascular events by preventing HF (4, 5). In addition, it has also been shown that inhibition of SGLT2 can reduce atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP) in zebrafish models of heart failure (6). Borghetti et al. showed that SGLT2 inhibitors prevent the deterioration of cardiac function in various models of HF (7). Further research revealed that SGLT2 inhibitors could reduce the risk of cardiovascular disease in both diabeticsand non-diabetic individuals (3). The Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure Trial (DAPA-HF trial) (ClinicalTrials.gov number, NCT03036124) demonstrated that treatment with dapagliflozin definitively reduced the risk of precipitated HF or death from cardiovascular causes in patients with HF. And with the development of clinical research, a risk association for AF was observed in the study of Shao et al. between dapagliflozin and empagliflozin (8).
In experimental research, empagliflozin showed a protective effect on diastolic function and cardiac hypertrophy in mice (9). In a rat model with DM, empagliflozin attenuated cardiac inflammation by activating AMPK signaling pathway and promoting autophagy in cardiomyocytes (10). On the other hand, dapagliflozin reduced inflammation by attenuation of NOD-like receptor 3 (NLRP3), interleukin 1β (IL1β), and interleukin-6 (IL6) through the activation of AMP-activated protein kinase (AMPK) pathways (11). Furthermore, dapagliflozin has demonstrated the ability to activate the mTOR pathway, slowing the progression of dilated cardiomyopathy (DCM) as a result (12). Empagliflozin ameliorated LV diastolic function in a DCM model by suppressing calcium/calmodulin-dependent protein kinase II (CaMKII) Thr286, a protein kinase which can retard the phosphorylation of ryanodine receptors (13). In a diabetic mouse model, empagliflozin stimulated sarcoplasmic ATPase2a (SERCA2a) function thus enabling the Ca2 + homeostasis required to improve diastolic function (14). A similar study revealed that empagliflozin can reduce the degree of myocardial fibrosis in diabetic mice by attenuating the transforming growth factor-Smad pathway (15). Yumei Ye et al. demonstrated that in cardiac fibroblasts, dapagliflozin down regulated the NLRP3 inflammasome (16).
From certain reports and studies, SGLT2 inhibitors appear to have an antiarrhythmic effect. Here, how SGLT2 inhibitors plays a protective effect in arrhythmia is discussed.
SGLT2 inhibitors in arrhythmia
As one of the most prevalent chronic diseases in the world, diabetes mellitus (DM) has been extensively investigated, from basic questions like prevention and pathogenesis to the challenging issues of pharmacological management and lifestyle modification (17). DM is associated with various complications including cardiovascular, cerebrovascular and renal disease (18). Therefore, it is imperative that improved cost-effective preventive and treatment solutions are continuously developed to combat the disease. The glucose-lowering function of SGLT2 inhibitors is a result of reduced glucose reabsorption due to SGLT2 cotransporter inhibition in the kidney (19). It has been established in past literature that T2DM is closely related to atrial fibrillation (AF) (20). Recently, some studies have revealed that SGLT2 inhibitors can significantly reduce the incidence rate of AF (21). The physiological mechanisms by which this reduction is achieved are related to myocardial remodeling arising from inflammation-induced cardiac fibrosis (22). Changes in atrial electrical remodeling have also been linked with reduced arrhythmogenesis (23).
Studies spanning decades have established that, regardless of the form and degree of electrical or structural remodeling in the ventricles, dysrhythmias can develop (24). The development and frequency of arrhythmias is closely related to various electrophysiological abnormalities, including ectopic automaticity, triggered by early after-depolarizations, and reentry in cardiac tissue (25). Ectopic beats of the atrium or ventricle usually stem from their respective ectopic automaticity. On the other hand, after depolarizations are usually triggered by an imbalance in Ca2 + homeostasis, resulting in early or delayed after-depolarizations (26). Reentrant arrhythmias have distinct mechanisms usually involving reentry circuits, an anatomical block and a slowed conduction pathway (27).
However, the risk of arrhythmias is generally inflated by fibrosis-induced cardiac remodeling, hypertrophy and dysfunction (28). As previously mentioned, electrical and morphological remodeling are two key forms of remodeling that contribute to arrhythmogenesis. This study reviews both forms and examines how SGLT2 inhibitors reverse them.
Clinical evidence
In past clinical studies, SGLT2 inhibitors have demonstrated outstanding cardiovascular benefits in patients with DMT2, particularly those with comorbid HF (4). In EMPA-REG OUTCOME study (29), empagliflozin reduced the incidence of HF by a staggering 35% (4). The CANVAS study (ClinicalTrials.gov, numbers, NCT01032629 and NCT01989754) confirmed that SGLT2 inhibitors could indeed significantly reduce the incidence of HF (hazard ratio: 0.67; 95% confidential interval: 0.52–0.87) (1). Dapagliflozin, one of the older SGLT2 inhibitors, has been shown to significantly reduce the morbidity and mortality of cardiovascular disease in HF patients with or without DM (hazard ratio: 0.75 and 0.73 respectively) (ClinicalTrials.gov numbers: NCT03036124) (30), improve biomarkers in HF and better patients’ quality of life (ClinicalTrials.gov numbers: NCT03036124) (31). For these reasons there is a consensus that SGLT2 inhibitors preserve cardiovascular function, keeping heart failure at bay. A recent study showed that empagliflozin could also reduce the blood pressure in DMT2 patients (32). Therefore, it is plausible that SGLT2 inhibitors slow the development of HF by lowering blood pressure to improve the load of heart. In these cases, improved diastolic function was observed, attributable to reduced preload. Similar studies revealed that only hemodynamic changes, and not physiological changes, contribute to the drug-induced improvement in diastolic function (33).
It has been established that patients with HF tend to develop atrial fibrillation (AF) (34), so it is clear that preventing HF would also retard the development of AF. Therefore, some studies suggested that SGLT2 inhibitors did not directly have protective effects on arrhythmias (35). SGLT2 inhibitors have indirect protective effects on arrhythmias, such as reducing the risk of AF suffering a myocardial function (36). A study suggested that SGLT2 inhibitors may reduce the risk of AF (35). Meanwhile, Zelniker et al. showed that compared with the group without dapagliflozin, the risk of atrial fibrillation in the group receiving dapagliflozin was reduced by 19%, and the total number of atrial fibrillation events was significantly reduced (37). According to the study from Li et al., SGLT2 inhibitors can significantly reduce the likelihood of serious adverse events (SAEs) in AF (risk ratio: 0.83; 95% confidence interval 0.71–0.96) (38). In addition, these agents were associated with a lower risk of new-onset AF in comparison to other hypoglycemic agents (hazard ratio: 0.61; 95% confidential interval: 0.50–0.73) (39). Dapagliflozin can also significantly reduce the risk of supraventricular and atrial tachycardia (37). On a positive note, the antiarrhythmic effect of dapagliflozin was independent of age, sex, BMI, HbA1c, and eGFR (37). Owing to the hypotensive abilities of SGLT2 inhibitors, it is conceivable that they reduce the risk of AF considerably (40). A meta-analysis published recently suggested that SGLT2 inhibitors specifically reduced the risk of ventricular tachycardia (VT). In the analysis report, only the group receiving empagliflozin was deemed to be at a lower risk of suffering VT. Therefore, SGLT2 inhibitors may in fact may notably protect these patients from VT (41). However, evidence from prospective trials showing how SGLT2 inhibitors primarily deter arrhythmia was lacking. Nonetheless, it can be safely stated that SGLT2 inhibitors have specific antiarrhythmic properties.
Obesity appears to be a another key risk factor for AF, a hypothesis backed by the Framingham Heart Study (42). Clinical studies have confirmed that SGLT2 inhibitors can reduce the risk of AF in patients with T2DM (40) which is associated with the loss of weight (43). Meanwhile, the Framingham study, a long term cohort study, has reported that the deposition of pericardial adipose tissue is associated with the arrhythmogenesis of atrial fibrillation (odds ratio: 1.28; 95% confidential interval: 1.03–1.58) (44). Notably, dapagliflozin and empagliflozin reduce body fat (45) and epicardial fat thickness (46, 47), consequently impeding arrhythmogenesis in diabetics. Accordingly, SGLT2 inhibitor-mediated reduction in epicardial fat volume staves off AF, although the exact mechanism thereof remains unknown. Table 1 summarizes related studies regarding the SGLT2 inhibitors and risk of arrhythmias.
TABLE 1
| References | Study design | Research population | Main finding |
| Curtain et al. (48) | Cross-sectional | Patients with heart failure | Dapagliflozin reduced the risk of any serious ventricular arrhythmia in patients with heart failure (HR 0.79) |
| Li et al. (41) | Meta-analysis | General population | SGLT2 inhibitor was associated with a lower risk of AF (RR 0.82, 95% CI 0.70–0.96) and VT (RR 0.73, 95% CI 0.53–0.99) |
| Zhou et al. (35) | Meta-analysis | General population | Canagliflozin has no direct effects on the incidence of AF (HR 0.76 [95% CI, 0.53–1.10]; P = 0.15) |
| Zhou et al. (49) | Cohort | T2DM patients | Canagliflozin did not significantly reduce the risk of AF (HR 0.84 [95% CI, 0.64–1.12]; P = 0.23) |
| Zelniker et al. (37) | Cross-sectional | T2DM patients | Dapagliflozin significantly reduce the risk of AF/AFL events [HR 0.81(95% CI, 0.68–0.95); P = 0.009) regardless of the patient’s previous history of AF |
| Kovesdy et al. (50) | Cohort | T2DM patients | Atrial fibrillation [HR 2.97 (CI 1.51, 5.84)] is related to adverse events of SGLT2 inhibitors in patients with diabetic kidney disease |
| Chen et al. (51) | Cohort | T2DM patients | SGLT2 inhibitors could significantly lower the risk of new-onset arrhythmias (aHR 0.830; 95% CI 0.751–0.916; P = 0.0002) |
| Bohm et al. (52) | Cross-sectional | T2DM patients | EMPA can reduce the risk of HF in patients with AF |
| Bonora et al. (53) | Cross-sectional | T2DM patients | Compared with other drugs, SGLT2 inhibitors can reduce the occurrence of AF more effectively |
| Wang et al. (54) | Meta-analysis | T2DM patients with HF | SGLT2 inhibitors can significantly reduce the incidence of AF (OR 0.8) and reduce the incidence of arrhythmia (OR 0.86) |
| Kwon et al. (55) | Cohort | T2DM patients with AF | SGLT2 inhibitors can significantly reduce the incidence of hospitalization for heart failure (HR 0.70; 95% CI 0.53 to 0.93; P = 0.012) |
The impact of SGLT2 inhibitors on the risk of arrhythmias.
T2DM, Type 2 diabetes; AF, atrial fibrillation; HF, heart failure; OR, odds ratio; HR, hazard ratio; CI, confidence interval.
Effects of SGLT2 inhibitors on ion homeostasis
Na+ and Ca2 + homeostasis are the key players in the cardiac cycle. The intracellular concentrations of Na+ and Ca2 + are influenced by the Na+ /H+ exchanger (NHE), Na+ /Ca2 + exchanger (NCX), Na+ /Ca2 + exchanger (NCLX), sarco/endoplasmic reticulum Ca2 + -ATPase (SERCA), and L-type calcium channel (ICa-L). SGLT2 inhibitors may directly affect the function of NHE, which could impact NCX and NCLX (56). SGLT2 inhibitors can enhance the phosphorylation of AMPK, thus suppressing the activity of NHE. SGLT2 inhibitors can increase the expression of SERCA2a, which improves contractility (57), subsequently decreasing the concentration of Ca2 + in cardiomyocytes. SGLT2 inhibitors can also improve the activity of phospholamban (PLN), resulting in the phosphorylation of calcium/calmodulin-dependent protein kinase II (CaMKII) and further affects sarcoplasmic release of Ca2 +. In this way, SGLT2 inhibitors prevent calcium overload and thus reducing the likelihood of arrhythmias. In this review, we have summarized several mechanisms of SGLT2 inhibitors acting on ion homeostasis and electrical remodeling in Figure 1.
FIGURE 1
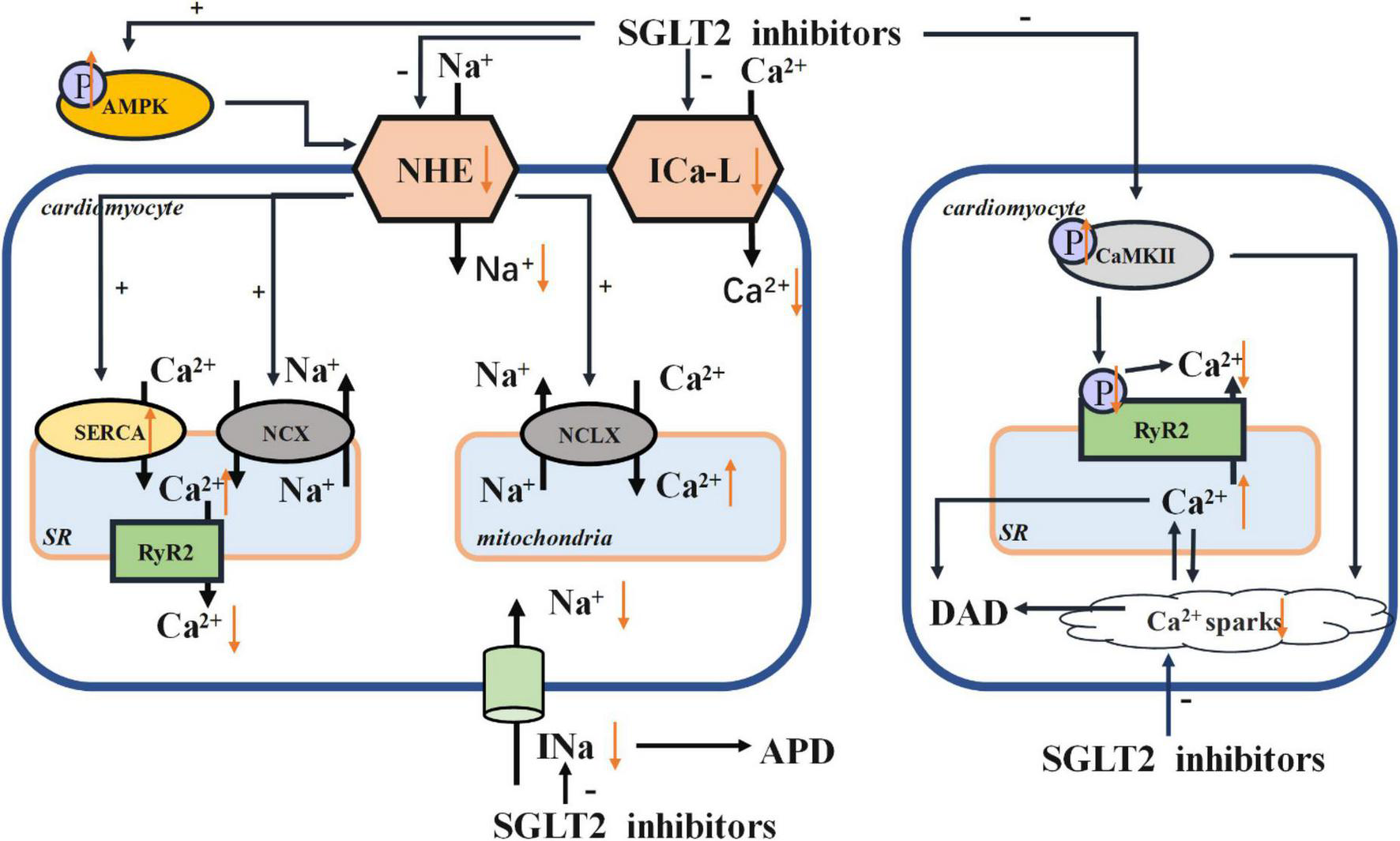
Electrical remodeling. AMPK, adenosine monophosphate kinase; NHE, Na+ /H+ exchanger; NCX, Na+ /Ca2 + exchanger; NCLX, Na+ /Ca2 + exchanger; SR, sarcoplasmic reticulum; SERCA, sarco/endoplasmic reticulum Ca2 + -ATPase; RyR, ryanodine receptor; ICa-L, L-type calcium channel; CaMKII, calcium/calmodulin-dependent protein kinase II; APD, action potential duration; DAD, delayed after depolarization.
Effects of SGLT2 inhibitors on sodium transient
Early after-depolarizations (EADs) and delayed after-depolarizations (DADs) are likely triggers of dysrhythmias. A decrease in repolarizing currents such as rectifier outward K+ currents or an increase in depolarizing currents such as late sodium current (INa) would trigger prolonged action potential duration (APD), which in turn induces EADs (58). In previous studies, the increase of late INa and Na+ influx solely accounted for upregulation of NHE activity (59). Moreover, by activating the activity of NHE1, the cytosolic concentrations of Na+ and Ca2 + were increased. As a result, the concentration of Ca2 + in mitochondria was decreased and eventually induced the cardiomyocyte apoptosis and the instigation of HF (60). Conversely, the NHE1 inhibitor cariporide has been shown to protect cardiomyocytes (61). Jiang et al. reported that empagliflozin could reduce the cytoplasmic concentrations of Na+ and Ca2 +, and also reverse the harmful effects of NHE1 overexpression in cardiomyocytes (60). This finding suggested that empagliflozin can protect myocardial function via a cardiac-specific NHE1. Both dapagliflozin and canagliflozin can also preserve cardiac function by reducing the concentration of cytosolic Na+ ([Na+]c) and the activity of cardiomyocyte specific NHE1 (62). Collectively, these studies point to SGLT2 inhibitors providing myocardial protective effects via regulation of intracellular Na+ ([Na+]i). In particular, a decrease in [Na+]i could prevent progression from ventricular tachycardia to life-threatening ventricular fibrillation. This phenomenon supports the claim increased Na+ concentration facilitates arrhythmogenesis of ventricular arrhythmias (63). Concurrently, At the same time, raised [Na+]i levels will eventually incite generation of reactive oxygen species (ROS) by mitochondria (64), compromising cardiac function as a result. Activation of late Na+ current will further exacerbate intracellular Na+ overload and disrupt contractile and electrical activity all the more. Some studies have suggested that empagliflozin can significantly attenuate late INa without affecting peak INa, thus imparting more protection to the heart (65). There is strong evidence that the induction of late INa is closely related to arrhythmias (61) and late INa could further fluctuate APD, which dictates the likelihood of arrhythmias (66). Worth noting, is that the dispersion of APD, the triggering of arrhythmias, and the induction of ventricular tachycardia can be attenuated by impeding the generation of late INa (67). SGLT2 inhibitors are potent late INa inhibitors and are highly selective for late INa (61). This signifies that SGLT2 inhibitors could markedly decrease the risk of arrhythmias.
Effects of SGLT2 inhibitors on calcium transients
It is clear that Ca2 + and Na+ homeostasis are the key point for heart rhythm and cardiac signal pathways (68, 69). Cardiomyocytes tightly link Ca2 + handling to Na+ handling by activating NCX and mitochondrial NCLX (70). Cardiac NCX, the central efflux pathway of Ca2 + from the mitochondria, extrudes intracellular Ca2 + into the extracellular space, ensuring levels of intracellular Ca2 + are regulated (71). Several studies have shown that inhibition of NCLX activity raises the concentration of intracellular Ca2 +, thereby preventing NAD(P)H oxidation and reducing mitochondrial ROS emissions (72). On this basis, NCLX can curb myocardial hypertrophy and preserve the normal contractile function, thereby reducing the occurrence of ventricular arrhythmias (72). NHEs and NCX often interact, meaning that Na+ concentration can have a direct impact on the Ca2 + concentration (10, 73).
In recent research, dapagliflozin decreased intracellular calcium transients, and the expression of voltage-dependent ICa-L, NCX, and NHE was reduced in dapagliflozin treatment (74). Some studies have shown that inhibition of ICa-L function can impede the entry of extracellular Ca2 +, resulting in a decreased levels of intracellular Ca2 + (75). It has also been learned that the density of ICa-L in ventricular myocytes of diabetics on other treatments was less than that in those receiving empagliflozin (76). The reversal of the density of ICa-L in cardiomyocytes treated with empagliflozin improved [Ca2 +]i transients and sarcoplasmic Ca2 + contents (76). Baartscheer et al. reported that empagliflozin has the ability to inhibit NHE activity, facilitating the reduction sodium and calcium levels within the cardiomyocytes (56). In light of these works, the protective effect of empagliflozin on the myocardium can be directly attributed to consistent ion homeostasis within cardiomyocytes (77). In this investigation, it was reported that empagliflozin prevented intracellular Na+ overload and myocardial oxidative stress by reducing [Na+]i and restoring mitochondrial Ca2 + handling (77). And the increase of [Ca2 +]m also reduced the incidence of ventricular arrhythmias and sudden cardiac death (72). Indeed, intracellular Na+ overload can facilitate cytoplasmic Ca2 + handling and activate the reverse mode Na+ /Ca2 + exchange function of NCX, thus promoting Ca2 + influx and cytosolic Ca2 + transient amplitude (78). Empagliflozin can revive the NCX and stimulate the reverse-mode function of NCX, playing its myocardial protective role as a result (76). However, empagliflozin can hinder leakage of Ca2 + into the SR, which may lead to Ca2 + deficit (76). Moreover, empagliflozin can also change the AP morphology (76). Empagliflozin can stimulate expression of the SERCA2a protein and then attenuate the decrease in Ca2 + stores with prolonged [Ca2 +]i decay (14).
Effects of SGLT2 inhibitors on calcium/calmodulin-dependent protein kinase II
Findings from past research show that DADs could induce arrhythmias. And this process is usually initiated by spontaneous release of Ca2 + from ryanodine receptors (RyR)2, resulting in calcium overload that induces arrhythmias (79). Mustroph et al. revealed that empagliflozin could reduce Ca2 + spark frequency and increase SR Ca2 + load, which are key conditions for setting arrhythmias in motion. It was also affirmed that, at a clinical dose, empagliflozin could reduce CaMKII activity (80). In past research, phosphorylation of PLN, one of the most important CaMKII phospho-sites, was significantly slowed by empagliflozin (5). Because CaMKII can phosphorylate RyR2, further SR Ca2 + release and Ca2 + sparks are also probable (81). Both changes in calcium concentrations generate a depolarizing Na+ influx (Iti), which may trigger DAD (82). On the other hand, it is possible that SGLT2 inhibitors indirectly suppress late INa by CaMKII (79). It is also worth noting that treatment with empagliflozin can suppress the Ca2 + sparks arising from transient release of Ca2 + (83).
Increased CaMKII expression and oxidation plays a critical role in the electrophysiological changes that characterize arrhythmogenesis. Therefore, the claim that SGLT2 inhibitors exert antiarrhythmic effects via inhibition of CaMKII can be safely made (84, 85). A similar study showed that dapagliflozin attenuated the upregulation of NHE through AMPK activation, validating the claim that SGLT2 inhibitors are similar to NHE inhibitors and have antiarrhythmic effects (86).
Effects of SGLT2 inhibitors on cardiac structure
Previous studies have shown that, via their diuretic and natriurtic effects, SGLT2 inhibitors consistently reduce preload, thus shielding the body from cardiovascular disease (87, 88). Moreover, remodeling of individual cardiomyocytes leads to the proliferation of fibroblasts and the production of extracellular matrix, culminating in morphological changes within the hearts (89). This manifests in rhythm irregularities such as atrial fibrillation (AF) (90, 91). Other studies report that SGLT2 inhibitors can reverse ventricular remodeling and reduce cardiac afterload by lowering arterial pressure (92, 93). Reduction in oxidative stress has also been attributed to SGLT inhibitors, an effect that impedes myocardial fibrosis, preserving cardiac contractility as a result (94). In this section, the mechanisms by which SGLT2 inhibitors mitigate structural remodeling of the heart are discussed. Figure 2 summarized the various structural remodeling mechanisms associated with SGLT2 inhibitors in inducing arrhythmias, including inflammation, fibrosis, oxidative stress and apoptosis.
FIGURE 2
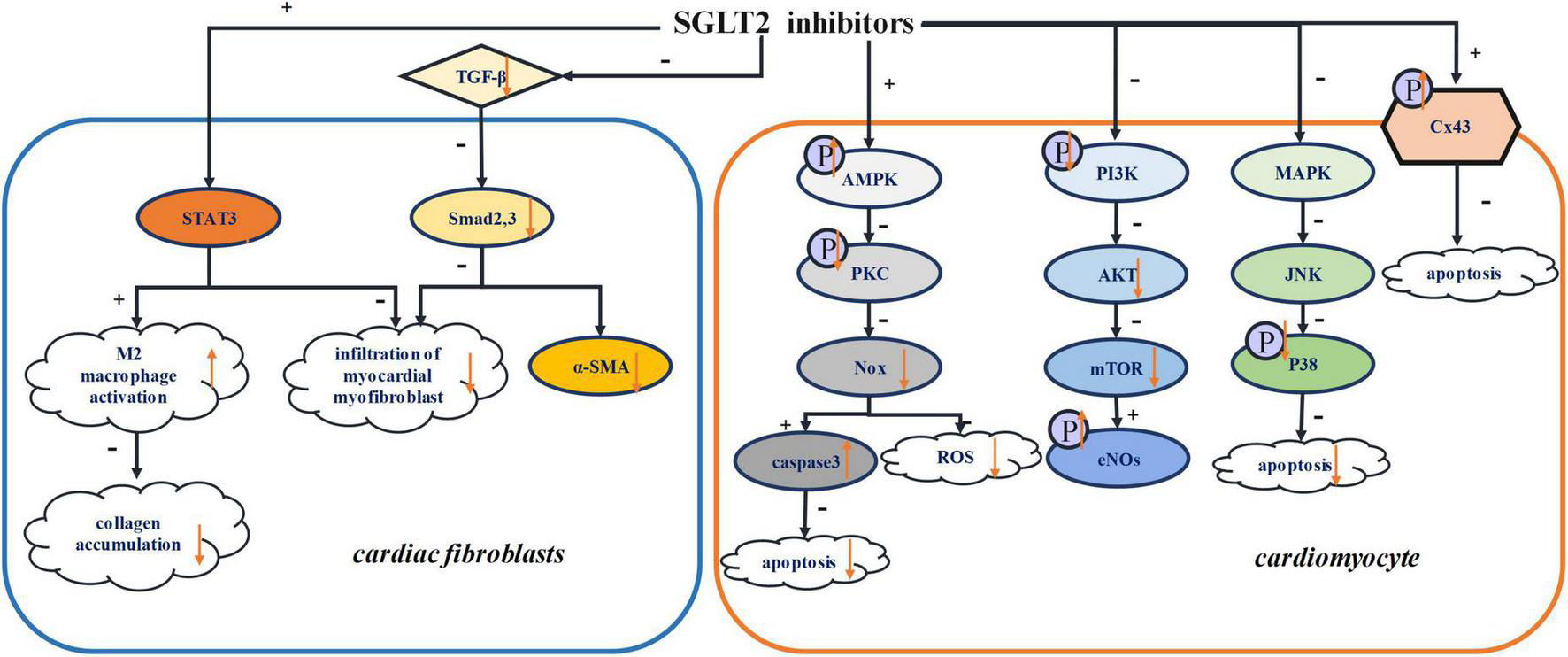
Structural remodeling. STAT3, signal transducer and activator of transcription 3; TGF-β, transforming growth factor-β; Smad2,3, mothers against decapentaplegic homolog 2,3; α-SMA, α-smooth muscle actin; AMPK, adenosine monophosphate kinase; PKC, protein kinase C; Nox, NADPH oxidase; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; Akt, serine/threonine-protein kinase; mTOR, mammalian target of rapamycin; eNOs, endothelial nitric oxide synthase; MAPK, Mitogen-activated protein kinase; JNK, Jun N-terminal kinase; Cx43, connexin 43.
Effects of SGLT2 inhibitors on cardiac fibrosis and inflammation
SGLT2 inhibitors can lower cardiac workload and improve left ventricular function (95). It has been put forward that SGLT2 inhibitors not only reduce fibrotic scars, but also increase LV wall thickness (60) and prevent AF inducibility in diabetic rats (96). Multiple studies have shown that AF is often closely linked with cardiac fibrosis in multiple animal models, affirming that myocardial fibrosis is indeed an important substrate in the development of AF (89). Further research reveals that myocardial fibrosis can trigger the generation of reentry circuits in myocardium, thus accelerating cardiac automaticity and hinder cation potential conduction, and then promote the occurrence of arrhythmias (97).
Tissue fibrosis and inflammation are instrumental in the progression of cardiac remodeling (98). According to a recent study, dapagliflozin activates M2 macrophage infiltration, inhibiting myocardial collagen synthesis and reducing proliferation of myofibroblasts in the border zone as result, which prevent the depolarization required for arrhythmogenesis. Further studies showed that dapagliflozin increases the activity of signal transducer and activator of transcription 3 (STAT3), which is a key transcription factor for minimizing myocardial myofibroblast infiltration (99). This evidence confirms that STAT3 signaling is critical for regulating myocardial fibroblast infiltration. By activating STAT3, the signal pathway of STAT3 would be attenuated, thereby promoting the activation of M2 macrophage and reducing the infiltration of myocardial myofibroblasts (100). Additionally, dapagliflozin was shown to promote M2 macrophage polarization via STAT3 signaling and then protect cardiomyocytes (99).
It has been indicated that empagliflozin markedly slows myocardial fibrosis in an animal model post myocardial infarction (101). Both fibrosis and the expression of fibrosis marker, such as collagen 1 and α-smooth muscle actin, were reduced by empagliflozin treatment. In addition, transforming growth factor-β (TGF-β) and extracellular matrix remodeling, which were significantly attenuated by empagliflozin, can induce the activation of fibroblasts (102). Dapagliflozin can attenuate cardiac remodeling, activation of the NLRP3 inflammasome, fibrotic activity, and LV impairment. However, the specific mechanism by which SGLT2 inhibitors influence NLRP3 inflammation and fibrosis is not clear. However, the anti-inflammatory and antifibrotic effects seem to be unrelated to glucose homeostasis and SGLT2 proteins (16). Ye et al. showed that dapagliflozin significantly increased AMPK levels and its increase could be blocked by AMPK inhibitors. Therefore, it can be concluded that the anti-inflammatory power of SGLT2 inhibitors is dependent on the AMPK pathway (16). This may also suggest that AMPK accounts for the protective effects of SGLT2 inhibitors. Several studies have shown that empagliflozin can promote the phosphorylation of AMPK, which would be less expressed in the model post myocardial injury (103). In addition, empagliflozin increased the AMP/ATP ratio, which would favor the activity of AMPK, ergo delivering its cardiovascular protective effects (104). For these reasons, an investigation of the direct or indirect effects of SGLT2 inhibitors on AMPK signaling pathways is of the essence.
Another experiment has shown that dapagliflozin could prevent cardiac remodeling by inhibiting c-Jun N-terminal kinase (JNK) and P38, two major proteins of mitogen-activated protein kinase (MAPK) pathways (100). It is known that the MAPK pathway plays a critical role the cellular processes that contribute to cardiac remodeling, particularly apoptosis and fibrosis (105). On the other hand, SGLT2 inhibitors can slow cardiac fibrosis by inhibiting NADPH oxidase enzyme family (99), which is related to oxidative stress. Moreover, empagliflozin could regulate the inflammation by reducing myocardial IL-6 and iNOS (106). Empagliflozin can significantly suppress the activity of TNF-α and iNOS (103). And dapagliflozin can inhibit inflammation by reducing the levels of inflammatory cytokines IL-6 and tumor necrosis factor α (TNF-α) and activating the NO-cGMP-PKG pathway (94). In other research, microvascular inflammation, involving the inflammatory mediators ICAM-1, VCAM-1, TNF-α, and IL-6, was higher in samples of myocardial tissue that showed cardiomyopathy with preserved ejection fraction. Empagliflozin significantly attenuated this phenomenon (107).
Effects of SGLT2 inhibitors on oxidative stress
Oxidative stress is a critical factor in cardiac remodeling (108). It has been reported that oxidative stress can induce arrhythmias by promoting cardiac fibrosis and constant changes in the electrophysiological properties of the heart (109, 110). It has also been claimed that oxidative stress could increase vulnerability to arrhythmias (111).
SGLT2 inhibitors can counteract oxidative stress and slow myocardial fibrosis (9, 112). In past research, AMPK inhibited ROS by suppressing the phosphorylation of protein kinase C (PKC) (107), thus inhibiting the activation of NADPH oxidase (Nox) (113). Nox is one of the most important sources of ROS (114), and empagliflozin can attenuate oxidative stress in that way. In some studies, dapagliflozin has been reported to mitigate oxidative stress through AMPK/PKC/Nox signaling pathways (115). In other research, with cardioprotective function, Nrf2/HO-1 has antagonistic effect on oxidative stress (116). And Li et al. suggested that empagliflozin could reduce the occurrence of oxidative stress by downregulating the expression of NOX4 and activating Nrf2/ARE signaling pathway (117). Dapagliflozin suppressed zinc transporter 7 (ZnT7), which led to Zn2 + efflux in cardiomyocytes. And it increased ZIP7, which caused Zn2 + to influx into cardiomyocytes (10). The upregulation of Zn2 + in cardiomyocytes had an antioxidative effects on cardiac muscles.
Effects of SGLT2 inhibitors on apoptotic pathways
It has been established that cardiomyocytes are terminally differentiated cells, and their massive death can lead to severe structural and functional defects. The death of cardiomyocyte usually occurs by apoptosis and necrosis. With extensive research, novel mechanisms of cell death have been discovered, such as necroptosis, pyroptosis, methuosis, and autophagy (118–120). Autophagy is a natural conservative process that promotes degradation of protein and the recycling of damaged cytoplasmic components in response to various stimuli such as stress, ischemic injury, and infection. However, the mechanisms of autophagy signals and regulation are still unclear (121–123). Similarly, the relationship between autophagy and acute myocardial infarctions is still controversial (124, 125). Both inhibition and activation of autophagy have been shown to reduce the size of myocardial infarction (60) and improve ventricular remodeling after myocardial infarction (126). In fact, autophagy is an essential process for maintaining the normal cardiac function. However, in abnormal conditions, excessive autophagy causes extreme destruction of cellular material and cardiomyocyte death (127). Recently, it has been suggested that empagliflozin may play a protective role by inducing cardiomyocyte autophagy (128, 129). In some research, empagliflozin reduced the detrimental effects of autosis (130). Matsui et al. suggested that, in response to glucose deprivation (GD), pretreatment with SGLT2 inhibitors improved cardiomyocyte survival and induced autophagy by activating the AMPK signaling pathway and inactivating the mechanistic target of rapamycin (mTOR) (130). AMPK-dependent signaling pathway can promote autophagy in ischemic cardiomyocytes (127). The maintenance of a high level of ATP/ADP ratio is essential for maintaining normal cardiomyocyte function (131). Jiang et al. indicated that, by playing a protective role in cardiomyocyte function, empagliflozin could mitigate GD-induced damage to cardiomyocytes. And it can also attenuate the cardiomyocyte apoptosis induced by GD (60). Moreover, dapagliflozin could suppress cardiomyocyte apoptosis by downregulating AMPK and upregulating caspase3 (115). The downregulation of PKC and Nox2 could activate caspase3, further linking dapagliflozin suppression of apoptosis and the AMPK/PKC/Nox signaling pathway (115). On the other hand, canagliflozin has been shown to accelerate the phosphorylation of AMPK (132). The phosphorylation of endothelial nitric oxide synthase (eNOS) and protein kinase B (Akt) (133), which could be related to apoptosis (132). As one of the causes of cardiovascular disease, eNOS can be observed in cardiomyocytes, where empagliflozin treatment triggers eNOS phosphorylation, confirming its cardioprotective abilities (104). The PI3K/Akt pathway is involved in cardiac remodeling, especially hypertrophic cardiomyopathy, via the mTOR pathway, and empagliflozin can suppress the cardiac hypertrophy caused by the PI3K/Akt/mTOR pathway (134). In other research, it is clear that empagliflozin can inhibit the PI3K/Akt/mTOR pathway by decreasing the phosphorylation of PI3K/Akt (135).
The relationship with connexin
Previous studies have demonstrated that dapagliflozin can delay the time of first VT, indicating that SGLT2 inhibitors exert antiarrhythmic effects mediated by Connexin 43 (Cx43) (136). Cx43 is a cardiomyocyte gap junction protein that facilitates cell communication by regulating the flow of electrical current (137). High phosphorylation of the Cx43 carboxyl terminal serine residue is essential for the residue of Cx43 (138). The mechanisms of Cx43 dephosphorylation, decreased Cx43 expression and Cx43 lateralization in the ventricle have not been defined. These processes have been reported to cause myocardial ischemia, arrhythmias and HF (139, 140). It has been found that cardiomyocyte death and electrical disorders are affected by Cx43 phosphorylation (141, 142). However, the mechanisms by which Cx43 phosphorylation triggers arrhythmias and the exact phosphorylation site(s) remain unclear (143). Lahnwong et al. showed that dapagliflozin significantly upregulated the phosphorylation of Cx43 and exerted anti-arrhythmia effect (136).
Effects of SGLT2 inhibitors on other organs
The mechanisms by which SGLT2 inhibitors regulate kidney function are diverse. Previous studies have shown that the direct action of these inhibitors on renal vessels mediates their effects on renal function (144). Clinical studies have shown that empagliflozin can decrease the risk of malignant adverse renal outcomes such as acute renal failure (145). However, it has been linked to side effects such as genital infections and urinary infections (145). Experiments have shown that empagliflozin reduces the reabsorption of sodium, which in turn leads decreases ultrafiltration of the blood (146). Empagliflozin can also modulate the progression of kidney disease by altering arterial stiffness and uric acid levels (147). In past research, it was found that empagliflozin improved the indicators of arterial stiffness (147). However, its specific mechanism has not been clarified. Some scholars have suggested that the aforementioned effects may be related to the reduction of body weight and the control of glycaemia as well as the reduction of oxidative stress triggered by SGLT2 inhibitors (148). There is evidence that empagliflozin reduces the glomerular pressure and thus improves the glomerular filtration function in patients with type 1 diabetes (145) but does not prevent proteinuria (29). Recent studies have shown that SGLT2 inhibitors can affect glomerular filtration rate (GFR) by increasing the tone of glomerular afferent arterioles, thereby reducing glomerular plasma flow and intraglomerular pressure. The SGLT2 inhibitors can also reduce the GFR by decreasing the effective filtration pressure through the osmotic effect of non-reabsorbed glucose (149). The decline in GFR induced by SGLT2 inhibitors slows the progression of renal function decline similar to angiotensin (149). These suggests that SGLT2 inhibitors decrease GFR which is beneficial to the renal function. Furthermore, it has been suggested that the protective effects of kidney from SGLT2 inhibitors may be due to the decrease in intraglomerular pressure caused by osmotic diuresis.
Compared with other hypoglycemic drugs, SGLT2 inhibitors induce hypoglycemia through a mechanism independent of insulin regulation. SGLT2 inhibitors reduce body weight and increase fatty acid oxidation and ketone (150). Moreover, SGLT2 inhibitors contribute to the development of ketoacidosis by enhancing ketogenesis (29). Although SGLT2 inhibitors can regulate the generation of ketone bodies, the specific mechanism is unclear. Some clinical studies have shown that canagliflozin can ameliorate hepatic steatosis and non-alcoholic fatty liver disease (151). It activates lipolysis, increase fatty acid oxidation, and reduce hepatic steatosis by promoting the activation of FGF21 (152). On the other hand, it can directly activate the synthesis of hepatic ketone bodies by regulating glycogen consumption, thereby reducing the possibility of hepatic steatosis (152).
Atherosclerosis is a disease characterized by the accumulation of lipids, fibrous elements, and calcium deposits in the intima of arteries (153). Previous studies have shown that SGLT2 inhibitors can prevent the development of atherosclerosis and improve prognosis (154). In a previous study, SGLT2 inhibitors reduced atheromatous plaque size and arterial macrophage infiltration in diabetic mouse. Other studies have demonstrated the inhibitors can prevent atherosclerosis by reducing the formation of arterial foam cells (155). On the other hand, dapagliflozin can reduce the level of NLRP3 in arteries to protect the vasculature (156), but the specific mechanism has not been studied and still needs to be further explored. In recent studies, NLRP3 was found to be involved in the formation of inflammation in atherosclerosis, especially the formation of cholesterol crystals (157), which in turn activates caspase-1 to promote the production of IL-1β and IL-18. Together, these effects promote the development of inflammatory response (158). It is still not clear how SGLT2 inhibitors regulate the production of NLRP3. However, they have been reported to prevent cholesterol crystal formation in arteries, demonstrating their antiatherogenic effects (156).
Future perspectives and conclusion
SGLT2 is mainly expressed in skeletal muscle and kidney, but not in heart (159). With the development of research, it has found that SGLT2 inhibitors can significantly reduce cardiovascular mortality (160, 161). SGLT1 is highly expressed in the heart (162). Therefore, SGLT2 inhibitors may regulate SGLT1 directly and attenuate the incidence of arrhythmia. However, the molecular mechanism by which SGLT2 inhibitors directly protect cardiac function remain unknown. Currently, studies have focused on investigating the indirect role of SGLT2 inhibitors in cardiovascular protection. In future, it will be interesting to determine whether SGLT2 inhibitors exert cardioprotective effects through SGLT1 receptors and the mechanism involved. In a recent study, SGLT2 inhibitors not only reduced the risk of HF hospitalization, but also reduced the odds of adverse event outcomes in HF (163).
In other reports, SGLT2 inhibitors significantly reduced the myocardial infarct size and reduced the risk of arrhythmia. Therefore, SGLT2 inhibitors are expected to play a key role in the protection against various cardiovascular diseases, especially HF, myocardial hypertrophy, myocardial infarction and arrhythmia. However, more studies are needed to elucidate its molecular mechanisms and targets of the cardioprotective effects of SGLT2 inhibitors, which may provide new therapeutic ideas for patients with cardiovascular risk factors. In addition, clarifying the cardiac protection mechanisms of SGLT2 inhibitors may be beneficial to bring cardiac protection into clinical life.
Statements
Author contributions
JG contributed to literature researches. GZ, XW, and JL contributed to data collection. JG and GX were involved in the original draft of the manuscript. XY and YX participated in the review and editing of the manuscript. All authors have read and approved the final manuscript.
Funding
This research was funded by National Natural Science Foundation of China (grant number: 81970286), Chang Jiang Scholars Program (grant number: T2017124), Dalian Talents Innovation Supporting Project (grant number: 2018RD09), and Liao Ning Revitalization Talents Program (grant number: XLYC2002096).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
NealBPerkovicVMahaffeyKWde ZeeuwDFulcherGEronduNet alCanagliflozin and cardiovascular and renal events in type 2 diabetes.N Engl J Med. (2017) 377:644–57. 10.1056/NEJMoa1611925
2.
ZelnikerTAWiviottSDRazIImKGoodrichELBonacaMPet alSGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials.Lancet. (2019) 393:31–9. 10.1016/s0140-6736(18)32590-x
3.
McMurrayJJVSolomonSDInzucchiSEKoberLKosiborodMNMartinezFAet alDapagliflozin in patients with heart failure and reduced ejection fraction.N Engl J Med. (2019) 381:1995–2008. 10.1056/NEJMoa1911303
4.
VermaSMcMurrayJJV. Sglt2 inhibitors and mechanisms of cardiovascular benefit: a state-of-the-art review.Diabetologia. (2018) 61:2108–17. 10.1007/s00125-018-4670-7
5.
VermaSMazerCDFitchettDInzucchiSEPfarrEGeorgeJTet alEmpagliflozin reduces cardiovascular events, mortality and renal events in participants with type 2 diabetes after coronary artery bypass graft surgery: subanalysis of the EMPA-REG OUTCOME§randomised trial.Diabetologia. (2018) 61:1712–23. 10.1007/s00125-018-4644-9
6.
ShiXVermaSYunJBrand-ArzamendiKSinghKKLiuXet alEffect of empagliflozin on cardiac biomarkers in a zebrafish model of heart failure: clues to the empa-reg outcome trial?Mol Cell Biochem. (2017) 433:97–102. 10.1007/s11010-017-3018-9
7.
BorghettiGvon LewinskiDEatonDMSourijHHouserSRWallnerM. Diabetic cardiomyopathy: current and future therapies. beyond glycemic control.Front Physiol. (2018) 9:1514. 10.3389/fphys.2018.01514
8.
ShaoSCChangKCHungMJYangNIChanYYChenHYet alComparative risk evaluation for cardiovascular events associated with dapagliflozin vs. empagliflozin in real-world type 2 diabetes patients: a multi-institutional cohort study.Cardiovasc Diabetol. (2019) 18:120. 10.1186/s12933-019-0919-9
9.
KusakaHKoibuchiNHasegawaYOgawaHKim-MitsuyamaS. Empagliflozin lessened cardiac injury and reduced visceral adipocyte hypertrophy in prediabetic rats with metabolic syndrome.Cardiovasc Diabetol. (2016) 15:157. 10.1186/s12933-016-0473-7
10.
LiNZhouH. SGLT2 inhibitors: a novel player in the treatment and prevention of diabetic cardiomyopathy.Drug Des Devel Ther. (2020) 14:4775–88. 10.2147/DDDT.S269514
11.
PalPBSonowalHShuklaKSrivastavaSKRamanaKV. Aldose reductase mediates NLRP3 inflammasome-initiated innate immune response in hyperglycemia-induced THP1 monocytes and male mice.Endocrinology. (2017) 158:3661–75. 10.1210/en.2017-00294
12.
Aragon-HerreraAFeijoo-BandinSOtero SantiagoMBarralLCampos-ToimilMGil-LongoJet alEmpagliflozin reduces the levels of CD36 and cardiotoxic lipids while improving autophagy in the hearts of Zucker diabetic fatty rats.Biochem Pharmacol. (2019) 170:113677. 10.1016/j.bcp.2019.113677
13.
MoellmannJKlinkhammerBMDrostePKappelBHaj-YehiaEMaxeinerSet alEmpagliflozin improves left ventricular diastolic function of DB/DB Mice.Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165807. 10.1016/j.bbadis.2020.165807
14.
HammoudiNJeongDSinghRFarhatAKomajdaMMayouxEet alEmpagliflozin improves left ventricular diastolic dysfunction in a genetic model of type 2 diabetes.Cardiovasc Drugs Ther. (2017) 31:233–46. 10.1007/s10557-017-6734-1
15.
AoyamaTMatsuiTNovikovMParkJHemmingsBRosenzweigA. Serum and glucocorticoid-responsive Kinase-1 regulates cardiomyocyte survival and hypertrophic response.Circulation. (2005) 111:1652–9. 10.1161/01.CIR.0000160352.58142.06
16.
YeYBajajMYangHCPerez-PoloJRBirnbaumY. SGLT-2 inhibition with dapagliflozin reduces the activation of the NLRP3/ASC inflammasome and attenuates the development of diabetic cardiomyopathy in mice with type 2 diabetes. Further augmentation of the effects with saxagliptin, a DPP4 inhibitor.Cardiovasc Drugs Ther. (2017) 31:119–32. 10.1007/s10557-017-6725-2
17.
KleinertMClemmensenCHofmannSMMooreMCRennerSWoodsSCet alAnimal models of obesity and diabetes mellitus.Nat Rev Endocrinol. (2018) 14:140–62. 10.1038/nrendo.2017.161
18.
UnnikrishnanRAnjanaRMMohanV. Diabetes mellitus and its complications in India.Nat Rev Endocrinol. (2016) 12:357–70. 10.1038/nrendo.2016.53
19.
GoergJSommerfeldMGreinerBLauerDSeckinYKulikovAet alLow-dose empagliflozin improves systolic heart function after myocardial infarction in rats: regulation of MMP9, NHE1, and SERCA2A.Int J Mol Sci. (2021) 22:5437. 10.3390/ijms22115437
20.
LiWJChenXQXuLLLiYQLuoBH. SGLT2 inhibitors and atrial fibrillation in type 2 diabetes: a systematic review with meta-analysis of 16 randomized controlled trials.Cardiovasc Diabetol. (2020) 19:130. 10.1186/s12933-020-01105-5
21.
Echouffo-TcheuguiJBShraderPThomasLGershBJKoweyPRMahaffeyKWet alCare patterns and outcomes in atrial fibrillation patients with and without diabetes: orbit-af registry.J Am Coll Cardiol. (2017) 70:1325–35. 10.1016/j.jacc.2017.07.755
22.
PackerM. Disease-treatment interactions in the management of patients with obesity and diabetes who have atrial fibrillation: the potential mediating influence of epicardial adipose tissue.Cardiovasc Diabetol. (2019) 18:121. 10.1186/s12933-019-0927-9
23.
ChanYHChangGJLaiYJChenWJChangSHHungLMet alAtrial fibrillation and its arrhythmogenesis associated with insulin resistance.Cardiovasc Diabetol. (2019) 18:125. 10.1186/s12933-019-0928-8
24.
ThomasDChristTFabritzLGoetteAHammwohnerMHeijmanJet alGerman cardiac society working group on cellular electrophysiology state-of-the-Art paper: impact of molecular mechanisms on clinical arrhythmia management.Clin Res Cardiol. (2019) 108:577–99. 10.1007/s00392-018-1377-1
25.
NguyenMNKiriazisHGaoXMDuXJ. Cardiac fibrosis and arrhythmogenesis.Compr Physiol. (2017) 7:1009–49. 10.1002/cphy.c160046
26.
ter BekkeRMVoldersPG. Arrhythmogenic mechano-electric heterogeneity in the long-QT syndrome.Prog Biophys Mol Biol. (2012) 110:347–58. 10.1016/j.pbiomolbio.2012.07.007
27.
NguyenTPQuZWeissJN. Cardiac fibrosis and arrhythmogenesis: the road to repair is paved with perils.J Mol Cell Cardiol. (2014) 70:83–91. 10.1016/j.yjmcc.2013.10.018
28.
CartledgeJEKaneCDiasPTesfomMClarkeLMcKeeBet alFunctional crosstalk between cardiac fibroblasts and adult cardiomyocytes by soluble mediators.Cardiovasc Res. (2015) 105:260–70. 10.1093/cvr/cvu264
29.
ZinmanBWannerCLachinJMFitchettDBluhmkiEHantelSet alEmpagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes.N Engl J Med. (2015) 373:2117–28. 10.1056/NEJMoa1504720
30.
PetrieMCVermaSDochertyKFInzucchiSEAnandIBelohlavekJet alEffect of dapagliflozin on worsening heart failure and cardiovascular death in patients with heart failure with and without diabetes.JAMA. (2020) 323:1353–68. 10.1001/jama.2020.1906
31.
KosiborodMNJhundPSDochertyKFDiezMPetrieMCVermaSet alEffects of dapagliflozin on symptoms, function, and quality of life in patients with heart failure and reduced ejection fraction: results from the Dapa-Hf trial.Circulation. (2020) 141:90–9. 10.1161/CIRCULATIONAHA.119.044138
32.
StriepeKJumarAOttCKargMVSchneiderMPKannenkerilDet alEffects of the selective sodium-glucose cotransporter 2 inhibitor empagliflozin on vascular function and central hemodynamics in patients with type 2 diabetes mellitus.Circulation. (2017) 136:1167–9. 10.1161/CIRCULATIONAHA.117.029529
33.
ShettySSKrumermanA. Putative protective effects of sodium-glucose cotransporter 2 inhibitors on atrial fibrillation through risk factor modulation and off-target actions: potential mechanisms and future directions.Cardiovasc Diabetol. (2022) 21:119. 10.1186/s12933-022-01552-2
34.
SanthanakrishnanRWangNLarsonMGMagnaniJWMcManusDDLubitzSAet alAtrial fibrillation begets heart failure and vice versa: temporal associations and differences in preserved versus reduced ejection fraction.Circulation. (2016) 133:484–92. 10.1161/CIRCULATIONAHA.115.018614
35.
ZhouZJardineMJLiQNeuenBLCannonCPde ZeeuwDet alEffect of SGLT2 inhibitors on stroke and atrial fibrillation in diabetic kidney disease: results from the credence trial and meta-analysis.Stroke. (2021) 52:1545–56. 10.1161/STROKEAHA.120.031623
36.
LeeHCShiouYLJhuoSJChangCYLiuPLJhuangWJet alThe sodium-glucose co-transporter 2 inhibitor empagliflozin attenuates cardiac fibrosis and improves ventricular hemodynamics in hypertensive heart failure rats.Cardiovasc Diabetol. (2019) 18:45. 10.1186/s12933-019-0849-6
37.
ZelnikerTABonacaMPFurtadoRHMMosenzonOKuderJFMurphySAet alEffect of dapagliflozin on atrial fibrillation in patients with type 2 diabetes mellitus: insights from the declare-timi 58 trial.Circulation. (2020) 141:1227–34. 10.1161/CIRCULATIONAHA.119.044183
38.
LiDLiuYHidruTHYangXWangYChenCet alProtective effects of sodium-glucose transporter 2 inhibitors on atrial fibrillation and atrial flutter: a systematic review and meta- analysis of randomized placebo-controlled trials.Front Endocrinol (Lausanne). (2021) 12:619586. 10.3389/fendo.2021.619586
39.
LingAWChanCCChenSWKaoYWHuangCYChanYHet alThe risk of new-onset atrial fibrillation in patients with type 2 diabetes mellitus treated with sodium glucose cotransporter 2 inhibitors versus dipeptidyl peptidase-4 inhibitors.Cardiovasc Diabetol. (2020) 19:188. 10.1186/s12933-020-01162-w
40.
TannerRMBaberUCarsonAPVoeksJBrownTMSolimanEZet alAssociation of the metabolic syndrome with atrial fibrillation among United States adults (from the reasons for geographic and racial differences in stroke [Regards] study).Am J Cardiol. (2011) 108:227–32. 10.1016/j.amjcard.2011.03.026
41.
LiHLLipGYHFengQFeiYTseYKWuMZet alSodium-glucose cotransporter 2 inhibitors (SGLT2I) and cardiac arrhythmias: a systematic review and meta-analysis.Cardiovasc Diabetol. (2021) 20:100. 10.1186/s12933-021-01293-8
42.
WangTJPariseHLevyDD’AgostinoRBWolfPAVasanRSet alObesity and the risk of new-onset atrial fibrillation.JAMA. (2004) 292:2471–7.
43.
ChanYHChenSWChaoTFKaoYWHuangCYChuPH. The impact of weight loss related to risk of new-onset atrial fibrillation in patients with type 2 diabetes mellitus treated with sodium-glucose cotransporter 2 inhibitor.Cardiovasc Diabetol. (2021) 20:93. 10.1186/s12933-021-01285-8
44.
ThanassoulisGMassaroJMO’DonnellCJHoffmannULevyDEllinorPTet alPericardial fat is associated with prevalent atrial fibrillation: the Framingham heart study.Circ Arrhythm Electrophysiol. (2010) 3:345–50. 10.1161/CIRCEP.109.912055
45.
SchorkASaynischJVosselerAJaghutrizBAHeyneNPeterAet alEffect of SGLT2 inhibitors on body composition, fluid status and renin-angiotensin-aldosterone system in type 2 diabetes: a prospective study using bioimpedance spectroscopy.Cardiovasc Diabetol. (2019) 18:46. 10.1186/s12933-019-0852-y
46.
Requena-IbanezJASantos-GallegoCGRodriguez-CorderoAVargas-DelgadoAPManciniDSartoriSet alMechanistic insights of empagliflozin in nondiabetic patients with HFREF: from the EMPA-Tropism study.JACC Heart Fail. (2021) 9:578–89. 10.1016/j.jchf.2021.04.014
47.
IacobellisGGra-MenendezS. Effects of dapagliflozin on epicardial fat thickness in patients with type 2 diabetes and obesity.Obesity (Silver Spring). (2020) 28:1068–74. 10.1002/oby.22798
48.
CurtainJPDochertyKFJhundPSPetrieMCInzucchiSEKoberLet alEffect of dapagliflozin on ventricular arrhythmias, resuscitated cardiac arrest, or sudden death in Dapa-HF.Eur Heart J. (2021) 42:3727–38. 10.1093/eurheartj/ehab560
49.
ZhouZLindleyRIRadholmKJenkinsBWatsonJPerkovicVet alCanagliflozin and stroke in type 2 diabetes mellitus.Stroke. (2019) 50:396–404. 10.1161/STROKEAHA.118.023009
50.
KovesdyCSchmedtNFolkertsKBowrinKRaadHBatechMet alPredictors of cardio-kidney complications and treatment failure in patients with chronic kidney disease and type 2 diabetes treated with SGLT2 inhibitors.BMC Med. (2022) 20:2. 10.1186/s12916-021-02191-2
51.
ChenHYHuangJYSiaoWZJongGP. The association between SGLT2 inhibitors and new-onset arrhythmias: a nationwide population-based longitudinal cohort study.Cardiovasc Diabetol. (2020) 19:73. 10.1186/s12933-020-01048-x
52.
BohmMSlawikJBrueckmannMMattheusMGeorgeJTOfstadAPet alEfficacy of empagliflozin on heart failure and renal outcomes in patients with atrial fibrillation: data from the Empa-reg outcome trial.Eur J Heart Fail. (2020) 22:126–35. 10.1002/ejhf.1663
53.
BonoraBMRaschiEAvogaroAFadiniGP. SGLT-2 inhibitors and atrial fibrillation in the food and drug administration adverse event reporting system.Cardiovasc Diabetol. (2021) 20:39. 10.1186/s12933-021-01243-4
54.
WangMZhangYWangZLiuDMaoSLiangB. The effectiveness of SGLT2 inhibitor in the incidence of atrial fibrillation/atrial flutter in patients with type 2 diabetes mellitus/heart failure: a systematic review and meta-analysis.J Thorac Dis. (2022) 14:1620–37. 10.21037/jtd-22-550
55.
KwonCHKimYJKimMJChaMJChoMSNamGBet alEffect of sodium-glucose cotransporter inhibitors on major adverse cardiovascular events and hospitalization for heart failure in patients with type 2 diabetes mellitus and atrial fibrillation.Am J Cardiol. (2022) 178:35–42. 10.1016/j.amjcard.2022.05.017
56.
BaartscheerASchumacherCAWustRCFioletJWStienenGJCoronelRet alEmpagliflozin decreases myocardial cytoplasmic Na(+) through inhibition of the cardiac Na(+)/H(+) exchanger in rats and rabbits.Diabetologia. (2017) 60:568–73. 10.1007/s00125-016-4134-x
57.
NeefSMaierLS. Novel aspects of excitation-contraction coupling in heart failure.Basic Res Cardiol. (2013) 108:360. 10.1007/s00395-013-0360-2
58.
ShryockJCSongYRajamaniSAntzelevitchCBelardinelliL. The arrhythmogenic consequences of increasing late ina in the cardiomyocyte.Cardiovasc Res. (2013) 99:600–11. 10.1093/cvr/cvt145
59.
GrandiEHerrenAW. Camkii-dependent regulation of cardiac Na(+) homeostasis.Front Pharmacol. (2014) 5:41. 10.3389/fphar.2014.00041
60.
JiangKXuYWangDChenFTuZQianJet alCardioprotective mechanism of SGLT2 inhibitor against myocardial infarction is through reduction of autosis.Protein Cell. (2022) 13:336–59. 10.1007/s13238-020-00809-4
61.
MakielskiJC. Late sodium current: a mechanism for angina, heart failure, and arrhythmia.Trends Cardiovasc Med. (2016) 26:115–22. 10.1016/j.tcm.2015.05.006
62.
UthmanLBaartscheerABleijlevensBSchumacherCAFioletJWTKoemanAet alClass effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: inhibition of Na(+)/H(+) exchanger, lowering of cytosolic Na(+) and vasodilation.Diabetologia. (2018) 61:722–6. 10.1007/s00125-017-4509-7
63.
AnzawaRBernardMTamareilleSBaetzDConfort-GounySGascardJPet alIntracellular sodium increase and susceptibility to ischaemia in hearts from type 2 diabetic DB/DB mice.Diabetologia. (2006) 49:598–606. 10.1007/s00125-005-0091-5
64.
KohlhaasMLiuTKnoppAZellerTOngMFBohmMet alElevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes.Circulation. (2010) 121:1606–13. 10.1161/CIRCULATIONAHA.109.914911
65.
PhilippaertKKalyaanamoorthySFatehiMLongWSoniSByrneNJet alCardiac late sodium channel current is a molecular target for the sodium/glucose cotransporter 2 inhibitor empagliflozin.Circulation. (2021) 143:2188–204. 10.1161/CIRCULATIONAHA.121.053350
66.
VarshneyaMMeiXSobieEA. Prediction of arrhythmia susceptibility through mathematical modeling and machine learning.Proc Natl Acad Sci U S A. (2021) 118:e2104019118. 10.1073/pnas.2104019118
67.
HowardTGreer-ShortASatroplusTPatelNNassalDMohlerPJet alCaMKII-dependent late Na(+) current increases electrical dispersion and arrhythmia in ischemia-reperfusion.Am J Physiol Heart Circ Physiol. (2018) 315:H794–801. 10.1152/ajpheart.00197.2018
68.
ShattockMJOttoliaMBersDMBlausteinMPBoguslavskyiABossuytJet alNa+/Ca2+ exchange and Na+/K+-atpase in the heart.J Physiol. (2015) 593:1361–82. 10.1113/jphysiol.2014.282319
69.
KhoCLeeAHajjarRJ. Altered sarcoplasmic reticulum calcium cycling–targets for heart failure therapy.Nat Rev Cardiol. (2012) 9:717–33. 10.1038/nrcardio.2012.145
70.
BerteroEPrates RomaLAmeriPMaackC. Cardiac effects of SGLT2 inhibitors: the sodium hypothesis.Cardiovasc Res. (2018) 114:12–8. 10.1093/cvr/cvx149
71.
WeberCRPiacentinoVIIIHouserSRBersDM. Dynamic regulation of sodium/calcium exchange function in human heart failure.Circulation. (2003) 108:2224–9. 10.1161/01.CIR.0000095274.72486.94
72.
LiuTTakimotoEDimaanoVLDeMazumderDKettlewellSSmithGet alInhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure.Circ Res. (2014) 115:44–54. 10.1161/CIRCRESAHA.115.303062
73.
DespaSBersDM. Na(+) transport in the normal and failing heart - remember the balance.J Mol Cell Cardiol. (2013) 61:2–10. 10.1016/j.yjmcc.2013.04.011
74.
ArowMWaldmanMYadinDNudelmanVShainbergAAbrahamNGet alSodium-glucose cotransporter 2 inhibitor dapagliflozin attenuates diabetic cardiomyopathy.Cardiovasc Diabetol. (2020) 19:7. 10.1186/s12933-019-0980-4
75.
LuZJiangY-PXuX-HBallouLMCohenISLinRZ. Decreased L-type Ca2+ current in cardiac myocytes of type 1 diabetic akita mice due to reduced phosphatidylinositol 3-kinase signaling.Diabetes. (2007) 56:2780–9. 10.2337/db06-1629
76.
LeeTIChenYCLinYKChungCCLuYYKaoYHet alEmpagliflozin attenuates myocardial sodium and calcium dysregulation and reverses cardiac remodeling in streptozotocin-induced diabetic rats.Int J Mol Sci. (2019) 20:1680. 10.3390/ijms20071680
77.
BayJKohlhaasMMaackC. Intracellular Na(+) and cardiac metabolism.J Mol Cell Cardiol. (2013) 61:20–7. 10.1016/j.yjmcc.2013.05.010
78.
BaartscheerA. [Na+]I and the driving force of the Na+/Ca2+-exchanger in heart failure.Cardiovasc Res. (2003) 57:986–95. 10.1016/s0008-6363(02)00848-9
79.
TrumMRiechelJWagnerS. Cardioprotection by SGLT2 inhibitors-does it all come down to Na(+)?Int J Mol Sci. (2021) 22:7976. 10.3390/ijms22157976
80.
MustrophJWagemannOLuchtCMTrumMHammerKPSagCMet alEmpagliflozin reduces ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes.ESC Heart Fail. (2018) 5:642–8. 10.1002/ehf2.12336
81.
MaierLSZhangTChenLDeSantiagoJBrownJHBersDM. Transgenic CaMKIIdeltac overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced Sr Ca2+ load and activated Sr Ca2+ release.Circ Res. (2003) 92:904–11. 10.1161/01.RES.0000069685.20258.F1
82.
PogwizdSMSchlotthauerKLiLYuanWBersDM. Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness.Circ Res. (2001) 88:1159–67. 10.1161/hh1101.091193
83.
DavidsonSMDuchenMR. Calcium microdomains and oxidative stress.Cell Calcium. (2006) 40:561–74. 10.1016/j.ceca.2006.08.017
84.
WagnerSRuffHMWeberSLBellmannSSowaTSchulteTet alReactive oxygen species-activated Ca/Calmodulin kinase IIdelta is required for late I(Na) augmentation leading to cellular Na and Ca overload.Circ Res. (2011) 108:555–65. 10.1161/CIRCRESAHA.110.221911
85.
Viatchenko-KarpinskiSKornyeyevDEl-BizriNBudasGFanPJiangZet alIntracellular Na+ overload causes oxidation of caMKII and leads to Ca2+ mishandling in isolated ventricular myocytes.J Mol Cell Cardiol. (2014) 76:247–56. 10.1016/j.yjmcc.2014.09.009
86.
YeYJiaXBajajMBirnbaumY. Dapagliflozin attenuates Na(+)/H(+) exchanger-1 in cardiofibroblasts via AMPK activation.Cardiovasc Drugs Ther. (2018) 32:553–8. 10.1007/s10557-018-6837-3
87.
VermaSMcMurrayJJVCherneyDZI. The metabolodiuretic promise of sodium-dependent glucose cotransporter 2 inhibition: the search for the sweet spot in heart failure.JAMA Cardiol. (2017) 2:939–40. 10.1001/jamacardio.2017.1891
88.
SattarNMcLarenJKristensenSLPreissDMcMurrayJJ. SGLT2 inhibition and cardiovascular events: why did empa-reg outcomes surprise and what were the likely mechanisms?Diabetologia. (2016) 59:1333–9. 10.1007/s00125-016-3956-x
89.
PiekASilljeHHWde BoerRA. The vicious cycle of arrhythmia and myocardial fibrosis.Eur J Heart Fail. (2019) 21:492–4. 10.1002/ejhf.1421
90.
PiekAde BoerRASilljeHH. The fibrosis-cell death axis in heart failure.Heart Fail Rev. (2016) 21:199–211. 10.1007/s10741-016-9536-9
91.
NattelS. How does fibrosis promote atrial fibrillation persistence: in silico findings, clinical observations, and experimental data.Cardiovasc Res. (2016) 110:295–7. 10.1093/cvr/cvw092
92.
KarioKOkadaKKatoMNishizawaMYoshidaTAsanoTet al24-hour blood pressure-lowering effect of an SGLT-2 inhibitor in patients with diabetes and uncontrolled nocturnal hypertension: results from the randomized, placebo-controlled sacra study.Circulation. (2018) 139:2089–97. 10.1161/CIRCULATIONAHA.118.037076
93.
WiviottSDRazIBonacaMPMosenzonOKatoETCahnAet alDapagliflozin and cardiovascular outcomes in type 2 diabetes.N Engl J Med. (2019) 380:347–57. 10.1056/NEJMoa1812389
94.
ZhangNFengBMaXSunKXuGZhouY. Dapagliflozin improves left ventricular remodeling and aorta sympathetic tone in a pig model of heart failure with preserved ejection fraction.Cardiovasc Diabetol. (2019) 18:107. 10.1186/s12933-019-0914-1
95.
SanoM. Hemodynamic effects of sodium-glucose cotransporter 2 inhibitors.J Clin Med Res. (2017) 9:457–60. 10.14740/jocmr3011w
96.
YanWZhangHLiuPWangHLiuJGaoCet alImpaired mitochondrial biogenesis due to dysfunctional Adiponectin-AMPK-PGC-1alpha signaling contributing to increased vulnerability in diabetic heart.Basic Res Cardiol. (2013) 108:329. 10.1007/s00395-013-0329-1
97.
ZhangPSuJMendeU. Cross talk between cardiac myocytes and fibroblasts: from multiscale investigative approaches to mechanisms and functional consequences.Am J Physiol Heart Circ Physiol. (2012) 303:H1385–96. 10.1152/ajpheart.01167.2011
98.
NakamuraMSadoshimaJ. Mechanisms of physiological and pathological cardiac hypertrophy.Nat Rev Cardiol. (2018) 15:387–407. 10.1038/s41569-018-0007-y
99.
LeeTMChangNCLinSZ. Dapagliflozin, a selective SGLT2 inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via Stat3 signaling in infarcted rat hearts.Free Radic Biol Med. (2017) 104:298–310. 10.1016/j.freeradbiomed.2017.01.035
100.
ShiLZhuDWangSJiangALiF. Dapagliflozin attenuates cardiac remodeling in mice model of cardiac pressure overload.Am J Hypertens. (2019) 32:452–9. 10.1093/ajh/hpz016
101.
YuristaSRSilljeHHWOberdorf-MaassSUSchoutenEMPavez GianiMGHillebrandsJLet alSodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction.Eur J Heart Fail. (2019) 21:862–73. 10.1002/ejhf.1473
102.
KangSVermaSTengGBelkeDSvystonyukDGuzzardiDet alDirect effects of empagliflozin on extracellular matrix remodeling in human cardiac fibroblasts: novel translational clues to empa-reg outcome.Can J Cardiol. (2017) 33:S169. 10.1016/j.cjca.2017.07.330
103.
KoyaniCNPlastiraISourijHHallstromSSchmidtARainerPPet alEmpagliflozin protects heart from inflammation and energy depletion via AMPK activation.Pharmacol Res. (2020) 158:104870. 10.1016/j.phrs.2020.104870
104.
ZhouHWangSZhuPHuSChenYRenJ. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission.Redox Biol. (2018) 15:335–46. 10.1016/j.redox.2017.12.019
105.
EspositoGPrasadSVRapacciuoloAMaoLKochWJRockmanHA. Cardiac overexpression of a GQ inhibitor blocks induction of extracellular signal–regulated kinase and C-Jun Nh2-terminal kinase activity in in vivo pressure overload.Circulation. (2001) 103:1453–8. 10.1161/01.cir.103.10.1453
106.
AndreadouIEfentakisPBalafasETogliattoGDavosCHVarelaAet alEmpagliflozin limits myocardial infarction in vivo and cell death in vitro: role of Stat3, mitochondria, and redox aspects.Front Physiol. (2017) 8:1077. 10.3389/fphys.2017.01077
107.
KolijnDPabelSTianYLodiMHerwigMCarrizzoAet alEmpagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase galpha oxidation.Cardiovasc Res. (2021) 117:495–507. 10.1093/cvr/cvaa123
108.
LeeTMLinSZChangNC. Antiarrhythmic effect of lithium in rats after myocardial infarction by activation of Nrf2/Ho-1 signaling.Free Radic Biol Med. (2014) 77:71–81. 10.1016/j.freeradbiomed.2014.08.022
109.
ZimaAVBlatterLA. Redox regulation of cardiac calcium channels and transporters.Cardiovasc Res. (2006) 71:310–21. 10.1016/j.cardiores.2006.02.019
110.
YooSAistrupGShiferawYNgJMohlerPJHundTJet alOxidative stress creates a unique, caMKII-mediated substrate for atrial fibrillation in heart failure.JCI Insight. (2018) 3:e120728. 10.1172/jci.insight.120728
111.
WagnerSRokitaAGAndersonMEMaierLS. Redox regulation of sodium and calcium handling.Antioxid Redox Signal. (2013) 18:1063–77. 10.1089/ars.2012.4818
112.
LinBKoibuchiNHasegawaYSuetaDToyamaKUekawaKet alGlycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice.Cardiovasc Diabetol. (2014) 13:148. 10.1186/s12933-014-0148-1
113.
KongSSLiuJJYuXJLuYZangWJ. Protection against ischemia-induced oxidative stress conferred by vagal stimulation in the rat heart: involvement of the AMPK-PKC pathway.Int J Mol Sci. (2012) 13:14311–25. 10.3390/ijms131114311
114.
DerekMYellonDJH. Myocardial reperfusion injury.New Engl J Med. (2007) 357:1121–35.
115.
TsaiKLHsiehPLChouWCChengHCHuangYTChanSH. Dapagliflozin attenuates hypoxia/reoxygenation-caused cardiac dysfunction and oxidative damage through modulation of AMPK.Cell Biosci. (2021) 11:44. 10.1186/s13578-021-00547-y
116.
SunXHanFLuQLiXRenDZhangJet alEmpagliflozin ameliorates obesity-related cardiac dysfunction by regulating Sestrin2-mediated AMPK-MTOR signaling and redox homeostasis in high-fat diet-induced obese mice.Diabetes. (2020) 69:1292–305. 10.2337/db19-0991
117.
LiCZhangJXueMLiXHanFLiuXet alSGLT2 inhibition with empagliflozin attenuates myocardial oxidative stress and fibrosis in diabetic mice heart.Cardiovasc Diabetol. (2019) 18:15. 10.1186/s12933-019-0816-2
118.
WhelanRSKaplinskiyVKitsisRN. Cell death in the pathogenesis of heart disease: mechanisms and significance.Annu Rev Physiol. (2010) 72:19–44. 10.1146/annurev.physiol.010908.163111
119.
LiuYShoji-KawataSSumpterRMJr.WeiYGinetVZhangLet alAutosis Is a Na+,K+-Atpase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia.Proc Natl Acad Sci U S A. (2013) 110:20364–71. 10.1073/pnas.1319661110
120.
GalluzziLVitaleIAaronsonSAAbramsJMAdamDAgostinisPet alMolecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018.Cell Death Differ. (2018) 25:486–541. 10.1038/s41418-017-0012-4
121.
Bravo-San PedroJMKroemerGGalluzziL. Autophagy and mitophagy in cardiovascular disease.Circ Res. (2017) 120:1812–24. 10.1161/CIRCRESAHA.117.311082
122.
SciarrettaSForteMFratiGSadoshimaJ. New insights into the role of MTOR signaling in the cardiovascular system.Circ Res. (2018) 122:489–505. 10.1161/CIRCRESAHA.117.311147
123.
SciarrettaSMaejimaYZablockiDSadoshimaJ. The role of autophagy in the heart.Annu Rev Physiol. (2018) 80:1–26. 10.1146/annurev-physiol-021317-121427
124.
LiuCYZhangYHLiRBZhouLYAnTZhangRCet alLncrna caif inhibits autophagy and attenuates myocardial infarction by blocking P53-mediated myocardin transcription.Nat Commun. (2018) 9:29. 10.1038/s41467-017-02280-y
125.
SantulliG. Stroke prevention: learning from the master (and Commander).Sci Transl Med. (2018) 10:eaav0340. 10.1126/scitranslmed.aav0340
126.
SciarrettaSYeeDNagarajanNBianchiFSaitoTValentiVet alTrehalose-induced activation of autophagy improves cardiac remodeling after myocardial infarction.J Am Coll Cardiol. (2018) 71:1999–2010. 10.1016/j.jacc.2018.02.066
127.
NahJZhaiPHuangCYFernandezAFMareeduSLevineBet alUpregulation of rubicon promotes autosis during myocardial ischemia/reperfusion injury.J Clin Invest. (2020) 130:2978–91. 10.1172/JCI132366
128.
AvogaroAFadiniGPDel PratoS. Reinterpreting cardiorenal protection of renal sodium-glucose cotransporter 2 inhibitors via cellular life history programming.Diabetes Care. (2020) 43:501–7. 10.2337/dc19-1410
129.
PackerM. SGLT2 inhibitors produce cardiorenal benefits by promoting adaptive cellular reprogramming to induce a state of fasting mimicry: a paradigm shift in understanding their mechanism of action.Diabetes Care. (2020) 43:508–11. 10.2337/dci19-0074
130.
MatsuiYTakagiHQuXAbdellatifMSakodaHAsanoTet alDistinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and beclin 1 in mediating autophagy.Circ Res. (2007) 100:914–22. 10.1161/01.RES.0000261924.76669.36
131.
GarciaDShawRJ. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance.Mol Cell. (2017) 66:789–800. 10.1016/j.molcel.2017.05.032
132.
SayourAAKorkmaz-IcozSLoganathanSRuppertMSayourVNOlahAet alAcute canagliflozin treatment protects against in vivo myocardial ischemia-reperfusion injury in non-diabetic male rats and enhances endothelium-dependent vasorelaxation.J Transl Med. (2019) 17:127. 10.1186/s12967-019-1881-8
133.
HawleySAFordRJSmithBKGowansGJManciniSJPittRDet alThe Na+/glucose cotransporter inhibitor canagliflozin activates AMPK by inhibiting mitochondrial function and increasing cellular AMP levels.Diabetes. (2016) 65:2784–94. 10.2337/db16-0058
134.
O’NeillBTAbelED. Akt1 in the cardiovascular system: friend or foe?J Clin Invest. (2005) 115:2059–64. 10.1172/JCI25900
135.
LuQLiuJLiXSunXZhangJRenDet alEmpagliflozin attenuates ischemia and reperfusion injury through LKB1/AMPK signaling pathway.Mol Cell Endocrinol. (2020) 501:110642. 10.1016/j.mce.2019.110642
136.
LahnwongSPaleeSApaijaiNSriwichaiinSKerdphooSJaiwongkamTet alAcute dapagliflozin administration exerts cardioprotective effects in rats with cardiac ischemia/reperfusion injury.Cardiovasc Diabetol. (2020) 19:91. 10.1186/s12933-020-01066-9
137.
LampePDTenBroekEMBurtJMKurataWEJohnsonRGLauAF. Phosphorylation of Connexin43 on Serine368 by protein kinase C regulates gap junctional communication.J Cell Biol. (2000) 149:1503–12.
138.
DeNicolaGFMartinEDChaikuadABassiRClarkJMartinoLet alMechanism and consequence of the autoactivation of P38alpha mitogen-activated protein kinase promoted by Tab1.Nat Struct Mol Biol. (2013) 20:1182–90. 10.1038/nsmb.2668
139.
SolanJLLampePD. Spatio-temporal regulation of Connexin43 phosphorylation and gap junction dynamics.Biochim Biophys Acta Biomembr. (2018) 1860:83–90. 10.1016/j.bbamem.2017.04.008
140.
SmythJWHongTTGaoDVoganJMJensenBCFongTSet alLimited forward trafficking of connexin 43 reduces cell-cell coupling in stressed human and mouse myocardium.J Clin Invest. (2010) 120:266–79. 10.1172/JCI39740
141.
MorelSChristoffersenCAxelsenLNMontecuccoFRochemontVFriasMAet alSphingosine-1-Phosphate reduces ischaemia-reperfusion injury by phosphorylating the gap junction protein Connexin43.Cardiovasc Res. (2016) 109:385–96. 10.1093/cvr/cvw004
142.
WangNDe VuystEPonsaertsRBoenglerKPalacios-PradoNWaumanJet alSelective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury.Basic Res Cardiol. (2013) 108:309. 10.1007/s00395-012-0309-x
143.
XueJYanXYangYChenMWuLGouZet alConnexin 43 dephosphorylation contributes to arrhythmias and cardiomyocyte apoptosis in ischemia/reperfusion hearts.Basic Res Cardiol. (2019) 114:40. 10.1007/s00395-019-0748-8
144.
CherneyDZPerkinsBASoleymanlouNMaioneMLaiVLeeAet alRenal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus.Circulation. (2014) 129:587–97. 10.1161/CIRCULATIONAHA.113.005081
145.
WannerCInzucchiSELachinJMFitchettDvon EynattenMMattheusMet alEmpagliflozin and progression of kidney disease in type 2 diabetes.N Engl J Med. (2016) 375:323–34. 10.1056/NEJMoa1515920
146.
VallonVGerasimovaMRoseMAMasudaTSatrianoJMayouxEet alSGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic akita mice.Am J Physiol Renal Physiol. (2014) 306:F194–204. 10.1152/ajprenal.00520.2013
147.
ChiltonRTikkanenICannonCPCroweSWoerleHJBroedlUCet alEffects of empagliflozin on blood pressure and markers of arterial stiffness and vascular resistance in patients with type 2 diabetes.Diabetes Obes Metab. (2015) 17:1180–93. 10.1111/dom.12572
148.
DengoALDennisEAOrrJSMarinikELEhrlichEDavyBMet alArterial destiffening with weight loss in overweight and obese middle-aged and older adults.Hypertension. (2010) 55:855–61. 10.1161/HYPERTENSIONAHA.109.147850
149.
NespouxJVallonV. SGLT2 inhibition and kidney protection.Clin Sci (Lond). (2018) 132:1329–39. 10.1042/CS20171298
150.
FralickMSchneeweissSPatornoE. Risk of diabetic ketoacidosis after initiation of an SGLT2 inhibitor.N Engl J Med. (2017) 376:2300–2.
151.
OhkiTIsogawaATodaNTagawaK. Effectiveness of ipragliflozin, a sodium-glucose co-transporter 2 inhibitor, as a second-line treatment for non-alcoholic fatty liver disease patients with type 2 diabetes mellitus who do not respond to incretin-based therapies including glucagon-like Peptide-1 analogs and Dipeptidyl Peptidase-4 inhibitors.Clin Drug Investig. (2016) 36:313–9. 10.1007/s40261-016-0383-1
152.
OsataphanSMacchiCSinghalGChimene-WeissJSalesVKozukaCet alSGLT2 inhibition reprograms systemic metabolism via FGF21-dependent and -independent mechanisms.JCI Insight. (2019) 4:e123130. 10.1172/jci.insight.123130
153.
MauriceRLFromageauJBrusseauEFinetGRioufolGCloutierG. On the potential of the lagrangian estimator for endovascular ultrasound elastography: in vivo human coronary artery study.Ultrasound Med Biol. (2007) 33:1199–205. 10.1016/j.ultrasmedbio.2007.01.018
154.
Pahud de MortangesASalvadorDJr.LaimerMMukaTWilhelmMBanoA. The role of SGLT2 inhibitors in atherosclerosis: a narrative mini-review.Front Pharmacol. (2021) 12:751214. 10.3389/fphar.2021.751214
155.
TerasakiMHiromuraMMoriYKohashiKNagashimaMKushimaHet alAmelioration of hyperglycemia with a sodium-glucose cotransporter 2 inhibitor prevents macrophage-driven atherosclerosis through macrophage foam cell formation suppression in type 1 and type 2 diabetic mice.PLoS One. (2015) 10:e0143396. 10.1371/journal.pone.0143396
156.
LengWOuyangXLeiXWuMChenLWuQet alThe SGLT-2 inhibitor dapagliflozin has a therapeutic effect on atherosclerosis in diabetic Apoe(−/−) mice.Mediators Inflamm. (2016) 2016:6305735. 10.1155/2016/6305735
157.
DuewellPKonoHRaynerKJSiroisCMVladimerGBauernfeindFGet alNLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals.Nature. (2010) 464:1357–61. 10.1038/nature08938
158.
CasselSLJolySSutterwalaFS. The NLRP3 inflammasome: a sensor of immune danger signals.Semin Immunol. (2009) 21:194–8. 10.1016/j.smim.2009.05.002
159.
WrightEMLooDDHirayamaBA. Biology of human sodium glucose transporters.Physiol Rev. (2011) 91:733–94. 10.1152/physrev.00055.2009
160.
BellRMYellonDM. SGLT2 inhibitors: hypotheses on the mechanism of cardiovascular protection.Lancet Diabetes Endocrinol. (2018) 6:435–7. 10.1016/s2213-8587(17)30314-5
161.
NassifMKosiborodM. Effect of glucose-lowering therapies on heart failure.Nat Rev Cardiol. (2018) 15:282–91. 10.1038/nrcardio.2017.211
162.
KashiwagiYNagoshiTYoshinoTTanakaTDItoKHaradaTet alExpression of SGLT1 in human hearts and impairment of cardiac glucose uptake by phlorizin during ischemia-reperfusion injury in mice.PLoS One. (2015) 10:e0130605. 10.1371/journal.pone.0130605
163.
AnkerSDButlerJFilippatosGFerreiraJPBocchiEBohmMet alEmpagliflozin in heart failure with a preserved ejection fraction.N Engl J Med. (2021) 385:1451–61. 10.1056/NEJMoa2107038
Summary
Keywords
SGLT2 inhibitors, arrhythmia, electrical remodeling, structural remodeling, mechanisms
Citation
Gao J, Xue G, Zhan G, Wang X, Li J, Yang X and Xia Y (2022) Benefits of SGLT2 inhibitors in arrhythmias. Front. Cardiovasc. Med. 9:1011429. doi: 10.3389/fcvm.2022.1011429
Received
04 August 2022
Accepted
04 October 2022
Published
20 October 2022
Volume
9 - 2022
Edited by
Yajing Wang, University of Alabama at Birmingham, United States
Reviewed by
Wuqiang Zhu, Mayo Clinic Arizona, United States; Fuyang Zhang, Fourth Military Medical University, China
Updates
Copyright
© 2022 Gao, Xue, Zhan, Wang, Li, Yang and Xia.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunlong Xia, yunlong_xia@126.com
†These authors have contributed equally to this work and share first authorship
This article was submitted to Cardiovascular Therapeutics, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.