Abstract
Takotsubo syndrome is a well-characterized cause of acute yet reversible heart failure associated with periods of intense emotional stress, often mimicking on presentation an acute coronary syndrome. Animal models of Takotsubo syndrome have been developed, either through the application of a stressor, or administration of exogenous catecholamine. We found that in a model of isoproterenol-induced Takotsubo syndrome in anesthetized rats hyperthermia (40–41°C) would occur after the administration of isoproterenol. Maintenance of this hyperthermia would result in an apical hypocontractility typical of the syndrome, whereas prevention of hyperthermia with active cooling to maintain a euthermic core body temperature prevented (but did not subsequently reverse) apical hypocontractility. In vitro experimentation with isolated cardiomyocytes showed no effect of hyperthermia on either baseline contractility or contractility change after beta-adrenoceptor stimulation. We suggest that the rise in body temperature that is characteristic of catecholamine storm may be a component in the development of Takotsubo syndrome.
Introduction
Takotsubo syndrome (TTS), previously known as Takotsubo cardiomyopathy or stress cardiomyopathy, is an acute and reversible heart failure that typically occurs after a period of intense emotional or physical stress; symptoms include chest pain and dyspnea and, as a result, can mimic an acute coronary syndrome (1). However, no culprit macrovascular pathology can be found on coronary angiography and a diagnosis of TTS is made due to the observation of the characteristic hypocontractile apex coupled to a hypercontractile base, although variants on this pattern have been observed, e.g., midventricular or basal hypocontractility and also right ventricular involvement (2, 3).
A number of models have been developed to study TTS, including rat in vivo and cellular models (4–6), and human induced pluripotent stem cells from Takotsubo syndrome patients (7), as well as healthy controls (8). These have produced insights on the various phases of Takotsubo syndrome, from the very acute response which includes adrenaline surge, lipotoxicity, nitric oxide generation [and a potential increase in nitrosative stress (9)] and endothelial damage, to the long-term effects through maintained inflammatory damage (10). Many models consist of the exogenous administration of catecholamines, such as the recently described induction of TTS-like contractile dysfunction using intraperitoneal (IP) 50 mg.kg−1 isoproterenol (5). We attempted to reproduce this model in our laboratory as the time course of dysfunction is similar to the human disease course, and with a prolonged period of apical hypokinesia there would be scope to test therapies during the establishment of TTS-like contractility rather than having to administer them before (4). However, the dysfunction observed in (5) could not be recreated in our laboratory.
It was noticed during initial experiments that body temperature tended to rise after isoprenaline administration. A rise in body temperature is known to be produced by high catecholamine levels (once known as “emotional fever”) (11), as is a suite of immunoinflammatory changes (12). In our experiments, the rise in body temperature was controlled by adjusting the external heating support for the anesthetized animal, which remained euthermic throughout. After discussion with the Omerovic group at the University of Gothenberg (the authors of the original IP isoprenaline study) a study was carried out to determine whether allowing this temperature rise in a controlled manner would also allow the apical dysfunction characteristic of TTS to occur. We found that hyperthermia was an essential component in the induction of TTS in this rat model, yet the removal of the hyperthermia after the initial induction phase did not change outcome. This may have relevance to the circumstances leading to the clinical presentation of Takotusbo syndrome.
Methods
All studies complied with Animals (Scientific Procedures) Act (1986), European Directive 2010/63/EU and with the 8th edition of the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. Animals were housed in groups of four and experiences a 12 h light/dark cycle. All rats were male and of the Sprague Dawley strain and obtained from Charles River Laboratories (UK). All animals were killed at the end of the procedure by an approved Schedule 1 method.
Induction of experimental Takotsubo syndrome model
Rats were anesthetized using IP ketamine-midazolam (50 and 5 mg.kg−1, respectively) with 25/2.5 mg.kg−1 administered PRN to maintain a sufficient plane of anesthesia for consistent echocardiographic measurement. 50 mg.kg−1 (-)-isoproterenol was administered IP and rats were monitored by two-dimensional parasternal long axis echocardiography (Visualsonics Vevo 770), with M-mode echocardiography used to assess regional cardiac function, compared to a baseline reading taken before catecholamine administration. A lead II ECG was taken using subdermal electrodes connected via a BioAmp to a PowerLab data acquisition system (AD Instruments).
Body temperature manipulation
Before catecholamine administration, body temperature was maintained between 37 and 38°C using a homeothermic temperature monitor, rectal temperature probe, and heating blanket (Harvard Apparatus). Heating was maintained at a similar level after the administration of isoprenaline, with a new set-point of 41°C used. Cooling, if required at either 60 or 90 min post-isoproterenol administration (Figure 1), was achieved through the removal of external heat sources and the application of ice packs to the abdomen until the rectal temperature was back in the euthermic zone, after which external heating was reapplied as required to maintain euthermia. For experiments shown in the Supplementary Figures, male rats were randomized to either of two groups and subsequently received 50 mg/kg isoprenaline intraperitoneally. Body temperature was maintained at 37.5 ± 0.5°C (normothermia) for the duration of the experiments in the first group whereas in the second group body temperature was first allowed to rise spontaneously and, starting at 15 min post isoprenaline, was maintained at 41 ± 0.5°C (hyperthermia) for the remainder of the experiment. Extent of akinesia was traced in the long axis and expressed as percentage of total LV endocardial length.
Figure 1
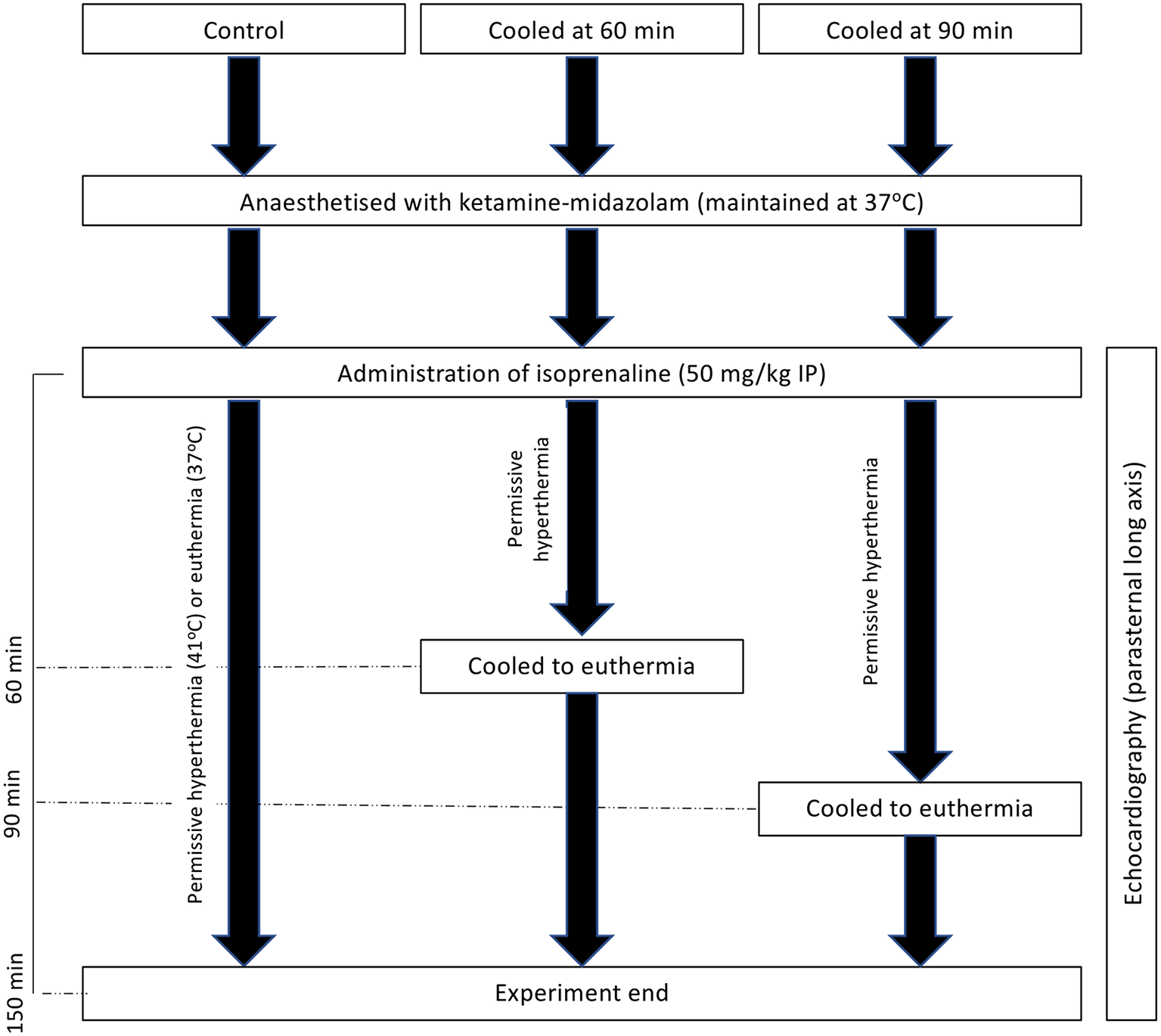
Experimental schematic of hyperthermic experiments with and without cooling to 150 min post-isoprenaline administration.
Rat cardiomyocyte isolation and in vitro contractility studies
Cardiomyocytes were isolated from adult male rats as previously described using a two-enzyme technique (13) and studied using a video edge detection and tracking system (IonOptix) as previously described (4). Cardiomyocytes were paced at 0.5 Hz throughout all studies and contraction was determined as shortening relative to the diastolic length. For studies assessing changes in baseline (i.e., without catecholamines present), cells were perfused with Krebs-Henseleit (KH) solution (1 mM Ca2+) at 37°C for 10 min, after which the temperature either remained constant or was elevated to 41°C. Contractility was measured at 10 and 20 min post-temperature change and changes are expressed relative to contractility at the end of the 10 min baseline period. For isoproterenol hyperthermia studies, cells were perfused with KH at either 37 or 41°C for 10 min, at the end of which a baseline recording was taken. 10−6 M isoproterenol was then perfused and contractility measurements taken at 10 and 20 min post-isoproterenol, and expressed as a relative change in contractility compared to the end of the 10 min baseline.
Statistical analysis
All in vivo contractility and temperature data was analyzed using two-way repeated measures ANOVA calculations with either Tukey's (for whole experiment analysis) or Dunnett's (individual time-point analysis) post-hoc tests for comparison to baseline or apex vs. base. One-way ANOVA with Sidak's post-hoc test was used to compare time-points with baseline for body temperature changes, and for cellular contractility studies. Statistical significance was defined as P < 0.05 and data was tested for normality using a Komogorov–Smirnov test. All statistical analyses were carried out using GraphPad Prism 6 and data is presented at Mean ± SEM unless otherwise stated. Intra-experimental mortality was not observed in any study and therefore not presented graphically.
Results
50 mg.kg–1 isoproterenol at euthermic temperatures does not cause a recapitulation of the Takotsubo-like contractility pattern
Rats were injected with 50 mg.kg−1 IP while under constant ketamine-midazolam anesthesia and serial M-mode measurements of apical and basal segment contractility were taken. Over the 100 min period following isoproterenol administration, no overall apical hypocontractility was seen, but rather a mild and sustained positive inotropy in both segments (Figure 2A). However, no information on right ventricular function was gathered. A mild yet significant increase in body temperature was seen (Figure 2B), which was attenuated by removing all heating and applying cooling as needed.
Figure 2
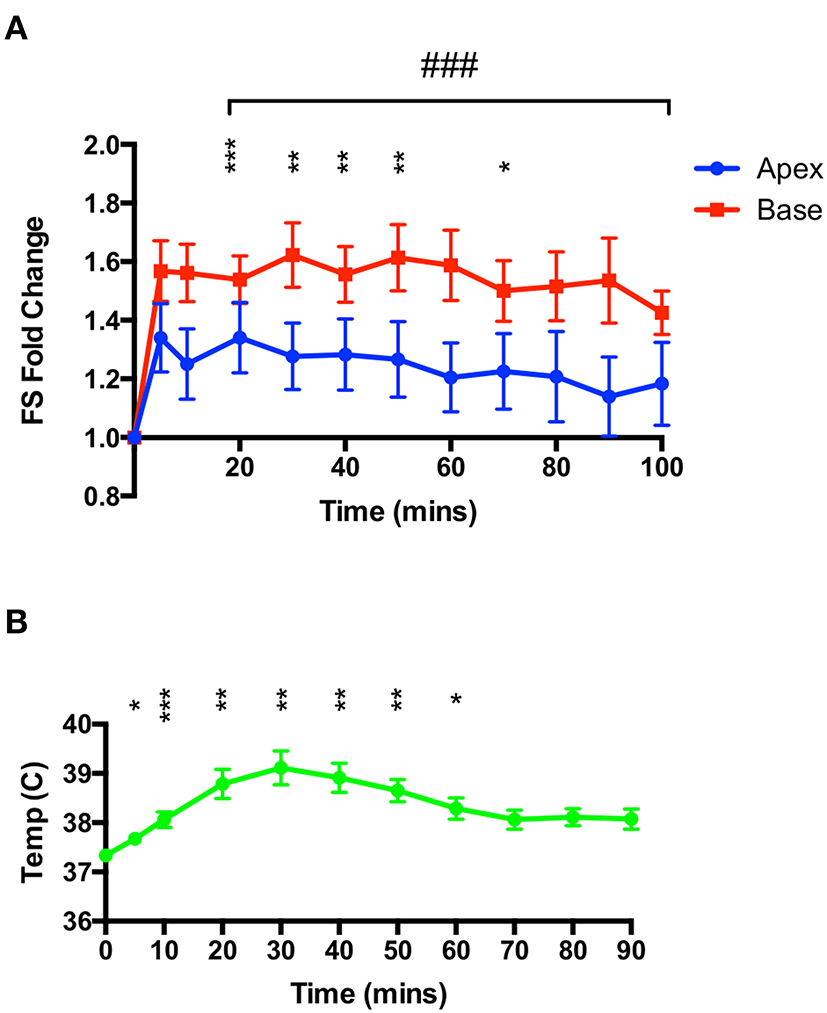
High-dose isoproterenol (50 mg.kg−1) under euthermic conditions does not result in TTS-like contractility. (A) 50 mg.kg−1 isoprenaline under ketamine-midazolam anesthesia resulted in a significant positive inotropic response in both apical and basal myocardial segments, although not for all time-points in the apex. No significant difference was seen between apical and basal contractility (n = 8). *P < 0.05, **P < 0.01, ***P < 0.001 vs. baseline (apex); ###P < 0.001 vs. baseline (base) two-way repeated measures ANOVA. (B) Rectal temperature increases after isoproterenol, although this is attenuated by removing heating from the animal. *P < 0.05, **P < 0.01, ***P < 0.001 relative to time 0, one-way ANOVA (n = 8 animals).
TTS-like contractility can be recapitulated with a 50 mg.kg–1 isoproterenol dose when the hyperthermic response can manifest
As shown in Figure 2, 50 mg.kg−1 isoproterenol, when administered under ketamine-midazolam anesthesia, causes a hyperthermia response that was, in initial studies, attenuated to maintain a euthermic temperature. In a second set of studies, animals were allowed to reach 41°C and maintained at that temperature throughout the 90 min of serial echocardiographic recording post-isoproterenol. This resulted in a negative inotropy in the apex at the 80 and 90 min timepoints compared to baseline, and a significant difference in inotropic change between apex and base at the 80 and 90 min timepoints (Figure 3). Heart rates for these animals can be found in the Supplementary Data.
Figure 3
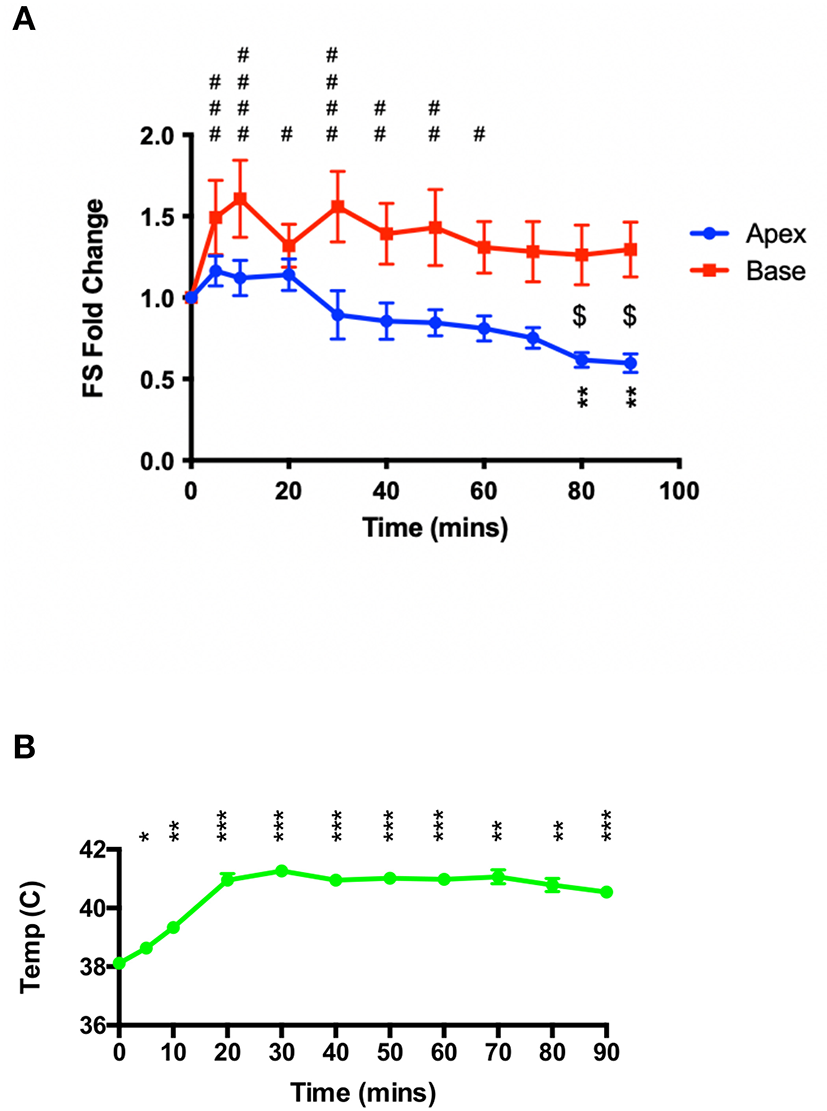
High-dose isoproterenol (50 mg.kg−1) administration under hyperthermic conditions (~41°C) results in apical-hypocontractility. (A) 50 mg.kg−1 isoprenaline under ketamine-midazolam anesthesia resulted a significant positive inotropic response was seen in the basal myocardial segment for the first 60 min, whereas the apical myocardial segment showed a significant negative inotropic response at 80 and 90 min post-isoprenaline. **P < 0.01 apex vs. baseline; $P < 0.05 apex vs. base; #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 base vs. baseline, two-way repeated measures ANOVA (n = 6). (B) Sustained heating of the animal maintains a significant elevated body temperature post-isoproterenol. *P < 0.05, **P < 0.01, ***P < 0.001 relative to baseline temperature, one-way ANOVA (n = 6 animals).
Cooling animals back to euthermia after the initial induction phase does not reverse or attenuate the apical dysfunction caused by high dose isoproterenol
As hyperthermia was essential to the induction of apical dysfunction, we hypothesized that restoration of a euthermic body temperature would either attenuate or reverse the dysfunction. In a separate set of studies shown in Figure 4, rats were randomized prior to study either to be cooled at 60 or 90 min post-isoproterenol, or be maintained at 41°C; all animals were studied to 150 min post-isoproterenol regardless of group. Through the application of cooling packs we were able to rapidly reduce the body temperature back into the euthermic range (Figure 4A). We found no significant change in apical hypocontractility in either cooled group compared to the maintained group; apical hypocontractility was sustained in all three groups (Figure 4B).
Figure 4
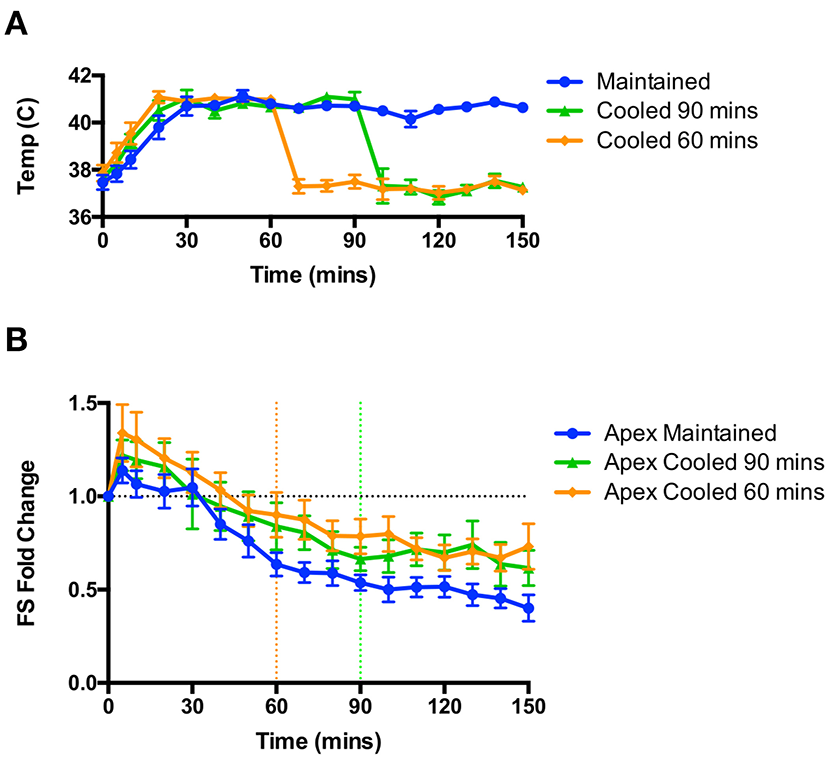
Rapid cooling of animals after the induction of apical dysfunction did not reverse or attenuate the hypocontractility. (A) Rapid changes in body temperature could be achieved through the application of cooling. (B) No effect of cooling was observed on apical dysfunction on rats cooled at 60 min (from orange dotted line) or 90 min (from green dotted line); n = 6 animals per group. Repeated measures ANOVA.
In vitro hyperthermia had no effect on baseline contractility or beta adrenoceptor responsiveness
Isolated rat left ventricular cardiomyocytes were perfused with Krebs-Henseleit (KH) solution at 37°C for 10 min, after which a measurement of cellular shortening was taken and perfusion temperature increased to either 41°C or maintained at 37°C (control). No significant changes in contractility were seen with this increase in temperature (Figure 5A). In a separate set of experiments, isolated cardiomyocytes were perfused with KH at either 37°C or 41°C, and after a 10 min stabilization period, a baseline measurement of cellular shortening was taken and isoproterenol (10−6 M) was added to the perfusate. Measurements were taken at 10 and 20 min post-isoproterenol addition, and no significant differences were seen between the two temperature groups at either time point (Figure 5B).
Figure 5
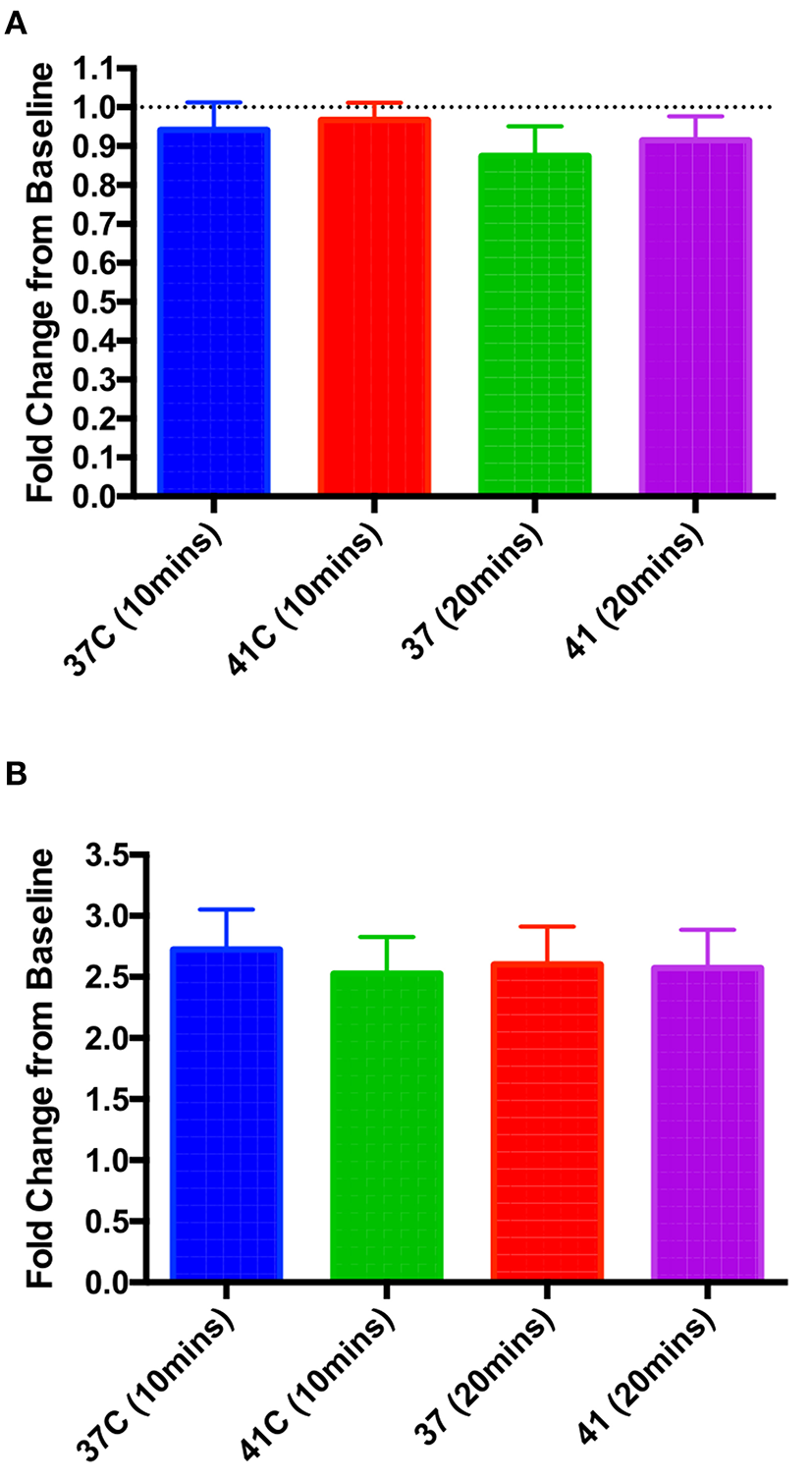
In vitro hyperthermia had no significant effect on either baseline contractility at 10 or 20 min post-temperature change (A) or the increase in contractility when 10−6 M isoproterenol was applied at either 10 or 20 min post-isoproterenol (B). One-way ANOVA, n = 9 and 10 cells (A), 8 and 7 (B), 37 and 41°C, respectively.
Discussion
TTS is a syndrome well-associated with an excess of catecholamines (14), and therefore pre-clinical models using excess catecholamines are useful to delineate the steps the lead to the induction of TTS. In a previous model of TTS developed by the Imperial laboratory (4) a single IV dose to produce a high epinephrine level (approximately equivalent to an Epi-Pen in human) was sufficient to induce a transient apical dysfunction in male rats, where contractility was at a nadir at 20–25 min post-epinephrine and recovered at 60 min post-epinephrine. While this is important to study immediate effects of a catecholamine spike, there are longer-term changes triggered which may underlie a persistent cardiac damage. Models using an intraperitoneal injection of isoproterenol, where animals are studied over hours to days/weeks, have shown an activation of immune responses (12) and an upregulation of pathways involved in inflammation (15). However, while attempting to reproduce the well-validated model of Shao et al. (5) we experienced difficulties, which we resolved in a collaborative effort with the Omerovic laboratory.
We noticed that after isoprenaline administration, animals tended to increase their body temperature, and after discussion between the two groups we determined that allowing the development of the natural transient isoproterenol-induced hyperthermia was essential for the consistent induction of TTS-like contractility in this animal model (Figure 6). Groups wishing to replicate this TTS model should pay close attention to body temperature and allow moderate hyperthermia to occur.
Figure 6
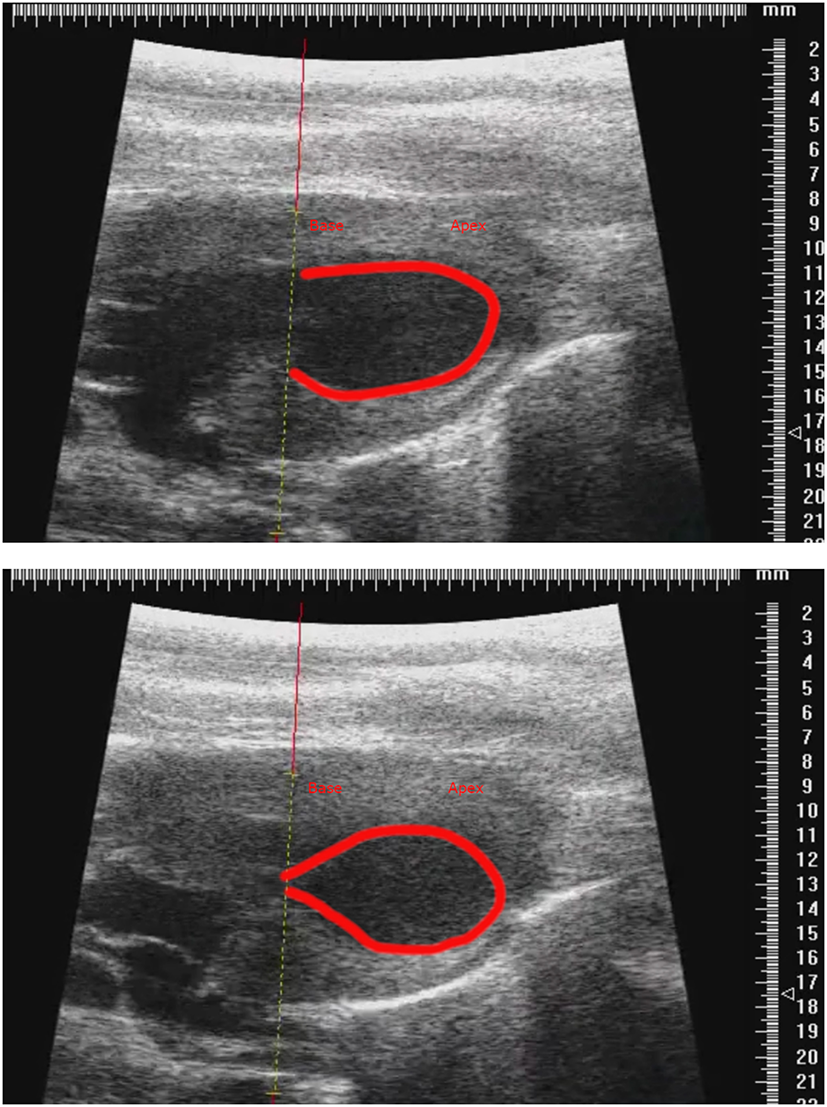
Representative parasternal long axis B-mode images of end-diastole (top) and end-systole (bottom) after induction of TTS-like contractility (base: left; apex: right), with trace (red line) drawn around endocardial border. Scale in mm.
There has been an anecdotal or correlative association between increased body temperature and Takotsubo syndrome in a number of circumstances. Takotsubo syndrome is statistically more likely to occur in summer or early autumn (unlike the winter presentation preference for most cardiac diseases) and is separately associated with higher ambient temperatures (16, 17). There has also been an afternoon predominance for TTS presentation compared to night-time (18) Fever and particularly sepsis have strong associations with Takotsubo syndrome (19), and high temperature was a predictor of worse outcomes in a series of 421 admissions for the syndrome (20). Hyperthyroidism, which is associated with raised body temperature, has been observed in a proportion of patients with Takotsubo syndrome (21). It is tempting to speculate that the temperature dysregulation and hot flushes in post-menopausal women may be an additional trigger for the syndrome. Furthermore, there have been increasing reports of TTS associated with COVID (22), and an early review has suggested that the morbidity and mortality of TTS may be exacerbated when superimposed on the hyper-inflammatory COVID background (23).
As hyperthermia was essential for the induction of TTS-like contractility changes, we sought to determine its importance in the maintenance of this contractility pattern. We found that cooling the animals back to a physiological body temperature at either 60 or 90 min post-isoproterenol had no effect on apical dysfunction, and changes in contraction followed a similar trend to animals maintained at 41°C. However, we do not know the effect of this cooling on long-term prognosis. Cellular experiments showed that hyperthermia alone did not increase contractility of cardiomyocytes, nor increase beta-adrenoceptor responsiveness, and therefore hyperthermia is affecting either the heart on an organ level, or at an integrated body level. The role of circulating immune cells was evident in the study on immune activation by single dose isoproterenol, and adoptive transfer of splenocytes from the isoproterenol-treated mice induced left ventricular dilation and impaired cardiac function in healthy recipients (12).
Mechanism of hyperthermia in TTS induction
Hyperthermia causes a wide range of physiological responses at enzymatic, cellular, organ, and whole-body levels. Through both pre-clinical models and clinical evidence, a number of hypotheses for the pathophysiology of TTS have been proposed. These include microvascular dysfunction (24), lipotoxicity within cardiomyocytes (5), and beta-adrenoceptor stimulus trafficking at the level of the cardiomyocyte (4). It may be that hyperthermia is acting on these, or through other mechanisms to increase apical dysfunction: there are a number of candidate underlying pathways, and their actions may not be mutually exclusive.
Increases in body temperature from 38 to 41°C in rats have been shown to increase sympathetic nerve discharge rate (11, 25), and may be supporting the hyper-catecholaminergic state present in the rat when isoproterenol is administered, particularly given the half-life of isoprenaline is <5 minu and a single dose of isoprenaline has been shown to induce persistent pathological cardiac changes (12). AMP-activated protein kinase (AMPK) is ubiquitously expressed throughout the body, and has been implicated in the pathophysiology of TTS as endomyocardial tissue biopsies taken during the acute TTS phase show increased PI3K/Akt pathway activity (26), which can be activated by AMPK (27). Hyperthermia has been shown to activate AMPK in cancer cells (28), and hypothermia shown to inhibit AMPK in the brain (29). If AMPK is activated by hyperthermia in the setting of TTS, this could be contributing to the protective mechanisms thought to be present in this disease as AMPK activation has been shown to reduce cardiomyocyte apoptosis (30, 31). Heat shock proteins are also likely to be activated by this thermal stress (32), and have been shown to reduce both isoproterenol-induced cardiac apoptosis (33) and remodeling (34). One study has demonstrated the inhibition of the late-sodium currents by hyperthermia in iPS-derived cardiomyocytes (35), which interestingly was also seen in an iPS-derived cardiomyocyte model of TTS (8). We know that nitrosative stress may play a role in TTS (36, 37), which has been shown to be induced by hyperthermia (38). It has also been shown that alpha-1 adrenoceptor signaling via reactive oxidative stress increases the arrhythmogenicity of the myocardium during high levels of catecholamines (39), although in our study isoproterenol was used, which does not act as an agonist on alpha-1 adrenoceptors. Given the increased incidence of a prolonged QT interval in TTS in both models and patient cohort analysis (8, 40), the role of hyperthermia on reactive oxygen species generation and its arrhythmogenicity in the setting of excess catecholamines should be further assessed.
The role of hyperthermia directly on beta-adrenoceptors at a molecular level is poorly understood: one conference abstract has demonstrated reduced beta-adrenoceptor responsiveness in bladder detrusor muscle between 37 and 42°C (41), whereas many studies have been carried out on beta-adrenoceptor responses at hypothermic temperatures. In isolated rat cardiomyocytes, decreases in temperature to 15°C showed an increase in beta-adrenoceptor affinity (using radioactive-labeled propranolol) compared to normothermia (37°C). Hypothermia also does not prevent a beta-adrenoceptor response, as shown in a porcine model of hypothermia, where 30 ng/kg/min infusion of epinephrine resulted in a positive inotropic response at 32°C (42). However, in another experiment at 25°C, isoprenaline resulted in no positive inotropic response (43). Therefore, there is conflicting evidence with regards to catecholamine stimulation with varying body temperature, and further work to elucidate the molecular changes in this model of hyperthermia-facilitated TTS is required.
In a recent computational model of TTS a failure of the Frank-Starling mechanism was shown to increase apical ballooning at end systole (44). In a study that assessed the effects of hyperthermia on the Frank-Starling mechanism in anesthetized dogs, it was shown that hyperthermia of a similar range to the animals presented in this paper (40–41°C) resulted in a significant decrease in the gradient of inotropy change with respect to preload, thereby showing a failure of the Frank-Starling mechanism (45). This could also partly explain the lack of effect seen in isolated cardiomyocytes as they are unloaded. Although a tempting proposition, and one which certainly fits with the aforementioned computational model of TTS (44), the necessary role of hyperthermia is likely multifactorial, particularly in the maintenance of a hypercateholaminergic state as would be present in human TTS precipitated by a stressful event.
Clinical perspective
We have shown that hyperthermia is essential for the induction of TTS in this isoproterenol-induced model, and therefore it may be prudent that patients at risk of TTS are monitored for pyrexia and anti-pyretics are prescribed with a lower threshold. Furthermore, early echocardiographic imaging in patients with severe hyperthermic states, such as malignant hyperthermia or ICU admissions for sepsis, could reveal cardiac dysfunction, and guide clinical care. Nevertheless, the role of hyperthermia in human TTS is currently unknown, and as we were able to remove the hyperthermic state within 60 min of the induction agent being given (equivalent to the emotional stressor in humans), we cannot conclude that a reduction in body temperature per se would be of use in the management of the acute syndrome.
Summary
In summary, we have shown that hyperthermia is essential in the induction of TTS in an isoproterenol-induced rat model, and the removal of hyperthermia around the time of the development of apical dysfunction does not attenuate or reverse it. Isolated cardiomyocyte studies showed that, at least in the unloaded state, baseline contractility, or isoproterenol-induced inotropic changes were not affected by hyperthermia. Further studies are needed to assess the role of hyperthermia in patients and, if so, whether this can be manipulated to improve patient outcome.
Funding
The study was financed by grants from the Swedish state under the agreement between the Swedish government and the county councils (the ALF-agreement), the Swedish Heart-Lung Foundation and the Swedish Scientific Council; by RG/17/13/33173, PG/17/3/32722; RG/17/6/32944; RG/12/18/30088; Medical Research Council UKMR/L006855/1; the National Heart and Lung Institute Foundation and British Heart Foundation BHF FS/16/52/32259.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by Central Animal Welfare and Ethical Review Board, Imperial College London Animal Ethics Committee, Gothenburg University.
Author contributions
MT, BR, and PW performed the experiments and analysis of data in this manuscript. MT wrote the initial draft of the manuscript, with contributions from PW, SH, and EO for the final draft. AL, EO, and SH supervised the research and had overall responsibility for the project. All authors contributed to the design of the experiments and analysis and interpretation of data. All authors contributed to the article and approved the submitted version.
Acknowledgments
The authors would like to thank Peter O'Gara for the expert isolation of cardiomyocytes used in the in vitro studies. MT would like to thank the National Heart and Lung Institute Foundation for the funding of his Ph.D.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.869585/full#supplementary-material
References
1.
TranterMHWrightPTSikkelMBLyonAR. Takotsubo cardiomyopathy: the pathophysiology. Heart Fail Clin. (2013) 9:187–96. 10.1016/j.hfc.2012.12.010
2.
LyonARBossoneESchneiderBSechtemUCitroRUnderwoodSRet al. Current state of knowledge on Takotsubo syndrome: a position statement from the taskforce on Takotsubo syndrome of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. (2016) 18:8–27. 10.1002/ejhf.424
3.
El-BattrawyISantoroFStiermaierTMöllerCGuastafierroFNovoGet al. Incidence and clinical impact of right ventricular involvement (biventricular ballooning) in Takotsubo syndrome: results from the GEIST registry. Chest. (2021) 160:1433–41. 10.1016/j.chest.2021.04.072
4.
PaurHWrightPTSikkelMBTranterMHMansfieldCO'GaraPet al. High levels of circulating epinephrine trigger apical cardiodepression in a β2-adrenergic receptor/Gi-dependent manner: a new model of Takotsubo cardiomyopathy. Circulation. (2012) 126:697–706. 10.1161/CIRCULATIONAHA.112.111591
5.
ShaoYRedforsBScharin TängMMöllmannHTroidlCSzardienSet al. Novel rat model reveals important roles of β-adrenoreceptors in stress-induced cardiomyopathy. Int J Cardiol. (2013) 168:1943–50. 10.1016/j.ijcard.2012.12.092
6.
UeyamaTKasamatsuKHanoTYamamotoKTsuruoYNishioI. Emotional stress induces transient left ventricular hypocontraction in the rat via activation of cardiac adrenoceptors: a possible animal model of “Tako-Tsubo” cardiomyopathy. Circ J Off J Jpn Circ Soc. (2002) 66:712–3. 10.1253/circj.66.712
7.
HübscherDRebsSMaurerWGhadriJRDresselRTemplinCet al. Generation of iPSC-lines from two independent Takotsubo syndrome patients with recurrent Takotsubo events. Stem Cell Res. (2020) 44:101746. 10.1016/j.scr.2020.101746
8.
El-BattrawyIZhaoZLanHSchünemannJDSattlerKBuljubasicFet al. Estradiol protection against toxic effects of catecholamine on electrical properties in human-induced pluripotent stem cell derived cardiomyocytes. Int J Cardiol. (2018) 254:195–202. 10.1016/j.ijcard.2017.11.007
9.
RidnourLAThomasDDMancardiDEspeyMGMirandaKMPaolocciNet al. The chemistry of nitrosative stress induced by nitric oxide and reactive nitrogen oxide species. Putting perspective on stressful biological situations. Biol Chem. (2004) 385:1–10. 10.1515/BC.2004.001
10.
FanXYangGKowitzJAkinIZhouXEl-BattrawyI. Takotsubo syndrome: translational implications and pathomechanisms. Int J Mol Sci. (2022) 23:1951. 10.3390/ijms23041951
11.
NakamuraKMorrisonSF. Central sympathetic network for thermoregulatory responses to psychological stress. Auton Neurosci Basic Clin. (2022) 237:102918. 10.1016/j.autneu.2021.102918
12.
ForteEPanahiMBaxanNNgFSBoyleJJBrancaJet al. Type 2 MI induced by a single high dose of isoproterenol in C57BL/6J mice triggers a persistent adaptive immune response against the heart. J Cell Mol Med. (2021) 25:229–43. 10.1111/jcmm.15937
13.
SatoMO'GaraPHardingSEFullerSJ. Enhancement of adenoviral gene transfer to adult rat cardiomyocytes in vivo by immobilization and ultrasound treatment of the heart. Gene Ther. (2005) 12:936–41. 10.1038/sj.gt.3302476
14.
WittsteinISThiemannDRLimaJACBaughmanKLSchulmanSPGerstenblithGet al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med. (2005) 352:539–48. 10.1056/NEJMoa043046
15.
OrasJRedforsBAliALundgrenJSihlbomCThorsellAet al. Anaesthetic-induced cardioprotection in an experimental model of the Takotsubo syndrome - isoflurane vs. propofol. Acta Anaesthesiol Scand. (2017) 61:309–21. 10.1111/aas.12857
16.
LooiJLLeeMGreyCWebsterMToAKerrAJ. Seasonal variation in Takotsubo syndrome compared with myocardial infarction: ANZACS-QI 16. N Z Med J. (2018) 131:21–9.
17.
KanaokaKOkayamaSTerasakiSNakanoTIshiiMNakaiMet al. Role of climatic factors in the incidence of Takotsubo syndrome: a nationwide study from 2012 to 2016. ESC Heart Fail. (2020) 7:2629–36. 10.1002/ehf2.12843
18.
El-BattrawyIAweimerALangSAnsariUGietzenTUllrichNet al. Impact of chronobiological variation in Takotsubo syndrome: prognosis and outcome. Front Cardiovasc Med. (2021) 8:676950. 10.3389/fcvm.2021.676950
19.
CappellettiSCiallellaCAromatarioMAshrafianHHardingSAthanasiouT. Takotsubo Cardiomyopathy and sepsis. Angiology. (2017) 68:288–303. 10.1177/0003319716653886
20.
KimuraAYoshikawaTIsogaiTTanakaHUedaTYamaguchiTet al. Impact of body temperature at admission on hospital outcomes in patients with Takotsubo syndrome: insights from the Tokyo cardiovascular care unit network registry. Eur Heart J Acute Cardiovasc Care. (2020) 9:703–10. 10.1177/2048872619886313
21.
AweimerAEl-BattrawyIAkinIBorggrefeMMüggeAPatsalisPCet al. Abnormal thyroid function is common in Takotsubo syndrome and depends on two distinct mechanisms: results of a multicentre observational study. J Intern Med. (2021) 289:675–87. 10.1111/joim.13189
22.
EftekharzadehPPatelASokolovaERodasAAhmedS. Takotsubo cardiomyopathy: a COVID-19 complication. Cureus. (2022) 14:e22803. 10.7759/cureus.22803
23.
TechasatianWNishimuraYNagamineTHaGHuangRShahPet al. Characteristics of Takotsubo cardiomyopathy in patients with COVID-19: systematic scoping review. Am Heart J Plus Cardiol Res Pract. (2022) 13:100092. 10.1016/j.ahjo.2022.100092
24.
GaliutoLDe CaterinaARPorfidiaAParaggioLBarchettaSLocorotondoGet al. Reversible coronary microvascular dysfunction: a common pathogenetic mechanism in apical ballooning or Tako-Tsubo syndrome. Eur Heart J. (2010) 31:1319–27. 10.1093/eurheartj/ehq039
25.
KenneyMJBarneyCCHiraiTGisolfiCV. Sympathetic nerve responses to hyperthermia in the anesthetized rat. J Appl Physiol. (1995) 78:881–9. 10.1152/jappl.1995.78.3.881
26.
NefHMMöllmannHHilpertPTroidlCVossSRolfAet al. Activated cell survival cascade protects cardiomyocytes from cell death in Tako-Tsubo cardiomyopathy. Eur J Heart Fail. (2009) 11:758–64. 10.1093/eurjhf/hfp076
27.
TaoRGongJLuoXZangMGuoWWenRet al. AMPK exerts dual regulatory effects on the PI3K pathway. J Mol Signal. (2010) 5:1. 10.1186/1750-2187-5-1
28.
LeeHParkHJParkCSOhETChoiBHWilliamsBet al. Response of breast cancer cells and cancer stem cells to metformin and hyperthermia alone or combined. PLoS ONE. (2014) 9:e87979. 10.1371/journal.pone.0087979
29.
LiJBenashskiSMcCulloughLD. Post-stroke hypothermia provides neuroprotection through inhibition of AMP-activated protein kinase. J Neurotrauma. (2011) 28:1281–8. 10.1089/neu.2011.1751
30.
DyckJRBLopaschukGD. AMPK alterations in cardiac physiology and pathology: enemy or ally?J Physiol. (2006) 574(Pt 1):95–112. 10.1113/jphysiol.2006.109389
31.
ZouMHXieZ. Regulation of interplay between autophagy and apoptosis in the diabetic heart: new role of AMPK. Autophagy. (2013) 9:624–5. 10.4161/auto.23577
32.
O'ConnorMJRemboldCM. Heat-induced force suppression and HSP20 phosphorylation in swine carotid media. J Appl Physiol. (2002) 93:484–8. 10.1152/japplphysiol.00009.2002
33.
FanGCChuGMittonBSongQYuanQKraniasEG. Small heat-shock protein HSP20 phosphorylation inhibits β-agonist-induced cardiac apoptosis. Circ Res. (2004) 94:1474–82. 10.1161/01.RES.0000129179.66631.00
34.
FanGCYuanQSongGWangYChenGQianJet al. Small heat-shock protein Hsp20 attenuates β-agonist-mediated cardiac remodeling through apoptosis signal-regulating kinase 1. Circ Res. (2006) 99:1233–42. 10.1161/01.RES.0000251074.19348.af
35.
El-BattrawyILangSZhaoZAkinIYücelGMeisterSet al. Hyperthermia influences the effects of sodium channel blocking drugs in human-induced pluripotent stem cell-derived cardiomyocytes. PLoS ONE. (2016) 11:e0166143. 10.1371/journal.pone.0166143
36.
SurikowSYNguyenTHStaffordIChapmanMChackoSSinghKet al. Nitrosative stress as a modulator of inflammatory change in a model of Takotsubo syndrome. JACC Basic Transl Sci. (2018) 3:213–26. 10.1016/j.jacbts.2017.10.002
37.
SurikowSYRamanBLicariJSinghKNguyenTHHorowitzJD. Evidence of nitrosative stress within hearts of patients dying of Tako-tsubo cardiomyopathy. Int J Cardiol. (2015) 189:112–4. 10.1016/j.ijcard.2015.03.416
38.
LambertGPGisolfiCVBergDJMoseleyPLOberleyLWKregelKC. Selected contribution: hyperthermia-induced intestinal permeability and the role of oxidative and nitrosative stress. J Appl Physiol. (2002) 92:1750–61; discussion: 1749. 10.1152/japplphysiol.00787.2001
39.
HuangMFanXYangZCyganekLLiXYuecelGet al. Alpha 1-adrenoceptor signalling contributes to toxic effects of catecholamine on electrical properties in cardiomyocytes. Europace. (2021) 23:1137–48. 10.1093/europace/euab008
40.
SyedFFAsirvathamSJFrancisJ. Arrhythmia occurrence with takotsubo cardiomyopathy: a literature review. Europace. (2011) 13:780–8. 10.1093/europace/euq435
41.
FarrSChess-WilliamsRMcDermottC. Hyperthermia Therapy Causes Depessed Detrusor Urothelial Responses With Muscarinic, Adrenergic Neurogenic Stimulation. Barcelona: International Continence Society (2013). Available online at: https://www.ics.org/Abstracts/Publish/180/000694.pdf
42.
DietrichsESSchancheTKondratievTGaustadSESagerGTveitaT. Negative inotropic effects of epinephrine in the presence of increased β-adrenoceptor sensitivity during hypothermia in a rat model. Cryobiology. (2015) 70:9–16. 10.1016/j.cryobiol.2014.10.012
43.
LauriT. Cardiovascular responses to beta-stimulation with isoproterenol in deep hypothermia. J Appl Physiol. (1996) 81:573–7. 10.1152/jappl.1996.81.2.573
44.
LandSNiedererSALouchWERøeÅTAronsenJMStuckeyDJet al. Computational modeling of Takotsubo cardiomyopathy: effect of spatially varying β-adrenergic stimulation in the rat left ventricle. Am J Physiol. (2014) 307:H1487–96. 10.1152/ajpheart.00443.2014
45.
SagachVFShimanskayaTV. Cardiodynamics and pumping function of the heart under conditions of hyperthermia. Bull Exp Biol Med. (1993) 116:893–5. 10.1007/BF00786048
Summary
Keywords
Takotsubo, stress, hyperthermia, catecholamine, isoprenaline
Citation
Tranter MH, Redfors B, Wright PT, Couch LS, Lyon AR, Omerovic E and Harding SE (2022) Hyperthermia as a trigger for Takotsubo syndrome in a rat model. Front. Cardiovasc. Med. 9:869585. doi: 10.3389/fcvm.2022.869585
Received
07 February 2022
Accepted
04 July 2022
Published
26 July 2022
Volume
9 - 2022
Edited by
Kiyotake Ishikawa, Icahn School of Medicine at Mount Sinai, United States
Reviewed by
Ibrahim El-Battrawy, Ruhr University Bochum, Germany; Marco Mongillo, University of Padua, Italy
Updates
Copyright
© 2022 Tranter, Redfors, Wright, Couch, Lyon, Omerovic and Harding.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew H. Tranter matthew.tranter@oriel.ox.ac.uk
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship
This article was submitted to Heart Failure and Transplantation, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.