Abstract
Pathogenic variants in the LMNA gene are known to cause laminopathies, a broad range of disorders with different clinical phenotypes. LMNA genetic variants lead to tissue-specific pathologies affecting various tissues and organs. Common manifestations of laminopathies include cardiovascular system abnormalities, in particular, cardiomyopathies and conduction disorders. In the present study, we used induced pluripotent stem cells from a patient carrying LMNA p.R249Q genetic variant to create an in vitro cardiac model of laminopathy. Induced pluripotent stem cell-derived cardiomyocytes with LMNA p.R249Q genetic variant showed a decreased sodium current density and an impaired sodium current kinetics alongside with changes in transcription levels of cardiac-specific genes. Thus, we obtained compelling in vitro evidence of an association between LMNA p.R249Q genetic variant and cardiac-related abnormalities.
Introduction
The nuclear lamins belong to the type V intermediate filament proteins. The type A lamins encoded by the LMNA gene encompass lamin A and C isoforms, resulting from the LMNA gene alternative splicing (1), as well as more rare lamin C2 (2) and lamin AD10 (3) isoforms. A-type lamins are predominantly expressed in differentiated cells, including cardiomyocytes (CMs) (4). Lamins A and C are central elements of the nuclear lamina and are responsible for the proper nucleus architecture. Besides the structural function, lamins play a role in the regulation of gene transcription by direct and indirect modulation of the chromatin organization, DNA replication, and intercellular signaling (5–8). More detailed information about the functions of A-type lamins can be found in our recent review (9).
Mutations in the LMNA gene cause severe genetic diseases called laminopathies. Laminopathies are characterized by a wide range of clinical manifestations, predominantly affecting tissues of mesenchymal origin (adipose, bone, and muscle tissues) (10, 11). According to ClinVar database, 777 missense genetic variants have been described in association with different phenotypes. Three of them are classified as benign or likely benign, 184 are pathogenic or likely pathogenic, 80 have a conflicting interpretation of pathogenicity, and 480 are the variants of uncertain significance. Clinical significances of the rest 30 missense variants are not provided. Cardiac structure and conduction abnormalities are common symptoms of some laminopathies, such as Emery-Dreifuss muscular dystrophy (EDMD), Limb-girdle muscular dystrophy 1B, and Hutchinson-Gilford Progeria Syndrome (12, 13). Despite the recent advances in iPSC-based laminopathies modeling and animal modeling, the molecular basis of these diseases and the origin of their high phenotypic variability remain unclear.
Most LMNA mutations affect the striated muscles and result in the development of muscular dystrophies. In 1955, EDMD was described for the first time as a disorder with cardiac and skeletal muscle involvement (14). The majority of EDMD cases are associated with genetic variants in LMNA, EMD, and FHL1 genes. Therefore, EDMD is a rare genetic disease characterized by progressive muscle weakness and atrophy, early contractures, cardiac conduction disturbances, and cardiomyopathies, eventually causing lethal arrhythmias (15, 16). The presence and severity of the clinical phenotypes are subtype-dependent and individual and could also include supraventricular and ventricular arrhythmias (15).
Currently, the detailed mechanism of LMNA genetic variants impact on the cardiac system is poorly understood. There are several hypotheses to explain the effect of the LMNA mutations on cardiac function. The structural hypothesis, proposed by Nikolova et al., suggests that LMNA mutations result in morphological abnormalities of a nucleus structure (17). According to the gene expression hypothesis, the main effect of LMNA mutations is mediated by the alterations in the chromatin organization, especially by the impaired formation of lamina-associated domains (LADs) (18, 19). In the mechanotransduction model, lamins are considered as a part of LINC complex (Linker of Nucleoskeleton and Cytoskeleton), which acts as regulatory machinery in contractile tissue, on the one hand, protecting the cell against mechanical stress (20) and, on the other, transmitting the extracellular mechanical signals to the nucleus (21).
Transgenic mouse models were widely used to study the role of LMNA genetic variants in cardiac conduction. Heterozygous lmna+/− mice developed cardiac conduction disease and ventricular dilatation and had a shorter lifespan (22). Lmna knockout and haploinsufficient mice demonstrated defective force transmission, increased levels of pro-adipogenic factors, and downregulation of Wnt signaling (23). Transgenic mice carrying Lmna-H222P and Lmna-N195K reconstituted the dilated cardiomyopathy phenotype (DCM) in mice (24, 25). Despite a large body of information obtained from murine models, these data could not be directly translated into humans due to the differences in cardiac physiology between species.
Up to date, the most relevant approaches for cardiovascular research are based on iPSC-derived cardiomyocytes (iPSC-CMs). These cells successfully recapitulated disease phenotypes specific to the patient's genetic background. Recently, iPSC-CMs models were used to study cardiomyopathy-associated changes in ion channel function (26–29). For example, sodium current reduction was found in various inherited cardiac diseases such as DCM and cardiac conduction disorder (30), Brugada syndrome (31, 32), and arrhythmogenic cardiomyopathy (33). Alterations in the biophysical properties of sodium channels were described in iPSC-CMs model of Myotonic dystrophy type 1 associated with the expansion of a CTG repeat in the 3′ untranslated region of a cAMP-dependent protein kinase (DMPK). The patient's iPSC-CMs showed the downregulation of adult SCN5A isoform and subsequent depolarizing shift of steady-state activation kinetics of the sodium current (34). Salvarani et al. (35) reported that the LMNA p.K219T genetic variant was associated with changes in electrophysiological properties of CMs, including altered action potential, reduced peak sodium current, and diminished conduction velocity.
In the present study, we analyzed patient-specific iPSC-CMs to estimate the impact of the LMNA p.R249Q (LMNA R249Q) genetic variant on the expression of cardiac-specific markers and sodium current parameters. Recently, we generated and characterized the iPSC line FAMRCi007-A from the patient carrying LMNA R249Q genetic variant. This patient was diagnosed with EDMD accompanied with an atrioventricular block and paroxysmal atrial fibrillation (36). Genetic variant R249Q has previously been identified as pathogenic and associated with cardiovascular dysfunction (37).
Here, we demonstrated that LMNA R249Q resulted in alterations of cardiac-specific gene expression and changes of sodium current parameters, including a decrease in sodium current density, delayed activation, and impaired inactivation. Overall, these results contribute to a better understanding of the phenotypic diversity of cardiomyopathies caused by the LMNA genetic variants.
Materials and Methods
Generation and cultivation of human IPSCs
iPSC line with LMNA R249Q (FAMRCi007-A) was generated from the patient's PBMCs using CytoTune-iPS 2.0 Sendai Reprogramming Kit (Invitrogen, USA) as described previously (36). Detailed information about FAMRCi007-A line biological characteristics could be found in hPSCreg database (https://hpscreg.eu/cell-line/FAMRCi007-A). Additionally, two control wild type lines were used: AD3 and WTSIi004-A.
AD3 line was generated from human neonatal fibroblasts (hNFs) using the lentiviral, nonintegrating CytoTune-iPS 2.0 Sendai Reprogramming kit (Invitrogen, USA) according to the manufacturer's instructions. hNFs were purchased from Lonza (Slough, UK) and were cultured as described (38). Generated AD3 iPSC line was characterized according to the protocol published before (39) and fulfilled all criteria of pluripotency (38, 40).
WTSIi004-A line was obtained from European Bank for Induced pluripotent Stem Cells (EBiSC) (https://www.sigmaaldrich.com/RU/en/product/sigma/66540074). The EBiSC Bank acknowledges Wellcome Trust Sanger Institute (WTSI) as the source of the hiPSC line WTSIi004-A (HPSI1113i-qolg_3), which was generated with support from EFPIA companies and the European Union (IMI-JU').
All iPSC lines were cultured on plates coated with Geltrex in Essential 8 medium (all from Thermo Fisher Scientific, USA). Cells were passaged upon reaching 70–80% confluence using ReLeSR (Stem Cell Technologies) as a detaching reagent. For the next 24 h after passaging, the culture media was supplemented with Rock kinase inhibitor Y-27632 (5 μM, Tocris). iPSCs were maintaned at 37°C with 5% CO2 in a humidified incubator.
Differentiation of IPSCs toward cardiomyocytes (CMs)
iPSC differentiation in CMs was performed by modulating Wnt pathway, applying small molecules (41) with modifications. In brief, iPSCs were seeded in density 6 × 105 cell/cm2 2 days before induction of cardiogenic differentiation. When cells reached 90–100% confluency differentiation was induced toward cardiogenic direction by changing growth medium (Essential 8) to differentiation medium [RPMI/B27ins-: Glutamax supplemented RPMI1640, B27 supplement without insulin, 100 U/ml penicillin, 100 μg/ml streptomycin (Thermo Fisher Scientific, USA)] with 6 μM CHIR99021 (Selleckchem, UK) (day 0). After 48 h previous medium was switched to RPMI/B27ins- medium containing 5 μM IWR1 (Stem Cell Technologies, Canada) (day 2). For the next 48 h, cells were cultured in RPMI/B27ins- medium. From day 6 to days 21–23 cells differentiated in RPMI/B27 medium [Glutamax supplemented RPMI1640, B27 supplement, 100 U/ml penicillin, 100 μg/ml streptomycin (Thermo Fisher Scientific, USA)]. RPMI/B27 medium was changed every 2 days. For electrophysiological analysis, from day 12 to day 15, metabolic selection of CMs was conducted as described previously (42). After the selection, cells were cultured in RPMI/B27 medium until seeding on Geltrex-coated coverslips for electrophysiological analysis of Nav1.5. Tryple Select reagent (Thermo Fisher Scientific, USA) was used to dissociate iPSC-CMs to single cell suspension.
Quantitative gene expression analysis and immunofluorescence staining were carried out at 0, 2, 5, 7, 14, and 21 days of cardiogenic differentiation.
A short scheme describing iPSC-CMs differentiation is presented in Figure 1A.
Figure 1
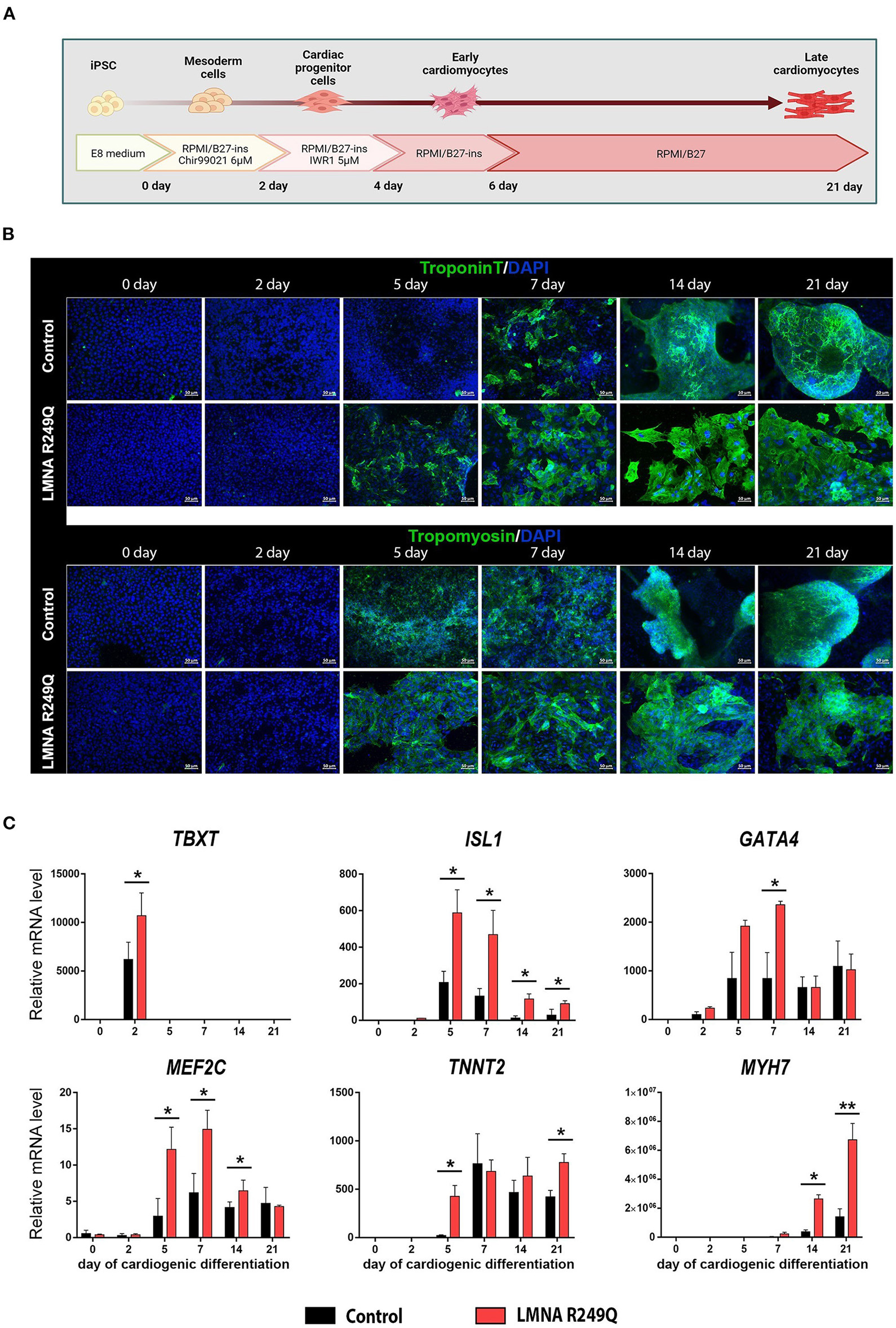
Cardiogenic differentiation of human induced pluripotent stem cells (iPSC) into cardiomyocytes. (A) Short scheme of cardiogenic differentiation consequent stages with descriptions of the medium content. (B) Immunofluorescence staining of iPSC (Control and LMNA R249Q) on a different stage of differentiation (0, 2, 5, 7, 14, 21 days) for cardiac-specific markers troponin T and tropomyosin. Scale bars, 50 μm. (C) Cardiac genes expression in Control iPSC-CMs and LMNA R249Q iPSC-CMs on different stages of cardiogenic differentiation as measured by qPCR. A value of 1 was given to mRNA levels for Control iPSC on day 0 of cardiogenic differentiation for all genes. The results are represented as mean ± SD; *p < 0.05, **p < 0.01.
Immunofluorescence staining
For immunofluorescence staining, cells were fixed with 4% paraformaldehyde for 12 min at room temperature, rinsed twice with phosphate-buffered saline (PBS). Using 0.5% Triton X-100 diluted in PBS, cells were permeabilized for 5 min. For blocking the non-specific binding, cells were incubated with 1% bovine serum albumin solution in PBS for 30 min, followed by washing with PBS. Then, the cells were incubated for 1 h with the primary antibodies: against human troponin T (Santa Cruz, USA) and human tropomyosin (Santa Cruz, USA). Secondary antibodies conjugated with Alexa488 (Invitrogen, USA) were used. For nuclei detection, cells were counterstained with DAPI (Sigma-Aldrich, USA) for 1 min. The coverslips with stained cells were mounted on glass slide with fluoromount (Sigma-Aldrich, USA). The staining was visualized using AxioObserver Z1 (Zeiss, Germany) microscope, and the image was processed using Zen Blue software (Zeiss, Germany).
Gene expression analysis
Gene expression studies were performed on iPSC-CMs on 0, 2, 5, 7, 14, and 21 days of differentiation using qPCR. Total RNA was isolated from cells with Extract RNA reagent (Evrogen, Russia) and was subsequently treated with DNase I (Thermo Fisher Scientific, USA). Reverse transcription reaction was performed using MMLV RT kit (Evrogen, Russia). QPCR measurement was conducted using the qPCR mix-HS SYBR+LowROX (Evrogen, Russia) with gene-specific primers (Supplementary File) on an LC480 platform (Roche, Basel, Switzerland). The relative expression of target genes was calculated by the 2−ΔΔCt method with the GADPH used as the reference gene.
Sodium current recording
Sodium current traces were obtained using whole-cell patch-clamp configuration. For electrophysiological recordings, single-cell CMs seeded on Geltrex-covered coverslips were used. The extracellular solution for sodium current recording contained 140 mM NaCl, 1 mM MgCl2, 1.8 mM CaCl2, 10 mM HEPES, 10 mM glucose, pH 7.4 (CsOH). Nifedipine 10 mkM was added to the extracellular solution to exclude calcium currents. The intracellular solution contained 130 mM CsCl, 10 mM NaCl, 10 mM, EGTA, 10 mM HEPES, pH 7.3 (CsOH). Microelectrodes were manufactured using a puller (P-1000, Sutter Instrument). The electrode resistance ranged from 1.8 to 3.2 MΩ. Data acquisition and junction potential correction were done using Axopatch 200B amplifier and Clampfit software version 10.3 (Molecular Devices Corporation). Currents were acquired at 20–50 kHz and low-pass filtered at 5 kHz using an analog-to-digital interface (Digidata 1440A acquisition system, Molecular Devices Corporation). The series resistance was compensated at 75–80%. To avoid the possible time-dependent shift of activation and steady state inactivation curves, all pulse protocols were applied more than 5 min after membrane rupture. Current densities at each test potential were obtained by dividing the INa by cell capacitance. Cell capacitance values did not differ significantly between experimental groups. All measurements were performed at room temperature.
Patch-clamp data analyses
Pulse protocols were applied with a holding potential of −100 mV. Current-voltage (I-V) curves were assessed by depolarizing voltage steps from −80 to 60 mV during 40 ms in 5 mV increments at 1 Hz frequency. The maximal INa at each voltage was obtained and the corresponding conductance (G) was calculated using equation G = INa/(V – Vrev), where V is the voltage test. The normalized G values were plotted against the voltage, and the G–V curves, which characterize the steady-state activation, were fitted to the Boltzmann function G/Gmax = 1/(1 + exp [(V1/2 – V)/k)], where Gmax is the maximal sodium conductance, V1/2 is the potential of half-maximal activation, and k is the slope factor. The voltage dependence of the steady-state inactivation was tested by measuring INa elicited by a 20 ms step to −15 mV after a prepulse of 500 ms ranging from −120 to 0 mV in 5 mV steps. The normalized INa was plotted against the prepulse voltage. The steady-state inactivation curves were fitted with the Boltzmann function.
Results
Generation of IPSC-based model of healthy CMs and LMNA R249Q CMs
iPSC-CMs carrying LMNA R249Q and control iPSC-CMs were generated according to the Wnt/β-catenin signaling pathway modulation protocol (Figure 1A). The duration of cardiogenic differentiation was 21–23 days for electrophysiological analysis. However, for immunofluorescence and qPCR analysis, the duration of differentiation varied (0, 2, 5, 7, 14, 21 days) to observe the expression of cardiac-specific markers in dynamic.
In the progress of differentiation, the iPSC-derived cardiomyocytes demonstrated gradually increasing levels of troponin T and tropomyosin, as was shown by immunostaining. We noted earlier expression increase of troponin T protein in iPSC-CMs with LMNA R249Q compared to control (Figure 1B). Any other changes related to structural abnormalities of iPSC-CMs carrying LMNA R249Q have not been observed.
To investigate whether the expression of cardiac-specific genes is altered in LMNA R249Q cardiomyocytes, we measured expression levels of the cardiac transcription factors TBXT, ISL1, GATA4, MEF2C and sarcomeric genes TNNT2 and MYH7 in control and LMNA R249Q iPSC-CMs by qPCR. Measurements were performed on different developmental stages of cardiogenic differentiation (0, 2, 5, 7, 14, 21 days) (Figure 1C). CHIR99021 treatment at the early stages of differentiation protocol activates mesoderm formation and thus markedly increases the expression of mesodermal marker TBXT (day 2). LMNA R249Q iPSC-CMs demonstrated significantly higher TBXT level compared to control iPSC-CMs. Expression of the essential early-stage cardiac genes (ISL1, GATA4, MEF2C) was detected after two days of differentiation. The LMNA R249Q was linked with elevated expression of these genes. In detail, increased ISL1 level was observed in the LMNA R249Q iPSC-CMs on 5, 7, 14, and 21 days of differentiation. The GATA4 level was increased in LMNA R249Q iPSC-CMs on day 7, and the MEF2C level was increased in LMNA R249Q iPSC-CMs on 5, 7, and 14 days of cardiogenic differentiation.
Expression of late-stage cardiac-specific markers (TNNT2 and MYH7) was detected starting from 5 to 7 days of differentiation. TNNT2 had a higher level in LMNA R249Q iPSC-CMs compared to control iPSC-CMs on day 5 and day 21 of differentiation. MYH7 expression was significantly increased in LMNA R249Q iPSC-CMs compared to control iPSC-CMs on day 14 and day 21 of differentiation.
Taking together, LMNA R249Q iPSC-CMs demonstrated increased expression levels of mesodermal and cardiac-specific transcription factors as well as sarcomeric genes, indicating overall dysregulation of gene expression profile in iPSC-CMs carrying LMNA R249Q genetic variant.
To elucidate whether the LMNA R249Q can affect gene expression in mature CMs we focused on the late-stage of cardiogenic differentiation (day 21) (Figure 2). We explored the expression of genes coding for sarcomeric proteins—ACTN2 and FLNC (Figure 2A), and found a significant upregulation of these genes in CMs carrying LMNA R249Q compared to control CMs. In addition, we observed a significant increase in the expression level of the DMPK gene, encoding myotonic dystrophy protein kinase (Figure 2B), and increased expression level of the SCN5A gene, encoding cardiac voltage-gated sodium channel Nav1.5. However, any changes in the expression of genes coding for ion channels SCN4B, TRPM4, and HCN4 have not been found (Figure 2C). Genes encoding components of intercellular contacts such as PKP2, DSP, and GJA5 were downregulated in LMNA R249Q CMs (Figure 2D). Thus, observed complex alterations in the expression level of the key structural genes could contribute to the cardiac manifestation of the laminopathies.
Figure 2
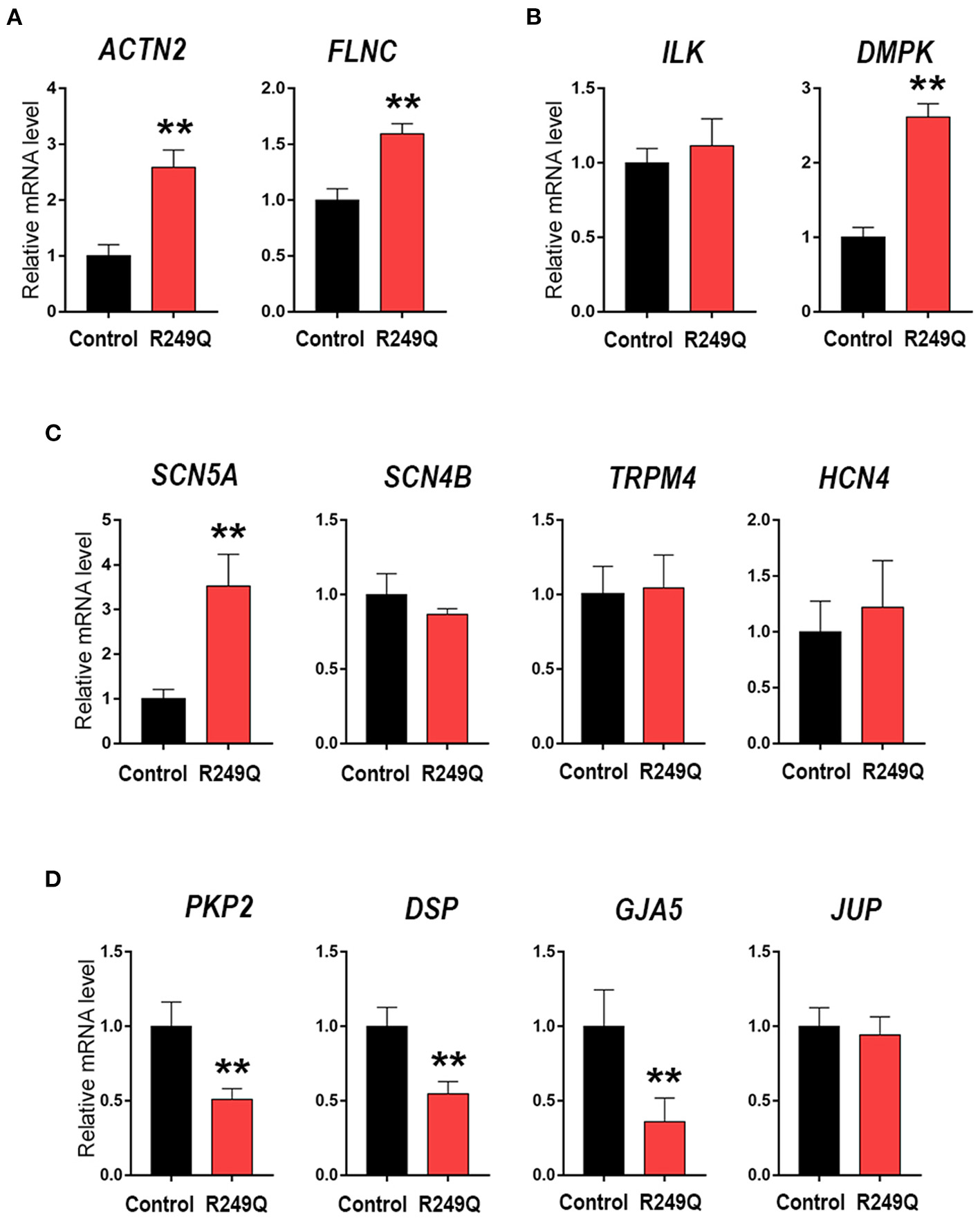
Cardiac genes expression in Control iPSC-CMs and LMNA R249Q iPSC-CMs on day 21 of the cardiogenic differentiation. mRNA level was measured by qPCR. The results are represented as mean ± SD; ** p < 0.01. (A) Level expression of the genes connected to sarcomeric cytoskeleton. (B) Genes encoding regulatory cardiac kinase. (C) Genes encoding ion channels. (D) Genes encoding proteins of an intercalated discs.
Functional characterization of voltage-gated sodium channel in the patient's cells
To define the functional effect of the LMNA genetic variant on the electrophysiological properties of iPSC-CMs, we performed sodium current recording via a patch-clamp technique. All experimental groups demonstrated typical sodium current traces (Figure 3A). The peak current density of iPSC-CMs from two different healthy controls did not differ significantly (Figure 3B; Table 1). However, patient's iPSC-CMs showed a marked reduction of peak current density compared with both controls. The analysis of the current-voltage relationship revealed that iPSC-CMs from both controls had their maximal current at −25 mV, whereas the patient's iPSC-CMs had a maximal current at a more depolarized potential of −20 mV (Figure 3C). It is well known that decreased current density may be caused by alterations in channel gating as well as protein trafficking disturbances.
Figure 3
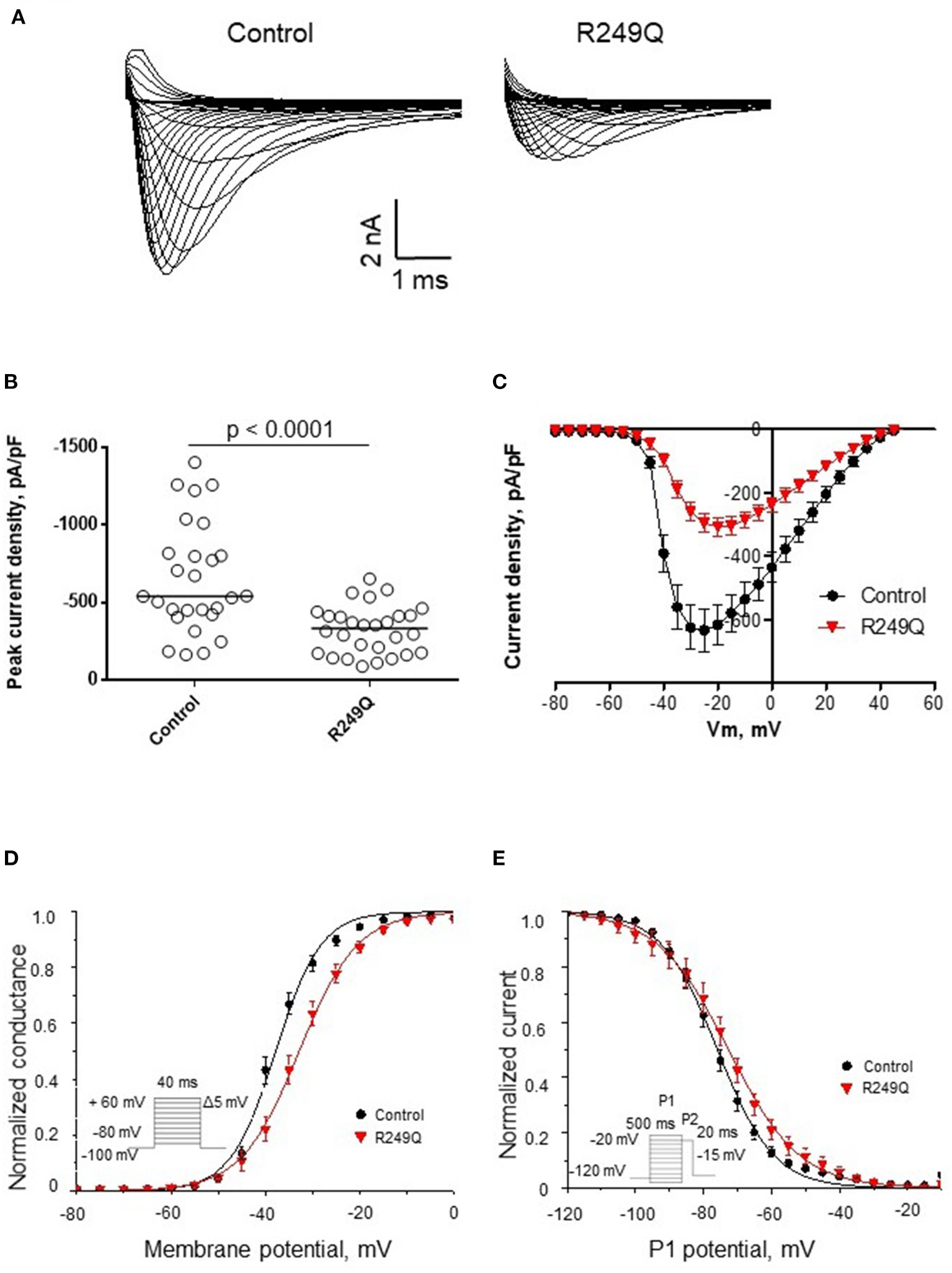
Functional characterization of sodium channel activity in iPSC-CMs carrying R249Q genetic variant in LMNA gene. (A) Typical sodium current traces in iPSC-CMs. (B) Peak sodium current densities for control and patient's iPSC-CMs. Data presented as medians. Patient's iPSC-CMs demonstrated marked decrease in sodium current density. (C) Current-voltage relationship of sodium channel in control and patient's iPSC-CMs. (D) Voltage-dependence of steady-state activation for control and patient's iPSC-CMs. Patient's iPSC-CMs showed delayed activation. (E) Voltage-dependence of steady-state inactivation for control and patient's iPSC-CMs. Patient's iPSC-CMs demonstrated impaired inactivation.
Table 1
| Control | n | R249Q | n | p | ||
|---|---|---|---|---|---|---|
| Current density at −20 mV | pA/pF | −652.9 ± 68.4 | 27 | −324.9 ± 29.3 | 28 | p < 0.0001 |
| Steady-state activation | V 1/2, mV | −37.7 ± 0.8 | 27 | −33.0 ± 1.2 | 28 | 0.002 |
| k | 4.0 ± 0.3 | 4.7 ± 0.2 | 0.02 | |||
| Steady-state inactivation | V 1/2, mV | −75.7 ± 1.3 | 23 | −73.4 ± 2.5 | 22 | 0.09 |
| k | 7.1 ± 0.3 | 8.0 ± 0.4 | 0.07 |
Biophysical parameters of sodium current in iPSC-CMs obtained from control and the patient's iPSC-CMs.
Capacitance. Control iPSC-CMs: 9.5 ± 0.85 pF; patient's iPSC-CMs: 11.4 ± 0.8 pF. p = 0.07.
To examine the reasons for sodium current density reduction, we explored the kinetic parameters of the channel steady-state activation and inactivation. LMNA R249Q iPSC-CMs demonstrated a significant depolarizing shift of the voltage-dependence of steady-state activation compared to control iPSC-CMs (Figure 3D; Table 1). These changes in channel gating properties indicate loss-of-function of the sodium channel in patient-derived iPSC-CMs. Surprisingly, the voltage-dependence of steady-state inactivation was shifted to depolarized potential (Figure 3E; Table 1), which is typical for gain-of-function phenotype.
Thus, LMNA R249Q expression resulted in altered sodium channel functional activity of iPSC-CMs, reflecting the complex influence of the LMNA variant on the electrophysiological properties of the patient's cardiac myocytes. Therefore, observed changes in sodium current characteristics can be considered as possible mechanism of arrhythmia development in this patient.
Discussion
Mutations in the LMNA gene, encoding A-type lamins, are known to cause heredity diseases with a wide range of phenotypes—laminopathies. One of the most frequent manifestations of these pathologies are heart rhythm and conduction abnormalities, including atrial fibrillation, ventricular arrhythmia, as well as sudden cardiac death. Dysfunction of a cardiac system can occur as a separate disease or as an accompanying pathology (22, 43–46). According to the gene transcription hypothesis (21), lamin A/C is considered a key regulator of gene expression. Therefore, we analyzed the expression profile of cardiac-specific genes in LMNA R249Q iPSC-CMs on the late-stage of cardiac differentiation. Obtained results showed the upregulation of the genes encoding the cytoskeletal proteins actin and filamin C in LMNA R249Q iPSC-CMs. The significant role of actin and filamin C in cardiomyocytes' physiology has previously been established (47). Both filamin C and actin were reported to contribute to the cortical cytoskeleton, thereby protecting the cardiomyocyte's structure from mechanical stress (48). Previously, DCM pathology was attributed to the disorganization of the intercalated discs and changes of cardiomyocyte shape. In particular, these changes refer to an increased expression of actin anchoring proteins suggesting an increased presence of filamentous actin (49). Considering that lamin A/C cooperates with actin via the LINC complex and participates in the regulation of the structural architecture of the contractile tissue (20), amino acid substitution in lamin A/C can impact sarcomere organization. In addition, lamin A/C, being involved in the regulation of the gene expression, can alter the proper transcriptional status of the cell because of LMNA genetic variants.
Furthermore, we demonstrated variation in cardiac genes expression in LMNA R249Q iPSC-CMs on the different stages of the cardiogenic differentiation. Since mutations in the LMNA gene lead to rearrangement of the spatial anchoring of genomic regions (referred to as LADs) to the nuclear lamina, its conformational changes are known to cause dysregulation of the gene expression (50). As a result, the earlier launch of the differentiation program can happen. In the present study, we found an enhanced cardiogenic differentiation capacity of the iPSC carrying the LMNA R249Q genetic variant. In the earlier report, an activation effect of the tissue-specific LMNA genetic variants on the adipogenic and osteogenic potential of human multipotent mesenchymal stromal cells was described (51). Another research established that both LMNA G232E/R482L variants caused dysregulation of muscle differentiation (52). From this perspective, we can assume that lamin A/C plays an essential role in the cell fate determination, and LMNA R249Q genetic variant leads to dysregulation of the differentiation program in the iPSC-CMs model.
Though clinical manifestations of cardiac arrhythmias and conduction abnormalities linked to LMNA variants have frequently been reported, there are only few reports of cardiac ion channels changes associated with LMNA genetic variants. In the present study, we determined that in LMNA R249Q iPSC-CMs, electrophysiological functions of cardiac sodium channel were markedly altered.
Recent work on iPSC-CMs revealed the effect of the LMNA K219T on the Nav1.5 function. LMNA K219T expression resulted in a decreased sodium current density and slower conduction velocity combined with reduced SCN5A, thus indicating an impaired activity of Nav1.5 channels (35). Our study found a similar effect of the LMNA R249Q variant on the Nav1.5 parameters, decreased sodium current density, and changes in sodium current kinetics. The genotype-phenotype correlation between LMNA genetic variant and resultant phenotype implies that pathologies caused by closely localized substitutions (both are localized in the rod domain of lamin A/C filament) share a common disease mechanism (53). However, in the present study, the expression of SCN5A in LMNA R249Q iPSC-CMs was increased, which could be a compensatory alteration. Probably even closely localized mutations in the LMNA do not share the completely identical pathogenic mechanisms via the epigenetically mediated regulation of SCN5A expression.
The question remains open whether the sodium current dysfunction is a primary or secondary consequence of the observed genetic variant in the LMNA gene. According to the expression data, LMNA R249Q iPSC-CMs had decreased levels of PKP2, DSP, and GJA5. The role of cell junction proteins in proper channel localization and function in intercalated discs is well established. Plakophilin and desmoplakin were reported to be integrated in the macromolecular complex of Nav1.5 in an intercalated discs pool of sodium channels (54). Nav1.5 was shown to be co-localized with plakophilin in intercalated discs, and the loss of PKP2 expression resulted in sodium current reduction in culture of cardiac myocytes (55). The iPSC-CMs based studies of the effect of PKP2 mutations on sodium current confirmed that PKP2 haploinsufficiency could lead to decreased sodium channel activity (33). Thus, in the current study, we further support the hypothesis that impaired expression of PKP2 can lead to Nav1.5 dysfunction.
Along with the previous electrophysiological characterization of the DSP H1684R variant in iPSC-CMs linked to the decreased sodium and calcium currents (56), the present results underscore the importance of intercellular contact proteins in proper channel functioning. The role of the impaired expression of GJA5 in sodium channel dysfunction remains unclear, despite the reported association of GJA5 mutations with atrial fibrillation (57). Taken together, the sodium current reduction can be considered a consequence of PKP2 and DSP deficiency in the LMNA patient cells. Precise molecular mechanisms underlying the electrophysiological defects in LMNA-associated cardiomyopathy remain unclear and demand further investigations.
To sum up, our results support the fundamental role of lamin A/C in cardiogenic differentiation, regulation of cardiogenic gene expression, and electrophysiology properties of the sodium ion channel.
Funding
This work was financially supported by the Ministry of Science and Higher Education of the Russian Federation (Agreement No. 075-15-2022-301).
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving human participants were reviewed and approved by Almazov National Medical Research Centre (Approval No. 13/19.06.2014). The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
KP and AZ performed the experiments, drafted, and contributed to the writing of the manuscript. AKh provided helpful advice and critical reading of the paper. IN participated in the generation and characterization of the iPSC lines. AKo, AM, and EV contributed to the conception or design of the work, final approval of the version to be published. AKo and EV provided funding acquisition. All authors approved the final version of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.932956/full#supplementary-material
- CMs
cardiomyocites
- DCM
dilated cardiomyopathy
- EDMD
Emery-Dreifuss muscular dystrophy
- iPSC
induced pluripotent stem cell
- LADs
lamina-associated domains
- LINC
linker of nucleoskeleton and cytoskeleton
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction.
Abbreviations
References
1.
Lin F Worman HJ . Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem. (1993) 268:16321–6. 10.1016/S0021-9258(19)85424-8
2.
Alsheimer M Von Glasenapp E Schnölzer M Heid H Benavente R . Meiotic lamin C2: The unique amino-terminal hexapeptide GNAEGR is essential for nuclear envelope association. Proc Natl Acad Sci USA. (2000) 97:13120–5. 10.1073/pnas.240466597
3.
Machiels BM Zorenc AHG Endert JM Kuijpers HJH Van Eys GJJM Ramaekers FCS et al . An alternative splicing product of the lamin A/C gene lacks exon 10. J Biol Chem. (1996) 271:9249–53. 10.1074/jbc.271.16.9249
4.
Schreiber KH Kennedy BK . When lamins go bad: Nuclear structure and disease. Cell. (2013) 152:1365–75. 10.1016/j.cell.2013.02.015
5.
Crasto S My I Di Pasquale E . The broad spectrum of LMNA cardiac diseases: from molecular mechanisms to clinical phenotype. Front Physiol. (2020) 11:1–11. 10.3389/fphys.2020.00761
6.
Dechat T Pfleghaar K Sengupta K Shimi T Shumaker DK Solimando L et al . Nuclear lamins: major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. (2008) 22:832–53. 10.1101/gad.1652708
7.
Dittmer T Misteli T . The lamin protein family. Genome Biol. (2011) 12. 10.1186/gb-2011-12-5-222
8.
Gruenbaum Y Margalit A Goldman RD Shumaker DK Wilson KL . The nuclear lamina comes of age. Nat Rev Mol Cell Biol. (2005) 6:21–31. 10.1038/nrm1550
9.
Malashicheva A Perepelina K . Diversity of nuclear lamin A/C action as a key to tissue-specific regulation of cellular identity in health and disease. Front Cell Dev Biol. (2021) 9:1–18. 10.3389/fcell.2021.761469
10.
de las Heras JI Meinke P Batrakou DG Srsen V Zuleger N Kerr ARW et al . Tissue specificity in the nuclear envelope supports its functional complexity. Nucleus. (2013) 4:460–77. 10.4161/nucl.26872
11.
Worman HJ . Nuclear lamins and laminopathies. J Pathol. (2012) 226:316–25. 10.1002/path.2999
12.
Arbustini E Pilotto A Repetto A Grasso M Negri A Diegoli M et al . Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease. J Am Coll Cardiol. (2002) 39:981–90. 10.1016/S0735-1097(02)01724-2
13.
van Tintelen JP Hofstra RMW Katerberg H Rossenbacker T Wiesfeld ACP du Marchie Sarvaas GJ et al . High yield of LMNA mutations in patients with dilated cardiomyopathy and/or conduction disease referred to cardiogenetics outpatient clinics. Am Heart J. (2007) 154:1130–9. 10.1016/j.ahj.2007.07.038
14.
Emery AE Dreifuss FE . Unusual type of benign X linked muscular dystrophy. J Neurol Neurosurg Psychiatry. (1996) 84:4–5. 10.1136/jnnp-2012-304198
15.
Heller SA Shih R Kalra R Kang PB . Emery-Dreifuss muscular dystrophy. Muscle and Nerve. (2020) 61:436–48. 10.1002/mus.26782
16.
Muchir A Worman HJ . Emery-Dreifuss muscular dystrophy. Curr Neurol Neurosci Rep. (2007) 7:78–83. 10.1007/s11910-007-0025-3
17.
Nikolova V Leimena C McMahon AC Tan JC Chandar S Jogia D et al . Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest. (2004) 113:357–69. 10.1172/JCI200419448
18.
Pickersgill H Kalverda B De Wit E Talhout W Fornerod M Van Steensel B et al . Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat Genet. (2006) 38:1005–14. 10.1038/ng1852
19.
Ye Q Worman HJ . Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins homologous to Drosophila HP1. J Biol Chem. (1996) 271:14653–6. 10.1074/jbc.271.25.14653
20.
Swift J Ivanovska IL Buxboim A Harada T Dingal PCDP Pinter J et al . Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science. (2013) 341. 10.1126/science.1240104
21.
Osmanagic-Myers S Foisner R . The structural and gene expression hypotheses in laminopathic diseases—not so different after all. Mol Biol Cell. (2019) 30:1786–90. 10.1091/mbc.E18-10-0672
22.
Chandar S Yeo LS Leimena C Tan JC Xiao XH Nikolova-Krstevski V et al . Effects of mechanical stress and carvedilol in lamin A/C-deficient dilated cardiomyopathy. Circ Res. (2010) 106:573–82. 10.1161/CIRCRESAHA.109.204388
23.
Tong J Li W Vidal C Yeo LS Fatkin D Duque G et al . Lamin A/C deficiency is associated with fat infiltration of muscle and bone. Mech Ageing Dev. (2011) 132:552–9. 10.1016/j.mad.2011.09.004
24.
Arimura T Helbling-Leclerc A Massart C Varnous S Niel F Lacène E et al . Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Hum Mol Genet. (2005) 14:155–69. 10.1093/hmg/ddi017
25.
Mounkes LC Kozlov SV Rottman JN Stewart CL . Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Hum Mol Genet. (2005) 14:2167–80. 10.1093/hmg/ddi221
26.
Crasto S De Pasquale E . Induced pluripotent stem cells to study mechanisms of laminopathies: Focus on epigenetics. Front Cell Dev Biol. (2018) 6:1–14. 10.3389/fcell.2018.00172
27.
Giacomelli E Meraviglia V Campostrini G Cochrane A Cao X van Helden RWJ et al . Human-iPSC-derived cardiac stromal cells enhance maturation in 3D cardiac microtissues and reveal non-cardiomyocyte contributions to heart disease. Cell Stem Cell. (2020) 26:862–79.e11. 10.1016/j.stem.2020.05.004
28.
Shah PP Lv W Rhoades JH Poleshko A Abbey D Caporizzo MA et al . Pathogenic LMNA variants disrupt cardiac lamina-chromatin interactions and de-repress alternative fate genes. Cell Stem Cell. (2021) 28:938–954.e9. 10.1016/j.stem.2020.12.016
29.
Steele-Stallard HB Pinton L Sarcar S Ozdemir T Maffioletti SM Zammit PS et al . Modeling skeletal muscle laminopathies using human induced pluripotent stem cells carrying pathogenic LMNA mutations. Front Physiol. (2018) 9:1–19. 10.3389/fphys.2018.01332
30.
Kamga MVK Reppel M Hescheler J Nguemo F . Modeling genetic cardiac channelopathies using induced pluripotent stem cells—Status quo from an electrophysiological perspective. Biochem Pharmacol. (2021) 192. 10.1016/j.bcp.2021.114746
31.
El-Battrawy I Müller J Zhao Z Cyganek L Zhong R Zhang F et al . Studying brugada syndrome with an SCN1B variants in human-induced pluripotent stem cell-derived cardiomyocytes. Front Cell Dev Biol. (2019) 7:1–14. 10.3389/fcell.2019.00261
32.
Zhu Y Wang L Cui C Qin H Chen H Chen S et al . Pathogenesis and drug response of iPSC-derived cardiomyocytes from two Brugada syndrome patients with different Nav1.5-subunit mutations. J Biomed Res. (2021) 35:395–407. 10.7555/JBR.35.20210045
33.
Khudiakov A Zaytseva A Perepelina K Smolina N Pervunina T Vasichkina E et al . Sodium current abnormalities and deregulation of Wnt/β-catenin signaling in iPSC-derived cardiomyocytes generated from patient with arrhythmogenic cardiomyopathy harboring compound genetic variants in plakophilin 2 gene. Mol Basis Dis. (2020) 1866:165915. 10.1016/j.bbadis.2020.165915
34.
Poulin H Mercier A Djemai M Pouliot V Deschenes I Boutjdir M et al . iPSC-derived cardiomyocytes from patients with myotonic dystrophy type 1 have abnormal ion channel functions and slower conduction velocities. Sci Rep. (2021) 11:1–13. 10.1038/s41598-021-82007-8
35.
Salvarani N Crasto S Miragoli M Bertero A Paulis M Kunderfranco P et al . The K219T-Lamin mutation induces conduction defects through epigenetic inhibition of SCN5A in human cardiac laminopathy. Nat Commun. (2019) 10:1–16. 10.1038/s41467-019-09929-w
36.
Perepelina K Kostina A Klauzen P Khudiakov A Rabino M Crasto S et al . Generation of two iPSC lines (FAMRCi007-A and FAMRCi007-B) from patient with Emery–Dreifuss muscular dystrophy and heart rhythm abnormalities carrying genetic variant LMNA p.Arg249Gln. Stem Cell Res. (2020). 47:101895. 10.1016/j.scr.2020.101895
37.
Di Barletta MR Ricci E Galluzzi G Tonali P Mora M Morandi L et al . Different mutations in the LMNA gene cause autosomal dominant autosomal recessive Emery-Dreifuss muscular dystrophy. Am J Hum Genet. (2000) 66:1407–12. 10.1086/302869
38.
Neganova I Chichagova V Armstrong L Lako M . A critical role for p38MAPK signalling pathway during reprogramming of human fibroblasts to iPSCs. Sci Rep. (2017) 7:1–13. 10.1038/srep41693
39.
Chichagova V Sanchez-Vera I Armstrong L Steel D Lako M . Generation of human induced pluripotent stem cells using rna-based sendai virus system and pluripotency validation of the resulting cell population. Methods Mol Biol. (2015) 1353:285–307. 10.1007/7651_2015_205
40.
Neganova I Shmeleva E Munkley J Chichagova V Anyfantis G Anderson R et al . JNK/SAPK signaling is essential for efficient reprogramming of human fibroblasts to induced pluripotent stem cells. Stem Cells. (2016) 34:1198–212. 10.1002/stem.2327
41.
Lian X Hsiao C Wilson G Zhu K Hazeltine LB Azarin SM et al . Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci U S A. (2012) 109. 10.1073/pnas.1200250109
42.
Tohyama S Hattori F Sano M Hishiki T Nagahata Y Matsuura T et al . Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell. (2013) 12:127–37. 10.1016/j.stem.2012.09.013
43.
Chaffee BR Shang F Chang ML Clement TM Eddy EM Wagner BD et al . Nuclear removal during terminal lens fiber cell differentiation requires CDK1 activity: appropriating mitosis-related nuclear disassembly. Dev. (2014) 141:3388–98. 10.1242/dev.106005
44.
Chen SN Lombardi R Karmouch J Tsai JY Czernuszewicz G Taylor MRG et al . DNA Damage response/TP53 pathway is activated and contributes to the pathogenesis of dilated cardiomyopathy associated with LMNA (Lamin A/C) mutations. Circ Res. (2019) 124:856–73. 10.1161/CIRCRESAHA.118.314238
45.
Corrigan DP Kuszczak D Rusinol AE Thewke DP Hrycyna CA Michaelis S et al . Prelamin A endoproteolytic processing in vitro by recombinant Zmpste24. Biochem J. (2005) 387:129–38. 10.1042/BJ20041359
46.
Fatkin D MacRae C Sasaki T Wolff MR Porcu M Frenneaux M et al . Missense mutations in the rod domain of the lamin a/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. (1999) 341:1715–24. 10.1056/NEJM199912023412302
47.
Ehler E . Actin-associated proteins and cardiomyopathy—the ‘unknown' beyond troponin and tropomyosin. Biophys Rev. (2018) 10:1121–8. 10.1007/s12551-018-0428-1
48.
Fujita M Mitsuhashi H Isogai S Nakata T Kawakami A Nonaka I et al . Filamin C plays an essential role in the maintenance of the structural integrity of cardiac and skeletal muscles, revealed by the medaka mutant zacro. Dev Biol. (2012) 361:79–89. 10.1016/j.ydbio.2011.10.008
49.
Ehler E Horowits R Zuppinger C Price RL Perriard E Leu M et al . Alterations at the intercalated disk associated with the absence of muscle LIM protein. J Cell Biol. (2001) 153:763–72. 10.1083/jcb.153.4.763
50.
Briand N Collas P . Lamina-associated domains: Peripheral matters and internal affairs. Genome Biol. (2020) 21:1–25. 10.1186/s13059-020-02003-5
51.
Malashicheva A Bogdanova M Zabirnyk A Smolina N Ignatieva E Freilikhman O et al . Various lamin A/C mutations alter expression profile of mesenchymal stem cells in mutation specific manner. Mol Genet Metab. (2015) 115:118–27. 10.1016/j.ymgme.2015.04.006
52.
Ignatieva EV Ivanova OA Komarova MY Khromova NV Polev DE Kostareva AA et al . Lmna mutations g232e and r482l cause dysregulation of skeletal muscle differentiation, bioenergetics, and metabolic gene expression profile. Genes (Basel). (2020) 11:1–20. 10.3390/genes11091057
53.
Scharner J Gnocchi VF Ellis JA Zammit PS . Genotype-phenotype correlations in laminopathies: how does fate translate? Biochem. Soc Trans. (2010) 38:257–62. 10.1042/BST0380257
54.
Shy D. Gillet L. Abriel H. (2013). Cardiac sodium channel NaV1.5 distribution in myocytes via interacting proteins: the multiple pool model. Mol. Cell Res. 1833:886–94. 10.1016/j.bbamcr.2012.10.026
55.
Sato PY Musa H Coombs W Guerrero-Serna G Patiño GA Taffet SM et al . Loss of plakophilin-2 expression leads to decreased sodium current and slower conduction velocity in cultured cardiac myocytes. Circ Res. (2009) 105:523–6. 10.1161/CIRCRESAHA.109.201418
56.
Gusev K Khudiakov A Zaytseva A Perepelina K Makeenok S Kaznacheyeva E et al . Impact of the DSP-H1684R genetic variant on ion channels activity in iPSC-derived cardiomyocytes. Cell Physiol Biochem. (2020) 54:696–706. 10.33594/000000249
57.
Bai D . Atrial fibrillation-linked GJA5/connexin40 mutants impaired gap junctions via different mechanisms. FEBS Lett. (2014) 588:1238–43. 10.1016/j.febslet.2014.02.064
Summary
Keywords
lamin A/C, LMNA mutation, cardiomyopathy, induced pluripotent stem cells, sodium ion channel, cell differentiation
Citation
Perepelina K, Zaytseva A, Khudiakov A, Neganova I, Vasichkina E, Malashicheva A and Kostareva A (2022) LMNA mutation leads to cardiac sodium channel dysfunction in the Emery-Dreifuss muscular dystrophy patient. Front. Cardiovasc. Med. 9:932956. doi: 10.3389/fcvm.2022.932956
Received
30 April 2022
Accepted
27 June 2022
Published
22 July 2022
Volume
9 - 2022
Edited by
Sakthivel Sadayappan, University of Cincinnati, United States
Reviewed by
Shiwei Yang, Children's Hospital of Nanjing Medical University, China; Amr Ghanam, Augusta University, United States
Updates
Copyright
© 2022 Perepelina, Zaytseva, Khudiakov, Neganova, Vasichkina, Malashicheva and Kostareva.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kseniya Perepelina perepelina.kseniya93@gmail.comAnastasia Zaytseva zaytseva.anastasia.zak@gmail.com
†These authors have contributed equally to this work and share first authorship
This article was submitted to Cardiovascular Genetics and Systems Medicine, a section of the journal Frontiers in Cardiovascular Medicine
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.