 Huang Rui
Huang Rui Fang Zhao1,2,3†
Fang Zhao1,2,3†- 1Department of Cardiology, Renmin Hospital of Wuhan University, Wuhan, China
- 2Cardiovascular Research Institute, Wuhan University, Wuhan, China
- 3Hubei Key Laboratory of Cardiology, Wuhan, China
- 4Department of Cardiology, The Central Hospital of Enshi Tujia and Miao Autonomous Prefecture, Enshi City, China
Background: Fibrosis of the myocardium is one of the main pathological changes of adverse cardiac remodeling, which is associated with unsatisfactory outcomes in patients with heart disease. Further investigations into the precise molecular mechanisms of cardiac fibrosis are urgently required to seek alternative therapeutic strategies for individuals suffering from heart failure. SMOC2 has been shown to be essential to exert key pathophysiological roles in various physiological processes in vivo, possibly contributing to the pathogenesis of fibrosis. A study investigating the relationship between SMOC2 and myocardial fibrosis has yet to be conducted.
Methods: Mice received a continuous ISO injection subcutaneously to induce cardiac fibrosis, and down-regulation of SMOC2 was achieved by adeno-associated virus-9 (AAV9)-mediated shRNA knockdown. Neonatal fibroblasts were separated and cultured in vitro with TGFβ to trigger fibrosis and infected with either sh-SMOC2 or sh-RNA as a control. The role and mechanisms of SMOC2 in myocardial fibrosis were further examined and analyzed.
Results: SMOC2 knockdown partially reversed cardiac functional impairment and cardiac fibrosis in vivo after 21 consecutive days of ISO injection. We further demonstrated that targeting SMOC2 expression effectively slowed down the trans-differentiation and collagen deposition of cardiac fibroblasts stimulated by TGFβ. Mechanistically, targeting SMOC2 expression inhibited the induction of ILK and p38 in vivo and in vitro, and ILK overexpression increased p38 phosphorylation activity and compromised the protective effects of sh-SMOC2-mediated cardiac fibrosis.
Conclusion: Therapeutic SMOC2 silencing alleviated cardiac fibrosis through inhibition of the ILK/p38 signaling, providing a preventative and control strategy for cardiac remodeling management in clinical practice.
Introduction
Adverse cardiac remodeling, defined as changes in left ventricular (LV) anatomical structure and function, is a common complication developed in the diseased heart as a result of an array of intra- and extra cardiac pathophysiological conditions and is correlated with a poor outcome and shortened survival in hospitalized individuals presenting with heart conditions (1–3). Implications from substantial evidence suggest that abnormal hypertrophy of existing cardiomyocytes and perpetual activation of cardiac fibroblasts are fundamental pathomechanisms of heart failure (4). Previous studies have demonstrated the involvement of AKT and MAPK pathways in myocardial remodeling and myocardial fibrosis, which has been widely recognized (5, 6). However, fibrosis of the myocardium is a complex pathological process with a multifactorial etiology, and further investigation into the specific in-depth molecular mechanisms is still imperative for improving prognoses (4, 7).
SMOC2, SPARC-related modular calcium-binding protein 2, which encodes a secreted modular protein containing a pair of FS domains and an EC domain and predominantly resides in the kidneys, lungs, myocardium, skeletal muscles, and ovaries, forms part of the BM-40/SPARC/osteonectin protein family as SMOC1 (8, 9). As a result of binding to cell surface receptors, cytokines, and proteases, SMOC2 regulates the cell-matrix interaction, facilitating ischemic myocardial microvascular regeneration and tumor cell growth (8, 10–12). SMOC2 is implicated in some preclinical studies as contributing to the regulation of fibrotic disorders (13–16). Study results previously suggested that SMOC2 could stimulate renal fibroblast-to-myofibroblast conversion or transition, facilitate extracellular matrix synthesis, and ultimately result in kidney fibrosis (13). As evidence for this finding, Xin et al. (14) disclosed that silencing SMOC2 could affect renal inflammation, fibrosis, and function in chronic kidney disease (CKD) mice. A subsequent study discovered that SMOC2 knockout was available to mitigate bleomycin-induced pulmonary fibrosis (15). In addition, a network analysis of expression profile data has identified that SMOC2 may be implicated in the pathogenesis of heart failure (17), which may allude to a possible connection between SMOC2 and cardiac fibrosis despite the lack of studies demonstrating this link.
Studies have shown that SMOC2 exerts its physiological effects in vivo in part by transducing integrin-linked kinase (ILK)-mediated intracellular signaling cascades (18, 19). SMOC2 is also involved in activating ILK directly, enabling it to propel the cell cycle forward (20). Overexpression of ILK increases the collagen type I expression in cardiac fibroblasts by activating nuclear factor-κB (NF-κB) while silencing ILK resulted in the reverse effect (21). Additionally, p38-MAPK, a generally accepted pathway involving fibrosis, can also be activated by ILK (22–24). Researchers found that ILK directly activated P38 MAPK to influence osteoblading effects, which could be reversed if ILK was targeted to be silenced (24).
As yet, it is unclear if ILK contributes to cardiac fibrosis by stimulating p38. Consequently, we designed and conceived this study to investigate the effect of SMOC2 on cardiac fibrosis and to understand the detailed mechanisms involved.
Materials and methods
Reagents
Transforming growth factor-β (TGF-β, 763104) was purchased from Biolegend (San Diego, CA, USA). Isoprenaline (ISO, I5627) was obtained from Sigma-Aldrich and prepared in DD water. The SMOC2 (SC-376104, 1:500 for western blot and 1:100 for satining) and ILK (SC-20019, 1:500) antibodies were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Antibodies against collagen type I (Col I, 1:1000), collagen type III (Col III, 1:1000), phospho-JNK (Tyr185,1:1000), JNK (1:2000), phospho-p38 MAPK (Thr180/Tyr182,1:1000), t-p38 (1:1000), phospho-AKT (1:5000), t-AKT (1:5000) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH,1:4000) were obtained from Proteintech (Wuhan, China). Anti-α smooth muscle actin (a-SMA, 1:100 for staining) antibody was purchased from Abcam (Cambridge, UK).
Animals and treatments
All animal experimental procedures were carried out under the supervision of the Animal Care and Use Committee of Renmin Hospital of Wuhan University and in compliance with established guidelines published by the National Institutes of Health of United States. Eight-week-old male C57BL/6 mice (body weight 23.5 ± 2 g) were provided by the Experimental Animal Center of the Three Gorges University (Hubei, China) for this study, and were acclimated to the experimental scenarios for 5–7 days before they were used. Under standard environmental conditions, all animals were fed a pellet diet and had access to water without restrictions. Mice were subcutaneously injected with ISO (10 mg/kg for 3 days and 5 mg/kg for 11 days) to induce cardiac fibrosis (25–27). For the control group, an equal volume of normal saline solution was administered. Four weeks before ISO injection, mice received a single intravenous injection of adeno-associated virus 9 (AAV9) carrying small hairpin RNA against SMOC2 (sh-SMOC2) and its corresponding negative control (shRNA) generated by Obio technology (Shanghai, China) to verify its role in vivo. Animals were randomly assigned to four groups: Control + sh-RNA, Control + sh-SMOC2, ISO + sh-RNA, and ISO + sh-SMOC2. Animals were sacrificed after an excessive inhalation of CO2, having their hearts and tibias harvested and measured to calculate the heart weight (HW)/body weight (BW) and HW/tibia length (TL).
Echocardiography
Following modeling, echocardiographic evaluations were conducted under light anesthesia with a Vinno 6th ultrasound device with Doppler imaging (VINNO6, Vinno Corporation, China). At the level of the left ventricular short-axis papillary muscle, M-mode tracking was used to measure the following parameters: left ventricular end-systolic diameter (LVEDs), left ventricular end-diastolic diameter (LVEDd), left ventricular posterior wall thickness (LVPW), interventricular septal thickness (IST), ejection fraction (EF), and left fractional ventricular shortening (FS).
Cell culture and treatment
A mixture of trypsin and collagenase II was used to extract CFs from the hearts of 1–3 days old mice. CFs were cultured in a medium with 10% FBS (fetal bovine serum) at 37°C with 5% CO2 after removing non-adherent cells after 1 h. Non-adherent cells were removed after 1 h, and the adherent CFs were cultured in DMEM/F12 medium containing 10% FBS (fetal bovine serum) at 37°C with 5% CO2. The following experiments were conducted with 2–3 passages of CFs. Following 8 h of serum-free DMEM/F12 culture, cells were stimulated with TGFβ. In order to knock down the SMOC2, CFs were transfected with short hairpin (sh)RNA adenovirus against SMOC2 performed by Shanghai Jikai Gene Chemical Technology Co., Ltd. As a negative control, adenovirus expressing short hairpin (sh)GFP was used. To confirm the role of ILK, CFs were transfected with adenovirus carrying ILK (Ad-ILK) or green fluorescent protein (Ad-GFP).
Determination of collagen contents
Commercial ELISA kits (collagen I and collagen III ELISA kits from Uscnlife, Wuhan, China) were used to measure collagen I and III content in the cell culture supernatant after cardiac fibroblasts were incubated with or without TGFβ.
Histological analysis and immunofluorescence
Heart tissue samples were fixed in 4% paraformaldehyde and embedded in paraffin before being cut into 5-micron sections. Heart tissue morphology and cardiac fibrosis were assessed with hematoxylin and eosin (HE) and Masson staining, respectively. Quantitative analysis was performed using Image J software.
CFs were stained with immunofluorescence in a well-cultured condition. Briefly, cell permeabilization was enhanced after fixation with 4% paraformaldehyde for 20 min, followed by incubation with 50–100 μl of disruption buffer for 10 min at room temperature. Cell coverslips were incubated with anti-α-SMA and anti-SMOC2 overnight at 4°C. The next day, cells were incubated with goat anti-mouse (SMOC2) pre-adsorbed or goat anti-rabbit (α-SMA) pre-adsorbed secondary antibodies for 1 h. Nuclei were subsequently stained with 4′,6-diamino-2-phenylindole (DAPI) and images were taken with a fluorescence microscope.
Western blot and RT-PCR
The heart tissues or cells were prepared to extract total proteins with RIPA lysis buffer (Servicebio, Wuhan, China). The extracts were resolved via 8 and 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF membranes (Millipore Corp. Bedford, MA, USA) using standard protocols. And then, the membranes were incubated with skim milk at room temperature for 1 h. Membranes were incubated with specific primary antibodies at 4°C overnight, followed by bio-tinylated secondary antibodies (goat anti-mouse HRP and goat anti-rabbit HRP) for 2 h at room temperature. The membranes were washed with TBST three times between each step mentioned above. Finally, the blots were detected by a hypersensitive ECL reagent (Biosharp, Beijing, China).
Total RNA was extracted using TRIzol reagent (Servicebio, Wuhan, China), and a cDNA synthesis kit was carried out to synthesize cDNA following the manufacturer’s protocol. Real-time PCR was carried out using EnTurboTM SYBR Green PCR SuperMix Kit (ELK Biotechnology). The mRNA levels were normalized to GAPDH. Molecular biological tests were conducted by individuals who were unaware of the treatment conditions of the animals and cells.
Statistics analysis
SPSS 26.0 software was used for analysis. All results are presented as mean ± standard error of the mean, and t-test was used for comparison between two groups, and one-way ANOVA was used for three or more groups, followed by post L-S-D test. Differences were considered statistically significant with p < 0.05.
Results
SMOC2 was highly expressed in ISO-induced mouse model
We first investigated whether SMOC2 expression was altered in ISO-induced mouse model. Mice were treated daily with ISO or an equal volume of saline as control for 21 days as described. It was found that SMOC2 protein levels increased markedly in the ISO group, as was the accumulation of collagen types I and III (Figures 1A–D). Notably, RT-PCR further revealed increased expression level of SMOC2 in the ISO group (Figure 1E). The above observations strongly implicate that SMOC2 may play a critical role in regulating ISO-induced myocardial remodeling.
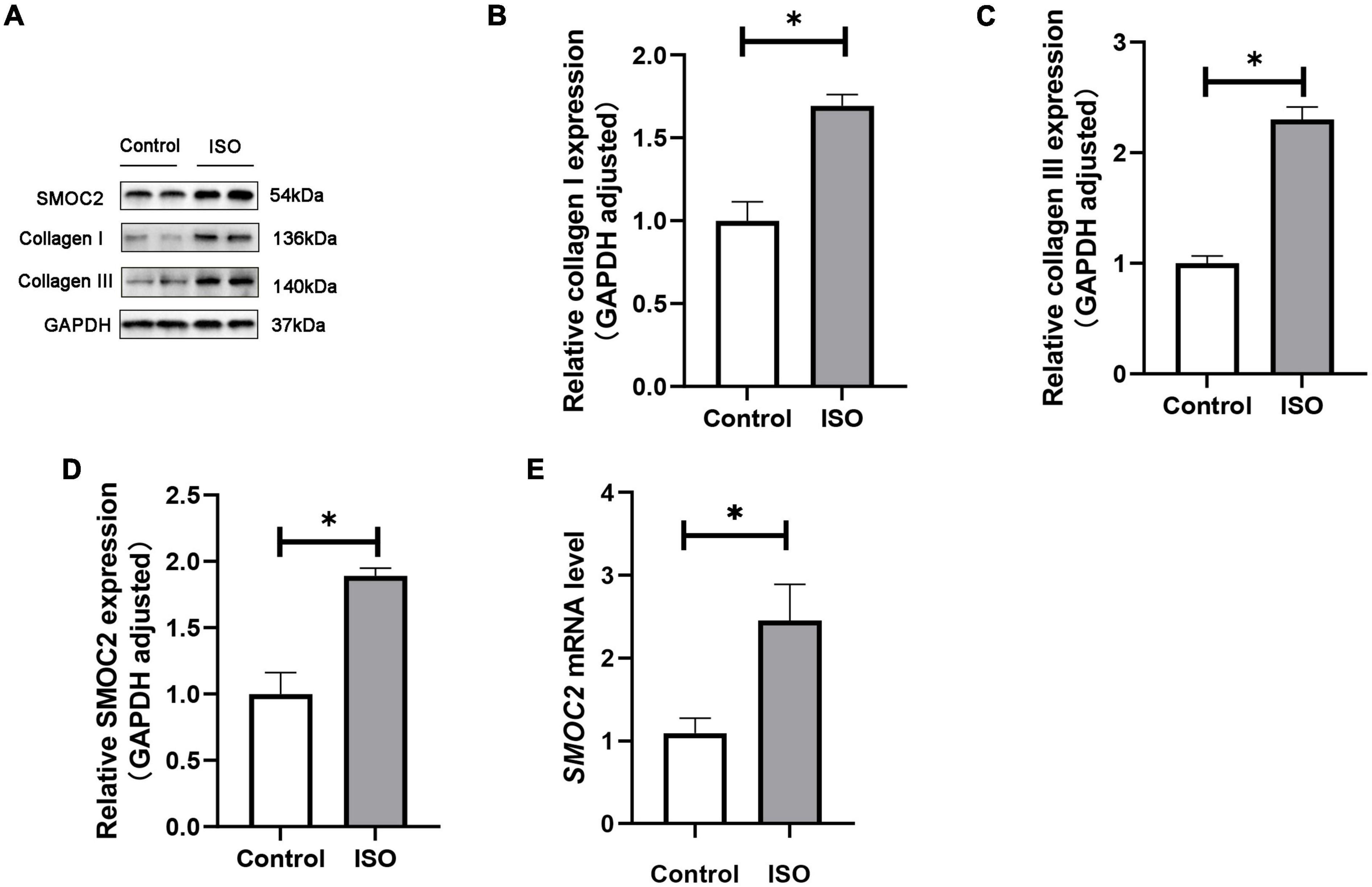
Figure 1. SMOC2 expression was increased in ISO-induced mice hearts. (A–D) Western blot image and quantitative results of the SMOC2, collagen I, and collagen III in the control group and the ISO group (n = 6). (E) RT-PCR analysis of SMOC2 mRNA levels in the two groups (n = 6). *p < 0.05.
SMOC2 knockdown partially reversed the myocardial function loss and myocardial fibrosis in response to ISO stimulation in vivo
We next sought to explore the specific effects of SMOC2 in ISO-induced cardiac fibrosis. Considering the up-regulation of SMOC2 expression in the ISO-induced mouse model, we constructed a SMOC2-knockdown AAV9 system to infect mice as previously described.
As shown in Figures 2A–C, hearts of animals administered sh-SMOC2 showed a significant decrease in SMOC2 mRNA and protein expression after 4 weeks compared to hearts of mice treated with sh-RNA. Mice infected with sh-RNA subjected to ISO injection for 3 weeks developed cardiac hypofunction, manifested as a decrease in EF, FS, and elevation in LVEDd, LVEDs, HW/TL, and HW/BW (Figures 2D–J). Notably, cardiac function was partially reversed in mice injected with sh-SMOC2 (Figures 2D–J). Neither ISO administration nor infection with sh-SMOC2 affected ventricular wall thickness, however (Figures 2K, L).
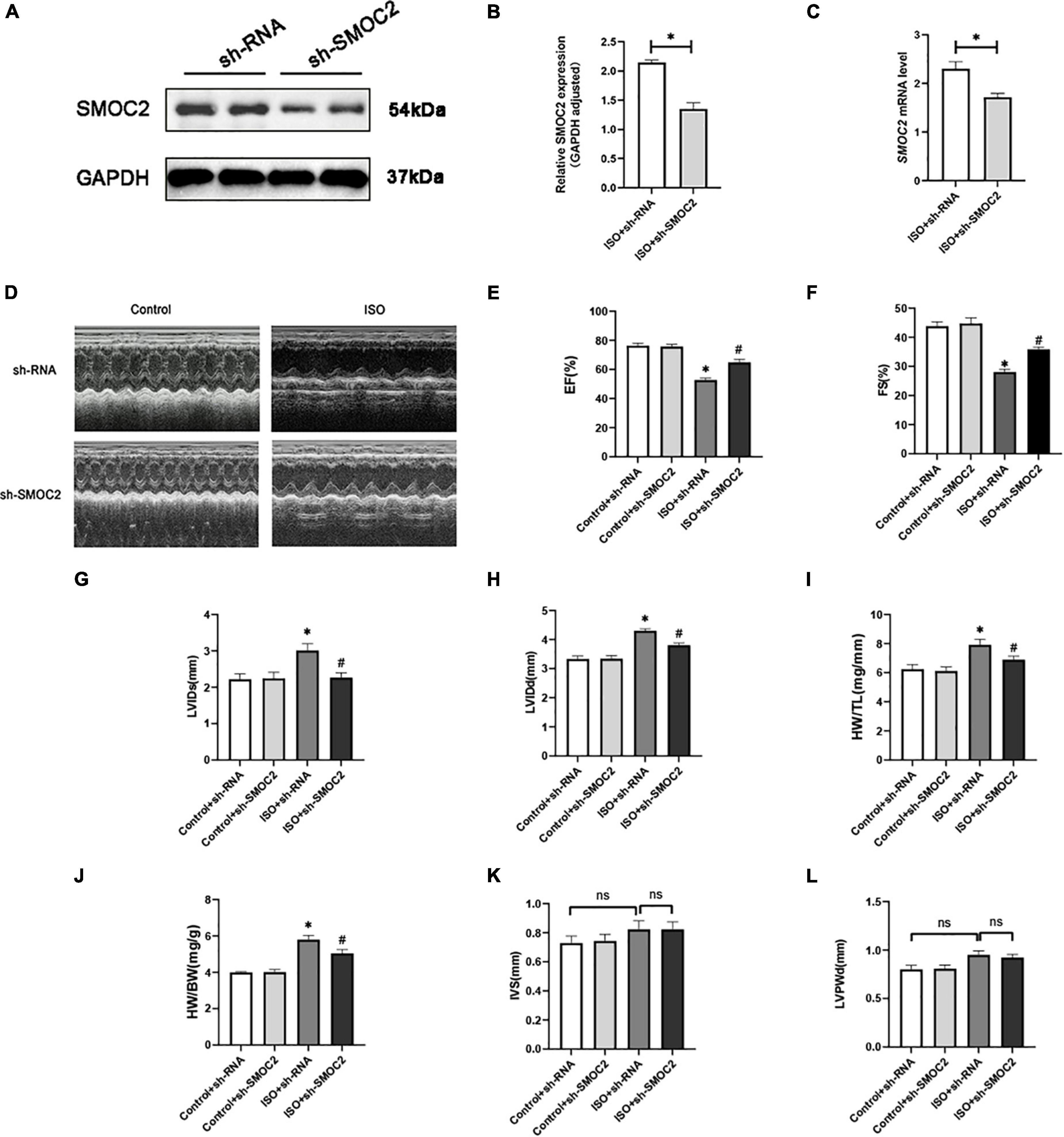
Figure 2. SMOC2 knockdown partially reversed the myocardial function loss. (A,B) SMOC2 expression after sh-RNA or sh-SMOC2 injection in mice heart (n = 6). (C) mRNA levels of SMOC2 after sh-RNA or sh-SMOC2 injection in mice heart (n = 6). (D) Representative images of echocardiographic measurement of cardiac function in mice. (E–L) Echocardiographic measurements of ejection fraction (EF), fractional shortening (FS), left ventricular internal diameter at end-systole (LVIDs), left ventricular internal diameter at end-diastole (LVIDd), HW/TL, HW/BW, interventricular septal thickness at diastole (IVS), and left ventricular posterior wall thickness at diastole (n = 8). *p < 0.05 vs. the control + sh-RNA group. #p < 0.05 vs. the ISO + sh-RNA group. n.s., non-significant.
In addition, morphological examination revealed that mice experienced more severe myofibrillar disarrangement and fibrosis after 3 weeks of ISO injection (Figures 3A, B). Moreover, these histological changes weakened after SMOC2 deficiency (Figures 3A, B). To substantiate this further, we detected biomarkers of myocardial fibrosis. As expected, collagen I and collagen III protein and mRNA levels were down-regulated post-sh-SMOC2 injection (Figures 3C–G). These findings implied an underlying physiopathological impact of SMOC2 in modulating cardiac fibrosis, and SMOC2 knockdown in mice can partially reverse the myocardial function reduction and myocardial fibrosis in response to ISO stimulation in vivo.
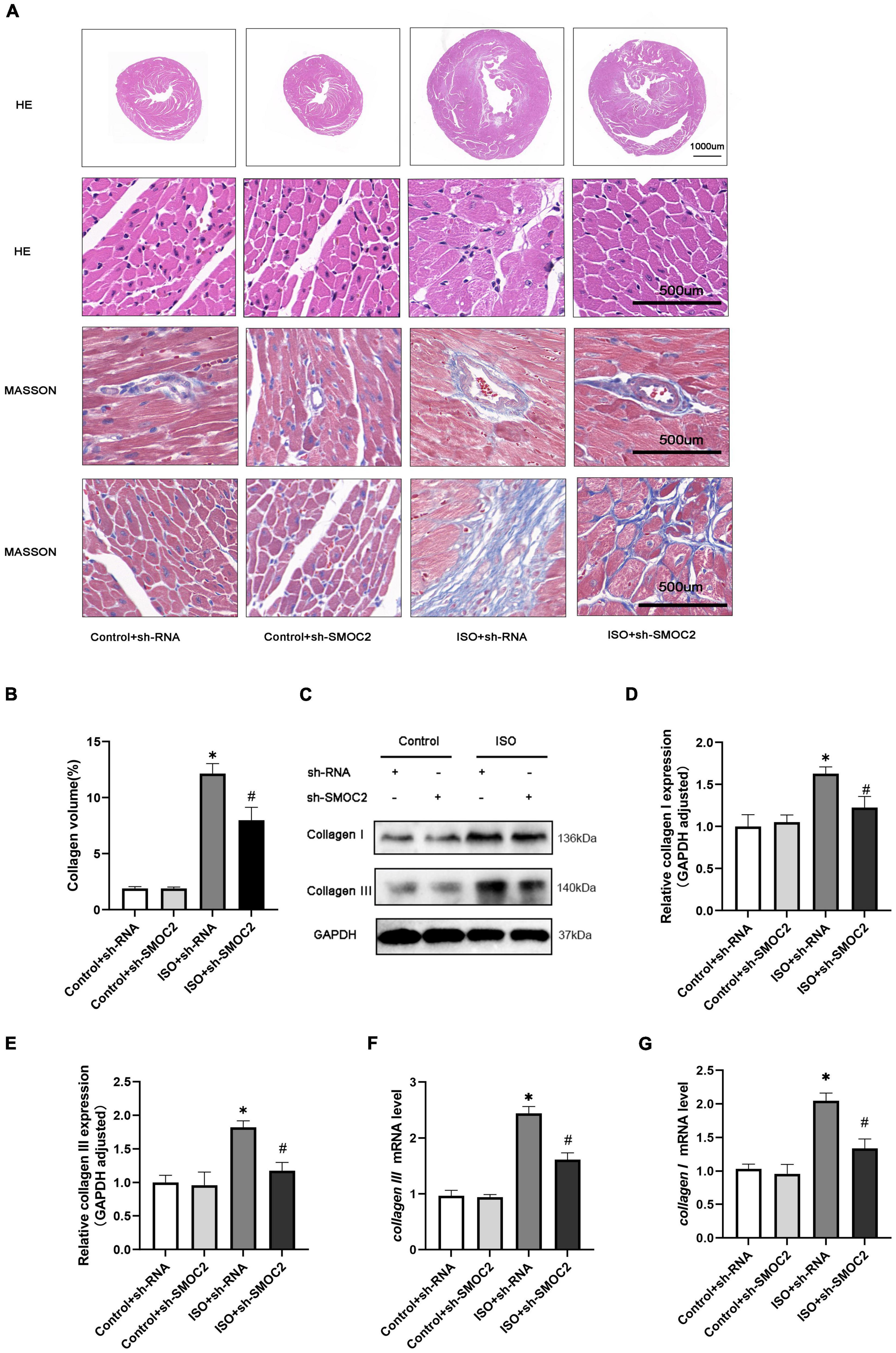
Figure 3. SMOC2 knockdown partially reversed the myocardial fibrosis in response to ISO Stimulation in vivo. (A,B) Representative images of HE and Masson staining and quantified analysis (n = 6). (C–E) Representative images of Western blots and the quantitative data (n = 6). (F,G) The relative mRNA levels of Collagen I, Collagen III (n = 3). *p < 0.05 vs. the control + sh-RNA group. #p < 0.05 vs. the ISO + sh-RNA group. n.s., non-significant.
Targeting SMOC2 expression attenuates myocardial fibrosis via the ILK and p38 pathway
Next, we went on to achieve further insight into the potential mechanisms accounting for our previous findings. Pre-existing research has identified that SMOC2 could activate downstream ILK (20, 28), which was reported to be involved in the fibrotic pathological process (21). Consequently, we focused on the expression of ILK. It was revealed that, compared with the control group, the ISO + shRNA injection group had a significantly higher ILK level, and, importantly, the effect of ILK production can be relieved by SMOC2 knockdown (Figures 4A, B). Given that the AKT and AMPK pathways have been implicated in the pathophysiology of fibrosis, we examined whether these pathways are involved in the process of SMOC2-medicated myocardial fibrosis. It was found that p-AKT, p-p38-MAPK, and p-JNK-MAPK were significantly elevated in the ISO + sh-RNA group, while the p-p38 was decreased in the ISO + sh-SMOC2 group (Figures 4A, D). Strikingly, knocking down SMOC2 did not affect the expression of p-JNK and p-AKT (Figures 4C, E and Supplementary Figures 1A–F). Collectively, these findings demonstrate that the ILK and p38 pathway is likely to participate in the ISO-induced cardiac fibrotic process in vivo.
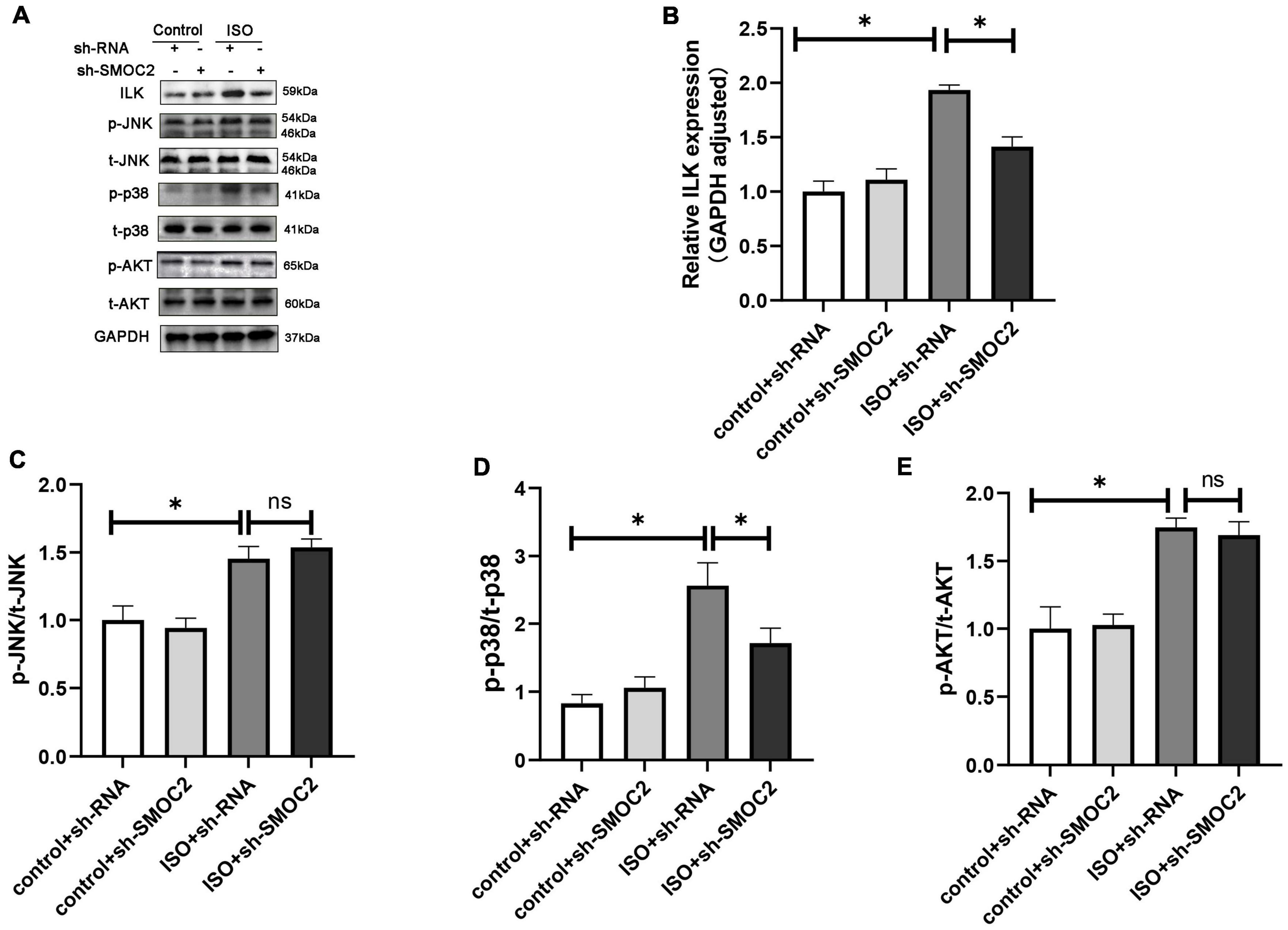
Figure 4. Targeting SMOC2 expression attenuates myocardial fibrosis via the ILK/p38 pathway. (A–E) Representative Western blots and quantitative results of ILK, t-AKT, p-AKT, t-JNK, p-JNK, t-p38, and p-p38 (n = 6). *p < 0.05. n.s., non-significant.
SMOC2 is up-regulated in TGFβ-induced cardiac fibroblasts
To gain further insight into the regulation of SMOC2 in cardiac fibrosis, we conducted in vitro experiments subsequently. Neonatal myocardial fibroblasts were separated by a differential adherent as described to explore the role of SMOC2 in TGFβ-induced fibroblast activation. The results showed TGFβ induced fibroblasts activation in a dose-dependent and time-dependent manner. After being treated with TGF β (10 ng/ml) for 24 h, the protein concentrations of SMOC2 reached the highest (Figures 5A–F). Consequently, the cells were treated with TGFβ at 10 ng/ml for 24 h before being harvested in subsequent analysis.
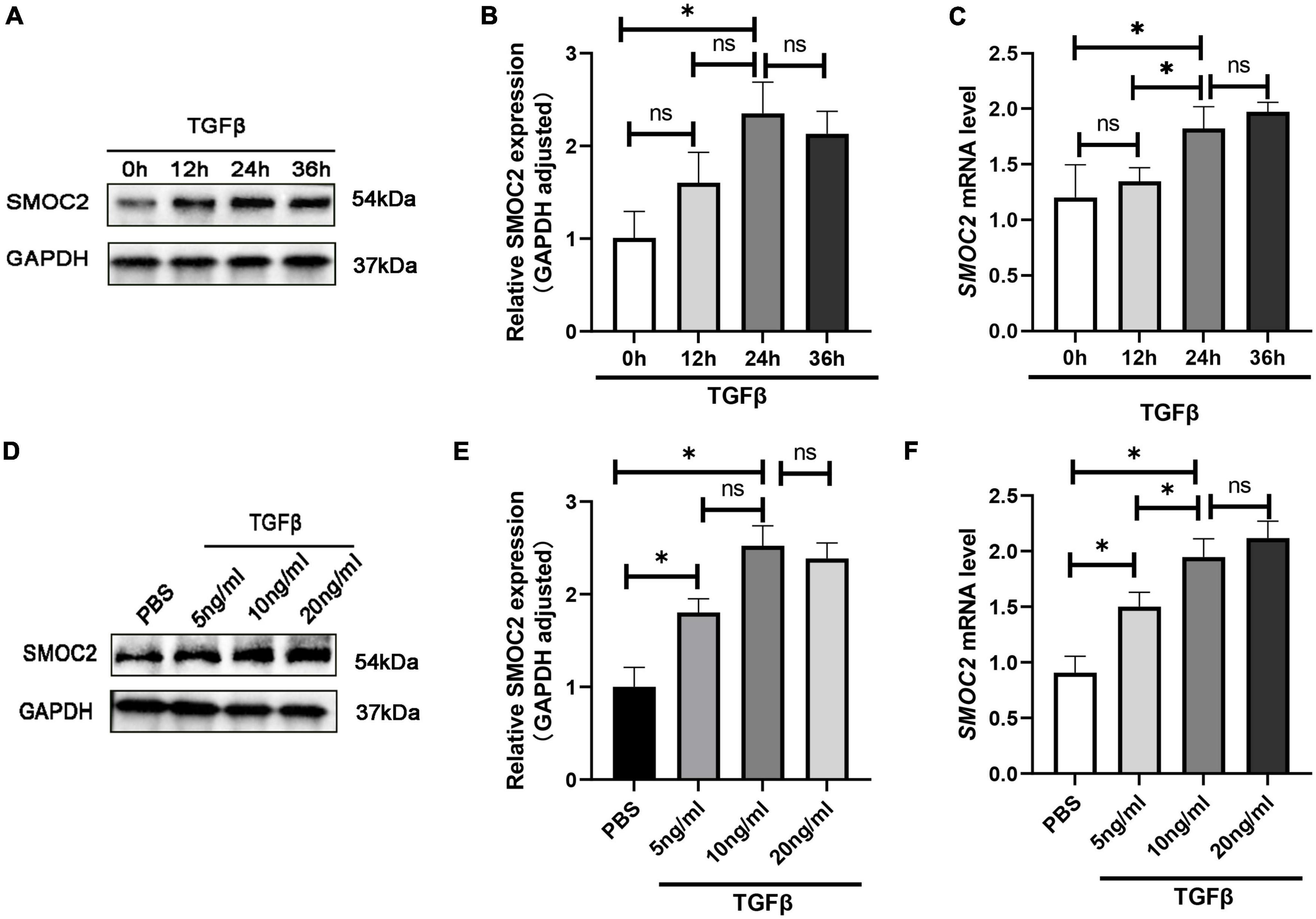
Figure 5. SMOC2 is up-regulated in TGFβ-induced cardiac fibroblasts. (A,B) Representative Western blots and quantitative results of SMOC2 in cardiac fibroblasts induced by different concentrations of TGFβ (n = 6). (C) mRNA levels of SMOC2 in cardiac fibroblasts induced by different concentrations of TGFβ (n = 3). (D,E) Representative Western blots and quantitative results of SMOC2 after induction of TGFβ at different times (n = 6). (F) mRNA levels of SMOC2 in cardiac fibroblasts after induction of TGFβ at different times (n = 3). *p < 0.05. n.s., non-significant.
Knocking SMOC2 improves the proliferation, migration, differentiation and exacerbates fibrosis of fibroblasts in vitro
In order to discuss the potential effects and mechanisms underlying the pro-fibrotic effect of SMOC2, we infected CFs with sh-SMOC2 or sh-RNA as control. As shown in Figures 6A–C, after being treated with TGFβ for 24 h, SMOC2 protein and mRNA levels increased. However, these changes could be relieved post sh-SMOC2 infection. Furthermore, these findings were confirmed by an immunofluorescence stain for SMOC2 (Figures 6D, F).
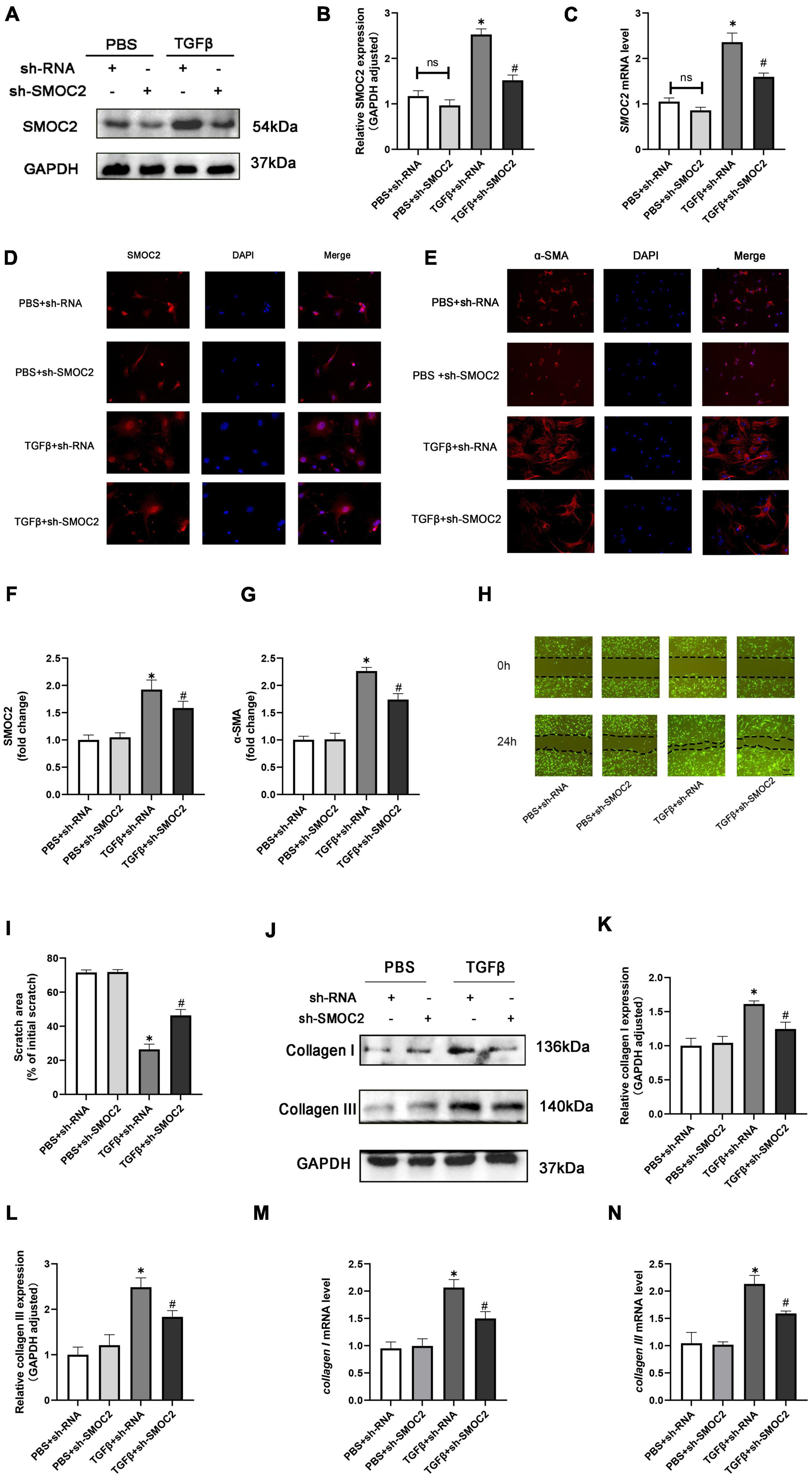
Figure 6. Targeting SMOC2 expression improves the proliferation, migration, and differentiation and exacerbates fibrosis of fibroblasts in vitro. (A,B) SMOC2 expression after sh-RNA or sh-SMOC2 infection (n = 6). (C) mRNA levels of SMOC2 after sh-RNA or sh-SMOC2 injection (n = 6). (D–G) Immunofluorescence staining of SMOC2 and α-SMA for the indicated groups (n = 6). (H,I) Representative images of the wound scratch assay at 0 and 24 h (n = 5). (J–N) Collagen I and collagen III expressions after sh-RNA or sh-SMOC2 infection in cardiac fibroblasts (n = 6). *p < 0.05 vs. the control + sh-RNA group. #p < 0.05 vs. the TGF β + sh-RNA group. n.s., non-significant.
The up-regulation of α-SMA expression, as a hallmark marker for differentiated myofibroblasts, was examined using immunofluorescence staining. As anticipated, the level of α-SMA was elevated in TGFβ + shRNA group, while SMOC2 down-regulation reduced the level of α-SMA (Figures 6E, G). Further analysis revealed that knocking SMOC2 could decrease the proliferation of TGFβ-induced CFs, as indicated by wound-healing scratch experiments (Figures 6H, I). Further analysis implicated that the protein and mRNA levels of collagen I and collagen III increased in the TGFβ + shRNA group, while knockdown of SMOC2 expression significantly inhibited type I and type III collagen production (Figures 6J–N).
Following that, we measured the collagen production of fibroblasts in the media. In agreement with the results above, the protein levels of fibrotic biomarkers such as type I and Type III collagen in the medium were elevated in the TGFβ group, whereas SMOC2 knockdown could relieve these changes (Supplementary Figures 2A, B).
Targeting SMOC2 expression ameliorates fibrosis via the ILK/p38 pathway in vitro
In order to explore the mechanisms of anti-fibrotic effects of sh-SMOC2, we also detected ILK, AKT, MAPK pathways. Consistent with the findings in vivo, the ILK level was elevated after being treated with TGFβ, which could be down-regulated by sh-SMOC2 administration (Figures 7A, B). In-depth studies also found that p-AKT, P-JNK, and p-P38 were significantly up-regulated, while the sh-SMOC2 infection significantly reduced p-p38 without affecting the phosphorylation levels of p-AKT and p-JNK (Figures 7A, C–E and Supplementary Figures 3A–F).
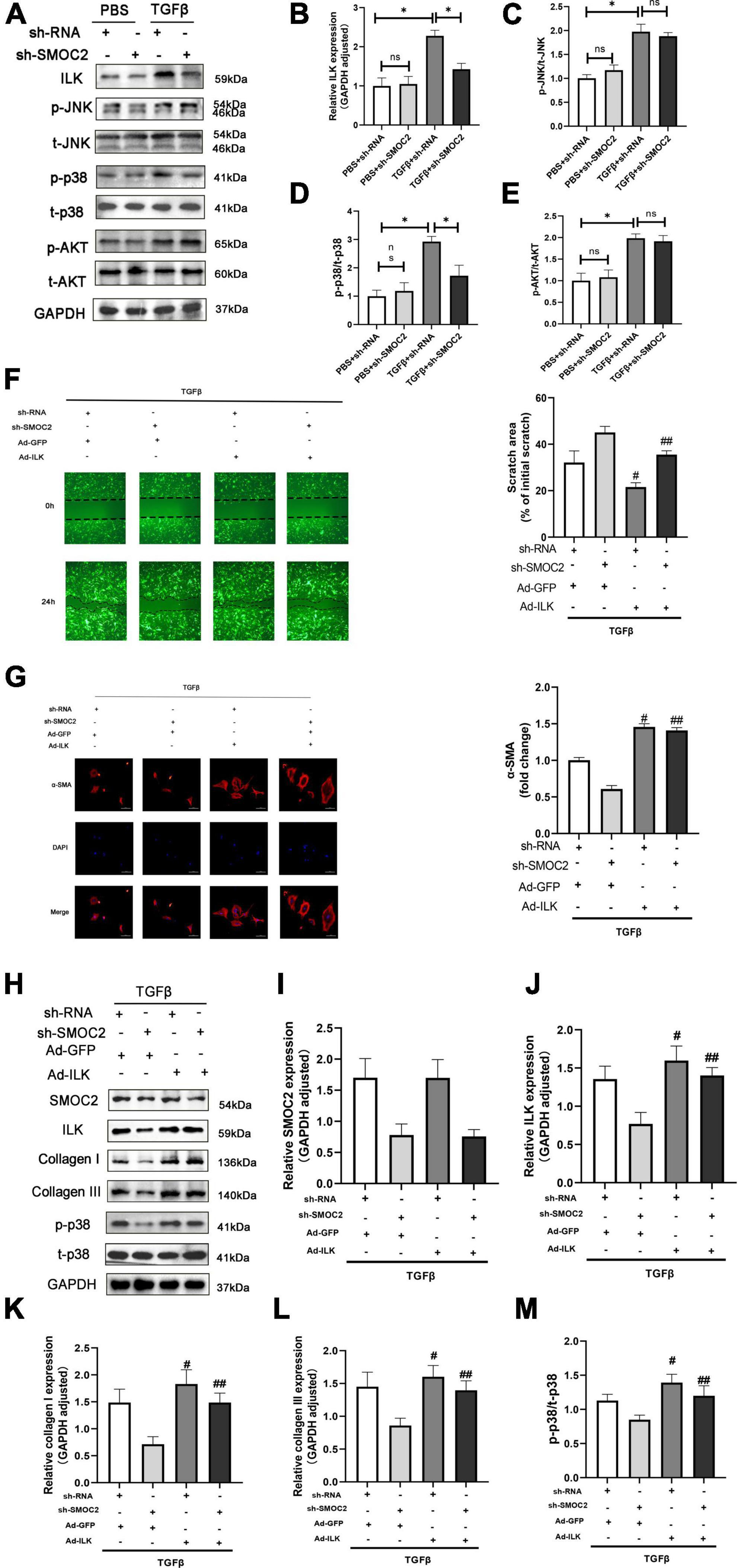
Figure 7. Targeting SMOC2 expression ameliorates fibrosis via the ILK/p38 pathway in vitro. (A–E) Representative Western blots and quantitative results of ILK, t-AKT, p-AKT, t-JNK, p-JNK, t-p38, and p-p38 (n = 6). *p < 0.05 vs. the control + sh-RNA group. #p < 0.05 vs. the ISO + sh-RNA group. n.s., non-significant. (F) Representative images of the wound scratch assay post Ad-ILK infection. (G) Representative images of immunohistochemical staining for α-SMA (n = 4). (H–M) Representative Western blots and quantitative results post Ad-ILK infection (n = 3). *p < 0.05. n.s., non-significant. #p < 0.05 vs. the sh-RNA + Ad-GFP group. ##p < 0.05 vs. the sh-SMOC2 + Ad-GFP group.
Next, we also verified whether ILK participated in the activation of p-p38 in CFs in vitro, and we infected CFs with Ad-ILK to overexpress ILK. In line with the above findings, the anti-fibrotic effect of sh-SMOC2 was knocked over after ILK overexpression, and the scratch experiment also suggested that ILK further promoted the migration of CFS and activated p-p38 (Figure 7F). To further confirm this notion, the expression of α-SMA by IHC staining was examined. The results also identified that Ad-ILK worsened the transformation of CFs (Figure 7G). Results of in vitro studies matched those of in vivo studies. The level of SMOC2 did not change due to overexpression of ILK (Figures 7H, I), but ILK, p-p38, collagen I and collagen III were significantly up-regulated (Figures 7H, J, K–M and Supplementary Figures 4A–D).
Discussion
To the best of our knowledge, this is the first report on the relationship between SMOC2 and myocardial fibrosis. Our study implied that the therapeutic silencing of SMOC2 can improve ISO-induced myocardial fibrosis and heart function reduction in vivo, and ameliorate proliferation and collagen deposition of CFs induced by TGFβ in vitro, and the down-regulation of ILK/p38 may be the underlying mechanism.
In addition to being associated with unfavorable outcomes, progressive cardiac fibrosis is one of the key hallmarks of heart failure (3, 29). Several etiopathogenic mechanisms contribute to myocardial fibrosis, a complex and multifactorial condition (5, 6). At present, the treatment of myocardial fibrosis is still an intractable clinical problem that demands to be further addressed (30, 31). Currently, only a few pharmacotherapy options, such as treatments targeting the increased activity of the renin-angiotensin-aldosterone system (RAAS) and sympathetic nervous system (SNS), are available to manage cardiac remodeling and fibrosis (31, 32). Despite exhibiting excellent efficacy, in some situations, the clinical application of these drugs has been restricted due to side effects or intolerance among patients (31). A better understanding of the molecular mechanisms of cardiac fibrosis is thus essential for developing novel therapeutic approaches.
SMOC2 is an encoded modular secretion protein that is known to influence cytokine activity, destabilize cell-substrate attachment, and modulate cell differentiation and cell cycle (8, 13, 16, 33, 34). A recently published study indicates that SMOC2 is an independent prognostic marker in patients with colorectal cancers (35). SMOC2 was shown to be a promising biomarker of kidney fibrosis in patients with CKD and was able to be used by researchers as a prognostic indicator (36). Previous studies have also demonstrated that SMOC2 is highly expressed upon kidney injury or in CKD models and stimulates the production of extracellular matrix, which can be ameliorated by infecting sh-RNA targeting SMOC2 (14, 16, 36). In addition, SMOC2 also participates in pulmonary fibrosis and hepatic steatosis caused by a high-fat diet targeting TGFβ1 pathways (13, 15). However, no research has examined the potential relationship between SMOC2 and cardiac fibrosis, nor have the relevant mechanisms been explored in more depth. The present study confirms for the first time that SMOC2 may contribute to the pathogenic process of cardiac fibrosis, which could be rescued at least partially by the deletion of SMOC2 expression in vivo and vitro.
ILK, a serine-threonine-protein kinase that exerts essential effects on cell transduction and molecular scaffolding, is known as an in vivo target of SMOC2 (18, 19, 37). Previously accumulation of evidence identified that ILK was associated with some cardiomyopathy phenotypes, as well as involved in cardiac remodeling (37). It was revealed that ILK adaptively increased in the pressure overloaded cardiac hypertrophy model and could further participate in the activation of CFs (21, 37–39). Overexpressed ILK promotes the cardiac fibrotic process by up-regulation of nuclear factor-κB (NF-κB) in cardiac fibroblasts that activate fibrotic-related genes such as CTGF and collagen I. On the other hand, ILK knockdown by siRNA could lead to attenuation of this fibrotic action (21). Therefore, it is estimated that ILK may contribute to pathological cardiac remodeling. In line with previous studies, we found that ILK was overactivated adaptively in sustained profibrogenic factors-induced mice fibrosis model to respond to the up-regulation of SMOC2.
However, other findings provided discrepant observations. ILK may be one of the key determinants that contributed to the protective effect of myocardial re-modeling after MI during Cardiac Shock Wave Therapy (40). What’s more, ILK overexpression also minimized cardiac remodeling and improved reperfusion after ischemia (41). And these were in agreement with the corresponding findings of another study (42). Researchers have recently published a study showing that targeting the regulatory ILK and relevant pathways ameliorates cardiac function in dilated myocardial animals induced by DOX, strongly implicating increased expression of ILK may ameliorate the prognosis in patients with HF (43). A mouse model of the ILK knockout was also shown to alter myocardial electrical properties, potentially resulting in fatal arrhythmias (37). Taken together, these studies suggest an intricate relationship between ILK and cardiac fibrosis and remodeling, and future research is still desperately warranted to shed light on this issue.
Evidence is mounting that the MAPK pathway, such as p38 and JNK1/2, functions in regulating cardiac apoptosis, hypertrophy, and fibrosis (44–47). And p38 was thought to be a dominant regulator in myocardial fibrosis (48). Additionally, it is interesting to note that several studies have identified the AKT pathway as being associated with fibrosis of the heart and collagen synthesis (5). The results of our present study are consistent with the previous observations, showing that AKT, p38, and JNK1/2 MAPKs were slightly activated following the pro-fibrotic stimuli in vivo and vitro. However, unexpectedly, SMOC2 knockdown impaired the expression of p38 without affecting AKT or JNK1/2 pathways, revealing p38 as a contributor to the development of SMOC2-mediated cardiac fibrosis. It was well-established that p38 phosphorylation in CFs could drive myofibroblasts to synthesize and formate collagen, which could be reversed when p38 was inhibited (6, 45). P38 has been implicated in a variety of pathogenetic processes: such as inflammation (49), osteogenic differentiation (50), apoptosis (51), and cancer differentiation (52). Specifically, p38 could be activated by several regulators thus exerting its physiological effects as a result. Zhang et al. (6) discovered that p38 was mediated by ribosomal protein S5 (RPS5) in press overload-induced cardiac fibrosis mice, whereas matrine administration alleviated this process. Additionally, Meijles et al. (53) revealed that p38 was regulated by apoptosis signal-regulating kinase 1 (AKS1) during the process of fibrosis and subsequent deterioration of hypertensive heart disease.
Interestingly, in a study, scholars found that ILK/p38 MAPK pathway can regulate the osteogenic effect on the surface of the a-C coated titanium (24). They found that ILK was significantly up-regulated on the surface of the C-Ti, and siRNA targeting ILK reduced p38 phosphorylation and osteogenic differentiation in the C-Ti surface, which shed light on the research into the relationship between ILK and p38 in the present study. As anticipated, overexpression of ILK in vivo or vitro would result in increased phosphorylation of p38 MAPK accompanied by elevated markers of cardiac fibrosis.
Conclusion and limitations
Our study identified that SMOC2 is involved in collagen deposition, cardiac fibroblasts activation, and, ultimately, cardiac fibrosis for the first time in response to pro-fibrotic stimuli such as ISO and TGFβ and ILK/p38 signaling pathway may be responsible for this important process (Figure 8). Furthermore, therapeutic SMOC2 silencing has protective effects against cardiac fibrosis by inhibiting ILK/p38 signaling, validating the critical role of SMOC2 in mediating cardiac fibrosis, and providing promising prevention and control strategies for cardiac remodeling management in clinical practice.
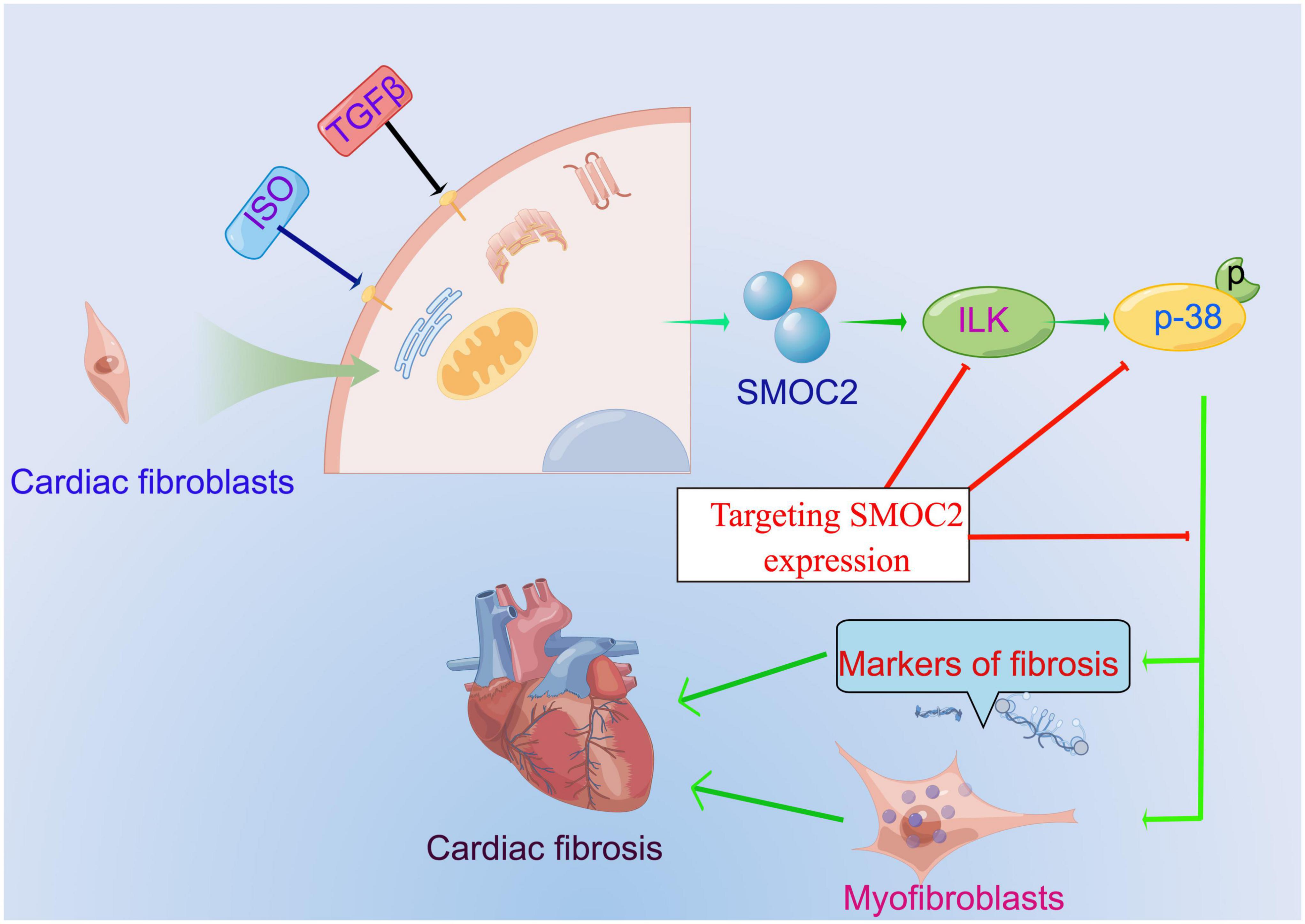
Figure 8. The proposed working model of targeting SMOC2 expression on pathological cardiac fibrosis.
There are several limitations to this study, despite its promising results. Firstly, we only applied the cardiac fibrosis model induced by ISO, and did not evaluate other cardiac fibrosis models. Secondly, a human validation of the findings was not performed. Moreover, an important limitation to this study is the use of neonatal cardiac fibroblasts. Finally, this study did not address the role of SMOC2 in cardiomyocytes and its role in cardiac hypertrophy remains to be defined. Therefore, the results of the study should be interpreted with caution.
Data availability statement
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was reviewed and approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University.
Author contributions
HR and FZ performed the experiments and analyzed the data. HR, LY, and JH designed the experiments, supervised and conceptualized the study, and wrote and edited the manuscript. HR and FZ wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the National Natural Science Foundation of China (No. 82160072) and the Science and Technology Support Project of Enshi Prefecture Science and Technology Bureau (No. D20210024).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.951704/full#supplementary-material
Supplementary Figure 1 | Total and phosphorylated protein levels in vivo. (A–F) Quantitative results of t-AKT, p-AKT, t-JNK, p-JNK, t-p38, and p-p38 (n = 6). *p < 0.05. n.s., non-significant.
Supplementary Figure 2 | Collagen I (A) and III (B) content in the cell culture supernatant. *p < 0.05. n.s., non-significant.
Supplementary Figure 3 | Total and phosphorylated protein levels in vitro. (A–F) Quantitative results of t-AKT, p-AKT, t-JNK, p-JNK, t-p38, and p-p38 (n = 6). *p < 0.05. n.s., non-significant.
Supplementary Figure 4 | Collagen I (C) and III (D) content in the cell culture supernatant and total (A) and phosphorylated (B) protein levels of p-38. n.s., non-significant. #p < 0.05 vs. the sh-RNA+Ad-GFP group. ##p < 0.05 vs. the sh-SMOC2+Ad-GFP group.
Supplementary Figure 5 | Original band of western blot in Figure 1A.
Supplementary Figure 6 | Original band of western blot in Figure 2A.
Supplementary Figure 7 | Original band of western blot in Figure 3C.
Supplementary Figure 8 | Original band of western blot in Figure 4A.
Supplementary Figure 9 | Original band of western blot in Figures 5A,B.
Supplementary Figure 10 | Original band of western blot in Figure 6A.
Supplementary Figure 11 | Original band of western blot in Figure 6J.
Supplementary Figure 12 | Original band of western blot in Figure 7A.
Supplementary Figure 13 | Original band of western blot in Figure 1H.
References
1. Kwong RY, Bax JJ. Unraveling the complex processes of adverse cardiac remodeling. Circ Cardiovasc Imaging. (2019) 12:e009086.
2. Curley D, Lavin Plaza B, Shah AM, Botnar RM. Molecular imaging of cardiac remodelling after myocardial infarction. Basic Res Cardiol. (2018) 113:10.
3. McLaughlin S, McNeill B, Podrebarac J, Hosoyama K, Sedlakova V, Cron G, et al. Injectable human recombinant collagen matrices limit adverse remodeling and improve cardiac function after myocardial infarction. Nat Commun. (2019) 10:4866. doi: 10.1038/s41467-019-12748-8
5. Ma ZG, Yuan YP, Zhang X, Xu SC, Wang SS, Tang QZ. Piperine attenuates pathological cardiac fibrosis via PPAR-γ/AKT pathways. EBioMedicine. (2017) 18:179–87. doi: 10.1016/j.ebiom.2017.03.021
6. Zhang X, Hu C, Zhang N, Wei WY, Li LL, Wu HM, et al. Matrine attenuates pathological cardiac fibrosis via RPS5/p38 in mice. Acta Pharmacol Sin. (2021) 42:573–84. doi: 10.1038/s41401-020-0473-8
7. Filippatos G, Angermann CE, Cleland JGF, Lam CSP, Dahlström U, Dickstein K, et al. Global differences in characteristics, precipitants, and initial management of patients presenting with acute heart failure. JAMA Cardiol. (2020) 5:401–10.
8. Maier S, Paulsson M, Hartmann U. The widely expressed extracellular matrix protein SMOC-2 promotes keratinocyte attachment and migration. Exp Cell Res. (2008) 314:2477–87. doi: 10.1016/j.yexcr.2008.05.020
9. Vannahme C, Gösling S, Paulsson M, Maurer P, Hartmann U. Characterization of SMOC-2, a modular extracellular calcium-binding protein. Biochem J. (2003) 373(Pt 3):805–14.
10. Rocnik EF, Liu P, Sato K, Walsh K, Vaziri C. The novel SPARC family member SMOC-2 potentiates angiogenic growth factor activity. J Biol Chem. (2006) 281:22855–64. doi: 10.1074/jbc.M513463200
11. Wong GS, Rustgi AK. Matricellular proteins: priming the tumour microenvironment for cancer development and metastasis. Br J Cancer. (2013) 108:755–61. doi: 10.1038/bjc.2012.592
12. Schellings MW, Pinto YM, Heymans S. Matricellular proteins in the heart: possible role during stress and remodeling. Cardiovasc Res. (2004) 64:24–31. doi: 10.1016/j.cardiores.2004.06.006
13. Yuting Y, Lifeng F, Qiwei H. Secreted modular calcium-binding protein 2 promotes high fat diet (HFD)-induced hepatic steatosis through enhancing lipid deposition, fibrosis and inflammation via targeting TGF-β1. Biochem Biophys Res Commun. (2019) 509:48–55. doi: 10.1016/j.bbrc.2018.12.006
14. Xin C, Lei J, Wang Q, Yin Y, Yang X, Moran Guerrero JA, et al. Therapeutic silencing of SMOC2 prevents kidney function loss in mouse model of chronic kidney disease. iScience. (2021) 24:103193. doi: 10.1016/j.isci.2021.103193
15. Luo L, Wang CC, Song XP, Wang HM, Zhou H, Sun Y, et al. Suppression of SMOC2 reduces bleomycin (BLM)-induced pulmonary fibrosis by inhibition of TGF-β1/SMADs pathway. Biomed Pharmacother. (2018) 105:841–7. doi: 10.1016/j.biopha.2018.03.058
16. Gerarduzzi C, Kumar RK, Trivedi P, Ajay AK, Iyer A, Boswell S, et al. Silencing SMOC2 ameliorates kidney fibrosis by inhibiting fibroblast to myofibroblast transformation. JCI Insight. (2017) 2:e90299. doi: 10.1172/jci.insight.90299
17. Li D, Lin H, Li L. Multiple feature selection strategies identified novel cardiac gene expression signature for heart failure. Front Physiol. (2020) 11:604241. doi: 10.3389/fphys.2020.604241
18. Shi Q, Bao S, Song L, Wu Q, Bigner DD, Hjelmeland AB, et al. Targeting SPARC expression decreases glioma cellular survival and invasion associated with reduced activities of FAK and ILK kinases. Oncogene. (2007) 26:4084–94. doi: 10.1038/sj.onc.1210181
19. Barker TH, Baneyx G, Cardó-Vila M, Workman GA, Weaver M, Menon PM, et al. SPARC regulates extracellular matrix organization through its modulation of integrin-linked kinase activity. J Biol Chem. (2005) 280:36483–93. doi: 10.1074/jbc.M504663200
20. Liu P, Lu J, Cardoso WV, Vaziri C. The SPARC-related factor SMOC-2 promotes growth factor-induced cyclin D1 expression and DNA synthesis via integrin-linked kinase. Mol Biol Cell. (2008) 19:248–61. doi: 10.1091/mbc.e07-05-0510
21. Thakur S, Li L, Gupta S. NF-κB-mediated integrin-linked kinase regulation in angiotensin II-induced pro-fibrotic process in cardiac fibroblasts. Life Sci. (2014) 107:68–75. doi: 10.1016/j.lfs.2014.04.030
22. Ying YT, Ren WJ, Tan X, Yang J, Liu R, Du AF. Annexin A2-mediated internalization of staphylococcus aureus into bovine mammary epithelial cells requires its interaction with clumping factor B. Microorganisms. (2021) 9:2090. doi: 10.3390/microorganisms9102090
23. Wang W, Liu Q, Zhang Y, Zhao L. Involvement of ILK/ERK1/2 and ILK/p38 pathways in mediating the enhanced osteoblast differentiation by micro/nanotopography. Acta Biomater. (2014) 10:3705–15. doi: 10.1016/j.actbio.2014.04.019
24. Yue G, Song W, Xu S, Sun Y, Wang Z. Role of ILK/p38 pathway in mediating the enhanced osteogenic differentiation of bone marrow mesenchymal stem cells on amorphous carbon coating. Biomater Sci. (2019) 7:975–84. doi: 10.1039/c8bm01151f
25. Jiang XH, Wu QQ, Xiao Y, Yuan Y, Yang Z, Bian ZY, et al. Evodiamine prevents isoproterenol-induced cardiac fibrosis by regulating endothelial-to-mesenchymal transition. Planta Med. (2017) 83:761–9. doi: 10.1055/s-0042-124044
26. Li N, Zhou H, Ma ZG, Zhu JX, Liu C, Song P, et al. Geniposide alleviates isoproterenol-induced cardiac fibrosis partially via SIRT1 activation in vivo and in vitro. Front Pharmacol. (2018) 9:854. doi: 10.3389/fphar.2018.00854
27. Li C, Ying S, Wu X, Zhu T, Zhou Q, Zhang Y, et al. CTRP1 aggravates cardiac fibrosis by regulating the NOX2/P38 pathway in macrophages. Cell J. (2022) 24:732–40. doi: 10.22074/cellj.2022.557327.1043
28. Liu P, Pazin DE, Merson RR, Albrecht KH, Vaziri C. The developmentally-regulated Smoc2 gene is repressed by Aryl-hydrocarbon receptor (Ahr) signaling. Gene. (2009) 433:72–80. doi: 10.1016/j.gene.2008.12.010
29. Aminzadeh MA, Tseliou E, Sun B, Cheng K, Malliaras K, Makkar RR, et al. Therapeutic efficacy of cardiosphere-derived cells in a transgenic mouse model of non-ischaemic dilated cardiomyopathy. Eur Heart J. (2015) 36:751–62. doi: 10.1093/eurheartj/ehu196
30. González A, Schelbert EB, Díez J, Butler J. Myocardial interstitial fibrosis in heart failure: biological and translational perspectives. J Am Coll Cardiol. (2018) 71:1696–706.
31. Park S, Nguyen NB, Pezhouman A, Ardehali R. Cardiac fibrosis: potential therapeutic targets. Transl Res. (2019) 209:121–37.
32. AlQudah M, Hale TM, Czubryt MP. Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol. (2020) 9:92–108.
33. Long F, Shi H, Li P, Guo S, Ma Y, Wei S, et al. A SMOC2 variant inhibits BMP signaling by competitively binding to BMPR1B and causes growth plate defects. Bone. (2021) 142:115686. doi: 10.1016/j.bone.2020.115686
34. Peeters T, Monteagudo S, Tylzanowski P, Luyten FP, Lories R, Cailotto F. SMOC2 inhibits calcification of osteoprogenitor and endothelial cells. PLoS One. (2018) 13:e0198104. doi: 10.1371/journal.pone.0198104
35. Jang BG, Kim HS, Bae JM, Kim WH, Kim HU, Kang GH. SMOC2, an intestinal stem cell marker, is an independent prognostic marker associated with better survival in colorectal cancers. Sci Rep. (2020) 10:14591. doi: 10.1038/s41598-020-71643-1
36. Schmidt IM, Colona MR, Kestenbaum BR, Alexopoulos LG, Palsson R, Srivastava A, et al. Cadherin-11, Sparc-related modular calcium binding protein-2, and Pigment epithelium-derived factor are promising non-invasive biomarkers of kidney fibrosis. Kidney Int. (2021) 100:672–83. doi: 10.1016/j.kint.2021.04.037
37. Quang KL, Maguy A, Qi XY, Naud P, Xiong F, Tadevosyan A, et al. Loss of cardiomyocyte integrin-linked kinase produces an arrhythmogenic cardiomyopathy in mice. Circ Arrhyth Electrophysiol. (2015) 8:921–32. doi: 10.1161/CIRCEP.115.001668
38. Sofia RR, Serra AJ, Silva JA Jr., Antonio EL, Manchini MT, Oliveira FA, et al. Gender-based differences in cardiac remodeling and ILK expression after myocardial infarction. Arquivos Bras Cardiol. (2014) 103:124–30. doi: 10.5935/abc.20140113
39. Xie J, Lu W, Gu R, Dai Q, Zong B, Ling L, et al. The impairment of ILK related angiogenesis involved in cardiac maladaptation after infarction. PLoS One. (2011) 6:e24115. doi: 10.1371/journal.pone.0024115
40. Yang W, He Y, Gan L, Zhang F, Hua B, Yang P, et al. Cardiac shock wave therapy promotes arteriogenesis of coronary micrangium, and ILK is involved in the biomechanical effects by proteomic analysis. Sci Rep. (2018) 8:1814. doi: 10.1038/s41598-018-19393-z
41. Mu D, Zhang XL, Xie J, Yuan HH, Wang K, Huang W, et al. Intracoronary transplantation of mesenchymal stem cells with overexpressed integrin-linked kinase improves cardiac function in porcine myocardial infarction. Sci Rep. (2016) 6:19155. doi: 10.1038/srep19155
42. Mao Q, Lin C, Gao J, Liang X, Gao W, Shen L, et al. Mesenchymal stem cells overexpressing integrin-linked kinase attenuate left ventricular remodeling and improve cardiac function after myocardial infarction. Mol Cell Biochem. (2014) 397:203–14.
43. Gu R, Bai J, Ling L, Ding L, Zhang N, Ye J, et al. Increased expression of integrin-linked kinase improves cardiac function and decreases mortality in dilated cardiomyopathy model of rats. PLoS One. (2012) 7:e31279. doi: 10.1371/journal.pone.0031279
44. Wang S, Ding L, Ji H, Xu Z, Liu Q, Zheng Y. The Role of p38 MAPK in the development of diabetic cardiomyopathy. Int J Mol Sci. (2016) 17:1037.
45. Aschar-Sobbi R, Izaddoustdar F, Korogyi AS, Wang Q, Farman GP, Yang F, et al. Increased atrial arrhythmia susceptibility induced by intense endurance exercise in mice requires TNFα. Nat Commun. (2015) 6:6018. doi: 10.1038/ncomms7018
46. Silva TA, Ferreira LFC, Pereira MCS, Calvet CM. Differential role of TGF-β in extracellular matrix regulation during trypanosoma cruzi-host cell interaction. Int J Mol Sci. (2019) 20:4836. doi: 10.3390/ijms20194836
47. Sun F, Duan W, Zhang Y, Zhang L, Qile M, Liu Z, et al. Simvastatin alleviates cardiac fibrosis induced by infarction via up-regulation of TGF-β receptor III expression. Br J Pharmacol. (2015) 172:3779–92. doi: 10.1111/bph.13166
48. Yang HX, Sun JH, Yao TT, Li Y, Xu GR, Zhang C, et al. Bellidifolin ameliorates isoprenaline-induced myocardial fibrosis by regulating TGF-β1/Smads and p38 signaling and preventing NR4A1 cytoplasmic localization. Front Pharmacol. (2021) 12:644886. doi: 10.3389/fphar.2021.644886
49. Lasola JJM, Cottingham AL, Scotland BL, Truong N, Hong CC, Shapiro P, et al. Immunomodulatory nanoparticles mitigate macrophage inflammation via inhibition of PAMP interactions and lactate-mediated functional reprogramming of NF-κB and p38 MAPK. Pharmaceutics. (2021) 13:1841. doi: 10.3390/pharmaceutics13111841
50. Meng L, Yuan L, Ni J, Fang M, Guo S, Cai H, et al. Mir24-2-5p suppresses the osteogenic differentiation with Gnai3 inhibition presenting a direct target via inactivating JNK-p38 MAPK signaling axis. Int J Biol Sci. (2021) 17:4238–53. doi: 10.7150/ijbs.60536
51. Chen Y, Chen Y, Tang C, Zhao Q, Xu T, Kang Q, et al. RPS4Y1 promotes high glucose-induced endothelial cell apoptosis and inflammation by activation of the p38 MAPK signaling. Diabetes Metab Synd Obesity. (2021) 14:4523–34. doi: 10.2147/DMSO.S329209
52. Bao S, Ji Z, Shi M, Liu X. EPB41L5 promotes EMT through the ERK/p38 MAPK signaling pathway in esophageal squamous cell carcinoma. Pathol Res Pract. (2021) 228:153682. doi: 10.1016/j.prp.2021.153682
53. Meijles DN, Cull JJ, Markou T, Cooper STE, Haines ZHR, Fuller SJ, et al. Redox regulation of cardiac ASK1 (apoptosis signal-regulating kinase 1) controls p38-MAPK (mitogen-activated protein kinase) and orchestrates cardiac remodeling to hypertension. Hypertension. (2020) 76:1208–18. doi: 10.1161/HYPERTENSIONAHA.119.14556
Keywords: SMOC2, cardiac fibrosis, cardiac remodeling, ILK, p38 MAPK
Citation: Rui H, Zhao F, Yuhua L and Hong J (2023) Suppression of SMOC2 alleviates myocardial fibrosis via the ILK/p38 pathway. Front. Cardiovasc. Med. 9:951704. doi: 10.3389/fcvm.2022.951704
Received: 24 May 2022; Accepted: 13 December 2022;
Published: 02 March 2023.
Edited by:
Muhammad Aslam, University of Giessen, GermanyReviewed by:
Daiana Escudero, Centro de Investigaciones Cardiovasculares Dr. Horacio Eugenio Cingolani, ArgentinaTao Yang, Shanghai University of Traditional Chinese Medicine, China
Scott Levick, West Virginia University, United States
Copyright © 2023 Rui, Zhao, Yuhua and Hong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiang Hong, aG9uZy1qaWFuZ0B3aHUuZWR1LmNu
†These authors have contributed equally to this work