 Xiafeng Yu1,†
Xiafeng Yu1,† Yu Tao2,†Xu Liu1Feng Yu2
Yu Tao2,†Xu Liu1Feng Yu2 Chuan Jiang1
Chuan Jiang1 Yingying Xiao1Haibo Zhang1Yongrui He3
Yingying Xiao1Haibo Zhang1Yongrui He3 Lincai Ye3Ying Wang2
Lincai Ye3Ying Wang2 Chunxia Zhou1*
Chunxia Zhou1* Jian Wang4*Zhengwen Jiang2*
Jian Wang4*Zhengwen Jiang2* Haifa Hong3*
Haifa Hong3*
- 1Department of Cardiothoracic Surgery, Shanghai Children’s Medical Center, Shanghai Jiaotong University School of Medicine, Shanghai, China
- 2Department of Genetics, Genesky Biotechnologies Inc., Shanghai, China
- 3Institute of Pediatric Congenital Heart Disease, Shanghai Children’s Medical Center, Shanghai Jiaotong University School of Medicine, Shanghai, China
- 4Department of Medical Genetics and Molecular Diagnostic Laboratory, Shanghai Children’s Medical Center, Shanghai Jiaotong University School of Medicine, Shanghai, China
Background: Copy number variations (CNVs) have been shown to be overrepresented in children with congenital heart disease (CHD). Genetic evaluation of CHD is currently underperformed in China. We sought to determine the occurrence of CNVs in CNV regions with disease-causing potential among a large cohort of Chinese pediatric CHD patients and investigate whether these CNVs could be the important critical modifiers of surgical intervention.
Methods: CNVs screenings were performed in 1,762 Chinese children who underwent at least one cardiac surgery. CNV status at over 200 CNV locus with disease-causing potential was analyzed with a high-throughput ligation-dependent probe amplification (HLPA) assay.
Results: We found 378 out of 1,762 samples (21.45%) to have at least one CNV and 2.38% of them were carrying multiple CNVs. The detection rates of ppCNVs (pathogenic and likely pathogenic CNVs) were 9.19% (162/1,762), significantly higher than that of the healthy Han Chinese individuals from The Database of Genomic Variants archive (9.19% vs. 3.63%; P = 0.0012). CHD cases with ppCNVs had a significantly higher proportion of complex surgeries compared to CHD patients with no ppCNVs (62.35% vs. 37.63%, P < 0.001). Duration of cardiopulmonary bypass and aortic cross clamp procedures were significantly longer in CHD cases with ppCNVs (all P < 0.05), while no group differences were identified for complications of surgery and one-month mortality after surgery. The detection rate of ppCNVs in the atrioventricular septal defect (AVSD) subgroup was significantly higher than that in other subgroups (23.10% vs. 9.70%, P = 0.002).
Conclusions: CNV burden is an important contributor to Chinese children with CHD. Our study demonstrated the robustness and diagnostic efficiency of HLPA method in the genetic screening of CNVs in CHD patients.
Introduction
Congenital heart disease (CHD), the most common type of birth-related structural and/or functional anomaly in the heart, affects nearly 1% of live births per year (1, 2). It has also been one of the leading causes of infant mortality, especially among children aged under 5 years in China (1). Although the rate of survival has been improved after the surgical intervention, the rate of long-term morbidity remains high. CHD encompasses a wide spectrum of congenital cardiac abnormalities, which greatly differ with respect to function, anatomy, and clinical outcomes (3).
The underlying mechanisms driving the gene mutation-associated CHD onset are complex and remain poorly understood, which include chromosomal aneuploidy (deletion or duplication of chromosomes), copy number variation (CNV), de novo mutation, and single-nucleotide polymorphism (SNP) (4, 5, 6, 7). Notably, any of these mechanisms can impact the function of certain gene products playing critical roles in healthy cardiac development and activity.
Approximately 12% of the human genome is believed to be regulated by CNV-mediated gene duplication or deletion mechanism (8), and pathogenic and/or large CNVs have been over-represented in a substantial proportion of CHD patients (9, 10). Subsequently, multiple recurrent CNV deletion loci, such as 22q11.2, 7q11.23, and 8p23.1, have been found to confer substantial risks for syndromic or isolated CHD (11, 12, 13). Additionally, large CNVs in CHD cases are associated with neurodevelopmental deficits, cognitive dysfunction, and poor clinical outcomes (14). For example, a genetic study of 422 cases with non-syndromic CHD found that the presence of pathogenic CNVs had a significantly negative impact on transplant-free survival and interventions (15). CNV-impacted diverse alterations in the chromosomal landscape have been extensively profiled by various genetic studies, such as fluorescence in situ hybridization (FISH) (16), array-based comparative genomic hybridization (aCGH) (17), single nucleotide polymorphism array (18), and next-generation sequencing (NGS) (19). However, most of the published reports included cohorts with limited sample sizes, and large-scale screening of CHD-related CNV was not performed in Chinese CHD patients. The high-throughput ligation-dependent probe amplification (HLPA) assay is an alternative CNV-screening tool with a great cost-benefit, minimum turn-around time, and high compatibility with multiplex PCR systems (20). HLPA has also shown high concordance with the aCGH-based CNV detection with respect to cost, time, and precision (21). Furthermore, the HLPA method has also been applied to identify the pathogenic CNV, 15q11.2 deletion, in a rare form of CHD, called total anomalous pulmonary venous connection (TAPVC) (22).
Thus, we sought to assess the frequency of occurrence of CNVs in over 200 recurrent CNV locus with potential pathogenic effect among a large cohort of Chinese pediatric patients with CHD using HLPA assay and investigate whether these CNVs could be the important critical modifiers of surgical intervention. We present the following article in accordance with the STROBE reporting checklist.
Methods
Study subjects and phenotype classification
A total of 1,762 patients (51% male, median age = 24.95 ± 30.78 months), who underwent at least one cardiac operation at Shanghai Children's Medical Center between January 2016 and January 2021, were recruited to this study. Past medical records, including admission notes and echocardiography (ECG) reports, were carefully reviewed during enrollment. This group of patients was evaluated using ECG, magnetic resonance imaging (MRI), cardiac catheterization, and/or surgical reports to diagnose specific CHD sub-type. Patients having only mild CHD abnormalities, such as isolated patent ductus arteriosus (PDA) and patent foramen ovale (PFO) or with a recognizable phenotypic syndrome, were excluded from this study.
CNV detection by HLPA methodology
Genomic DNA was isolated from the 2 ml peripheral blood samples, collected from each subject during admission, using QIAamp DNA Blood kit (Qiagen, Hilden, Germany), following the manufacturer's procedures. CNV detection was performed with HLPA methodology. (D04T1006, Genesky, Suzhou, China),.HLPA is a modified version of the multiplex LPA (MLPA) method for the quantification of gene copy numbers in a multiplex PCR setup (23, 25). The validity and reliability of this method can be found elsewhere. Based on an earlier HLPA assay that used 170 and 341 pairs of probes to target each of the 24 chromosomes for the detection of aneuploidies (20, 23, 24)., the technique was enhanced by the addition of several hundred of pairs of probes to achieve a higher resolution for the detection of CNVs. This led to the inclusion of 1602 pairs of probes in the current assay to identify aneuploidies and CNVs. Briefly, a mixture of amplified fragments with various amplicon lengths, subsequently labeled with fluorophores, was prepared, which was then subjected to capillary electrophoresis for size-based separation and quantification of each labelled-amplicon. Peaks were retrieved with Genemapper V5.0 (Applied biosystems) and data analysis was performed by CNV Reader 1.0 (Genesky, Suzhou, China) (25). The relative value for each probe was then compared with the matching values obtained in all reference samples (inter-sample normalization). The final probe ratio was around 2.0 if the region of interest was unaffected by CNV, whereas an increased or decreased value indicated the duplication or deletion, respectively. Like MLPA, each probe contained a specific sequence complementary to the genomic target sequence as well as an adapter sequence that enabled exponential amplification of the template DNA using universal primers. HLPA method exploited a “lengthening” ligation system using a ligation template and a pair of elongated ligation probes to further increase the length of the downstream probe, further enabling the size-based simultaneous sorting of amplification products (20). MLPA, on the other hand, requires a stuffer sequence in the downstream probe that can reach a length of 200 bp, making it costly and challenging to chemically synthesis. Each lengthening probe had a sequence complementary to the ligation template and was hybridized in immediate proximity to each other. After hybridization, the probes specific to genomic DNA and the ligation template were ligated simultaneously by a thermally stable DNA ligase. The universal PCR primers were used to subsequently amplify the single/double ligated probes to obtain a contiguous DNA fragment. To further enhance the throughput, we used four types of 5′ universal primers labeled with different fluorophores and two types of 3′ primers that corresponded to the conventional 3′ probe and the lengthening ligation probe to amplify the ligated probes. A total of 1,602 probes were designed to pick up over 200 known recurrent CNV loci (Figure 1, Supplementary Table S1), including 200 core CNV regions and a range of regions flanking the core regions in the mitotic chromosome, genomic regions at about 0 Mb, 10 Mb, and 20 Mb distances from the telomeric end, and 3 gene-mutation loci (RBM8A: c.67 + 32G > C, c.−21delG; FGFR3: c.1138G > A), allowing the identification of CNV regions corresponding to 58 syndromes of CHD defined by DECIPHER (Supplementary Table S2). These regions were covered by at least 4 CNV segments and related to high-risk predisposition to congenital phylogenetic abnormalities as well as intellectual disability, according to the International Standards for Cytogenomic Array (ISCA) database of chromosomal aneuploidy, duplication, and deletion of the telomeric ends.
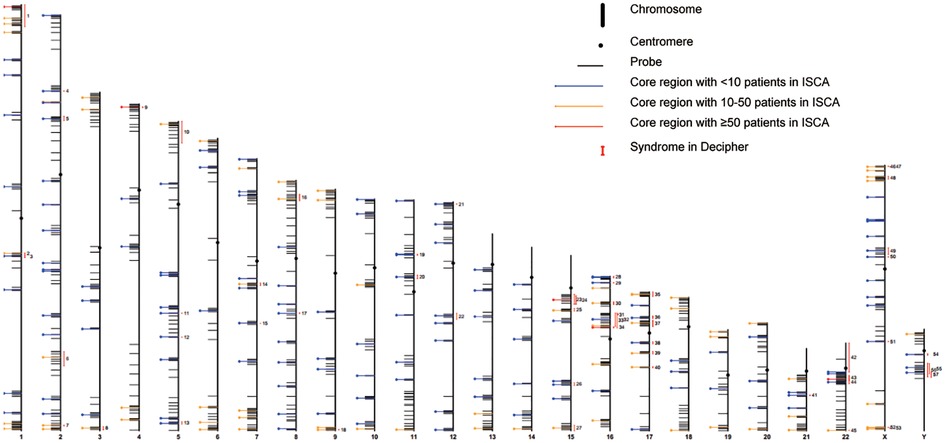
Figure 1. Genome-wide distribution of 1,602 probes on human chromosomes for CNV detection in CHD.
Definition of CNV categories
Each CNV was classified into one of the following categories: (1) benign, (2) likely benign, (3) variants of uncertain significance, (4) likely pathogenic, or (5) pathogenic. Benign CNVs were defined as CNVs: (i) with an incidence of >1% in the Database of Genomic Variants (DGV); (ii) reported as benign in the ISCA or DECIFER database, and/or (iii) listed in the DGV with incidence below 1%, and do not contain any genes. Likely benign CNVs were defined as gene-containing variants detected multiple times in normal populations with <1% incidence rate. Variants of uncertain significance were defined as CNVs which were: (i) not eligible for the definition of pathogenic/likely pathogenic CNV nor benign CNV, (ii) not reported in the literature, and/or (iii) lacking sufficient evidence to make a definitive classification. Likely pathogenic CNVs were defined as deletions or duplications that: (i) partially overlapped with reported pathogenic deletions or duplications; (ii) affected genes that were suspected, but not functionally confirmed, in the pathogenesis of the disease, and/or (iii) affected genes for which there was evidence to support double- or triple-dose sensitivity, but not enough to draw firm conclusions. Pathogenic CNVs were defined as deletions or duplications that matched in location and size to the previously identified pathogenic regions in established microdeletion/microduplication syndromes. Likely pathogenic and pathogenic CNVs were grouped together as “potentially pathogenic” CNVs (ppCNVs).
Statistical analysis
SPSS 20.0 software was used for statistical analysis. Student t-test was applied for the continuous variables, while Chi-squared (χ2) test or the Fisher's exact test was applied to compare groups whenever appropriate. A P-value of <0.05 was considered statistically significant.
Results
Diagnostic yield of CNV testing for children with CHD
Between January 2016 and January 2021, about 17,708 patients received CHD correction surgeries at Shanghai Children's Medical Center and a total of 1,762 samples were eligible for the CNV analysis. Among the subjects, 51.0% were male with a mean age of 24.95 ± 30.78 months. Out of these patients, 703 individuals (39.90%) underwent complex surgeries and 159 (9.02%) individuals had multiple surgeries.
The overall diagnostic yield of CNV testing for children with CHD was 21.45% (378/1,762), while 2.38% (42/1,762) of these patients carried multiple CNV. A summary of chromosomal abnormalities in 378 CHD patients is provided in Table 1 and Figure 2, and detailed information is provided in Supplementary Table S3. Clinically, ppCNV abnormalities were detected in 162 of 1,762 samples (9.19%), including 36 cases with whole chromosome aneuploidies (2.04%), 42 cases with 22q11 deletions or duplications (2.38%), and 84 cases with microdeletions or duplications (4.77%) related to other known chromosomal disease syndromes.
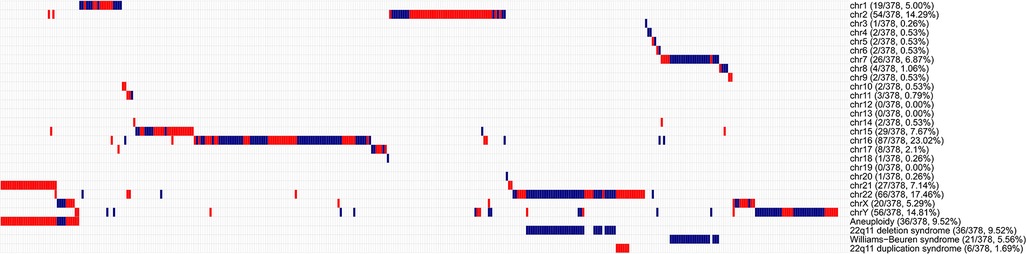
Figure 2. Overview of chromosomal aberrations detected in 378 patients by the HLPA assay A plot shows the CNV status of each of the 378 individuals on each chromosome, as labeled on the right. Each column represents a sample. Duplications are depicted by red bars, and deletions are depicted by blue bars. High proportions of CNVs are found in genomic regions correlated with aneuploidy, 22q11 deletion syndrome, Williams-Beuren syndrome, and 22q11 duplication syndrome, as labeled on the right.
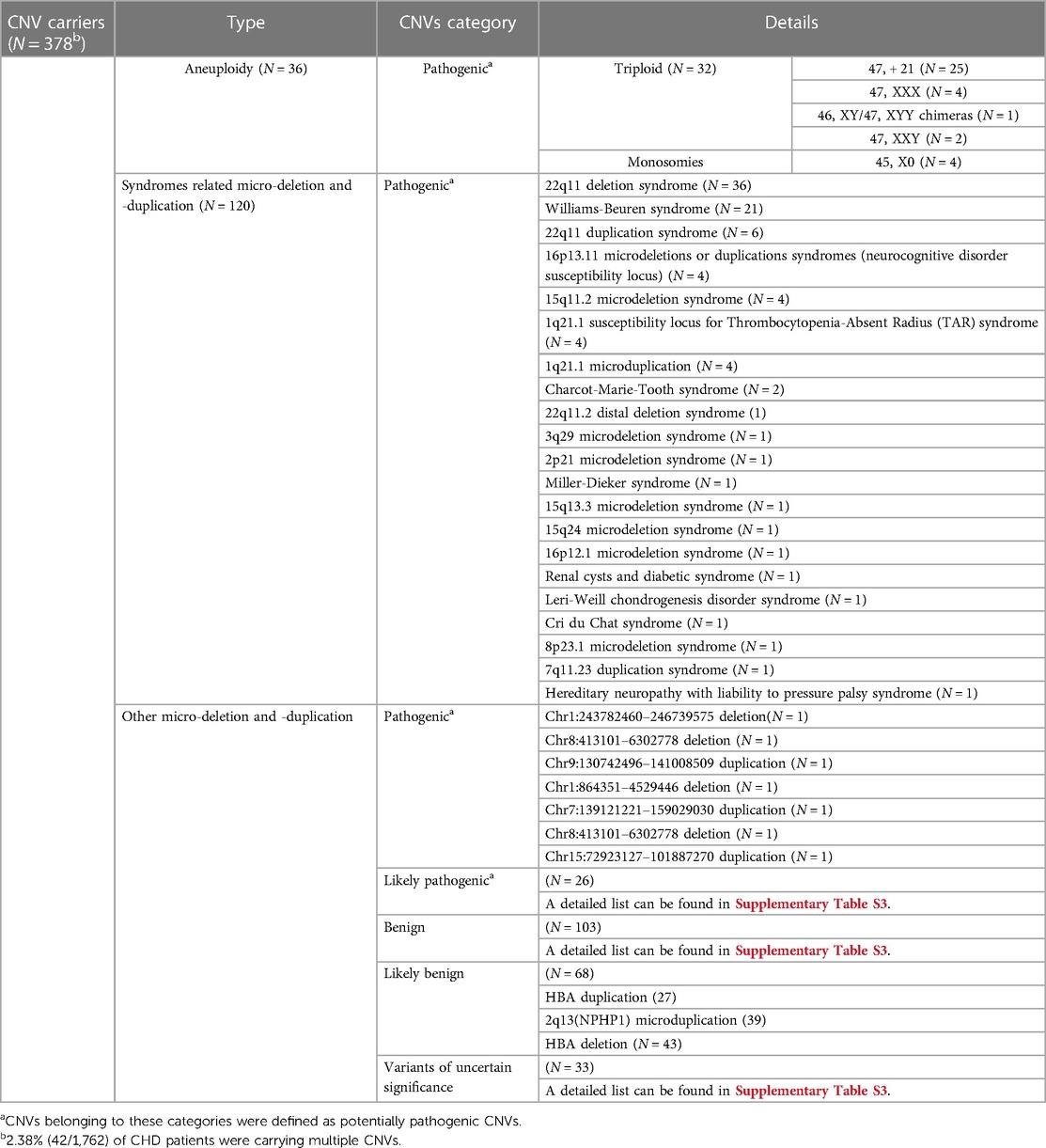
Table 1. Summary of chromosomal abnormalities of CHD patients.
In comparison, 11 individuals with 4 such potentially pathogenic CNVs were identified in 303 healthy Han Chinese individuals from DGV database (http://dgv.tcag.ca/dgv/docs/GRCh37_hg19_variants_2020-02-25.txt), according to 90% reciprocal overlap criterion (Supplementary Table S4). Significant difference in the proportion of ppCNV events between the CHD cases and the controls (9.19% vs. 3.63%; P = 0.0012) was presented. Results from a subset of CHD cases and three control individuals showed that a reliable 24 chromosome copy number profile could be obtained using the HLPA assay (Figure 3).
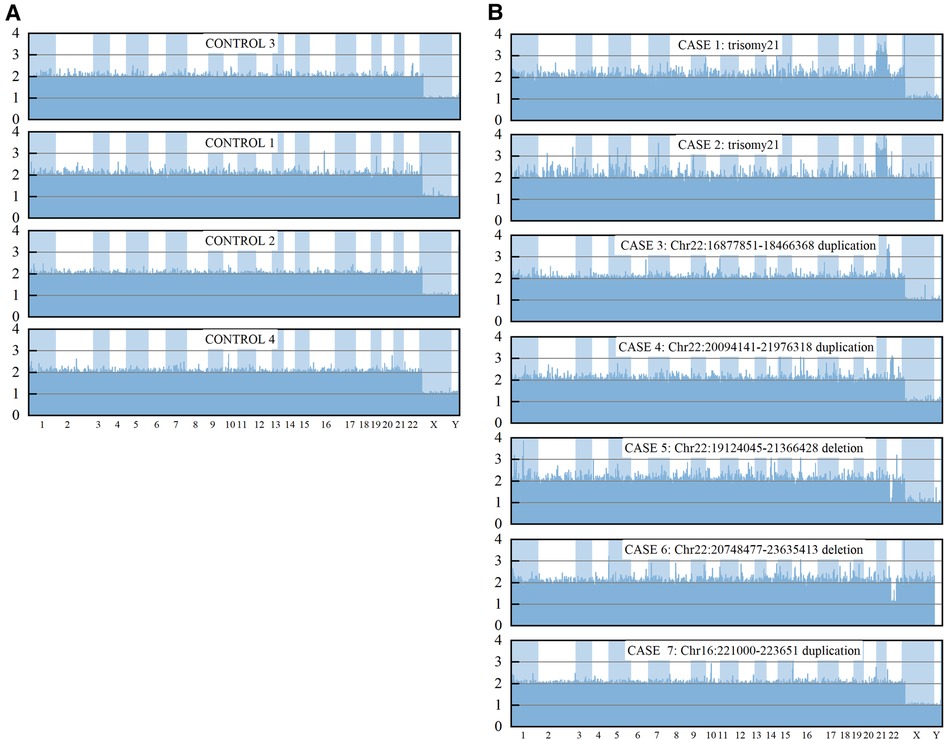
Figure 3. CNV measurements for control samples and a subset of CHD by HLPA. The final probe ratio depicted in the Y axis was around 2.0 if the region of interest was unaffected by CNV, whereas an increased or decreased value indicated the duplication or deletion, respectively.
Baseline characteristics and clinical data related to the first surgery of the CHD cases stratified by presence or absence of CNV or ppCNVs are presented in Supplementary Table S5 and Table 2. CHD cases with CNV exhibited a significantly higher proportion of complex surgeries (48.68% vs. 37.50%, P < 0.001). Duration of surgery time for cardiopulmonary bypass (CPBT), aortic cross clamp (ACCT) and mechanical ventilation procedures (MVT) were significantly longer in CHD cases with CNV (all P < 0.05). There were no significant differences in terms of age, weight and gender distribution between the two groups. No significant differences were identified between the groups for incidences of delayed sternal closure, continuous positive airway pressure, open-chest hemostasis, use of extracorporeal membrane oxygenation (ECMO) or left ventricular assist device (LVAD) systems, complications of surgery or one-month mortality after surgery.
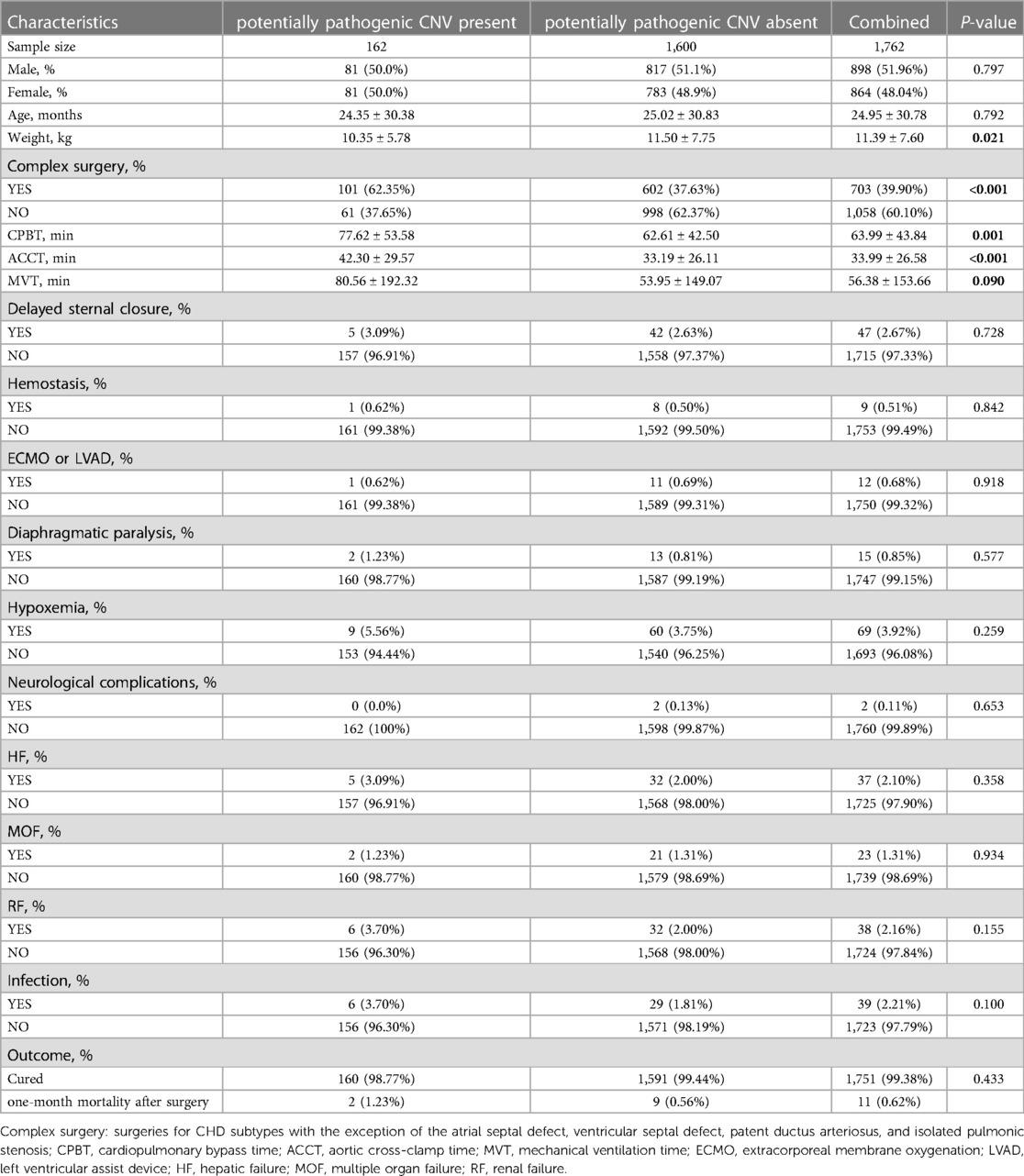
Table 2. Baseline characteristics of CHD patients (stratified by the presence or absence of ppCNVs).
In total, 162 CHD cases were identified with ppCNVs and had significantly higher incidences of complex surgeries compared to that of non-carriers (62.35% vs. 37.63%, P < 0.001). Durations for cardiopulmonary bypass and aortic cross clamp procedures were significantly longer in CHD cases with ppCNVs (all P < 0.05). There were no significant differences with respect to age, gender distribution or ventilation times between the two groups. No significant differences were identified between the groups for incidences of delayed sternal closure, continuous positive airway pressure, open-chest hemostasis, use of ECMO or LVAD systems, surgical complications, and one-month mortality after surgery.
Sub-group analysis of CHD sub-types
We categorized all children with CHD into 12 sub-groups. Detection number and rates of ppCNV findings in different CHD sub-groups are listed in Table 3 and Figure 4. Overall, the septal defect, defined as ventricular septal defect and atrial septal defect were the most observed heart malformation affecting 981 cases (55.67%), followed by conotruncal defects in 247 cases (14.02%). Patients with atrioventricular septal defect (AVSD) (23.10%) and left ventricular outflow tract obstruction (LVOTO) (20.90%) were diagnosed with high detection rates of ppCNV abnormalities, while patients with other CHD (0.00%) and AVSD (5.90%) symptoms exhibited low detection rates. The detection rate of patients with ppCNVs in the AVSD sub-group was significantly higher than that in the other sub-groups (23.10% vs. 9.70%, P = 0.002).
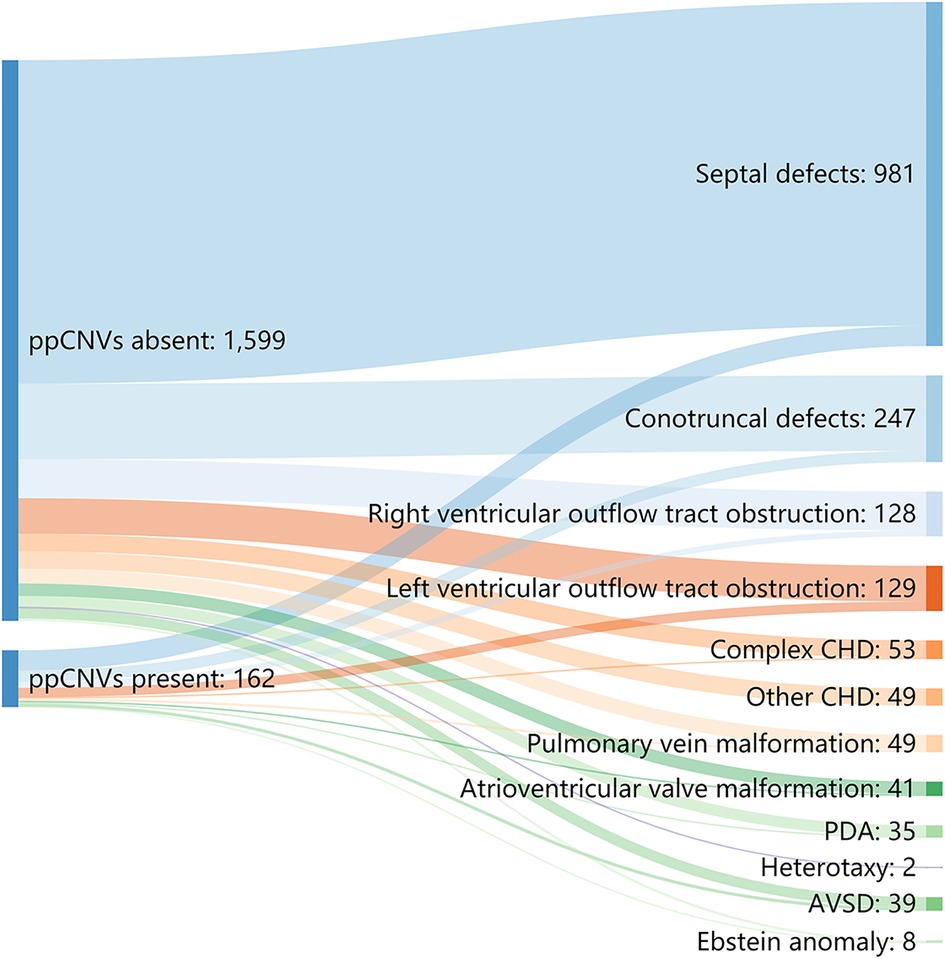
Figure 4. Sankey diagram describing the relative flow of CHD sub-groups according to the presence or absence of ppCNVs. CHD, congenital heart disease; PDA, patent ductus arteriosus; AVSD, atrioventricular septal defect.
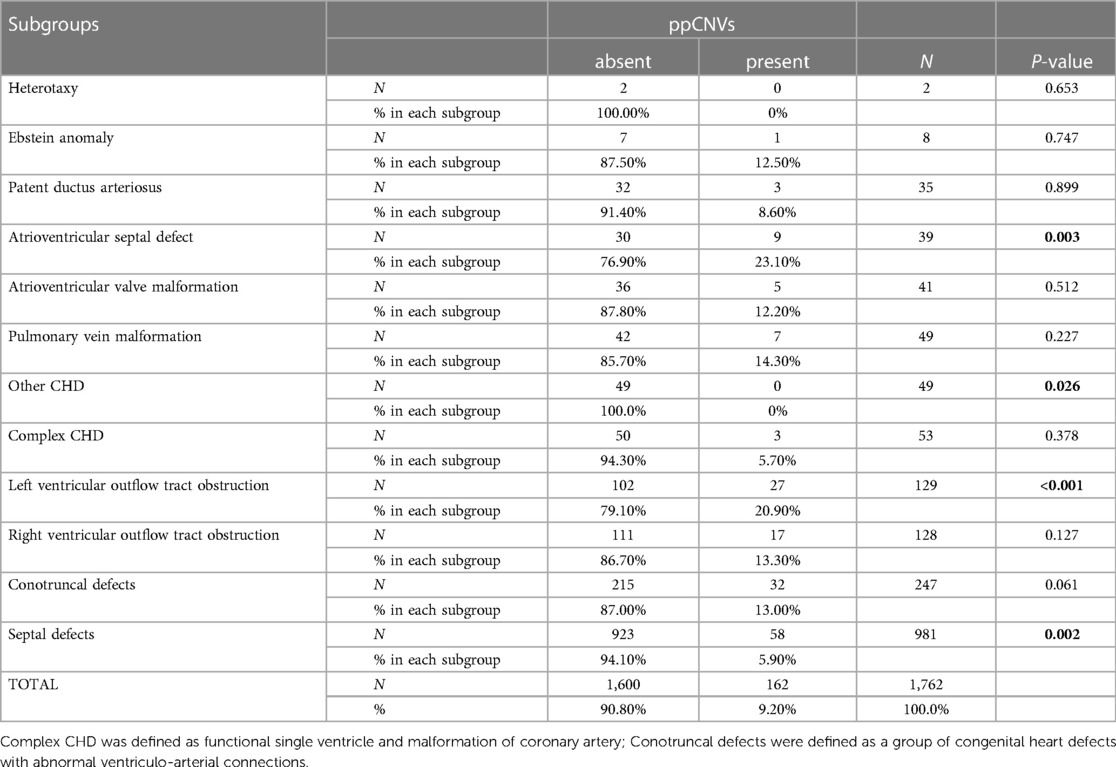
Table 3. Rates of chromosomal abnormalities in different CHD sub-groups.
Discussion
Genome-wide association studies of pathogenic and/or rare CNVs have highlighted the significant contribution of genetic variations toward CHD susceptibility (26, 27, 28, 29). In this study, 1,762 CHD patients, who underwent corrective surgeries for cardiac defects, were genetically screened for rare CNVs. The overall detection rate of ppCNVs was 9.19%, indicating a substantial genetic heterogeneity among the patients and the detection rate was slightly lower in accordance with previous studies (19, 30).
Notably, 2.04% of the CNV anomalies were aneuploidies, of which trisomy 21 was the most common chromosomal anomaly in these patients. Since most babies with Down syndrome present CHD, early screening and surgical interventions can greatly increase their survival rate and the life quality (31). Multiple deletion and duplication syndromes have been described both clinically and mechanistically (32). In many cases, genetic analysis of these syndromes has provided insights into the etiology of associated congenital anomalies. For example, the recognition that the majority of patients with DiGeorge syndrome carry a 22q11.2 deletion led to the discovery that a large number of cardiac patients with a subset of syndromic phenotypes might have 22q11.2 deletion (33). Further molecular studies have demonstrated that the T-Box transcription factor 1 (TBX1) gene, located within the 22q11.2 region, plays a critical role in the development of the abnormal cardiac phenotypes (34). Thus, we hypothesized that screening for known CNVs associated with the aberrant chromosomal disease might facilitate the genetic diagnosis of CHD in pediatric patients. Interestingly, we found 126 individuals with CNVs implicated in the known microdeletion or duplication syndromes, including 42 individuals with 22q11 microdeletions or duplications (2.38%) and 87 (4.77%) related to other chromosomal disease syndromes. Since the clinical manifestations of certain chromosomal diseases syndromes occur later than cardiac abnormalities, for example, abnormal neurodevelopmental phenotypes, genetic screening for CHD patients is highly recommended.
While CNV burden has previously been reported to be an important modifier of survival outcomes following CHD surgeries (15), our study did not identify correlation between CNV status and one-month survival after surgery. Further exploration with longer follow-up time is warranted. Our study was the first to investigate the contribution of CNVs to the complexity of corrective surgeries for cardiac defects. As described earlier, CHD cases with ppCNVs had a significantly higher proportion of complex surgeries. Time of turnover and blocking were also significantly longer in CHD cases with ppCNVs. Considering the long-term and complex impact of CNV burden, CHD patients should be genetically screened to evaluate the necessity of correction surgeries prior to surgical interventions for better disease management (35). Further research in this area is highly recommended to improve the treatment plan.
Consistent with the findings by Wang et al. (30), the septal defect was the most common type of heart malformation in our cohort, followed by conotruncal defects, with similar detection rates. Although Wang et al. demonstrated that the detection rate of pathogenic chromosomal abnormalities in fetuses with AVSD was significantly higher than those in the other sub-groups, the rate of incidence in these study sub-groups was lower- 73.7% in Wang et al. vs. 23.10% in this study. Furthermore, LVOTO (20.90%) was revealed to harbor a higher rate of ppCNV abnormalities compared with other studies (13.30% (19) and 0% (30)). These differences in rates could be due to differences in sample sizes and/or the proportions of isolated CHD cases between these studies.
Several high-throughput techniques, including SNP arrays, CGH arrays, and NGS-based assays are now widely used in genome-wide association studies (GWAS) to investigate the broader implications of chromosomal abnormalities in human diseases. These techniques, however, are expensive and, thus, may not be suitable for the preliminary rapid screenings in the clinics. Alternatively, other techniques, including FISH and MLPA, have also been used in recent studies. Despite FISH being the gold standard method for detecting chromosomal abnormalities, the technique has some key limitations, such as the time-consuming, laborious, requirement for experienced personnel and low-throughput efficiency, preventing its routine use in diagnostics. MLPA, a clinically effective targeted test, provides easily interpretable results also has a constraint of probe number (36). On the other hand, HLPA shares several advantages of MLPA, including high target specificity, since DNA probes require perfect complementarity to the target sequences for efficient ligation. Additionally, HLPA employs optimized lengthening ligation steps, allowing the use of a large number of short probes compared to MLPA. Notably, the use of short probes increases both annealing and amplification efficiencies, thereby, reducing the assay variability. In this study, we demonstrated that HLPA method could detect more than 200 genomic loci in a single reaction, using probes separated with an average chromosomal distance of 10 Mb, suggesting that CNVs with the sizes larger than 10 Mb can be readily detected. The turnaround time was within 24 h and, importantly, the cost for HLPA was substantially lower than that of other techniques (e.g., 1/10 of the cost of aCGH). Thus, the HLPA assay method was found to be the most suitable one with respect to higher genome coverage, accuracy, and cost-effectiveness.
Some limitations of this study should be considered. First, by concentrating on the overall burden of identified ppCNVs in healthy Han Chinese people from public datasets, we were only able to address the rarity of each particular CNV, and statistical power was constrained due to the absence of comparable cohorts. Second, due to the limited number of genomic samples of parents of the affected CHD cases, we were not able to determine whether the ppCNVs reported in our study were de novo or inherited. Third, like MLPA and aCGH techniques, which depend upon comparisons with reference chromosomes to determine the copy number, HLPA was unable to detect polyploidies. Moreover, due to the limited screening resolution, it could not detect small size microdeletions/duplications or complex structural rearrangements, such as balanced translocations and inversions. Therefore, the detection rates in this study might underestimate the actual rate of incidence of chromosome abnormalities. Finally, as the prevalence of individuals living with CHD has been increasing due to advances in pediatric surgery, more attention is needed concerning the contributions of CNV abnormalities on long-term surgical outcomes. We are currently conducting a 5-year follow-up study to evaluate the impact of CNVs on patients' outcomes.
In summary, this study focuses on the CHD pathology in children provides significant insights into the genomic landscape of Chinese CHD patients. Our data suggest that CNV burden could be an important contributor to the Chinese pediatric CHD patients. Our study also demonstrated the robustness of HLPA method for profiling CNVs in CHD patients, indicating the clinical potential of this technique toward its routine application in the clinical setting prior to cardiac surgery.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author/s.
Ethics statement
Subjects were enrolled at Shanghai Children’s Medical Center under a protocol approved by the Institutional Review Boards of Shanghai Children’s Medical Center, affiliated to Shanghai Jiao Tong University School of Medicine (SCMCIRB—K2021036-1), with written informed consent from each parent or guardian of the subjects. No identifiable information was used in this manuscript. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
(I) Conception and design: HFH and ZWJ (II) Administrative support: CXZ and JW (III) Provision of study materials or patients: YRH., CJ., YYX., HBZ., XL and LCY (IV) Collection and assembly of data: FY and YW (V) Data analysis and interpretation: YT and XFY (VI) Manuscript writing: All authors. (VII) Final approval of manuscript: All authors. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the Guided Project Fund of Shanghai Science and Technology Commission(20Y11910400)ÿNational Natural Science Foundation of China (81570281), The Clinical Science and Technology Innovation Project of Shanghai Shenkang Hospital Development Center (SHDC12018128).
Acknowledgments
The authors thank all subjects for their participation in this study and the Genesky Biotechnologies Inc., Shanghai for their experimental assistance.
Conflict of interest
YT, FY, YW and ZWJ are employees of Genesky Biotechnologies Inc., Shanghai.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1164577/full#supplementary-material.
References
1. Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. (2002) 39:1890–900. doi: 10.1016/S0735-1097(02)01886-7
2. Calzolari E, Garani G, Cocchi G, Magnani C, Rivieri F, Neville A, et al. Congenital heart defects: 15 years of experience of the Emilia-Romagna registry (Italy). Eur J Epidemiol. (2003) 18:773–80. doi: 10.1023/A:1025312603880
3. Hoang TT, Goldmuntz E, Roberts AE, Chung WK, Kline JK, Deanfield JE, et al. The congenital heart disease genetic network study: cohort description. PloS one. (2018) 13:e0191319–e0191319. doi: 10.1371/journal.pone.0191319
4. Thienpont B, Mertens L, de Ravel T, Eyskens B, Boshoff D, Maas N, et al. Submicroscopic chromosomal imbalances detected by array-CGH are a frequent cause of congenital heart defects in selected patients. Eur Heart J. (2007) 28:2778–84. doi: 10.1093/eurheartj/ehl560
5. Erdogan F, Larsen LA, Zhang L, Tümer Z, Tommerup N, Chen W, et al. High frequency of submicroscopic genomic aberrations detected by tiling path array comparative genome hybridisation in patients with isolated congenital heart disease. J Med Genet. (2008) 45:704–9. doi: 10.1136/jmg.2008.058776
6. Southard AE, Edelmann LJ, Gelb BD. Role of copy number variants in structural birth defects. Pediatrics. (2012) 129:755–63. doi: 10.1542/peds.2011-2337
7. Gelb BD, Chung WK. Complex genetics and the etiology of human congenital heart disease. Cold Spring Harbor Perspect Med. (2014) 4:a013953. doi: 10.1101/cshperspect.a013953
8. Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. (2006) 444:444–54. doi: 10.1038/nature05329
9. Fahed AC, Gelb BD, Seidman JG, Seidman CE. Genetics of congenital heart disease: the glass half empty. Circ Res. (2013) 112:707–20. doi: 10.1161/CIRCRESAHA.112.300853
10. Soemedi R, Wilson IJ, Bentham J, Darlay R, Töpf A, Zelenika D, et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am J Hum Genet. (2012) 91:489–501. doi: 10.1016/j.ajhg.2012.08.003
11. Pierpont ME, Basson CT, Benson DW Jr., Gelb BD, Giglia TM, Goldmuntz E, et al. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American heart association congenital cardiac defects committee, council on cardiovascular disease in the young: endorsed by the American academy of pediatrics. Circulation. (2007) 115:3015–38. doi: 10.1161/CIRCULATIONAHA.106.183056
12. Soemedi R, Topf A, Wilson IJ, Darlay R, Rahman T, Glen E, et al. Phenotype-specific effect of chromosome 1q21.1 rearrangements and GJA5 duplications in 2436 congenital heart disease patients and 6760 controls. Hum Mol Genet. (2012) 21:1513–20. doi: 10.1093/hmg/ddr589
13. Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, et al. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of fallot. Nat Genet. (2009) 41:931–5. doi: 10.1038/ng.415
14. Carey AS, Liang L, Edwards J, Brandt T, Mei H, Sharp AJ, et al. Effect of copy number variants on outcomes for infants with single ventricle heart defects. Circulation. Cardiovascular Genetics. (2013) 6:444–51. doi: 10.1161/CIRCGENETICS.113.000189
15. Kim DS, Kim JH, Burt AA, Crosslin DR, Burnham N, Kim CE, et al. Burden of potentially pathologic copy number variants is higher in children with isolated congenital heart disease and significantly impairs covariate-adjusted transplant-free survival. J Thorac Cardiovasc Surg. (2016) 151:1147–51. e4. doi: 10.1016/j.jtcvs.2015.09.136
16. Geddes GC, Butterly M, Sajan I. FISH For 22q11.2 deletion not cost-effective for infants with congenital heart disease with microarray. Pediatr Cardiol. (2015) 36:531–6. doi: 10.1007/s00246-014-1045-9
17. Jansen FA, Blumenfeld YJ, Fisher A, Cobben JM, Odibo AO, Borrell A, et al. Array comparative genomic hybridization and fetal congenital heart defects: a systematic review and meta-analysis. Ultrasound Obstet Gynecol. (2015) 45:27–35. doi: 10.1002/uog.14695
18. Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D, et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. (2009) 84(2):148–61. doi: 10.1016/j.ajhg.2008.12.014
19. Zhu X, Li J, Ru T, Wang Y, Xu Y, Yang Y, et al. Identification of copy number variations associated with congenital heart disease by chromosomal microarray analysis and next-generation sequencing. Prenat Diagn. (2016) 36:321–7. doi: 10.1002/pd.4782
20. Chen S, Liu D, Zhang J, Li S, Zhang L, Fan J, et al. A copy number variation genotyping method for aneuploidy detection in spontaneous abortion specimens. Prenat Diagn. (2017) 37:176–83. doi: 10.1002/pd.4986
21. Ruitenberg A, Ott A, van Swieten JC, Hofman A, Breteler MM. Incidence of dementia: does gender make a difference? Neurobiol Aging. (2001) 22:575–80. doi: 10.1016/S0197-4580(01)00231-7
22. Li X, Shi G, Li Y, Zhang X, Xiang Y, Wang T, et al. 15q11.2 Deletion is enriched in patients with total anomalous pulmonary venous connection. J Med Genet. (2021) 58:116–24. doi: 10.1136/jmedgenet-2019-106608
23. Wang Y, Zhou R, Jiang L, Meng L, Tan J, Qiao F, et al. Identification of chromosomal abnormalities in early pregnancy loss using a high-throughput ligation-dependent probe amplification-based assay. J Mol Diagn. (2021) 23:38–45. doi: 10.1016/j.jmoldx.2020.10.002
24. Yang L, Tang Y, Lu M, Yang Y, Xiao J, Wang Q, et al. Novel rapid molecular diagnosis of fetal chromosomal abnormalities associated with recurrent pregnancy loss. Acta Obstet Gynecol Scand. (2016) 95:1433–40. doi: 10.1111/aogs.13026
25. Mao J, Wang H, Li H, Song X, Wang T, Xiang J, et al. Genetic analysis of products of conception using a HLPA/SNP-array strategy. Mol Cytogenet. (2019) 12:40. doi: 10.1186/s13039-019-0452-2
26. Helm B, Landis B, Ware S. Genetic evaluation of inpatient neonatal and infantile congenital heart defects: new findings and review of the literature. Genes (Basel). (2021) 12(8):1244. doi: 10.3390/genes12081244
27. Meerschaut I, Vergult S, Dheedene A, Menten B, De Groote K, De Wilde H, et al. A reassessment of copy number variations in congenital heart defects: picturing the whole genome. Genes (Basel). (2021) 12(7):1048. doi: 10.3390/genes12071048
28. Audain E, Wilsdon A, Breckpot J, Izarzugaza J, Fitzgerald T, Kahlert A, et al. Integrative analysis of genomic variants reveals new associations of candidate haploinsufficient genes with congenital heart disease. PLoS Genet. (2021) 17:e1009679. doi: 10.1371/journal.pgen.1009679
29. Pineda T, Zarante I, Paredes A, Rozo J, Reyes M, Moreno-Niño O. CNVs in the 22q11.2 chromosomal region should be an early suspect in infants with congenital cardiac disease. Clinical Medicine Insights. (2021) 15:11795468211016870. doi: 10.1177/11795468211016870
30. Wang Y, Cao L, Liang D, Meng L, Wu Y, Qiao F, et al. Prenatal chromosomal microarray analysis in fetuses with congenital heart disease: a prospective cohort study. Am J Obstet Gynecol. (2018) 218:244.e1–244.e17. doi: 10.1016/j.ajog.2017.10.225
31. Morales-Demori R. Congenital heart disease and cardiac procedural outcomes in patients with trisomy 21 and turner syndrome. Congenit Heart Dis. (2017) 12:820–7. doi: 10.1111/chd.12521
32. Emanuel BS, Shaikh TH. Segmental duplications: an “expanding” role in genomic instability and disease. Nat Rev Genet. (2001) 2:791–800. doi: 10.1038/35093500
33. Goldmuntz E, Paluru P, Glessner J, Hakonarson H, Biegel JA, White PS, et al. Microdeletions and microduplications in patients with congenital heart disease and multiple congenital anomalies. Congenit Heart Dis. (2011) 6:592–602. doi: 10.1111/j.1747-0803.2011.00582.x
34. Vitelli F, Morishima M, Taddei I, Lindsay EA, Baldini A. Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum Mol Genet. (2002) 11:915–22. doi: 10.1093/hmg/11.8.915
35. Wang M-H, Friton JJ, Raffals LE, Leighton JA, Pasha SF, Picco MF, et al. Novel genetic variant predicts surgical recurrence risk in Crohn's Disease patients. Inflamm Bowel Dis. (2021) 27(12):1968–74. doi: 10.1093/ibd/izaa362
36. Capkova P, Srovnal J, Capkova Z, Staffova K, Becvarova V, Trkova M, et al. MLPA Is a practical and complementary alternative to CMA for diagnostic testing in patients with autism spectrum disorders and identifying new candidate CNVs associated with autism. PeerJ. (2019) 6:e6183–e6183. doi: 10.7717/peerj.6183
Keywords: congenital heart disease, pediatric, copy number variation, ligation-dependent probe amplification, surgery
Citation: Yu X, Tao Y, Liu X, Yu F, Jiang C, Xiao Y, Zhang H, He Y, Ye L, Wang Y, Zhou C, Wang J, Jiang Z and Hong H (2023) The implication of chromosomal abnormalities in the surgical outcomes of Chinese pediatric patients with congenital heart disease. Front. Cardiovasc. Med. 10:1164577. doi: 10.3389/fcvm.2023.1164577
Received: 13 February 2023; Accepted: 5 April 2023;
Published: 24 May 2023.
Edited by:
Yoshihide Mitani, Mie University, JapanReviewed by:
Toshio Nakanishi, Tokyo Women's Medical University, JapanJuan Carlos Núñez-Enríquez, Instituto Mexicano del Seguro Social, Mexico
Ola Eid, National Research Centre, Egypt
© 2023 Yu, Tao, Liu, Yu, Jiang, Xiao, Zhang, He, Ye, Wang, Zhou, Wang, Jiang and Hong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunxia Zhou emN4YXBhcmlzQDE2My5jb20= Jian Wang bGFid2FuZ2ppYW5Ac2hzbXUuZWR1LmNu Zhengwen Jiang emhlbmd3ZW5qQGdlbmVza2llcy5jb20= Haifa Hong aG9uZ2hmc2NtY0AxNjMuY29t
†These authors have contributed equally to this work
Abbreviations CHD, congenital heart disease; CNV, copy number variation; HLPA, high-throughput ligation-dependent probe amplification; PDA, patent ductus arteriosus; PFO, patent foramen ovale; CPBT, time for cardiopulmonary bypass; ACCT, time for aortic cross clamp; MVT, time for mechanical ventilation procedures; ECMO, extracorporeal membrane oxygenation; LVAD, left ventricular assist device; AVSD, atrioventricular septal defect; LVOTO, left ventricular outflow tract obstruction.