 Laura J. den Hartigh
Laura J. den Hartigh Karolline S. May
Karolline S. May Xue-Song Zhang3
Xue-Song Zhang3 Alan Chait
Alan Chait Martin J. Blaser
Martin J. Blaser- 1Department of Medicine, Division of Metabolism, Endocrinology, and Nutrition, University of Washington, Seattle, WA, United States
- 2Diabetes Institute, University of Washington, Seattle, WA, United States
- 3Center for Advanced Biotechnology and Medicine, Rutgers University, Piscataway, NJ, United States
Serum amyloid A (SAA) subtypes 1–3 are well-described acute phase reactants that are elevated in acute inflammatory conditions such as infection, tissue injury, and trauma, while SAA4 is constitutively expressed. SAA subtypes also have been implicated as playing roles in chronic metabolic diseases including obesity, diabetes, and cardiovascular disease, and possibly in autoimmune diseases such as systemic lupus erythematosis, rheumatoid arthritis, and inflammatory bowel disease. Distinctions between the expression kinetics of SAA in acute inflammatory responses and chronic disease states suggest the potential for differentiating SAA functions. Although circulating SAA levels can rise up to 1,000-fold during an acute inflammatory event, elevations are more modest (∼5-fold) in chronic metabolic conditions. The majority of acute-phase SAA derives from the liver, while in chronic inflammatory conditions SAA also derives from adipose tissue, the intestine, and elsewhere. In this review, roles for SAA subtypes in chronic metabolic disease states are contrasted to current knowledge about acute phase SAA. Investigations show distinct differences between SAA expression and function in human and animal models of metabolic disease, as well as sexual dimorphism of SAA subtype responses.
1. Introduction
Members of the serum amyloid A (SAA) family are acute phase reactants and chemokines that are elevated in acute inflammatory conditions such as infection (1, 2), as well as chronic inflammatory conditions including autoimmune disorders (3–8), obesity (9–13), type 2 diabetes (T2D) (14, 15), and cardiovascular disease (CVD) (16–19) (reviewed extensively in 20, 21). Several SAA subtypes are present across diverse animal species (22), including invertebrates (23), suggesting important conserved functions. Since SAA is poorly soluble in aqueous solutions, it circulates associated with lipoproteins, in particular high density lipoprotein (HDL), and is considered an apolipoprotein (24, 25). Functions of particular SAA subtypes include roles in host defense (26–30), chemoattraction (31–34), lipid metabolism (35–37), and inflammation (38). We now review the emerging knowledge about distinctive functions of the different SAA subtypes.
1.1. SAA subtypes and receptors
Of the 4 known SAA subtypes, SAA1 and SAA2 are highly expressed in the liver in mammals including humans in response to inflammatory stimuli, and can circulate at high concentrations, usually bound to HDL (39). SAA1 and SAA2 are highly homologous, differing in only a few amino acids. In contrast, SAA3 is more highly expressed in extrahepatic tissues in particular animal species (40, 41). SAA3 is not known to circulate under most conditions, with the exception of high dose lipopolysaccharide (LPS) injection (42). SAA3 is considered to be a pseudogene in humans due to a premature stop codon (43), leading to a frame shift in codon 31, thereby deleting the last ten amino acids (44). SAA3 is only ∼40% homologous to SAA1/2. Since in humans SAA1 and SAA2 are expressed from both liver and extrahepatic tissues, it has been difficult to conclusively distinguish hepatic from extrahepatic SAA functions in humans. However, phenotypic distinctions between hepatic and extra-hepatic SAA subtypes in mice, due to the predominance of extrahepatic Saa3, allow sharper definition (18, 38, 44). SAA3 protein has been detected in human mammary gland epithelial cell lines (45), although its expression is more commonly found in non-human mammals. SAA4 is constitutively expressed by most cell types and responds only minimally to inflammatory stimuli (46, 47). In many prior studies, distinctions between specific SAA subtypes were not reported, perhaps due to the lack of available antibodies capable of distinguishing them. This is unfortunate, as it is possible that different SAA subtypes exert different functions in the context of metabolic disease. In this review, we use the term “SAA” to refer to SAA1/2, or to reflect that the authors of work described did not specify particular SAA subtypes. In addition, in accordance with scientific nomenclature standards, “SAA” will refer to humans, while “Saa” corresponds to mouse.
The major identified SAA receptors are listed in Table 1. SAA binds to formyl peptide like receptors 1 and 2 (FPLR1 and FPLR2) in human monocytes, neutrophils, human embryonic kidney (HEK293) cells, and human umbilical vein endothelial cells (HUVECs), thus promoting chemotaxis and increased calcium flux. In response to varied stimuli (Table 1), mitogen-activated protein kinases (MAPKs) and nuclear factor kappa B (NFκB) pathways are further activated, which leads to secretion of tumor necrosis factor alpha (TNFα), interleukin-8 (IL-8), and monocyte chemotactic protein-1 (MCP-1) (32, 33, 48–51, 69, 70). The receptor for advanced glycation end products (RAGE) is another known SAA receptor present on several tissues and cell types. SAA mediates the activation of the AGE/RAGE axis and NFκB pathways, with subsequent transcription of interleukin-6 (IL-6), heme oxygenase type-1 (HO-1) and monocyte colony stimulating factor (M-CSF) (52–55). Moreover, SAA induces signal transducer and activator of transcription 1 (STAT1)-mediated high mobility group box 1 (HMGB1) expression and protein kinase R (PKR) activation, potentially through RAGE and toll-like receptors (TLRs) (52). SAA has affinity for TLR2 and TLR4 (56–61, 71, 72), and to the oxidized low-density lipoprotein receptor (LOX-1) (62) and scavenger receptor class B type 1 (SRB1), thus mainly signaling via the MAPK pathway in both immune and epithelial cells. A recently described SAA receptor is Selenoprotein S/Tanis [SELS in humans (63, 65, 67, 68), Tanis in animal models (68)]. Tanis/SELS is highly expressed in liver, skeletal muscle, and adipose tissue (68), which may distinguish SAA effects mediated by this receptor from those found primarily on immune cells. SELS expression on adipose tissue is highly correlated to circulating SAA levels, suggesting a potential feed-forward mechanism (68, 73). Importantly, most of these potential SAA receptors respond to multiple ligands, with SELS having the highest degree of SAA-specificity. Collectively, varied SAA receptor expression patterns on different cell and tissue types could indicate different SAA functions.
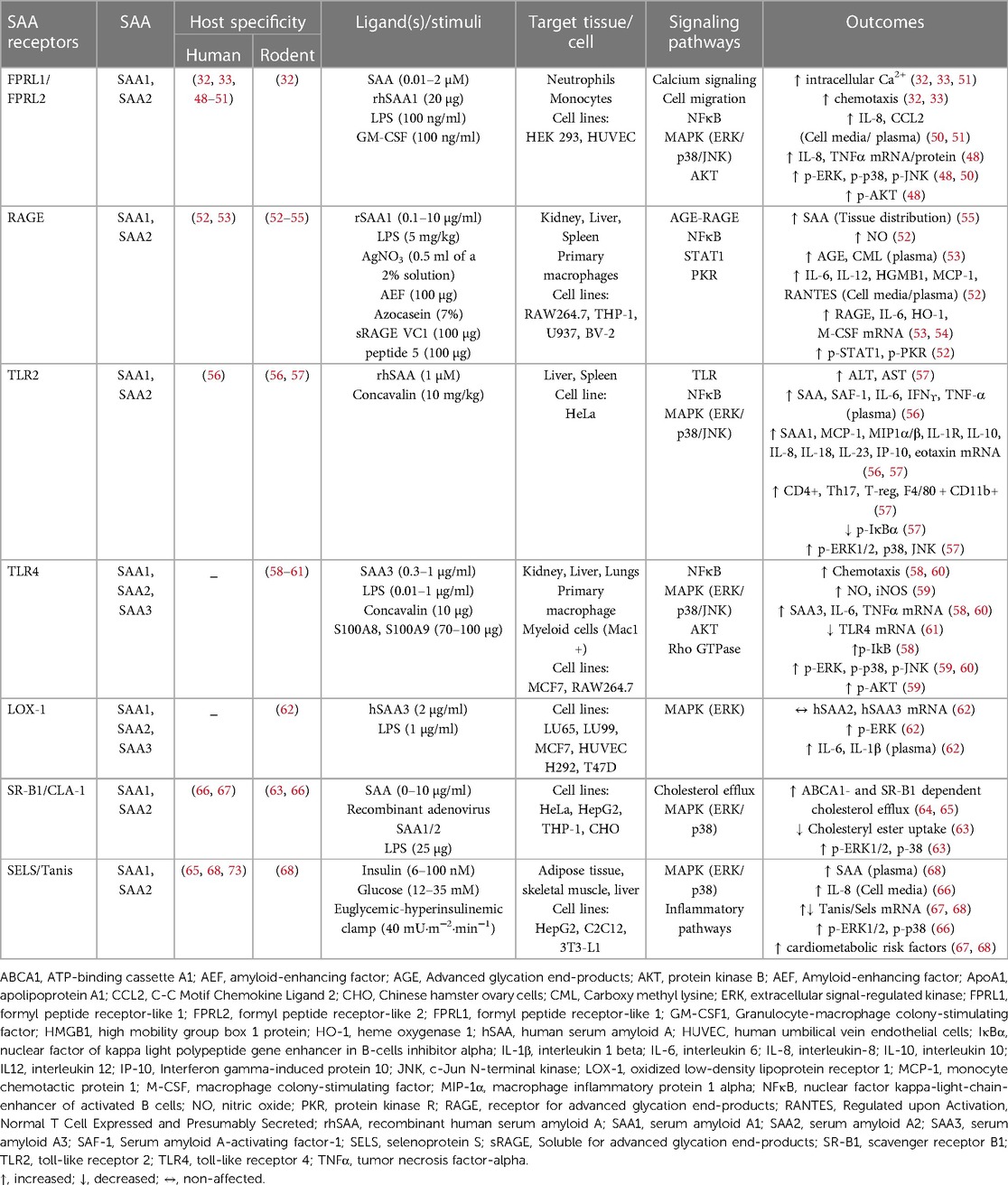
Table 1. SAA receptors and downstream signaling pathways.
1.2. SAA regulation in the acute phase response
Considerable research has been focused on the kinetics of hepatic SAA expression and secretion during an acute inflammatory response [reviewed in (20, 74)]. The mechanics of SAA expression and secretion vary with the stimulus type. Systemic levels of SAA can be 1,000-fold higher than baseline during an acute inflammatory response to sepsis (75, 76), viral infections including COVID-19 (1, 77, 78), vaccinations (79), or tissue trauma (80). The immediate systemic levels of SAA are primarily hepatic in origin during infection (44), with contributions from extra-hepatic sources following tissue trauma (81). Hepatic SAA production is triggered by bacterial products such as endotoxin or inflammatory cytokines interleukin-1 beta (IL-1β), interleukin-6 (IL-6), and TNFα that reach the liver (74). While much prior work has focused on the hepatic acute-phase SAA1 and SAA2 subtypes, important roles for extra-hepatic SAA in the chronic inflammatory processes associated with metabolic diseases are now emerging (82, 83).
1.3. SAA vs. CRP
Since its discovery nearly 100 years ago, C-reactive protein (CRP) has been used in clinical practice as a marker of acute inflammation (84). CRP is known to rapidly increase in response to infection or trauma, and has a short half-life that enables a rapid decrease when the stimulus ceases (85). However, SAA rises in parallel with CRP in the same acute inflammatory conditions, and may be a more sensitive marker for acute events (19, 86–88). Similar to CRP, hepatic SAA is regulated by the above inflammatory cytokines (IL-1β, IL-6, and TNFα 89, 90), although CRP can be induced by pathways related to interleukin-17 (IL-17) and hepatocyte nuclear factor (HNF), in contrast to SAA (91). In addition to inflammatory cytokines, hormones including glucocorticoids, leptin, and thyroid hormone also regulate SAA expression (92, 93). Indeed, SAA levels may be better predictors of coronary artery disease (CAD), cancer, and of related poor outcomes than CRP (19, 94). However, CRP levels more accurately predict poor outcome in elderly populations (95). SAA as a biomarker of acute infection or traumatic injury remains less widely used clinically due to a lack of robust calibration reagents and routine assays. There would be great value to developing reliable, robust, and cost-effective SAA clinical assays.
2. SAA in chronic metabolic diseases
Chronic inflammatory conditions tend to promote much lower elevations in systemic SAA (∼3 to 10-fold) than acute inflammatory conditions and may be sustained, deriving from diverse tissues such as the liver, adipose tissue, lung, small and large intestines, and hematopoietic cells such as macrophages (9, 11, 18, 96–100). The markedly different systemic SAA levels observed in acute vs. chronic inflammatory conditions suggests the potential for different mechanisms (91), prompting speculation that SAA is an important concentration-dependent effector of innate and adaptive immune responses (44). Aging has been associated with increased SAA levels (83, 101, 102), as have aging-related metabolic conditions. Evidence for potential roles of SAA in several metabolic diseases are discussed in the sections that follow, with an emphasis on obesity, diabetes, non-alcoholic fatty liver disease (NAFLD), CVD, autoimmune conditions such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA), and inflammatory bowel diseases (IBD) ulcerative colitis (UC) and Crohn's disease (CD) (Figure 1).
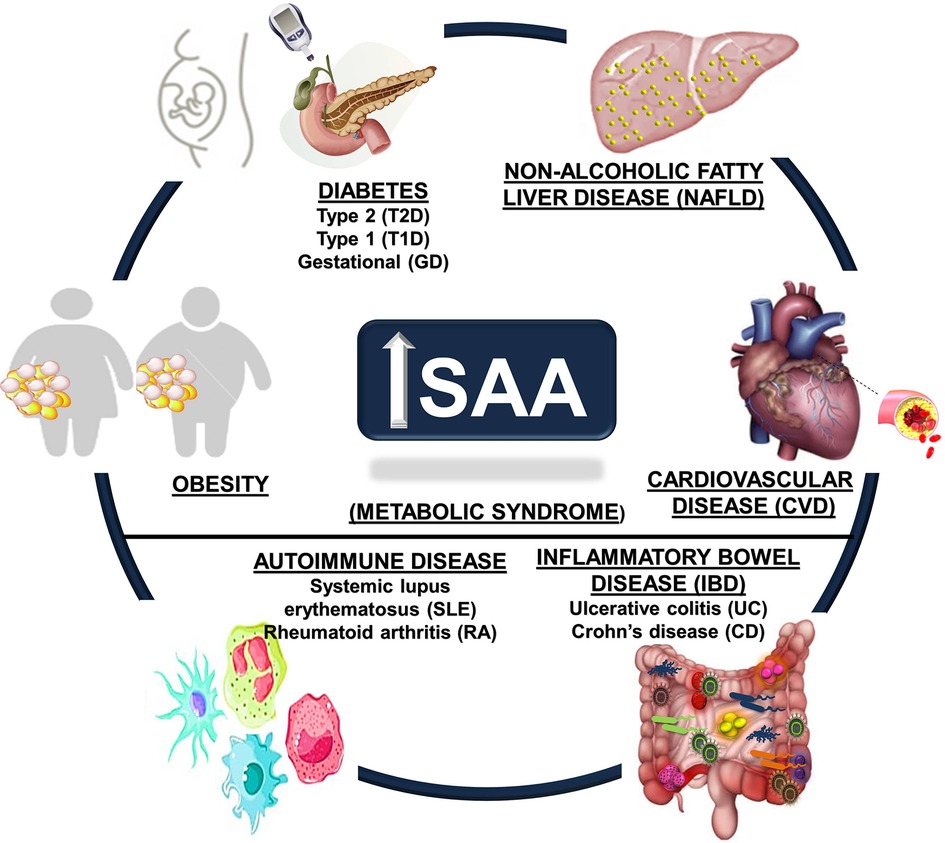
Figure 1. Metabolic disease states associated with increased circulating SAA. Obesity, cardiovascular disease (CVD), autoimmune diseases (including systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA)), diabetes (Type 1, Type 2, and gestational), inflammatory bowel disease (IBD, including Crohn's disease (CD) and ulcerative colitis (UC)), and non-alcoholic fatty liver disease (NAFLD) are chronic metabolic conditions that are all associated with increased circulating SAA levels.
2.1. Obesity and metabolic syndrome
The increased circulating SAA levels observed in individuals with obesity are highly correlated with body mass index (BMI), body weight, adiposity, and SAA1 and SAA2 mRNA expression in white adipose tissue (WAT) (11, 96), and are not related to hepatic SAA1 or SAA2 expression (9, 97, 98, 103). Circulating SAA levels have been positively associated with visceral adiposity (104), suggesting visceral fat as a potential source. However, the relative contributions of subcutaneous and visceral WAT to SAA production are not known, nor has it been determined that WAT-derived SAA contributes to the circulating SAA pool in obesity (105), or whether WAT-derived SAA induces local cytokine production that stimulates hepatic SAA expression. SELS, a major SAA receptor, is expressed in adipose tissue and directly associates with adiposity and BMI (65), suggesting a potential feed-forward mechanism that contributes to the sustained adipose tissue inflammatory state in obesity (71). Whether increased SAA expression in WAT plays a local or systemic role in obesity pathogenesis, or whether it is merely a biomarker of disease severity, is unknown. The extent to which WAT, liver, or both contribute to systemic SAA levels has not been resolved.
An initial study of 34 subjects with obesity showed a 6-fold increase in SAA expression in subcutaneous WAT compared with 27 lean controls; this was associated with 20-fold higher expression from adipocytes than the WAT stromal vascular fraction (96), which contains pre-adipocytes, immune cells, and vasculature. A meta-analysis confirmed a strong positive association between BMI and circulating SAA levels (13), and showed that SAA1 and SAA2 expression was higher in subcutaneous WAT in people with overweight and/or obesity (97, 106). In addition, serum SAA levels are positively associated with adipocyte diameter (106, 107). Distinctions between SAA1 and SAA2 were generally not made in these early studies due to the lack of distinguishing primers and subtype-specific antibodies that persists.
Conversely, weight loss can reduce circulating and adipose tissue-derived SAA levels in humans. A meta-analysis of 10 studies showed that weight loss significantly reduced circulating SAA levels (13). Weight loss following a low-fat (LFD) (n = 19) or very low carbohydrate diet (VLCD) (n = 22) led to reduced circulating SAA levels proportional to the amount of weight lost and also associated with insulin resistance (88). Several independent studies showed that weight loss due to a VLCD in women (n = 33–48) was strongly associated with reduced plasma SAA and adipocyte-derived SAA (9, 10, 96, 108), while insulin sensitivity was not consistently affected (9, 108). These divergent phenotypes could reflect different subject characteristics, with postmenopausal women showing a metabolic benefit from SAA reduction (9, 10) while premenopausal women did not (108). Another study in 439 women reported similar reductions in plasma SAA with weight loss due to dietary intervention, but not exercise (109). Importantly, other inflammatory markers including MCP1 and CRP also decreased during weight loss (109). Roux-en-Y gastric bypass significantly reduced circulating SAA levels in women with obesity (n = 20) (110). Additional studies are required to determine whether specifically reducing SAA in the context of weight loss is beneficial.
Mouse studies parallel the observation that SAA levels are increased in humans with obesity, and that adipose tissue mRNA expression of Saa is similarly increased in the obese state. Initial studies identified Saa3 as the specific subtype expressed in murine adipocytes (111) and macrophages (112, 113), both essential for development of obesity. Ob/ob mice, which spontaneously develop obesity due to increased food consumption subsequent to leptin deficiency, have elevated circulating and adipose tissue Saa levels (114, 115). Further, diet-induced obese mice consistently have elevated Saa3 mRNA levels in adipose tissue (82, 116–121). However, obesity-associated adipose-derived Saa3 does not contribute to circulating Saa levels in mice (105). Mice engineered to express luciferase via the Saa3 promotor only show luciferase activity in adipose tissue following long-term high fat diet (HFD)-fed conditions, with no detectable luciferase in any tissue examined after one week of HFD or after acute injection with LPS, providing temporal data about Saa3 expression kinetics (121). However, using more sensitive mass spectrometry, we have shown that a single high dose LPS injection is sufficient to induce Saa3 expression in adipose tissue, associated with increases circulating Saa (42), an effect supported by identifying Saa3 in LPS-stimulated plasma using isoelectric focusing gels and ELISA (122).
Sleep deprivation has been associated with sharp increases in SAA. Circulating SAA levels increased by more than 4-fold in mice experiencing paradoxical sleep deprivation for 72 h, an effect coincident with increased adipose tissue Saa3 mRNA expression, but not Saa1/2 (123). Circulating Saa and Saa3 mRNA returned to basal levels when sleep was restored. Importantly, increased circulating SAA also has been observed in humans deprived of sleep for either 24 or 48 h (123). In another study, SAA levels were 2-fold elevated in 17 adults who regularly experienced obstructive sleep apnea, which disrupts sleep, compared to weight-matched controls (124). Obstructive sleep apnea is strongly associated with the metabolic syndrome (125), also associated with SAA levels, which may confound interpretation of these studies. Because sleep deprivation and disrupted sleep schedules increase risk for obesity and its complications, disrupted sleep-induced SAA could be considered a novel risk factor for metabolic disease.
Studies in which mouse Saa is perturbed genetically have yielded ambiguous results. Mice engineered to express human SAA1 from WAT had elevated circulating human SAA1 mirroring obesity levels even without an obesogenic stimulus (126), providing evidence that WAT-derived SAA circulates. However, overexpressing SAA1 from WAT had no observed effects on body weight, WAT inflammation, or glucose or insulin tolerance (127). Loss of extrahepatic Saa3 in obese mice led to improved local WAT inflammation and systemic lipoprotein profiles and to resistance to high fat diet (HFD)-induced obesity, particularly in female mice (82). By contrast, subsequent Saa3 knock out mice were more prone to HFD-induced obesity with increased adiposity (128). Further, triple knock-out mice (Saa1, Saa2, and Saa3-deficient) showed no effect of a HFD on body weight or adiposity, but had worsened glucose and insulin tolerance (129). These divergent results suggest that the distinct metabolic characteristics of the models used, such as the inclusion of dietary sucrose/cholesterol, which particular Saa subtypes are perturbed, or gut microbiota composition and function, could have major impacts on observed phenotypes related to Saa.
Despite such phenotypic differences in obesity when SAA subtypes were perturbed, several studies point towards SAA promoting adipose tissue expansion. Silencing Saa3 in cultured pre-adipocytes reduced their adipogenic potential, leading to smaller adipose tissue depots when injected into NUDE mice (130). Similarly, targeting Saa using anti-sense oligonucleotides reduced adipose tissue expansion and inflammation as well as circulating endotoxin levels in male Swiss Webster mice (131), suggesting that disrupting Saa signaling also improved intestinal barrier integrity. Increased Saa3 expression in visceral adipose tissue from obese mice is highly correlated with macrophage number and inflammatory expression profile (121), suggesting that interaction with macrophages may drive adipocyte Saa3 expression. Thus, the crosstalk between adipocytes and macrophages that promotes adipose tissue inflammation and subsequent insulin resistance in obesity may require SAA (121).
2.2. Type 2 diabetes and gestational diabetes
Excess visceral adiposity and increased systemic inflammation are associated with insulin resistance (132, 133), which is the reduced capacity for insulin-stimulated glucose uptake in metabolically active tissues such as adipose tissue and skeletal muscle. Pancreatic insulin secretion subsequently increases to compensate for reduced insulin sensitivity to maintain euglycemia. If the pancreatic beta cells are unable to secrete sufficient insulin to compensate for the reduced insulin sensitivity (termed beta cell dysfunction), hyperglycemia ensues, leading to glucose intolerance and eventually T2D (134). Cross-sectional studies in men of European, Asian Indian, and American descent have shown that total, visceral, and subcutaneous adiposity, BMI, and waist circumference are all negatively associated with insulin sensitivity (135, 136). In addition to its association with obesity, with a key contribution from adipose tissue, SAA is similarly associated with T2D in humans and in animal models. In 134 patients with T2D, circulating SAA levels strongly correlated with hemoglobin A1c (HbA1c) and homeostatic model assessment for insulin resistance (HOMA-IR) after controlling for age, sex, and BMI status (14), suggesting a relationship between SAA and insulin resistance.
In humans, diabetes and circulating SAA levels are strongly related (11, 107, 137–140), and a prospective association between SAA and incident T2D has been reported (15). In a study of 765 older men (mean age 77), 112 with T2D, serum SAA strongly correlated with diabetes status, an association lost when adjusted for BMI, waist circumference, or fasting insulin levels (141). In a small study, omental adipose tissue from subjects with diabetes (n = 6) had a 3-fold increase in SAA mRNA expression compared with non-diabetic controls (n = 10), and omental SAA expression strongly correlated with fasting glucose levels and total body fat mass (142). In 134 subjects with T2D, HbA1c and HOMA-IR strongly correlated with circulating SAA levels after controlling for age, sex, and BMI (14); the effect was reduced with adjustment for parameters related to glucose metabolism (15), suggesting linkage between SAA and insulin resistance. In subjects with both obesity and T2D, SAA is bound to apoB-containing lipoproteins including very low-density lipoproteins (VLDL) and low-density lipoproteins (LDL), in addition to HDL [its usual transport partner in plasma (37)], similar to observations in mice (143). The mechanism for SAA binding to these lipoproteins in people with diabetes is unknown. Evidence exists that a truncated form of SAA1, which is missing an N-terminal arginine, is reduced in subjects with T2D and is negatively associated with glycemic control (144). Adipose tissue SELS was positively associated with measures of glycemic control in both lean and obese subjects (65, 73), as well as in age- and weight-matched subjects with diabetes (145). Moreover, insulin increases SELS expression in cultured adipocytes (65), suggesting a potential feed-forward mechanism for increased SAA expression in insulin resistance. SAA disrupts insulin signaling in cultured adipocytes (120, 146), suggesting a potential mechanism for its association with T2D. Most T2D subjects also have abdominal obesity, making it difficult to tease apart obesity-specific and T2D-specific contributions of SAA.
However, a strong association exists between diabetes and SAA that is independent of obesity. One study of 182 T2D subjects showed elevated serum SAA levels compared to healthy weight-matched controls (n = 180), with mean BMI of 24 in both groups (147). A small study similarly showed that SAA levels were elevated in age- and weight-matched subjects with T2D compared with normoglycemic controls (73). Controlling for age, sex, and BMI revealed a sustained correlation between indices of glucose dysregulation (i.e., HbA1c, HOMA-IR) and SAA, suggesting an effect specific to the diabetic state (14). However, another study found no differences in SAA levels between weight-matched subjects with obesity or T2D (110). To our knowledge, only a single study has reported no differences in SAA between healthy insulin-sensitive subjects and those with T2D (148). Emerging evidence suggests that improving insulin sensitivity drives the reduction in SAA levels following weight loss. In a small study in which subjects with overweight or obesity were given rosiglitazone for 12 weeks, circulating SAA levels were reduced by 37% despite the absence of weight loss, and WAT explants from these subjects showed lower SAA secretion post-treatment (9). Pharmacotherapy for T2D (i.e., metformin, glipizide, rosiglitazone, insulin, or acarbose) reduces serum SAA levels in T2D subjects (9, 139, 149). Thus, while the diabetic state and SAA levels are directly associated, whether SAA plays a distinct role in T2D pathology independent of a role in obesity remains to be determined.
SAA levels are further elevated in subjects with T2D and nephropathy (147, 150) and retinopathy (151). SAA may be an important predictor for end-stage renal disease and death in patients with diabetic kidney disease, with elevated intra-renal SAA expression (152). SAA is elevated in T2D patients with proteinuria, with serum SAA levels positively associated with albumin excretion rate and glomerular membrane thickening (140, 153), consistent with a potential causal role.
Similar links between Saa and T2D have been observed in animal models. In mice, a HFD promotes early increases in Saa3 expression in white adipose tissue, with subsequently elevated hepatic levels of Saa1 and Saa2 (120). In these models, insulin resistance is highly correlated with circulating Saa levels (120). In hepatocytes, overexpression of the Saa receptor, Tanis, led to decreased insulin-stimulated glucose uptake and glycogen synthesis, indicating increased insulin resistance (73). Db/db mice, which lack the leptin receptor and spontaneously develop features resembling obesity and T2D, express high levels of Saa3 from adipocytes, but not the liver (114). In a common rodent model of T2D in which obesity is initiated by consumption of a HFD and hyperglycemia is triggered by the administration of low-dose streptozotocin (STZ), a beta cell toxin that promotes hyperglycemia, renal Saa3 is increased (154).
Systemic SAA levels are elevated in pregnancy, especially in women with gestational diabetes (GD) (101). Serum SAA levels were 14% higher in 39 pregnant women with GD than in 25 healthy controls, and SAA was positively associated with BMI, age, oral glucose tolerance test, and HbA1c levels (155). It is unknown whether GD itself increases systemic SAA levels, or whether increased SAA simply reflects gestational weight gain (156). While one study did not observe increased SAA levels in GD patients, decreased variability in SAA levels was observed (157). Further studies are required to conclusively determine if SAA plays a detrimental role in GD. Indeed, a prospective clinical trial (NCT04238936) aims to compare SAA levels between women diagnosed with GD and healthy controls.
2.3. Polycystic ovary syndrome (PCOS)
PCOS is a chronic inflammatory condition that impacts ∼5%–10% of women of reproductive age in industrialized countries and is associated with an increased incidence of obesity, diabetes, and atherosclerosis (158, 159). In a study of 83 subjects with PCOS, serum SAA levels were double those of 39 age-matched controls (160). Omental and subcutaneous WAT biopsies showed increased SAA mRNA and protein expression, suggesting that the circulating SAA derived at least in part from adipose tissue. Incubation of adipose tissue explants with glucose increased SAA production, providing evidence that SAA secretion may be regulated by hyperglycemia. PCOS subjects were insulin-resistant, and a 6-month treatment regimen with metformin reduced circulating SAA levels, suggesting a possible link between SAA and adipose tissue insulin sensitivity (160). Because PCOS is associated with enhanced WAT lipolysis (161), and WAT-derived SAA also augments lipolysis (9), we speculate that WAT-derived SAA may play a causal role in PCOS-mediated metabolic dysfunction.
2.4. Non-alcoholic fatty liver disease (NAFLD)
NAFLD is commonly present as part of the metabolic syndrome (162), a constellation of disorders that increase the risk for CVD and diabetes, including abdominal obesity, hyperglycemia/insulin resistance, hypertension, and dyslipidemia (163). NAFLD is characterized by triglyceride accumulation in hepatocytes (steatosis), which can progress to steatohepatitis, characterized by the accumulation of inflammatory cells. SAA levels often are elevated in patients with the metabolic syndrome (164, 165). SAA was found to be 2–3-fold higher in patients with non-alcoholic steatohepatitis relative to age-matched healthy controls (166). Because liver biopsy, the gold standard diagnostic test for the presence of NAFLD, is an invasive procedure, non-invasive biomarkers for this condition would be highly desirable. However, although SAA could potentially be a useful biomarker for NAFLD, it is too non-specific to justify its use for this purpose.
Mechanisms linking SAA and NAFLD remain speculative. In the Cohort on Diabetes and Atherosclerosis Maastricht (CODAM) study, in which alanine amino transferase (ALT) was used as a surrogate measure of NAFLD, multiple linear regression analysis was used to investigate the association between ALT and several metabolic syndrome components as potential mediators of the liver disease. Their findings suggest that insulin resistance is the key pathophysiological mechanism to explain the association between the metabolic syndrome and NAFLD, with adipose tissue inflammation, endothelial dysfunction and free fatty acid levels likely playing lesser roles (167). However, ALT is an imperfect biomarker for NAFLD. Cytokines produced by liver-resident and infiltrating inflammatory cells may play important roles in liver inflammation and NAFLD. SAA may exacerbate hepatic steatosis via the TLR4-mediated NFκB signaling pathway (168). Hepatocyte-derived SAA1 promotes intrahepatic platelet aggregation and aggravates liver inflammation in NAFLD (169). Studies using hypercholesterolemic mice deficient in IL-1α or IL-1β showed the importance of these two cytokines in transforming steatosis to steatohepatitis and liver fibrosis (170). Given the well-documented link between SAA and IL-1β, SAA may also be important for liver disease progression. However, this requires additional study.
2.5. Cardiovascular disease (CVD)
Inflammation is a hallmark of atherosclerosis (171), and a recent clinical trial, The Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS), for the first time showed in a proof-of-concept trial that inhibiting inflammation using an antibody against Il-1β decreased cardiovascular events (172). The relationship between inflammation and CVD has been extensively studied by measurement of the inflammatory marker, CRP, which consistently has been shown to be modestly and chronically elevated in CVD patients and to predict the risk of cardiovascular events in a similar manner to SAA (19, 173, 174), although SAA has not been studied as extensively as CRP. As noted earlier, acute phase SAA is a good predictor of coronary artery disease outcomes (19, 94).
SAA could simply be a biomarker of the chronic inflammatory state that is present in CVD, similar to CRP; alternatively, it may play pathogenic roles. As described below, considerable evidence points to its role as a mediator rather than simply being a marker of atherosclerotic CVD. In considering its possible mediating role, potential differences between effects of lipoprotein-bound SAA and free SAA derived from extrahepatic cells in the artery wall must be distinguished.
SAA mRNA is present in macrophages, smooth muscle cells and endothelial cells in human atherosclerotic lesions (18), findings that suggest an immune response within the atherosclerotic artery wall, in which locally generated SAA is unlikely to be associated with lipoproteins. However, other studies showed immunohistochemical colocalization of Saa with apolipoproteins, including apoA1, the major apolipoprotein of HDL, in murine atherosclerotic lesions (175), consistent with SAA being transported to the artery wall by plasma lipoproteins.
Several studies in mice provide evidence for Saa being an atherosclerosis mediator. LDL receptor (Ldlr)-deficient mice fed a pro-inflammatory diet with or without added cholesterol showed marked increases in plasma Saa levels, which correlated with atherosclerosis extent (116). Mice in which Saa was either overexpressed or silenced suggest Saa roles in atherosclerosis pathogenesis, although the data are not uniform. Chow-fed Apoe-deficient mice in whom Saa was overexpressed using a lentiviral vector had increased en face and aortic root lesions compared to control-fed mice, although differences were not observed with an atherogenic diet (176). Plasma levels of IL-6 and TNFα and expression of vascular cell adhesion molecule 1 (VCAM-1) and monocyte chemotactic protein-1 (MCP-1), and lesion macrophage content all increased with Saa overexpression (176). In another experimental approach, a single injection of a human Saa-containing adenovirus in Apoe-deficient mice increased plasma Saa levels for ∼10 days, leading to increased atherosclerosis (177). When repeated injections of the human SAA-containing adenovirus were administered to immune-deficient mice to prevent an antibody response to the human protein, brachiocephalic lesions and aortic lesion area were markedly increased (177). The authors postulated that the increase in atherosclerosis was due to SAA-mediated induction of transforming growth factor-β (TGFβ), which increased vascular biglycan expression and led to increased LDL retention (see later). Deficiency of Saa in Ldlr-deficient mice led to reduced atherosclerosis in the ascending aortic arch but not in the aortic root or innominate artery at 6 weeks, although this difference was lost by 12 weeks (178). Parallel findings were observed in male Ldlr-deficient mice also deficient in FPLR2, one of the major Saa receptors, although the effect was more prolonged than in the Saa/Ldlr double knockout mice (179). In both studies, transplantation of Saa-deficient bone marrow-derived cells replicated the findings, suggesting that the reduced atherosclerosis may have resulted from the absence of free Saa in lesions rather than in the circulation. However, in Apoe/Saa double knockout mice, no difference in lesion area was observed at ∼50 weeks (180), although no early time points were examined. A subsequent study in Apoe-deficient male mice also lacking Saa1 and Saa2 using Saa3 suppression with an anti-sense oligonucleotide showed significantly reduced atherosclerosis (181). These results imply that all acute phase Saa isoforms have pro-atherogenic properties, and that deficiency/suppression of all 3 acute phase isoforms is required for atheroprotection in mice. Saa3 effects on atherosclerosis were not reported in female mice, despite sexually dimorphic Saa3 expression (182) (see below). Saa transgenic rabbits failed to show an increase in atherosclerotic lesions (183). Therefore, in summary, while most mouse studies suggest that Saa contributes to the development of early atherosclerotic lesions, results in Saa-deficient models are not consistent, possibly related to the nature of the model and the timing of observations. Nevertheless, such studies raise the question of how Saa might affect the atherogenic process. Several potential mechanisms are plausible.
Since SAA can be expressed by several cells of the artery wall (18), including perivascular adipocytes (184) and macrophages (18, 112, 113, 182–187), the locally produced SAA in lesions unattached to lipoproteins could have signaling functions that might be atherogenic. These include activation of the NFkB and MAPK signaling pathways via interaction with receptors such as class B scavenger receptor CD36, TLR4, TLR2, FPLR2 and RAGE (176, 188, 189). Activation of monocytes/macrophages and perivascular adipocytes can generate chemoattractant molecules such as MCP-1, which could lead to the recruitment of additional inflammatory cells and vascular smooth muscle cells. SAA also can be a direct chemoattractant (190). Moreover, direct activation of the chemoattractant receptor, FPLR2, by free SAA could further attract inflammatory cells into developing vascular lesions. Free SAA also has been shown to induce a phenotypic switch in vascular smooth muscle cells towards a more proliferative type of cell that synthesizes more matrix molecules (188). However, in vitro studies using free SAA should be interpreted cautiously, since minor contamination with endotoxin could lead to similar effects.
HDL-bound SAA also may play a role in atherogenesis. When SAA is secreted by the liver as part of the acute or chronic inflammatory response, it circulates in plasma bound to HDL, although it can associate with less dense lipoproteins under certain circumstances (24, 25, 37, 143). HDL particles that carry SAA, so-called “inflammatory HDL”, is less atheroprotective than normal HDL, with reduced inhibition of inflammation in cells due to its being trapped by cell surface proteoglycans (191), versican in the case of adipocytes and biglycan produced by macrophages (118). Trapping of SAA-containing HDL at the cell surface prevents it from adequately promoting reverse cholesterol transport (192). HDL derived from inflamed mice devoid of Saa1 and Saa2 functioned normally, as it did when the proteoglycans were removed from the cell surface either chemically or by genetic manipulation (118). Humans treated with low levels of endotoxin also had impaired cholesterol efflux capacity from macrophages, despite no change in circulating HDL-cholesterol levels. Proteomic analyses showed that the cholesterol efflux capacity of HDL correlated inversely with Saa1 and Saa2 content (193). Binding of SAA-containing HDL by extracellular proteoglycans such as biglycan in humans (194) and perlecan in mice (174) may lead to HDL retention in the vascular intima, increasing susceptibility to oxidative and enzymatic damage similarly to trapped LDL (195). Retained HDL could thus be pro-atherogenic, compared to its more widely accepted anti-atherogenic properties. The products of oxidative and enzymatic damage to retained lipoproteins may play important roles in atherogenesis (196).
Finally, SAA might stimulate thrombosis, which often precipitates clinical events. SAA can induce tissue factor production by monocyte/macrophages (197) and platelet activation (198). Thus, SAA could play multiple roles in the atherosclerotic process from monocyte adhesion, inflammatory and smooth muscle cell chemotaxis, cellular inflammation, HDL function, retention of atherogenic lipoproteins in the artery wall, and thrombogenesis. The net effect is that SAA is likely to play a causative role in atherogenesis, although the extensive data are not fully consistent.
2.6. Type 1 diabetes
In contrast to T2D, type 1 diabetes (T1D) develops as a result of autoimmune destruction of pancreatic beta cells, reducing insulin production capacity; subjects with T1D thus require exogenous insulin to maintain euglycemia. Little is known regarding SAA and T1D. One study has shown that SAA levels were elevated in 1,139 subjects with T1D compared with 848 healthy controls (199); however, these plasma donors were not age-matched, and the T1D subjects tended to be older. However, SAA increased specifically in HDL in subjects with T1D compared to age-, sex-, and BMI-matched controls, an effect much stronger when subjects were stratified by HbA1c and was not observed for CRP (200). A common T1D model can be generated in mice by injecting them with the pancreatic beta cell toxin STZ, leading to beta cell apoptosis (201, 202). In such STZ-treated mice, circulating SAA levels increased (203), with increased Saa3 expression specifically from adipocytes (114). Whether hyperglycemia or STZ itself stimulated adipose Saa3 was not determined. However, treating cultured 3T3-L1 adipocytes with 12–25 mM glucose induces Saa3 mRNA expression (114, 190, 204), an effect replicated by hyperglycemic clamps in mice (114), suggesting that hyperglycemia is the critical factor. Whether glucose-stimulated SAA expression changes systemic SAA levels or performs local functions is not known. As with studies related to T2M, mechanistic studies are needed in mice to determine whether SAA is sufficient or required for the pathology of T1D.
2.7. Autoimmune diseases: systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA)
SLE and RA are chronic diseases in which a person's immune system attacks its own tissues, resulting in inflammation and tissue damage in affected organs. While RA can be physically debilitating but typically is not life-threatening, SLE can lead to severe complications such as kidney failure, seizures, and increased risk of thrombosis. SAA may be a biomarker for both conditions (3, 6, 205–207). SAA promotes T-helper 17 (Th17) differentiation (208, 209), which plays important immunologic roles. However, excessive Th17 responses also can promote autoimmune conditions including SLE and RA (210). In patients with RA, their joints contain elevated SAA (6, 205), with levels correlating with plasma SAA levels and disease progression (211, 212). Rather than simply diffusing into joints from the bloodstream, SAA itself may be expressed in synoviocytes, macrophages, and endothelial cells within synovial tissues in RA patients (6, 213). Computational modeling identified Saa3 as the gene most strongly correlated with the severity of collagen-induced arthritis (214). Moreover, synovial fibroblasts isolated and cultured from patients with RA produced 2–4 times more SAA than those from healthy subjects (213). Whether SAA directly contributes to these autoimmune diseases remains to be elucidated. A potential mechanism is that in RA patients, SAA and associated cytokines potently induce matrix degrading enzymes in synovial fibroblasts (213, 215, 216), which if left unchecked could contribute to disease pathogenesis and joint destruction. Treatment with the TNF antagonist etanercept reduces RA disease severity while simultaneously reducing circulating SAA levels (217), providing one linkage between SAA and RA, but the causal direction is unknown.
2.8. Inflammatory bowel disease (IBD)
The two major types of IBD include ulcerative colitis (UC) and Crohn's disease (CD). Both are complex conditions that result from chronic dysregulated immune function in the gastrointestinal tract (218). UC is limited to the colon; however, CD can involve any part of the gastrointestinal tract, but usually affects the distal small intestine and/or the colon (219). Previous work suggests that SAA may be a more sensitive biomarker for IBD than CRP (8, 220, 221), as SAA levels remain elevated while CRP disappears in patients who are in clinical remission (222).
Patients presenting with either UC or CD consistently show elevated serum SAA levels (220, 223). In humans, intestinal biopsies from CD patients showed significantly increased colonic SAA1/2 expression levels (224). From an extensive panel of inflammatory markers, including CRP, IL-22, and IL-6, SAA had among the highest positive associations with a Simplified Endoscopy Score for CD (SES-CD, r = 0.4), fecal calprotectin (r = 0.39), Crohn's Disease Activity Index (CDAI, r = 0.14), and stool frequency (r = 0.18) (223). Such studies link intestinal SAA to potential roles in disease development or protection. Subjects with CD without mucosal healing had higher SAA levels than subjects in clinical remission (225), suggesting SAA as a marker for CD severity. In patients with UC who were in remission, consumption of a low-fat, high-fiber diet improved quality of life, in conjunction with reduced circulating SAA levels (226). In one clinical trial, SAA was a highly significant predictor of CD severity, and treatment with filgotinib, a selective JAK1/STAT inhibitor, improved CD symptoms while simultaneously reducing circulating SAA levels (223).
Mouse models of IBD similarly display elevated circulating SAA levels. Systemic SAA as well as local Saa3 expression levels become elevated within days of administration of dextran sodium sulfate (DSS) in drinking water in a mouse model of colitis (227–229), an effect that may function to protect colonic epithelium from acute injury by recruiting IL-22-producing neutrophils (228). This does not appear to be specific to that model, as mice given trinitrobenzone sulfonic acid (TNBS) via colonic catheter, in another well-studied colitis model, also responded with increased systemic SAA (230, 231). Pharmacological treatments including 6-thioguanine and cyclosporine A, utilized to improve colitis outcomes in mice, effectively reduced circulating SAA levels (229), as did administration of Bacillus subtilis spores as a probiotic (230). To date, only a few studies have indirectly examined IBD phenotypes with concurrent SAA genetic perturbation. One study showed that mice concurrently deficient in Saa1, Saa2, and Saa3 had attenuated colitis as assessed by histology (209). However, in the proximal colon of the mouse, Saa1 and Saa2 expression is confined to the epithelium, while Saa3 expression is found in immune cells including monocytes, macrophages, and dendritic cells (209). These data suggest that Saa1/2 exert system-wide functions of mucosal sensing and defense, while Saa3 drives local function, due in part to the differential potentiation of Th17 responses by these subtypes (209). Similarly, mice that were deficient in Saa1 and Saa2 that had colitis-associated colon cancer showed attenuated weight loss, gut histological damage, and gut inflammation (232), findings that suggest that Saa1/2 may augment colitis severity. Saa1/2 deficiency resulted in reduced Saa3 colonic expression (232). Conversely, specific deletion of only Saa3 rendered mice more susceptible to dextran sulfate sodium (DSS)-induced colitis (228), implying that Saa3 may be protective against IBD. Collectively, the precise roles for different SAA subtypes in IBD remain unknown, but emerging evidence suggests that Saa1/2 and Saa3 have different triggers and functions.
3. Tissue- and stimulus-specific SAA effects
Expression kinetics for each SAA subtype varies greatly by tissue source and stimulus type. While SAA1 and SAA2 are primary players in the acute phase response, in mice Saa3 may play a more prominent role in local inflammation. Mice express Saa3 in many extra-hepatic tissues including adipose tissue, lung, macrophages, and small/large intestine, with the liver predominantly producing Saa1 and Saa2 (40). This distinct division in murine subtype expression patterns enables the study of extra-hepatic Saa in metabolic disease. Extra-hepatic Saa expression appears predominant in chronic inflammatory conditions, while Saa derives largely from the liver in more acute inflammation (9, 18, 20, 91, 96, 98, 100). In humans, it is much more difficult to separate the contribution of extra-hepatic SAA to metabolic disease phenotypes in humans, because SAA1 and SAA2 are expressed both from liver and extra-hepatic tissues. Thus, much of our knowledge of extra-hepatic SAA originates from mouse models. In this section, the various SAA subtypes and their expression patterns in response to particular stimuli from various tissue and cell types will be discussed (Table 2).
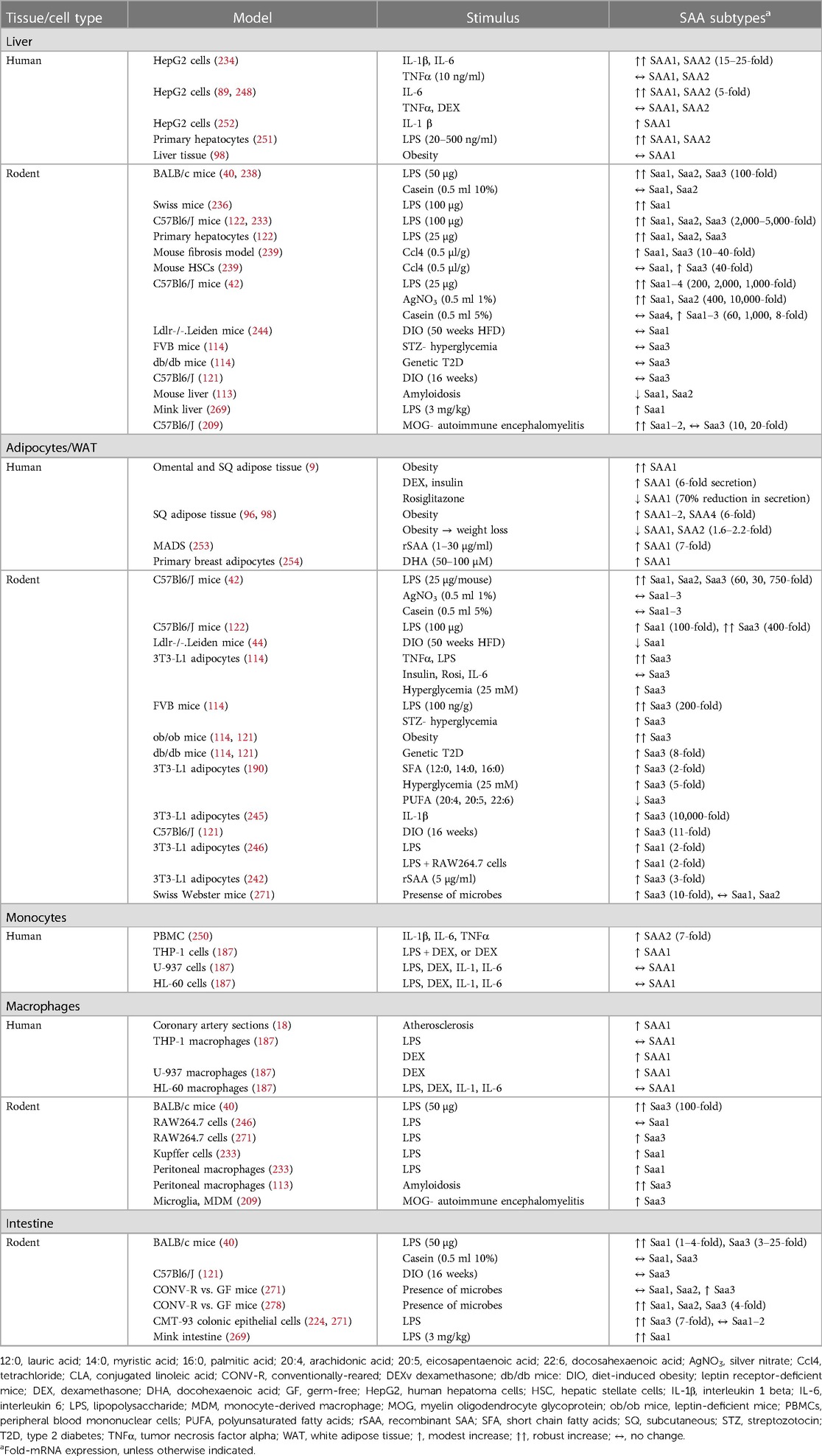
Table 2. Tissue- and stimulus-specific SAA effects in humans and rodents.
3.1. Liver
The liver is perhaps the most frequently studied SAA-expressing tissue, wherein hepatic resident macrophages (i.e., Kupffer cells) produce Saa3 (in mice) and hepatocytes make SAA1/2 (233, 239). As such, an influx of immune cells could specifically increase Saa3 expression in the liver in mice. In cultured hepatocytes, particular combinations of cytokines predictably increase SAA1 and SAA2 gene and protein expression (234). Hepatocytes secrete high levels of SAA during an acute inflammatory insult in mice and humans (22). HepG2 cells, a human hepatocyte cell line, can express SAA1 and SAA2 in response to IL-1β and IL-6 in a dose-dependent manner, an effect augmented by pre-treatment with dexamethasone or TNFα (89, 248, 250). Primary human Kupffer cells co-cultured with hepatocytes secrete high levels of SAA2 following treatment with IL-1β and IL-6 (237), suggesting a potential paracrine signaling mechanism.
As discussed above, potent inflammatory stimuli initiate robust, rapid, but short-lived (∼24 h) SAA1 and SAA2 expression from the liver. LPS at dosages ranging from 0.25 to 100 µg/mouse increases murine hepatic mRNA expression of Saa1 (up to 2,000-fold), Saa2 (up to 200-fold), and to a lesser extent Saa3 (up to 40-fold) in an NFκB-dependent manner, with circulating SAA levels subsequently increasing to 3,000 µg/ml (42, 236, 238). All three SAA subtypes reach peak hepatic mRNA expression 12 h after LPS administration (238). Only high-dose LPS (25 µg) increases circulating Saa3 in mice (42, 122). Similarly, LPS activates SAA1 and SAA2 mRNA expression and secretion in human primary hepatocytes (251). Patients with sepsis have elevated SAA levels (235), which are stronger predictive markers of sepsis severity (76). SAA was a more sensitive and earlier predictor of neonatal sepsis than the more traditional CRP (75).
Other models of sterile inflammation in mice also have been shown to increase hepatic Saa levels. Silver nitrate (AgNO3), administered by subcutaneous injection of 0.5 ml of a 1% solution, increases hepatic Saa1 (40-fold), Saa2 (1,000-fold), and Saa3 (200-fold), and leads to circulating Saa levels equivalent to that observed with high doses of LPS (42). However, in contrast to findings after LPS, we did not find evidence of Saa3 in plasma following AgNO3 injection (42). Injection with casein (administered by subcutaneous injection of 0.5 ml of a 5% solution) modestly increased hepatic Saa1 (6-fold), Saa2 (100-fold), and Saa3 (5-fold) mRNA expression, resulting in much smaller increases in plasma Saa levels (42). Collectively, acute inflammatory stimuli differ in the resulting hepatic expression levels of SAA1–3, leading to varied systemic SAA concentrations, suggesting differential regulation.
In metabolic disease states such as obesity and T2D, hepatic SAA expression likely results from cytokine signaling from extra-hepatic tissues such as WAT (240, 252). A recent study identified SAA1 protein from both WAT and liver as a candidate biomarker associated with low-grade inflammation. There was a much stronger correlation of SAA1 with inflammation in the liver than with WAT inflammation (244), suggesting a more dominant hepatic role of SAA1. However, this particular study mined gene ontology datasets using general inflammatory search terms, so the particular metabolic conditions (i.e., obesity) of the original study subjects were not indicated.
3.2. Adipocytes
The acute inflammatory studies cited above showed effects on SAA subtypes expressed in the liver, but there also were strong SAA responses in adipose tissue. While reported hepatic SAA responses to LPS in mice are largely due to Saa1 and Saa2, adipose tissue responds to LPS with massive (∼500-fold) increases in Saa3 mRNA compared with 40-fold Saa3 increases in the liver (42). This effect appears to be LPS-specific, as neither AgNO3 or casein altered Saa1, Saa2, or Saa3 mRNA levels in adipose tissue (42). Thus, we speculate that LPS can induce expression of all three Saa subtypes in both liver and adipose tissue that all contribute to circulating levels, while AgNO3 and casein primarily target hepatic Saa. In this section, we present evidence for differential SAA subtype expression in response to several inflammatory mediators and metabolic factors.
Many stimuli have been shown to increase Saa3 mRNA and protein expression in cultured adipocytes. These include high levels of glucose (114, 190, 204), saturated fatty acids (190, 204), conjugated linoleic acids (204), pro-inflammatory cytokines including TNFα and IL-1β (114, 190, 245), and LPS (114). Conversely, anti-inflammatory stimuli such as polyunsaturated fatty acids (190) and rosiglitazone (114) reduce adipocyte Saa3 expression. In addition to chemical activation, 3T3-L1 adipocytes also express Saa3 in response to macrophage-derived components (121, 246), suggesting an important role in cell-cell communication, with similar effects observed in cultured human adipocytes and in mice. Human SGBS cells treated with saturated fatty acids display increased Saa1 expression, while polyunsaturated fatty acids decreased glucose-induced Saa1 (190), suggesting that the major adipose SAA subtype in humans is SAA1. Mice injected with LPS robustly increased Saa3 expression in visceral WAT comparable to hepatic Saa1/2 expression levels in the same mice (42); using mass spectrometry methods, Saa3 was identified in their plasma (42), suggesting that Saa3 can circulate under particular inflammatory conditions.
Recombinant (i.e., exogenous) SAA can directly impact adipocyte metabolism. In cultured 3T3-L1 adipocytes, recombinant SAA (rSAA, 5 µg/ml) reduced adipogenesis, accompanied by reduced adipogenic transcription factors and proteins including peroxisome proliferator-activated receptor gamma (PPARγ), CCAAT enhancer binding protein beta (C/EBPβ), and GLUT4 (242). rSAA also reduced lipid accumulation, increased lipolysis, prevented glucose uptake, triggered secretion of inflammatory cytokines IL6 and TNFα and increased mRNA expression of Saa3. In multipotent adipose-derived stem (MADS) cells isolated from human subcutaneous adipose tissue induced to differentiate into primary adipocytes in vitro, free- and HDL-associated rSAA increased MCP-1, IL-6, and IL-8 secretion in a dose-dependent manner (253). This pro-inflammatory phenotype was dependent on NFkB, not due to endotoxin contamination (243, 253). Moreover, rSAA treatment reduced mRNA expression of adiponectin, fatty acid synthase (FAS), C/EBPα, PPARγ, and GLUT4 (253, 254), suggesting impaired adipogenesis capacity. A propensity for rSAA to increase lipolysis also has been reported in human adipose tissue (9). The pro-inflammatory, pro-lipolytic, and anti-adipogenic effects of SAA also have been shown in primary porcine adipocytes (243).
Recent technical advances have enabled the study of adipose tissue down to the single-cell level (255, 256). Spatial transcriptomics on human subcutaneous abdominal adipose tissue sections has revealed 3 distinct subsets of adipocytes, including those rich in genes for leptin (AdipoLEP), the lipid droplet-associated proteins perilipin1 and −4 (AdipoPLIN), and SAA1/2 (AdipoSAA) (257). AdipoLEP was enriched in genes encoding matrix metabolism, AdipoPLIN in genes associated with lipid and glucose metabolism, and AdipoSAA in multiple retinol-binding adipokines (i.e., RBP4) (257). These have been linked with obesity co-morbidities including T2D, hepatic steatosis, inflammation, and metabolic syndrome (258, 259). Approximately 8% of the adipocytes examined were AdipoSAA, with similar proportions in donors with or without obesity, but there was high variability among donors (from 2%–18% of all adipocyte populations) (257). Whether or not the proportion of AdipoSAA cells differs in omental WAT, or is related to sex, is of interest.
In addition to secreting adipokines and nutrients into the circulation, adipocytes also secrete extracellular vesicles (EVs), including microvesicles, exosomes, and apoptotic bodies (260). EVs are heterogeneous membrane vesicles secreted by many cell types, including adipocytes, and function to facilitate intercellular communication within and between tissues via protein signaling, immune responses, and nutrient transport (261). EVs contain diverse cargo including proteins, lipids, and miRNAs. Adipocyte-derived exosomes can be identified by their adipocyte-specific protein cargo, chiefly adiponectin and perilipin (262). EVs differing in cellular origins possess unique biological properties, enabling cell- or tissue-specific effects. EV production derived from WAT is increased during obesity (263–266), and is correlated with insulin resistance in both humans and in animal models. SAA1 and SAA2 have been identified in EVs isolated from human adipose tissue (262), and Saa3 observed within vesicle-like structures within murine adipose tissue (121). These findings raise the possibility that adipose tissue-derived SAA communicates systemically with other target tissues, in addition to its local effects.
3.3. Macrophages
Macrophages are present in all peripheral tissues and contribute to systemic metabolism. Macrophage classification schema are emerging, but largely revolve around their functional potential, including the capacity to elicit an inflammatory response and ability to phagocytose pathogens and cellular debris (267). As such, macrophages can either contribute to or resolve inflammation. Moreover, macrophages that only reside within particular tissues often receive their own classification, such as hepatic Kupffer cells or central microglia. All tissues from which Saa3 expression can be detected have a dynamic macrophage population, suggesting a potential common source of Saa3.
In obesity, adipose tissue exhibits both increased SAA expression (SAA1 and SAA2 in humans and Saa3 in mice), as well as increased macrophage infiltration. Importantly, all SAA subtypes are expressed from macrophages (187). Initial studies showed that acute inflammatory stimuli, including LPS and casein, induced only Saa3 mRNA in murine macrophages (40, 113). Saa3 mRNA also increases in activated RAW264.7 macrophages (121), murine bone marrow-derived macrophages (121), murine J774.1 macrophages (18, 112), and murine foam cells within atherosclerotic lesions (18), but not in the human THP-1 cell line (187). Saa3 protein co-localizes with F4/80+ macrophages in obese adipose tissue (121).
That macrophages express SAA subtypes as well as SAA receptors, including TLR2, TLR4, RAGE, and SRB1, suggests autocrine activities that likely contribute to local effects (20, 268). Deletion of putative SAA receptors yields a blunted macrophage response to SAA. BMDMs from mice deficient in TLR2 exhibit a blunted inflammatory response to SAA (1 µM) (56), and neutralizing antibodies to TLR2 blunted SAA-mediated activation of THP-1 macrophages (20). Similar effects have been observed in peritoneal macrophages from TLR4-deficient mice (59). SAAs may bind to macrophage-produced extracellular matrix (ECM) components, including proteoglycans and glycoproteins (195). Collectively, an increasing body of work connects SAA and macrophages.
Monocytes freshly isolated from humans or monocytic cell lines consistently respond to SAA with potent pro-inflammatory responses. Within an hour of treating with rSAA, peripheral human blood mononuclear cells (PBMCs), THP-1 monocytic cells, and monocyte-derived macrophages (MDMs) all exhibit rapid expression of IL-1β, MCP1, IL-6, IL-8, TNFα, and macrophage inflammatory protein 1 alpha (MIP-1α), an effect that is sustained for 8–24 h and is similar to LPS (241). Similar effects were observed in RAW264 monocytes treated with rSAA, which yielded a pro-inflammatory phenotype characterized by increased MCP-1, IL-6, IL-8, and TNFα secretion (9). While a potent inflammatory stimulus (i.e., LPS or casein) initiates a robust, rapid, but short-lived (∼24 h) hepatic Saa response, and from macrophages directly treated in culture, a similarly rapid but more prolonged Saa3 response (72 h) has been observed in isolated peripheral macrophages, indicating markedly different hepatic expression kinetics (40, 113). Whether such different expression kinetics reflect a more prolonged response that is cell-type specific or is an effect secondary to the acute phase response remains to be determined.
3.4. Intestine
Intestinal SAA can be induced by several mechanisms, which are complicated by the potential for differing SAA subtype expression from varying intestinal cells. SAA1/2 are highly expressed in intestinal epithelium and in the endothelium lining the intestinal submucosal blood vessels in rabbits, rodents, and humans (224, 269, 270). Conversely, Saa3 has been detected at low levels in mouse colonic epithelium (224), but is more prevalent in intestinal immune cells (209). Moreover, in mice, Saa3 expression is more strongly induced by LPS and microbes in colonic epithelium than Saa1/2 (224, 247, 271). Induction of SAA1 and SAA2 in small intestinal epithelial cells by commensal microbes requires both IL-23 and IL-22 in a STAT3-dependent manner (272). Male Syrian hamsters injected with LPS (100 µg/g body weight) also expressed high Saa levels (unknown subtypes) in the duodenum, jejunum, and ileum (273). Mouse intestinal Saa3 is most closely related to human SAA1 with 70% amino acid homology (271), and may serve local gut functions (247).
SAA expression differs markedly throughout the intestinal tract, with SAA2 having the most variable expression between the ileum and rectum in subjects with IBD (274). Germ-free mice have very low ileal levels of Saa1 and Saa2, but higher expression in the colon than in conventional mice (275). These findings are consistent with an anti-bacterial SAA role in relation to an omnipresent colonic microbiota, and a much more variable ileal microbiota. As conventional mice mature, intestinal Saa rises in the ileum, reflecting the increasing bacterial load, but do not change in the colon. Perturbing early-in-life gut microbiome affected intestinal Saa expression. With pulsed therapeutic-level antibiotic (PAT) exposures at early ages after weaning, non-obese diabetic (NOD) mice have consistently decreased Saa1/2 and Saa3 expression in the ileum but not in the colon (249, 276, 277). Younger mice (P12) had significantly increased Saa1/2 and Saa3 expression in both ileum and colon two days after antibiotic exposure ended, indicating that intestinal Saa can biphasically respond to gut microbiome changes in patterns that are both age- and microbiome context-dependent during this critical period for host immune development. The early-life antibiotic-exposed mice showed significantly increased Saa1/2 and Saa3 expression in the ileum but not in the colon at P17 days (277). These studies further confirmed that early-life intestinal SAA expression is subject to regulation linked to gut microbiota composition, potentially reflecting an ancient evolutionary strategy to regulate the establishment of immune responses or tolerance in the developing animal.
Mono-colonization of germ-free mice with segmented filamentous bacteria (SFB) rapidly induces expression of Saa1, Saa2, and Saa3 in the terminal ileum, consistent with the unique spatial expression patterns of SAA in the gut. Induction of ileal Saa is further increased by conventionalization using fecal microbial transplant (FMT) from specific pathogen-free (SPF) mice (278). Induction of ileal Saa1 and Saa2 by SFB is mediated through the IL-23/IL-22 circuit in ileal epithelial cells (272). The SFB-induced ileal Saa proteins promote Th17 cell differentiation from ileal lamina propria dendritic cells and contribute to protective immune responses in the ileal mucosa (278).
Conversely, as anti-bacterial molecules, SAA may modulate gut bacterial growth and composition either directly or through downstream intestinal immune responses. Consistent with observations in mice, in vitro studies showed that overexpression of Saa1/2 in intestinal epithelial cell lines reduces growth of co-cultured bacterial cells (224). Similarly, in zebrafish SAA in intestinal epithelial cells derived via transgene expression constrains the bactericidal activity of neutrophils, and promotes neutrophil recruitment to the intestine that is functionally distinct from hepatic SAA expression (279). In a mouse model of DSS-induced colitis, Saa induction in the large intestine was required to dampen local inflammation, while SAA1/2 overexpression in cultured epithelial cells reduced the viability of co-cultured E.coli (224), suggesting a potential bactericidal function of SAA that may contribute to barrier integrity. Transgenic mice engineered to overexpress Saa1 are partially protected against inflammatory responses to cecal ligation and puncture (280), suggesting an inverse relationship between gut-derived SAA and inflammation.
The anti-inflammatory properties of intestinal Saa1 are most specific to LPS-induced inflammation, an effect that could be dosage-dependent. Saa1 has the ability to bind LPS and form a complex, which then facilitates the clearance of LPS by macrophages (280). The transition of Saa1 from exerting pro-inflammatory effects to anti-inflammatory effects may reflect the proteolysis of the Saa1 protein. The N-terminal and C-terminal domains of Saa1 are crucial for its pro-inflammatory activity, and their removal via proteolysis can transform Saa1 into an anti-inflammatory agent (280, 281). Whether other SAA proteins also are capable of switching from pro-inflammatory to anti-inflammatory functions is unknown. The precise functions of intestinal SAAs deserve further investigation.
4. Sexual dimorphism of SAA
Circulating SAA is positively associated with BMI and adiposity, with a propensity to also associate with fasting glucose, insulin, HbA1C, and HOMA-IR. An emerging literature describes unique sexual dimorphic relationships between SAA and several metabolic disease states. Fully characterizing sex differences in SAA expression kinetics and functional potential is thus of great importance.
Large-scale RNA-sequencing studies of healthy humans showed that adipose tissue contains ∼3,000 sexually differentiated genes, one of the highest levels of all tissues examined (282). There was higher expression in women of all known SAA subtypes (SAA1, SAA2, and SAA3(p) (the SAA3 pseudogene)), which were among the most highly sex-differential genes (283). In contrast, with the exception of breast and skin, no other SAA-expressing tissues (i.e., liver, lung, blood) show SAA subtypes in their lists of sex-biased genes (283). These findings have been replicated in several large-scale sequencing studies spanning dozens of tissues in healthy men and women (284, 285), and in mice (286). SELS, a major SAA receptor, is elevated in the adipose tissue of subjects with T2DM and correlated with measures of glycemic control (73), but sex was not investigated in these studies. Collectively, many studies indicate that adipose tissue from female mice and humans expresses higher SAA than tissue from males, but the involvement of sex differences in the pathophysiology of obesity or associated metabolic disorders is not known.
Healthy women (with BMI < 25) have higher circulating SAA than age-matched men, despite the men having a slightly higher average BMI (287). SAA positively correlates with BMI, waist circumference, waist-to-hip ratio, insulin, and HOMA-IR in both sexes. After adjusting for BMI, only the correlations with insulin and HOMA-IR remained significant for men, but not women.
One of the first studies to address potential sex differences in SAA kinetics characterized the association between adipocyte size and circulating SAA levels in men and women over a large range in BMIs, with the additional aim to examine potential associations with measures of glycemic control (107). Women generally had higher circulating SAA levels than men, and stronger correlations with BMI, adiposity, subcutaneous adipocyte diameter, fasting insulin, HOMA-IR, and leptin (107). This could relate to the higher proportion of subcutaneous WAT in women than men.
By contrast, the liver, the source of most acute-phase SAA, has rarely been implicated in sex differential SAA expression. In contrast to several studies that have not found SAA to be differentially expressed by sex in the liver (282, 283), one study has shown that males tend to express slightly higher levels of SAA subtypes than female mice. Male CD-1 mice have modestly higher hepatic mRNA expression levels of Saa1, Saa2, and Saa3 than females (288). Adipose tissue-derived SAA may be impacted by sex steroids, as WAT is highly enriched in these molecules (289), with levels widely varying in metabolic disease. In experiments using cultured murine peritoneal macrophages and BMDMs, testosterone and 17β-estradiol directly impacted Saa3 gene expression (182). Saa3-deleted macrophages show sexually dimorphic responses to sex steroids. After estradiol exposure, Saa3-deficient BMDMs harvested from male mice showed a massive increase in inflammatory gene expression compared to wild-type macrophages, with concurrent elevation of the estrogen receptor (182). Thus, a relationship between macrophages, sex steroid signaling, SAA, and metabolic disease is present but needs further definition.
Our prior studies have supported a potential sexual dimorphic role of Saa3 in a mouse model with global Saa3 deficiency (82). When given a high fat high sucrose (HFHS) diet, female mice, but not male mice, were protected from body weight gain and associated insulin resistance. To determine whether there was similar sexually dimorphic protection against atherosclerosis in female mice, we crossed our global Saa3-KO mice with mice deficient in LDLR, which promotes hypercholesterolemia and is a common model for studying atherosclerosis. In that study, male Saa3−/− Ldlr−/− mice were protected from atherosclerosis, while female Saa3−/− Ldlr−/− mice were not (182). We speculate that in these models, Saa3 modulates effects via pathways that could be tissue-specific. In the obese state, Saa3 is expressed primarily from hypertrophic adipocytes, and also expressed from adipose tissue macrophages (96). Conversely, in the setting of hypercholesterolemic atherosclerosis, Saa3 expression likely originates from aortic and/or hepatic macrophages in addition to adipose tissue. Thus, in these different models, Saa3 deficiency leads to divergent phenotypes in males and females.
Other studies also suggest a potential interaction between sex hormones and SAA. Women with RA had higher SAA levels than men with RA (211, 212, 290). In a linear regression model involving the ratio of estradiol to testosterone (E2:T), sex and the E2:T ratio were highly significant and independent predictors of circulating SAA (290). Women with BMI < 25 have also been reported to have higher SAA levels than men (287), and SAA correlates more strongly with BMI and adiposity in women than in men (11). These observations suggest that sex hormones play roles in regulating SAA expression. Circulating SAA is higher in women taking oral estrogen-containing contraceptives (291, 292) and in women undergoing estrogen replacement therapy (287, 293). The apparent estradiol-mediated increase in SAA observed in these studies was secondary to elevations in CRP. More work is required to determine the mechanisms linking sex hormones and SAA.
Phenotypic responses to pro-inflammatory stimuli have differed in macrophages harvested from male or female mice (182). Compared to male mice, bone marrow-derived macrophages (BMDMs) isolated from female mice and treated with pro-inflammatory fatty acids or LPS showed lower levels of inflammatory cytokine expression (294). This effect appears to be cell-autonomous, since sex hormones were not present. Transplanting male bone marrow into donor female mice led to a phenotypically male pattern of obesity-associated adipose tissue inflammation (294). However, the absence of Saa3 in BMDMs negated this inherent sex-specific effect (182). The specific interactions between Saa3 and sex hormones remains to be characterized, but could explain the sexually dimorphic observations related to SAA expression in metabolic disease.
5. SAA-targeting therapies
Targeting SAA may be a potential therapeutic avenue for dampening inflammation. One approach is to target pathways that will reduce SAA expression. Tocilizumab, a monoclonal antibody that targets IL-6 and reduces SAA levels (295), has been effective in treating a small number of patients with amyloidosis involving the gastrointestinal tract (296) and kidneys associated with Familial Mediterranean Fever (297, 298), and amyloidosis associated with rheumatoid arthritis (299), but this approach could potentially also be developed for use in other chronic inflammatory conditions. Anakinra and canakinumab, monoclonal antibodies that target IL-1β, have been used to reduce SAA levels in inflammatory conditions such as Familial Mediterranean Fever (300) and gouty arthritis (301). Moreover, the CANTOS trial, for the first time, showed that inhibition of inflammation using an antibody against Il-1β decreased cardiovascular events (172), providing further evidence for the importance of inflammation in atherosclerosis. Since SAA appears to play a role in the pathogenesis of atherosclerosis (see previous sections), it is possible an approach that inhibits Il-1β could be more widely adapted for preventing atherosclerosis, as well as rheumatic diseases and even in hyperinflammatory states associated with COVID-19 (302).
SAA contains binding sites that are specific for heparin and heparin sulfate, which have been postulated to be useful for preventing amyloidogenic conformation of SAA (303). SAA also inhibits acyl coenzyme A cholesterol acyltransferase and enhances cholesterol esterase activities shifting stored intracellular cholesteryl esters to free cholesterol, which can be transported from cells. Liposomal preparations of small synthetic peptides of SAA can bind and neutralize SAA, facilitating reverse cholesterol transport and preventing and reversing aortic lesions in mouse models of atherosclerosis (304). Eprodisate, which binds to the glycosaminoglycan binding site on amyloid fibrils, thus preventing polymerization and tissue deposition, may slow the progression of AA amyloidosis-related renal disease (64, 305), and also may be applicable to other amyloid related conditions.
All these approaches are still in experimental phases, but demonstrate potential proof-of-concept mechanisms for future SAA-targeted therapies.
6. Concluding remarks and perspectives
Elevations of SAA subtypes have been consistently associated with metabolic diseases such as obesity, diabetes, CVD, and autoimmune conditions in humans and in animal models. After 40 years of investigation, evidence is not yet sufficient to determine whether SAA plays causal roles in metabolic disease development and progression, or is merely a biomarker of broader phenomena akin to CRP. In this review, we have presented evidence that associations with several metabolic disease states differ in expression kinetics and dominant SAA subtypes, as well as tissue, cellular, and spatial expression patterns, implicating the tissue microenvironment as crucial to SAA function. In particular, while evidence suggests that WAT SAA expression increases in obesity, whether such increases contribute to the circulating SAA pool is not known. Due to distinct subtype expression patterns in mice vs. humans, it could be possible for WAT-SAA to circulate in humans, but not in mice. As such, we propose that the SAA functions associated with metabolic disease are physiologically distinct from those in acute-phase reactions. Moreover, accumulating evidence suggests that different SAA subtypes, long considered to be pro-inflammatory molecules, may play beneficial roles in conditions like IBD, highlighting the importance of the microenvironment for particular SAA-mediated phenotypes. Finally, we speculate that SAA could play important roles in the differential progression of sexually dimorphic metabolic conditions.
Author contributions
LJD, KSM, XSZ, AC, and MJB reviewed the literature and contributed to the preparation of this manuscript. LJdH and KSM prepared the figures and tables. All authors contributed to the article and approved the submitted version.
Funding
The authors are thankful for research support from the United States Department of Agriculture (USDA) National Institute of Food and Agriculture (NIFA) award #2020-67001-30716. This work was also supported by the National Institutes of Health training program in Diabetes, Obesity, and Metabolism (T32 DK007247) and the training program in Nutrition, Obesity, and Atherosclerosis (T32 HL007028), both at the University of Washington; and U01 AI22285 from NIH, Transatlantic Program of the Fondation Leducq, and the Emch Foundation, at Rutgers.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ABCA1, ATP binding cassette subfamily A member 1; AGE, advanced glycation end products; AgNO3, silver nitrate; AKT, protein kinase B; ALT, alanine aminotransferase; apoA1, apolipoprotein-1; apoB, apolipoprotein B; BMDM, bone marrow-derived macrophages; BMI, body mass index; C/EBPβ, CCAAT enhancer binding protein beta; CD, Chron's disease; CD36, class B scavenger receptor; CDAI, Crohn's disease activity index; CHO, Chinese Ovary Cells; COVID-19, Coronavirus disease 19; CML, carboxy methyl lysine; CRP, C-reactive protein; CVD, cardiovascular disease; DSS, dextran sodium sulfate; ECM, extracellular matrix; ERK, extracellular signal-regulated kinase; EVs, extracellular vesicles; FMT, fecal microbial transplant' FPR1, formyl peptide receptor like-1; FPR2, formyl peptide receptor like-2; GD, gestational diabetes; GLUT4, glucose transporter 4; GM-CSF, granulocyte-macrophage colony stimulating factor; HbA1c, hemoglobin A1c; HDL, high density lipoprotein; HFD, high fat diet; HMGB1, high mobility group box 1; HNF, hepatocyte nuclear factor; HO-1, heme oxygenase type-1; HOMA-IR, homeostatic model assessment for insulin resistance; hSAA, human serum amyloid A; HUVEC, human umbilical vein endothelial cells; IBD, inflammatory bowel disease; IBS, irritable bowel syndrome; IκBα, nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor alpha; IL-1, interleukin-1; IL-1α, interleukin-1 alpha; IL-1β, interleukin-1 beta; IL-6, interleukin-6; IL-8, interleukin-8; IL-10, interleukin-10; IL-12, interleukin 12; IL-17, interleukin-17; IL-22, interleukin-22; IL-23, interleukin-23; iNOS, inducible nitric oxide synthase; IP-10, interferon γ-induced protein 10; JNK, c-Jun N-terminal kinase; LDL, low-density lipoprotein; LDLR, low-density protein receptor; LFD, low fat diet; LOX-1, oxidized low-density lipoprotein; LPS, lipopolysaccharide; Mɸ, macrophage; MAPK, mitogen-activated protein kinase; MCP-1, monocyte chemotactic protein-1; M-CSF, monocyte colony stimulating factor; MDMs, monocyte-derived macrophages; MIP1α, macrophage inflammatory protein-1 alpha; NAFLD, non-alcoholic fatty liver disease; NFκB, nuclear factor kappa B; NO, nitric oxide; PAT, pulsed therapeutic-level antibiotic; PBMCs, peripheral blood mononuclear cells; PCOS, polycystic ovary syndrome; PKA, protein kinase A; PKR, protein kinase R; PLIN, perilipin; PPARγ, peroxisome proliferator-activated receptor gamma; RA, rheumatoid arthritis; RAGE, receptor for advanced glycation end-products; RANTES, regulated on activation, normal T cell expressed and secreted; RBP4, retinol-binding protein 4; rSAA, recombinant serum amyloid A; SAA, serum amyloid A, SAA1, serum amyloid A1; SAA2, serum amyloid A2; SAA3, serum amyloid A3; SAA4, serum amyloid A4; SAF-1, serum amyloid A-activating factor 1; SELS, selenoprotein S; SES-CD, simplified endoscopy score for Crohn's disease; SLE, Systemic lupus erythematosus; SPF, specific pathogen-free; SRB1, scavenger receptor class B type 1; STZ, streptozotocin; T1D, type 1 diabetes; T2D, type 2 diabetes; TGFβ, transforming growth factor β; Th17, T-helper 17 cells; THP-1, human leukemia monocytic cell line; TLR-2, toll-like receptor 2; TLR-4, toll-like receptor 4; TNBS, trinitrobenzone sulfonic acid; TNFα, tumor necrosis factor alpha; UC, ulcerative colitis; VCAM-1, vascular cell adhesion molecule 1; VLCD, very low carbohydrate diet; VLDL, very low-density lipoprotein; WAT, white adipose tissue.
References
1. Miwata H, Yamada T, Okada M, Kudo T, Kimura H, Morishima T. Serum amyloid A protein in acute viral infections. Arch Dis Child. (1993) 68:210–4. doi: 10.1136/adc.68.2.210
2. Pizzini C, Mussap M, Plebani M, Fanos V. C-reactive protein and serum amyloid A protein in neonatal infections. Scand J Infect Dis. (2000) 32:229–35. doi: 10.1080/00365540050165848
3. Cunnane G, Grehan S, Geoghegan S, McCormack C, Shields D, Whitehead AS, et al. Serum amyloid A in the assessment of early inflammatory arthritis. J Rheumatol. (2000) 27:58–63.10648018
4. Lange U, Boss B, Teichmann J, Klör HU, Neeck G. Serum amyloid A--an indicator of inflammation in ankylosing spondylitis. Rheumatol Int. (2000) 19:119–22. doi: 10.1007/s002960050114
5. Gang N, Drenth JP, Langevitz P, Zemer D, Brezniak N, Pras M, et al. Activation of the cytokine network in familial Mediterranean fever. J Rheumatol. (1999) 26:890–7.10229412
6. O’Hara R, Murphy EP, Whitehead AS, FitzGerald O, Bresnihan B. Acute-phase serum amyloid A production by rheumatoid arthritis synovial tissue. Arthritis Res. (2000) 2:142–4. doi: 10.1186/ar78
7. Vallon R, Freuler F, Desta-Tsedu N, Robeva A, Dawson J, Wenner P, et al. Serum amyloid A (apoSAA) expression is up-regulated in rheumatoid arthritis and induces transcription of matrix metalloproteinases. J Immunol. (2001) 166:2801–7. doi: 10.4049/jimmunol.166.4.2801
8. Niederau C, Backmerhoff F, Schumacher B. Inflammatory mediators and acute phase proteins in patients with Crohn’s disease and ulcerative colitis. Hepatogastroenterology. (1997) 44:90–107.9058126
9. Yang RZ, Lee MJ, Hu H, Pollin TI, Ryan AS, Nicklas BJ, et al. Acute-phase serum amyloid A: an inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Med. (2006) 3:e287. doi: 10.1371/journal.pmed.0030287
10. Yang RZ, Blumenthal JB, Glynn NM, Lee MJ, Goldberg AP, Gong DW, et al. Decrease of circulating SAA is correlated with reduction of abdominal SAA secretion during weight loss. Obesity (Silver Spring). (2014) 22:1085–90. doi: 10.1002/oby.20657
11. Lappalainen T, Kolehmainen M, Schwab U, Pulkkinen L, Laaksonen DE, Rauramaa R, et al. Serum concentrations and expressions of serum amyloid A and leptin in adipose tissue are interrelated: the Genobin study. Eur J Endocrinol. (2008) 158:333–41. doi: 10.1530/EJE-07-0598
12. Santana AB, Gurgel MS, de Oliveira Montanari JF, Bonini FM, de Barros-Mazon S. Serum amyloid a is associated with obesity and estrogen receptor-negative tumors in postmenopausal women with breast cancer. Cancer Epidemiol Biomarkers Prev. (2013) 22:270–4. doi: 10.1158/1055-9965.EPI-12-1020
13. Zhao Y, He X, Shi X, Huang C, Liu J, Zhou S, et al. Association between serum amyloid A and obesity: a meta-analysis and systematic review. Inflamm Res. (2010) 59:323–34. doi: 10.1007/s00011-010-0163-y
14. Leinonen E, Hurt-Camejo E, Wiklund O, Hultén LM, Hiukka A, Taskinen MR. Insulin resistance and adiposity correlate with acute-phase reaction and soluble cell adhesion molecules in type 2 diabetes. Atherosclerosis. (2003) 166:387–94. doi: 10.1016/s0021-9150(02)00371-4
15. Marzi C, Huth C, Herder C, Baumert J, Thorand B, Rathmann W, et al. Acute-phase serum amyloid A protein and its implication in the development of type 2 diabetes in the KORA S4/F4 study. Diabetes Care. (2013) 36:1321–6. doi: 10.2337/dc12-1514
16. Fyfe AI, Rothenberg LS, DeBeer FC, Cantor RM, Rotter JI, Lusis AJ. Association between serum amyloid A proteins and coronary artery disease: evidence from two distinct arteriosclerotic processes. Circulation. (1997) 96:2914–9. doi: 10.1161/01.CIR.96.9.2914
17. Kosuge M, Ebina T, Ishikawa T, Hibi K, Tsukahara K, Okuda J, et al. Serum amyloid A is a better predictor of clinical outcomes than C-reactive protein in non-ST-segment elevation acute coronary syndromes. Circ J. (2007) 71:186–90. doi: 10.1253/circj.71.186
18. Meek RL, Urieli-Shoval S, Benditt EP. Expression of apolipoprotein serum amyloid A mRNA in human atherosclerotic lesions and cultured vascular cells: implications for serum amyloid A function. Proc Natl Acad Sci U S A. (1994) 91:3186–90. doi: 10.1073/pnas.91.8.3186
19. Johnson BD, Kip KE, Marroquin OC, Ridker PM, Kelsey SF, Shaw LJ, et al. Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women: the national heart, lung, and blood institute-sponsored women’s ischemia syndrome evaluation (WISE). Circulation. (2004) 109:726–32. doi: 10.1161/01.CIR.0000115516.54550.B1
21. Getz GS, Krishack PA, Reardon CA. Serum amyloid A and atherosclerosis. Curr Opin Lipidol. (2016) 27:531–5. doi: 10.1097/MOL.0000000000000331
22. Uhlar CM, Burgess CJ, Sharp PM, Whitehead AS. Evolution of the serum amyloid A (SAA) protein superfamily. Genomics. (1994) 19:228–35. doi: 10.1006/geno.1994.1052
23. Wang J, Yang Y, Zhang A, Zeng L, Xiao S, Ma H, et al. Serum amyloid protein (SAA) as a healthy marker for immune function in Tridacna crocea. Fish Shellfish Immunol. (2022) 122:495–500. doi: 10.1016/j.fsi.2022.02.038
24. Benditt EP, Eriksen N. Amyloid protein SAA is associated with high density lipoprotein from human serum. Proc Natl Acad Sci U S A. (1977) 74:4025–8. doi: 10.1073/pnas.74.9.4025
25. Benditt EP, Eriksen N, Hanson RH. Amyloid protein SAA is an apoprotein of mouse plasma high density lipoprotein. Proc Natl Acad Sci U S A. (1979) 76:4092–6. doi: 10.1073/pnas.76.8.4092
26. Badolato R, Johnston JA, Wang JM, McVicar D, Xu LL, Oppenheim JJ, et al. Serum amyloid A induces calcium mobilization and chemotaxis of human monocytes by activating a pertussis toxin-sensitive signaling pathway. J Immunol. (1995) 155:4004–10. doi: 10.4049/jimmunol.155.8.4004
27. Zheng H, Li H, Zhang J, Fan H, Jia L, Ma W, et al. Serum amyloid A exhibits pH dependent antibacterial action and contributes to host defense against. J Biol Chem. (2020) 295:2570–81. doi: 10.1074/jbc.RA119.010626
28. Cai Z, Cai L, Jiang J, Chang KS, van der Westhuyzen DR, Luo G. Human serum amyloid A protein inhibits hepatitis C virus entry into cells. J Virol. (2007) 81:6128–33. doi: 10.1128/JVI.02627-06
29. Hirakura Y, Carreras I, Sipe JD, Kagan BL. Channel formation by serum amyloid A: a potential mechanism for amyloid pathogenesis and host defense. Amyloid. (2002) 9:13–23. doi: 10.3109/13506120209072440
30. Badolato R, Wang JM, Stornello SL, Ponzi AN, Duse M, Musso T. Serum amyloid A is an activator of PMN antimicrobial functions: induction of degranulation, phagocytosis, and enhancement of anti-Candida activity. J Leukoc Biol. (2000) 67:381–6. doi: 10.1002/jlb.67.3.381
31. Badolato R, Wang JM, Murphy WJ, Lloyd AR, Michiel DF, Bausserman LL, et al. Serum amyloid A is a chemoattractant: induction of migration, adhesion, and tissue infiltration of monocytes and polymorphonuclear leukocytes. J Exp Med. (1994) 180:203–9. doi: 10.1084/jem.180.1.203
32. Liang TS, Wang JM, Murphy PM, Gao JL. Serum amyloid A is a chemotactic agonist at FPR2, a low-affinity N-formylpeptide receptor on mouse neutrophils. Biochem Biophys Res Commun. (2000) 270:331–5. doi: 10.1006/bbrc.2000.2416
33. Su SB, Gong W, Gao JL, Shen W, Murphy PM, Oppenheim JJ, et al. A seven-transmembrane, G protein-coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J Exp Med. (1999) 189:395–402. doi: 10.1084/jem.189.2.395
34. Olsson N, Siegbahn A, Nilsson G. Serum amyloid A induces chemotaxis of human mast cells by activating a pertussis toxin-sensitive signal transduction pathway. Biochem Biophys Res Commun. (1999) 254:143–6. doi: 10.1006/bbrc.1998.9911
35. Coetzee GA, Strachan AF, van der Westhuyzen DR, Hoppe HC, Jeenah MS, de Beer FC. Serum amyloid A-containing human high density lipoprotein 3. Density, size, and apolipoprotein composition. J Biol Chem. (1986) 261:9644–51. doi: 10.1016/S0021-9258(18)67562-3
36. Benditt EP, Hoffman JS, Eriksen N, Parmelee DC, Walsh KA. SAA, an apoprotein of HDL: its structure and function. Ann N Y Acad Sci. (1982) 389:183–9. doi: 10.1111/j.1749-6632.1982.tb22136.x
37. Jahangiri A, Wilson PG, Hou T, Brown A, King VL, Tannock LR. Serum amyloid A is found on ApoB-containing lipoproteins in obese humans with diabetes. Obesity (Silver Spring). (2013) 21:993–6. doi: 10.1002/oby.20126
38. Ye RD, Sun L. Emerging functions of serum amyloid A in inflammation. J Leukoc Biol. (2015) 98:923–9. doi: 10.1189/jlb.3VMR0315-080R
39. Webb NR. High-density lipoproteins and serum amyloid A (SAA). Curr Atheroscler Rep. (2021) 23:7. doi: 10.1007/s11883-020-00901-4
40. Meek RL, Benditt EP. Amyloid A gene family expression in different mouse tissues. J Exp Med. (1986) 164:2006–17. doi: 10.1084/jem.164.6.2006
41. Upragarin N, Landman WJ, Gaastra W, Gruys E. Extrahepatic production of acute phase serum amyloid A. Histol Histopathol. (2005) 20:1295–307. doi: 10.14670/HH-20.1295
42. Chait A, den Hartigh LJ, Wang S, Goodspeed L, Babenko I, Altemeier WA, et al. Presence of serum amyloid A3 in mouse plasma is dependent on the nature and extent of the inflammatory stimulus. Sci Rep. (2020) 10:10397. doi: 10.1038/s41598-020-66898-7
43. Kluve-Beckerman B, Drumm ML, Benson MD. Nonexpression of the human serum amyloid A three (SAA3) gene. DNA Cell Biol. (1991) 10:651–61. doi: 10.1089/dna.1991.10.651
44. De Buck M, Gouwy M, Wang JM, Van Snick J, Opdenakker G, Struyf S, et al. Structure and expression of different Serum amyloid A (SAA) variants and their concentration-dependent functions during host insults. Curr Med Chem. (2016) 23:1725–55. doi: 10.2174/0929867323666160418114600
45. Larson MA, Wei SH, Weber A, Weber AT, McDonald TL. Induction of human mammary-associated serum amyloid A3 expression by prolactin or lipopolysaccharide. Biochem Biophys Res Commun. (2003) 301:1030–7. doi: 10.1016/S0006-291X(03)00045-7
46. Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate acute-phase reactant. Eur J Biochem. (1999) 265:501–23. doi: 10.1046/j.1432-1327.1999.00657.x
47. de Beer MC, de Beer FC, Gerardot CJ, Cecil DR, Webb NR, Goodson ML, et al. Structure of the mouse Saa4 gene and its linkage to the serum amyloid A gene family. Genomics. (1996) 34:139–42. doi: 10.1006/geno.1996.0253
48. Lee HY, Kim MK, Park KS, Shin EH, Jo SH, Kim SD, et al. Serum amyloid A induces contrary immune responses via formyl peptide receptor-like 1 in human monocytes. Mol Pharmacol. (2006) 70:241–8. doi: 10.1124/mol.105.022103
49. He R, Sang H, Ye RD. Serum amyloid A induces IL-8 secretion through a G protein-coupled receptor, FPRL1/LXA4R. Blood. (2003) 101:1572–81. doi: 10.1182/blood-2002-05-1431
50. Lee HY, Kim SD, Shim JW, Kim HJ, Yun J, Baek SH, et al. A pertussis toxin sensitive G-protein-independent pathway is involved in serum amyloid A-induced formyl peptide receptor 2-mediated CCL2 production. Exp Mol Med. (2010) 42:302–9. doi: 10.3858/emm.2010.42.4.029
51. Christenson K, Björkman L, Tängemo C, Bylund J. Serum amyloid A inhibits apoptosis of human neutrophils via a P2X7-sensitive pathway independent of formyl peptide receptor-like 1. J Leukoc Biol. (2008) 83:139–48. doi: 10.1189/jlb.0507276
52. Li W, Zhu S, Li J, D’Amore J, D’Angelo J, Yang H, et al. Serum amyloid A stimulates PKR expression and HMGB1 release possibly through TLR4/RAGE receptors. Mol Med. (2015) 21:515–25. doi: 10.2119/molmed.2015.00109
53. Röcken C, Kientsch-Engel R, Mansfeld S, Stix B, Stubenrauch K, Weigle B, et al. Advanced glycation end products and receptor for advanced glycation end products in AA amyloidosis. Am J Pathol. (2003) 162:1213–20. doi: 10.1016/S0002-9440(10)63917-X
54. Yan SD, Zhu H, Zhu A, Golabek A, Du H, Roher A, et al. Receptor-dependent cell stress and amyloid accumulation in systemic amyloidosis. Nat Med. (2000) 6:643–51. doi: 10.1038/76216
55. Kennel SJ, Williams A, Stuckey A, Richey T, Wooliver C, Chazin W, et al. The pattern recognition reagents RAGE VC1 and peptide p5 share common binding sites and exhibit specific reactivity with AA amyloid in mice. Amyloid. (2016) 23:8–16. doi: 10.3109/13506129.2015.1112782
56. Cheng N, He R, Tian J, Ye PP, Ye RD. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J Immunol. (2008) 181:22–6. doi: 10.4049/jimmunol.181.1.22
57. Ji YR, Kim HJ, Bae KB, Lee S, Kim MO, Ryoo ZY. Hepatic serum amyloid A1 aggravates T cell-mediated hepatitis by inducing chemokines via Toll-like receptor 2 in mice. J Biol Chem. (2015) 290:12804–11. doi: 10.1074/jbc.M114.635763
58. Hiratsuka S, Watanabe A, Sakurai Y, Akashi-Takamura S, Ishibashi S, Miyake K, et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. (2008) 10:1349–55. doi: 10.1038/ncb1794
59. Sandri S, Rodriguez D, Gomes E, Monteiro HP, Russo M, Campa A. Is serum amyloid A an endogenous TLR4 agonist? J Leukoc Biol. (2008) 83:1174–80. doi: 10.1189/jlb.0407203
60. Deguchi A, Tomita T, Omori T, Komatsu A, Ohto U, Takahashi S, et al. Serum amyloid A3 binds MD-2 to activate p38 and NF-κB pathways in a MyD88-dependent manner. J Immunol. (2013) 191:1856–64. doi: 10.4049/jimmunol.1201996
61. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. (1998) 282:2085–8. doi: 10.1126/science.282.5396.2085
62. Tomita T, Ieguchi K, Sawamura T, Maru Y. Human serum amyloid A3 (SAA3) protein, expressed as a fusion protein with SAA2, binds the oxidized low density lipoprotein receptor. PLoS One. (2015) 10:e0118835. doi: 10.1371/journal.pone.0118835
63. Cai L, de Beer MC, de Beer FC, van der Westhuyzen DR. Serum amyloid A is a ligand for scavenger receptor class B type I and inhibits high density lipoprotein binding and selective lipid uptake. J Biol Chem. (2005) 280:2954–61. doi: 10.1074/jbc.M411555200
64. Dember LM, Hawkins PN, Hazenberg BP, Gorevic PD, Merlini G, Butrimiene I, et al. Eprodisate for the treatment of renal disease in AA amyloidosis. N Engl J Med. (2007) 356:2349–60. doi: 10.1056/NEJMoa065644
65. Olsson M, Olsson B, Jacobson P, Thelle DS, Björkegren J, Walley A, et al. Expression of the selenoprotein S (SELS) gene in subcutaneous adipose tissue and SELS genotype are associated with metabolic risk factors. Metab Clin Exp. (2011) 60:114–20. doi: 10.1016/j.metabol.2010.05.011
66. van der Westhuyzen DR, Cai L, de Beer MC, de Beer FC. Serum amyloid A promotes cholesterol efflux mediated by scavenger receptor B-I. J Biol Chem. (2005) 280:35890–5. doi: 10.1074/jbc.M505685200
67. Baranova IN, Vishnyakova TG, Bocharov AV, Kurlander R, Chen Z, Kimelman ML, et al. Serum amyloid A binding to CLA-1 (CD36 and LIMPII analogous-1) mediates serum amyloid A protein-induced activation of ERK1/2 and p38 mitogen-activated protein kinases. J Biol Chem. (2005) 280:8031–40. doi: 10.1074/jbc.M405009200
68. Walder K, Kantham L, McMillan JS, Trevaskis J, Kerr L, De Silva A, et al. Tanis: a link between type 2 diabetes and inflammation? Diabetes. (2002) 51:1859–66. doi: 10.2337/diabetes.51.6.1859
69. Gao JL, Murphy PM. Species and subtype variants of the N-formyl peptide chemotactic receptor reveal multiple important functional domains. J Biol Chem. (1993) 268:25395–401. doi: 10.1016/S0021-9258(19)74405-6
70. Abouelasrar Salama S, Gouwy M, Van Damme J, Struyf S. Acute-serum amyloid A and A-SAA-derived peptides as formyl peptide receptor (FPR) 2 ligands. Front Endocrinol (Lausanne). (2023) 14:1119227. doi: 10.3389/fendo.2023.1119227
71. De Buck M, Gouwy M, Wang JM, Van Snick J, Proost P, Struyf S, et al. The cytokine-serum amyloid A-chemokine network. Cytokine Growth Factor Rev. (2016) 30:55–69. doi: 10.1016/j.cytogfr.2015.12.010
72. Ray A, Shakya A, Kumar D, Benson MD, Ray BK. Inflammation-responsive transcription factor SAF-1 activity is linked to the development of amyloid A amyloidosis. J Immunol. (2006) 177:2601–9. doi: 10.4049/jimmunol.177.4.2601
73. Karlsson HK, Tsuchida H, Lake S, Koistinen HA, Krook A. Relationship between serum amyloid A level and Tanis/SelS mRNA expression in skeletal muscle and adipose tissue from healthy and type 2 diabetic subjects. Diabetes. (2004) 53:1424–8. doi: 10.2337/diabetes.53.6.1424
74. Jensen LE, Whitehead AS. Regulation of serum amyloid A protein expression during the acute-phase response. Biochem J. (1998) 334(Pt 3):489–503. doi: 10.1042/bj3340489
75. Arnon S, Litmanovitz I, Regev RH, Bauer S, Shainkin-Kestenbaum R, Dolfin T. Serum amyloid A: an early and accurate marker of neonatal early-onset sepsis. J Perinatol. (2007) 27:297–302. doi: 10.1038/sj.jp.7211682
76. Yu MH, Chen MH, Han F, Li Q, Sun RH, Tu YX. Prognostic value of the biomarkers serum amyloid A and nitric oxide in patients with sepsis. Int Immunopharmacol. (2018) 62:287–92. doi: 10.1016/j.intimp.2018.07.024
77. Li Y, Xiaojing H, Zhuanyun L, Li D, Yang J. Prognostic value of serum amyloid A in COVID-19: a meta-analysis. Medicine (Baltimore). (2022) 101:e28880. doi: 10.1097/MD.0000000000028880
78. Nakayama T, Sonoda S, Urano T, Yamada T, Okada M. Monitoring both serum amyloid protein A and C-reactive protein as inflammatory markers in infectious diseases. Clin Chem. (1993) 39:293–7. doi: 10.1093/clinchem/39.2.293
79. Perez L. Acute phase protein response to viral infection and vaccination. Arch Biochem Biophys. (2019) 671:196–202. doi: 10.1016/j.abb.2019.07.013
80. Villapol S, Kryndushkin D, Balarezo MG, Campbell AM, Saavedra JM, Shewmaker FP, et al. Hepatic expression of serum amyloid A1 is induced by traumatic brain injury and modulated by telmisartan. Am J Pathol. (2015) 185:2641–52. doi: 10.1016/j.ajpath.2015.06.016
81. Soriano S, Moffet B, Wicker E, Villapol S. Serum amyloid A is expressed in the brain after traumatic brain injury in a sex-dependent manner. Cell Mol Neurobiol. (2020) 40:1199–211. doi: 10.1007/s10571-020-00808-3
82. den Hartigh LJ, Wang S, Goodspeed L, Ding Y, Averill M, Subramanian S, et al. Deletion of serum amyloid A3 improves high fat high sucrose diet-induced adipose tissue inflammation and hyperlipidemia in female mice. PLoS One. (2014) 9:e108564. doi: 10.1371/journal.pone.0108564
83. Anuurad E, Enkhmaa B, Gungor Z, Zhang W, Tracy RP, Pearson TA, et al. Age as a modulator of inflammatory cardiovascular risk factors. Arterioscler Thromb Vasc Biol. (2011) 31:2151–6. doi: 10.1161/ATVBAHA.111.232348
84. Sproston NR, Ashworth JJ. Role of C-reactive protein at sites of inflammation and infection. Front Immunol. (2018) 9:754. doi: 10.3389/fimmu.2018.00754
85. Pepys MB, Baltz ML. Acute phase proteins with special reference to C-reactive protein and related proteins (pentaxins) and serum amyloid A protein. Adv Immunol. (1983) 34:141–212. doi: 10.1016/s0065-2776(08)60379-x
86. Maury CP. Comparative study of serum amyloid A protein and C-reactive protein in disease. Clin Sci (Lond). (1985) 68:233–8. doi: 10.1042/cs0680233
87. Chambers RE, Hutton CW, Dieppe PA, Whicher JT. Comparative study of C reactive protein and serum amyloid A protein in experimental inflammation. Ann Rheum Dis. (1991) 50:677–9. doi: 10.1136/ard.50.10.677
88. O’Brien KD, Brehm BJ, Seeley RJ, Bean J, Wener MH, Daniels S, et al. Diet-induced weight loss is associated with decreases in plasma serum amyloid a and C-reactive protein independent of dietary macronutrient composition in obese subjects. J Clin Endocrinol Metab. (2005) 90:2244–9. doi: 10.1210/jc.2004-1011
89. Thorn CF, Lu ZY, Whitehead AS. Regulation of the human acute phase serum amyloid A genes by tumour necrosis factor-alpha, interleukin-6 and glucocorticoids in hepatic and epithelial cell lines. Scand J Immunol. (2004) 59:152–8. doi: 10.1111/j.0300-9475.2004.01369.x
90. Speelman T, Dale L, Louw A, Verhoog NJD. The association of acute phase proteins in stress and inflammation-induced T2D. Cells. (2022) 11(14):2163. doi: 10.3390/cells11142163
91. Ehlting C, Wolf SD, Bode JG. Acute-phase protein synthesis: a key feature of innate immune functions of the liver. Biol Chem. (2021) 402:1129–45. doi: 10.1515/hsz-2021-0209
92. Moura Neto A, Parisi MC, Tambascia MA, Pavin EJ, Alegre SM, Zantut-Wittmann DE. Relationship of thyroid hormone levels and cardiovascular events in patients with type 2 diabetes. Endocrine. (2014) 45:84–91. doi: 10.1007/s12020-013-9938-6
93. Moura Neto A, Parisi MC, Alegre SM, Pavin EJ, Tambascia MA, Zantut-Wittmann DE. Relation of thyroid hormone abnormalities with subclinical inflammatory activity in patients with type 1 and type 2 diabetes mellitus. Endocrine. (2016) 51:63–71. doi: 10.1007/s12020-015-0651-5
94. Raynes JG, Cooper EH. Comparison of serum amyloid A protein and C-reactive protein concentrations in cancer and non-malignant disease. J Clin Pathol. (1983) 36:798–803. doi: 10.1136/jcp.36.7.798
95. Hogarth MB, Gallimore R, Savage P, Palmer AJ, Starr JM, Bulpitt CJ, et al. Acute phase proteins, C-reactive protein and serum amyloid A protein, as prognostic markers in the elderly inpatient. Age Ageing. (1997) 26:153–8. doi: 10.1093/ageing/26.2.153
96. Poitou C, Viguerie N, Cancello R, De Matteis R, Cinti S, Stich V, et al. Serum amyloid A: production by human white adipocyte and regulation by obesity and nutrition. Diabetologia. (2005) 48:519–28. doi: 10.1007/s00125-004-1654-6
97. Sjöholm K, Palming J, Olofsson LE, Gummesson A, Svensson PA, Lystig TC, et al. A microarray search for genes predominantly expressed in human omental adipocytes: adipose tissue as a major production site of serum amyloid A. J Clin Endocrinol Metab. (2005) 90:2233–9. doi: 10.1210/jc.2004-1830
98. Poitou C, Coussieu C, Rouault C, Coupaye M, Cancello R, Bedel JF, et al. Serum amyloid A: a marker of adiposity-induced low-grade inflammation but not of metabolic status. Obesity (Silver Spring). (2006) 14:309–18. doi: 10.1038/oby.2006.40
99. Maier W, Altwegg LA, Corti R, Gay S, Hersberger M, Maly FE, et al. Inflammatory markers at the site of ruptured plaque in acute myocardial infarction: locally increased interleukin-6 and serum amyloid A but decreased C-reactive protein. Circulation. (2005) 111:1355–61. doi: 10.1161/01.CIR.0000158479.58589.0A
100. Yamada T, Kakihara T, Kamishima T, Fukuda T, Kawai T. Both acute phase and constitutive serum amyloid A are present in atherosclerotic lesions. Pathol Int. (1996) 46:797–800. doi: 10.1111/j.1440-1827.1996.tb03552.x
101. Rosenthal CJ, Franklin EC. Variation with age and disease of an amyloid A protein-related serum component. J Clin Invest. (1975) 55:746–53. doi: 10.1172/JCI107985
102. Holzer M, Trieb M, Konya V, Wadsack C, Heinemann A, Marsche G. Aging affects high-density lipoprotein composition and function. Biochim Biophys Acta. (2013) 1831:1442–8. doi: 10.1016/j.bbalip.2013.06.004
103. Danesh J, Muir J, Wong YK, Ward M, Gallimore JR, Pepys MB. Risk factors for coronary heart disease and acute-phase proteins. A population-based study. Eur Heart J. (1999) 20:954–9. doi: 10.1053/euhj.1998.1309
104. Lee CG, Carr MC, Murdoch SJ, Mitchell E, Woods NF, Wener MH, et al. Adipokines, inflammation, and visceral adiposity across the menopausal transition: a prospective study. J Clin Endocrinol Metab. (2009) 94:1104–10. doi: 10.1210/jc.2008-0701
105. Chiba T, Han CY, Vaisar T, Shimokado K, Kargi A, Chen MH, et al. Serum amyloid A3 does not contribute to circulating SAA levels. J Lipid Res. (2009) 50:1353–62. doi: 10.1194/jlr.M900089-JLR200
106. Jernås M, Palming J, Sjöholm K, Jennische E, Svensson PA, Gabrielsson BG, et al. Separation of human adipocytes by size: hypertrophic fat cells display distinct gene expression. FASEB J. (2006) 20:1540–2. doi: 10.1096/fj.05-5678fje
107. Sjöholm K, Lundgren M, Olsson M, Eriksson JW. Association of serum amyloid A levels with adipocyte size and serum levels of adipokines: differences between men and women. Cytokine. (2009) 48:260–6. doi: 10.1016/j.cyto.2009.08.005
108. Siklova-Vitkova M, Klimcakova E, Polak J, Kovacova Z, Tencerova M, Rossmeislova L, et al. Adipose tissue secretion and expression of adipocyte-produced and stromavascular fraction-produced adipokines vary during multiple phases of weight-reducing dietary intervention in obese women. J Clin Endocrinol Metab. (2012) 97:E1176–81. doi: 10.1210/jc.2011-2380
109. Imayama I, Ulrich CM, Alfano CM, Wang C, Xiao L, Wener MH, et al. Effects of a caloric restriction weight loss diet and exercise on inflammatory biomarkers in overweight/obese postmenopausal women: a randomized controlled trial. Cancer Res. (2012) 72:2314–26. doi: 10.1158/0008-5472.CAN-11-3092
110. Catalán V, Gómez-Ambrosi J, Ramirez B, Rotellar F, Pastor C, Silva C, et al. Proinflammatory cytokines in obesity: impact of type 2 diabetes mellitus and gastric bypass. Obes Surg. (2007) 17:1464–74. doi: 10.1007/s11695-008-9424-z
111. Benditt EP, Meek RL. Expression of the third member of the serum amyloid A gene family in mouse adipocytes. J Exp Med. (1989) 169:1841–6. doi: 10.1084/jem.169.5.1841
112. Meek RL, Eriksen N, Benditt EP. Murine serum amyloid A3 is a high density apolipoprotein and is secreted by macrophages. Proc Natl Acad Sci U S A. (1992) 89:7949–52. doi: 10.1073/pnas.89.17.7949
113. Rokita H, Shirahama T, Cohen AS, Meek RL, Benditt EP, Sipe JD. Differential expression of the amyloid SAA 3 gene in liver and peritoneal macrophages of mice undergoing dissimilar inflammatory episodes. J Immunol. (1987) 139:3849–53. doi: 10.4049/jimmunol.139.11.3849
114. Lin Y, Rajala MW, Berger JP, Moller DE, Barzilai N, Scherer PE. Hyperglycemia-induced production of acute phase reactants in adipose tissue. J Biol Chem. (2001) 276:42077–83. doi: 10.1074/jbc.M107101200
115. Soukas A, Cohen P, Socci ND, Friedman JM. Leptin-specific patterns of gene expression in white adipose tissue. Genes Dev. (2000) 14:963–80. doi: 10.1101/gad.14.8.963
116. Subramanian S, Han C, Chiba T, McMillen T, Wang S, Haw AR, et al. Dietary cholesterol worsens adipose tissue macrophage accumulation and atherosclerosis in obese LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. (2008) 28:685–91. doi: 10.1161/ATVBAHA.107.157685
117. Subramanian S, Goodspeed L, Wang S, Kim J, Zeng L, Ioannou GN, et al. Dietary cholesterol exacerbates hepatic steatosis and inflammation in obese LDL receptor-deficient mice. J Lipid Res. (2011) 52:1626–35. doi: 10.1194/jlr.M016246
118. Han CY, Kang I, Harten IA, Gebe JA, Chan CK, Omer M, et al. Adipocyte-derived versican and macrophage-derived biglycan control adipose tissue inflammation in obesity. Cell Rep. (2020) 31:107818. doi: 10.1016/j.celrep.2020.107818
119. Den Hartigh LJ, Omer M, Goodspeed L, Wang S, Wietecha T, O’Brien KD, et al. Adipocyte-specific deficiency of NADPH oxidase 4 delays the onset of insulin resistance and attenuates adipose tissue inflammation in obesity. Arterioscler Thromb Vasc Biol. (2017) 37:466–75. doi: 10.1161/ATVBAHA.116.308749
120. Scheja L, Heese B, Zitzer H, Michael MD, Siesky AM, Pospisil H, et al. Acute-phase serum amyloid A as a marker of insulin resistance in mice. Exp Diabetes Res. (2008) 2008:230837. doi: 10.1155/2008/230837
121. Sanada Y, Yamamoto T, Satake R, Yamashita A, Kanai S, Kato N, et al. Serum amyloid A3 gene expression in adipocytes is an indicator of the interaction with macrophages. Sci Rep. (2016) 6:38697. doi: 10.1038/srep38697
122. Tannock LR, De Beer MC, Ji A, Shridas P, Noffsinger VP, den Hartigh L, et al. Serum amyloid A3 is a high density lipoprotein-associated acute-phase protein. J Lipid Res. (2018) 59:339–47. doi: 10.1194/jlr.M080887
123. de Oliveira EM, Visniauskas B, Tufik S, Andersen ML, Chagas JR, Campa A. Serum amyloid A production is triggered by sleep deprivation in mice and humans: is that the link between sleep loss and associated comorbidities? Nutrients. (2017) 9(3):311. doi: 10.3390/nu9030311
124. Svatikova A, Wolk R, Shamsuzzaman AS, Kara T, Olson EJ, Somers VK. Serum amyloid a in obstructive sleep apnea. Circulation. (2003) 108:1451–4. doi: 10.1161/01.CIR.0000089091.09527.B8
125. Parish JM, Adam T, Facchiano L. Relationship of metabolic syndrome and obstructive sleep apnea. J Clin Sleep Med. (2007) 3:467–72. doi: 10.5664/jcsm.26910
126. Olsson M, Ahlin S, Olsson B, Svensson PA, Ståhlman M, Borén J, et al. Establishment of a transgenic mouse model specifically expressing human serum amyloid A in adipose tissue. PLoS One. (2011) 6:e19609. doi: 10.1371/journal.pone.0019609
127. Ahlin S, Olsson M, Olsson B, Svensson PA, Sjöholm K. No evidence for a role of adipose tissue-derived Serum amyloid A in the development of insulin resistance or obesity-related inflammation in hSAA1(+/-) transgenic mice. PLoS One. (2013) 8:e72204. doi: 10.1371/journal.pone.0072204
128. Ather JL, Poynter ME. Serum amyloid A3 is required for normal weight and immunometabolic function in mice. PLoS One. (2018) 13:e0192352. doi: 10.1371/journal.pone.0192352
129. Ji A, Trumbauer AC, Noffsinger VP, Jeon H, Patrick AC, De Beer FC, et al. Serum amyloid A is not obligatory for high-fat, high-sucrose, cholesterol-fed diet-induced obesity and its metabolic and inflammatory complications. PLoS One. (2022) 17:e0266688. doi: 10.1371/journal.pone.0266688
130. Vercalsteren E, Vranckx C, Vermeire I, Gooijen M, Lijnen R, Scroyen I. Serum amyloid A3 deficiency impairs in vitro and in vivo adipocyte differentiation. Adipocyte. (2021) 10:242–50. doi: 10.1080/21623945.2021.1916220
131. de Oliveira EM, Ascar TP, Silva JC, Sandri S, Migliorini S, Fock RA, et al. Serum amyloid A links endotoxaemia to weight gain and insulin resistance in mice. Diabetologia. (2016) 59:1760–8. doi: 10.1007/s00125-016-3970-z
132. Qatanani M, Lazar MA. Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev. (2007) 21:1443–55. doi: 10.1101/gad.1550907
133. Burhans MS, Hagman DK, Kuzma JN, Schmidt KA, Kratz M. Contribution of adipose tissue inflammation to the development of type 2 diabetes Mellitus. Compr Physiol. (2018) 9:1–58. doi: 10.1002/cphy.c170040
134. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. (2006) 444:840–6. doi: 10.1038/nature05482
135. Raji A, Seely EW, Arky RA, Simonson DC. Body fat distribution and insulin resistance in healthy Asian Indians and Caucasians. J Clin Endocrinol Metab. (2001) 86:5366–71. doi: 10.1210/jcem.86.11.7992
136. Paradisi G, Smith L, Burtner C, Leaming R, Garvey WT, Hook G, et al. Dual energy x-ray absorptiometry assessment of fat mass distribution and its association with the insulin resistance syndrome. Diabetes Care. (1999) 22:1310–7. doi: 10.2337/diacare.22.8.1310
137. Griffiths K, Pazderska A, Ahmed M, McGowan A, Maxwell AP, McEneny J, et al. Type 2 diabetes in young females results in increased Serum amyloid A and changes to features of high density lipoproteins in both HDL2 and HDL3. J Diabetes Res. (2017) 2017:1314864. doi: 10.1155/2017/1314864
138. Tsun JG, Shiu SW, Wong Y, Yung S, Chan TM, Tan KC. Impact of serum amyloid A on cellular cholesterol efflux to serum in type 2 diabetes mellitus. Atherosclerosis. (2013) 231:405–10. doi: 10.1016/j.atherosclerosis.2013.10.008
139. Du JL, Liu JF, Men LL, Yao JJ, Sun LP, Sun GH, et al. Effects of five-year intensive multifactorial intervention on the serum amyloid A and macroangiopathy in patients with short-duration type 2 diabetes mellitus. Chin Med J (Engl). (2009) 122:2560–6. doi: 10.3760/cma.j.issn.0366-6999.2009.21.007
140. Kumon Y, Suehiro T, Itahara T, Ikeda Y, Hashimoto K. Serum amyloid A protein in patients with non-insulin-dependent diabetes mellitus. Clin Biochem. (1994) 27:469–73. doi: 10.1016/0009-9120(94)00044-v
141. Helmersson J, Vessby B, Larsson A, Basu S. Association of type 2 diabetes with cyclooxygenase-mediated inflammation and oxidative stress in an elderly population. Circulation. (2004) 109:1729–34. doi: 10.1161/01.CIR.0000124718.99562.91
142. Samaras K, Botelho NK, Chisholm DJ, Lord RV. Subcutaneous and visceral adipose tissue gene expression of serum adipokines that predict type 2 diabetes. Obesity (Silver Spring). (2010) 18:884–9. doi: 10.1038/oby.2009.443
143. Lewis KE, Kirk EA, McDonald TO, Wang S, Wight TN, O’Brien KD, et al. Increase in serum amyloid a evoked by dietary cholesterol is associated with increased atherosclerosis in mice. Circulation. (2004) 110:540–5. doi: 10.1161/01.CIR.0000136819.93989.E1
144. Yassine HN, Trenchevska O, He H, Borges CR, Nedelkov D, Mack W, et al. Serum amyloid a truncations in type 2 diabetes mellitus. PLoS One. (2015) 10:e0115320. doi: 10.1371/journal.pone.0115320
145. Du JL, Sun CK, Lü B, Men LL, Yao JJ, An LJ, et al. Association of SelS mRNA expression in omental adipose tissue with Homa-IR and serum amyloid A in patients with type 2 diabetes mellitus. Chin Med J (Engl). (2008) 121:1165–8. doi: 10.1097/00029330-200807010-00003
146. Ye XY, Xue YM, Sha JP, Li CZ, Zhen ZJ. Serum amyloid A attenuates cellular insulin sensitivity by increasing JNK activity in 3T3-L1 adipocytes. J Endocrinol Invest. (2009) 32:568–75. doi: 10.1007/BF03346510
147. Liu Q, Sun J, Xu T, Bian G, Yang F. Associations of serum amyloid A and 25-hydroxyvitamin D with diabetic nephropathy: a cross-sectional study. J Clin Lab Anal. (2022) 36:e24283. doi: 10.1002/jcla.24283
148. Pitsavos C, Tampourlou M, Panagiotakos DB, Skoumas Y, Chrysohoou C, Nomikos T, et al. Association between low-grade systemic inflammation and type 2 diabetes mellitus among men and women from the ATTICA study. Rev Diabet Stud. (2007) 4:98–104. doi: 10.1900/RDS.2007.4.98
149. Ebeling P, Teppo AM, Koistinen HA, Viikari J, Rönnemaa T, Nissén M, et al. Troglitazone reduces hyperglycaemia and selectively acute-phase serum proteins in patients with type II diabetes. Diabetologia. (1999) 42:1433–8. doi: 10.1007/s001250051315
150. Azabdaftari A, van der Giet M, Schuchardt M, Hennermann JB, Plöckinger U, Querfeld U. The cardiovascular phenotype of adult patients with phenylketonuria. Orphanet J Rare Dis. (2019) 14:213. doi: 10.1186/s13023-019-1188-0
151. Loporchio DF, Tam EK, Cho J, Chung J, Jun GR, Xia W, et al. Cytokine levels in human vitreous in proliferative diabetic retinopathy. Cells. (2021) 10(5):1069. doi: 10.3390/cells10051069
152. Dieter BP, McPherson SM, Afkarian M, de Boer IH, Mehrotra R, Short R, et al. Serum amyloid a and risk of death and end-stage renal disease in diabetic kidney disease. J Diabetes Complications. (2016) 30:1467–72. doi: 10.1016/j.jdiacomp.2016.07.018
153. Dalla Vestra M, Mussap M, Gallina P, Bruseghin M, Cernigoi AM, Saller A, et al. Acute-phase markers of inflammation and glomerular structure in patients with type 2 diabetes. J Am Soc Nephrol. (2005) 16(Suppl 1):S78–82. doi: 10.1681/asn.2004110961
154. Saliu TP, Yazawa N, Hashimoto K, Miyata K, Kudo A, Horii M, et al. Serum amyloid A3 promoter-driven luciferase activity enables visualization of diabetic kidney disease. Int J Mol Sci. (2022) 23(2):899. doi: 10.3390/ijms23020899
155. Eren MA, Vural M, Cece H, Camuzcuoglu H, Yildiz S, Toy H, et al. Association of serum amyloid A with subclinical atherosclerosis in women with gestational diabetes. Gynecol Endocrinol. (2012) 28:1010–3. doi: 10.3109/09513590.2012.705371
156. Hrolfsdottir L, Schalkwijk CG, Birgisdottir BE, Gunnarsdottir I, Maslova E, Granström C, et al. Maternal diet, gestational weight gain, and inflammatory markers during pregnancy. Obesity (Silver Spring). (2016) 24:2133–9. doi: 10.1002/oby.21617
157. Pöyhönen-Alho M, Ebeling P, Saarinen A, Kaaja R. Decreased variation of inflammatory markers in gestational diabetes. Diabetes Metab Res Rev. (2011) 27:269–76. doi: 10.1002/dmrr.1170
158. Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev. (1997) 18:774–800. doi: 10.1210/edrv.18.6.0318
159. Diamanti-Kandarakis E. Insulin resistance in PCOS. Endocrine. (2006) 30:13–7. doi: 10.1385/ENDO:30:1:13
160. Tan BK, Adya R, Shan X, Aghilla M, Lehnert H, Keay SD, et al. The anti-atherogenic aspect of metformin treatment in insulin resistant women with the polycystic ovary syndrome: role of the newly established pro-inflammatory adipokine acute-phase serum amyloid A; evidence of an adipose tissue-monocyte axis. Atherosclerosis. (2011) 216:402–8. doi: 10.1016/j.atherosclerosis.2010.08.069
161. Arner P. Effects of testosterone on fat cell lipolysis. Species differences and possible role in polycystic ovarian syndrome. Biochimie. (2005) 87:39–43. doi: 10.1016/j.biochi.2004.11.012
162. Yki-Järvinen H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. (2014) 2:901–10. doi: 10.1016/S2213-8587(14)70032-4
163. Grundy SM, Brewer HB, Cleeman JI, Smith SC, Lenfant C, Association AH, et al. Definition of metabolic syndrome: report of the national heart, lung, and blood institute/American heart association conference on scientific issues related to definition. Circulation. (2004) 109:433–8. doi: 10.1161/01.CIR.0000111245.75752.C6
164. Reddy P, Lent-Schochet D, Ramakrishnan N, McLaughlin M, Jialal I. Metabolic syndrome is an inflammatory disorder: a conspiracy between adipose tissue and phagocytes. Clin Chim Acta. (2019) 496:35–44. doi: 10.1016/j.cca.2019.06.019
165. Kappelle PJ, Bijzet J, Hazenberg BP, Dullaart RP. Lower serum paraoxonase-1 activity is related to higher serum amyloid a levels in metabolic syndrome. Arch Med Res. (2011) 42:219–25. doi: 10.1016/j.arcmed.2011.05.002
166. Yuan ZY, Zhang XX, Wu YJ, Zeng ZP, She WM, Chen SY, et al. Serum amyloid A levels in patients with liver diseases. World J Gastroenterol. (2019) 25:6440–50. doi: 10.3748/wjg.v25.i43.6440
167. Jacobs M, van Greevenbroek MM, van der Kallen CJ, Ferreira I, Feskens EJ, Jansen EH, et al. The association between the metabolic syndrome and alanine amino transferase is mediated by insulin resistance via related metabolic intermediates (the cohort on diabetes and atherosclerosis Maastricht [CODAM] study). Metab Clin Exp. (2011) 60:969–75. doi: 10.1016/j.metabol.2010.09.006
168. Jiang B, Wang D, Hu Y, Li W, Liu F, Zhu X, et al. Serum amyloid A1 exacerbates hepatic steatosis via TLR4-mediated NF-κB signaling pathway. Mol Metab. (2022) 59:101462. doi: 10.1016/j.molmet.2022.101462
169. Li D, Xie P, Zhao S, Zhao J, Yao Y, Zhao Y, et al. Hepatocytes derived increased SAA1 promotes intrahepatic platelet aggregation and aggravates liver inflammation in NAFLD. Biochem Biophys Res Commun. (2021) 555:54–60. doi: 10.1016/j.bbrc.2021.02.124
170. Sultan M, Ben-Ari Z, Masoud R, Pappo O, Harats D, Kamari Y, et al. Interleukin-1α and interleukin-1β play a central role in the pathogenesis of fulminant hepatic failure in mice. PLoS One. (2017) 12:e0184084. doi: 10.1371/journal.pone.0184084
171. Ross R. The pathogenesis of atherosclerosis--an update. N Engl J Med. (1986) 314:488–500. doi: 10.1056/NEJM198602203140806
172. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
173. Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. (2000) 342:836–43. doi: 10.1056/NEJM200003233421202
174. Jousilahti P, Salomaa V, Rasi V, Vahtera E, Palosuo T. The association of c-reactive protein, serum amyloid a and fibrinogen with prevalent coronary heart disease--baseline findings of the PAIS project. Atherosclerosis. (2001) 156:451–6. doi: 10.1016/s0021-9150(00)00681-x
175. O’Brien KD, McDonald TO, Kunjathoor V, Eng K, Knopp EA, Lewis K, et al. Serum amyloid A and lipoprotein retention in murine models of atherosclerosis. Arterioscler Thromb Vasc Biol. (2005) 25:785–90. doi: 10.1161/01.ATV.0000158383.65277.2b
176. Dong Z, Wu T, Qin W, An C, Wang Z, Zhang M, et al. Serum amyloid A directly accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Mol Med. (2011) 17:1357–64. doi: 10.2119/molmed.2011.00186
177. Thompson JC, Jayne C, Thompson J, Wilson PG, Yoder MH, Webb N, et al. A brief elevation of serum amyloid A is sufficient to increase atherosclerosis. J Lipid Res. (2015) 56:286–93. doi: 10.1194/jlr.M054015
178. Krishack PA, Bhanvadia CV, Lukens J, Sontag TJ, De Beer MC, Getz GS, et al. Serum amyloid A facilitates early lesion development in ldlr-/- mice. J Am Heart Assoc. (2015) 4(7):e001858. doi: 10.1161/JAHA.115.001858
179. Petri MH, Laguna-Fernández A, Gonzalez-Diez M, Paulsson-Berne G, Hansson GK, Bäck M. The role of the FPR2/ALX receptor in atherosclerosis development and plaque stability. Cardiovasc Res. (2015) 105:65–74. doi: 10.1093/cvr/cvu224
180. Webb NR, De Beer MC, Wroblewski JM, Ji A, Bailey W, Shridas P, et al. Deficiency of endogenous acute-phase Serum amyloid A protects apoE-/- mice from angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. (2015) 35:1156–65. doi: 10.1161/ATVBAHA.114.304776
181. Thompson JC, Wilson PG, Shridas P, Ji A, de Beer M, de Beer FC, et al. Serum amyloid A3 is pro-atherogenic. Atherosclerosis. (2017) 268:32–5. doi: 10.1016/j.atherosclerosis.2017.11.011
182. Chait A, Wang S, Goodspeed L, Gomes D, Turk KE, Wietecha T, et al. Sexually dimorphic relationships among Saa3 (Serum amyloid A3) inflammation, and cholesterol metabolism modulate atherosclerosis in mice. Arterioscler Thromb Vasc Biol. (2021):ATVBAHA121316066. doi: 10.1161/ATVBAHA.121.316066
183. Koike T, Kitajima S, Yu Y, Nishijima K, Zhang J, Ozaki Y, et al. Human C-reactive protein does not promote atherosclerosis in transgenic rabbits. Circulation. (2009) 120:2088–94. doi: 10.1161/CIRCULATIONAHA.109.872796
184. Shridas P, Ji A, Trumbauer AC, Noffsinger VP, Leung SW, Dugan AJ, et al. Adipocyte-Derived Serum amyloid A promotes angiotensin II-induced abdominal aortic aneurysms in obese C57BL/6J mice. Arterioscler Thromb Vasc Biol. (2022) 42:632–43. doi: 10.1161/ATVBAHA.121.317225
185. Yamada T, Wada A, Itoh K, Igari J. Serum amyloid A secretion from monocytic leukaemia cell line THP-1 and cultured human peripheral monocytes. Scand J Immunol. (2000) 52:7–12. doi: 10.1046/j.1365-3083.2000.00734.x
186. Ray BK, Ray A. Involvement of an SAF-like transcription factor in the activation of serum amyloid A gene in monocyte/macrophage cells by lipopolysaccharide. Biochemistry. (1997) 36:4662–8. doi: 10.1021/bi9624595
187. Urieli-Shoval S, Meek RL, Hanson RH, Eriksen N, Benditt EP. Human serum amyloid A genes are expressed in monocyte/macrophage cell lines. Am J Pathol. (1994) 145:650–60.8080047
188. Zhang X, Chen J, Wang S. Serum amyloid A induces a vascular smooth muscle cell phenotype switch through the p38 MAPK signaling pathway. Biomed Res Int. (2017) 2017:4941379. doi: 10.1155/2017/4941379
189. Lv M, Xia YF, Li B, Liu H, Pan JY, Li BB, et al. Serum amyloid A stimulates vascular endothelial growth factor receptor 2 expression and angiogenesis. J Physiol Biochem. (2016) 72:71–81. doi: 10.1007/s13105-015-0462-4
190. Yeop Han C, Kargi A, Omer M, Chan C, Wabitsch M, O’Brien K, et al. Differential effect of saturated and unsaturated free fatty acids on the generation of monocyte adhesion and chemotactic factors by adipocytes: dissociation of adipocyte hypertrophy from inflammation. Diabetes. (2010) 59:386–96. doi: 10.2337/db09-0925
191. Han CY, Tang C, Guevara ME, Wei H, Wietecha T, Shao B, et al. Serum amyloid A impairs the antiinflammatory properties of HDL. J Clin Invest. (2016) 126:266–81. doi: 10.1172/JCI83475
192. Han CY, Kang I, Omer M, Wang S, Wietecha T, Wight TN, et al. Serum amyloid A-containing HDL binds adipocyte-derived versican and macrophage-derived biglycan, reducing its antiinflammatory properties. JCI Insight. (2020) 5(20):e142635. doi: 10.1172/jci.insight.142635
193. Vaisar T, Tang C, Babenko I, Hutchins P, Wimberger J, Suffredini AF, et al. Inflammatory remodeling of the HDL proteome impairs cholesterol efflux capacity. J Lipid Res. (2015) 56:1519–30. doi: 10.1194/jlr.M059089
194. O’Brien KD, Olin KL, Alpers CE, Chiu W, Ferguson M, Hudkins K, et al. Comparison of apolipoprotein and proteoglycan deposits in human coronary atherosclerotic plaques: colocalization of biglycan with apolipoproteins. Circulation. (1998) 98:519–27. doi: 10.1161/01.cir.98.6.519
195. Chiba T, Chang MY, Wang S, Wight TN, McMillen TS, Oram JF, et al. Serum amyloid A facilitates the binding of high-density lipoprotein from mice injected with lipopolysaccharide to vascular proteoglycans. Arterioscler Thromb Vasc Biol. (2011) 31:1326–32. doi: 10.1161/ATVBAHA.111.226159
196. Williams KJ, Tabas I. The response-to-retention hypothesis of atherogenesis reinforced. Curr Opin Lipidol. (1998) 9:471–4. doi: 10.1097/00041433-199810000-00012
197. Song C, Shen Y, Yamen E, Hsu K, Yan W, Witting PK, et al. Serum amyloid A may potentiate prothrombotic and proinflammatory events in acute coronary syndromes. Atherosclerosis. (2009) 202:596–604. doi: 10.1016/j.atherosclerosis.2008.04.049
198. Page MJ, Thomson GJA, Nunes JM, Engelbrecht AM, Nell TA, de Villiers WJS, et al. Serum amyloid A binds to fibrin(ogen), promoting fibrin amyloid formation. Sci Rep. (2019) 9:3102. doi: 10.1038/s41598-019-39056-x
199. Zhi W, Sharma A, Purohit S, Miller E, Bode B, Anderson SW, et al. Discovery and validation of serum protein changes in type 1 diabetes patients using high throughput two dimensional liquid chromatography-mass spectrometry and immunoassays. Mol Cell Proteomics. (2011) 10:M111.012203. doi: 10.1074/mcp.M111.012203
200. McEneny J, Daniels JA, McGowan A, Gunness A, Moore K, Stevenson M, et al. A cross-sectional study demonstrating increased Serum amyloid A related inflammation in high-density lipoproteins from subjects with type 1 diabetes Mellitus and how this association was augmented by poor glycaemic control. J Diabetes Res. (2015) 2015:351601. doi: 10.1155/2015/351601
201. Lenzen S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia. (2008) 51:216–26. doi: 10.1007/s00125-007-0886-7
202. Rakieten N, Rakieten ML, Nadkarni MR. Studies on the diabetogenic action of streptozotocin (NSC-37917). Cancer Chemother Rep. (1963) 29:91–8.
203. Pahwa R, Balderas M, Jialal I, Chen X, Luna RA, Devaraj S. Gut microbiome and inflammation: a study of diabetic inflammasome-knockout mice. J Diabetes Res. (2017) 2017:6519785. doi: 10.1155/2017/6519785
204. den Hartigh LJ, Han CY, Wang S, Omer M, Chait A. 10E,12Z-conjugated Linoleic acid impairs adipocyte triglyceride storage by enhancing fatty acid oxidation, lipolysis, and mitochondrial reactive oxygen species. J Lipid Res. (2013) 54:2964–78. doi: 10.1194/jlr.M035188
205. Shen C, Sun XG, Liu N, Mu Y, Hong CC, Wei W, et al. Increased serum amyloid A and its association with autoantibodies, acute phase reactants and disease activity in patients with rheumatoid arthritis. Mol Med Rep. (2015) 11:1528–34. doi: 10.3892/mmr.2014.2804
206. Hilliquin P. Biological markers in inflammatory rheumatic diseases. Cell Mol Biol (Noisy-le-Grand). (1995) 41:993–1006.8747080
207. Maury CP, Teppo AM, Wegelius O. Relationship between urinary sialylated saccharides, serum amyloid A protein, and C-reactive protein in rheumatoid arthritis and systemic lupus erythematosus. Ann Rheum Dis. (1982) 41:268–71. doi: 10.1136/ard.41.3.268
208. Kasahara K, Tanoue T, Yamashita T, Yodoi K, Matsumoto T, Emoto T, et al. Commensal bacteria at the crossroad between cholesterol homeostasis and chronic inflammation in atherosclerosis. J Lipid Res. (2017) 58:519–28. doi: 10.1194/jlr.M072165
209. Lee JY, Hall JA, Kroehling L, Wu L, Najar T, Nguyen HH, et al. Serum amyloid A proteins induce pathogenic Th17 cells and promote inflammatory disease. Cell. (2020) 180:79–91.e16. doi: 10.1016/j.cell.2019.11.026
210. Furuzawa-Carballeda J, Vargas-Rojas MI, Cabral AR. Autoimmune inflammation from the Th17 perspective. Autoimmun Rev. (2007) 6:169–75. doi: 10.1016/j.autrev.2006.10.002
211. Chambers RE, MacFarlane DG, Whicher JT, Dieppe PA. Serum amyloid-A protein concentration in rheumatoid arthritis and its role in monitoring disease activity. Ann Rheum Dis. (1983) 42:665–7. doi: 10.1136/ard.42.6.665
212. de Seny D, Cobraiville G, Charlier E, Neuville S, Esser N, Malaise D, et al. Acute-phase serum amyloid a in osteoarthritis: regulatory mechanism and proinflammatory properties. PLoS One. (2013) 8:e66769. doi: 10.1371/journal.pone.0066769
213. O’Hara R, Murphy EP, Whitehead AS, FitzGerald O, Bresnihan B. Local expression of the serum amyloid A and formyl peptide receptor-like 1 genes in synovial tissue is associated with matrix metalloproteinase production in patients with inflammatory arthritis. Arthritis Rheum. (2004) 50:1788–99. doi: 10.1002/art.20301
214. Geurts J, Joosten LA, Takahashi N, Arntz OJ, Glück A, Bennink MB, et al. Computational design and application of endogenous promoters for transcriptionally targeted gene therapy for rheumatoid arthritis. Mol Ther. (2009) 17:1877–87. doi: 10.1038/mt.2009.182
215. Connolly M, Mullan RH, McCormick J, Matthews C, Sullivan O, Kennedy A, et al. Acute-phase serum amyloid A regulates tumor necrosis factor α and matrix turnover and predicts disease progression in patients with inflammatory arthritis before and after biologic therapy. Arthritis Rheum. (2012) 64:1035–45. doi: 10.1002/art.33455
216. Migita K, Kawabe Y, Tominaga M, Origuchi T, Aoyagi T, Eguchi K. Serum amyloid A protein induces production of matrix metalloproteinases by human synovial fibroblasts. Lab Invest. (1998) 78:535–9.9605178
217. Hwang YG, Balasubramani GK, Metes ID, Levesque MC, Bridges SL, Moreland LW. Differential response of serum amyloid A to different therapies in early rheumatoid arthritis and its potential value as a disease activity biomarker. Arthritis Res Ther. (2016) 18:108. doi: 10.1186/s13075-016-1009-y
218. Eichele DD, Kharbanda KK. Dextran sodium sulfate colitis murine model: an indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J Gastroenterol. (2017) 23:6016–29. doi: 10.3748/wjg.v23.i33.6016
219. Roda G, Chien Ng S, Kotze PG, Argollo M, Panaccione R, Spinelli A, et al. Crohn’s disease. Nat Rev Dis Primers. (2020) 6:22. doi: 10.1038/s41572-020-0156-2
220. Bourgonje AR, von Martels JZH, Gabriëls RY, Blokzijl T, Buist-Homan M, Heegsma J, et al. A combined set of four serum inflammatory biomarkers reliably predicts endoscopic disease activity in inflammatory bowel disease. Front Med (Lausanne). (2019) 6:251. doi: 10.3389/fmed.2019.00251
221. Chambers RE, Stross P, Barry RE, Whicher JT. Serum amyloid A protein compared with C-reactive protein, alpha 1-antichymotrypsin and alpha 1-acid glycoprotein as a monitor of inflammatory bowel disease. Eur J Clin Invest. (1987) 17:460–7. doi: 10.1111/j.1365-2362.1987.tb01143.x
222. Wakai M, Hayashi R, Tanaka S, Naito T, Kumada J, Nomura M, et al. Serum amyloid A is a better predictive biomarker of mucosal healing than C-reactive protein in ulcerative colitis in clinical remission. BMC Gastroenterol. (2020) 20:85. doi: 10.1186/s12876-020-01229-8
223. Roblin X, Serone A, Yoon OK, Zhuo L, Grant E, Woo J, et al. Effects of JAK1-preferential inhibitor filgotinib on circulating biomarkers and whole blood genes/pathways of patients with moderately to severely active Crohn’s disease. Inflamm Bowel Dis. (2022) 28:1207–18. doi: 10.1093/ibd/izab253
224. Eckhardt ER, Witta J, Zhong J, Arsenescu R, Arsenescu V, Wang Y, et al. Intestinal epithelial serum amyloid A modulates bacterial growth in vitro and pro-inflammatory responses in mouse experimental colitis. BMC Gastroenterol. (2010) 10:133. doi: 10.1186/1471-230X-10-133
225. Yarur AJ, Quintero MA, Jain A, Czul F, Barkin JS, Abreu MT. Serum amyloid A as a surrogate marker for mucosal and histologic inflammation in patients with Crohn’s disease. Inflamm Bowel Dis. (2017) 23:158–64. doi: 10.1097/MIB.0000000000000991
226. Fritsch J, Garces L, Quintero MA, Pignac-Kobinger J, Santander AM, Fernández I, et al. Low-Fat, high-fiber diet reduces markers of inflammation and dysbiosis and improves quality of life in patients with ulcerative colitis. Clin Gastroenterol Hepatol. (2021) 19:1189–99.e30. doi: 10.1016/j.cgh.2020.05.026
227. Vidal-Lletjós S, Andriamihaja M, Blais A, Grauso M, Lepage P, Davila AM, et al. Mucosal healing progression after acute colitis in mice. World J Gastroenterol. (2019) 25:3572–89. doi: 10.3748/wjg.v25.i27.3572
228. Zhang G, Liu J, Wu L, Fan Y, Sun L, Qian F, et al. Elevated expression of serum amyloid A 3 protects colon epithelium against acute injury through TLR2-dependent induction of neutrophil IL-22 expression in a mouse model of colitis. Front Immunol. (2018) 9:1503. doi: 10.3389/fimmu.2018.01503
229. Sann H, Erichsen J, Hessmann M, Pahl A, Hoffmeyer A. Efficacy of drugs used in the treatment of IBD and combinations thereof in acute DSS-induced colitis in mice. Life Sci. (2013) 92:708–18. doi: 10.1016/j.lfs.2013.01.028
230. Foligné B, Peys E, Vandenkerckhove J, Van Hemel J, Dewulf J, Breton J, et al. Spores from two distinct colony types of the strain Bacillus subtilis PB6 substantiate anti-inflammatory probiotic effects in mice. Clin Nutr. (2012) 31:987–94. doi: 10.1016/j.clnu.2012.05.016
231. Kono T, Kaneko A, Hira Y, Suzuki T, Chisato N, Ohtake N, et al. Anti-colitis and -adhesion effects of daikenchuto via endogenous adrenomedullin enhancement in Crohn’s disease mouse model. J Crohns Colitis. (2010) 4:161–70. doi: 10.1016/j.crohns.2009.09.006
232. Davis TA, Conradie D, Shridas P, de Beer FC, Engelbrecht AM, de Villiers WJS. Serum amyloid A promotes inflammation-associated damage and tumorigenesis in a mouse model of colitis-associated cancer. Cell Mol Gastroenterol Hepatol. (2021) 12:1329–41. doi: 10.1016/j.jcmgh.2021.06.016
233. Ramadori G, Rieder H, Sipe J, Shirahama T, Meyer zum Büschenfelde KH. Murine tissue macrophages synthesize and secrete amyloid proteins different to amyloid A (AA). Eur J Clin Invest. (1989) 19:316–22. doi: 10.1111/j.1365-2362.1989.tb00236.x
234. Hagihara K, Nishikawa T, Isobe T, Song J, Sugamata Y, Yoshizaki K. IL-6 plays a critical role in the synergistic induction of human serum amyloid A (SAA) gene when stimulated with proinflammatory cytokines as analyzed with an SAA isoform real-time quantitative RT-PCR assay system. Biochem Biophys Res Commun. (2004) 314:363–9. doi: 10.1016/j.bbrc.2003.12.096
235. Cicarelli DD, Vieira JE, Benseñor FE. Comparison of C-reactive protein and serum amyloid a protein in septic shock patients. Mediators Inflamm. (2008) 2008:631414. doi: 10.1155/2008/631414
236. Morrow JF, Stearman RS, Peltzman CG, Potter DA. Induction of hepatic synthesis of serum amyloid A protein and actin. Proc Natl Acad Sci U S A. (1981) 78:4718–22. doi: 10.1073/pnas.78.8.4718
237. Nguyen TV, Ukairo O, Khetani SR, McVay M, Kanchagar C, Seghezzi W, et al. Establishment of a hepatocyte-kupffer cell coculture model for assessment of proinflammatory cytokine effects on metabolizing enzymes and drug transporters. Drug Metab Dispos. (2015) 43:774–85. doi: 10.1124/dmd.114.061317
238. Lowell CA, Stearman RS, Morrow JF. Transcriptional regulation of serum amyloid A gene expression. J Biol Chem. (1986) 261:8453–61. doi: 10.1016/S0021-9258(19)83933-9
239. Siegmund SV, Schlosser M, Schildberg FA, Seki E, De Minicis S, Uchinami H, et al. Serum amyloid A induces inflammation, proliferation and cell death in activated hepatic stellate cells. PLoS One. (2016) 11:e0150893. doi: 10.1371/journal.pone.0150893
240. Betts JC, Cheshire JK, Akira S, Kishimoto T, Woo P. The role of NF-kappa B and NF-IL6 transactivating factors in the synergistic activation of human serum amyloid A gene expression by interleukin-1 and interleukin-6. J Biol Chem. (1993) 268:25624–31. doi: 10.1016/S0021-9258(19)74435-4
241. Song C, Hsu K, Yamen E, Yan W, Fock J, Witting PK, et al. Serum amyloid A induction of cytokines in monocytes/macrophages and lymphocytes. Atherosclerosis. (2009) 207:374–83. doi: 10.1016/j.atherosclerosis.2009.05.007
242. Filippin-Monteiro FB, de Oliveira EM, Sandri S, Knebel FH, Albuquerque RC, Campa A. Serum amyloid A is a growth factor for 3T3-L1 adipocytes, inhibits differentiation and promotes insulin resistance. Int J Obes (Lond). (2012) 36:1032–9. doi: 10.1038/ijo.2011.193
243. Liu LR, Lin SP, Chen CC, Chen YJ, Tai CC, Chang SC, et al. Serum amyloid A induces lipolysis by downregulating perilipin through ERK1/2 and PKA signaling pathways. Obesity (Silver Spring). (2011) 19:2301–9. doi: 10.1038/oby.2011.176
244. van Bilsen JHM, van den Brink W, van den Hoek AM, Dulos R, Caspers MPM, Kleemann R, et al. Mechanism-based biomarker prediction for low-grade inflammation in liver and adipose tissue. Front Physiol. (2021) 12:703370. doi: 10.3389/fphys.2021.703370
245. Sommer G, Weise S, Kralisch S, Scherer PE, Lössner U, Blüher M, et al. The adipokine SAA3 is induced by interleukin-1beta in mouse adipocytes. J Cell Biochem. (2008) 104:2241–7. doi: 10.1002/jcb.21782
246. Nakarai H, Yamashita A, Nagayasu S, Iwashita M, Kumamoto S, Ohyama H, et al. Adipocyte-macrophage interaction may mediate LPS-induced low-grade inflammation: potential link with metabolic complications. Innate Immun. (2012) 18:164–70. doi: 10.1177/1753425910393370
247. Reigstad CS, Bäckhed F. Microbial regulation of SAA3 expression in mouse colon and adipose tissue. Gut Microbes. (2010) 1:55–7. doi: 10.4161/gmic.1.1.10514
248. Thorn CF, Whitehead AS. Differential glucocorticoid enhancement of the cytokine-driven transcriptional activation of the human acute phase serum amyloid A genes, SAA1 and SAA2. J Immunol. (2002) 169:399–406. doi: 10.4049/jimmunol.169.1.399
249. Zhang XS, Yin YS, Wang J, Battaglia T, Krautkramer K, Li WV, et al. Maternal cecal microbiota transfer rescues early-life antibiotic-induced enhancement of type 1 diabetes in mice. Cell Host Microbe. (2021) 29:1249–65.9. doi: 10.1016/j.chom.2021.06.014
250. Uhlar CM, Grehan S, Steel DM, Steinkasserer A, Whitehead AS. Use of the acute phase serum amyloid A2 (SAA2) gene promoter in the analysis of pro- and anti-inflammatory mediators: differential kinetics of SAA2 promoter induction by IL-1 beta and TNF-alpha compared to IL-6. J Immunol Methods. (1997) 203:123–30. doi: 10.1016/s0022-1759(96)00220-7
251. Migita K, Abiru S, Nakamura M, Komori A, Yoshida Y, Yokoyama T, et al. Lipopolysaccharide signaling induces serum amyloid A (SAA) synthesis in human hepatocytes in vitro. FEBS Lett. (2004) 569:235–9. doi: 10.1016/j.febslet.2004.05.072
252. Edbrooke MR, Foldi J, Cheshire JK, Li F, Faulkes DJ, Woo P. Constitutive and NF-kappa B-like proteins in the regulation of the serum amyloid A gene by interleukin 1. Cytokine. (1991) 3:380–8. doi: 10.1016/1043-4666(91)90041-b
253. Faty A, Ferré P, Commans S. The acute phase protein Serum amyloid A induces lipolysis and inflammation in human adipocytes through distinct pathways. PLoS One. (2012) 7:e34031. doi: 10.1371/journal.pone.0034031
254. Wang YC, Kuo WH, Chen CY, Lin HY, Wu HT, Liu BH, et al. Docosahexaenoic acid regulates serum amyloid A protein to promote lipolysis through down regulation of perilipin. J Nutr Biochem. (2010) 21:317–24. doi: 10.1016/j.jnutbio.2009.01.004
255. Sun W. Analysis of single-cell/nucleus transcriptome data in adipose tissue. Methods Mol Biol. (2022) 2448:291–306. doi: 10.1007/978-1-0716-2087-8_19
256. Deutsch A, Feng D, Pessin JE, Shinoda K. The impact of single-cell genomics on adipose tissue research. Int J Mol Sci. (2020) 21(13):4773. doi: 10.3390/ijms21134773
257. Bäckdahl J, Franzén L, Massier L, Li Q, Jalkanen J, Gao H, et al. Spatial mapping reveals human adipocyte subpopulations with distinct sensitivities to insulin. Cell Metab. (2021) 33:2301. doi: 10.1016/j.cmet.2021.10.012
258. Kotnik P, Fischer-Posovszky P, Wabitsch M. RBP4: a controversial adipokine. Eur J Endocrinol. (2011) 165:703–11. doi: 10.1530/EJE-11-0431
259. Funcke JB, Scherer PE. Beyond adiponectin and leptin: adipose tissue-derived mediators of inter-organ communication. J Lipid Res. (2019) 60:1648–84. doi: 10.1194/jlr.R094060
260. Doyle LM, Wang MZ. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells. (2019) 8(7):727. doi: 10.3390/cells8070727
261. Zhao R, Zhao T, He Z, Cai R, Pang W. Composition, isolation, identification and function of adipose tissue-derived exosomes. Adipocyte. (2021) 10:587–604. doi: 10.1080/21623945.2021.1983242
262. Hartwig S, De Filippo E, Göddeke S, Knebel B, Kotzka J, Al-Hasani H, et al. Exosomal proteins constitute an essential part of the human adipose tissue secretome. Biochim Biophys Acta Proteins Proteom. (2019) 1867:140172. doi: 10.1016/j.bbapap.2018.11.009
263. Lazar I, Clement E, Dauvillier S, Milhas D, Ducoux-Petit M, LeGonidec S, et al. Adipocyte exosomes promote melanoma aggressiveness through fatty acid oxidation: a novel mechanism linking obesity and cancer. Cancer Res. (2016) 76:4051–7. doi: 10.1158/0008-5472.CAN-16-0651
264. Eguchi A, Lazic M, Armando AM, Phillips SA, Katebian R, Maraka S, et al. Circulating adipocyte-derived extracellular vesicles are novel markers of metabolic stress. J Mol Med (Berl). (2016) 94:1241–53. doi: 10.1007/s00109-016-1446-8
265. Dang SY, Leng Y, Wang ZX, Xiao X, Zhang X, Wen T, et al. Exosomal transfer of obesity adipose tissue for decreased miR-141-3p mediate insulin resistance of hepatocytes. Int J Biol Sci. (2019) 15:351–68. doi: 10.7150/ijbs.28522
266. Pardo F, Villalobos-Labra R, Sobrevia B, Toledo F, Sobrevia L. Extracellular vesicles in obesity and diabetes mellitus. Mol Aspects Med. (2018) 60:81–91. doi: 10.1016/j.mam.2017.11.010
267. Kanter JE, Hsu CC, Bornfeldt KE. Monocytes and macrophages as protagonists in vascular complications of diabetes. Front Cardiovasc Med. (2020) 7:10. doi: 10.3389/fcvm.2020.00010
268. Kisilevsky R, Subrahmanyan L. Serum amyloid A changes high density lipoprotein’s cellular affinity. A clue to serum amyloid A’s principal function. Lab Invest. (1992) 66:778–85.1602745
269. Marhaug G, Hackett B, Dowton SB. Serum amyloid A gene expression in rabbit, mink and mouse. Clin Exp Immunol. (1997) 107:425–34. doi: 10.1111/j.1365-2249.1997.287-c31178.x
270. Gutfeld O, Prus D, Ackerman Z, Dishon S, Linke RP, Levin M, et al. Expression of serum amyloid A, in normal, dysplastic, and neoplastic human colonic mucosa: implication for a role in colonic tumorigenesis. J Histochem Cytochem. (2006) 54:63–73. doi: 10.1369/jhc.5A6645.2005
271. Reigstad CS, Lundén GO, Felin J, Bäckhed F. Regulation of serum amyloid A3 (SAA3) in mouse colonic epithelium and adipose tissue by the intestinal microbiota. PLoS One. (2009) 4:e5842. doi: 10.1371/journal.pone.0005842
272. Sano T, Huang W, Hall JA, Yang Y, Chen A, Gavzy SJ, et al. An IL-23R/IL-22 circuit regulates epithelial Serum amyloid A to promote local effector Th17 responses. Cell. (2015) 163:381–93. doi: 10.1016/j.cell.2015.08.061
273. Hardardóttir I, Sipe J, Moser AH, Fielding CJ, Feingold KR, Grünfeld C. LPS And cytokines regulate extra hepatic mRNA levels of apolipoproteins during the acute phase response in Syrian hamsters. Biochim Biophys Acta. (1997) 1344:210–20. doi: 10.1016/s0005-2760(96)00143-9
274. Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, Poon TW, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. (2019) 569:655–62. doi: 10.1038/s41586-019-1237-9
275. Larsson E, Tremaroli V, Lee YS, Koren O, Nookaew I, Fricker A, et al. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut. (2012) 61:1124–31. doi: 10.1136/gutjnl-2011-301104
276. Livanos AE, Greiner TU, Vangay P, Pathmasiri W, Stewart D, McRitchie S, et al. Antibiotic-mediated gut microbiome perturbation accelerates development of type 1 diabetes in mice. Nat Microbiol. (2016) 1:16140. doi: 10.1038/nmicrobiol.2016.140
277. Zhang XS, Li J, Krautkramer KA, Badri M, Battaglia T, Borbet TC, et al. Antibiotic-induced acceleration of type 1 diabetes alters maturation of innate intestinal immunity. Elife. (2018) 7:e37816. doi: 10.7554/eLife.37816
278. Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. (2009) 139:485–98. doi: 10.1016/j.cell.2009.09.033
279. Murdoch CC, Espenschied ST, Matty MA, Mueller O, Tobin DM, Rawls JF. Intestinal Serum amyloid A suppresses systemic neutrophil activation and bactericidal activity in response to microbiota colonization. PLoS Pathog. (2019) 15:e1007381. doi: 10.1371/journal.ppat.1007381
280. Cheng N, Liang Y, Du X, Ye RD. Serum amyloid A promotes LPS clearance and suppresses LPS-induced inflammation and tissue injury. EMBO Rep. (2018) 19(10):e45517. doi: 10.15252/embr.201745517
281. Zhou H, Chen M, Zhang G, Ye RD. Suppression of lipopolysaccharide-induced inflammatory response by fragments from Serum amyloid A. J Immunol. (2017) 199:1105–12. doi: 10.4049/jimmunol.1700470
282. Gershoni M, Pietrokovski S. The landscape of sex-differential transcriptome and its consequent selection in human adults. BMC Biol. (2017) 15:7. doi: 10.1186/s12915-017-0352-z
283. Oliva M, Muñoz-Aguirre M, Kim-Hellmuth S, Wucher V, Gewirtz ADH, Cotter DJ, et al. The impact of sex on gene expression across human tissues. Science. (2020) 369(6509):eaba3066. doi: 10.1126/science.aba3066
284. Lopes-Ramos CM, Chen CY, Kuijjer ML, Paulson JN, Sonawane AR, Fagny M, et al. Sex differences in gene expression and regulatory networks across 29 human tissues. Cell Rep. (2020) 31:107795. doi: 10.1016/j.celrep.2020.107795
285. Hartman RJG, Mokry M, Pasterkamp G, den Ruijter HM. Sex-dependent gene co-expression in the human body. Sci Rep. (2021) 11:18758. doi: 10.1038/s41598-021-98059-9
286. Lamason R, Zhao P, Rawat R, Davis A, Hall JC, Chae JJ, et al. Sexual dimorphism in immune response genes as a function of puberty. BMC Immunol. (2006) 7:2. doi: 10.1186/1471-2172-7-2
287. Jylhävä J, Haarala A, Eklund C, Pertovaara M, Kähönen M, Hutri-Kähönen N, et al. Serum amyloid A is independently associated with metabolic risk factors but not with early atherosclerosis: the cardiovascular risk in young finns study. J Intern Med. (2009) 266:286–95. doi: 10.1111/j.1365-2796.2009.02120.x
288. Lau-Corona D, Suvorov A, Waxman DJ. Feminization of male mouse liver by persistent growth hormone stimulation: activation of sex-biased transcriptional networks and dynamic changes in chromatin states. Mol Cell Biol. (2017) 37(19):e00301–17. doi: 10.1128/MCB.00301-17
289. Bélanger C, Luu-The V, Dupont P, Tchernof A. Adipose tissue intracrinology: potential importance of local androgen/estrogen metabolism in the regulation of adiposity. Horm Metab Res. (2002) 34:737–45. doi: 10.1055/s-2002-38265
290. Masi AT, Rehman AA, Jorgenson LC, Smith JM, Aldag JC. Sexual dimorphisms of adrenal steroids, sex hormones, and immunological biomarkers and possible risk factors for developing rheumatoid arthritis. Int J Endocrinol. (2015) 2015:929246. doi: 10.1155/2015/929246
291. Abbas A, Fadel PJ, Wang Z, Arbique D, Jialal I, Vongpatanasin W. Contrasting effects of oral versus transdermal estrogen on serum amyloid A (SAA) and high-density lipoprotein-SAA in postmenopausal women. Arterioscler Thromb Vasc Biol. (2004) 24:e164–167. doi: 10.1161/01.ATV.0000140198.16664.8e
292. van Rooijen M, Hansson LO, Frostegård J, Silveira A, Hamsten A, Bremme K. Treatment with combined oral contraceptives induces a rise in serum C-reactive protein in the absence of a general inflammatory response. J Thromb Haemost. (2006) 4:77–82. doi: 10.1111/j.1538-7836.2005.01690.x
293. Wakatsuki A, Okatani Y, Ikenoue N, Fukaya T. Effect of medroxyprogesterone acetate on vascular inflammatory markers in postmenopausal women receiving estrogen. Circulation. (2002) 105:1436–9. doi: 10.1161/hc1202.105945
294. Singer K, Maley N, Mergian T, DelProposto J, Cho KW, Zamarron BF, et al. Differences in hematopoietic stem cells contribute to sexually dimorphic inflammatory responses to high fat diet-induced obesity. J Biol Chem. (2015) 290:13250–62. doi: 10.1074/jbc.M114.634568
295. Henes JC, Saur S, Kofler DM, Kedor C, Meisner C, Schuett M, et al. Tocilizumab for the treatment of familial Mediterranean fever-A randomized, double-blind, placebo-controlled phase II study. J Clin Med. (2022) 11(18):5360. doi: 10.3390/jcm11185360
296. Hamanoue S, Suwabe T, Hoshino J, Sumida K, Mise K, Hayami N, et al. Successful treatment with humanized anti-interleukin-6 receptor antibody (tocilizumab) in a case of AA amyloidosis complicated by familial Mediterranean fever. Mod Rheumatol. (2016) 26:610–3. doi: 10.3109/14397595.2014.908810
297. Inui K, Sawa N, Suwabe T, Mizuno H, Yamanouchi M, Hiramatsu R, et al. Long term administration of tocilizumab improves renal amyloid A (AA) amyloidosis deposition in Familial Mediterranean fever. Mod Rheumatol Case Rep. (2020) 4:310–1. doi: 10.1080/24725625.2020.1739193
298. Yamada Y, Ueno T, Irifuku T, Nakashima A, Doi S, Ichinohe T, et al. Tocilizumab histologically improved AA renal amyloidosis in a patient with multicentric castleman disease: a case report. Clin Nephrol. (2018) 90:232–6. doi: 10.5414/CN109273
299. Okuda Y, Takasugi K. Successful use of a humanized anti-interleukin-6 receptor antibody, tocilizumab, to treat amyloid A amyloidosis complicating juvenile idiopathic arthritis. Arthritis Rheum. (2006) 54:2997–3000. doi: 10.1002/art.22118
300. Siligato R, Gembillo G, Calabrese V, Conti G, Santoro D. Amyloidosis and glomerular diseases in familial Mediterranean fever. Medicina (Kaunas). (2021) 57(10):1049. doi: 10.3390/medicina57101049
301. Jena M, Tripathy A, Mishra A, Maiti R. Effect of canakinumab on clinical and biochemical parameters in acute gouty arthritis: a meta-analysis. Inflammopharmacology. (2021) 29:35–47. doi: 10.1007/s10787-020-00753-z
302. Almusalami EM, Lockett A, Ferro A, Posner J. Serum amyloid A-A potential therapeutic target for hyper-inflammatory syndrome associated with COVID-19. Front Med (Lausanne). (2023) 10:1135695. doi: 10.3389/fmed.2023.1135695
303. Ancsin JB, Kisilevsky R. The heparin/heparan sulfate-binding site on apo-serum amyloid A. Implications for the therapeutic intervention of amyloidosis. J Biol Chem. (1999) 274:7172–81. doi: 10.1074/jbc.274.11.7172
304. Tam SP, Ancsin JB, Tan R, Kisilevsky R. Peptides derived from serum amyloid A prevent, and reverse, aortic lipid lesions in apoE-/- mice. J Lipid Res. (2005) 46:2091–101. doi: 10.1194/jlr.M500191-JLR200
Keywords: obesity, diabetes, cardiovascular disease, SAA, intestine, liver, adipocytes, macrophages
Citation: den Hartigh LJ, May KS, Zhang XS, Chait A and Blaser MJ (2023) Serum amyloid A and metabolic disease: evidence for a critical role in chronic inflammatory conditions. Front. Cardiovasc. Med. 10:1197432. doi: 10.3389/fcvm.2023.1197432
Received: 31 March 2023; Accepted: 15 May 2023;
Published: 15 June 2023.
Edited by:
Catherine A. Reardon, The University of Chicago, United StatesReviewed by:
George Sack, Johns Hopkins Medicine, United StatesXuchu Que, University of California, San Diego, United States
© 2023 den Hartigh, May, Zhang, Chait and Blaser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura J. den Hartigh bGF1cmFkaEB1Lndhc2hpbmd0b24uZWR1