Abstract
Introduction:
Mitochondria are central energy generators for the heart, producing adenosine triphosphate (ATP) through the oxidative phosphorylation (OXPHOS) system. However, mitochondria also guide critical cell decisions and responses to the environmental stressors.
Methods:
This study evaluated whether prolonged electromagnetic stress affects the mitochondrial OXPHOS system and structural modifications of the myocardium. To induce prolonged electromagnetic stress, mice were exposed to 915 MHz electromagnetic fields (EMFs) for 28 days.
Results:
Analysis of mitochondrial OXPHOS capacity in EMF-exposed mice pointed to a significant increase in cardiac protein expression of the Complex I, II, III and IV subunits, while expression level of α-subunit of ATP synthase (Complex V) was stable among groups. Furthermore, measurement of respiratory function in isolated cardiac mitochondria using the Seahorse XF24 analyzer demonstrated that prolonged electromagnetic stress modifies the mitochondrial respiratory capacity. However, the plasma level of malondialdehyde, an indicator of oxidative stress, and myocardial expression of mitochondria-resident antioxidant enzyme superoxide dismutase 2 remained unchanged in EMF-exposed mice as compared to controls. At the structural and functional state of left ventricles, no abnormalities were identified in the heart of mice subjected to electromagnetic stress.
Discussion:
Taken together, these data suggest that prolonged exposure to EMFs could affect mitochondrial oxidative metabolism through modulating cardiac OXPHOS system.
1. Introduction
Mitochondria are exuberantly present in cardiac muscle and serve as the capital source of energy production. In cardiac cells, the primary function of the mitochondria is to federate the high energy requirement of the beating heart by generating energy in the form of adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS) machinery (1, 2). The OXPHOS system requires an orchestrated transfer of electrons via five multi-subunit enzymes: Complex I (NADH dehydrogenase or NADH: ubiquinone oxidoreductase), Complex II (succinate dehydrogenase or succinate:ubiquinone oxidoreductase), Complex III (the bc1 complex or ubiquinone: cytochrome c oxidoreductase), Complex IV (cytochrome c oxidase, cyclooxygenase or reduced cytochrome c: oxygen oxidoreductase), and Complex V (ATP synthase), which are localized on the inner mitochondrial membrane (3–5). Furthermore, OXPHOS machinery also comprises two electron transport carriers, ubiquinone or coenzyme Q10 and cytochrome c (6, 7). In the course of electron transport, Complexes I, III, and IV affiliate protons from the mitochondrial matrix to the intermembrane space culminating in enhanced membrane potential (8). In the presence of adenosine diphosphate (ADP), Complex V favors flow of protons to the matrix culminating in ATP formation (3, 4). Mitochondrial OXPHOS complexes are not only responsible for energy generation but also connected to the multifaceted cell events, including reactive oxygen species (ROS) formation, inflammation, and apoptosis (9, 10). Therefore, the defects in the OXPHOS system, originating from genetic or environmental factors, have been reported in multiple diseases including heart failure, arrhythmia, and hypertension (10).
Electromagnetic fields (EMFs) depict one of the most prevalent growing environmental counterinfluences on biological systems. The exceeding common exposure to EMFs occurs from telecommunications networks, public broadcast infrastructure, wireless technologies such as Wi-Fi, and the operation of mobile devices. Several sources of EMFs are also used in medical and industrial applications and security scanning equipment. Therefore, exposure to EMFs, particularly those emitted by mobile communications transmitting at 900 MHz, remain a much controversial topic, which is related to their potential effects on human health. Both elevated cardiovascular risk and marked cardioprotection have been attributed to EMF exposure (11, 12). It has been reported that these effects are mediated by mitochondrial ROS, which are involved in vital cell decisions and can be capital or excessively toxic to cellular homeostasis (13). To alleviate ROS-mediated cellular damage, living organisms have anti-oxidative mechanisms including superoxide dismutase (SOD), glutathione peroxidase, and catalase (14). Previous research has mainly concerned the short-term biological effects of EMFs on oxidative stress and antioxidant mechanisms; however, long-term impact of EMFs has been a relatively minor focus.
Considering the central place of mitochondrial metabolism in the acquisition and maintenance of cardiac cell integrity, in this study, we examined the prolonged exposure to 915 MHz EMFs on mitochondrial respiratory function and oxidative stress status in relation to structural integrity of the myocardium.
2. Materials and methods
2.1. Animal studies
Animal experiments were performed in accordance with the guidelines established by the European Communities Council Directive (2010/63/EU Council Directive Decree) and approved by the local ethical committee (project N 20/1048/14/02, national agreement 2020072717253128). Adult Swiss Webster male mice 9 months old were used for these investigations (Envigo RMS, France). Animals were given free access to standard food and water under controlled conditions. Two days prior to experiments, mice were adapted to new environmental conditions (light was on a 12 h light–12 h dark cycle) as previously described (15). Mobile communication technology operates at 900 MHz frequency; thus, we focus on the effects of frequency close to 900 MHz. Mice were randomly divided into two groups: control sham (n = 7) and EMFs (n = 8) groups, respectively. The mice from EMF group were exposed to a 915 MHz EMFs for 28 days (9 h/day). Control groups of animals did not exposure of EMF. Animals were sacrificed and hearts were removed and rinsed in 4°C phosphate-buffered saline (PBS). Heart weight and body weight were measured the day of sacrifice. Heart weight to body weight ratio in grams was calculated by dividing the weight of the heart by the weight of the whole animal. Harvested tissues were divided into three parts: (1) embedded into optimal cutting temperature compound (OCT) (Sigma-Aldrich, Saint-Quentin-Fallavier, France) under ice-cold 2-methylbutane for cryo-section, (2) for Western blot, and (3) the remaining portion was used for quantitative polymerase chain reaction (qRT-PCR).
2.2. Radio frequency approach to an in vivo model
To induce electromagnetic stress, in vivo experiments were performed in a Giga-TEM (GTEM) cell. For animal adaptation, 24 h prior to tests mice were placed in the GTEM cell. A solid-state radiofrequency generator with a fixed frequency of 915 MHz (WSPS-915–1000) (Chengdu Wattsine Electronics Technology, Chengdu, Sichuan, China) was used for in vivo experiments. The Webster was modeled by the sphere of equivalent volume in these simulations. Three spheres were implanted along the wave propagation axis of the GTEM cell. The relative permittivity value was 55, the conductivity of 3 S/m were applied for the experimental protocol. The estimated specific absorption rate (SAR) values were around 40 W/kg. Using MATLAB codes, the calculation of whole body SAR was taken by volume integration of absorption power as previously described (15, 16). In radio frequency (RF) testing, the input power was 4 W in the high frequency structure simulator (HFSS) simulation and in the experimentation, as previously described (15, 16).
2.3. Western blot
Extraction of proteins from cardiac tissues was performed as previously described (15, 17). Briefly, for protein extraction, the RIPA buffer (50 mM Tris–HCl, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100, 1 mM EDTA, completed with protease and phosphatase inhibitor cocktails) was used. To determine the proteins concentration of the lysate, we used the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA, United States). Equivalent protein amounts were loaded in each lane of gels. Proteins were loaded in the Laemmli sample buffer, denaturated, and were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting. Furthermore, the proteins were separated by electrophoresis and transferred to a nitrocellulose membrane (Amersham Protran, GE Healthcare, Germany) using electroblotting apparatus (Bio-Rad, Hercules, CA, United States). Then, membranes were incubated with 5% bovine serum albumin (BSA) in Tris-buffered saline tween-20 buffer (TBST; 25 mM Tris, pH 7.5, 150 mM NaCl, and 0.1% Tween20) for 1 h to prevent nonspecific binding sites and incubated overnight at 4°C with the primary antibodies. In final steps, immunoreactive bands were detected by chemiluminescence with the Clarity Western ECL Substrate (Bio-Rad, Hercules, CA, United States) using the ChemiDoc MP Acquisition system (Bio-Rad, Hercules, CA, United States). The antibodies used in this study were Anti-SOD2 (ab137037), MitoProfile® Total OXPHOS Rodent WB Antibody Cocktail (ab110413) and Anti-β-Actin (sc-47778). For quantification, β-Actin was used as a loading control. The intensity of the individual bands was evaluated by ImageJ and normalized to the corresponding input control bands.
2.4. Oxygen consumption rate measurement
To determine mitochondrial respiratory capacity, we measured the oxygen consumption rate (OCR) using the Seahorse XF24 extracellular flux analyzer. We measured OCR following a previously established protocol designed for analyzing mitochondrial oxidative function in frozen tissues (18). Cardiac tissues were homogenized in a cold MAS buffer containing 70 mM sucrose, 220 mM mannitol, 5 mM KH2PO4, 5 mM MgCl2, 1 mM EGTA, and 2 mM HEPES, pH 7.4. The homogenates were centrifuged at 1,000 × g for 5 min at 4°C, and the supernatants were collected. The supernatants were further processed for mitochondrial isolation following the protocol by Osto et al. (18). Protein concentration was then determined using the Pierce BCA protein assay kit (Thermo Fisher Scientific). The samples containing 40 μg of total protein were loaded to each well of a Seahorse XF24 cell culture plate (Agilent).
2.5. Malondialdehyde determination
Blood samples were centrifuged and the plasma was isolated, aliquoted, and stored at −80°C until further analysis. Plasma level of malondialdehyde (MDA) was measured using the thiobarbituric acid-based procedure described by Wasowicz et al. (19). Briefly, 100 μl of plasma are added to the solution containing 29 mmol/L thiobarbituric acid in 8.75 mol/L acetic acid. Samples are heated for 60 min at 95°C and cooled down. The level of MDA was measured spectrophotometrically at 535 nm and expressed as µM concentrations using calibrating curves.
2.6. Morphology
The mice were sacrificed after 28 days of experimental protocol. Briefly, picrosirius red staining of cardiac cryosections (10 µm thick) was performed according to the standard protocol to visualize interstitial collagen. In stains for collagen content, we used a 0.1% solution of Sirius red in saturated aqueous solution of picric acid for 1 h at room temperature, followed by quickly washing in two changes of acidified water (0.5% acetic acid in water). Then, slides were dehydrated in ascending concentrations of ethanol and cleared in two stages in xylene. The slides were examined using light microscope equipped with a camera. The quantification of cardiac fibrosis was performed using ImageJ software.
2.7. Quantitative RT–PCR analysis
The expression level of genes was assessed using qRT-PCR. The total RNAs were isolated from cardiac muscle using RNeasy mini kit (Qiagen, Hilden, Germany). Total RNAs (300 ng) were reverse transcribed as previously described using Superscript II reverse transcriptase (Invitrogen, Waltham, MA, United States) (15). The sequences of the primers used are as follow and given in the 5′-3′ orientation: SOD2: GGACAAACCTGAGCCCTAAG (forward) and CAAAAGACCCAAAGTCACGC (reverse); Collagen type I: TGTGTGCGATGACGTGCAAT (forward) and GGGTCCCTCGACTCCTACA (reverse); Collagen type III: AAGGCGAATTCAAGGCTGAA (forward) and TGTGTTTAGTACAGCCATCCTCTAGAA (reverse); transforming growth factor beta-1 (TGFβ-1): GAGCCCGAAGCGGACTACTA (forward) and CACTGCTTCCCGAATGTCTGA (reverse); atrial natriuretic peptide (ANP): AGAGTGGGCAGAGACAGCAAA (forward) and AAGGCCAAGACGAGGAAGAAG (reverse); brain natriuretic peptide (BNP): GCACAAGATAGACCGGATCG (forward) and CCCAGGCAGAGTCAGAAAC (reverse); hypoxanthine phosphorybosyl transferase (HPRT): TGAAAGACTTGCTCGAGATGTCAT (forward) and TCCAGCAGGTCAGCAAAGAA (reverse). The validated PCR parameters were 5 min at 95°C followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. The relative amount of target mRNA was calculated by the 2-ΔΔCt method and expression of target gene was normalized to HPRT housekeeping gene expression.
2.8. Statistical analysis
Data were expressed as mean ± standard error of the mean (SEM). Statistical analysis between experimental groups was performed using unpaired Student’s t-test (GraphPad Prism version 9.3.1). A P-value of <0.05 was considered statistically significant.
3. Results
Defects in the mitochondrial OXPHOS system, a terminal biochemical pathway in energy production, result in deleterious, frequently multisystem disorders (3, 7). To determine changes in mitochondrial energy metabolism in conditions of prolonged electromagnetic stress, we examined the effects of EMFs on the expression level of OXPHOS proteins after 28 days. Total protein extracts from heart tissues of mice subjected to 915 MHz frequency EMFs were subjected to immunoblotting with the Total OXPHOS Rodent Antibody Cocktail kit to detect all five complexes simultaneously (Figure 1). The densitometric quantification of the bands from immunoblots demonstrated a significant increase in the expression level of Complex I and II subunits in EMF-exposed mice compared to control animals. Moreover, myocardial expression of Complex III and Complex IV subunits increased significantly in mice subjected to electromagnetic stress (Figure 1). However, the cardiac level of the α-subunit of ATP synthase (Complex V) remained stable among groups (Figure 1).
Figure 1
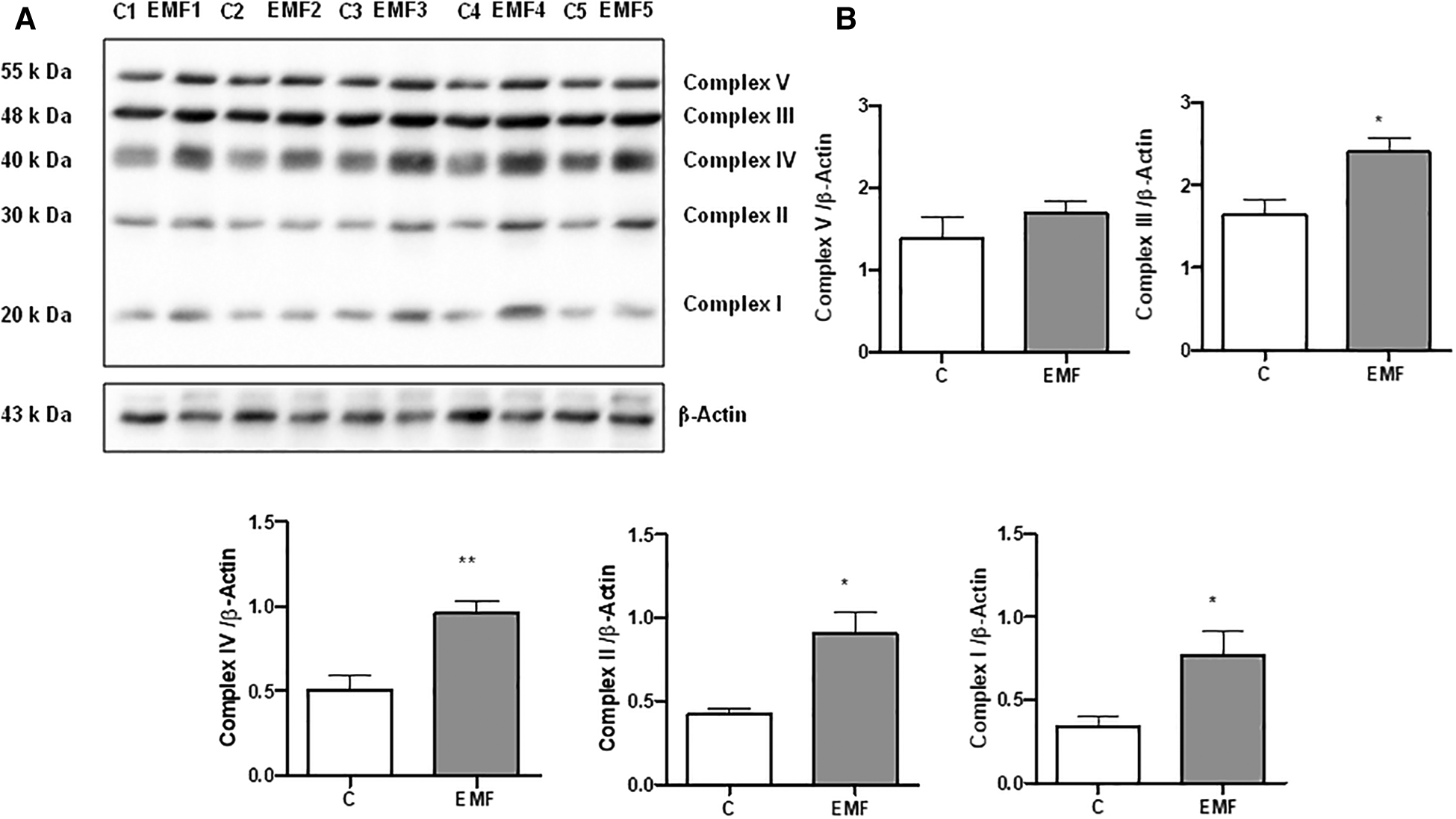
Myocardial expression of MitoProfile Total OXPHOS in mice exposed to 915 MHz EMFs for 28 days. (A) Representative Western blot image and (B) quantification of MitoProfile Total OXPHOS protein expression levels in control and EMF-exposed mice. The results present the mean ± SEM. *P < 0.05 vs. control, **P < 0.01 vs. control. EMFs, electromagnetic fields; OXPHOS, oxidative phosphorylation; SEM, standard error of the mean.
To determine if the observed modifications in protein expression translate into altered mitochondrial function, we next examined mitochondrial respiration in isolated mitochondria obtained from control and EMF-exposed mouse hearts using the Agilent Seahorse XF Cell Mito Stress test. As shown in Figure 2, no significant changes in mitochondrial basal respiration were noted between control and EMF-exposed groups; however, mitochondrial maximal respiration and the spare respiratory capacity in EMF-challenged mitochondria were significantly elevated compared to the control group. Because oxygen consumption can be non-mitochondrial, respiration specific to the cellular non-mitochondrial component was assessed by inhibiting mitochondrial Complex I and Complex III with rotenone and antimycin A. As shown in Figure 2, the electromagnetic stress also significantly increased the non-mitochondrial oxygen consumption compared to the control (Figure 2). However, ATP production and coupling efficiency were unchanged between control and EMF-challenged mice (Figure 2). Representative plot showing OCRs of cardiac mitochondria isolated from control and EMF-exposed mice is presented in Supplementary Figure S1.
Figure 2
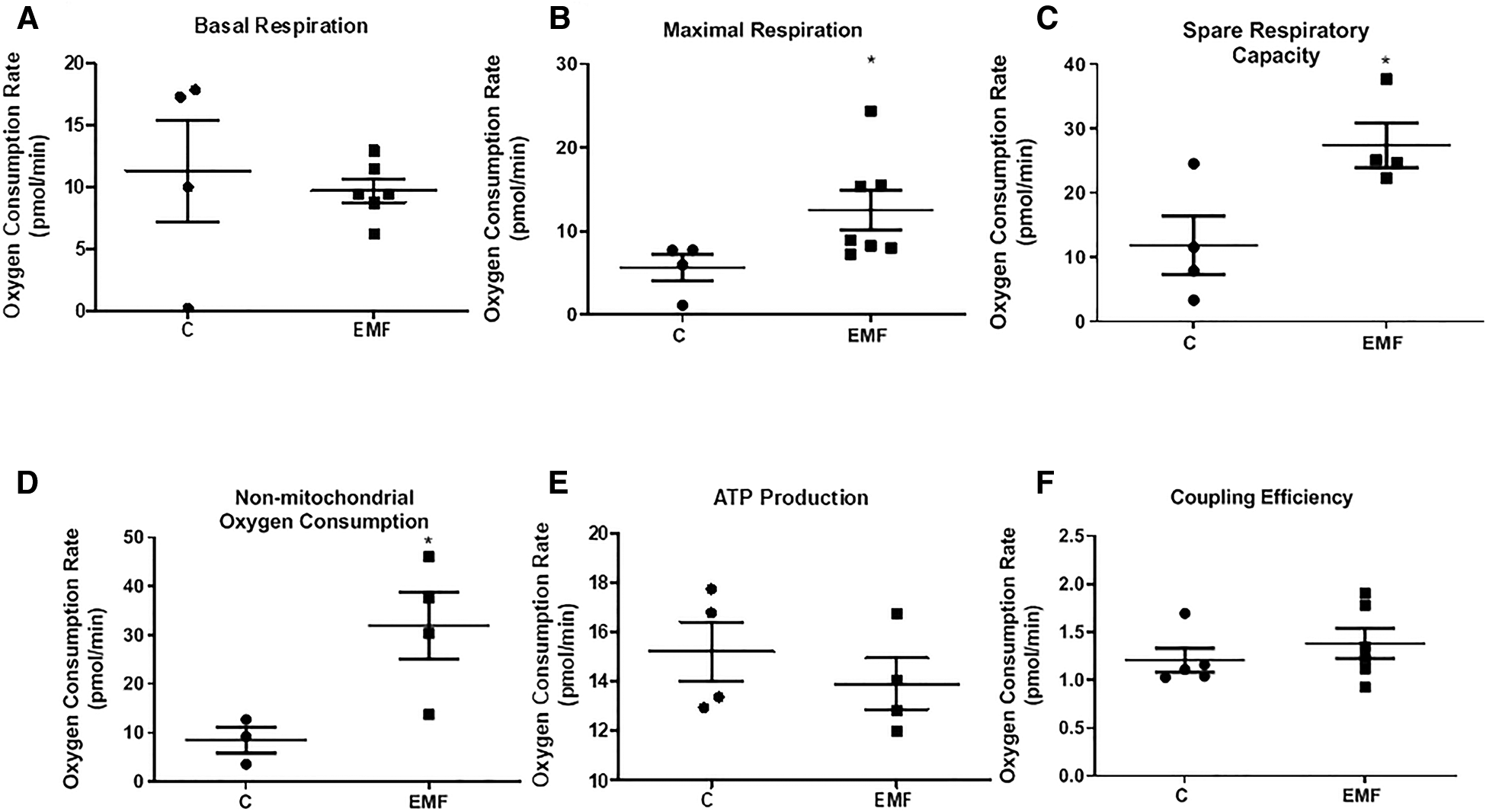
Mitochondrial respiratory function in mice exposed to 915 MHz EMFs for 28 days. (A) Basal respiration, (B) maximal respiration, (C) spare respiratory capacity, (D) non-mitochondrial oxygen consumption, (E) ATP production, and (F) coupling efficiency in control and EMF-exposed mice. The results present the mean ± SEM. *P < 0.05 vs. control. EMFs, electromagnetic fields; ATP, adenosine triphosphate; SEM, standard error of the mean.
To elucidate whether the elevated state respiration in EMF-challenged cardiac tissue was associated with the alterations of antioxidant system, we assessed gene and protein expressions of SOD2, a key component of the mitochondrial antioxidant defense. As shown in Figures 3A–C, myocardial expression levels of mRNA and SOD2 protein expression were unchanged between control and EMF-exposed mice.
Figure 3
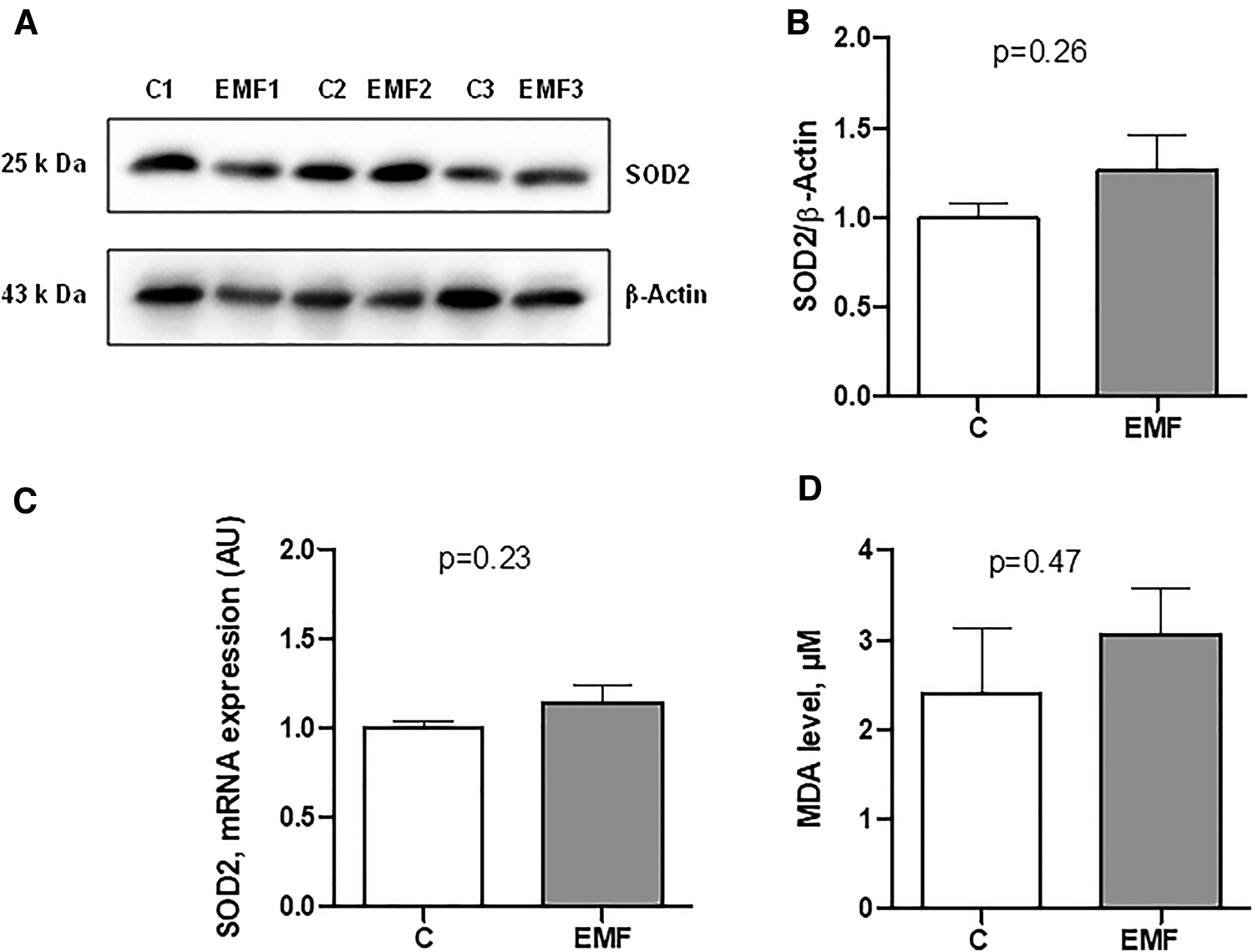
Myocardial expression of SOD2 in mice exposed to 915 MHz EMFs for 28 days. (A) Representative Western blot image, (B) quantification of SOD2 protein expression levels, (C) qRT-PCR analysis of SOD2 mRNA expression level and (D) plasma MDA level in control and EMF-exposed mice. The results present the mean ± SEM. EMFs, electromagnetic fields; SOD2, superoxide dismutase 2; qRT-PCR, quantitative polymerase chain reaction; MDA, malondialdehyde; SEM, standard error of the mean.
We next measured the plasma level of MDA, a lipid peroxidation product that is used for the evaluation of oxidative stress (20, 21). As shown in Figure 3D, no statistical difference was observed in terms of the MDA levels in EMF-exposed mice compared to the controls.
Mitochondrial metabolism and structural tissue integrity are critical links in response to stressors. We next examined the total content of collagen, the most abundant component of extracellular matrix (ECM) in the heart. As shown in Figure 4, no statistically significant differences in the total collagen level were observed in the cardiac tissue between the control and EMF-exposed groups.
Figure 4
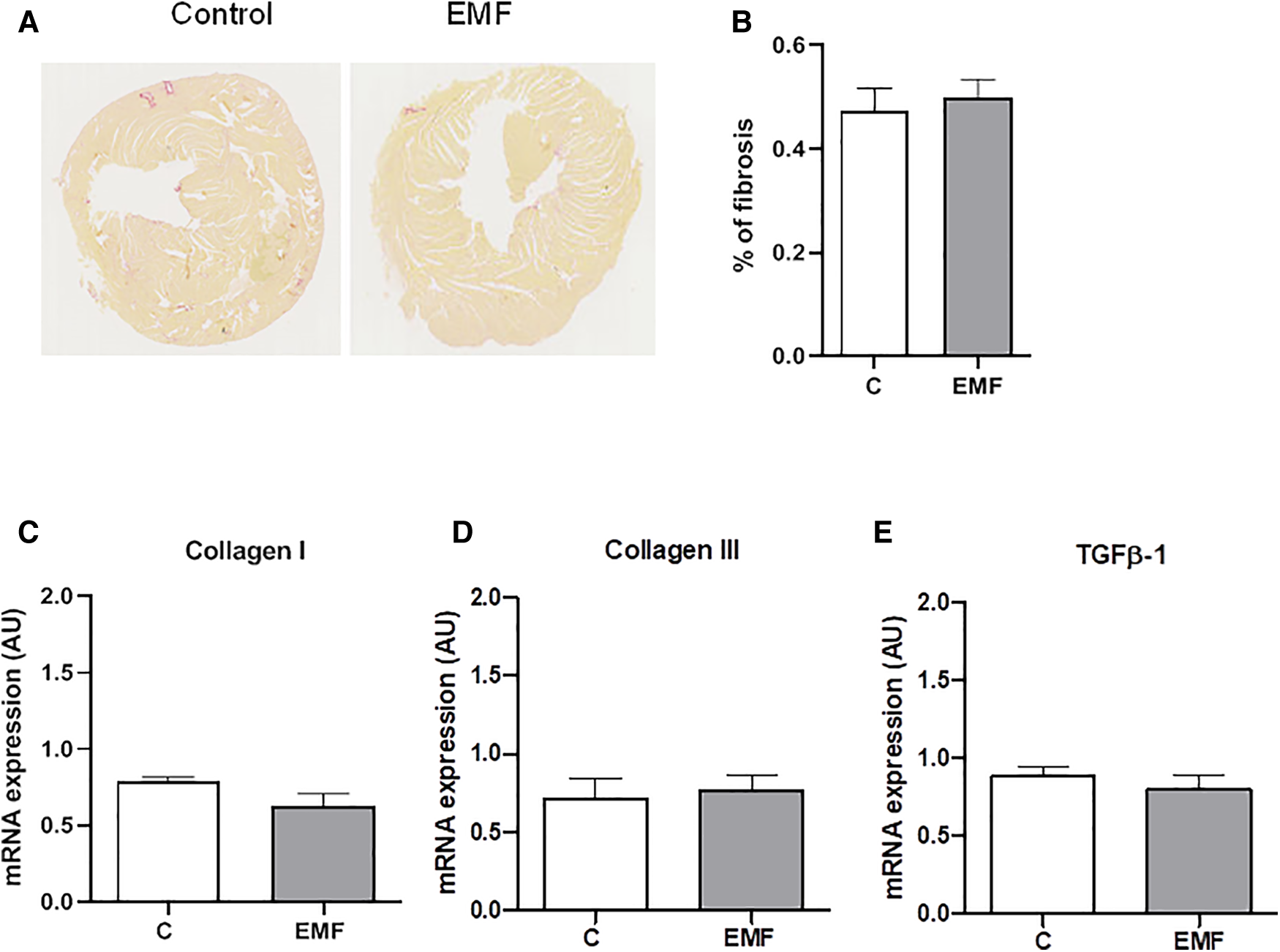
Prolonged effect of EMFs on cardiac collagen content in mice subjected to 915 MHz EMFs for 28 days. (A) Representative image of Sirius red-stained cardiac sections, (B) quantification of (A), (C) qRT-PCR analysis of mRNA expression levels of Collagen type I, (D) Collagen type III, and (E) TGFβ-1. The results present the mean ± SEM. EMFs, electromagnetic fields; qRT-PCR, quantitative polymerase chain reaction; TGFβ-1, transforming growth factor beta-1; SEM, standard error of the mean.
To confirm the histomorphological observation, the gene expression levels of Collagen I and Collagen III, the major structural components of the myocardial ECM, were examined via qRT-PCR. As shown in Figure 4, mRNA expression of Collagen types I and III were not significantly different between the control and EMF-exposed groups. We also found that mRNA expression of TGFβ-1, a cardiac fibrotic factor, was comparable among control and EMF-challenged mice.
To examine whether EMFs could cause the abnormalities of left ventricular function and hypertrophy in mice, we next measured the level of ANP and BNP. As shown in Figures 5A,B, no statistically significant differences in ANP and BNP levels were observed in the cardiac tissue between the control and EMF-challenged groups. In addition, the average heart weight/body weight ratio was not significantly different between control and EMF-exposed mice (Figure 5C).
Figure 5
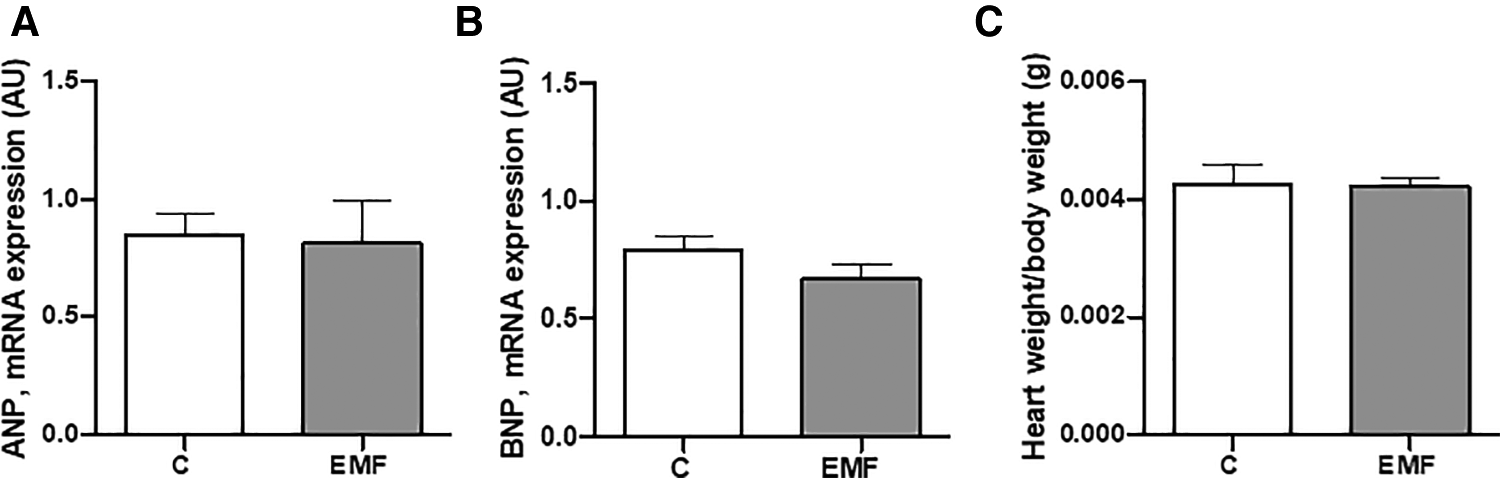
The effects of prolonged exposure to EMFs on structural integrity of cardiac tissue. qRT-PCR analysis of mRNA expression level of ANP (A) and BNP (B), and (C) ratio of heart weight to body weight in control group and in mice subjected to 915 MHz EMFs for 28 days. The results present the mean ± SEM. EMFs, electromagnetic fields; qRT-PCR, quantitative polymerase chain reaction; ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; SEM, standard error of the mean.
4. Discussion
Mitochondrial metabolic reprogramming is a distinctive characteristic of cardiac cells and occurs as a consequence of adaptation or maladaptation to the environmental stress (22, 23). In a healthy heart, mitochondrial oxidative metabolism provides ATP and precursors for macromolecular biosynthesis to meet the energy requirements for cardiac cell survival (24, 25). The predominant mechanisms of cardiac damage are the defects in OXPHOS capacity and oxidative stress. Metabolic reprogramming enables cardiac cells to adapt and orchestrate oxidative stress responses (26–28). The precise picture of how electromagnetic stress calibrates metabolic changes in cardiac tissue via OXPHOS system is obscure. Here, we demonstrate for the first time that prolonged exposure to 915 MHz EMFs for 28 days affects the respiratory capacity of mitochondria without compromising the structural integrity of the heart. These findings provide new insights into the essential role of mitochondrial respiration in cardiac adaptation to prolonged electromagnetic stress.
There is a long-running debate on whether EMFs impact mitochondrial bioenergetics in living systems. In cardiac tissue, mitochondrial oxidative metabolism is the principal source of energy, which underscores the essential role of mitochondria in heart’s performance (26). Mitochondria compose 30% of cardiomyocyte volume and supply cardiac energy status through OXPHOS machinery (29). A healthy heart is metabolically flexible, as it is able to shift between OXPHOS components to provide adequate ATP generation in response to stress (30–32). However, defects in the OXPHOS system can modify metabolic homeostasis in cardiac cells and contribute to heart failure (33–35). In the present study, analysis of the mitochondrial OXPHOS system in EMF-challenged hearts after 28 days pointed to increased protein level expression of Complex I, the largest component of the respiratory chain. Mitochondrial Complex I subunit is capital for maintaining the functional integrity of the respiratory complexes and to provide efficient transfer of electrons between electron transport chain (ETC) complexes. In a failing heart, mitochondrial respiratory Complex I is highly sensitive to structural and functional damage, which can contribute to postischemic mitochondrial ETC disorders, decline in respiratory function, and energy supply deficits (36). We also demonstrated that cardiac Complex II and III were upregulated in the group of mice subjected to EMFs for 28 days. Large quantities of evidence state the respiratory Complex II subunit as a fondement and calibrator of mitochondrial ROS. Both functional loss of Complex II and pharmacological inhibition can result in ROS generation in cardiac cells, with a relevant impact on the development of pathophysiological manifestations. Respiratory Complexes I, II, and III are the capital sites for ROS generation (37). Mitochondrial Complex I inhibition by rotenone can trigger ROS generation in submitochondrial modifications. Several studies reported that oxidation of either Complex I or Complex II substrates in the presence of Complex III suppression with antimycin A favors ROS formation (38, 39). In myocardial infarction, rotenone reduces ROS overproduction and limits mitochondrial damage in the isolated rat heart (38). Recent evidence points to the respiratory Complex II as a source and calibrator of mitochondrial ROS. Both pharmacological inhibition and functional loss of Complex II can lead to ROS formation in cardiac cells, with a relevant impact on the development of pathophysiological conditions including heart disease (37, 40). In the cardiovascular system, Complex II can contribute notably to ROS production both directly and indirectly (via reverse electron transfer), with physiological and pathological impacts (37, 41). Abnormal mitochondrial ROS generation is involved in cardiac ischemia-reperfusion (I/R) injury, and Complex II inhibitors exert protective effects in I/R models by suppressing reverse electron transport (RET) (42, 43). We found that in mice exposed to EMFs for 28 days, the cardiac expression level of Complex IV was upregulated, while Complex V level expression remained relatively stable. In OXPHOS machinery, Complex IV catalyzes the mitochondrial ETC and transformation of oxygen to water, linked to proton translocation (44–46). Recent studies suggest that mitochondrial Complex IV dysfunction drives cell adaptive signaling and metabolic perturbations (47). In cancer cells, dysregulation of mitochondrial Complex IV may stimulate the metastatic potential and reflect the positive correlation to metastatic spread of cancer (47–49). Our findings suggest that prolonged exposure to EMFs alters mitochondrial oxidative metabolism through modulating the OXPHOS system in the heart. Furthermore, using the Seahorse Extracellular Flux Analyzer, we examined mitochondrial respiration in isolated mitochondria obtained from control and EMF-exposed mouse hearts. We found that prolonged electromagnetic stress increases mitochondrial maximal respiration and spare respiratory capacity. In addition, we demonstrated the electromagnetic stress also significantly enhances non-mitochondrial oxygen consumption.
The increased mitochondrial respiration in conditions of electromagnetic stress may be an adaptive response on the cardiac level, illustrating a fundamental link between environmental influence and myocardial energy metabolism. Interestingly, cardiac adoption of metabolic upregulation of Complexes I, II, III, and IV was uncoupled to cardiac expression of SOD2, a first-line component of mitochondrial antioxidant defense against superoxide produced by respiration. Indeed, we did not observe significant differences in SOD2 expression among control and EMF-exposed mice for 28 days. However, further studies are needed to evaluate the total myocardial antioxidant potential in cardiac tissue under prolonged electromagnetic stress.
In addition, we evaluated the effect of prolonged exposition to EMFs on the plasma MDA level, one of known indices of oxidative stress. Free oxygen radicals enhance lipid peroxidation, and MDA is a lipid peroxidation product that is used for the evaluation of oxidative stress (50, 51). In our experiments, no significant difference was found between plasma MDA level in EMF-exposed and control groups suggesting that prolonged exposure to electromagnetic stress does not alter lipid peroxidation.
To address the structural and functional state of left ventricles, we evaluated the myocardial level of ANP and BNP in mice subjected to prolonged electromagnetic stress. Currently, ANP and BNP are widely used as significant indicators for the cardiac dysfunction and clinical diagnosis of heart failure in clinical medicine (52, 53). Our findings point to a constant level of ANP and BNP in EMF-challenged hearts isolated from mice subjected to EMFs for 28 days, suggesting that prolonged electromagnetic stress does not induce cardiac abnormalities. We have recently demonstrated that short-term exposure to EMF for 48 and 72 h in cardiac tissues did not alter the cardiac structural integrity in terms of collagen deposition, necrotic myofibers, and cardiomyocyte size (15). In the present study, we also evaluated the cardiac collagen level, the main structural element forming the ECM. Analysis of cardiac sections from EMF-exposed mice exposed for 28 days demonstrated that collagen content was not affected suggesting that prolonged electromagnetic stress did not induce cardiac fibrosis. Several studies reported abnormalities in collagen synthesis and deposition under electromagnetic stress (54, 55). Soda et al. demonstrated that exposure to low-frequency EMFs (3 mT, 60 Hz) increases the collagen synthesis in mouse osteoblasts—like MC3T3-E1 cells (55). A previous study has demonstrated that EMFs can accelerate collagen production in bone and cartilage due to effects of K, Ca, and Mg ion transport (54). In conditions of myofibroblast activation linked to the diabetic wound healing, Choi et al. reported elevated collagen fibers in response to electromagnetic stress (56). These observations clearly suggest a wide variability of EMF-related effects on the structural components of ECM. Further research into the mechanisms that regulate ECM integrity in cardiac tissue under electromagnetic environment is required.
5. Conclusion
The ability of cardiac cells to adjust the mitochondrial oxidative metabolism in response to environmental stress is a well-recognized signature of the cardiovascular system. Persistent metabolic changes may result in tissue abnormalities or may trigger cell maladaptive responses. Our study points at notable differences between control and EMF-challenged mice hearts at the level of mitochondrial respiration after prolonged EMF exposure. As the mouse heart imitates the mammalian cardiovascular phenotype, our data suggest that metabolic oxidative status changes dynamically in response to EMFs in the myocardium, challenging our quest to better understand cardiac cell biology under conditions of electromagnetic environment.
Statements
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by project N 20/1048/14/02, national agreement 2020072717253128.
Author contributions
Conceptualization, methodology, and resources: OK and JT. Software: JT and OK. Validation: IM and LS. Formal analysis and writing—original draft preparation: OK and LS. Investigation: LS, IM, and DM. Data curation, supervision, project administration, and funding acquisition: OK. Writing—review and editing: OK, LS, IM, and VZ. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by ANSES (grant number EST-2016 RF-01) and by INSERM.
Acknowledgments
We are grateful to Angelo Parini for administrative support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2023.1205893/full#supplementary-material
References
1.
TahrirFGLangfordDAminiSMohseni AhooyiTKhaliliK. Mitochondrial quality control in cardiac cells: mechanisms and role in cardiac cell injury and disease. J Cell Physiol. (2019) 234(6):8122–33. 10.1002/jcp.27597
2.
Vakifahmetoglu-NorbergHOuchidaATNorbergE. The role of mitochondria in metabolism and cell death. Biochem Biophys Res Commun. (2017) 482(3):426–31. 10.1016/j.bbrc.2016.11.088
3.
SharmaLKLuJBaiY. Mitochondrial respiratory complex I: structure, function and implication in human diseases. Curr Med Chem. (2009) 16(10):1266–77. 10.2174/092986709787846578
4.
Fernandez-VizarraEZevianiM. Mitochondrial disorders of the OXPHOS system. FEBS Lett. (2021) 595(8):1062–106. 10.1002/1873-3468.13995
5.
YinMO’NeillLAJ. The role of the electron transport chain in immunity. FASEB J. (2021) 35(12):e21974. 10.1096/fj.202101161R
6.
XuYXueD3rdBankheadANeamatiN. Why all the fuss about oxidative phosphorylation (OXPHOS)?J Med Chem. (2020) 63(23):14276–307. 10.1021/acs.jmedchem.0c01013
7.
Van den HeuvelLSmeitinkJ. The oxidative phosphorylation (OXPHOS) system: nuclear genes and human genetic diseases. Bioessays. (2001) 23(6):518–25. 10.1002/bies.1071
8.
KotrasováVKeresztesováBOndrovičováGBauerJAHavalováHPevalaVet alMitochondrial kinases and the role of mitochondrial protein phosphorylation in health and disease. Life (Basel). (2021) 11(2):82. 10.3390/life11020082
9.
CalbetJALMartín-RodríguezSMartin-RinconMMorales-AlamoD. An integrative approach to the regulation of mitochondrial respiration during exercise: focus on high-intensity exercise. Redox Biol. (2020) 35:101478. 10.1016/j.redox.2020.101478
10.
SchottlenderNGottfriedIAsheryU. Hyperbaric oxygen treatment: effects on mitochondrial function and oxidative stress. Biomolecules. (2021) 11(12):1827. 10.3390/biom11121827
11.
BiałyDWawrzyńskaMBil-LulaIKrzywonos-ZawadzkaASapa-WojciechowskaAArkowskiJet alLow frequency electromagnetic field decreases ischemia-reperfusion injury of human cardiomyocytes and supports their metabolic function. Exp Biol Med (Maywood). (2018) 243(10):809–16. 10.1177/1535370218779773
12.
SavitzDALiaoDSastreAKlecknerRCKavetR. Magnetic field exposure and cardiovascular disease mortality among electric utility workers. Am J Epidemiol. (1999) 149(2):135–42. 10.1093/oxfordjournals.aje.a009779
13.
KıvrakEGYurtKKKaplanAAAlkanIAltunG. Effects of electromagnetic fields exposure on the antioxidant defense system. J Microsc Ultrastruct. (2017) 5(4):167–76. 10.1016/j.jmau.2017.07.003
14.
SantiniSJCordoneVFaloneSMijitMTatoneCAmicarelliFet alRole of mitochondria in the oxidative stress induced by electromagnetic fields: focus on reproductive systems. Oxidative Med Cel Long. (2018) 2018:5076271. 10.1155/2018/5076271
15.
MartinelliICinatoMKeitaSMarsalDAntoszewskiVTaoJet alCardiac cell exposure to electromagnetic fields: focus on oxidative stress and apoptosis. Biomedicines. (2022) 10(5):929. 10.3390/biomedicines10050929
16.
TaoJLiZ-GVuongT-HKunduzovaO. GTEM cell design for controlled exposure to electromagnetic fields in biological system. bioRxiv. (2022). 10.1101/2022.03.03.482821
17.
MartinelliITimotinAMoreno-CorchadoPMarsalDKramarSLoyHet alGalanin promotes autophagy and alleviates apoptosis in the hypertrophied heart through FoxO1 pathway. Redox Biol. (2021) 40:101866. 10.1016/j.redox.2021.101866
18.
OstoCBenadorIYNgoJLiesaMStilesLAcin-PerezRet alMeasuring mitochondrial respiration in previously frozen biological samples. Curr Protoc Cell Biol. (2020) 89(1):e116. 10.1002/cpcb.116
19.
WasowiczWNèveJPeretzA. Optimized steps in fluorometric determination of thiobarbituric acid-reactive substances in serum: importance of extraction pH and influence of sample preservation and storage. Clin Chem. (1993), 39(12):2522–6. 10.1093/clinchem/39.12.2522
20.
HagemanJJBastAVermeulenNP. Monitoring of oxidative free radical damage in vivo: analytical aspects. Chem Biol Interact. (1992) 82(3):243–93. 10.1016/0009-2797(92)90001-2
21.
CuiXGongJHanHHeLTengYTetleyTet alRelationship between free and total malondialdehyde, a well-established marker of oxidative stress, in various types of human biospecimens. J Thorac Dis. (2018) 10(5):3088–97. 10.21037/jtd.2018.05.92
22.
RobeyRBWeiszJKuemmerleNBSalzbergACBergABrownDGet alMetabolic reprogramming and dysregulated metabolism: cause, consequence and/or enabler of environmental carcinogenesis?Carcinogenesis. (2015) 36(1):203–31. 10.1093/carcin/bgv037
23.
CadeteVJJVasamGMenziesKJBurelleY. Mitochondrial quality control in the cardiac system: an integrative view. Biochim Biophys Acta Mol Basis Dis. (2019) 1865(4):782–96. 10.1016/j.bbadis.2018.11.018
24.
LuntSYVander HeidenMG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. (2011) 27(1):441–64. 10.1146/annurev-cellbio-092910-154237
25.
KuhnARvan BilsenM. Oncometabolism: a paradigm for the metabolic remodeling of the failing heart. Int J Mol Sci. (2022) 23(22):13902. 10.3390/ijms232213902
26.
PeoplesJNSarafAGhazalNPhamTTKwongJQ. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med. (2019) 51(12):1–13. 10.1038/s12276-019-0355-7
27.
D'OriaRSchipaniRLeonardiniANatalicchioAPerriniSCignarelliAet alThe role of oxidative stress in cardiac disease: from physiological response to injury factor. Oxid Med Cell Longev. (2020) 2020:5732956. 10.1155/2020/5732956
28.
TsutsuiHKinugawaSMatsushimaS. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. (2011) 301(6):2181–90. 10.1152/ajpheart.00554.2011
29.
ChenYLiuYDornGW. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. (2011) 109(12):1327–31. 10.1161/CIRCRESAHA.111.258723
30.
NarendraDPYouleRJ. Targeting mitochondrial dysfunction: role for PINK1 and parkin in mitochondrial quality control. Antioxid Redox Signal. (2011) 14(10):1929–38. 10.1089/ars.2010.3799
31.
KoeneSSmeitinkJ. Mitochondrial medicine: entering the era of treatment. J Intern Med. (2009) 265(2):193–209. 10.1111/j.1365-2796.2008.02058.x
32.
JonckheereAISmeitinkJARodenburgRJ. Mitochondrial ATP synthase: architecture, function and pathology. J Inherit Metab Dis. (2012) 35(2):211–25. 10.1007/s10545-011-9382-9
33.
OsterholtMNguyenTDSchwarzerMDoenstT. Alterations in mitochondrial function in cardiac hypertrophy and heart failure. Heart Fail Rev. (2013) 18(5):645–56. 10.1007/s10741-012-9346-7
34.
RoscaMGTandlerBHoppelCL. Mitochondria in cardiac hypertrophy and heart failure. J Mol Cell Cardiol. (2013) 55:31–41. 10.1016/j.yjmcc.2012.09.002
35.
ZhouBTianR. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. (2018) 128(9):3716–26. 10.1172/JCI120849
36.
KangPTChenCLLinPZhangLZweierJLChenYR. Mitochondrial complex I in the post-ischemic heart reperfusion-mediated oxidative injury and protein cysteine sulfonation. J Mol Cell Cardiol. (2018) 121:190–204. 10.1016/j.yjmcc.2018.07.244
37.
Hadrava VanovaKKrausMNeuzilJRohlenaJ. Mitochondrial complex II and reactive oxygen species in disease and therapy. Redox Rep. (2020) 25(1):26–32. 10.1080/13510002.2020.1752002
38.
ChenQVazquezEJMoghaddasSHoppelCLLesnefskyEJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. (2003) 278(38):36027–31. 10.1074/jbc.M304854200
39.
St-PierreJBuckinghamJARoebuckSJBrandMD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem. (2002) 277(47):44784–90. 10.1074/jbc.M207217200
40.
DröseS. Differential effects of complex II on mitochondrial ROS production and their relation to cardioprotective pre- and postconditioning. Biochim Biophys Acta. (2013) 1827(5):578–87. 10.1016/j.bbabio.2013.01.004
41.
RalphSJMoreno-SánchezRNeuzilJRodríguez-EnríquezS. Inhibitors of succinate: quinone reductase/complex II regulate production of mitochondrial reactive oxygen species and protect normal cells from ischemic damage but induce specific cancer cell death. Pharm Res. (2011) 28(11):2695–730. 10.1007/s11095-011-0566-7
42.
Valls-LacalleLBarbaIMiró-CasasERuiz-MeanaMRodríguez-SinovasAGarcía-DoradoD. Selective inhibition of succinate dehydrogenase in reperfused myocardium with intracoronary malonate reduces infarct size. Sci Rep. (2018) 8(1):2442. 10.1038/s41598-018-20866-4
43.
WojtovichAPBrookesPS. The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res Cardiol. (2009) 104(2):121–9. 10.1007/s00395-009-0001-y
44.
LiYParkJSDengHBaiY. Cytochrome c oxidase subunit IV is essential for assembly and respiratory function of the enzyme complex. J Bioenerg Biomembr. (2006) 38(5–6):283–91. 10.1007/s10863-006-9052-z
45.
Fernández-VizarraETirantiVZevianiM. Assembly of the oxidative phosphorylation system in humans: what we have learned by studying its defects. Biochim Biophys Acta. (2009) 1793(1):200–11. 10.1016/j.bbamcr.2008.05.028
46.
WikströmMKrabKSharmaV. Oxygen activation and energy conservation by cytochrome c oxidase. Chem Rev. (2018) 118(5):2469–90. 10.1021/acs.chemrev.7b00664
47.
UchenunuOZhdanovAVHuttonPJovanovicPWangYAndreevDEet alMitochondrial complex IV defects induce metabolic and signaling perturbations that expose potential vulnerabilities in HCT116 cells. FEBS Open Bio. (2022) 12(5):959–82. 10.1002/2211-5463.13398
48.
De CraeneBBerxG. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. (2013) 13(2):97–110. 10.1038/nrc3447
49.
YangJWeinbergRA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. (2008) 14(6):818–29. 10.1016/j.devcel.2008.05.009
50.
CuiXGongJHanHHeLTengYet alRelationship between free and total malondialdehyde, a well-established marker of oxidative stress, in various types of human biospecimens. J Thorac Dis. (2018) 10(5):3088–97. 10.21037/jtd.2018.05.92
51.
HagemanJJBastAVermeulenNP. Monitoring of oxidative free radical damage in vivo: analytical aspects. Chem Biol Interact. (1992) 82:243–93. 10.1016/0009-2797(92)90001-2
52.
MaaloufRBaileyS. A review on B-type natriuretic peptide monitoring: assays and biosensors. Heart Fail Rev. (2016) 21(5):567–78. 10.1007/s10741-016-9544-9
53.
ChowSLMaiselASAnandIBozkurtBde BoerRAFelkerGMet alRole of biomarkers for the prevention, assessment, and management of heart failure: a scientific statement from the American heart association. Circulation. (2017) 135(22):e1054–91. 10.1161/CIR.0000000000000490
54.
AaronRKCiomborDMSimonBJ. Treatment of nonunions with electric and electromagnetic fields. Clin Orthop Relat Res. (2004) 419:21–9. 10.1097/00003086-200402000-00005
55.
SodaAIkeharaTKinouchiYYoshizakiK. Effect of exposure to an extremely low frequency-electromagnetic field on the cellular collagen with respect to signaling pathways in osteoblast-like cells. J Med Invest. (2008) 55(3–4):267–78. 10.2152/jmi.55.267
56.
ChoiM-CCheungK-KLiXCheingGL-Y. Pulsed electromagnetic field (PEMF) promotes collagen fibre deposition associated with increased myofibroblast population in the early healing phase of diabetic wound. Arch Dermatol Res. (2016) 308(1):21–9. 10.1007/s00403-015-1604-9
Summary
Keywords
electromagnetic stress, heart, metabolism, oxidative phosphorylation, superoxide dismutase
Citation
Savchenko L, Martinelli I, Marsal D, Zhdan V, Tao J and Kunduzova O (2023) Myocardial capacity of mitochondrial oxidative phosphorylation in response to prolonged electromagnetic stress. Front. Cardiovasc. Med. 10:1205893. doi: 10.3389/fcvm.2023.1205893
Received
14 April 2023
Accepted
22 May 2023
Published
07 June 2023
Volume
10 - 2023
Edited by
Ngan F. Huang, Stanford University, United States
Reviewed by
Qun Chen, Virginia Commonwealth University, United States Jason Bazil, Michigan State University, United States
Updates
Copyright
© 2023 Savchenko, Martinelli, Marsal, Zhdan, Tao and Kunduzova.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oksana Kunduzova oxana.koundouzova@inserm.fr
†These authors have contributed equally to this work
Abbreviations ADP, adenosine diphosphate; ANP, atrial natriuretic peptide; ATP, adenosine triphosphate; BNP, brain natriuretic peptide; BSA, bovine serum albumin; ECM, extracellular matrix; EMFs, electromagnetic fields; ETC, electron transport chain; HPRT, hypoxanthine phosphorybosyl transferase; HRP, horseradish peroxidase; I/R, ischemia-reperfusion; MDA, malondialdehyde; OCR, oxygen consumption rate; OCT, optimal cutting temperature compound; OXPHOS, oxidative phosphorylation; PBS, phosphate-buffered saline; qRT-PCR, quantitative polymerase chain reaction; RET, reverse electron transport; ROS, reactive oxygen species; SAR, specific absorption rate; SOD2, superoxide dismutase 2; TGFβ-1, transforming growth factor beta-1.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.