Abstract
Background:
The aim of the study was to determine the prevalence of rare disease-causing variants in cardiomyopathy-associated genes in a cohort of patients with ischemic and non-ischemic dilated cardiomyopathy undergoing heart transplant.
Methods:
We conducted a single-center cohort study of 60 adult patients with left ventricular ejection fraction ≤50% and left ventricular end-diastolic dimension ≥95th percentile for sex/height who underwent heart transplant between January 2017 and December 2023 and consented to participate in a cardiac tissue biobank. We evaluated the prevalence of rare (minor allele frequency <0.1%) disease-causing (pathogenic or likely pathogenic by American College of Genetics and Genomics criteria) variants in cardiomyopathy-associated genes.
Results:
A total of 60 individuals fulfilled the inclusion criteria: 16 with ischemic dilated cardiomyopathy [88% men, median age 65 years, interquartile range (IQR) 64–68 years] and 44 with non-ischemic dilated cardiomyopathy (80% men, median age 53 years, IQR 39–65 years). We found that the prevalence of disease-causing variants was similar between patients with ischemic dilated cardiomyopathy (3/16 or 19%; 95% credible interval 6%–36%) and those with non-ischemic dilated cardiomyopathy (10/44 or 23%; 95% credible interval 12%–33%). Variants in the ischemic dilated cardiomyopathy group were found in the TTN and DMD genes. Variants in the non-ischemic dilated cardiomyopathy group were found in the TTN, FLNC, LMNA, MYH7, and RBM20 genes.
Conclusions:
Patients with ischemic dilated cardiomyopathy undergoing heart transplant possessed a similar burden of rare disease-causing variants as those with non-ischemic dilated cardiomyopathy. Our results suggest that genetic testing may be beneficial in patients with advanced heart failure requiring heart transplant due to ischemic dilated cardiomyopathy to detect disease-causing variants in cardiomyopathy-associated genes.
Background
Ischemic and non-ischemic cardiomyopathy are the most common indications for heart transplantation, comprising approximately 30% and 50% of heart transplants in both Europe and the United States, respectively (1). Although definitions vary, a standardized definition for ischemic cardiomyopathy is symptomatic heart failure with left ventricular ejection fraction (LVEF) ≤40% and a history of myocardial infarction or revascularization, ≥75% stenosis of the left main or proximal left anterior descending coronary arteries, or ≥75% stenosis of at least two epicardial arteries (2). In contrast, non-ischemic dilated cardiomyopathy (NIDCM) is defined as LVEF ≤50% and left ventricular end-diastolic diameter (LVEDD) ≥95th percentile for sex and height while excluding coronary artery disease (CAD), abnormal loading conditions, or cardiac toxins (3).
Imaging studies suggest that ischemic cardiomyopathy results from acute ischemic injury to the myocardium and that the severity of the resulting left ventricular dysfunction is proportional to the degree of ischemic injury (4, 5). The pathophysiology of non-ischemic dilated cardiomyopathy is generally considered to be distinct, and rare disease-causing genetic variants, those classified by the American College of Genetics and Genomics criteria as likely pathogenic or pathogenic (LP/P), can be identified in 20% of patients (6, 7). However, a subset of patients with ischemic cardiomyopathy develops ventricular dilation, referred to as ischemic dilated cardiomyopathy (IDCM). It is difficult to distinguish IDCM from NIDCM because patients can have risk factors or features of both diseases. When evaluated by cardiac magnetic resonance imaging (CMRI), patients with IDCM and DCM exhibit overlap in ventricular volume and late gadolinium enhancement patterns (8). These observations suggest that IDCM and NIDCM may share pathophysiologic features.
We hypothesized that patients with IDCM harbor disease-causing variants in cardiomyopathy-associated genes at a prevalence similar to that of patients with NIDCM. Thus, we sought to determine the prevalence of rare disease-causing variants in cardiomyopathy-associated genes in a cohort of patients with IDCM and NIDCM undergoing heart transplant. Therefore, this study tests our hypothesis that rare disease-causing genetic variants contribute to the development of advanced heart failure due to IDCM.
Methods
Study cohort
We identified 60 patients who consented to participate in a cardiac tissue biobank (Cedars-Sinai IRB protocols Pro00010979 and Study00001617) with dilated cardiomyopathy [defined as LVEF ≤ 50% and LVEDD ≥ 95th percentile for sex and height (3)] and underwent heart transplant at Cedars-Sinai Medical Center in Los Angeles, California, between 1 January 2017 and 31 December 2023 for ischemic or non-ischemic cardiomyopathy. Patients with other forms of cardiomyopathy, including amyloidosis, sarcoidosis, and peripartum, were excluded (Figure 1). Patients with a history of myocardial infarction or revascularization, ≥75% stenosis of the left main or proximal left anterior descending coronary arteries, or ≥75% stenosis of at least two epicardial arteries were classified as having IDCM per the Felker definition (2). Echocardiography was performed after coronary revascularization but before heart transplantation.
Figure 1
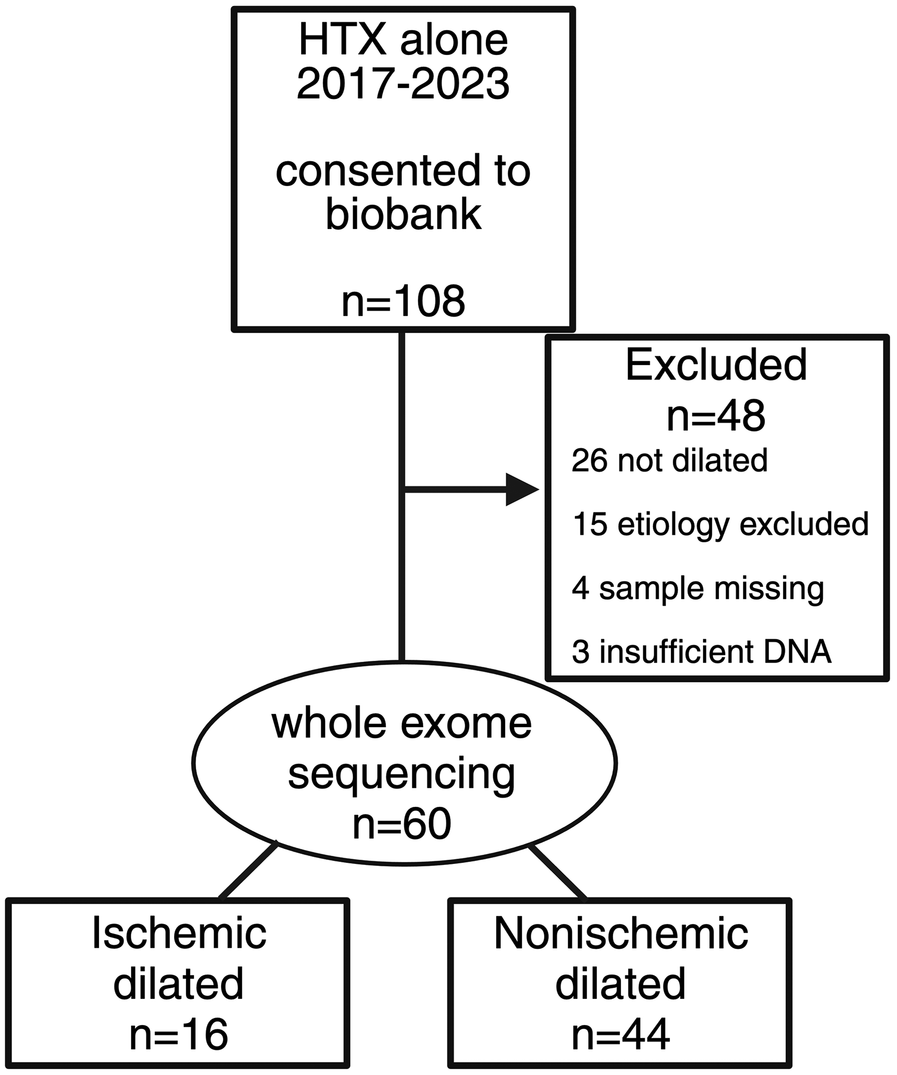
CONSORT diagram of patient flow for the study cohort. A total of 108 patients underwent heart transplant alone between 1 January 2017 and 31 December 2023 who were consented to a cardiac tissue biobank. Of them, 48 patients were excluded for reasons indicated in the figure. Whole exome sequencing was performed on 60 patients, 16 with ischemic dilated cardiomyopathy and 44 with non-ischemic dilated cardiomyopathy.
Exome sequencing, gene selection, variant filtering, and variant interpretation
For a full description of methods, see the online Supplementary Material. Briefly, genomic DNA was isolated from explanted hearts for whole exome sequencing. We assessed for variants in 36 genes associated with DCM, as previously selected in the DCM Precision Medicine Study (7) and the DMD given its association with DCM (9, 10). Variants were assigned as benign/likely benign or LP/P if the variant had that classification in ClinVar with a two-star rating or above. For variants not present in ClinVar, variant interpretation was performed using the ClinGen guidelines for DCM (11) with modifications from the DCM Precision Medicine Study (7). For variants in TTN, criterion PVS1_strong was only applied if the variants were in the A-band or an exon with percent splice in >90%. The variant interpretation criteria we used are detailed in the online Supplementary Material.
Statistical analysis
Statistical analysis was performed using R version 4.2.2 (12). Variables were compared using the Kruskal–Wallis test (non-Gaussian continuous variables), t-test (Gaussian continuous variables), and the chi-square test (categorical variables). A power analysis was performed using the R package “pwrss” (13), which revealed that our study cohort size (n = 60) provided a power of 0.51 to detect a twofold difference in the odds of a patient in the IDCM group possessing a disease-causing variant (given a baseline probability of 0.2 and type I error rate of 0.05). Therefore, we used Bayesian methods implemented in the R packages “BayesFactor” (14) and “bayestestR” (15) to calculate the 95% credible interval of the proportion of patients in each group carrying a disease-carrying variant.
Results
Of the 60 patients with dilated cardiomyopathy, 16 were classified as IDCM (27%) and 44 as NIDCM (73%). Patients with IDCM were older (median age 65 vs 53 years, IQR 64–68 vs. 39–65 years; p < 0.001) but similar in sex, race, ethnicity, and body mass index (Table 1). Patients with IDCM more frequently had diabetes mellitus (81% vs. 23%; p < 0.001) and hyperlipidemia (100% vs. 41%; p < 0.001). There was no difference between patients with IDCM and NIDCM with regard to the frequency of myocarditis, aortic or mitral valve disease requiring replacement, atrial or ventricular arrhythmias, or the presence of a cardiac implantable electronic device (Table 2). Most patients with IDCM had a history of coronary revascularization (94% vs. 0%; p < 0.001) or myocardial infarction (94% vs. 0%; p < 0.001). There was no difference between patients with IDCM and NIDCM with regard to a family history of cardiomyopathy or sudden cardiac arrest. Finally, there was no difference in mean left ventricular ejection fraction (17% vs. 17%; p = 0.849) or mean LVEDD (7.1 vs. 7.1 cm; p = 0.733) between the groups on the echocardiogram performed closest before transplant.
Table 1
| Factor | IDCM n = 16 | NIDCM N = 44 | P-value |
|---|---|---|---|
| Age at HTX | 65 [64–68] | 53 [39–65] | <0.001 |
| Male | 14 (88%) | 35 (79.5%) | 0.710 |
| Race | 0.091 | ||
| White | 11 (69%) | 24 (55%) | |
| Black | 0 | 10 (23%) | |
| Asian/Other | 5 (31%) | 10 (23%) | |
| Hispanic | 5 (31%) | 14 (32%) | 1.000 |
| BMI | 26 [24–28] | 26 [23–28] | 0.828 |
Characteristics of patients in the study cohort with ischemic dilated versus non-ischemic dilated cardiomyopathy.
BMI, body mass index.
Age and BMI are presented as mean [interquartile range].
Table 2
| Factor | IDCM n = 16 | NIDCM N = 44 | P-value |
|---|---|---|---|
| Medical history | |||
| Heavy alcohol use | 0 | 3 (7%, 0%–14%) | 0.558 |
| Diabetes mellitus | 13 (81%, 62%–100%) | 10 (23%, 10%–35%) | <0.001 |
| Hyperlipidemia | 16 (100%) | 18 (41%, 26%–55%) | <0.001 |
| Hx myocarditis | 0 | 5 (11%, 2%–21%) | 0.311 |
| Hx MVR | 4 (25%, 4%–46%) | 4 (9%, 1%–18%) | 0.192 |
| Hx AVR | 0 | 4 (9%, 1%–18%) | 0.565 |
| Hx atrial arrhythmias | 9 (56%, 32%–81%) | 28 (64%, 49%–78%) | 0.826 |
| Hx ventricular arrhythmias | 8 (50%, 26%–74%) | 24 (55%, 40%–69%) | 0.984 |
| CIED | 12 (75%, 54%–96%) | 32 (73%, 60%–86%) | 1.000 |
| Hx PCI or CABG | 15 (94%, 82%–100%) | 0 | <0.001 |
| Hx MI | 15 (94%, 82%–100%) | 0 | <0.001 |
| Family history of CM | 4 (25%, 4%–46%) | 18 (41%, 26%–55%) | 0.408 |
| Family history of SCD | 0 | 2 (5%, 0%–11%) | 1.000 |
| Echocardiography | |||
| EF | 16.4 (5.43) | 16.7 (5.65) | 0.849 |
| LVEDD | 7.12 (0.65) | 7.06 (0.73) | 0.733 |
Medical history of patients in the study cohort with ischemic dilated versus non-ischemic dilated cardiomyopathy.
AVR, aortic valve replacement; CABG, coronary artery bypass grafting; CIED, cardiac implantable electronic device; CM, cardiomyopathy; EF, ejection fraction; Hx, history; LVEDD, left ventricular end-diastolic diameter; MI, history of myocardial infarction; MVR, mitral valve replacement; PCI, percutaneous coronary intervention; SCD, sudden cardiac death; SVT, supraventricular tachycardia; VF, ventricular fibrillation; VT, ventricular tachycardia.
(1) Medical history variables are presented as number (proportion, 95% confidence intervals). (2) Echocardiographic variables EF and LVEDD are presented as mean (standard deviation).
We identified 13 P/LP variants in six cardiomyopathy-associated genes within the 60 patients in the study cohort (variants are described in Table 3 and Supplementary Table S1). Of the 13 variants, 10 were previously identified in patients with dilated cardiomyopathy (Supplementary Table S1) and three were novel. The proportion of patients with P/LP variants was similar between the groups, with 3/16 (19%; 95% credible interval 6%–36%) patients in the IDCM group and 10/44 (23%; 95% credible interval 12%–33%) patients in the NIDCM group (Figure 2). Most (8/13 or 62%) P/LP variants were found in TTN, with no difference in the proportion of patients with TTN variants between the IDCM (2/16 or 13%; 95% credible interval 3%–26%) and NIDCM groups (6/44 or 14%; 95% credible interval 5%–22%).
Table 3
| Varianta | Gene | Coding | Protein | Class |
|---|---|---|---|---|
| Ischemic dilated cardiomyopathy | ||||
| chr2-178548829-GTT-G | TTN | ENST00000589042 c.92795_92796del | ENSP00000467141 p.Lys30932ThrfsTer6 | LP |
| chr2-178577785-G-A | TTN | ENST00000589042 c.68641C>T | ENSP00000467141 p.Arg22881Ter | LP |
| chrX-32614303-CT-C | DMD | ENST00000357033 c.1481del | ENSP00000354923 p.Lys494ArgfsTer7 | LP |
| Non-ischemic dilated cardiomyopathy | ||||
| chr1-156134838-C-T | LMNA | ENST00000368300 c.673C>T | ENSP00000357283 p.Arg225Ter | P |
| chr2-178547890-TG-T | TTN | ENST00000589042 c.93735del | ENSP00000467141 p.Val31247Ter | LP |
| chr2-178560772-GT-G | TTN | ENST00000589042 c.85359del | ENSP00000467141 p.Pro28454GlnfsTer7 | LP |
| chr2-178567508-TC-T | TTN | ENST00000589042 c.78623del | ENSP00000467141 p.Gly26208GlufsTer21 | LP |
| chr2-178570990-ATTCT-A | TTN | ENST00000589042 c.75138_75141del | ENSP00000467141 p.Lys25046AsnfsTer8 | P/LP |
| chr2-178612453-CTCTTTTCCACAATG-C | TTN | ENST00000589042 c.50058_50071del | ENSP00000467141 p.Tyr16686Ter | LP |
| chr2-178738152-G-T | TTN | ENST00000589042 c.14301C>A | ENSP00000467141 p.Cys4767Ter | LP |
| chr7-128844045-C-T | FLNC | ENST00000325888 c.2971C>T | ENSP00000327145 p.Arg991Ter | P/LP |
| chr10-110812298-G-A | RBM20 | ENST00000369519 c.1901G>A | ENSP00000358532 p.Arg634Gln | P/LP |
| chr14-23425970-C-T | MYH7 | ENST00000355349 c.2156G>A | ENSP00000347507 p.Arg719Gln | P |
Pathogenic/likely pathogenic variants identified in the study cohort.
LP, likely pathogenic; P, pathogenic.
Variants are described as chromosome-position-reference-alternate, with position provided per GRCh38.
Figure 2
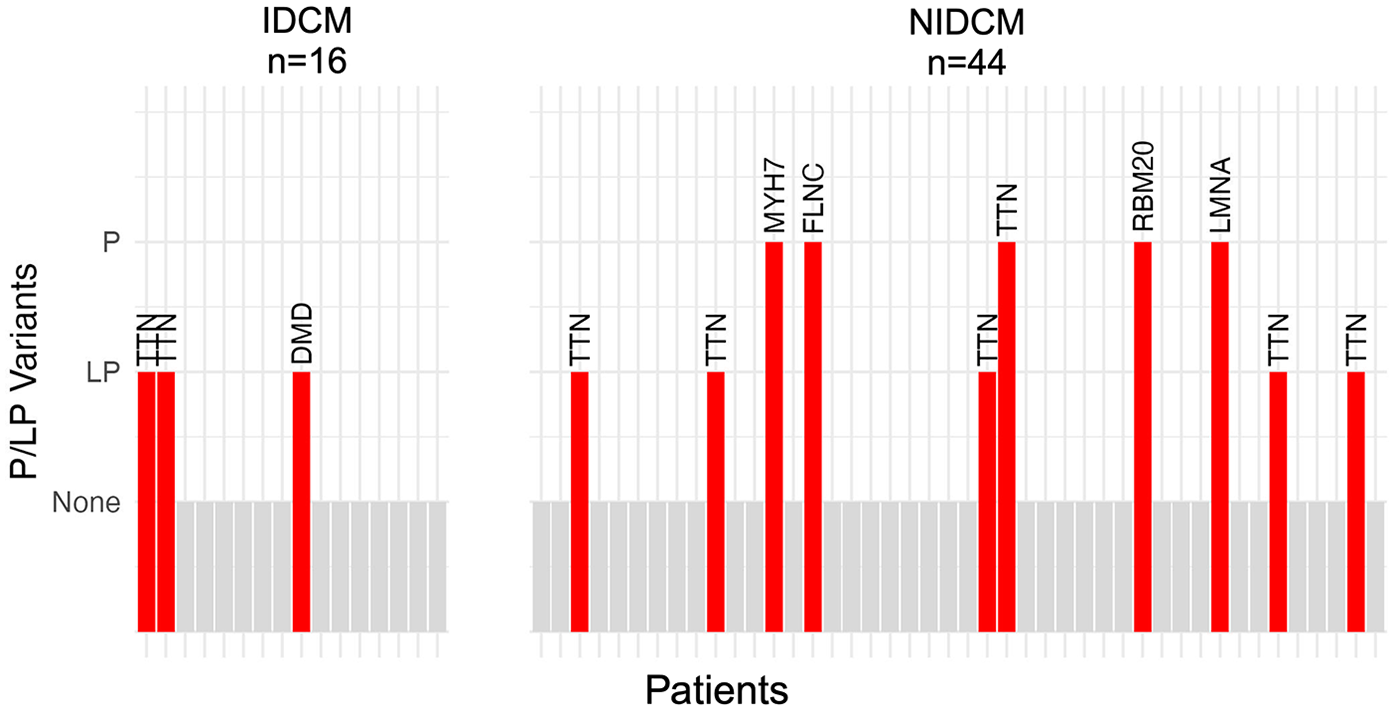
Bar plot of patients with rare disease-causing (likely pathogenic or pathogenic) variants in the study cohort. Each patient is depicted by a vertical bar; patients without a disease-causing variant are depicted by gray bars and patients with a disease-causing variant are depicted by red bars. The name of the gene in which the variant was found is indicated above the bar. The prevalence of disease-causing variants was similar between patients with ischemic dilated cardiomyopathy (bars on left; 3/16 or 19%; 95% credible interval 6%–36%) and those with non-ischemic dilated cardiomyopathy (bars on right; 10/44 or 23%; 95% credible interval 12%–33%).
Discussion
In a cohort of patients with dilated cardiomyopathy who underwent heart transplant, patients with IDCM or NIDCM had a similar prevalence of disease-causing variants in cardiomyopathy-associated genes compared to NIDCM patients. Our results are consistent with previous studies suggesting a superimposed non-ischemic component in ischemic cardiomyopathy. Using cardiac magnetic resonance imaging, approximately 15% of patients with CAD have a superimposed non-ischemic scar pattern (16, 17). Disease-causing variants contribute to lower EF and worse outcomes in patients with CAD (16).
Two previous studies have investigated the interplay between CAD, heart failure, and disease-causing genetic variants in cardiomyopathy-associated genes. In 2021, Povysil et al. reported the identification of disease-causing variants in ICM patients from the Candesartan in Heart Failure-Assessment of Reduction in Mortality and Morbidity (CHARM) and Controlled Rosuvastatin Multinational Trial in Heart Failure (CORONA) clinical trials; however, the prevalence of disease-causing variants was substantially lower than the IDCM group in our study at 3% (18). More recently, Jones et al. reported the identification of disease-causing variants in cardiomyopathy-associated genes in patients with CAD in the UK Biobank, also with a lower prevalence of disease-causing variants than the IDCM group in our study at 0.6% (16). We identified disease-causing variants in 19% of the patients with IDCM in our cohort of patients with advanced heart failure requiring heart transplant. Although we utilized a larger panel of DCM-associated genes, TTN was the most prevalent gene with disease-causing variants in all three studies. We posit that the higher prevalence of disease-causing variants we observed in the IDCM group in our study may be due to the increased severity of left ventricular dysfunction, given that all patients in our study cohort had undergone heart transplant. Further prospective investigation is needed to test this hypothesis.
Genetic testing and cascade screening is recommended in NIDCM (19); however, no guidance exists on genetic testing in patients with IDCM. Our results suggest that it may be prudent to extend this recommendation to patients similar to our study cohort: those with advanced heart failure due to IDCM requiring heart transplant. Our findings may also explain why some patients with ICM do not benefit from revascularization (20–23), i.e., in these cases, extant left ventricular dilation and dysfunction may be driven, at least in part, by genetic mechanisms.
Although our study is limited by a small cohort size compared to heart failure clinical trials, a cohort of 60 heart transplant recipients is substantial and would represent 3–6 times the annual volume of most heart transplant centers in Europe and the United States, where the median volume is 10–20 heart transplants per year (1). Although women were underrepresented in our study cohort, it was ethnically diverse, and it was also representative with respect to the proportion of heart transplants due to ICM and NICM. We were only able to enroll a portion of patients undergoing heart transplantation at our center during the study period in the cardiac tissue biobank used for this study, yielding a potential selection bias. A second potential selection bias is that given that the study cohort was composed of patients with advanced heart failure requiring heart transplant, the frequency of disease-causing variants in the IDCM group may be higher than in patients with less severe cardiomyopathy.
Conclusions
In a cohort of heart transplant recipients with DCM, we found a similar prevalence of rare disease-causing variants in cardiomyopathy-associated genes in patients with IDCM as compared to NIDCM. Our results suggest that genetic testing may be beneficial in patients with advanced heart failure requiring heart transplant due to IDCM to detect disease-causing variants in cardiomyopathy-associated genes.
Statements
Data availability statement
The datasets presented in this article are not readily available because the aforementioned IRB protocols did not provide for public release of genetic data. Requests to access the datasets can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Cedars-Sinai Medical Center Institutional Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
LCa: Conceptualization, Data curation, Formal analysis, Investigation, Writing – original draft, Writing – review & editing. JR: Data curation, Investigation, Writing – review & editing. IW: Investigation, Project administration, Resources, Writing – review & editing. AK: Methodology, Project administration, Resources, Writing – review & editing. MK: Project administration, Resources, Writing – review & editing. LCz: Conceptualization, Data curation, Investigation, Methodology, Project administration, Resources, Writing – review & editing. EK: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by a philanthropic donation by Joel Freedman. This work was also supported by the National Institutes of Health (NIH) to AK (R00-HL-141702).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2025.1542653/full#supplementary-material
References
1.
KhushKKCherikhWSChambersDCHarhayMOHayesDJrHsichEet alThe International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: thirty-sixth adult heart transplantation report - 2019; focus theme: donor and recipient size match. J Heart Lung Transplant. (2019) 38(10):1056–66. 10.1016/j.healun.2019.08.004
2.
FelkerGMShawLKO'ConnorCM. A standardized definition of ischemic cardiomyopathy for use in clinical research. J Am Coll Cardiol. (2002) 39(2):210–8. 10.1016/S0735-1097(01)01738-7
3.
KinnamonDDMoralesABowenDJBurkeWHershbergerRE, Consortium* DCM. Toward genetics-driven early intervention in dilated cardiomyopathy: design and implementation of the DCM precision medicine study. Circ Cardiovasc Genet. (2017) 10(6):e001826. 10.1161/CIRCGENETICS.117.001826
4.
MasciPGGanameJFranconeMDesmetWLorenzoniVIacucciIet alRelationship between location and size of myocardial infarction and their reciprocal influences on post-infarction left ventricular remodelling. Eur Heart J. (2011) 32(13):1640–8. 10.1093/eurheartj/ehr064
5.
FeistritzerHJNanosMEitelIJobsAde Waha-ThieleSMeyer-SaraeiRet alDeterminants and prognostic value of cardiac magnetic resonance imaging-derived infarct characteristics in non-ST-elevation myocardial infarction. Eur Heart J Cardiovasc Imaging. (2020) 21(1):67–76. 10.1093/ehjci/jez165
6.
VerdonschotJAJHazebroekMRKrapelsIPCHenkensMRaafsAWangPet alImplications of genetic testing in dilated cardiomyopathy. Circ Genom Precis Med. (2020) 13(5):476–87. 10.1161/CIRCGEN.120.003031
7.
JordanEKinnamonDDHaasGJHofmeyerMKransdorfEEwaldGAet alGenetic architecture of dilated cardiomyopathy in individuals of African and European ancestry. JAMA. (2023) 330(5):432–41. 10.1001/jama.2023.11970
8.
WonEDonninoRSrichaiMBSedlisSPFeitFRolnitzkyLet alDiagnostic accuracy of cardiac magnetic resonance imaging in the evaluation of newly diagnosed heart failure with reduced left ventricular ejection fraction. Am J Cardiol. (2015) 116(7):1082–7. 10.1016/j.amjcard.2015.06.032
9.
Restrepo-CordobaMAWahbiKFlorianARJimenez-JaimezJPolitanoLAradMet alPrevalence and clinical outcomes of dystrophin-associated dilated cardiomyopathy without severe skeletal myopathy. Eur J Heart Fail. (2021) 23(8):1276–86. 10.1002/ejhf.2250
10.
JohnsonROtwayRChinEHorvatCOhanianMWilcoxJALet alDMD-associated dilated cardiomyopathy: genotypes, phenotypes, and phenocopies. Circ Genom Precis Med. (2023) 16(5):421–30. 10.1161/CIRCGEN.123.004221
11.
MoralesAKinnamonDDPlattJEVattaJDorschnerMOMet alVariant interpretation for dilated cardiomyopathy: refinement of the American college of medical genetics and genomics/ClinGen guidelines for the DCM precision medicine study. Circ Genom Precis Med. (2020) 13(2):e002480. 10.1161/CIRCGEN.119.002480
12.
R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria (2023). Available at:https://www.R-project.org/(Accessed March 20, 2025).
13.
BulusM. pwrss: Statistical Power and Sample Size Calculation Tools. R package version 0.3.1 (2023). Available at:https://CRAN.R-project.org/package=pwrss(Accessed March 20, 2025).
14.
MoreyRRouderJ. BayesFactor: Computation of Bayes Factors for Common Designs. R package version 0.9 (2023). Available at:https://richarddmorey.github.io/BayesFactor/(Accessed March 20, 2025).
15.
MakowskiDBen-ShacharMSLüdeckeDBayestestR. Describing effects and their uncertainty, existence and significance within the Bayesian framework. J Open Source Softw. (2019) 4(40):1541. 10.21105/joss.01541
16.
JonesREHammersleyDJZhengSMcGurkKAde MarvaoATheotokisPIet alAssessing the association between genetic and phenotypic features of dilated cardiomyopathy and outcome in patients with coronary artery disease. Eur J Heart Fail. (2024) 26(1):46–55. 10.1002/ejhf.3033
17.
BawaskarPThomasNIsmailKGuoYChhikaraSAthwalPSSet alNonischemic or dual cardiomyopathy in patients with coronary artery disease. Circulation. (2024) 149(11):807–21. 10.1161/CIRCULATIONAHA.123.067032
18.
PovysilGChazaraOCarssKJDeeviSVVWangQArmisenJet alAssessing the role of rare genetic variation in patients with heart failure. JAMA Cardiol. (2021) 6(4):379–86. 10.1001/jamacardio.2020.6500
19.
HershbergerREGivertzMMHoCYJudgeDPKantorPFMcBrideKLet alGenetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. (2018) 20(9):899–909. 10.1038/s41436-018-0039-z
20.
VelazquezEJLeeKLDejaMAJainASopkoGMarchenkoAet alCoronary-artery bypass surgery in patients with left ventricular dysfunction. N Engl J Med. (2011) 364(17):1607–16. 10.1056/NEJMoa1100356
21.
PereraDClaytonTO'KanePDGreenwoodJPWeerackodyRRyanMet alPercutaneous revascularization for ischemic left ventricular dysfunction. N Engl J Med. (2022) 387(15):1351–60. 10.1056/NEJMoa2206606
22.
IaconelliAPellicoriPDolcePBustiMRuggioAAspromonteNet alCoronary revascularization for heart failure with coronary artery disease: a systematic review and meta-analysis of randomized trials. Eur J Heart Fail. (2023) 25(7):1094–104. 10.1002/ejhf.2911
23.
Moliner-AbosCCalvo-BarceloMSole-GonzalezEBorrellas MartinAFluvia-BruguesPSanchez-VegaJet alRevascularization and outcomes in ischaemic left ventricular dysfunction after heart failure admission: the RevascHeart study. Eur J Heart Fail. (2025) 27(3):598–605. 10.1002/ejhf.3463
Summary
Keywords
ischemic cardiomyopathy, dilated cardiomyopathy, heart transplantation, genetic testing, ischemic dilated cardiomyopathy
Citation
Cao L, Rushakoff J, Williamson I, Karlstaedt A, Kittleson M, Czer L and Kransdorf EP (2025) Similar burden of rare genetic variants in ischemic and non-ischemic dilated cardiomyopathy. Front. Cardiovasc. Med. 12:1542653. doi: 10.3389/fcvm.2025.1542653
Received
10 December 2024
Accepted
03 April 2025
Published
29 April 2025
Volume
12 - 2025
Edited by
Francesco Gentile, Sant'Anna School of Advanced Studies, Italy
Reviewed by
Mahsima Shabani, Vanderbilt University Medical Center, United States
Nirmal Vadgama, Stanford University, United States
Updates
Copyright
© 2025 Cao, Rushakoff, Williamson, Karlstaedt, Kittleson, Czer and Kransdorf.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Evan P. Kransdorf evan.kransdorf@cshs.org
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.