 Xiangyu Fei1,2,3,4,5,†
Xiangyu Fei1,2,3,4,5,† Chao Song2,3,4,5,†Jian Cui2,3,4,5,†Yuqing Li1,2,3,4,5,†
Chao Song2,3,4,5,†Jian Cui2,3,4,5,†Yuqing Li1,2,3,4,5,† Xiaoyong Lei1*
Xiaoyong Lei1* Huifang Tang1,2,3,4,5*
Huifang Tang1,2,3,4,5*
- 1School of Pharmacy, Hengyang Medical College, University of South China, Hengyang, Hunan, China
- 2Hunan Provincial Key Laboratory of Multi-omics And Artificial Intelligence of Cardiovascular Diseases, University of South China, Hengyang, Hunan, China
- 3Department of Cardiology, Hengyang Medical School, The First Affiliated Hospital, University of South China, Hengyang, Hunan, China
- 4Clinical Research Center for Myocardial Injury in Hunan Province, The First Affiliated Hospital, Hengyang, Hunan, China
- 5Institute of Cardiovascular Disease, Hengyang Medical School, The First Affiliated Hospital, University of South China, Hengyang, Hunan, China
Cardiovascular diseases (CVDs) have become the leading cause of death globally, surpassing infectious diseases and other chronic illnesses. The incidence and mortality rates of CVDs are rising worldwide, posing a key challenge in public health. The ubiquitination system is a vast and complex. It is an important post-translational modification that plays a crucial role in various cellular processes. Deubiquitination is catalyzed by deubiquitinases (DUBs), which remove ubiquitin (Ub) from ubiquitinated proteins, thereby reversing the ubiquitination process. DUBs play an important role in many biological processes, such as DNA repair, cell metabolism, differentiation, epigenetic regulation, and protein stability control. They also participate in the regulation of many signaling pathways associated with the development and progression of CVDs. In this review, we primarily focus on the role of DUBs in various key pathological mechanisms of atherosclerosis (AS), such as foam cell formation, vascular remodeling (VR), endothelial-to-mesenchymal transition (End-MT), and clonal hematopoiesis (CH). In the heart, we summarize the involvement of DUBs in diseases and pathological processes, including heart failure (HF), myocardial infarction (MI), myocardial hypertrophy (MH) and ischemia/reperfusion (I/R) injury. Additionally, we also explore the diabetic cardiomyopathy (DCM) and the use of doxorubicin-induced cardiotoxicity in clinical settings. A comprehensive understanding of deubiquitination may provide new insights for the treatment and drug design of CVDs.
1 Introduction
Despite the increasing depth of understanding and research into CVDs, their mortality rate remains high (1). The ubiquitin-proteasome system (UPS) plays a significant role in CVDs due to its unique functions. As one of the key protein degradation systems within cells, the UPS is primarily responsible for the degradation and regulation of target proteins. It is involved in the degradation of over 80% of the proteins within cellular proteins (2). The system consists of Ub, ubiquitin-activating enzymes (E1s), ubiquitin-conjugating enzymes (E2s), ubiquitin ligases (E3s), and DUBs. The ubiquitination process, mediated by E1s, E2s, and E3s, is referred to as ubiquitination (3). Protein homeostasis imbalance is a key factor that disrupts cellular homeostasis. These enzymes typically mark their specific target proteins using Ub, leading to their degradation by the proteasome, or further mediate signaling based on the protein's modification sites and the number of attached Ub molecules, thus participating in various physiological processes (4) (Figure 1). The reverse process of ubiquitination is called deubiquitination. Deubiquitination is primarily catalyzed by DUBs, which are classified into seven families based on sequence and structural similarity: ubiquitin-specific proteases (USPs), ovarian tumor proteases (OTUs), ubiquitin c-terminal hydrolases (UCHs), Machado-Josephin domain proteases (MJDs), JAB1/MPN+/MOV34 (JAMM) domain proteases, monocyte chemoattractant protein-Induced protein (MCPIP), and a novel family of DUBs associated with ubiquitin-binding, motif Interacting with Ub-containing novel DUBs (MINDYs) (5). DUBs are a group of proteins that recognize and cleave Ub chains. They interact with substrate proteins, preventing their degradation by the UPS. DUBs contain ubiquitin-binding domains (6), which enable that they perform specific and precise regulation through the recognition and recruitment of ubiquitinated proteins (7). By cleaving the peptide or isopeptide bonds between Ub and its substrate, DUBs remove Ub from ubiquitinated proteins, thereby stabilizing the substrate and reversing ubiquitination (8). Additionally, DUBs regulate signaling pathways to prevent their overactivation, maintaining UPS homeostasis (9). Through Ub removal, DUBs rescue target proteins from degradation signals, preserving their stability (10). The dynamic balance between ubiquitination and deubiquitination is intricately linked to various cellular functions and is critical in disease pathogenesis and therapeutic interventions (11).
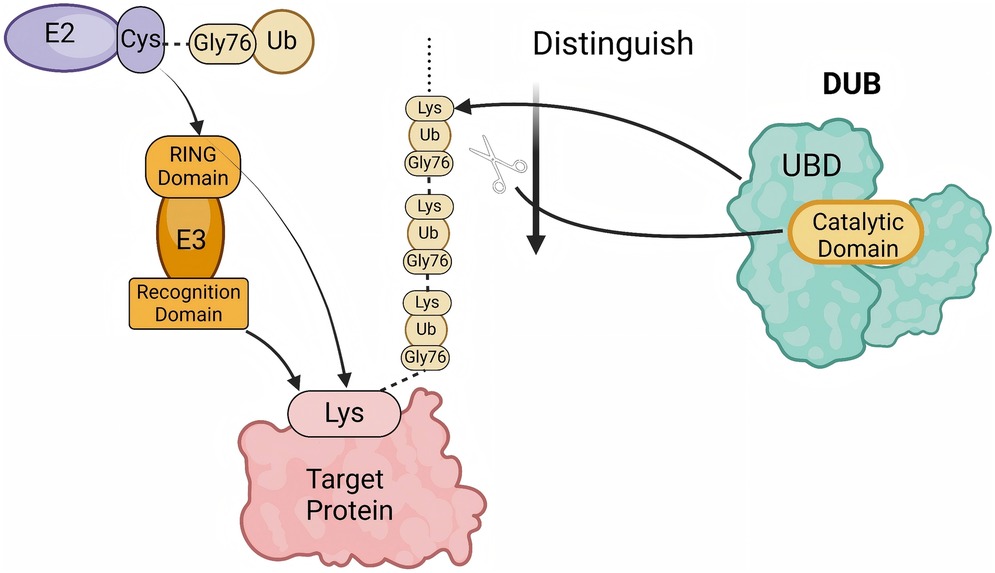
Figure 1. The process of polyubiquitination and deubiquitination. The E2 enzyme carries an activated ubiquitin (Ub) molecule via its cysteine (Cys) residue and transfers it to the target protein under the action of the E3 ubiquitin ligase. The E3 ligase, composed of a RING domain and a recognition domain, facilitates the attachment of ubiquitin to the lysine (Lys) residue of the target protein. Subsequently, a polyubiquitin chain is formed through the linkage between the glycine residue at position 76 (Gly76) of one ubiquitin molecule and the Lys residue of the preceding ubiquitin. Deubiquitinases (DUBs) recognize and cleave ubiquitin chains at specific sites. The ubiquitin-binding domain (UBD) of DUBs identifies the ubiquitin chain, while the catalytic domain performs the cleavage, thereby regulating the ubiquitination levels of proteins. (created with BioRender.com).
To date, more than 100 DUBs have been identified (12). Recent studies have demonstrated that DUBs play crucial regulatory roles in various CVDs and control the onset and progression of disease through multiple mechanisms. This review will systematically examine the role of DUBs in CVDs, categorized by different disease types (Figure 2).
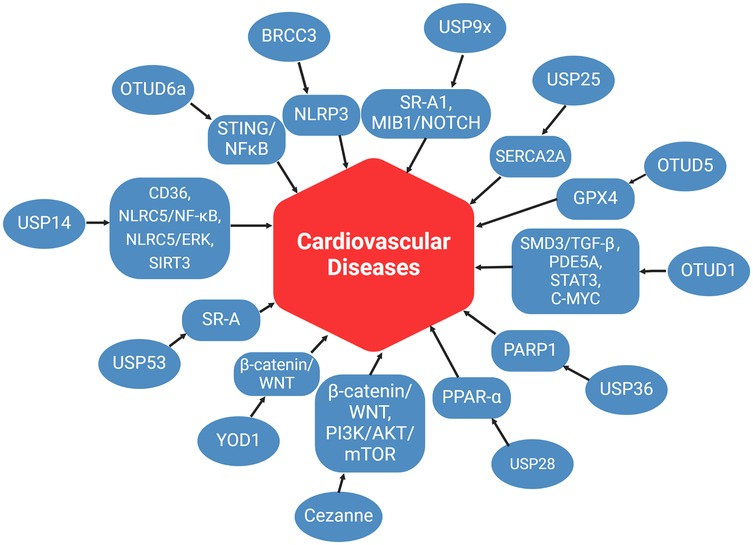
Figure 2. The role of DUBs in CVD. This figure summarizes the mechanisms of DUBs in various pathways associated with CVD, as discussed in this review article. (created with BioRender.com).
2 Atherosclerosis
AS is a multifocal, immune-mediated chronic inflammatory disease that predominantly affects large and medium-sized arteries and is closely associated with lipid deposition. The hallmark pathological feature of AS is excessive cholesterol accumulation in arterial walls, leading to progressive thickening and hardening (13). AS a leading contributor to acute cardiovascular events including MI and stroke, AS represents a complex disease process involving multiple pathophysiological mechanisms such as dysregulated lipid metabolism, aberrant immune responses, and VR (14). uring the onset and progression of AS, DUBs play crucial roles in regulating several processes, including endothelial cell function, immune cell activation, inflammation, and lipid uptake. These findings suggest that DUBs may serve as potential therapeutic targets for AS treatment. We will systematically elucidate the mechanistic role of DUBs in the initiation and progression of atherosclerosis across its different developmental stages.
2.1 Endothelial dysfunction and inflammatory regulation
Atherosclerosis can be broadly divided into several stages: lipid deposition, persistent inflammatory response, foam cell formation, vascular smooth muscle cell (VSMC) proliferation, and VR (15). In the early stage of atherosclerosis, no apparent atherosclerotic plaques have formed in the vascular wall; however, a series of pathophysiological changes have already begun to take place. The earliest alterations occur in endothelial cells, with endothelial cell injury being defined as the initiating event of atherosclerosis (16). Endothelial cells, positioned at the interface between solid and semi-solid states, serve as the outermost protective barrier of blood vessels. Under physiological conditions, they maintain vascular homeostasis by regulating vascular tone, inhibiting thrombosis, and modulating inflammatory responses (17). However, when exposed to pathological stimuli such as from disturbed blood flow (18), Oxidative stress (19), inflammatory factors and elevated lipid levels, endothelial cells undergo an inflammatory response, leading to structural alterations and functional impairment, ultimately disrupting vascular homeostasis, by suppressing the inflammatory response to reduce endothelial cell injury, the progression and development of atherosclerosis can be effectively alleviated. A study has shown that USP14 expression is significantly downregulated in atherosclerotic plaques. Further experiments revealed that overexpression of USP14 alleviates ox-LDL-induced endothelial inflammation, primarily by deubiquitinating NLRC5, thereby inhibiting NF-κB activation and reducing the release of pro-inflammatory cytokines (20). Interestingly, another study by Zhang et al. reported that USP14 expression is upregulated in the serum of atherosclerosis patients, and overexpression of USP14 promotes the activation of the Smad2/3 signaling pathway via NLRC5 deubiquitination (21), leading to increased secretion of pro-inflammatory cytokines. These seemingly contradictory findings, however, can be reasonably explained. Firstly, NLRC5 itself exhibits dual functions. While it is generally considered an inhibitor of inflammation, it can also act as a pro-inflammatory regulator under certain conditions (22). Secondly, USP14's deubiquitination activity may result in different biological effects depending on the type of ubiquitin chains removed. Lastly, USP14 exerts distinct functions in different cell types and pathological conditions. For instance, in macrophages, downregulation of USP14 suppresses inflammation (23). In THP-1 and RAW264.7 cells, inhibition of USP14 inactivates the ERK signaling pathway, thereby reducing LPS-induced inflammation (24). In TCMK-1 cells, USP14 inhibition downregulates TFAP2A while upregulating TBK1, leading to reduced oxidative stress and inflammation (25). Undoubtedly, USP14 plays a crucial role in the pathogenesis of atherosclerosis. Although its precise mechanisms require further investigation, current evidence strongly suggests that USP14 is closely linked to inflammation regulation. Specific USP14 inhibitors, such as IU1 and other compounds, have been identified (26). Unfortunately, these inhibitors remain in the experimental stage and have yet to be translated into clinical applications. In the future, USP14 may emerge as a promising therapeutic target for controlling inflammation and treating atherosclerosis, offering new avenues for disease intervention. End-MT is a critical phenotypic alteration associated with endothelial dysfunction (27). Under maladaptive and pathological conditions, newly transformed mesenchymal cells undergo fibrosis, leading to excessive extracellular matrix deposition, increased tissue stiffness, and ultimately impaired vascular function, thereby promoting disease progression. OTUD1, a deubiquitinase belonging to the OTUD family, has been identified as a key regulator of End-MT. Huang et al. Demonstrated (28) that OTUD1 deubiquitinates SMAD3, thereby exacerbating AngII-induced VR and collagen deposition by promoting End-MT. SMAD3 is a crucial signal transduction protein involved in multiple cardiovascular diseases (29–31). As a member of the SMAD family, SMAD3 forms a complex with other family proteins to regulate gene expression (32). OTUD1 stabilizes SMAD3 and enhances the formation of the SMAD3/SMAD4 complex, thereby modulating the transcription of genes associated with End-MT and VR. β-catenin is a central regulatory factor in the Wnt signaling pathway, playing a pivotal role in cellular proliferation and differentiation. It has been extensively investigated as a therapeutic target in cardiovascular and other diseases (33, 34). The deubiquitinase YOD1 interacts with β-catenin via its OTU domain, removing its K48-linked ubiquitin chains, thereby preventing its degradation. This process leads to the aberrant nuclear accumulation of β--catenin, which subsequently enhances the transcription of EndMT-related genes, further promoting endothelial dysfunction, VR, and pathological progression. Collectively, these molecular mechanisms highlight the intricate regulatory network governing End-MT and its role in endothelial dysfunction and VR. The pathological alterations induced by End-MT not only compromise endothelial integrity but also create a pro-inflammatory and pro-oxidative microenvironment within the vasculature. These changes, in turn, facilitate the recruitment and infiltration of immune cells, further amplifying vascular damage and contributing to the progression of atherosclerosis.
2.2 Lipid deposition and foam cell formation
Following endothelial cell injury, structural alterations and functional impairment lead to a marked increase in vascular permeability, facilitating the penetration of large amounts of LDL into the subendothelial space. Under the influence of reactive ROS, LDL undergoes oxidation, forming ox-LDL (35). Simultaneously, the expression of ICAM-1 and VCAM-1 is upregulated, further enhancing monocyte adhesion and enabling their transendothelial migration into the intimal layer. Upon stimulation by MCP-1 and TNF-α, monocytes differentiate into macrophages. Macrophages, through scavenger receptors such as SR-A1, CD36, and LOX-1, indiscriminately uptake ox-LDL (36). Since these receptors are not subject to negative feedback regulation by intracellular cholesterol levels, the continuous accumulation of ox-LDL within macrophages results in excessive intracellular lipid overload, ultimately driving their transformation into foam cells (36). Foam cells not only exacerbate the inflammatory response but also significantly increase the risk of plaque rupture. As early as 2006, studies reported that heightened activity of the UPS is closely associated with increased plaque inflammation and vulnerability (37), suggesting that UPS plays a crucial role in the pathogenesis and progression of atherosclerosis. Furthermore, deubiquitination may serve as a key regulatory factor in foam cell formation, warranting further investigation into its underlying mechanisms. In macrophages and mouse models with USP9X deficiency, studies have shown a significant increase in ox-LDL uptake, lipid accumulation, lesion macrophage content, and necrotic core expansion. Mechanistically, USP9X inhibits foam cell formation by removing K63-linked polyubiquitination of SR-A1 at the K27 site (38). Additionally, USP14 has been identified as a deubiquitinase for CD36, promoting foam cell formation by deubiquitinating CD36 in THP-1 and RAW264.7 macrophages (39). Similarly, VSMC serve as another major source of foam cells, and the deubiquitinase USP53 facilitates lipid uptake and accelerates foam cell formation by stabilizing SR-A through deubiquitination (40). Beyond lipid uptake, macrophage polarization plays a critical role in the pathogenesis and progression of atherosclerosis (41). In the early stages of atherosclerosis, M1 macrophages secrete pro-inflammatory cytokines such as TNF-α and IL-6, exacerbating local inflammation and arterial wall damage, thereby accelerating lesion development (42). In contrast, during disease progression, M2 macrophages contribute to lesion repair and plaque stabilization by secreting anti-inflammatory cytokines like IL-10, thus slowing the progression of atherosclerosis Modulating macrophage polarization may offer a novel therapeutic approach for atherosclerosis (43). Studies have demonstrated that the deubiquitinase Mysm1 plays a crucial role in macrophage survival and polarization. Its deficiency leads to accelerated proliferation and increased production of pro-inflammatory factors, promoting an M1-like phenotype (44). In another study, Rui et al. revealed that USP14 stabilizes cGAS, thereby enhancing cGAS-STING pathway activation and promoting ox-LDL/4-HNE-induced pro-inflammatory M1 macrophage polarization. Conversely, inhibiting USP14 facilitates IL-4/IL-13-induced anti-inflammatory M2 macrophage polarization, alleviating inflammation in atherosclerosis (23). However, whether USP14 can directly deubiquitinate cGAS to stabilize the cGAS protein remains unclear.Collectively, DUBs play a pivotal role in regulating macrophage lipid uptake and polarization. Further exploration of their specific mechanisms may contribute to the development of novel therapeutic strategies for atherosclerosis.
2.3 VSMC proliferation, migration, and VR
As inflammation persists and foam cells accumulate, structural changes gradually occur in the vascular wall, characterized by VSMC proliferation, migration, and extracellular matrix remodeling. This process, known as vascular remodeling, is a critical hallmark of atherosclerosis progression (45). The phenotypic transition, proliferation, and migration of VSMC not only influence the stability of atherosclerotic plaques but also directly determine the severity of arterial stenosis. Additionally, End-MT and macrophage polarization collectively regulate vascular remodeling, further promoting vascular stiffening and fibrosis, thereby exacerbating pathological vascular remodeling (46). Cezanne is a DUBs belonging to the OTUD family. It was first characterized in 2001 as a negative regulator of the NF-κB signaling pathway (47). Subsequent studies revealed that it can be induced by various pro-inflammatory cytokines and functions as an inhibitor of the NF-κB signaling pathway, thereby forming a negative feedback loop in inflammatory cytokine signaling (48). Recent studies have revealed that the DUBs Cezanne plays a pivotal role in VSMC proliferation, migration, and arterial remodeling. Specifically, Cezanne regulates the Wnt/β-catenin signaling pathway by deubiquitinating β-catenin, thereby modulating the expression of cysteine-rich protein 61. Furthermore, analyses of human atherosclerotic tissue samples have provided additional evidence supporting the role of Cezanne in disease progression (49). The Wnt/β-catenin signaling pathway plays a crucial role in the development and pathological remodeling of the cardiovascular system. Dysregulation of this pathway is closely associated with the onset and progression of various cardiovascular diseases (50). Targeting β-catenin for therapeutic intervention has shown promise in cancer research (51), and is gaining attention for its potential in cardiovascular diseases. However, as an intrinsically disordered protein, β-catenin lacks well-defined drug-binding pockets and exhibits poor metabolic stability, posing significant challenges for direct targeting (52). Consequently, indirect regulatory strategies have emerged as a promising alternative. DUBs play a crucial role in cardiovascular diseases and offer new avenues for intervention. For instance, Cezanne modulates β-catenin stability and signaling by regulating its ubiquitination status. Developing therapeutics targeting upstream DUBs such as Cezanne may overcome the limitations of direct β-catenin targeting and expand treatment strategies for cardiovascular diseases. Notably, USP10 (53) and USP14 (54) have also been implicated in the regulation of VSMC proliferation and migration, highlighting the broader involvement of DUBs in vascular remodeling. DUBs play a critical regulatory role in both vascular remodeling and vascular calcification during atherosclerosis. These discoveries not only enhance our understanding of the pathological mechanisms underlying atherosclerosis but also provide potential therapeutic targets for anti-atherosclerotic and anti-calcification strategies.
2.4 Clonal hematopoiesis
In recent years, CH has been recognized as an independent risk factor for cardiovascular diseases, alongside traditional risk factors such as smoking and low-density lipoproteins (55). CH is characterized by the presence of mutant hematopoietic cell clones with selective proliferative advantages in peripheral blood (56). It is closely associated with atherosclerosis, primarily by promoting chronic inflammation and exacerbating vascular injury (57). CH is typically driven by somatic mutations in HSCs, such as TET2, DNMT3A, and ASXL1 mutations, which enhance the pro-inflammatory properties of immune cells, including monocytes and macrophages, thereby intensifying vascular inflammation. Among these mutations, TET2 deficiency promotes NLRP3 inflammasome activation, leading to increased secretion of IL-1β and IL-18, which in turn induces endothelial cell injury, foam cell accumulation, and aberrant proliferation and migration of VSMC, thereby accelerating the initiation and progression of atherosclerotic lesions (58). BRCC3 plays a critical role in CH-driven atherosclerosis. As a DUBs, BRCC3 directly deubiquitinates NLRP3, enhancing its stability and sustaining inflammasome activation. Consequently, aberrant BRCC3 activation amplifies pro-inflammatory signaling, exacerbating vascular inflammation and atherosclerosis (59). Targeting BRCC3 represents a promising strategy to mitigate CH-induced atherosclerosis. Inhibiting BRCC3 prevents NLRP3 deubiquitination, thereby reducing excessive inflammasome activation and suppressing IL-1β secretion, ultimately alleviating vascular inflammation associated with CH. Unlike traditional NLRP3 inhibitors such as MCC950 (60), which block NLRP3 assembly and activation by targeting the NACHT domain and have demonstrated efficacy in reducing atherosclerotic plaque formation in murine models (61), BRCC3 inhibition offers distinct advantages. While MCC950 has shown promising anti-inflammatory effects in various disease models (62, 63), its clinical application has been hindered by hepatotoxicity. Moreover, traditional anti-inflammatory therapies typically target key inflammatory cytokines or signaling pathways directly. While effective in reducing inflammation, they may also compromise baseline immune function, increasing the risk of infections and other adverse effects. In contrast, BRCC3 inhibition provides a more refined regulatory mechanism. By selectively modulating NLRP3 stability, BRCC3 inhibition specifically attenuates excessive inflammasome activation driven by TET2 deficiency while preserving baseline inflammasome function. This targeted approach not only minimizes adverse effects but also maintains essential immune defense mechanisms, positioning BRCC3 inhibition as a safer and more precise therapeutic strategy compared to conventional anti-inflammatory treatments.
3 Heart failure
HF is a clinical syndrome characterized by the heart's inability to pump blood effectively to meet the body's metabolic demands or maintain normal blood flow. The causes of HF are diverse, typically involving structural and functional changes in the heart, along with the interactions of systemic factors. Additionally, DUBs play a crucial regulatory role in the onset and progression of HF. The following discussion will explore the mechanisms through which several DUBs contribute to this process.
3.1 Myocardial hypertrophy
The primary function of the heart is to ensure adequate perfusion of peripheral organs, meeting their metabolic demands under both normal and stressed conditions. To accomplish this under increased preload or afterload, the heart and individual cardiomyocytes typically undergo hypertrophy. This hypertrophic response initially develops as an adaptive mechanism to both physiological and pathological stimuli. However, pathological hypertrophy often progresses to HF over time (64). Each form of hypertrophy is initiated and regulated by a variety of cellular signaling pathways. Over the past decade, many previously unrecognized mechanisms have been identified that regulate MH from both positive and negative aspects, including cellular metabolic proliferation (65, 66), immune responses (67, 68), and epigenetic modifications (69). Currently, classic medications, including β-adrenergic blockers and renin-angiotensin-aldosterone system inhibitors, are used to improve pathological ventricular hypertrophy. However, the incidence of HF remains high, suggesting that other signaling pathways or regulatory proteins also play crucial roles in pathological hypertrophy.
Calcium ion (Ca2+) cycling plays a crucial role in the contraction and relaxation of cardiomyocytes. The sarcoplasmic reticulum (SR), as the organelle that stores Ca2+, is primarily responsible for mediating the uptake and release of Ca2+ during the contraction-relaxation process. Dysregulation of sarcoplasmic reticulum function can severely impair Ca2+ cycling, leading to abnormalities in cardiomyocyte function (70). Sarcoplasmic/endoplasmic reticulum Calcium ATPase 2a (SERCA2a) is the cardiac-specific SERCA isoform, responsible for mediating the re-uptake of Ca2+ into the SR during cardiomyocyte diastole. SERCA2a regulates cardiac contractility and relaxation by controlling Ca2+ uptake, thus influencing cardiac function. In HF, the expression and activity of SERCA2a are reduced. Gene therapy aimed at increasing SERCA2a expression in the heart has been shown to be effective (71). USP25 is a product of the 21q11.2 gene, consisting of 25 exons, and can produce three isoforms through alternative splicing: USP25a, USP25b, and USP25m. USP25m is specifically expressed in skeletal muscle and the heart, and it is upregulated during myogenesis, suggesting a potential role for USP25 in cardiac biology. Studies have shown that the deletion of USP25 exacerbates MH and cardiac dysfunction induced by Ang II and TAC. In contrast, restoring USP25 expression significantly improves pathological cardiac hypertrophy. Mechanistically, USP25 regulates the uptake of Ca2+ by deubiquitinating and stabilizing SERCA2a via K48-linked deubiquitination (72), thereby influencing cardiac function.Another member of the OTUD family, OTUD6a, has also been found to be involved in the regulation of MH (73). It primarily regulates the STING-NF-κB inflammatory axis by deubiquitinating the K48-linked Ub chains on STING, thereby maintaining STING stability. Inhibiting OTUD6a significantly reduces the activation of the STING signaling pathway, thus slowing down the progression of MH. Targeting DUBs to regulate MY could become a new direction for the treatment of HF.
3.2 Myocardial infarction
MI is a severe cardiovascular event characterized by ischemic necrosis of cardiomyocytes due to a sudden reduction or complete cessation of coronary blood flow. The underlying pathophysiological mechanisms primarily involve atherosclerotic plaque rupture, subsequent thrombus formation, and coronary artery spasm, ultimately leading to myocardial perfusion impairment and irreversible cardiomyocyte injury. The occurrence of MI can trigger a range of severe cardiovascular complications, including HF (74). OTUD1 plays a crucial role not only in VR but also in cardiac diseases. Wang et al. (75) established MI and HF mouse models by performing MI surgery and administering isoproterenol, respectively, to investigate the role of OTUD1. The study found that the expression levels of OTUD1 were significantly elevated in both models. Knockdown of OTUD1 resulted in substantial structural improvements, alleviation of cardiac damage, and restoration of cardiac function. The mechanism primarily involves OTUD1 deubiquitinating phosphodiesterase 5A (PDE5A) in cardiomyocytes, inhibiting its degradation, and subsequently suppressing the activation of the cGMP-PKG pathway. This process may be a key factor leading to the disruption of calcium homeostasis and MH. cGMP and its kinase PKG play critical roles in regulating cardiomyocytes and cardiac function (76). PDE5A, a member of the phosphodiesterase family, plays an important regulatory role in the cardiovascular system (77). Numerous studies have demonstrated that inhibiting PDE5A can effectively alleviate the progression of HF (78, 79). Therefore, investigating the upstream regulatory proteins of PDE5A and their underlying mechanisms holds significant clinical implications for the treatment of HF. Additionally, another study revealed that OTUD1 can regulate pathological cardiac remodeling and HF by removing K63-linked ubiquitination from STAT3, thereby promoting phosphorylation at the Y705 site, increasing p-STAT3 levels, and facilitating its nuclear translocation (80). Liu et al. identified that OTUD5, another member of the OTUD family closely related to OTUD1, acts as a deubiquitinase for glutathione peroxidase 4 (GPX4) in cardiomyocytes (81), 4-hydroxy-2-nonenal (4-HNE), a reactive aldehyde derived from lipid peroxidation, is commonly produced during oxidative stress. Numerous studies have confirmed that 4-HNE is significantly elevated during MI and is a primary contributor to cell death (82, 83). In this study (81), the authors demonstrated that 4-HNE induces carbonylation of GPX4, impairing its normal function and promoting K48-linked ubiquitination, which leads to proteasomal degradation and triggers ferroptosis in cardiomyocytes. OTUD5 counteracts this process by removing Ub chains from GPX4, thereby protecting it from 4-HNE-mediated damage and degradation. This mechanism effectively prevents ferroptosis and mitigates ischemia-reperfusion-induced cardiac injury (84), Preventing cardiomyocyte damage represents a critical strategy in treating cardiac diseases. As a deubiquitinase for GPX4, OTUD5 plays a vital role in maintaining cellular homeostasis. Its depletion has been shown to exacerbate ferroptosis, evidenced by a significant reduction in the SLC7A11/SLC3A2 complex in OTUD5-silenced cardiomyocytes. This suggests that OTUD5 may also regulate ferroptosis through transcriptional pathways, providing a broader understanding of its cardioprotective mechanisms.Targeting OTUD5 in clinical practice could represent a promising therapeutic strategy for patients undergoing reperfusion therapy after MI.
In CVDs, DUBs critically regulate cardiomyocyte stability and function. They contribute to HF pathogenesis by modulating pathological MH, post-myocardial infarction remodeling, and maladaptive responses. DUBs mediate cardiac injury and remodeling through transcriptional regulation, inflammatory signaling, and cell survival mechanisms. Current research provides novel insights into HF progression mechanisms and identifies DUBs as potential therapeutic targets.
4 Cardiomyopathy
Based on etiology and pathophysiological characteristics, cardiomyopathy can be classified into genetic and acquired types, including dilated cardiomyopathy, hypertrophic cardiomyopathy, restrictive cardiomyopathy, and secondary cardiomyopathy caused by specific underlying conditions (85). The main features of cardiomyopathy include myocardial structural remodeling, ventricular dysfunction, and the potential progression to HF and arrhythmias (86). Among acquired cardiomyopathies, toxic and metabolic factors can significantly affect myocardial function. Doxorubicin (DOX), due to its potent anticancer properties, is widely used in clinical practice. However, its severe cardiotoxicity limits the clinical doses that can be administered. DOX-induced cardiotoxicity is a common form of drug-induced cardiomyopathy in clinical practice, primarily characterized by oxidative stress injury in cardiomyocytes, mitochondrial dysfunction, and impaired cardiac contractility. Therefore, studying its toxic mechanisms and developing targeted prevention or treatment strategies are currently key focuses of both clinical and basic research (87). OTUD1 has been found to deubiquitinate c-MYC, and inhibiting OTUD1 can effectively protect mice from DOX-induced cardiac dysfunction (88). Moreover, DUBs such as USP36 (89), USP14 (90), and Cezanne (91) play significant roles in DOX-induced cardiac dysfunction. Although their specific targets and the signaling pathways they modulate differ, the cellular processes ultimately regulated by these DUBs, including apoptosis and oxidative stress, are closely associated with their toxic effects. These mechanisms are key contributors to the cardiac toxicity observed (92). Targeting DUBs to mitigate the cardiac toxicity they induce offers a promising new direction for research and potential therapeutic strategies.
In addition to toxic factors, metabolic abnormalities also play a crucial role in the development of cardiomyopathy. DCM is a specific form of cardiomyopathy caused by diabetes, characterized by cardiomyocyte hypertrophy, fibrosis, cardiac dysfunction, and arrhythmias (93). DUBs play a crucial role in DCM. In the db/db mouse model of diabetes, differences in UPS expression in diabetic myocardial tissue were already observed in young db/db mice, with a significant reduction in the levels of certain DUBs (94). Recent studies have indicated that the primary cause of HF in DCM is the impairment of cardiomyocyte function. Mitochondrial dysfunction is a critical and often overlooked aspect of the pathology and pathogenesis of DCM. Xie et al. Discovered (95), that the expression of USP28 was significantly reduced in the hearts of diabetic patients and db/db mice. Furthermore, compared to the control group, Myh6-Cre+/USP28fl/fl mice showed more severe, progressive heart dysfunction, lipid accumulation, and mitochondrial disruption. Further investigation revealed that USP28 stabilizes peroxisome proliferator-activated receptor α through deubiquitination, which in turn promotes the transcription of mitofusin 2, thus protecting mitochondrial morphology and function and alleviating diabetic HF. Protein C (PC), a natural anticoagulant, is activated to form activated protein C (aPC), which has been found to induce the key deubiquitinase OTUB1 through PAR1 and EPCR signaling pathways. This induction helps maintain the expression of YB-1. The sustained levels of YB-1 inhibit the transcription of MEF2B, thereby protecting the heart from the effects of DCM (96). It is worth noting that Xigris, a drug approved in 2001 for the treatment of sepsis, contains the active ingredient recombinant activated protein C (rAPC). However, it was withdrawn by the FDA due to severe bleeding complications. Recent studies, however, have indicated that aPC plays an important role in I/R injury as well (97). In conclusion, although research on the role of DUBs in DCM is still limited, it is undeniable that targeting DUBs offers a novel approach for treating DCM.
5 Conclusions
The process of deubiquitination plays a critical regulatory role in the onset and progression of CVDs, particularly in regulating key signaling pathways and maintaining cellular homeostasis (Table 1). Clinically, the application of DUBs is primarily focused on cancer, neurodegenerative diseases, inflammation, and immunity. These enzymes mainly regulate protein homeostasis, the cell cycle, DNA repair, and signal transduction, thus participating in many key biological processes. In cancer treatment, deubiquitinase inhibition strategies can generally be divided into two types: stabilization of the enzymatic active site conformation and allosteric inhibition of non-catalytic sites (101). Many newly developed DUB inhibitors, such as Novartis's CSN5i-3 (102) or the already marketed drug pimozide (103), have been found to exhibit significant functions in the field of cancer therapy.
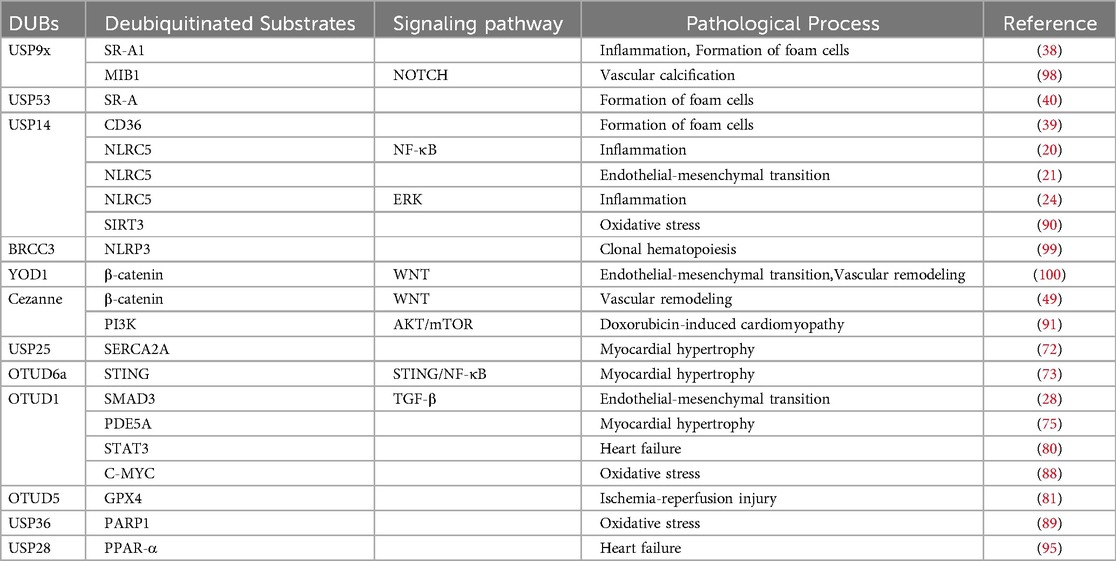
Table 1. Deubiquitinases are involved in cardiovascular pathways.
In cardiovascular research, there has been significant interest in the development of DUB-targeting drugs, but most of these studies remain in the predictive or experimental stages, with no drugs successfully translated for clinical use. Targeting DUBs offers significant advantages over traditional small molecule drugs. Instead of merely inhibiting the active sites of proteins and reducing their activity, DUB inhibitors can completely degrade the target proteins, effectively eliminating the residual activity that often leads to side effects with traditional drugs. Although the role of DUBs in CVDs is gradually being revealed, the specific mechanisms remain incompletely understood. Future research should focus on exploring the relationships between DUBs, signaling pathways, transcription factors, and other biomarkers relevant to CVDs, to further investigate their multifaceted roles in disease progression.
Author contributions
XF: Writing – original draft. CS: Writing – original draft. JC: Writing – review & editing. YL: Writing – review & editing. XL: Writing – review & editing. HT: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from the Hunan Provincial Health High-Level Talent Scientific Research Project (No. R2023131), Natural Science Foundation of Hunan Province (No. 2023JJ30547, 2023JJ60051), Health Research Project of Hunan Provincial Health Commission (No. W20241008, W20243224), Innovation Platform and Talent Program (No. 2023TP1047), Clinical Research 4310 Program of the First Affiliated Hospital of the University of South China (No. 20214310NHYCG03), National Natural Science Foundation of China (No. 62302206); Provincial Key Laboratory of Multi-omics And Artificial Intelligence of Cardiovascular Diseases (No. 2023TP1047); Natural Science Foundation of Hunan Province (No. 2023JJ40594).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
CVDs, cardiovascular diseases; DUBs, deubiquitinases; Ub, ubiquitin; AS, atherosclerosis; VR, vascular remodeling; End-MT, endothelial-to-mesenchymal transition; HF, heart failure; MI, myocardial infarction; MH, myocardial hypertrophy; I/R, ischemia/reperfusion; DCM, diabetic cardiomyopathy; UPS, ubiquitin-proteasome system; E1s, ubiquitin-activating enzymes; E2s, ubiquitin-conjugating enzymes; E3s, ubiquitin ligases; USPs, ubiquitin-specific proteases; OTUs, ovarian tumor proteases; UCHs, ubiquitin C-terminal hydrolases; MJDs, machado-josephin domain proteases; JAMM, JAB1/MPN+/MOV34; MCPIP, monocyte chemoattractant protein-induced protein; MINDYs, motif interacting with Ub-containing novel DUBs; ox-LDL, oxidized low-density lipoprotein; SR-A1, scavenger receptor A1; ALDH2, aldehyde dehydrogenase 2; cGAS, cyclic GMP-AMP synthase; 4-HNE, 4-hydroxy-2-nonenal; LDL, low-density lipoprotein; NF-κB, nuclear factor kappa-B; NLRC5, NOD-like receptor family CARD domain containing 5; ERK, extracellular signal-regulated kinase; CH, clonal hematopoiesis; BRCC3, BRCA1/BRCA2-containing complex subunit 3; NLRP3: NOD-like receptor family pyrin domain containing 3; TGF-β, transforming growth factor-beta; SMAD3, SMAD family member 3; VSMC, vascular smooth muscle cell; DOX, doxorubicin; Ca2+, calcium ion; SR, sarcoplasmic reticulum; SERCA2a, sarcoplasmic/endoplasmic reticulum calcium ATPase 2a; PDE5A, phosphodiesterase 5A; GPX4, glutathione peroxidase 4.
References
1. Zhao D, Liu J, Wang M, Zhang X, Zhou M. Epidemiology of cardiovascular disease in China: current features and implications. Nat Rev Cardiol. (2019) 16:203–12. doi: 10.1038/s41569-018-0119-4
2. Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. (2002) 82:373–428. doi: 10.1152/physrev.00027.2001
3. Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. (1998) 67:425–79. doi: 10.1146/annurev.biochem.67.1.425
4. Swatek KN, Komander D. Ubiquitin modifications. Cell Res. (2016) 26:399–422. doi: 10.1038/cr.2016.39
5. Han S, Wang R, Zhang Y, Li X, Gan Y, Gao F, et al. The role of ubiquitination and deubiquitination in tumor invasion and metastasis. Int J Biol Sci. (2022) 18:2292–303. doi: 10.7150/ijbs.69411
6. Komander D, Clague MJ, Urbé S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. (2009) 10:550–63. doi: 10.1038/nrm2731
7. Clague MJ, Urbé S, Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Biol. (2019) 20:338–52. doi: 10.1038/s41580-019-0099-1
8. Mennerich D, Kubaichuk K, Kietzmann T. DUBs, hypoxia, and cancer. Trends Cancer. (2019) 5:632–53. doi: 10.1016/j.trecan.2019.08.005
9. Li Y, Reverter D. Molecular mechanisms of DUBs regulation in signaling and disease. Int J Mol Sci. (2021) 22:986. doi: 10.3390/IJMS22030986
10. Lange SM, Armstrong LA, Kulathu Y. Deubiquitinases: from mechanisms to their inhibition by small molecules. Mol Cell. (2022) 82:15–29. doi: 10.1016/j.molcel.2021.10.027
11. Cai J, Culley MK, Zhao Y, Zhao J. The role of ubiquitination and deubiquitination in the regulation of cell junctions. Protein Cell. (2018) 9:754–69. doi: 10.1007/s13238-017-0486-3
12. Grabbe C, Husnjak K, Dikic I. The spatial and temporal organization of ubiquitin networks. Nat Rev Mol Cell Biol. (2011) 12:295–307. doi: 10.1038/nrm3099
13. Falk E. Pathogenesis of atherosclerosis. J Am Coll Cardiol. (2006) 47:C7–C12. doi: 10.1016/j.jacc.2005.09.068
14. Ajoolabady A, Pratico D, Lin L, Mantzoros CS, Bahijri S, Tuomilehto J, et al. Inflammation in atherosclerosis: pathophysiology and mechanisms. Cell Death Dis. (2024) 15:817. doi: 10.1038/s41419-024-07166-8
15. Libby P. The changing landscape of atherosclerosis. Nature. (2021) 592:524–33. doi: 10.1038/s41586-021-03392-8
16. Bu LL, Yuan HH, Xie LL, Guo MH, Liao DF, Zheng XL. New dawn for atherosclerosis: vascular endothelial cell senescence and death. Int J Mol Sci. (2023) 24:1–54. doi: 10.3390/ijms242015160
17. Wang X, Shen Y, Shang M, Liu X, Munn LL. Endothelial mechanobiology in atherosclerosis. Cardiovasc Res. (2023) 119:1656–75. doi: 10.1093/cvr/cvad076
18. Cheng H, Zhong W, Wang L, Zhang Q, Ma X, Wang Y, et al. Effects of shear stress on vascular endothelial functions in atherosclerosis and potential therapeutic approaches. Biomed Pharmacother. (2023) 158:114198. doi: 10.1016/j.biopha.2022.114198
19. Zheng D, Liu J, Piao H, Zhu Z, Wei R, Liu K. ROS-triggered endothelial cell death mechanisms: focus on pyroptosis, parthanatos, and ferroptosis. Front Immunol. (2022) 13:1039241. doi: 10.3389/fimmu.2022.1039241
20. Fu Y, Qiu J, Wu J, Zhang L, Wei F, Lu L, et al. USP14-mediated NLRC5 upregulation inhibits endothelial cell activation and inflammation in atherosclerosis. Biochim Biophys Acta Mol Cell Biol Lipids. (2023) 1868:159258. doi: 10.1016/j.bbalip.2022.159258
21. Zhang Z, Guo Q, Ma C, Zhao Z, Shi Q, Yu H, et al. USF1 Transcriptionally activates USP14 to drive atherosclerosis by promoting EndMT through NLRC5/Smad2/3 axis. Mol Med. (2024) 30:32. doi: 10.1186/s10020-024-00798-8
22. Wu Y, Shi T, Li J. NLRC5: a paradigm for NLRs in immunological and inflammatory reaction. Cancer Lett. (2019) 451:92–9. doi: 10.1016/j.canlet.2019.03.005
23. Rui H, Yu H, Chi K, Han Z, Zhu W, Zhang J, et al. ALDH2 Deficiency augments atherosclerosis through the USP14-cGAS-dependent polarization of proinflammatory macrophages. Redox Biol. (2024) 76:103318. doi: 10.1016/j.redox.2024.103318
24. Liu N, Kong T, Chen X, Hu H, Gu H, Liu S, et al. Ubiquitin-specific protease 14 regulates LPS-induced inflammation by increasing ERK1/2 phosphorylation and NF-κB activation. Mol Cell Biochem. (2017) 431:87–96. doi: 10.1007/s11010-017-2978-0
25. Li Y, Dong B, Wang Y, Bi H, Zhang J, Ding C, et al. Inhibition of Usp14 ameliorates renal ischemia-reperfusion injury by reducing Tfap2a stabilization and facilitating mitophagy. Transl Res. (2024) 270:94–103. doi: 10.1016/j.trsl.2024.04.002
26. Moon S, Muniyappan S, Lee SB, Lee BH. Small-Molecule inhibitors targeting proteasome-associated deubiquitinases. Int J Mol Sci. (2021) 22:6213. doi: 10.3390/ijms22126213
27. Alvandi Z, Bischoff J. Endothelial-mesenchymal transition in cardiovascular disease. Arterioscler Thromb Vasc Biol. (2021) 41:2357–69. doi: 10.1161/ATVBAHA.121.313788
28. Huang Z, Shen S, Wang M, Li W, Wu G, Huang W, et al. Mouse endothelial OTUD1 promotes angiotensin II-induced vascular remodeling by deubiquitinating SMAD3. EMBO Rep. (2023) 24:e56135. doi: 10.15252/embr.202256135
29. Humeres C, Venugopal H, Frangogiannis NG. Smad-dependent pathways in the infarcted and failing heart. Curr Opin Pharmacol. (2022) 64:102207. doi: 10.1016/j.coph.2022.102207
30. Graf K, Schaefer-Graf UM. Is Smad3 the key to inflammation and fibrosis in hypertensive heart disease? Hypertension. (2010) 55:1088–9. doi: 10.1161/HYPERTENSIONAHA.110.150466
31. Wang M, Wang M, Zhao J, Xu H, Xi Y, Yang H. Dengzhan Shengmai capsule attenuates cardiac fibrosis in post-myocardial infarction rats by regulating LTBP2 and TGF-β1/Smad3 pathway. Phytomedicine. (2023) 116:154849. doi: 10.1016/j.phymed.2023.154849
32. Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. (2005) 19:2783–810. doi: 10.1101/gad.1350705
33. Akoumianakis I, Polkinghorne M, Antoniades C. Non-canonical WNT signalling in cardiovascular disease: mechanisms and therapeutic implications. Nat Rev Cardiol. (2022) 19:783–97. doi: 10.1038/s41569-022-00718-5
34. Moncla LM, Briend M, Bossé Y, Mathieu P. Calcific aortic valve disease: mechanisms, prevention and treatment. Nat Rev Cardiol. (2023) 20:546–59. doi: 10.1038/s41569-023-00845-7
35. Jiang H, Zhou Y, Nabavi SM, Sahebkar A, Little PJ, Xu S, et al. Mechanisms of oxidized LDL-mediated endothelial dysfunction and its consequences for the development of atherosclerosis. Front Cardiovasc Med. (2022) 9:925923. doi: 10.3389/fcvm.2022.925923
36. Manning-Tobin JJ, Moore KJ, Seimon TA, Bell SA, Sharuk M, Alvarez-Leite JI, et al. Loss of SR-A and CD36 activity reduces atherosclerotic lesion complexity without abrogating foam cell formation in hyperlipidemic mice. Arterioscler Thromb Vasc Biol. (2009) 29:19–26. doi: 10.1161/ATVBAHA.108.176644
37. Marfella R, D'Amico M, Di Filippo C, Baldi A, Siniscalchi M, Sasso FC, et al. Increased activity of the ubiquitin-proteasome system in patients with symptomatic carotid disease is associated with enhanced inflammation and may destabilize the atherosclerotic plaque. J Am Coll Cardiol. (2006) 47:2444–55. doi: 10.1016/j.jacc.2006.01.073
38. Wang B, Tang X, Yao L, Wang Y, Chen Z, Li M, et al. Disruption of USP9X in macrophages promotes foam cell formation and atherosclerosis. J Clin Invest. (2022) 132:1–15. doi: 10.1172/JCI154217
39. Zhang F, Xia X, Chai R, Xu R, Xu Q, Liu M, et al. Inhibition of USP14 suppresses the formation of foam cell by promoting CD36 degradation. J Cell Mol Med. (2020) 24:3292–302. doi: 10.1111/jcmm.15002
40. Liu X, Zheng T, Zhang Y, Zhao Y, Liu F, Dai S, et al. Endothelial dickkopf-1 promotes smooth muscle cell-derived foam cell formation via USP53-mediated deubiquitination of SR-A during atherosclerosis. Int J Biol Sci. (2024) 20:2943–64. doi: 10.7150/ijbs.91957
41. Wu J, He S, Song Z, Chen S, Lin X, Sun H, et al. Macrophage polarization states in atherosclerosis. Front Immunol. (2023) 14:1185587. doi: 10.3389/fimmu.2023.1185587
42. Wan Y, Mo L, Huang H, Mo L, Zhu W, Li W, et al. Cadmium contributes to atherosclerosis by affecting macrophage polarization. Food Chem Toxicol. (2023) 173:113603. doi: 10.1016/j.fct.2023.113603
43. Hou P, Fang J, Liu Z, Shi Y, Agostini M, Bernassola F, et al. Macrophage polarization and metabolism in atherosclerosis. Cell Death Dis. (2023) 14:691. doi: 10.1038/s41419-023-06206-z
44. Zhao X, Huang XH, Dong XH, Wang YH, Yang HX, Wang Y, et al. Deubiquitinase Mysm1 regulates macrophage survival and polarization. Mol Biol Rep. (2018) 45:2393–401. doi: 10.1007/s11033-018-4405-3
45. Humphrey JD. Mechanisms of vascular remodeling in hypertension. Am J Hypertens. (2021) 34:432–41. doi: 10.1093/ajh/hpaa195
46. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. (2016) 118:692–702. doi: 10.1161/CIRCRESAHA.115.306361
47. Evans PC, Taylor ER, Coadwell J, Heyninck K, Beyaert R, Kilshaw PJ. Isolation and characterization of two novel A20-like proteins. Biochem J. (2001) 357:617–23. doi: 10.1042/bj3570617
48. Enesa K, Zakkar M, Chaudhury H, Luong Le A, Rawlinson L, Mason JC, et al. NF-kappaB suppression by the deubiquitinating enzyme Cezanne: a novel negative feedback loop in pro-inflammatory signaling. J Biol Chem. (2008) 283:7036–45. doi: 10.1074/jbc.M708690200
49. An W, Luong LA, Bowden NP, Yang M, Wu W, Zhou X, et al. Cezanne is a critical regulator of pathological arterial remodelling by targeting β-catenin signalling. Cardiovasc Res. (2022) 118:638–53. doi: 10.1093/cvr/cvab056
50. Liu J, Xiao Q, Xiao J, Niu C, Li Y, Zhang X, et al. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct Target Ther. (2022) 7:3. doi: 10.1038/s41392-021-00762-6
51. Song P, Gao Z, Bao Y, Chen L, Huang Y, Liu Y, et al. Wnt/β-catenin signaling pathway in carcinogenesis and cancer therapy. J Hematol Oncol. (2024) 17:46. doi: 10.1186/s13045-024-01563-4
52. Biesaga M, Frigolé-Vivas M, Salvatella X. Intrinsically disordered proteins and biomolecular condensates as drug targets. Curr Opin Chem Biol. (2021) 62:90–100. doi: 10.1016/j.cbpa.2021.02.009
53. Xia X, Liu X, Chai R, Xu Q, Luo Z, Gu J, et al. USP10 Exacerbates neointima formation by stabilizing Skp2 protein in vascular smooth muscle cells. J Biol Chem. (2021) 297:101258. doi: 10.1016/j.jbc.2021.101258
54. Xia X, Liu X, Xu Q, Gu J, Ling S, Liu Y, et al. USP14 Deficiency inhibits neointima formation following vascular injury via degradation of Skp2 protein. Cell Death Discov. (2024) 10:295. doi: 10.1038/s41420-024-02069-1
55. Dorsheimer L, Assmus B, Rasper T, Ortmann CA, Ecke A, Abou-El-Ardat K, et al. Association of mutations contributing to clonal hematopoiesis with prognosis in chronic ischemic heart failure. JAMA Cardiol. (2019) 4:25–33. doi: 10.1001/jamacardio.2018.3965
56. Jaiswal S, Ebert BL. Clonal hematopoiesis in human aging and disease. Science. (2019) 366:4673. doi: 10.1126/science.aan4673
57. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. (2017) 377:111–21. doi: 10.1056/NEJMoa1701719
58. Fuster JJ, Walsh K. Somatic mutations and clonal hematopoiesis. Circ Res. (2018) 122:523–32. doi: 10.1161/CIRCRESAHA.117.312115
59. Yalcinkaya M, Liu W, Thomas LA, Olszewska M, Xiao T, Abramowicz S, et al. BRCC3-mediated NLRP3 deubiquitylation promotes inflammasome activation and atherosclerosis in Tet2 clonal hematopoiesis. Circulation. (2023) 148:1764–77. doi: 10.1161/CIRCULATIONAHA.123.065344
60. Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. (2015) 21:248–55. doi: 10.1038/nm.3806
61. Zeng W, Wu D, Sun Y, Suo Y, Yu Q, Zeng M, et al. The selective NLRP3 inhibitor MCC950 hinders atherosclerosis development by attenuating inflammation and pyroptosis in macrophages. Sci Rep. (2021) 11:19305. doi: 10.1038/s41598-021-98437-3
62. Ward R, Li W, Abdul Y, Jackson L, Dong G, Jamil S, et al. NLRP3 Inflammasome inhibition with MCC950 improves diabetes-mediated cognitive impairment and vasoneuronal remodeling after ischemia. Pharmacol Res. (2019) 142:237–50. doi: 10.1016/j.phrs.2019.01.035
63. Naeem A, Prakash R, Kumari N, Ali Khan M, Quaiyoom Khan A, Uddin S, et al. MCC950 Reduces autophagy and improves cognitive function by inhibiting NLRP3-dependent neuroinflammation in a rat model of Alzheimer’s disease. Brain Behav Immun. (2024) 116:70–84. doi: 10.1016/j.bbi.2023.11.031
64. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. (2018) 15:387–407. doi: 10.1038/s41569-018-0007-y
65. Zhang Z, Tang J, Song J, Xie M, Liu Y, Dong Z, et al. Elabela alleviates ferroptosis, myocardial remodeling, fibrosis and heart dysfunction in hypertensive mice by modulating the IL-6/STAT3/GPX4 signaling. Free Radic Biol Med. (2022) 181:130–42. doi: 10.1016/j.freeradbiomed.2022.01.020
66. Foglia MJ, Poss KD. Building and re-building the heart by cardiomyocyte proliferation. Development. (2016) 143:729–40. doi: 10.1242/dev.132910
67. Dingar D, Merlen C, Grandy S, Gillis MA, Villeneuve LR, Mamarbachi AM, et al. Effect of pressure overload-induced hypertrophy on the expression and localization of p38 MAP kinase isoforms in the mouse heart. Cell Signal. (2010) 22:1634–44. doi: 10.1016/j.cellsig.2010.06.002
68. Nawaito SA, Dingar D, Sahadevan P, Hussein B, Sahmi F, Shi Y, et al. MK5 Haplodeficiency attenuates hypertrophy and preserves diastolic function during remodeling induced by chronic pressure overload in the mouse heart. Am J Physiol Heart Circ Physiol. (2017) 313:H46–58. doi: 10.1152/ajpheart.00597.2016
69. Tingare A, Thienpont B, Roderick HL. Epigenetics in the heart: the role of histone modifications in cardiac remodelling. Biochem Soc Trans. (2013) 41:789–96. doi: 10.1042/BST20130012
70. Zhihao L, Jingyu N, Lan L, Michael S, Rui G, Xiyun B, et al. SERCA2a: a key protein in the Ca(2+) cycle of the heart failure. Heart Fail Rev. (2020) 25:523–35. doi: 10.1007/s10741-019-09873-3
71. Shareef MA, Anwer LA, Poizat C. Cardiac SERCA2A/B: therapeutic targets for heart failure. Eur J Pharmacol. (2014) 724:1–8. doi: 10.1016/j.ejphar.2013.12.018
72. Ye B, Zhou H, Chen Y, Luo W, Lin W, Zhao Y, et al. USP25 Ameliorates pathological cardiac hypertrophy by stabilizing SERCA2a in cardiomyocytes. Circ Res. (2023) 132:465–80. doi: 10.1161/CIRCRESAHA.122.321849
73. Fang Z, Han J, Lin L, Ye B, Qu X, Zhang Y, et al. Deubiquitinase OTUD6a drives cardiac inflammation and hypertrophy by deubiquitination of STING. Biochim Biophys Acta Mol Basis Dis. (2024) 1870:167061. doi: 10.1016/j.bbadis.2024.167061
74. Thygesen K, Alpert JS, White HD. Universal definition of myocardial infarction. J Am Coll Cardiol. (2007) 50:2173–95. doi: 10.1016/j.jacc.2007.09.011
75. Wang Q, Liang S, Qian J, Xu J, Zheng Q, Wang M, et al. OTUD1 Promotes isoprenaline- and myocardial infarction-induced heart failure by targeting PDE5A in cardiomyocytes. Biochim Biophys Acta Mol Basis Dis. (2024) 1870:167018. doi: 10.1016/j.bbadis.2024.167018
76. Inserte J, Garcia-Dorado D. The cGMP/PKG pathway as a common mediator of cardioprotection: translatability and mechanism. Br J Pharmacol. (2015) 172:1996–2009. doi: 10.1111/bph.12959
77. Kass DA, Champion HC, Beavo JA. Phosphodiesterase type 5. Circ Res. (2007) 101:1084–95. doi: 10.1161/CIRCRESAHA.107.162511
78. Liao M, Xie Q, Zhao Y, Yang C, Lin C, Wang G, et al. Main active components of Si-Miao-Yong-An decoction (SMYAD) attenuate autophagy and apoptosis via the PDE5A-AKT and TLR4-NOX4 pathways in isoproterenol (ISO)-induced heart failure models. Pharmacol Res. (2022) 176:106077. doi: 10.1016/j.phrs.2022.106077
79. Kukreja RC, Ockaili R, Salloum F, Yin C, Hawkins J, Das A, et al. Cardioprotection with phosphodiesterase-5 inhibition–a novel preconditioning strategy. J Mol Cell Cardiol. (2004) 36:165–73. doi: 10.1016/j.yjmcc.2003.11.001
80. Wang M, Han X, Yu T, Wang M, Luo W, Zou C, et al. OTUD1 Promotes pathological cardiac remodeling and heart failure by targeting STAT3 in cardiomyocytes. Theranostics. (2023) 13:2263–80. doi: 10.7150/thno.83340
81. Liu L, Pang J, Qin D, Li R, Zou D, Chi K, et al. Deubiquitinase OTUD5 as a novel protector against 4-HNE-triggered ferroptosis in myocardial ischemia/reperfusion injury. Adv Sci. (2023) 10:e2301852. doi: 10.1002/advs.202301852
82. Zhai X, Wang W, Sun S, Han Y, Li J, Cao S, et al. 4-Hydroxy-2-nonenal promotes cardiomyocyte necroptosis via stabilizing receptor-interacting serine/threonine-protein kinase 1. Front Cell Dev Biol. (2021) 9:721795. doi: 10.3389/fcell.2021.721795
83. Mali VR, Palaniyandi SS. Regulation and therapeutic strategies of 4-hydroxy-2-nonenal metabolism in heart disease. Free Radic Res. (2014) 48:251–63. doi: 10.3109/10715762.2013.864761
84. Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. (2019) 99:1765–817. doi: 10.1152/physrev.00022.2018
85. Brieler J, Breeden MA, Tucker J. Cardiomyopathy: an overview. Am Fam Physician. (2017) 96:640–6.29431384
86. Lukas Laws J, Lancaster MC, Ben Shoemaker M, Stevenson WG, Hung RR, Wells Q, et al. Arrhythmias as presentation of genetic cardiomyopathy. Circ Res. (2022) 130:1698–722. doi: 10.1161/CIRCRESAHA.122.319835
87. Carvalho C, Santos RX, Cardoso S, Correia S, Oliveira PJ, Santos MS, et al. Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem. (2009) 16:3267–85. doi: 10.2174/092986709788803312
88. Xu F, Zang T, Chen H, Zhou C, Wang R, Yu Y, et al. Deubiquitinase OTUB1 regulates doxorubicin-induced cardiotoxicity via deubiquitinating c-MYC. Cell Signal. (2024) 113:110937. doi: 10.1016/j.cellsig.2023.110937
89. Wang D, Jiang Z, Kan J, Jiang X, Pan C, You S, et al. USP36-mediated PARP1 deubiquitination in doxorubicin-induced cardiomyopathy. Cell Signal. (2024) 117:111070. doi: 10.1016/j.cellsig.2024.111070
90. Zhang Z, Jin B, Zhang Y, Yang M, Wang C, Zhu Y, et al. USP14 Modulates cell pyroptosis and ameliorates doxorubicin-induced cardiotoxicity by deubiquitinating and stabilizing SIRT3. Free Radic Biol Med. (2024) 225:741–57. doi: 10.1016/j.freeradbiomed.2024.10.302
91. Zhang J, Zha Y, Jiao Y, Li Y, Zhang S. Protective role of cezanne in doxorubicin-induced cardiotoxicity by inhibiting autophagy, apoptosis and oxidative stress. Toxicology. (2023) 485:153426. doi: 10.1016/j.tox.2023.153426
92. Kong CY, Guo Z, Song P, Zhang X, Yuan YP, Teng T, et al. Underlying the mechanisms of doxorubicin-induced acute cardiotoxicity: oxidative stress and cell death. Int J Biol Sci. (2022) 18:760–70. doi: 10.7150/ijbs.65258
93. Dillmann WH. Diabetic cardiomyopathy. Circ Res. (2019) 124:1160–2. doi: 10.1161/CIRCRESAHA.118.314665
94. Nahum-Ankonina O, Kurtzwald-Josefson E, Ciechanover A, Waldman M, Shwartz-Rohaker O, Hochhauser E, et al. Ubiquitin proteasome system role in diabetes-induced cardiomyopathy. Int J Mol Sci. (2023) 24:1–15. doi: 10.3390/ijms242015376
95. Xie SY, Liu SQ, Zhang T, Shi WK, Xing Y, Fang WX, et al. USP28 serves as a key suppressor of mitochondrial morphofunctional defects and cardiac dysfunction in the diabetic heart. Circulation. (2024) 149:684–706. doi: 10.1161/CIRCULATIONAHA.123.065603
96. Zhong X, Wang T, Xie Y, Wang M, Zhang W, Dai L, et al. Activated protein C ameliorates diabetic cardiomyopathy via modulating OTUB1/YB-1/MEF2B axis. Front Cardiovasc Med. (2021) 8:758158. doi: 10.3389/fcvm.2021.758158
97. Nazir S, Gadi I, Al-Dabet MM, Elwakiel A, Kohli S, Ghosh S, et al. Cytoprotective activated protein C averts Nlrp3 inflammasome-induced ischemia-reperfusion injury via mTORC1 inhibition. Blood. (2017) 130:2664–77. doi: 10.1182/blood-2017-05-782102
98. Majumdar U, Manivannan S, Basu M, Ueyama Y, Blaser MC, Cameron E, et al. Nitric oxide prevents aortic valve calcification by S-nitrosylation of USP9X to activate NOTCH signaling. Sci Adv. (2021) 7:1–15. doi: 10.1126/sciadv.abe3706
99. Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell. (2013) 49:331–8. doi: 10.1016/j.molcel.2012.11.009
100. Lin WT, Jiang YC, Mei YL, Chen YH, Zheng ZZ, Han X, et al. Endothelial deubiquinatase YOD1 mediates ang II-induced vascular endothelial-mesenchymal transition and remodeling by regulating β-catenin. Acta Pharmacol Sin. (2024) 45:1618–31. doi: 10.1038/s41401-024-01278-9
101. Ren J, Yu P, Liu S, Li R, Niu X, Chen Y, et al. Deubiquitylating enzymes in cancer and immunity. Adv Sci. (2023) 10:e2303807. doi: 10.1002/advs.202303807
102. Schlierf A, Altmann E, Quancard J, Jefferson AB, Assenberg R, Renatus M, et al. Targeted inhibition of the COP9 signalosome for treatment of cancer. Nat Commun. (2016) 7:13166. doi: 10.1038/ncomms13166
Keywords: deubiquitinases, deubiquitination, ubiquitin-proteasome system, signaling pathways, proteostasis, cardiovascular disease
Citation: Fei X, Song C, Cui J, Li Y, Lei X and Tang H (2025) The role of deubiquitinases in cardiovascular diseases: mechanisms and therapeutic implications. Front. Cardiovasc. Med. 12:1582049. doi: 10.3389/fcvm.2025.1582049
Received: 23 February 2025; Accepted: 22 April 2025;
Published: 1 May 2025.
Edited by:
Tamer M. A. Mohamed, Baylor College of Medicine, United StatesReviewed by:
Jianjun Jiang, Zhejiang Taizhou Hospital, ChinaZhiming Zhang, Wenzhou Medical University, China, in collaboration with reviewer JJ
Ismawati Ismawati, Riau University, Indonesia
Copyright: © 2025 Fei, Song, Cui, Li, Lei and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huifang Tang, dGFuZ2h1aWZhbmc5OTlAMTYzLmNvbQ==; Xiaoyong Lei, TGVpeF95b25nQDE2My5jb20=
†These authors have contributed equally to this work