 Alexandra Jimenez-Armijo1†
Alexandra Jimenez-Armijo1† Khadja Oumensour2†
Khadja Oumensour2† Bouchra Bousfiha2
Bouchra Bousfiha2 Tristan Rey1,3
Tristan Rey1,3 Virginie Laugel-Haushalter1,4
Virginie Laugel-Haushalter1,4 Agnès Bloch-Zupan1,4,5*‡Samira El Arabi2‡
Agnès Bloch-Zupan1,4,5*‡Samira El Arabi2‡- 1Université de Strasbourg, Institut de Génétique et de Biologie Moléculaire et Cellulaire, INSERM U1258, CNRS-UMR7104, Illkirch-Graffenstaden, France
- 2Faculté de médecine dentaire de l'Université Hassan II, Service de Pédodontie - Centre de Consultations et Traitements Dentaires, Casablanca, Morocco
- 3Hôpitaux Universitaires de Strasbourg, Laboratoires de diagnostic génétique, Institut de Génétique Médicale d'Alsace, Strasbourg, France
- 4Université de Strasbourg, Faculté de Chirurgie Dentaire, Strasbourg, France
- 5Hôpitaux Universitaires de Strasbourg, Pôle de Médecine et Chirurgie Bucco-Dentaires, Centre de référence des maladies rares orales et dentaires, CRMR O-Rares, Filière Santé Maladies rares TETE COU, European Reference Network ERN CRANIO, Strasbourg, France
This paper reports the case of a Moroccan girl with a phenotype within the clinical spectrum of both Hallermann-Streiff (HSS, OMIM 234100) and Oculodentodigital Dysplasia (ODDD, OMIM 164200) syndromes. The patient presented with repeated dental abscesses and severe early childhood caries. She had no learning deficit nor psychomotor regression; however, a language delay was noted. She also presented with obstructive sleep apnea syndrome and specific craniofacial features pathognomonic of HSS. Radiographic examination showed enamel and dentin defects, giving a ghost-like tooth appearance. Several clinical features of ODDD overlap those of HSS and may confuse diagnosis, considering that the inheritance of HSS is not described yet. The diagnostic odyssey of this patient ended with the identification by exome sequencing of a novel homozygous alteration in the GJA1 gene. A missense substitution in exon 2 [Chr6(GRCh37): g.121768554C>G NM_000165.4: c.561C>G p.Cys187Trp] was identified by whole-exome sequencing (WES), suggesting a diagnosis of ODDD. This is the first report of a homozygous mutation affecting the second extracellular loop of the CX43 protein.
Introduction
Hallermann-Streiff syndrome (HSS, OMIM 234100) is a highly recognizable rare disease characterized by developmental delay, proportionate short stature, skeletal, chest, respiratory and skin defects, as well as distinct craniofacial features such as sparse hair, skin atrophy over scalp and nose, brachycephaly, frontal bossing, micrognathia, low set ears, microphthalmia, small pointed nose, microstomia, and developmental tooth anomalies. The inheritance pattern of this rare disease remains unsolved, with most cases having been described as isolated (1).
Several clinical features of HSS, such as microphthalmia, small nose, hypotrichosis and dental anomalies, overlap those found in oculodentodigital dysplasia (ODDD) confusing differential diagnosis. Alterations in Gap Junction protein Alpha 1 gene (GJA1) (OMIM 121014, HGNC ID: 4274, NM_000165.5), which is located on chromosome 6q22.31 and organized in two exons and one intron, have been reported as causative for ODDD. ODDD mode of inheritance has mostly been autosomal dominant (AD, OMIM 164200) but also occasionally autosomal recessive (AR, OMIM 257850) (2–4). With AD transmission, one affected parent transmits the variant to an affected child, and the variant may lead either to the complete spectrum of malformations or to milder manifestations. Only few individuals with recessive variants in GJA1 have been reported (4–7), and a single one was described as presenting an overlapping HSS/ODDD spectrum associated to variant c.227G>A, p.R76H (8). In general, more severe clinical features are observed in AR ODDD (9).
GJA1 encodes the transmembrane, gap junction protein connexin 43 (CX43) that allows the exchange of ions and small molecules between cells (9, 10). This protein is composed of four α-helical transmembrane domains, two extracellular loops, and a cytoplasmic loop with the amino and carboxyl termini in the cytoplasm (11). Alterations in GJA1 could produce protein products with a proper size but with different ways of assembling into functional gap junction plaques. A reduction in CX43 cell-to-cell communication caused by these variants contributes to the many symptoms associated with ODDD (12).
AD ODDD individuals present with a variable phenotype, including a characteristic facial appearance, and variable eye and digital anomalies (bilateral syndactyly, camptodactyly, and clinodactyly) (9). Other features such as hearing loss and neurologic abnormalities like seizures can occur as well (6). The craniofacial dysmorphic features (see Table 1) include microphthalmia, hypertelorism, thin nose, hypotrichosis, and dental anomalies like enamel dysplasia, tooth agenesis, and microdontia. A high prevalence of caries is also reported.
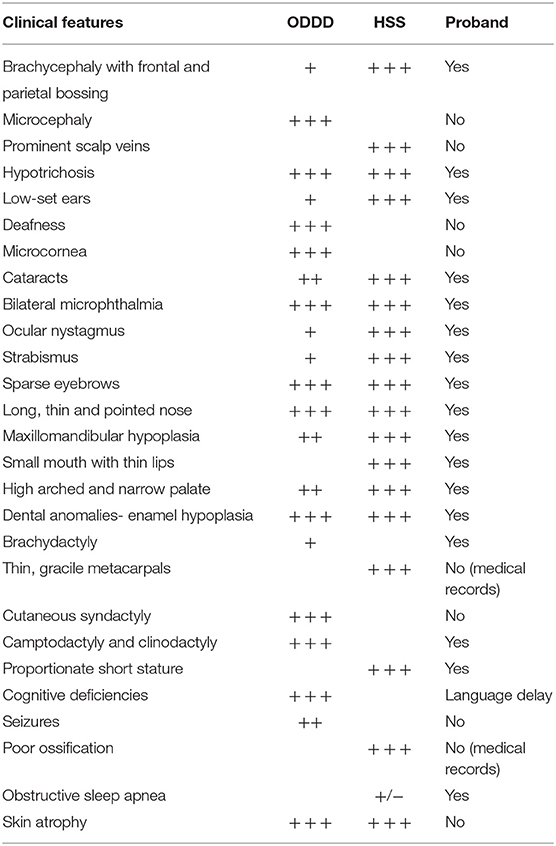
Table 1. Clinical features in HSS, ODDD, and the proband.
In this paper, we report the case of a Moroccan girl with a phenotype within the spectrum of both HSS and ODDD, whose diagnostic odyssey ended with the identification of a novel homozygous GJA1 variant.
Case Presentation
This clinical case was conducted in compliance with the CARE guidelines.
Parents gave written informed consent for the transfer of clinical data in D4/phenodent database (www.phenodent.org), genetic testing (GenoDENT), DNA biobanking (https://clinicaltrials.gov: NCT01746121 et NCT02397824; DC-2012-1677 et DC-2012-1002), and the current publication.
A 3-year-old girl was examined in the pediatric dentistry unit because of repeated dental abscesses. She was born from second-degree consanguineous parents and has two siblings with no reported health problems. The medical history showed a normal pregnancy and a neonatal period without difficulties. The patient had no learning deficit nor psychomotor regression; however, a language delay was noted. She also presented obstructive sleep apnea syndrome.
Extraoral examination showed specific craniofacial features (Table 1, Figure 1) pathognomonic of HSS. Intraoral examination revealed poor oral hygiene with the presence of abundant dental plaque and gingival inflammation. Parents experienced great difficulties in brushing the child teeth.
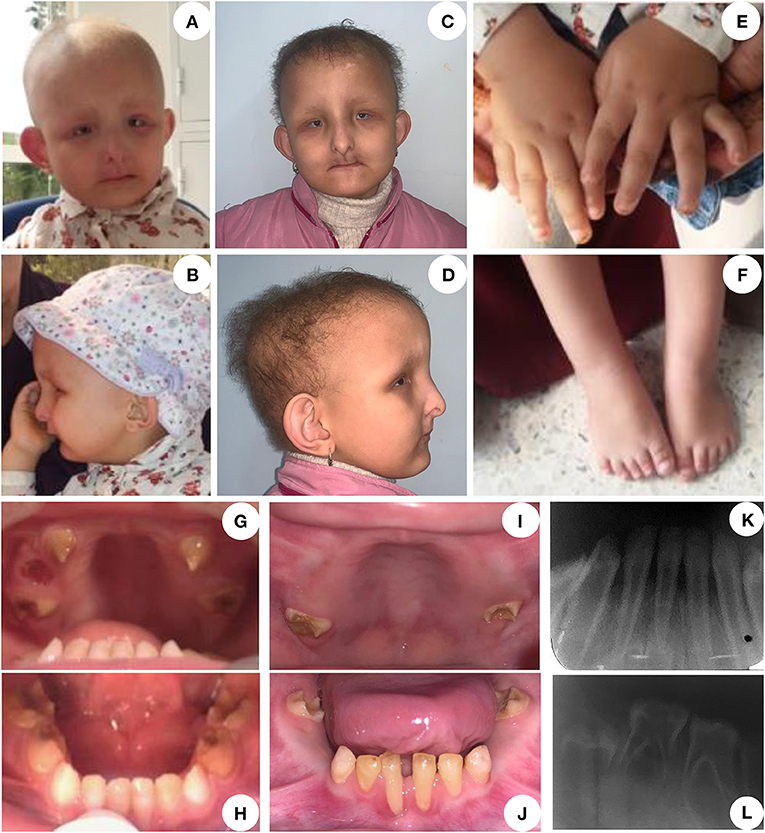
Figure 1. Clinical features of the proband including intraoral examination and x-rays of primary and permanent dentitions. (A,B) First medical visit at 3-year-old. (A) Hypotrichosis, bilateral microphthalmia with nystagmus, hypertelorism. (B) A specific facial gestalt with convex nasal ridge, low set ears and mandibular retrognathia. (C,D) Patient at 6-year-old. Lack of lip support and accentuation of the nasolabial folds due to multiple teeth extractions. (E,F) The patient has brachydactyly, hand bilateral fifth finger clinodactyly, no signs of syndactyly. (G,H) Oral cavity at 3 years old. Primary teeth are affected by a severe form of early childhood caries. Teeth have a brownish yellow color and show significant brittleness of the dental structures. Temporary maxillary canines (G) are thin and conoid. (I,J) Intraoral images at 6-year-old. All primary teeth with dental abscess were extracted. A gingival recession was visible on 81 and 71. (K,L) Intraoral radiographs taken at 3 years of age confirmed hard tissue defects, thin dentin walls, short roots with gaping apices and large pulps giving the appearance of phantom teeth.
All primary teeth presented brownish-yellow dyschromia, and a diagnosis of severe early childhood caries was proposed. However, significant fragility of dental structures suggested underlying mineralized tissue developmental anomalies (Figures 1G–J). Primary canines showed clearly hypoplastic enamel (Figure 1G).
Radiographic examination at 3 and 6 years of age confirmed the presence of enamel and dentin defects, with the teeth having a ghost-like appearance (very thin, under-mineralized hard tissues, short roots, and large pulps) (Figures 1K,L). Hypomineralized enamel and dentin were also clearly visible in first unerupted permanent molars (Figure 1L).
Before patient's oral management, hemostasis tests and a complete blood count were requested. Due to recurrent abscesses, primary tooth extractions were performed. Given the patient's anxiety, prosthetic rehabilitation was postponed until she was more cooperative. Regular follow-up was scheduled after 1 month and then every 3 months for preventive treatment to avoid appearance of new carious lesions on the remaining teeth. After 2 years, the child's cooperation allowed a prosthetic rehabilitation with removable denture, to be carried out.
To clarify the genetic etiology causing the HSS phenotype and associated dental anomalies, whole- exome sequencing (WES) analysis was carried out. Genomic DNA was isolated from the saliva of the proband and her parents, using the Oragene® DNA OG-250 commercial kits (DNA Genotek Inc., Ottawa, Ont, Canada) according to the manufacturer's protocol. WES was performed by Integragen (Evry, France). WES and bioinformatic analysis were done following the protocol described in (13). The variants were filtered excluding (1) variants represented with an allele frequency of more than 1% in public variation databases-including the 1,000 Genomes (14), the gnomAD database (15), (2) variants in the 5' or 3' UTR, (3) variants with intronic locations and no prediction of local splice effect, and (4) synonymous variants without prediction of local splice effect. The analysis was focused on homozygous and compound heterozygous variants consistant with a recessive mode of inheritance and/or on de novo variants. After variant filtration, 15 homozygous, 5 compound heterozygous and 2 de novo variants in 21 genes were left for investigation. None of them, excepting variants in GJA1 were consistent with the patient's phenotype see Supplementary Tables 1, 2.
A novel homozygous variant in the GJA1 gene, with a missense substitution in exon 2 [Chr6(GRCh37): g.121768554C>G NM_000165.4: c.561C>G p.Cys187Trp], was therefore identified. Parents were both heterozygous for the variant. Sanger sequencing was used to confirm WES data (Figure 2). Primer pair for exon 2 of GJA1 was designed using AmplifX 1.5.4 (sense: TCTATCTTTGAGGTGGCCTTCT; antisense: CCACAATGGCTAGTGGCTGTAA). The amplification of the region of interest was performed on genomic DNA template, from both parents and proband, followed by bidirectional Sanger sequencing. This analysis comforted the exome sequencing results.
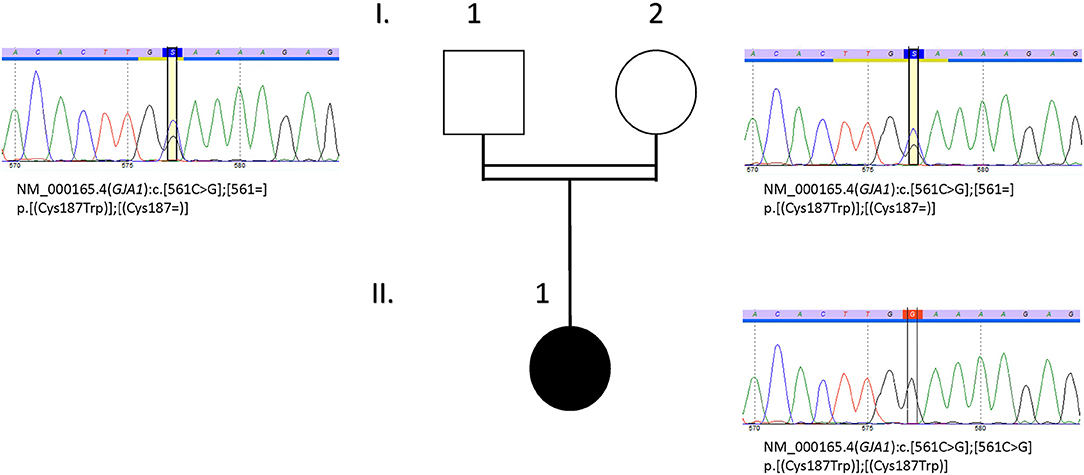
Figure 2. Pedigree and DNA sequencing chromatograms of the family. Sanger sequencing was done to confirm familial segregation of the variant NM_000165.4 (GJA1):c.[561C>G] identified by exome sequencing. Parents (I.1 and I.2) were heterozygous and the index case (II.1) was homozygous for this variant.
The variant (c.561C>G p.Cys187Trp) affected the second extracellular loop of the CX43 protein (Figure 3). The alignment of this domain sequence with sequences of other species showed that it was largely conserved between species, with the altered cysteine (Cys187) being highly conserved (Figure 3C). This variant was also predicted to be deleterious by SIFT (16) and by Polyphen2 (Polymorphism Phenotyping v2) (PPH2) (17). The variant is also not present in healthy people gnomAD database (15). Collectively, these findings suggested that this amino acid had an important function. This variant was therefore classified as class 4, likely pathogenic, according to ACMG criteria (18).
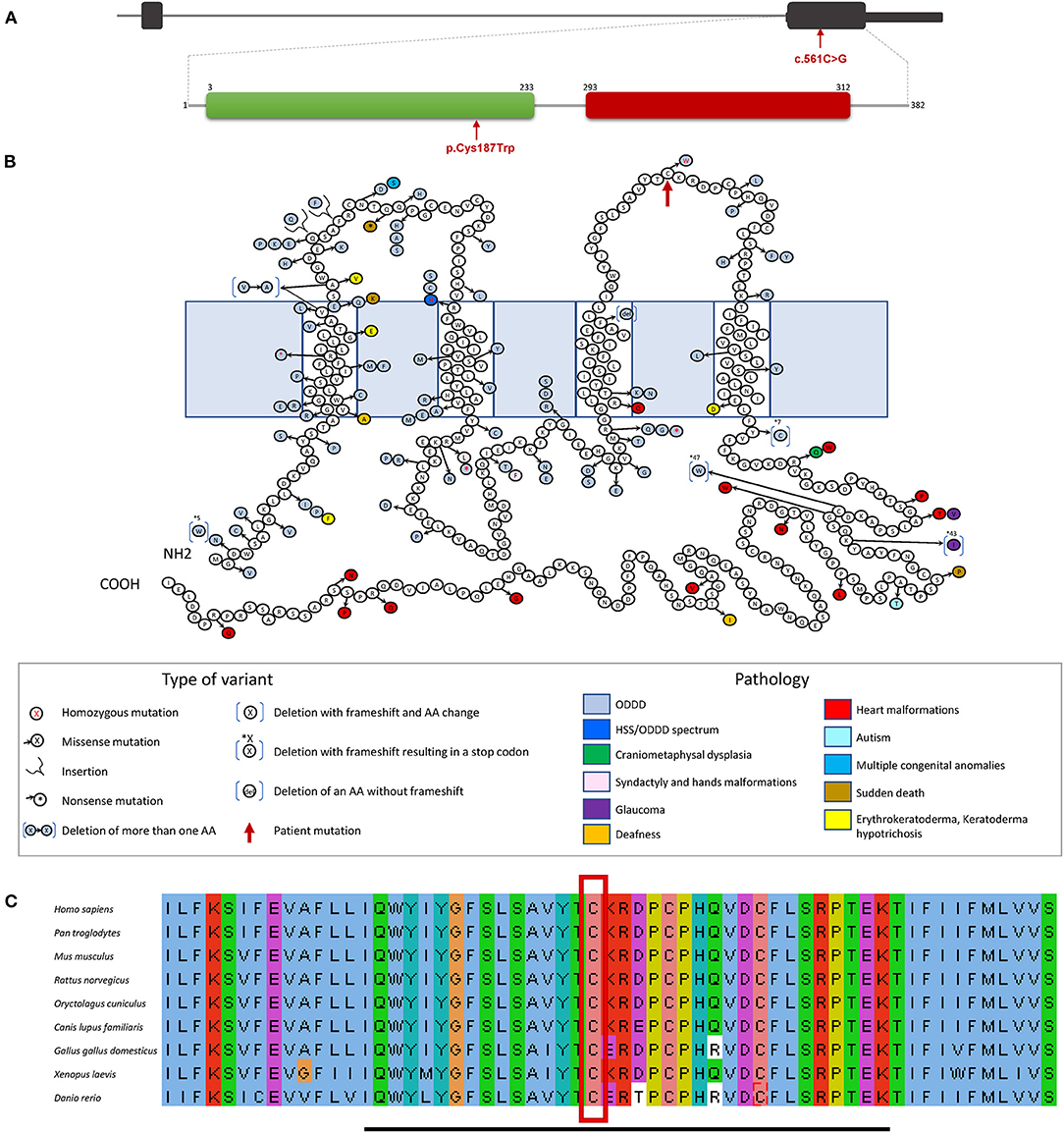
Figure 3. GJA1 gene and protein structure. (A) GJA1 is a two exons gene (black boxes) with the entire protein coding sequence included within exon 2. The protein contains two domains, a connexin domain (green box), and a connexin43 domain (red box). The variant identified in our patient is in exon 2 (red arrow), encoding the connexin domain (green box) of the protein modifying an amino acid in the second extracellular loop. (B) The protein structure shows the different amino acid changes and subsequent corresponding diseases reported so far, ODDD is represented in light blue. The new homozygous variant (c.561C>G p.Cys187Trp) discovered in our patient (red arrow) is located in the sequence area coding for the protein second extracellular loop. (C) Multiple sequence alignment of the second extracellular loop of the CX43 is indicated by the black bar. The amino acid, a cysteine (C), becoming tryptophan (W), as a consequence of the missense alteration (red square) is conserved, through evolution, from Homo sapiens to Danio rerio.
Discussion
Oculodentodigital dysplasia (ODDD) and Hallermann-Streiff syndrome (HSS) share several clinical features. ODDD is caused by alterations in GJA1 gene and mostly inherited in a dominant manner. The molecular etiology of HSS is not yet fully understood. Individuals diagnosed with ODDD or HSS/ODDD spectrum disorder, have been reported with a recessive pattern of inheritance (4, 8, 9).
Our patient initially diagnosed as suffering from HSS presented symptoms overlapping with both HSS and ODDD phenotypes (Table 1). In contrast to HSS, ODDD is characterized by specific digital anomalies (clinodactyly, camptodactyly, and syndactyly) (9). The girl presented brachydactyly in both hands and feet, and bilateral fifth finger clinodactyly in her hands, all pathognomonic signs of ODDD. She also had other symptoms matching both HSS and ODDD, such as hypotrichosis, and microphthalmia, as well as other traits more common in HSS like strabismus, high arched palate, and sleep apnea (Table 1). Whole-exome sequencing analysis excluded unlikely pathogenic alterations and pointed toward homozygous missense variants in GJA1. It provided a potential molecular diagnosis orienting toward a clinical diagnosis of ODDD. The proband's consanguineous parents, neither of whom reported clinical findings consistent with either syndromes, were both heterozygous for the selected GJA1 variant.
The c.561C>G p.Cys187Trp detected variant affected a highly conserved amino acid (Figure 3) located in the second extracellular loop of CX43. Many modifications in the amino acid sequence of CX43 are disease causing, indicating that CX43 has a low tolerance for change to maintain its cellular function (19). Eighty-five percentage of ODDD causing genetic alterations are located within the first half of the protein, prior to amino acid 192. The extracellular cysteines are particularly important for the maintenance of the protein structure (9). This is the first reported variant affecting a cysteine residue of the extracellular loops. Functional studies of alterations, in the first and second extracellular loops, causing ODDD have shown that the protein product does not form homomeric junctional plaques, resulting in an abnormal intracellular space localization linked to the endoplasmic reticulum instead of a normal extracellular space localization (12).
We therefore considered the variant as disease causing and likely pathogenic and classified it as class 4, according to ACMG criteria (18). The proband's genotype was consistent with the HSS/ODDD spectrum phenotype and homozygous variants had been reported once before in a patient presenting similar clinical traits i.e., a HSS/ODDD spectrum phenotype (8).
The enamel phenotype was however intriguing suggesting an important role of CX43 during amelogenesis. Indeed, CX43 contributes to ion transport enabling passage of ions directly from cells of the papillary layer into the ameloblast layer. Inactivation of Cx43 in mouse induced distinct skeletal abnormalities and also affected amelogenesis, suggesting that gap junctions are required to fully mineralize enamel at maturation stage (20, 21).
Understanding the underlying disease pathophysiology, explaining the origin of the enamel defects, strengthened the need for special oral care. Multidisciplinary management is indispensable for these rare disease patients. For the pediatric dentist, the management of tooth mineralized tissues abnormalities, present in both HSS and ODDD, constitutes a challenge, especially in young children. It is recommended to protect, isolate (stainless steel crowns, glass ionomers, composite…) and maintain on the arch the dysplastic teeth until skeletal growth is complete. If infectious complications occur, tooth extraction, especially in the primary dentition, should be performed (22). This was the case for our patient. The use of general anesthesia was not recommended due to the deformation of the airways that could lead to fatal complications.
Recognizing orodental anomalies (i.e., enamel, dentin structural defects) as part of a rare disease is crucial to implement early preventive and appropriate management aiming at avoiding secondary burden such as infection and definite loss of teeth. These developmental anomalies could provide important clues facilitating rare disease diagnosis. Reaching an accurate diagnosis may require both precise phenotyping (clinical description) and genotyping.
Genetic testing is used to orientate, confirm a diagnosis. It can change the medical care. It also provides families with more information about long-term and health care needs, and future family planning. This may entitle patients to benefit from special multidisciplinary healthcare and management in dedicated rare diseases centers, to receive appropriate treatment according to international existing guidelines. In the family mentioned in this paper, searching for the gene responsible for an initial diagnosis of HSS pointed toward a related syndrome, ODDD. The molecular basis of HSS remains unknown, but this new discovered homozygous variant strengthens the hypothesis of an overlapping phenotype/genotype correlation in the intermingled HSS/ODDD spectrum. HSS/ODDD may actually be a single syndrome with clinical features spanning both HSS and ODDD and homozygous variants in specific locations of the GJA1 gene sequence.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
Parents gave written informed consent for the transfer of clinical data in D4/phenodent database (www.phenodent.org), genetic testing (GenoDENT) and DNA biobanking (https://clinicaltrials.gov: NCT01746121 et NCT02397824; DC-2012-1677 et DC-2012-1002), and the current publication. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
AB-Z and SE conceived the ideas. KO and BB collected the data. TR prepared the genetic material. VL-H analyzed the exome data. AJ-A and VL-H analyzed the clinical data. AJ-A, VL-H, and AB-Z led the writing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the project No. 1.7 RARENET: a trinational network for education, research and management of complex and rare disorders in the Upper Rhine co-financed by the European Regional Development Fund (ERDF) of the European Union in the framework of the INTERREG V Upper Rhine program. AB-Z was a USIAS 2015 Fellow of the Institute of Advanced Studies (Institut d'Etudes Avancées) de l'Université de Strasbourg, France as well as a member of the ERN (European reference network) CRANIO initiative. This work was also supported by grants from the French Ministry of Health (National Program for Clinical Research, PHRC 2008 N°4266 Amelogenesis imperfecta), the University Hospital of Strasbourg (HUS, API, 2009–2012, Development of the oral cavity: from gene to clinical phenotype in Human) and the grant ANR-10-LABX-0030-INRT, a French State fund managed by the Agence Nationale de la Recherche under the frame programme Investissements d'Avenir labelled ANR-10-IDEX-0002-02. AJ-A received a CONYCIT/ANID, PhD Fellowship from Chile.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the family for their participation and invaluable contribution. We thank Mrs. Marzena Kawczynski for continuous support and help with patient data management as well as Pr O. Klein (UCSF School of Dentistry) for critical reading of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fdmed.2021.675130/full#supplementary-material
Supplementary Table 1. Whole-exome sequencing data. https://doi.org/10.6084/m9.figshare.14387909.v4.
Supplementary Table 2. Homozygous, compound heterozygous and de novo variants after filtration. https://doi.org/10.6084/m9.figshare.14387909.v4.
References
1. Schmidt J, and Wollnik B. Hallermann-Streiff syndrome: a missing molecular link for a highly recognizable syndrome. Am J Med Genet Part C Semin Med Genet. (2018) 178:398–406. doi: 10.1002/ajmg.c.31668
2. Abrams CK, and Scherer SS. Gap junctions in inherited human disorders of the central nervous system. Biochim Biophys Acta Biomembr. (2012) 1818:2030–47. doi: 10.1016/j.bbamem.2011.08.015
3. Porntaveetus T, Srichomthong C, Ohazama A, Suphapeetiporn K, and Shotelersuk V. A novel GJA1 mutation in oculodentodigital dysplasia with extensive loss of enamel. Oral Dis. (2017) 23:795–800. doi: 10.1111/odi.12663
4. Taşdelen E, Durmaz CD, and Karabulut HG. Autosomal recessive oculodentodigital dysplasia: a case report and review of the literature. Cytogenet Genome Res. (2018) 154:181–6. doi: 10.1159/000489000
5. Hu Y, Chen I.-P., de Almeida S, Tiziani V, Do Amaral CMR, et al. A novel autosomal recessive GJA1 missense mutation linked to craniometaphyseal dysplasia. PLoS ONE. (2013) 8:e73576. doi: 10.1371/journal.pone.0073576
6. Joss SK, Ghazawy S, Tomkins S, Ahmed M, Bradbury J, and Sheridan E. Variable expression of neurological phenotype in autosomal recessive oculodentodigital dysplasia of two sibs and review of the literature. Eur J Pediatr. (2008) 167:341–5. doi: 10.1007/s00431-007-0468-1
7. Richardson RJ, Joss S, Tomkin S, Ahmed M, Sheridan E, and Dixon MJ. A nonsense mutation in the first transmembrane domain of connexin 43 underlies autosomal recessive oculodentodigital syndrome. J Med Genet. (2006) 43:e37. doi: 10.1136/jmg.2005.037655
8. Pizzuti A, Flex E, Mingarelli R, Salpietro C, Zelante L, and Dallapiccola B. A homozygous GJA1 gene mutation causes a Hallermann-Streiff/ODDD spectrum phenotype. Hum Mutat. (2004) 23:286. doi: 10.1002/humu.9220
9. Paznekas WA, Karczeski B, Vermeer S, Lowry RB, Delatycki M, Laurence F, et al. GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum Mutat. (2009) 30:724–33. doi: 10.1002/humu.20958
10. Evans WH, and Martin PEM. Gap junctions: structure and function (review). Mol Membr Biol. (2002) 19:121–36. doi: 10.1080/09687680210139839
11. Laird DW, and Revel JP. Biochemical and immunochemical analysis of the arrangement of connexin43 in rat heart gap junction membranes. J Cell Sci. (1990) 97:109–17. doi: 10.1242/jcs.97.1.109
12. Shibayama J, Paznekas W, Seki A, Taffet S, Jabs EW, Delmar M, et al. Functional characterization of connexin43 mutations found in patients with oculodentodigital dysplasia. Circ Res. (2005) 96:e83–91. doi: 10.1161/01.RES.0000168369.79972.d2
13. Laugel-Haushalter V, Morkmued S, Stoetzel C, Geoffroy V, Muller J, Boland A, et al. Genetic evidence supporting the role of the calcium channel, CACNA1S, in tooth cusp and root patterning. Front Physiol. (2018) 9:1329. doi: 10.3389/fphys.2018.01329
14. The 1000 Genomes Project Consortium Auton A Brooks LD Durbin RM Garrison EP Kang HM. A global reference for human genetic variation. Nature. (2015) 526:68–74. doi: 10.1038/nature15393
15. Exome Aggregation Consortium, Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. (2016) 536:285–91. doi: 10.1038/nature19057
16. Vaser R, Adusumalli S, Leng SN, Sikic M, and Ng PC. SIFT missense predictions for genomes. Nat Protoc. (2016) 11:1–9. doi: 10.1038/nprot.2015.123
17. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
18. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
19. Laird DW. Syndromic and non-syndromic disease-linked Cx43 mutations. FEBS Lett. (2014) 588:1339–48. doi: 10.1016/j.febslet.2013.12.022
20. Al-Ansari S, Jalali R, Plotkin LI, Bronckers ALJJ, DenBesten P, Zhang Y, et al. The importance of connexin 43 in enamel development and mineralization. Front Physiol. (2018) 9:750. doi: 10.3389/fphys.2018.00750
21. Toth K, Shao Q, Lorentz R, and Laird DW. Decreased levels of Cx43 gap junctions result in ameloblast dysregulation and enamel hypoplasia in Gja1Jrt/+ mice. J Cell Physiol. (2010) 223:601–9. doi: 10.1002/jcp.22046
Keywords: case report, rare disease, oculodentodigital dysplasia, Hallermann Streiff, dental anomalies, connexin 43, syndrome
Citation: Jimenez-Armijo A, Oumensour K, Bousfiha B, Rey T, Laugel-Haushalter V, Bloch-Zupan A and El Arabi S (2021) A Novel Homozygous Variant in GJA1 Causing a Hallermann-Streiff/Oculodentodigital Dysplasia Overlapping Phenotype: A Clinical Report. Front. Dent. Med. 2:675130. doi: 10.3389/fdmed.2021.675130
Received: 02 March 2021; Accepted: 28 April 2021;
Published: 04 June 2021.
Edited by:
Maisa Hanna-Maija Seppala, King's College London, United KingdomReviewed by:
Dong Han, Peking University Hospital of Stomatology, ChinaDobrawa Napierala, University of Pittsburgh, United States
Copyright © 2021 Jimenez-Armijo, Oumensour, Bousfiha, Rey, Laugel-Haushalter, Bloch-Zupan and El Arabi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Agnès Bloch-Zupan, YWduZXMuYmxvY2gtenVwYW5AdW5pc3RyYS5mcg==
†These authors share first authorship
‡These authors share senior authorship